DOI:
10.1039/D4DT01929F
(Paper)
Dalton Trans., 2024,
53, 18974-18982
Carbon–phosphorus stapled Au(I) anticancer agents via bisphosphine induced reductive elimination†
Received
4th July 2024
, Accepted 7th August 2024
First published on 14th August 2024
Abstract
Towards the goal of generating new stabilized gold complexes as potent anticancer agents, we report here a novel class of Au(I) agents from Au(III)-mediated Caryl–P bond formation captured within the same complex by reacting a C^N cyclometalated Au(III) complex with bisphosphines. Cyclometalated Au(III) complexes of the type [Au(C^N)Cl2], where C^N represent different aryl pyridine framework reacted with bis(2-diphenylphosphino)phenyl ether in refluxing methanol to access an unsymmetrical gold complex featuring C–P coupling and Au(I)-phosphine. The complexes were characterized by 1H-NMR, 13C-NMR, and 31P-NMR and mass spectrometry. The structures of the complexes were characterized by X-ray crystallography and purity ascertained by HPLC and elemental analysis. The complexes demonstrate promising anticancer activity in a broad panel of cancer cell lines of different tumor origin. Mechanistically, the complexes induce apoptosis, generate mitochondrial ROS, depolarize mitochondrial membrane potential and modulate mitochondrial respiration in cancer cells. Overall, we developed a new structural class of Au(I) complexes with promising anticancer potential with potential utility in other applications.
Introduction
Therapeutic gold (Au)-phosphine complexes have garnered enormous attention over the past decades due to the clinical utility of the oral FDA approved gold-containing drug, auranofin.1–4 Auranofin possesses an Au(I) center coordinated to a thiosugar (3,4,5-triacetyloxy-6-(acetyloxymethyl)oxane-2-thiolate) and triethylphosphine in a linear geometry. The approval of auranofin in 1985 for the treatment of rheumatoid arthritis was accompanied by high remission rates and well tolerability with patients.5 Recent repurposing efforts of auranofin in different disease indications such as cancer,6–8 microbial,9,10 parasitic,11,12 and HIV,13,14 has spurned new research directions in the discovery and development of gold-derived complexes for disease treatment. New gold-containing structural scaffolds with sufficient stability and electrochemical character has the potential to drive new biological mechanisms and attain therapeutic impact.15,16 To this end, several Au(I)-phosphines have been synthesized as anticancer agents.17–19 The use of monophosphine and bisphosphines to access neutral, cationic and multinuclear Au(I) complexes is well precedented.20–22
Synthetic strategies to generate new Au(I) scaffolds is crucial for improved anticancer activity.23 One such approach to obtain Au(I) complexes is reductive elimination from Au(III) salts or organogold(III) complexes.24 Initial work by Vicente and coworkers showed the formation of biaryls via C–C coupling with the release of Au(I)-phosphines from a diaryldiazonium Au(III) complex.25 Toste et al. illustrated Au(III)-mediated C–P reductive elimination from several phosphine-supported organogold(III) complexes of the archetype [(R3P)Au(aryl)Cl2].26 The work employed the reaction of organogold(III) with silver salts or Lewis bases to generate Caryl–P adducts by reductive elimination. Seminal work involving the use of aryldiazonium salts and H-phosphonates by dual gold and photoredox catalysis leads to the irreversible formation of Caryl–P bonds.27 In 2019, work from our laboratory utilized phosphines to promote intramolecular C–N bond formation with the concomitant formation of a four-coordinate chiral Au(I)-phosphine complex.24 Later work by Casini and coworkers demonstrated direct C–P bond formation by reductive elimination from the reaction of [C^N]-cyclometalated Au(III) complex containing benzoyl pyridine with excess 1,3,5-triaza-7-phosphaadamantane (PTA) and KPF6.28 Whereas these examples of C–X (X = C, N, P) coupling involving phosphine ligands may be associated with the decomposition pathway in aryl Au(III)-mediated reaction methods and catalytic transformations,29 we posit that reductive elimination strategies can be leveraged to diversify Au(I) complexes bearing phosphine ligands with unique spatial and geometric structures.
In this work, we report on the synthesis and characterization of novel unsymmetrical Au(I) complexes bearing bis(2-diphenylphosphino)phenyl ether (DPEphos) ligands and evaluated their anticancer potential in vitro. Structure elucidation revealed that these complexes possess an arylated phosphorus, often known as C–P coupling by the arylpyridine of the C^N-cyclometalated Au(III) in one vertical plane and linear phosphine-Au(I)-Cl in another. The complexes demonstrate optimal stability in reducing L-GSH conditions over 24 h. We tested bioactivity of the complexes in several cancer cell lines from breast, ovary, and lung origin and found significant cell killing potential in these aggressive cancers that need more effective therapies in the clinic. Further, the complexes induce apoptosis; generate mitochondrial ROS, and perturb mitochondria bioenergetics and function. Taken together, a new class of Au(I) phosphines with potent anticancer activity is reported. The synthetic strategy proposed has the potential to generate molecules of broad utility in probe, therapeutic, materials, and catalytic applications.
Results and discussion
Rationale and approach
The unique reactivity of gold is derived from its unusual relativistic effects because of an increased atomic nuclear charge, penetrable 6s electron velocity into the nucleus, and expanded mass leading to the separation of 6s and 5d orbitals.30 Applied gold complexes exists predominantly in the +1 or +3 oxidation states.31 Making useful gold-derived complexes for biological applications require innovative reaction methods to generate functionally stable gold complexes due to the relatively high electrochemical potential of the Au3+/Au1+ redox couple (i.e. Au3+ + 2e− → Au1+, E° = 1.41 V and Au1+ + e− → Au(s), E° = 1.69 V).32 Several strategies to stabilize the Au(I) center include the use of strong σ-donating ligands such as carbenes,33 N-donor chelates,34–40 and phosphines.41–43 In fact, auranofin uses a thiolate and phosphine to stabilize the Au(I) center. To develop Au(I) complexes with improved therapeutic potential beyond auranofin, descriptors such redox, geometry, coordination number and ligand tuning need optimization. We posit that structural composition and modification of Au(I) complexes dictate biological function. Thus, we enlisted a phosphine ligand, DPEphos that offers a wide bite angle upon chelation to gold and leveraged its bidentate character to facilitate simultaneous C–P coupling and linear Au(I) center in one molecule.44–46 This strategy allows for electrochemical tuning, complex stability, and expanded ligand sphere around the metal center. Here, we rationalized that wide bisphosphine ligands promote dual reductive elimination from [Au(C^N)Cl2] and Au(I) phosphine trapping to afford arylpyridine stapled phosphine Au(I) complex. We hypothesize that this new structural class will impart distinct biological properties in the context of gold-derived probe/drug discovery.
Synthesis and characterization
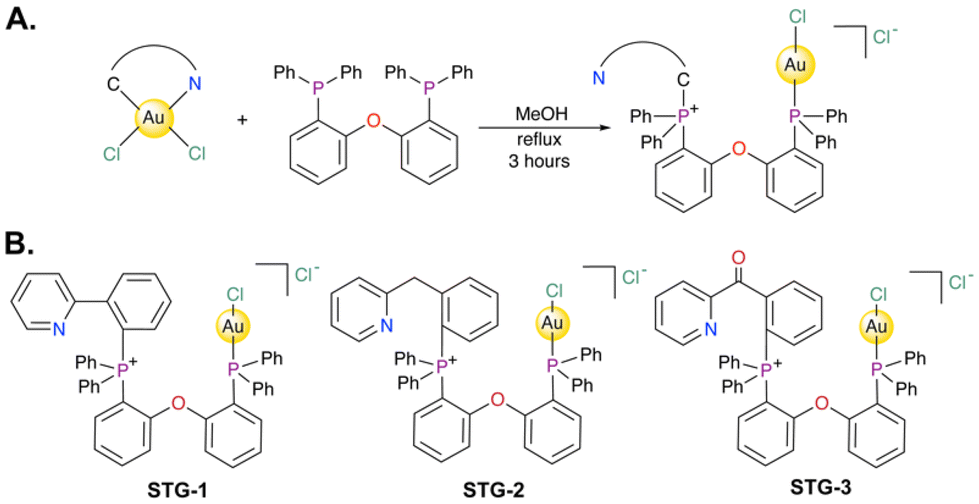
The reaction to generate the novel carbon-phosphine stapled Au(I) complexes under investigation in this report is described in Fig. 1A. Different [C^N]-cyclometalated Au(III)Cl2 complexes where C^N were phenylpyridine (STG-1), benzylpyridine (STG-2), or benzoylpyridine (STG-3) were reacted with bis(2-diphenylphosphino)phenyl ether (DPEphos) in methanol at 60 °C for 3 h. The reaction solution turned clear from an initial slight yellow color. At the end of the reaction, the solvent was removed in vacuo and subsequent recrystallization from dichloromethane/ether resulted in compounds STG-1, STG-2, and STG-3 (Fig. 1B). The strong σ-donating character of the phosphine ligand and high energetic conditions promote reductive elimination from the Au(III) starting material, yielding a C–P coupled product. However, the wide bidentate feature of the phosphine ligand used allows for Au(I) coordination to the other phosphine to form an overall unsymmetrical neutral complex. It is conceivable that the first step of the reaction involves ligand association of the bisphosphine ligand to the Au(III) center in a cis-coordination fashion with a fifth axial chlorido ligand coordinated to the Au(III) center. Subsequent C–P coupling at the phosphorus site cis to the arylpyridine Caryl bonded to Au(III) facilitates reduction to Au(I) with concomitant bonding to the other phosphine cis to Npyridine.
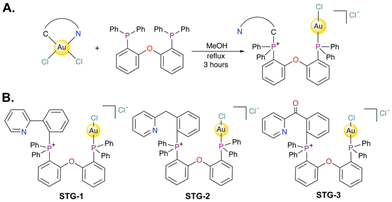 |
| | Fig. 1 (A) Synthetic scheme to access novel C–P stapled Au(I) bisphosphine complexes studied in this work. (B) Structures of STG-1, STG-2, STG-3. | |
We characterized the three (3) complexes by 1H, 13C, and 31P{1H} NMR spectroscopy (Fig. S1–S9†), and high-performance liquid chromatography electrospray ionization mass spectrometry (HPLC-ESI-MS) (Fig. S11–S16†). The purity of the complexes was confirmed by elemental analysis and HPLC-ESI-MS. The 31P{1H} of DPEphos have a single resonance at −17.93 ppm. Upon coordination with the gold center, the resonance shifts downfield to 20.26 ppm. The 1H-NMR and 13C{1H} NMR of all complexes show signals that are consistent with the compound resonances. Taken together, the complexes reported in this study was synthesized with detailed characterization by spectroscopy and their purity ascertained by both EA and HPLC establishing rigor of the science put forth.
X-ray crystallography
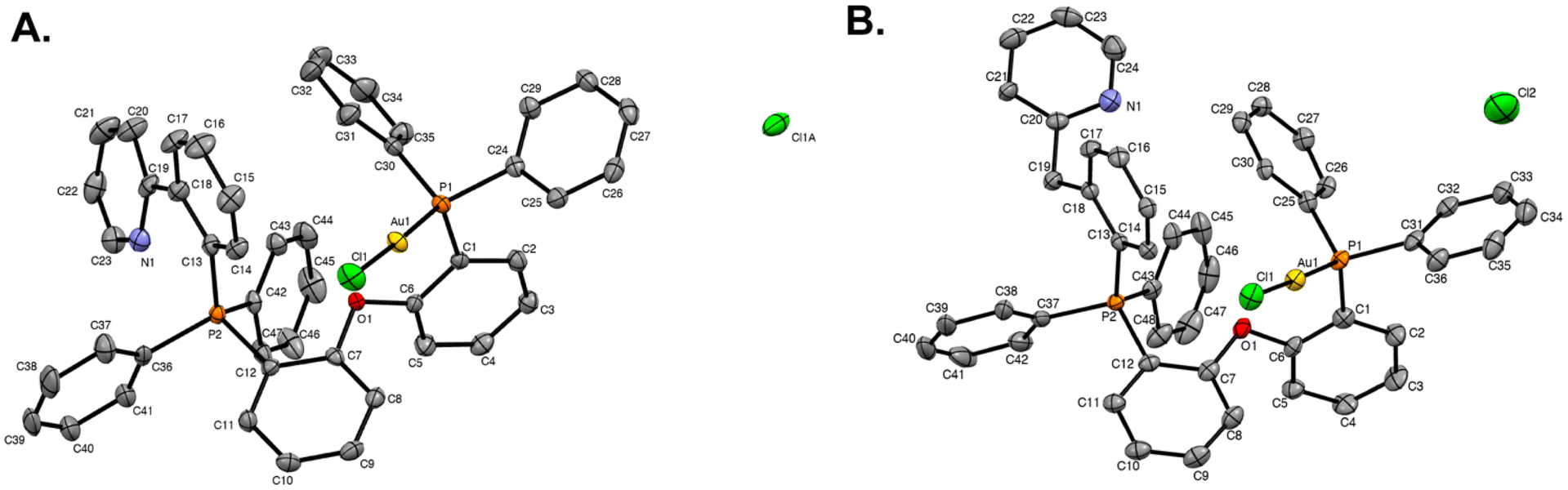
Single crystals of STG-1–2 were grown by vapor diffusion of diethyl ether into concentrated solutions of each compound in dichloromethane at room temperature. X-ray diffraction was used to elucidate the molecular structures of STG-1 (Fig. 2A) and STG-2 (Fig. 2B). Formation of a Caryl–P bond is observed in both structures and has consistent bond lengths of 1.809 Å in STG-1 and 1.804 Å in STG-2 (Table 1). The adjacent phosphorous atom is bound to gold at lengths of 2.2234 Å (STG-1) and 2.2254 Å (STG-2). Gold is linearly coordinated with P–Au(I)–Cl bond angles of 177.81° (STG-1) and 177.41° (STG-2). In all, both structures are consistent in their geometric parameters.
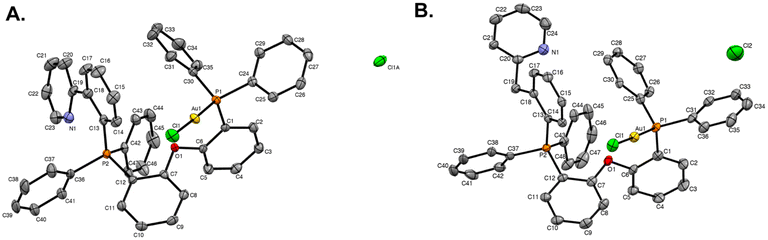 |
| | Fig. 2 X-ray crystal structures of (A) STG-1 and (B) STG-2. Ellipsoids are drawn at 50%. Hydrogens and solvent molecules have been omitted for clarity. | |
Table 1 Selected bond lengths and angles for STG complexes
| |
P1–Au1 |
Au1–Cl1 |
P2–C13 |
P1–Au1–Cl1 |
|
STG-1
|
2.2234(18) Å |
2.2778(18) Å |
1.809(4) Å |
177.81(9)° |
|
STG-2
|
2.2254(10) Å |
2.2771(10) Å |
1.803(4) Å |
177.41(4)° |
Electrochemistry
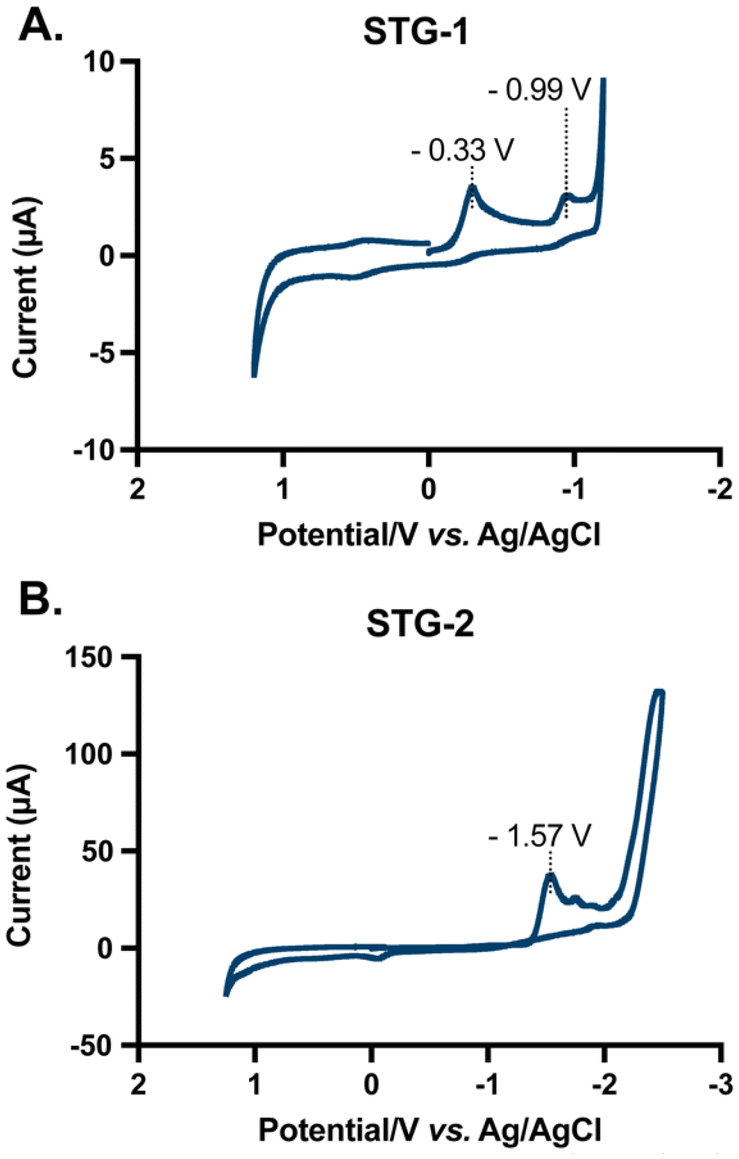
The electrochemical characteristics of STG-1 and STG-2 was ascertained by cyclic voltammetry in dimethyl sulfoxide (DMSO) using tetrabutylammonium hexafluorophosphate [N(Bu)4PF6] as the supporting electrolyte. The cyclic voltammogram of STG-1 reveals two major reduction events at −0.33 V and −0.99 V versus the Ag/AgCl reference electrode (Fig. 3A). Free DPEphos ligand did not show redox activity (Fig. S18 and S21†), suggesting that both reduction events are due to the Au(I) complex, STG-1. The reduction events may be attributable to metal–ligand transfer or ligand–metal transfer events. It is within reason that Au(I)/Au(0) reduction is possible. However, no colloidal gold formation was observed. The other reduction event could be attributed to ligand centered events due to arylation of the adjacent phosphorous, a phenomenon previously observed by Toste in C–P reductive eliminations.26 The voltammogram of STG-2 has one major reduction event at −1.57 V which likely corresponds to the Au(I)/Au(0) reduction potential of this molecule (Fig. 3B). The 500 mV difference between STG-1 and STG-2 reduction potentials highlights how choice of ligand can confer distinct electrochemical properties to a molecule. We believe that the benzylpyridine that is Caryl–P stapled destabilizes the Au(I) center, which results in the more negative reduction potential observed when compared to STG-1. Laguna has explained some of the intricate ligand effects on the electrochemical nature of Au(III) and Au(I) complexes,47 and we believe the choice of arylpyridine ligand used in the Caryl–P coupling could add an interesting layer of complexity that warrants further investigation. The results of this electrochemical study demonstrate the intriguing electrochemical behavior of this class of compounds. More importantly, these complexes appear to be relatively stable towards reduction in solution as evident by their negative reduction potentials of −0.99 V (STG-1) and −1.57 V (STG-2). This electrochemical stability may impart distinct biological effects and utility. Of note, auranofin does not convey redox properties.
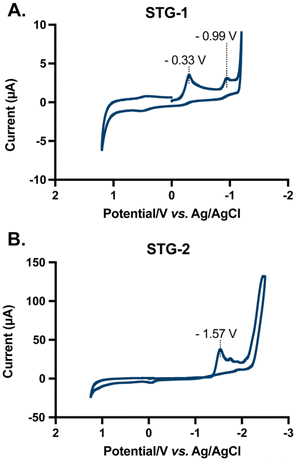 |
| | Fig. 3 Cyclic voltammograms of STG compounds (5 mM) in DMSO with [N(Bu)4PF6] as supporting electrolyte. Potentials are shown vs. Ag/AgCl. (A) Voltammogram of STG-1 showing two reduction events at −0.33 V and −0.99 V. (B) Voltammogram of STG-2 showing one reduction event at −1.57 V. Data collected with a glassy carbon working electrode and Pt counter electrode. | |
Stability against biological reductant L-GSH
A key player in cellular defense against oxidative stress is glutathione (L-GSH) and it is one of the most abundant biomolecules in cells.48,49 Glutathione contains an electron donating thiol (–SH) group, allowing it to scavenge harmful reactive oxygen species in cells. Mechanistic studies into Au(I) complexes in biological environments demonstrate the propensity for Au(I) to participate in ligand exchange reactions with biological nucleophiles, such as thiols.50 Because of this, we deemed it necessary to examine the solution stability of STG-1 towards biological nucleophiles such as L-GSH. We incubated STG-1 with an excess of L-GSH at 37 °C in DMSO-d6 and monitored for changes in the 1H NMR spectra that would be evident of GSH oxidation (Fig. S11†). In all 1H-NMR spectra recorded, there is an observation of proton resonances near peaks of L-GSH and at 6.81 ppm, which can be attributed to exchange interactions between the Au(I) atom of STG-1 and thiol of L-GSH. Spectra recorded from 0 to 6 hours show intact glutathione cysteine α-CH (4.36 ppm) and β-CH2 (2.66 ppm and 2.80 ppm) proton resonances, indicating that L-GSH remains unoxidized and the Au(I) center in STG-1 may be resistant towards rapid reduction by biological nucleophiles. Starting at 12 hours, new proton resonances at 3.11 ppm and 4.52 ppm are observed and increase in intensity in the spectrum taken at 24 hours. The new peaks are likely due to shifts from the cysteine α-CH and β-CH2 proton resonances upon oxidation of L-GSH to GSSG. It is of note that no major changes in the proton resonances of STG-1 are observed after 24 hours incubation with excess L-GSH. These results demonstrate that STG-1 and this class of compounds may maintain solution stability in the presence of cellular nucleophiles, an important quality that could have positive implications in vitro or in vivo.
In vitro cytotoxicity
Biological activity of the three compounds was first assessed in a panel of cell lines with the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. We used the cancer cell lines including breast, MDA-MB-468, MDA-MB-231, SUM-159, were generous gifts from Dr Kathleen O'Connor at the University of Kentucky and MCF-7 was graciously provided by Dr Yadi Wu at the University of Kentucky; ovarian (OVCAR-3, A2780), and lung cancers (A549, H460, H1299) were purchased from American Type Culture Collection, ATCC. In this assay, MTT is reduced by metabolically active cells and forms a purple-colored formazan product. After treatment with compounds for 72 hours, cell survival was assessed by measuring absorbance of solubilized formazan product, and IC50 values were determined. We found that all three compounds demonstrated potency in cell lines tested, with IC50 values in the low μM range (Table 2). The compound STG-1 showed the highest potency across most cell lines. Hence, we selected STG-1 as our lead candidate for further studies.
Table 2 IC50 values determined by performing the MTT cell viability assay in a panel of cancerous cell lines. Data is the mean ± S.D
| |
STG-1
|
STG-2
|
STG-3
|
| MDA-MB-231 |
2.7 ± 0.39 |
3.0 ± 0.36 |
4.2 ± 0.92 |
| MDA-MB-468 |
0.37 ± 0.10 |
0.86 ± 0.08 |
0.68 ± 0.19 |
| A549 |
0.48 ± 0.05 |
1.0 ± 0.07 |
0.65 ± 0.07 |
| H460 |
0.79 ± 0.13 |
1.7 ± 0.17 |
1.3 ± 0.34 |
| SUM-159 |
0.34 ± 0.17 |
0.97 ± 0.20 |
0.37 ± 0.26 |
| OVCAR-3 |
0.45 ± 0.05 |
0.67 ± 0.04 |
0.38 ± 0.04 |
| H1299 |
3.5 ± 2.4 |
1.1 ± 0.23 |
0.28 ± 0.01 |
| A2780 |
0.66 ± 0.09 |
0.11 ± 0.09 |
0.33 ± 0.08 |
| MCF-7 |
1.3 ± 0.22 |
0.76 ± 0.08 |
0.55 ± 0.15 |
STG-1 induces apoptosis in SUM-159 cells
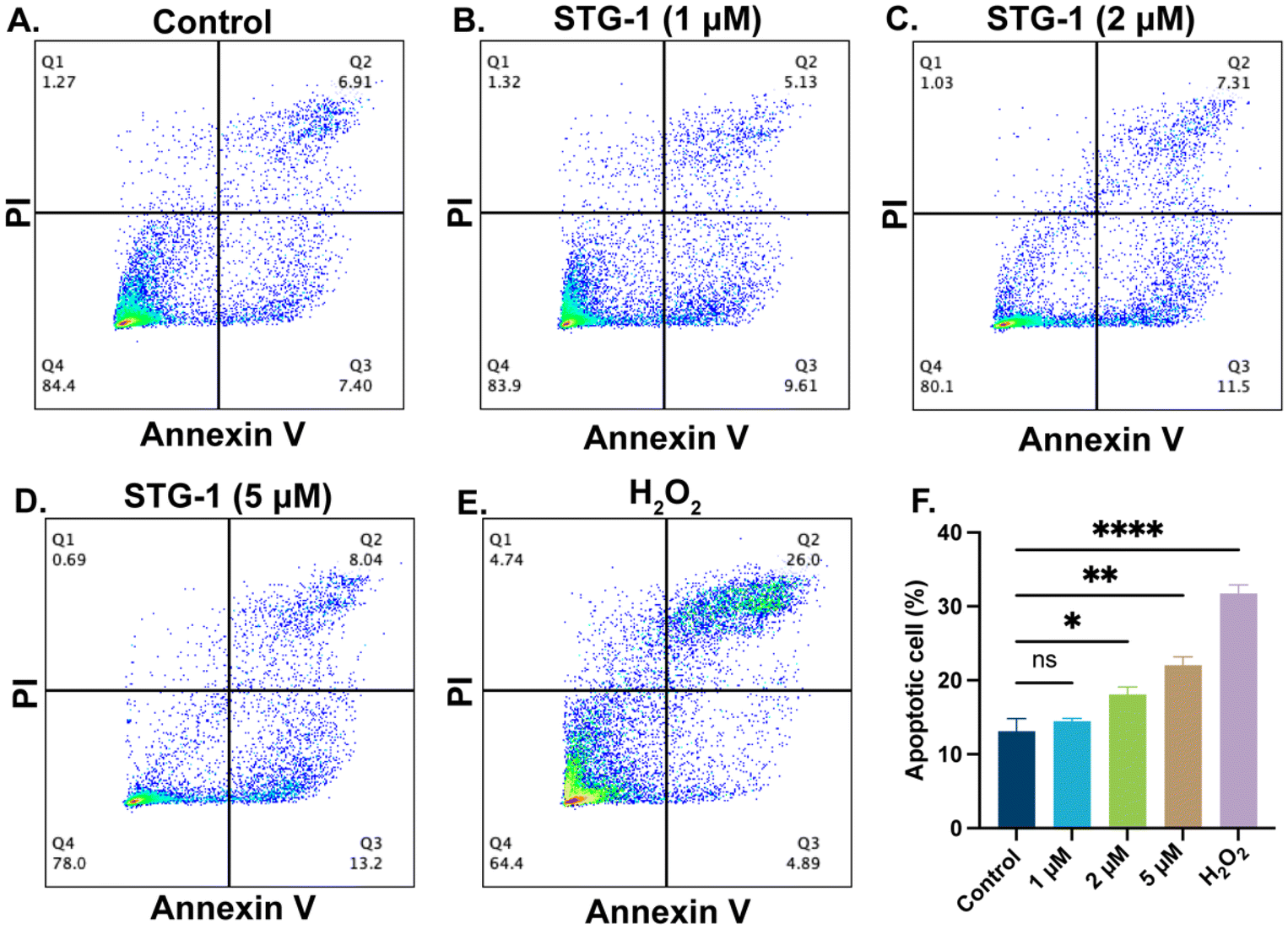
Au(I) agents have been reported to induce apoptosis,51 thus we investigated the potential of our novel Au(I) complexes to induce apoptosis as a mechanism of cell death. By utilizing the dual-staining FITC-Annexin V/PI flow cytometry assay, we observed that STG-1 promotes apoptosis in a dose-dependent manner. SUM-159 cells treated with STG-1 for 24 hours show a dose-dependent response in the increase of apoptotic cell populations (Fig. 4A–E), which is marked by the increase of Annexin V fluorescence intensity due to Annexin V engagement with phosphatidyl serine flipped to the outer leaflet of cells undergoing apoptosis. Quantification of the apoptotic quadrants show that there was a 1.38% increase of apoptotic cells at a treatment of 1 μM, and significant increases of 4.99% (2 μM) and 8.94% (5 μM) compared to control (Fig. 4F). It is likely that this class of compounds behaves in a manner consistent with other Au(I) anticancer agents by inducing apoptosis, but its mechanism of action may be different.
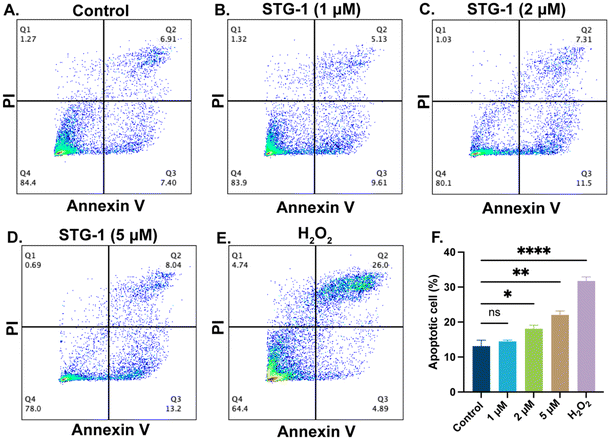 |
| | Fig. 4 Results from the FITC-Annexin V/PI assay. (A–E) Dot plots of representative populations of cells with different treatment of compounds. (F) Bar chart of percentage of apoptotic cells determined by the assay. Data analyzed via one-way ANOVA with Dunnett's multiple comparisons test (****p < 0.0001, **p < 0.01, *p < 0.05, ns – not significant). | |
STG-1 depolarizes mitochondrial membrane in cancer cells
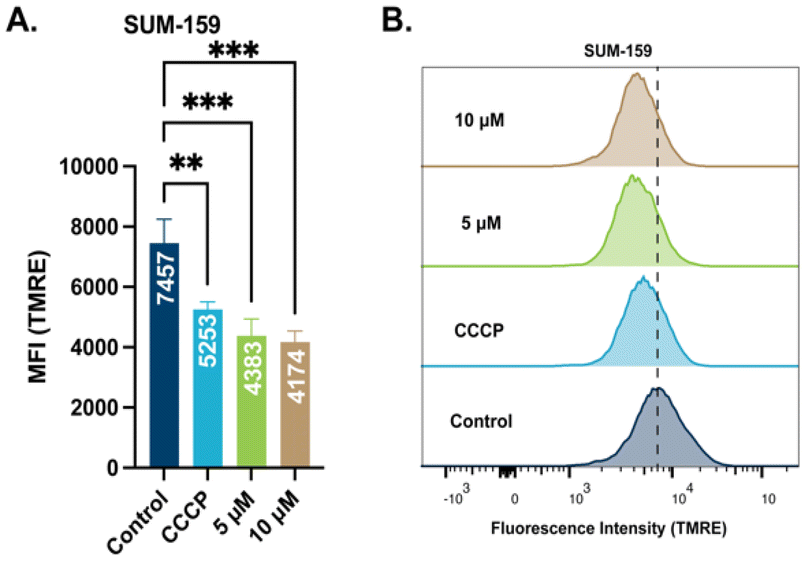
To elucidate the mechanism of action of this class of Au(I) complexes that leads to cell death, we sought to examine the effect of the complexes on mitochondria. Mitochondrial membrane potential (Δψm) is a valuable descriptor of mitochondrial health and the metabolic state of the cell. In healthy cells, Δψm is maintained by the mitochondrial respiratory chain that promote redox events for anabolic and catabolic processes.52 Impaired Δψm is observed in cancer cells with a more negatively charged membrane potential leading to mitochondria dysfunction. By utilizing the cationic dye tetramethyl rhodamine methyl ester (TMRE) we examined the effect of STG-1 on mitochondrial membrane potential. TMRE is taken up by mitochondria proportional to its membrane potential. SUM-159 cells were treated with STG-1 at concentrations of 5 μM or 10 μM for 2 hours and analyzed by fluorescence activated cell sorting (FACS) (Fig. 5A and B). A known uncoupler of oxidative phosphorylation (CCCP) that induces mitochondrial membrane potential depolarization was used as a positive control. The mean fluorescence intensity (MFI) of TMRE significantly decreased in treated cells compared to the controls. This result indicates the ability of this class of compound to depolarize the mitochondrial membrane potential in cancer cells.
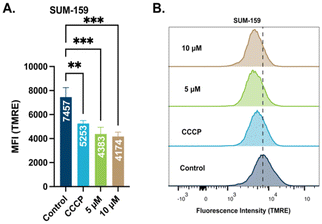 |
| | Fig. 5 Results from TMRE assay to determine mitochondrial membrane potential. (A) Plotted mean fluorescence intensity (MFI) of cells treated with STG-1 at different concentrations. (B) Histograms of fluorescence intensity of TMRE amongst populations. Data analyzed via one-way ANOVA with Dunnett's multiple comparisons test (***p < 0.001, **p < 0.01, ns – not significant). | |
STG-1 increases mitochondrial ROS
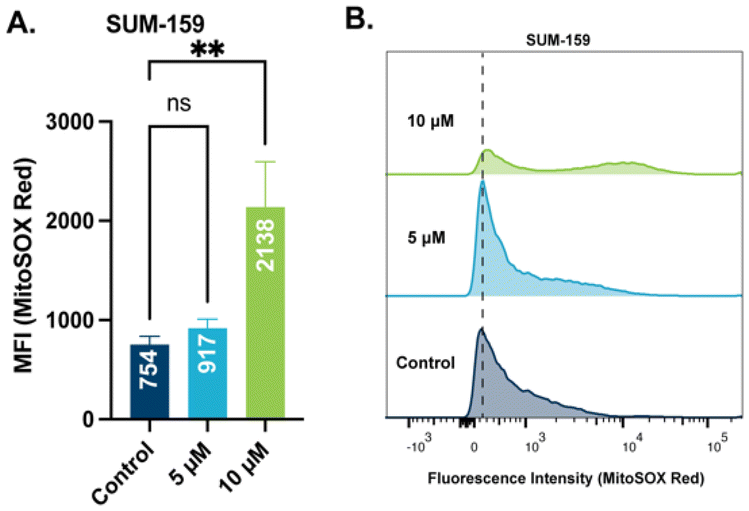
Mitochondrial membrane potential is used by ATP synthase to facilitate the production of ATP.53 When mitochondrial membrane potential is perturbed, a leak of electrons can occur followed by an increase in mitochondrial reactive oxygen species (ROS).54 With observed depolarization of the mitochondrial membrane by STG-1, an examination of ROS production is warranted. We chose to quantify mitochondrial ROS levels with the hydroethidine based probe, MitoSOX Red, which has been validated to selectively target mitochondria. SUM-159 treated with STG-1 at a concentration 10 μM (Fig. 6A and B) showed significantly higher MFI than the controls. This result further indicates that this class of compounds targets the mitochondria and promotes the generation of significant ROS that may be lethal to cancer cells.
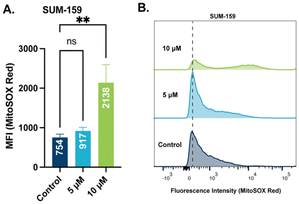 |
| | Fig. 6 Results from mitochondrial ROS assay with MitoSOX Red. (A) Plotted mean fluorescence activity (MFI) of MitoSOX Red in SUM-159 cells treated with STG-1. Significance plotted from one-way ANOVA with Dunnett's multiple comparisons test. (B) Histograms of fluorescence intensity of MitoSOX Red at different concentrations of STG-1. Data analyzed via one-way ANOVA with Dunnett's multiple comparisons test (**p < 0.01, ns – not significant). | |
STG-1 affects mitochondrial bioenergetics
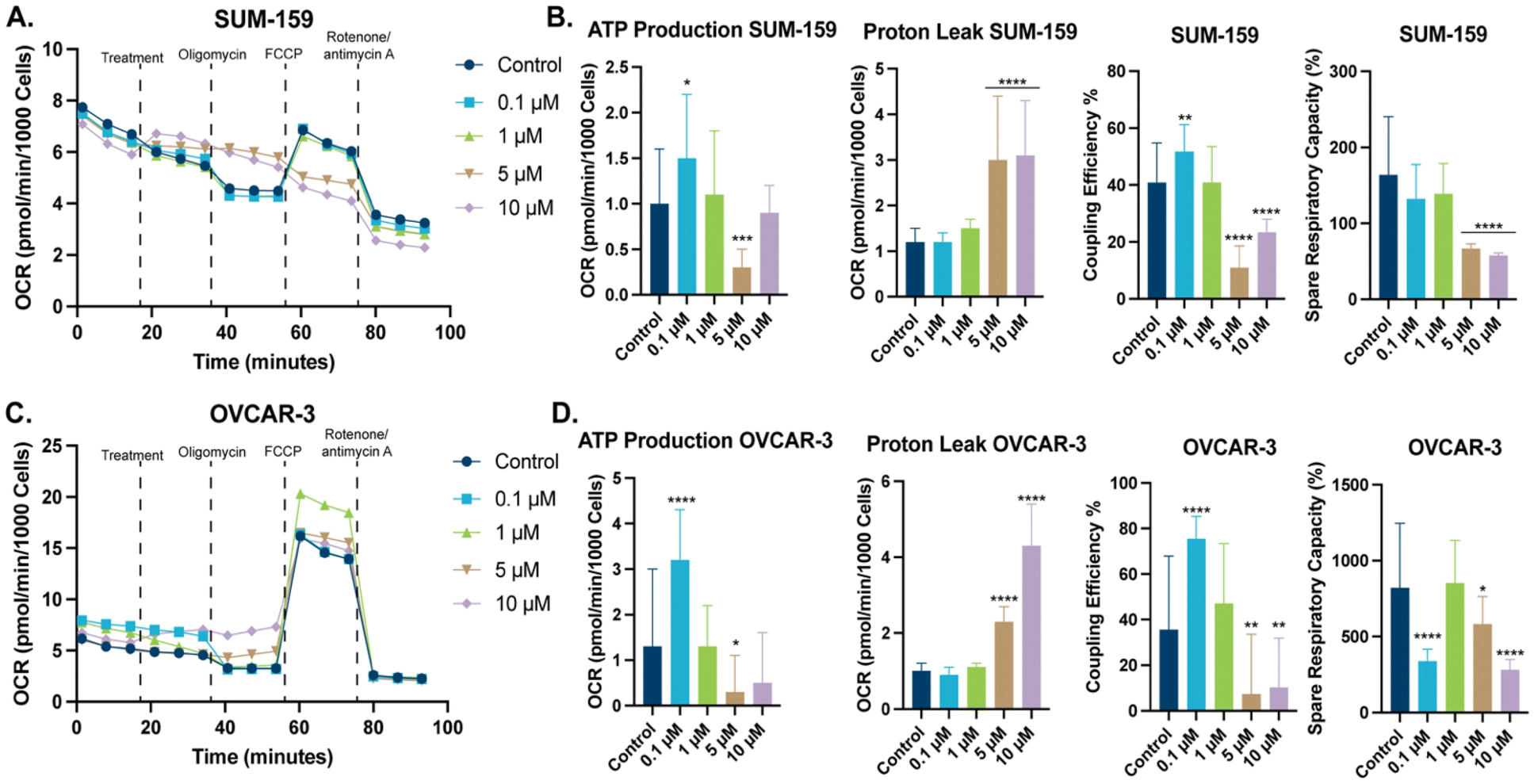
To better our understanding of this class of compounds effects on mitochondria, we examined mitochondrial bioenergetics with a Seahorse XF Cell Mito Stress Test. In this test, examination of the oxygen consumption rate (OCR) of cells is measured, when various inhibitors of the electron transport chain (ETC) are added. The change in OCR allows for the determination of key mitochondrial energetic parameters. These parameters include proton leak, ATP respiration, and coupling efficiency. The initial OCR of SUM-159 and OVCAR-3 cells was recorded (Fig. 7A and C), addition of STG-1 in a dose-dependent manner modulated OCR, specifically, maximal respiration in both cell lines. Subsequent addition of the ATP synthase inhibitor, oligomycin resulted in a decrease of OCR, which can be attributed to inhibition of ATP production. Taking the difference between the OCR before and after addition of oligomycin, we can see that STG-1 significantly decreases ATP production at sufficient concentrations (Fig. 7B and D). Further comparison of the OCR with oligomycin and that with complex I and III inhibitors (rotenone and antimycin A respectively) shows that proton leak was significantly increased at those concentrations as well. Decreased ATP production and increased proton leak was observed in both cell lines. We can take the ratio between ATP production and proton leak to deduce the coupling efficiency of the mitochondria when treated with STG-1, which unsurprisingly showed significant decrease in both cell lines. This result points towards STG-1 mitochondrial uncoupling properties, and explains the effects seen on mitochondrial membrane potential and the increased mitochondrial ROS. Additionally, FCCP was added after oligomycin to determine maximal respiration since FCCP allows protons to flow freely across the inner mitochondrial membrane. The ratio between the OCR of maximal and basal respiration is the percent spare respiratory capacity, indicative of the ATP production a cell can utilize, if necessary. In both cell lines, spare respiratory capacity was diminished at all concentrations of STG-1 treatment. This result taken together with effects on ATP production and proton leak, indicates this class of compounds functionally perturb the mitochondria.
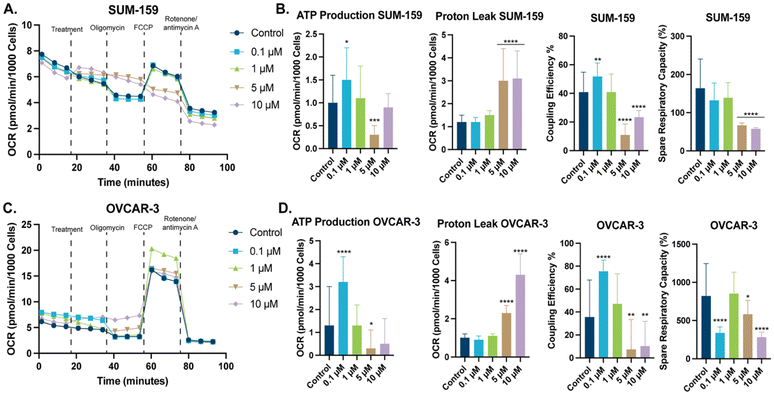 |
| | Fig. 7 Results from the Seahorse XF Mito Cell Stress Test. (A) OCR graph of SUM-159 cells treated with STG-1. (B) Key mitochondrial parameters extrapolated from the OCR of SUM-159 cells after addition of ETC inhibitors. (C) OCR graph of OVCAR-3 cells treated with STG-1. (D) Key mitochondrial parameters extrapolated from the OCR of OVCAR-3 cells after addition of ETC inhibitors. Data plotted as the mean ± S.D. with GraphPad Prism 10.2.0. Data analyzed via one-way ANOVA with Dunnett's multiple comparisons test (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, ns – not significant). | |
Conclusions
Gold has been extensively studied in the fields of catalysis,55–57 materials,58,59 and medicinal uses.60,61 Recent repurposing of the Au(I) containing auranofin in different disease implications,62–64 has led to the emergence of many novel gold agents for treating disease.65–68 To this end, we demonstrate the diversification of current Au(I) scaffolds by generating a class of Caryl–P stapled Au(I) bisphosphines complexes with potent cytotoxicity. The novel arrangement of Au(I) and aryl-pyridine on the bisphosphine ligand was confirmed by X-ray crystallography. The formation of Caryl–P bonds from reductive elimination of Au(III) has been demonstrated previously by Toste26,27 and Casini.28 To our knowledge, this is the first report of this transformation occurring with retention of gold within the same molecule. We believe this development will provide a platform for new synthetic materials beneficial to areas of catalysis, biological probe development, and therapeutics. Further, the novel complexes demonstrate structural stability towards the biological nucleophile, L-GSH as evident by 1H NMR. The compounds also impart potent cytotoxicity and mildly induce apoptosis. The ability of this class of compounds to decrease mitochondrial membrane potential, increase reactive oxygen species, and perturb mitochondrial energetics implicate mitochondria as a potential target of these complexes. In summary, our work presents a novel Au(I) scaffold foundational new areas of catalysis, biological probes, and gold-based drug design.
Author contributions
Conceptualization, S. T. G. and S. G. A.; methodology, synthesis and characterization, biological assay S. T. G. and O. A. O., X-ray crystallography S. P., writing – original draft preparation, S. T. G. and S. G. A.; writing – review and editing, S. T. G. and S. G. A.; supervision, S. G. A.; funding acquisition, S. G. A.
Data availability
Data will be available to share upon request.
Conflicts of interest
The authors declare the following competing financial interest(s): S. G. A. has patents pending to the University of Kentucky Research Foundation.
Acknowledgements
This work and S. G. A. was supported by grant R01CA258421-01 from the National Cancer Institute. The authors would like to appreciate the following research centres and facilities at the University of Kentucky for their help towards the completion of the research described in this article. The UK NMR Center supported by NSF (CHE-997738) and the UK X-ray facility supported by the MRI program from NSF (CHE-1625732). Mitostress experiments were carried out by support from the shared resource(s) of the University of Kentucky Markey Cancer Center (P30CA177558). The authors also acknowledge support of the Center for Pharmaceutical Research and Innovation (NIH P20GM130456) and UK Research Leadership Academy (RLA), Emerging Themes for Research Program.
References
- R. T. Mertens, S. Gukathasan, A. S. Arojojoye, C. Olelewe and S. G. Awuah, Chem. Rev., 2023, 123, 6612–6667 CrossRef PubMed.
- F. H. Abdalbari and C. M. Telleria, Discover Oncol., 2021, 12, 42 CrossRef PubMed.
- J. H. Kim, E. Reeder, S. Parkin and S. G. Awuah, Sci. Rep., 2019, 9, 12335 CrossRef PubMed.
- A. Kumar Singh, A. Kumar, H. Singh, P. Sonawane, P. Pathak, M. Grishina, J. Pal Yadav, A. Verma and P. Kumar, Chem. Biodiversity, 2023, 20, e202300061 CrossRef PubMed.
- D. E. Furst, Pharmacotherapy, 1983, 3, 284–296 CrossRef CAS PubMed.
- W. Fiskus, N. Saba, M. Shen, M. Ghias, J. Liu, S. D. Gupta, L. Chauhan, R. Rao, S. Gunewardena, K. Schorno, C. P. Austin, K. Maddocks, J. Byrd, A. Melnick, P. Huang, A. Wiestner and K. N. Bhalla, Cancer Res., 2014, 74, 2520–2532 CrossRef CAS PubMed.
- X. Chen, X. Shi, C. Zhao, X. Li, X. Lan, S. Liu, H. Huang, N. Liu, S. Liao, D. Zang, W. Song, Q. Liu, B. Z. Carter, Q. P. Dou, X. Wang and J. Liu, Oncotarget, 2014, 5, 9118–9132 CrossRef PubMed.
- C. Marzano, V. Gandin, A. Folda, G. Scutari, A. Bindoli and M. P. Rigobello, Free Radicals Biol. Med., 2007, 42, 872–881 CrossRef CAS.
- Y. Hokai, B. Jurkowicz, J. Fernández-Gallardo, N. Zakirkhodjaev, M. Sanaú, T. R. Muth and M. Contel, J. Inorg. Biochem., 2014, 138, 81–88 CrossRef CAS PubMed.
- S. Jackson-Rosario, D. Cowart, A. Myers, R. Tarrien, R. L. Levine, R. A. Scott and W. T. Self, J. Biol. Inorg. Chem., 2009, 14, 507–519 CrossRef CAS.
- E. R. Sharlow, S. Leimgruber, S. Murray, A. Lira, R. J. Sciotti, M. Hickman, T. Hudson, S. Leed, D. Caridha, A. M. Barrios, D. Close, M. Grögl and J. S. Lazo, ACS Chem. Biol., 2014, 9, 663–672 CrossRef CAS.
- A. N. Kuntz, E. Davioud-Charvet, A. A. Sayed, L. L. Califf, J. Dessolin, E. S. Arnér and D. L. Williams, PLoS Med., 2007, 4, e206 CrossRef PubMed.
- B. Chirullo, R. Sgarbanti, D. Limongi, I. L. Shytaj, D. Alvarez, B. Das, A. Boe, S. DaFonseca, N. Chomont, L. Liotta, E. I. Petricoin, S. Norelli, E. Pelosi, E. Garaci, A. Savarino and A. T. Palamara, Cell Death Dis., 2013, 4, e944 CrossRef CAS.
- I. L. Shytaj, B. Chirullo, W. Wagner, M. G. Ferrari, R. Sgarbanti, A. D. Corte, C. LaBranche, L. Lopalco, A. T. Palamara, D. Montefiori, M. G. Lewis, E. Garaci and A. Savarino, Retrovirology, 2013, 10, 71 CrossRef CAS.
- J. Oberkofler, B. Aikman, R. Bonsignore, A. Pöthig, J. Platts, A. Casini and F. E. Kühn, Eur. J. Inorg. Chem., 2020, 1040–1051 CrossRef CAS.
- M. D. Đurović, Ž. D. Bugarčić, F. W. Heinemann and R. van Eldik, Dalton Trans., 2014, 43, 3911–3921 RSC.
- T. S. Reddy, S. H. Privér, N. Mirzadeh and S. K. Bhargava, J. Inorg. Biochem., 2017, 175, 1–8 CrossRef CAS PubMed.
- R. Hayashi, K. Nakatsui, D. Sugiyama, T. Kitajima, N. Oohara, M. Sugiya, S. Osada and H. Kodama, J. Inorg. Biochem., 2014, 137, 109–114 CrossRef CAS.
- C. E. Greif, R. T. Mertens, G. Berger, S. Parkin and S. G. Awuah, RSC Chem. Biol., 2023, 4, 592–599 RSC.
- L. M. González-Barcia, S. Fernández-Fariña, L. Rodríguez-Silva, M. R. Bermejo, A. M. González-Noya and R. Pedrido, J. Inorg. Biochem., 2020, 203, 110931 CrossRef PubMed.
- W. F. Gabrielli, S. D. Nogai, M. Nell, S. Cronje and H. G. Raubenheimer, Polyhedron, 2012, 34, 188–197 CrossRef CAS.
- N. S. Townsend, M. Green and C. A. Russell, Organometallics, 2012, 31, 2543–2545 CrossRef CAS.
-
S. Gukathasan and S. G. Awuah, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2022, pp. 1–32 Search PubMed.
- J. H. Kim, R. T. Mertens, A. Agarwal, S. Parkin, G. Berger and S. G. Awuah, Dalton Trans., 2019, 48, 6273–6282 RSC.
- J. Vicente, M. Dolores Bermudez and J. Escribano, Organometallics, 1991, 10, 3380–3384 CrossRef.
- H. Kawai, W. J. Wolf, A. G. DiPasquale, M. S. Winston and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 587–593 CrossRef.
- Y. He, H. Wu and F. D. Toste, Chem. Sci., 2015, 6, 1194–1198 RSC.
- R. Bonsignore, S. R. Thomas, W. T. Klooster, S. J. Coles, R. L. Jenkins, D. Bourissou, G. Barone and A. Casini, Chem. – Eur. J., 2020, 26, 4226–4231 CrossRef PubMed.
-
R. T. Mertens and S. G. Awuah, Catalysis by Metal Complexes and Nanomaterials: Fundamentals and Applications, American Chemical Society, 2019, ch. 2, vol. 1317, pp. 19–55 Search PubMed.
- N. Bartlett, Gold Bull., 1998, 31, 22–25 CrossRef.
- T. J. Bergendahl, J. Chem. Educ., 1975, 52, 731 CrossRef.
-
D. C. Harris and C. A. Lucy, Quantitative Chemical Analysis, Freeman Custom Publishing, 2016 Search PubMed.
- R. J. Harris and R. A. Widenhoefer, Chem. Soc. Rev., 2016, 45, 4533–4551 RSC.
- P. A. Bonnardel, R. V. Parish and R. G. Pritchard, J. Chem. Soc., Dalton Trans., 1996, 3185–3193, 10.1039/DT9960003185.
- N. S. Radulović, N. M. Stojanović, B. Đ. Glišić, P. J. Randjelović, Z. Z. Stojanović-Radić, K. V. Mitić, M. G. Nikolić and M. I. Djuran, Polyhedron, 2018, 141, 164–180 CrossRef.
- S. Gukathasan, O. A. Obisesan, S. Saryazdi, L. Ratliff, S. Parkin, R. B. Grossman and S. G. Awuah, Inorg. Chem., 2023, 62, 13118–13129 CrossRef PubMed.
- S. Gukathasan, S. Parkin and S. G. Awuah, Inorg. Chem., 2019, 58, 9326–9340 CrossRef PubMed.
- J. Hyun Kim, S. Ofori, R. T. Mertens, S. Parkin and S. G. Awuah, ChemMedChem, 2021, 16, 3222–3230 CrossRef CAS PubMed.
- M. V. Babak, K. R. Chong, P. Rapta, M. Zannikou, H. M. Tang, L. Reichert, M. R. Chang, V. Kushnarev, P. Heffeter, S. M. Meier-Menches, Z. C. Lim, J. Y. Yap, A. Casini, I. V. Balyasnikova and W. H. Ang, Angew. Chem., Int. Ed., 2021, 60, 13405–13413 CrossRef CAS PubMed.
- J.-J. Zhang, R. W.-Y. Sun and C.-M. Che, Chem. Commun., 2012, 48, 3388–3390 RSC.
- T. Srinivasa Reddy, S. H. Privér, V. V. Rao, N. Mirzadeh and S. K. Bhargava, Dalton Trans., 2018, 47, 15312–15323 RSC.
- R. T. Mertens, W. C. Jennings, S. Ofori, J. H. Kim, S. Parkin, G. F. Kwakye and S. G. Awuah, JACS Au, 2021, 1, 439–449 CrossRef PubMed.
- O. Rackham, S. J. Nichols, P. J. Leedman, S. J. Berners-Price and A. Filipovska, Biochem. Pharmacol., 2007, 74, 992–1002 CrossRef.
- P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS.
- P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519–1530, 10.1039/A807799A.
- P. W. N. M. van Leeuwen, P. C. J. Kamer and J. N. H. Reek, Pure Appl. Chem., 1999, 71, 1443–1452 CAS.
- U. Koelle and A. Laguna, Inorg. Chim. Acta, 1999, 290, 44–50 CrossRef CAS.
- R. Franco and J. A. Cidlowski, Cell Death Differ., 2009, 16, 1303–1314 CrossRef CAS PubMed.
- S. Raj Rai, C. Bhattacharyya, A. Sarkar, S. Chakraborty, E. Sircar, S. Dutta and R. Sengupta, ChemistrySelect, 2021, 6, 4566–4590 CrossRef CAS.
- C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS.
- J. F. Schlagintweit, C. H. G. Jakob, N. L. Wilke, M. Ahrweiler, C. Frias, J. Frias, M. König, E.-M. H. J. Esslinger, F. Marques, J. F. Machado, R. M. Reich, T. S. Morais, J. D. G. Correia, A. Prokop and F. E. Kühn, J. Med. Chem., 2021, 64, 15747–15757 CrossRef CAS.
- L. D. Zorova, V. A. Popkov, E. Y. Plotnikov, D. N. Silachev, I. B. Pevzner, S. S. Jankauskas, V. A. Babenko, S. D. Zorov, A. V. Balakireva, M. Juhaszova, S. J. Sollott and D. B. Zorov, Anal. Biochem., 2018, 552, 50–59 CrossRef CAS PubMed.
- A. E. Senior, S. Nadanaciva and J. Weber, Biochim. Biophys. Acta, Bioenerg., 2002, 1553, 188–211 CrossRef CAS.
- S. Papa and V. P. Skulachev, Mol. Cell. Biochem., 1997, 174, 305–319 CrossRef CAS.
- N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994–2009 CrossRef CAS.
- A. C. H. Jans, X. Caumes and J. N. H. Reek, ChemCatChem, 2019, 11, 287–297 CrossRef CAS.
- A. M. Asiri and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 4471–4503 RSC.
- G. D. Wilk, R. E. Martinez, J. F. Chervinsky, F. Spaepen and J. A. Golovchenko, Appl. Phys. Lett., 1994, 65, 866–868 CrossRef CAS.
- Y. Takahashi, Y. Furukawa, T. Ishida and S. Yamada, Nanoscale, 2016, 8, 8520–8524 RSC.
- B. M. Sutton, E. McGusty, D. T. Walz and M. J. DiMartino, J. Med. Chem., 1972, 15, 1095–1098 CrossRef PubMed.
- V. Gandin, A. P. Fernandes, M. P. Rigobello, B. Dani, F. Sorrentino, F. Tisato, M. Björnstedt, A. Bindoli, A. Sturaro, R. Rella and C. Marzano, Biochem. Pharmacol., 2010, 79, 90–101 CrossRef PubMed.
- C. Fan, W. Zheng, X. Fu, X. Li, Y. S. Wong and T. Chen, Cell Death Dis., 2014, 5, e1191 CrossRef.
- N. Liu, X. Li, H. Huang, C. Zhao, S. Liao, C. Yang, S. Liu, W. Song, X. Lu, X. Lan, X. Chen, S. Yi, L. Xu, L. Jiang, C. Zhao, X. Dong, P. Zhou, S. Li, S. Wang, X. Shi, P. Q. Dou, X. Wang and J. Liu, Oncotarget, 2014, 5(14), 5453–5471 CrossRef PubMed.
- T. Okada, B. K. Patterson, S.-Q. Ye and M. E. Gurney, Virology, 1993, 192, 631–642 CrossRef CAS.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS.
- K.-B. Huang, F.-Y. Wang, X.-M. Tang, H.-W. Feng, Z.-F. Chen, Y.-C. Liu, Y.-N. Liu and H. Liang, J. Med. Chem., 2018, 61, 3478–3490 CrossRef CAS PubMed.
- I. Mármol, M. Virumbrales-Muñoz, J. Quero, C. Sánchez-de-Diego, L. Fernández, I. Ochoa, E. Cerrada and M. J. R. Yoldi, J. Inorg. Biochem., 2017, 176, 123–133 CrossRef.
- S. Radisavljević, I. Bratsos, A. Scheurer, J. Korzekwa, R. Masnikosa, A. Tot, N. Gligorijević, S. Radulović and A. Rilak Simović, Dalton Trans., 2018, 47, 13696–13712 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Table S1 and Fig. S1–S25. CCDC 2366025 and 2366026. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01929f |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Oluwatosin A.
Obisesan
a,
Sean
Parkin
a,
Oluwatosin A.
Obisesan
a,
Sean
Parkin