
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA noble nexus: a phosphino-phen ligand for tethering precious metals†
Paul D.
Newman
 *,
James A.
Platts
,
Basheer
Alrashidi
,
Simon J. A.
Pope
and
Benson M.
Kariuki
*,
James A.
Platts
,
Basheer
Alrashidi
,
Simon J. A.
Pope
and
Benson M.
Kariuki
School of Chemistry, Cardiff University, Cardiff, Wales, UK CF10 3AT. E-mail: newmanp1@cardiff.ac.uk
First published on 20th August 2024
Abstract
Controlled formation of mixed-metal bimetallics was achieved via two derivatised 1,10-phenanthroline ligands bearing an imino- or amino-phosphine appendage at the 5-position. Selective coordination of the phen group to the [Re(CO)3Cl] core was achieved enabling precise construction of bimetallic complexes with a second rhenium centre or with gold. The mixed Ru/Au complex was similarly obtained with the imino-phosphine but access to the heterobimetallic iridium systems required prior formation of the P-bound gold complexes subsequent to the introduction of the [Ir(Ppy)2]+ fragment. The Re/Pd, Re/Pt, Ir/Pd and Ir/Pt compounds were prepared from the combination of κ-N′′,P-Pd(Pt)Cl2 and the appropriate rhenium or iridium precursors. Spectroscopic and theoretical analyses have been employed to investigate the structural and electronic impact of the second metal.
Introduction
Bimetallic complexes, especially those containing disparate metal ions, are becoming increasingly important molecular platforms in an array of applications including catalysis1 and medical imaging and therapy.2 Formation of homo-bimetallic systems is generally easier as there is no necessity to include disparate donor groups to improve (or reduce) metal-ion selectivity. The generation of hetero-bimetallics, on the other hand, requires careful consideration of ligand design for selective, sequential coordination of each unique metal ion. To discriminate between the two metals of choice, the ligand superstructure should contain separate donor units that are sufficiently different in character to allow sequential introduction of the metal ions. While numerous examples in the literature exemplify this approach, phosphino-functionalised systems are relatively rare,3 and those where the P-donor is attached directly or remotely to one or more of the positions of a 1,10-phenanthroline (phen) ring remain underreported4 and form the basis of the current study.The area of metallaphotoredox catalysis has expanded dramatically over the last decade or so since its significant potential has been realised.5 For all its many successes, most reported examples employ separate photo- and redox-active catalysts and thus rely on the formation of encounter complexes at one or more stages of the catalytic cycle. Rather than being beholden to such processes, improvements can be achieved by combining each active catalyst into a single molecule to obviate the need for diffusion-controlled contact. Before any downstream application of new bimetallics, it is important to fully understand the character and behaviour of the systems notably regarding electronic communication and potential energy/electron transfer pathways across the ligand superstructure. We are interested in bpy and phen-based platforms and have designed and prepared two novel phenanthroline ligands appended with an imino- or amino-phosphine donor set to explore their ability to accommodate two disparate metal ions. This facility is implicit in the ligand design as the phenanthroline unit is strategically included for attachment to a photo-active centre allowing the introduction of a second, ideally redox or medicinally active, metal at the N′′,P donor. High levels of selectivity are critical for the successful preparation of the desired bimetallics and we report here our initial studies on the preparation of N,N′-M1/N′′,P-M2 complexes where M1 is rhenium, ruthenium or iridium and depending upon the nature of M1, M2 is gold, rhenium, palladium or platinum.
Results and discussion
Ligand synthesis and characterisation
The ditopic ligands consist of a 1,10-phenanthroline unit (N,N′ donor) with an imino- (L1) or amino-phosphine (L2) group (N′′,P donor) tethered to the central ring of phen. The distinction in donor set coupled with different chelate ring sizes, five versus six, was considered sufficient to allow discriminative coordination through initial selective binding of one metal prior to the introduction of the second metal ion. Part of the current remit was to explore the differences in the molecular make-up and structure of the two ligands and their complexes as L1 might be expected to have an extended pi-network involving the phen and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups while L2 has an insulating NHCH2 link between phen and phosphine. The phen group is well suited to the coordination of photo-active Re(I), Ru(II) or Ir(III) cores whereas the imino(amino)phosphine function serves to bind a second metal. The arrangement of the two donor sites does not allow for any direct or close contact between the two metal centres so that any communication between the metals can only occur through the ligand. The synthetic protocol for the formation of L1 and L2 is shown in Scheme 1. The initial step was high yielding and gave the desired L1 as a yellow solid which was air-stable in the solid state but susceptible to slow oxidation in solution. There is a possibility of geometric isomerism about the imine nitrogen but only a single species was observed by NMR spectroscopy with a singlet at −11.6 ppm in the 31P{1H} NMR spectrum and peaks consistent with one isomer being observed in the 1H and 13C{1H} spectra. Assignment of the E isomer shown in the scheme, was determined by an observable NOESY contact between the hydrogen at the 4-position of phen and the imine hydrogen (see the ESI†).
N groups while L2 has an insulating NHCH2 link between phen and phosphine. The phen group is well suited to the coordination of photo-active Re(I), Ru(II) or Ir(III) cores whereas the imino(amino)phosphine function serves to bind a second metal. The arrangement of the two donor sites does not allow for any direct or close contact between the two metal centres so that any communication between the metals can only occur through the ligand. The synthetic protocol for the formation of L1 and L2 is shown in Scheme 1. The initial step was high yielding and gave the desired L1 as a yellow solid which was air-stable in the solid state but susceptible to slow oxidation in solution. There is a possibility of geometric isomerism about the imine nitrogen but only a single species was observed by NMR spectroscopy with a singlet at −11.6 ppm in the 31P{1H} NMR spectrum and peaks consistent with one isomer being observed in the 1H and 13C{1H} spectra. Assignment of the E isomer shown in the scheme, was determined by an observable NOESY contact between the hydrogen at the 4-position of phen and the imine hydrogen (see the ESI†).
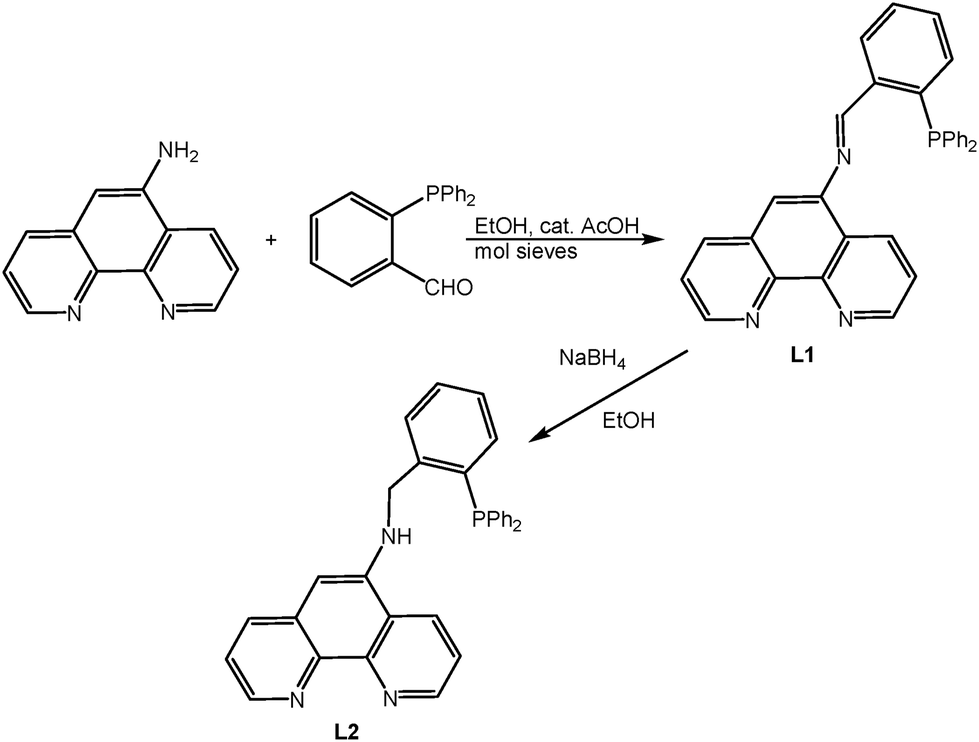 | ||
| Scheme 1 Synthesis of the L1 and L2 ligands. | ||
The subsequent reduction of L1 was performed with NaBH4 in MeOH. Although a yellow suspension was observed throughout the reduction (even upon heating the mixture), the isolated yellow solid proved to be L2 when analysed by NMR spectroscopy and HRMS. The most obvious differences between the 1H NMR spectrum of L2 and L1 were the appearance of broad singlets at 4.72 ppm for the CH2 hydrogens and 4.54 ppm for the NH hydrogen. The aromatic region is similar to that of L1 except that the imine hydrogen is necessarily absent and the only observable NOESY contact is between the CH2 hydrogens and H4.
Synthesis and characterisation of the monometallic rhenium complexes ReL1 and ReL2
The preparation of the rhenium complex fac-[Re(CO)3(κ2-N,N′-L1)Cl], ReL1, was achieved through heating a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [Re(CO)5Cl] and L1 in degassed PhCl for 3–4 hours under nitrogen. The complex precipitated out of solution upon cooling and was isolated by filtration in air. A second crop was obtained from the mother liquor after removal of all volatiles and trituration with toluene. Potential competitive coordination of the P- (and possibly imine) donor was not evident from the 31P{1H} NMR spectrum of the acquired complex as no significant change in the chemical shift was observed compared to the free ligand. There was also no evidence of any cis–trans isomerism about the imine group upon coordination. Compared to L1, small but noticeable downfield coordination shifts for the hydrogens of the ligand are seen in the 1H NMR spectrum of ReL1 including the singlet for the hydrogen at the 4-position. The presence of a NOESY contact between the imine hydrogen and the hydrogen at the 4-position of the phenanthroline confirmed the E isomer as noted for the free ligand. Repeat preparations revealed variable amounts (up to 10%) of a second species and although the nature of this by-product was not established unequivocally, it was not the result of oxidation of the phosphine as the chemical shift (16 ppm) does not accord with a Ar3PO unit. This was supported by the lack of any observable peak for an oxidised species in the mass spectrum. A more likely explanation is that the phosphine has displaced the chloride ligand in the second rhenium centre to give a dimer in solution. It is not recommended to leave solutions of the complex for extended periods of time as, although the conversion was moderately slow, the dimeric species proliferated over time.
:1 mixture of [Re(CO)5Cl] and L1 in degassed PhCl for 3–4 hours under nitrogen. The complex precipitated out of solution upon cooling and was isolated by filtration in air. A second crop was obtained from the mother liquor after removal of all volatiles and trituration with toluene. Potential competitive coordination of the P- (and possibly imine) donor was not evident from the 31P{1H} NMR spectrum of the acquired complex as no significant change in the chemical shift was observed compared to the free ligand. There was also no evidence of any cis–trans isomerism about the imine group upon coordination. Compared to L1, small but noticeable downfield coordination shifts for the hydrogens of the ligand are seen in the 1H NMR spectrum of ReL1 including the singlet for the hydrogen at the 4-position. The presence of a NOESY contact between the imine hydrogen and the hydrogen at the 4-position of the phenanthroline confirmed the E isomer as noted for the free ligand. Repeat preparations revealed variable amounts (up to 10%) of a second species and although the nature of this by-product was not established unequivocally, it was not the result of oxidation of the phosphine as the chemical shift (16 ppm) does not accord with a Ar3PO unit. This was supported by the lack of any observable peak for an oxidised species in the mass spectrum. A more likely explanation is that the phosphine has displaced the chloride ligand in the second rhenium centre to give a dimer in solution. It is not recommended to leave solutions of the complex for extended periods of time as, although the conversion was moderately slow, the dimeric species proliferated over time.
The rhenium complex of the reduced ligand, fac-[Re(CO)3(κ2-N,N′-L2)Cl] (ReL2) was prepared in a similar manner except using toluene as the solvent. The isolated bright yellow solid was only sparingly soluble in hydrochlorocarbons and acetone but freely soluble in DMSO. The complex showed typical coordination shifts in the 1H NMR spectrum whilst the 31P{1H} NMR spectrum remained largely unaltered. Akin to the free ligand, the CH2 group was seen as a broad singlet in the 1H NMR spectrum. As noted for the rhenium complex with L1, the analogous complex with L2 was prone to dimer formation in solution. In addition, ReL2 was, surprisingly, much more susceptible to oxidation than the uncoordinated ligand with substantial R3PO formation being observed within 20–30 minutes of dissolution upon exposure to air.
Synthesis and characterisation of the dimetallic rhenium complexes 2ReL1 and 2ReL2
When the reaction between L1 and [Re(CO)5Cl] was repeated in a 1:2 ratio, a dimeric species [{Re(CO)3(κ2-N,N′-L1)Cl}{Re′(CO)3(κ2-N′′,P-L1)(CO)3Cl}] (2ReL1) was isolated as a yellow solid. Analysis of the NMR spectra obtained immediately after dissolution showed a major species at 15.0 ppm and two (very) minor species at 12.5 and 12.4 ppm in the 31P{1H} NMR spectrum. Monitoring of this sample showed a conversion from the major to the minor species to the extent that the prevalence was ultimately inverted. This occurred irrespective of the nature of the NMR solvent but never resulted in complete dominance of any one species. Inspection of the HRMS showed peaks at 1042.0038 and 1085.0327 amu corresponding to molecular formulae of C37H22N3O6PClRe and C39H25N4O6PClRe, respectively. Both formulations are compliant with [Re(CO)3] cores with the loss of a chloride from one rhenium in the former case and the replacement of this lost chloride with CH3CN in the species with a higher mass. Although CO loss in the gas phase cannot be ruled out, these formulations suggest bidentate N′′,P coordination to the second rhenium.
To further shed light on the nature of the dirhenium dimer, crystals were obtained by vapour diffusion of diethyl ether into a dichloromethane solution of the complex. The molecular structure of the complex determined by SCXRD is shown in Fig. 1 with selected metrics in Table 1. As anticipated from the NMR and MS results, the structure is a dimer with one rhenium bound through the phen site and the other through both the P and N of the iminophosphine group. The Re–Nphen bond lengths of 2.176(4) and 2.177(4) Å are typical of a [Re(CO)3(phen)Cl] species while the Re–Nimine bond is slightly longer at 2.212(4) Å but shorter than 2.238(7) Å reported for a related monomeric system.6 The Re–P bond length of 2.4335(10) Å is unremarkable and compares to 2.446(2) Å in the related complex.6 The imine nitrogen is distorted slightly from planarity with a CN–C bond angle of 115.6(4)° and CN and C–N bond lengths of 1.285(6) and 1.439(5) Å, respectively. The six-membered chelate ring adopts an envelope conformation with the rhenium atom being out of plane. It is noteworthy that the CN bond vector of the imine adopts an orientation almost perpendicular to the phenanthroline plane, which contrasts with the alignment calculated for the free ligand (see later); this is necessary to allow coordination of the imine nitrogen to the P-bound rhenium. The remaining atoms/groups on non-phen rhenium are on the same side of the phen plane as the axial CO group trans to the chloride of the [Re(CO)3((κ2-N,N′-L1)Cl)] fragment. There is a second possible isomer where the [Re′(κ2-N′′,P)(CO)4Cl] unit projects towards the chloride of [Re(CO)3((κ2-N,N′-L1)Cl)]. Furthermore, the Cl ligand in [Re′(κ2-N′′,P)(CO)3Cl] is seen to point away from the other rhenium in the solid-state structure but this could also orientate towards rhenium and these isomeric possibilities may explain the changes observed in the NMR spectra noted above (similar NMR changes were observed irrespective of the source of 2ReL1 including the crystals used for SCXRD).
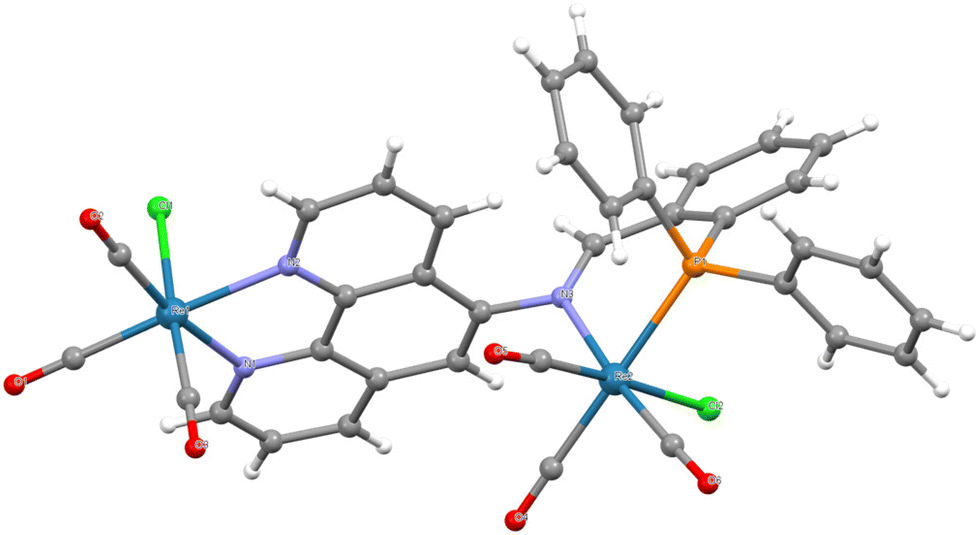 | ||
| Fig. 1 Molecular structure of 2ReL1 with lattice solvent molecules removed for clarity. Pertinent metrics and labelling are provided in the ESI.† | ||
| N–Cphen | N–Cim/am | C–N–Cphen | C–N–C(5)–C(4) | |
|---|---|---|---|---|
| AuL1 | 1.415 | 1.268 | 119.36 | 51.0 |
| 2ReL1 | 1.439 | 1.286 | 115.55 | 82.7 |
| 2ReL2 | 1.460 | 1.530 | 111.84 | 36.7 |
| ReAuL2 | 1.362 | 1.446 | 122.51 | 2.8 |
A similar 1:2 reaction with L2 produced an analogous result with the crude product being an isomeric mixture of two species with the major isomer prevailing at around 85%. Crystals of this 2ReL2 dimer were obtained from acetone by vapour diffusion of diethyl ether and the molecular structure determined by SCXRD is shown in Fig. 2. Unlike the 2ReL1 structure only the C–C–C–P atoms of the six-membered chelate are coplanar with the nitrogen and rhenium out-of-plane forming a two-atom flap of an envelope conformation. The N–Camine vector attains an angle of 36.7° to the phen plane which is closer to planar than perpendicular with N–CH2R and N–Cphen being 1.529(9) and 1.459(8) Å, respectively, and C–N–C = 111.9(6)°. As the amine nitrogen is tetrahedral with four different substituents, it is necessarily chiral and adopts the S configuration in the solid state with the NH hydrogen directed towards H6 of the phen ring rather than H4. The remaining metrics are unexceptional, although it is noteworthy that the Re–Namine bond length is longer at 2.315(6) Å than that seen in 2ReL1 suggesting weaker donation of this anilinic nitrogen compared to the imine.
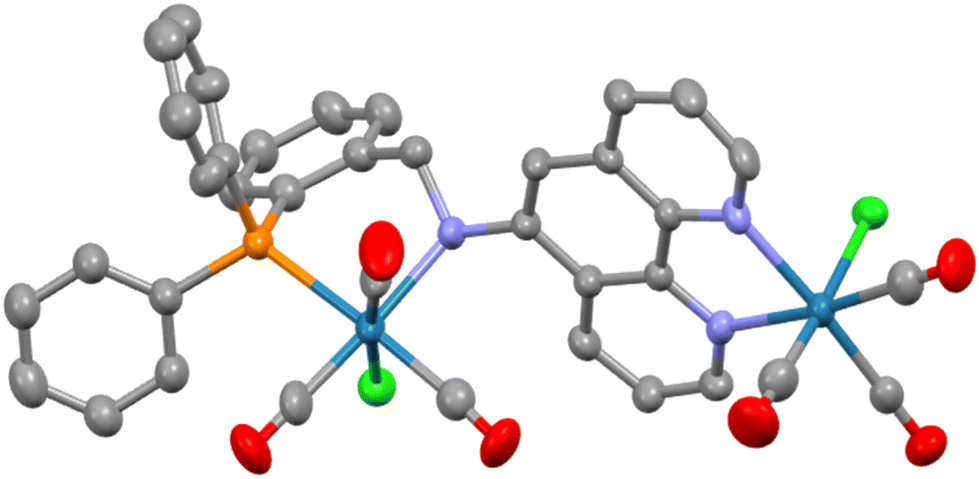 | ||
| Fig. 2 Molecular structure of 2ReL2 with hydrogens and lattice solvent molecules removed for clarity. Pertinent metrics and labelling are provided in the ESI.† | ||
Dissolution of the crystals of 2ReL2 in CDCl3 showed the presence of two isomers in the 1H and 31P{1H} NMR spectra in an ∼9:1 ratio. As expected from the solid-state structure, the CH2 hydrogens are inequivalent and present as a doublet of doublets at 4.60 ppm and a virtual triplet at 5.13 ppm in the 1H NMR spectrum. The NH hydrogen is seen as a doublet at 5.51 ppm revealing coupling to just one of the CH2 hydrogens in agreement with the dihedral angles of around 90° and 180° seen in the solid state. Through-space contacts between the CH2 hydrogens and the H4 hydrogen of the phen and selected ortho-hydrogens of the PAr2 groups are evident for the major isomer in the NOESY spectrum, in agreement with the structure determined by SCXRD. There are five separate signals for the CO ligands in the 13C{1H} NMR spectrum with three showing 1JCP coupling, reflecting the inequivalence of all but two of the carbonyls. Unlike 2ReL1 isomeric redistribution was not noted for 2ReL2 in solution, with the initial major isomer always being predominant. Aside from the isomeric possibilities described for 2ReL1, there is a further potential source of isomerism in 2ReL2 as the secondary nitrogen can be R or S (this is the stereochemistry in the crystal). This can lead to spectroscopically discrete species in each case as the NH projects towards Cl in one case and CO in the other.
Synthesis and characterisation of the dimetallic rhenium/gold complexes ReAuL1 and ReAuL2
Once it was established that selective coordination of the bipy group occurred upon synthesis of the fac-[Re(CO)3(κ2-N,N′-L1)Cl] complex, we sought to explore the introduction of second (non-rhenium) metal fragments through P- and/or P,N′′-donation. The choice of AuCl as the second metal unit was judicious as it is sterically small, redox stable and expected to bind only through the P-donor. Access was synthetically simple through the room temperature reaction of the two reagents in dichloromethane. The resultant yellow solid obtained after the removal of volatiles, fac-[Re(CO)3(κ2-N,N′-L1)Cl{(κ-P-L1)AuCl}] (ReAuL1), was air-stable and could be recrystallised from an acetone solution by vapour diffusion of diethyl ether. The 31P{1H} NMR spectrum showed the anticipated coordination shift of around 40 ppm to give a singlet at 29.7 ppm.7 An obvious change in the 1H NMR spectrum was seen for the resonance of the H4 hydrogen which occurred at 7.51 ppm, a shift of ∼0.9 ppm downfield of its position in the parent rhenium complex. Slight upfield shifts for the H1 and H7 hydrogens and a significant downfield shift of 0.32 ppm for the H3 hydrogen were also observed along with a decrease in the 4JHP coupling constant from 4.6 to 1.6 Hz for the NCH imine hydrogen. A single isomeric species was evident and equivalent to that observed for both the free ligand and the rhenium complex as determined by 2D NMR experiments. The HRMS shows the parent peak minus one chloride at 970.0291 amu.The ReAuL2 complex was prepared in an analogous fashion to the above and isolated as a yellow solid. The ΔδP change was very similar to the previous complex at 40.2 ppm. The most noticeable change in the 1H NMR spectrum was the obvious non-equivalence of the CH2 hydrogens which manifest as an AB pattern at 4.69 and 3.86 ppm (2JH–H = 16.0 Hz), respectively. This is a consequence of the hydrogens being diastereotopic as is evident from the solid-state molecular structure determined by SCXRD techniques and shown in Fig. 3. As expected, the gold atom is linear {P–Au–Cl = 177.42(6)°}, two-coordinate and bound only by the P atom of the organic ligand with an unexceptional Au–P bond length of 2.2364(15) Å.8 The N–Cphen bond of 1.362(7) Å is unusually short compared to the other crystallographically characterised complexes (Table 1) with an expanded C–N–Cphen bond angle of 122.5(5)° reflecting a sp2 hybridised nitrogen atom and conjugation with the phen ring. The nitrogen and carbon of the NHCH2 link are coplanar with the phen unit whereas the H2C–CPh bond projects to one side of the phen plane with a C–N–C–CPhen torsion angle of 67.7°. This orientation directs the AuCl unit to the Re–Cl side of the phenanthroline plane in the solid state structure. This raises the possibility of rotamers where the opposing orientation has the AuCl located towards the axial rhenium carbonyl; there is no evidence of two species in solution indicating easy conversion between the two positions.
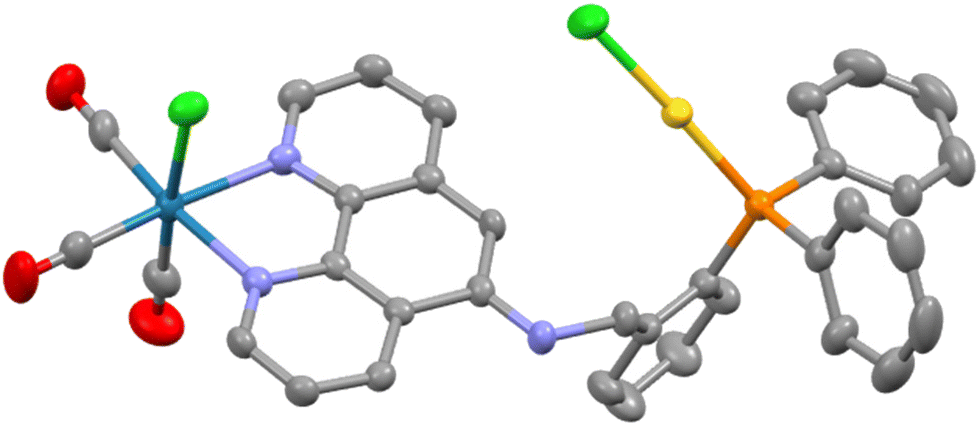 | ||
| Fig. 3 Molecular structure of ReAuL2 with hydrogens and lattice solvent molecules removed for clarity. Pertinent metrics and labelling are provided in the ESI.† | ||
Synthesis and characterisation of the dimetallic iridium/gold and ruthenium/gold complexes IrAuL1, IrAuL2 and RuAuL1
Preparation of the mononuclear iridium complex [Ir(PPy)2(L1)]BF4 was not possible from either [Ir(PPy)2Cl]2 or [Ir(PPy)2(MeCN)2]BF4 as competitive coordination between the κ2-N,N′ and the κ2-P,N′′ sites was observed during the attempted formation of the desired complex. Given the sensitivity of the phosphine group to oxidation, separation of the mixture proved onerous and hence a different synthetic pathway to the mixed Ir/Au complex was sought through prior coordination of the P-donor to Au(I) and subsequent introduction of the [Ir(PPy)2]+ core; preferential P-coordination to Au(I) has been noted in related pyridyl- and amino-derivatised phosphine lgands.9 The [Au(κ-P-L1)Cl] complex was easily prepared from a 1:1 mixture of L1 and [Au(THT)Cl] in CH2Cl2 under ambient conditions. The white solid obtained after removal of the volatiles was crystallised from CHCl3 by vapour diffusion of diethyl ether and the molecular structure determined by SCXRD is shown in Fig. 4.
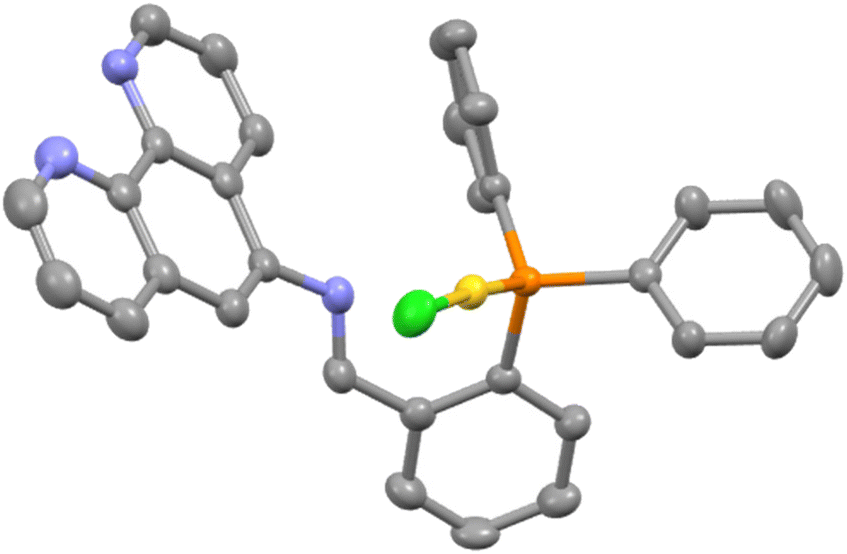 | ||
| Fig. 4 Molecular structure of AuL1 with lattice solvent molecules removed for clarity. Pertinent metrics and labelling are provided in the ESI.† | ||
The metrics around the NCH link are in agreement with both the carbon and the nitrogen being sp2 hybridised and there is no evidence to suggest any interaction between the imine nitrogen and the gold as the separation is 3.00 Å and the lone pair of the nitrogen is directed away from gold. Both the Au–P bond length of 2.300(10) Å and the P–Au–Cl angle of 175.80(4) are unremarkable. The 1H NMR spectrum of the gold complex shows some significant differences from that of the free ligand, most notably for the hydrogen atoms proximate to the AuCl fragment. The resonances assigned as H4 and H5 (see the ESI† for NMR assignments) are shifted downfield and upfield by 0.59 and 0.28 ppm respectively.
With AuL1 in hand, introduction of the [Ir(Ppy)2]+ core was readily achieved through the reaction of AuL1 with [Ir(Ppy)2(MeCN)2]BF4 at room temperature. Although the reaction took several days for completion, the desired complex, IrAuL1, was obtained in quantitative yield as a golden-orange solid. The 31P{1H} spectrum of the complex is, unsurprisingly, largely unchanged compared to AuL1 with a singlet at δP = 30.1 ppm being observed. Both H4 and the NCH hydrogens are shifted downfield (Table S1†) in the 1H NMR spectrum of IrAuL1 suggesting contact deshielding between the two.
The [Au(κ-P-L2)Cl] (AuL2) complex was prepared in a similar manner to that detailed above for AuL1 and subsequently reacted with [Ir(PPy)2(MeCN)2]BF4 in CH2Cl2 at RT for 7 days. Removal of the volatiles gave the desired [Ir(PPy)2(κ2-N,N′-L2){(κ-P-L2)AuCl}] (IrAuL2) as a rust-brown solid. The coordination shift of 41.6 ppm in the 31P{1H} NMR spectrum is typical and the 1H spectrum shows features akin to those of ReAuL2, notably the AB pattern for the CH2 hydrogens at 4.90 ppm. Relatively minor shifts are observed for the phen protons in the 1H NMR spectrum of ReAuL2 compared to AuL2 (Table S1†).
The reaction of L1 with [Ru(bpy)2(MeCN)2](BF4)2 proved to be very capricious as competition from P-coordination was evident during synthesis and the resultant complex, [Ru(bpy)2(L1)](BF4)2 (RuL1), was air-sensitive making separation of the reaction mixture difficult. Although frustrating, it was possible on occasions to isolate the desired complex sufficiently pure for characterisation. NMR spectroscopic analysis of [Ru(bpy)2(L1)](BF4)2 showed the expected number of peaks with the majority of the phen signals moving upfield relative to their position in the spectrum of L1 and the related parent [Ru(bpy)2(phen)]2+ complex.10 This reflects the shielding effect of the bpy groups on ruthenium. As anticipated, the 31P{1H} NMR spectrum was little different from that of L1 and the HRMS showed the presence of both the di-cation and the mono-cationic ion-pair with one BF4− molecule. Due to the sensitive nature of RuL1, it was crucial to use it immediately for the preparation of the mixed ruthenium/gold complex RuAuL1. As noted for the bimetallic rhenium and iridium complexes with gold the NMR spectra were consistent with the formation of a single isomer and there was no evidence of restricted rotation at room temperature. The 31P{1H} NMR spectrum gave a singlet at 30.1 ppm and the 1H NMR spectrum was sufficiently well-resolved to enable assignment of some of the hydrogens on L1 (Fig. S22 and Table S1†).
Despite numerous attempts, it was not possible to isolate [Ru(bpy)2(L2)](BF4)2 in a pure form. The preparations suffered from the phen/phosphine competition noted above and the resultant [Ru(bpy)2(L2)](BF4)2 complex was, to our surprise, aggressively air-sensitive.
Synthesis and characterisation of the dimetallic rhenium and iridium complexes with platinum and palladium RePtL1, RePdL1, IrPtL1 and IrPdL1
The mixed metal complexes of rhenium with platinum and palladium could be accessed by prior coordination of the iminophosphine donor to PtCl2 and PdCl2 and subsequent introduction of the [Re(CO)3Cl] core. Although isolated in high yield, the yellow [(κ2-N′′,P-L1)MCl2] complexes proved difficult to analyse largely as a consequence of their poor solubility. However, this did not prevent access to [{Re(CO)3(κ2-N,N′-L1)Cl}{Pt(κ2-N′′,P-L1)Cl2}], RePtL1 and [{Re(CO)3(κ2-N,N′-L1)Cl}{ Pd(κ2-N′′,P-L1)Cl2}], RePdL1. Heating a suspension of Pd(Pt)L1 with [Re(CO)5Cl] in chlorobenzene gave, on cooling, yellow (Pt) or yellowish-orange (Pd) precipitates. 31P{1H} NMR analysis of the mixed rhenium/platinum precipitate showed the presence of two species as indicated by two singlets with associated satellites (1JP–Pt ∼ 3630 Hz) in an ∼1:2 ratio at 1.3 and 1.0 ppm, respectively. The 1H NMR spectrum was broadened at room temperature but better resolution for most of the peaks was observed at +80 °C (Fig. S92 and S93†). Although the 1H spectrum appeared to represent primarily one species, the 31P{1H} NMR at high temperatures did not achieve coalescence. As noted for the dirhenium complexes, the presence of isomers can be explained by restricted rotation about the C–Nphen bond where the PPh2 fragment can be orientated on the side of the phen ring where the Re-bound chloride resides or on the opposite side towards the CO ligand that is trans to this chloride. The barrier to interconversion between these two forms is not accessible at room temperature.
The RePdL1 complex was prepared in a similar manner to platinum but gave two products distinguished by their solubility in the reaction solvent. The precipitate obtained from the PhCl solvent proved to be a mixture of at least three compounds whereas the PhCl soluble fraction was pure albeit as a mixture of two isomers. The two isomers were observed at 19.3 (major) and 14.0 (minor) ppm in the 31P{1H} spectrum, upfield of the 36.3 ppm reported for a related phosphine-imine complex.11 The expected duplication of peaks was obvious for some signals in the 1H NMR spectrum which enabled the isomer ratio of 63:37 to be determined.
The mixed IrPtL1 and IrPdL1 complexes were prepared from [Ir(PPy)2(MeCN)2]BF4 by the room temperature reaction with one equivalent of [(κ2-N′′,P-L1)MCl2]. The isolated orange (Pt) and yellow (Pd) solids were sparingly soluble in CDCl3 or d6-acetone, more soluble in CD2Cl2 and freely soluble in d6-dmso. Two peaks were observed in the 31P{1H} NMR spectrum of the platinum complex at 1.7 and 1.1 ppm in a close to 1:1 ratio with broad, poorly defined satellites. Broadened satellite peaks were also evident in the 31P{1H} NMR spectrum of RePtL1 and is likely the result of chemical shift anisotropy; efforts to observe a 195Pt NMR signal were unsuccessful in both instances presumably for the same reason. Although there was no observable coalescence of the peaks upon heating to 80 °C, the peak separation narrowed from 94 to 77 Hz. The 1H NMR spectrum did show some coalescence of peaks at the higher temperature as exemplified by the Nimine hydrogen that appears as two separate singlets separated by 12 Hz at RT but one singlet at higher temperature. Similar to that observed for RePd(Pt)L1, two isomers are present in solution due to atropisomerism where the Cphen–N single bond acts as the axis connecting the phen and N′′PPtCl2 planes with their relative orientation determining the Ra and Sa stereoisomers. Atropisomerism with square planar metal complexes has been reported previously.12 The major peak in the HRMS of IrPtL1 is for the dicationic [M − Cl]2+ species at 599.5925 amu with less intense peaks for the [M]+ (1233.1541 amu) and the [M − Cl + HCO2]+ (1244.1818 amu) cations.
The atropisomerism extended to the IrPdL1 complex with major and minor isomers being present in an ∼83:17 ratio. The coordination shifts in the 31P{1H} NMR spectrum were remarkably small at ΔδP = 8.6 ppm for the minor and 8.2 ppm for the major isomer. It is well-established that coordination shifts are sensitive to both electronic and steric parameters,13 and, while it is accepted that chemical shifts for P-atoms in chelate rings have a further contribution,14 it is noteworthy that the coordination shifts are significantly smaller for IrPdL1 than for RePdL1. Inspection of the 1D and 2D 1H NMR spectra confirmed the non-planar arrangement of the CNimine bond to the phen ring as there was no discernible through-space contact between the imine NCH and the H4 hydrogen of the phen ring. Heating a solution of the complex in d6-dmso to 80 °C produced no discernible change in either the 31P{1H} or 1H NMR spectra.
Attempts to prepare the analogous complexes with L2 proved unsuccessful as the initial complexation with PdCl2 and PtCl2 was completely unselective leading to a mixture of compounds prohibiting the formation of the desired bimetallics.
Electronic properties
The UV-vis absorption properties were assessed on dilute CH2Cl2 solutions of the ligands and complexes with extrapolated data given in Table 2 and Tables S114–S116 in the ESI.† The electronic manifolds are a combination of both ligand-centred and metal-to-ligand charge transfer (MLCT) type transitions. For the ReL1 and ReL2 complexes, the intense absorption maxima below 280 nm mimic those observed for the uncoordinated ligands and are assigned to intra-ligand π → π* transitions (Fig. S114 and S115†). There is very little difference in the absorption spectra of L1 and ReL1 although there is evidence of an MLCT band to a longer wavelength (>385 nm) for the latter. This trend is also evident on comparing L2 and ReL2 although here the low energy absorption is more evident. Unlike ReL1, the electronic spectrum of 2ReL1 does not resemble that of the uncoordinated ligand. The ligand-based peaks below 270 nm are no longer evident and there is only a single maximum observed below 285 nm. Aside from an apparent shoulder at around 320 nm the only other obvious feature is a maximum of relatively low intensity at 397 nm. This is likely a result of a structural change in L1 upon coordination of the second rhenium centre as noted in the SCXRD structure (Fig. 1) where the imine bond orientates perpendicular to the phenanthroline ring as opposed to being approximately coplanar in the ligand itself and in ReL1. This reflects minimal conjugation of the CNimine bond with the phenanthroline moiety in 2ReL1. The electronic spectrum of 2ReL2 is closely similar to that of 2ReL1 which is anticipated as communication through the saturated link is expected to be minimal. This extends to the ReAuL1 and ReAuL2 complexes where the electronic spectra are virtually indistinguishable but distinct from those of ReL1 and ReL2. As noted for all the bimetallic systems, the electronic spectra of the RePdL1 and RePtL1 complexes do not show the high energy fine structure associated with the ligands themselves and are characterised by an obvious maxima below 280 nm and a broad, shallow band at longer wavelengths (Fig. S116†).
| Compounda | λ max (104 M−1 cm−1)b/nm |
|---|---|
| a Recorded in aerated CH2Cl2 at 293 K. b 1 × 10−5 M. | |
| L1 | 339 (1.17), 278sh (4.48), 270 (8.64), 263 (8.88), 256sh (7.16), 250sh (5.61) |
| ReL1 | 337 (1.55), 285sh (3.61), 270 (7.70), 263 (8.48), 258sh (7.25), 250sh (5.77) |
| 2ReL1 | 397 (0.69), 270 (3.66) |
| ReAuL1 | 427sh (0.36), 357 (0.99), 300 (1.96), 269 (2.45) |
| RuL1 | 456 (1.05), 427sh (0.90), 289 (5.55) |
| RuAuL1 | 456 (1.25), 426sh (1.04), 289 (6.19) |
| IrAuL1 | 387sh (1.15), 337sh (1.81), 272 (5.94), 252 (6.68) |
| L2 | 339 (0.60), 285sh (2.56), 270 (6.61), 263 (7.35), 257sh (5.88), 250sh (4.07) |
| ReL2 | 363 (1.04), 299sh (2.02), 270 (6.84), 263 (7.63), 258 (6.48), 250sh (5.41) |
| 2ReL2 | 395 (0.44), 272 (2.9) |
| ReAuL2 | 430 (0.34), 358 (0.96), 300 (1.89), 270 (2.35) |
| IrAuL2 | 338sh (1.37), 288sh (3.59), 268sh (5.23), 248 (5.95) |
| PdL1 | 336 (1.47), 276 (4.05) |
| PtL1 | 302sh (1.84), 270 (5.87) |
| RePdL1 | 393sh (1.37), 349 (2.01), 270 (5.97) |
| RePtL1 | 393 (0.84), 270 (4.15) |
| IrPdL1 | 369sh (1.29), 263 (7.87), 242sh (9.97) |
| IrPtL1 | 379sh (1.75), 269 (9.29), 253 (9.90) |
The addition of the AuCl fragment has little influence on the electronic profile of RuL1 as the spectra of both RuL1 and RuAuL1 complexes are virtually identical with a manifold characteristics of a [Ru(bpy)2(phen)]2+ system.15 MLCT features are evident between 410 and 480 nm with a more intense ligand-based maximum at 289 nm in each case. Subtle differences are apparent in the UV/vis spectra of IrAuL1 and IrAuL2 with a more pronounced shoulder at ∼390 nm being evident in the former.
DFT studies
To examine electronic and structural properties in more detail, we turned to DFT studies of selected complexes. Optimisation at the B3LYP/defSVP level recovered all key properties of L1 and its Re, Pd and RePd complexes. Frontier molecular orbitals calculated at B3LYP/def2TZVP for L1, ReL1, PdL1 and RePdL1 are shown in Fig. 5. For the free ligand, the HOMO is mainly localised on the phen ring while the LUMO is on the imine and attached phenyl ring (see the ESI†). Binding of Re(CO)3Cl alters the HOMO to being metal-centred, while the LUMO becomes phen centred as shown in Fig. 5 (i.e. a classical MLCT representation). The HOMO in [N′′,P-PdCl2] is phen and one chloride ligand based while the LUMO extends across the imine bond and the directly bound phenyl ring with a contribution from the metal. In the RePdL1 bimetallic the HOMO resembles the ReL1 complex and the LUMO the PdL1 complex. The calculated C(4)–C(5)–N–Cimine torsion angles for the low energy conformations of L1, ReL1 and RePdL1 are 49°, 49° and 69°, respectively, accordant with the structural changes induced on bimetallic formation noted above (at its extreme this angle is 83° in 2ReL1). DFT calculation of absorption spectra indicates strong HOMO–LUMO absorption in L1, and the relatively small changes in the position of absorption bands that arise following coordination of Re and/or Pd. Analysis of molecular orbitals involved in transitions indicate that intra-ligand charge transfer dominates in free L1 and in the Pd and Au complexes, switching to MLCT when Re is coordinated to the phen moiety (Fig. S122†).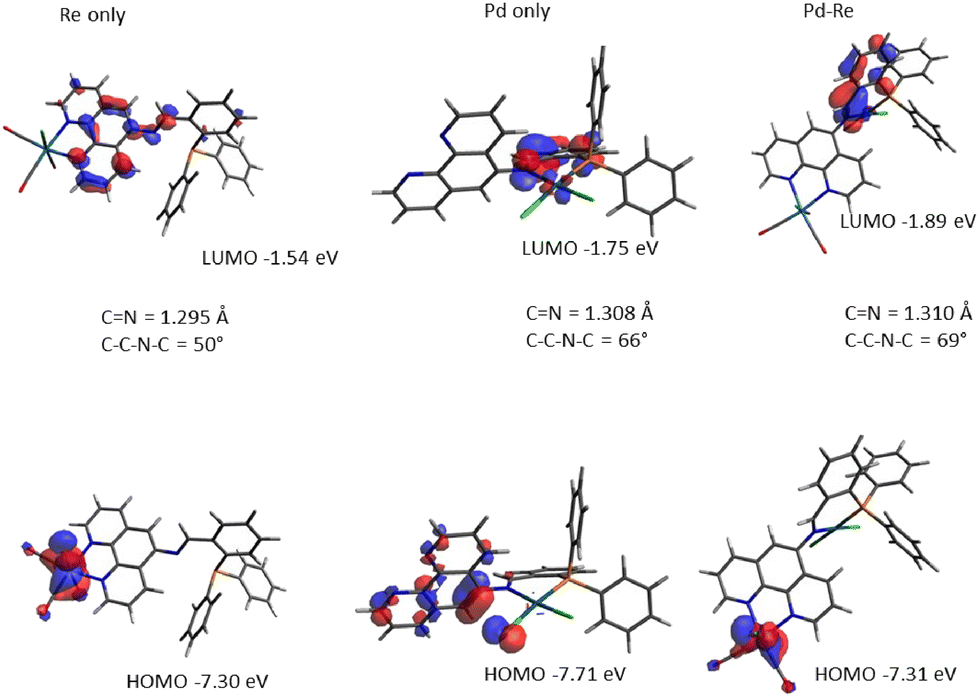 | ||
| Fig. 5 Frontier orbital representations of selected complexes of L1. | ||
Conclusions
Two novel ditopic phen-phosphine ligands L1 and L2 have been prepared and selectively coordinated to rhenium and, to a lesser extent Ru, through the phen function. The subsequent reaction of these complexes with Au(THT)Cl led to the formation of bimetallic Re/Au and Ru/Au species. Homo-bimetallic dirhenium complexes were also obtained from a 2:1 ligand:[Re(CO)5Cl] reaction. Competitive binding prevented access to mono-metallic Ir complexes by this approach, but the mixed metal Ir/Au complexes could be obtained by prior binding of the AuCl fragment at the phosphorus donor before introducing the [Ir(Ppy)2]+ core. A similar synthetic approach utilising N′′,P-κ2 Pd and Pt complexes gave access to the mixed Ir/Pd(Pt) and RePd(Pt) bimetallics. N,N′-Binding of [Re(CO)3Cl] had minimal impact on the electronic spectra and gross structures of L1 and L2 whereas significant changes occurred when a second metal was introduced at the P-atom or N′′,P binding site of L1. These result from conformational changes within L1 upon bimetallic formation exemplified by an increase in pertinent torsion angles. We are continuing our studies on these ligands and will report our findings in due course.
Experimental
All chemicals were purchased from commercial sources and used without further purification unless otherwise stated. All reactions and manipulations involving phosphines were performed under nitrogen using standard Schlenk techniques and previously dried, degassed solvents. NMR spectra were recorded on Bruker Fourier 300, DPX 400, and Avance 500 or 600 MHz NMR spectrometers. 1H and 13C{1H} NMR chemical shifts were referenced relative to the residual solvent resonances in the deuterated solvent. Mass spectra (ESI) were recorded on a Waters LCT premier XE spectrometer. UV/Vis spectra were obtained on a Cary 60 spectrophotometer and recorded over the range of 800 to 250 nm, with a 600 nm min−1 scan rate using a 1 cm path length quartz cuvette.Single-crystal X-ray diffraction data were collected on an Agilent SuperNova Dual Atlas diffractometer equipped with a mirror monochromator. Cu radiation was used for 2ReL1 and 2ReL2 whereas Mo radiation was used for AuL1 and ReL2Au. An Oxford Cryosystems cooling apparatus was used to regulate the sample temperature. CrysAlisPro16 software was used for data processing, SHELXT17 for structure solution and SHELXL18 for least-squares structure refinement. Anisotropic displacement parameters (ADPs) were used for non-hydrogen atoms. For 2ReL2, the ADPs for the triphenylphosphine group indicated possible disorder and this was modelled as two components of occupancies 0.412(5) and 0.588(5). In the final cycles of least-squares refinement for all structures, the geometry of the hydrogen atom locations was idealized, and a riding model was applied with Uiso set at 1.2 or 1.5 times the value of Ueq for the atom to which the hydrogen atoms are bound. CCDC 2372883–2372886† contain the supplementary crystallographic data for this paper. A table of pertinent details of the data collection and refinement is included in the ESI.† All calculations used the Gaussian09 package.19 Geometry optimisation was carried out using BLYP20 with the def2-SVP basis set,21 harmonic frequency calculations confirming the status as minima. Orbitals and electronic spectra were calculated using CAM-B3LYP22 with the def2-TZVP basis set in the SCRF model of DCM.23
Synthesis of L1·EtOH
A mixture of 2-(diphenylphosphino)benzaldehyde (1.49 g, 5.1 mmol) and 5-aminophenanthroline (1.00 g, 5.1 mmol) was refluxed in dry, degassed EtOH containing a few drops of acetic acid in a Dean–Stark apparatus with molecular sieves in the receiving arm for three hours under nitrogen. After cooling, the volatiles were removed in vacuo to give a yellow solid. Yield = quant. 1H (CDCl3, 400 MHz): 9.09 (m, 2H, H7, H8), 9.01 (dd, 4.3, 1.7 Hz, 1H, H1), 8.25 (dd, 8.3, 1.7 Hz, 1H, H5), 8.21 (ddd, 7.8, 3.8, 1.3 Hz, 1H, Ph), 7.99 (dd, 8.1, 1.7 Hz, 1H, H3), 7.49 (dd, 8.1, 4.4 Hz, 1H, H2), 7.46 (obs, 1H, Ph), 7.44 (dd, 8.2, 4.3 Hz, 1H, H6), 7.37–7.25 (m, 10H), 6.92 (ddd, 7.8, 4.6, 1.0 Hz, 1H, Ph), 6.64 (s, 1H, H4) ppm. 13C{1H} (CDCl3, 75 MHz): 160.4 (d, 17.9 Hz, CH), 150.5 (CH), 149.2 (CH), 147.7 (C), 146.2 (C), 145.2 (C), 139.2 (d, 21.6 Hz, C), 138.5 (d, 16.2 Hz, C), 135.6 (d, 9.3 Hz, C), 135.7 (CH), 134.4 (CH), 134.1 (CH), 133.8 (CH), 132.9 (CH), 131.4 (CH), 129.5 (d, 3.7 Hz, CH), 129.1 (CH), 128.9 (d, 2.6 Hz, CH), 129.8 (CH), 128.8 (CH), 126.0 (C), 123.1 (CH), 122.8 (CH), 110.8 (CH) ppm. 31P{1H} (CDCl3, 162 MHz): −11.6 ppm. HRMS (ES): m/z 468.1637 (calc. 468.1630) [L]+, 100%.Synthesis of L2
To a stirred suspension of L1·EtOH (1 g) in EtOH (50 ml) was added excess solid NaBH4 (0.5 g) in portions over 2 h. The reaction mixture was refluxed for 1 h and allowed to cool. The yellow ppt was filtered under N2, washed with water (2 × 5 ml) and dried at the pump. Yield = 690 mg (68%). A second crop was obtained after concentrating the mother liquor and isolated as above. Yield = 285 mg (28%). 1H (CDCl3, 400 MHz): 8.99 (dd, 4.3, 1.4 Hz, 1H, H7), 8.83 (dd, 4.3, 1.5 Hz, 1H, H1), 7.92 (dd, 8.2, 1.5 Hz, 1H, H3), 7.51 (dd, 7.5, 4.5 Hz, 1H, Ph), 7.38 (dd, 8.0, 4.3 Hz, 1H, H2), 7.32 (td, 7.5, 1.2 Hz, 2H), 7.28–7.10 (m, 12H), 6.96 (ddd, 7.8, 4.3, 1.1 Hz, 1H, Ph), 6.61 (s, 1H, H4), 4.72 (d, 5.1 Hz, 2H, CH2), 4.36 (t, 5.1 Hz, 1H, NH) ppm. 13C{1H} (CDCl3, 100 MHz): 149.6 (CH), 146.6 (C), 146.4 (CH), 142.4 (d, 24.7 Hz, C), 141.8 (C), 140.2 (C), 136.7 (d, 14.7 Hz, C), 136.5 (d, 9.6 Hz, C), 134.5 (CH), 133.9 (CH), 133.8 (CH), 133.7 (CH), 130.3 (C), 129.5 (d, 5.1 Hz, CH), 129.4 (CH), 128.9 (CH), 128.8 (CH), 128.7 (CH), 128.3 (d, 9.8 Hz, CH), 123.2 (CH), 121.9 (CH), 100.4 (CH), 47.9 (d, 20.8 Hz, CH2) ppm. 31P{1H} (CDCl3, 162 MHz): −16.5 ppm. HRMS (ES): m/z 470.1791 (calc. 470.1786) [L + H]+, 100%.Synthesis of ReL1
A mixture of L1·EtOH (100 mg, 0.195 mmol) and Re(CO)5Cl (70 mg, 0.195 mmol) was heated at ∼100 °C in degassed chlorobenzene (8 ml) for 3 h under nitrogen. After cooling, the yellow precipitate was filtered off and air-dried. Yield = 40 mg (25%). The filtrate was taken to dryness in vacuo to give a yellow solid that was triturated with degassed toluene (5 ml) overnight and filtered to give a further crop of the yellow solid. Yield = 60 mg (38%). 1H (CDCl3, 400 MHz): 9.26 (dd, 5.1, 1.4 Hz, 1H, H1/7), 9.20 (dd, 5.1, 1.3 Hz, 1H, H1/7), 9.01 (d, 4.6 Hz, 1H, NCH), 8.28 (dt, 8.3, 0.9 Hz, 2H, H3/5), 8.14 (ddd, 7.8, 3.8, 1.1 Hz, 1H), 7.71 (dd, 8.3, 5.1 Hz, 1H, H2/6), 7.60 (dd, 8.4, 5.0 Hz, 1H, H2/6), 7.50 (td, 7.5, 1.0 Hz, 1H), 7.40 (td, 7.6, 1.2 Hz, 1H), 7.34–7.22 (m, 10H), 6.96 (ddd, 7.8, 4.5, 1.1 Hz, Hz, 1H, Ph), 6.90 (s, 1H, H4) ppm. 13C{1H} (CDCl3, 100 MHz): 197.1 (CO), 197.0 (CO), 189.5 (CO), 162.7 (d, 14.6 Hz, CH), 153.1 (CH), 151.6 (CH), 149.0 (C), 147.1 (C), 145.7 (C), 139.8 (d, 23.1 Hz, C), 137.8 (d, 16.3 Hz, C), 137.6 (CH), 136.8 (d, 9.0 Hz, C), 136.6 (d, 9.4 Hz, C), 135.7 (d, 2.1 Hz, CH), 134.3 (CH), 134.2 (CH), 134.1 (CH), 134.0 (CH), 132.1 (CH), 131.0 (C), 130.9 (d, 3.2 Hz, CH), 129.7 (C), 129.2 (CH), 129.1 (CH), 129.0 (CH), 128.9 (CH), 128.6 (C), 128.3 (C), 126.4 (C), 125.8 (CH), 125.5 (CH), 111.2 (CH) ppm. 31P{1H} (CDCl3, 162 MHz): −10.7 ppm. HRMS (ES): m/z 774.0706 (calc. 774.0693) [M]+, 100%.Synthesis of ReL2
A mixture of L2 (100 mg, 0.214 mmol) and Re(CO)5Cl (77 mg, 0.214 mmol) was heated at ∼100 °C in degassed toluene (12 ml) for 3 h under nitrogen. After cooling, the bright yellow precipitate was filtered off and air-dried. Yield = 130 mg (75%). The solid was poorly soluble in CDCl3 so NMR spectra were recorded in d6-dmso. 1H (d6-dmso, 400 MHz): 9.25 (d, 4.6 Hz, 1H, H7), 8.85 (dd, 5.1, 0.9 Hz, 1H, H1), 8.80 (d, 8.5 Hz, 1H, H5), 8.21 (d, 8.5 Hz, 1H, H3), 7.84 (dd, 8.5, 5.2 Hz, 1H, H6), 7.68 (dd, 8.3, 4.9 Hz, 1H, H2), 7.51 (dd, 7.2, 4.5 Hz, 1H, PPh2), 7.30–7.05 (m, 12H), 6.77 (dd, 7.1, 4.7 Hz, 1H, Ph), 6.50 (s, 1H, H4), 4.64 (s br, 2H, CH2) ppm. 13C{1H} (d6-dmso, 100 MHz): 198.4 (CO), 198.3 (CO), 190.6 (CO), 153.4 (CH), 148.5 (CH), 147.0 (C), 143.3 (C), 142.2 (d, 23.1 Hz, C), 140.5 (C), 136.4 (d obs, C), 136.3 (CH), 136.2 (d, 9.9 Hz, C), 135.7 (d, 15.6 Hz, C), 134.2 (CH), 134.0 (CH), 133.9 (CH), 133.8 (CH), 132.8 (C), 129.7 (CH), 129.5 (CH), 129.4 (CH), 129.3 (CH), 129.2 (CH), 129.1 (CH), 128.3 (d, 5.0 Hz, CH), 128.2 (CH), 126.8 (CH), 125.5 (CH), 124.4 (C), 98.6 (CH), 46.5 (d, 23.2 Hz, CH2) ppm. 31P{1H} (d6-dmso, 162 MHz): −16.9 ppm. HRMS (ES): m/z 740.1115 (calc. 740.1113) [M − Cl]+, 50%.Synthesis of 2ReL1
A mixture of L1·EtOH (100 mg, 0.195 mmol) and Re(CO)5Cl (140 mg, 0.390 mmol) was heated at ∼100 °C in degassed chlorobenzene (8 ml) for 3 h under nitrogen. After cooling, the yellow precipitate was filtered off and air-dried. Yield = 128 mg (42%). 1H (d6-acetone, 400 MHz): 9.52 (dd, 5.2, 1.3 Hz, 1H, H1), 9.44 (dd, 4.7, 1.6 Hz, 1H, H7), 9.25 (d, 1.5 Hz, 1H, NCH), 9.17 (dd, 8.3, 1.0 Hz, 2H, H3), 8.23 (dd, 8.4, 5.1 Hz, 1H, H2), 8.13 (m, 1H, Ph), 8.07 (s, 1H, H4), 7.98 (m, 2H), 7.91–7.75 (m, 5H), 7.68 (m, 4H), 7.59–7.44 (m, 4H, H6), 7.19 (m, 1H, Ph) ppm. 31P{1H} (d6-acetone, 162 MHz): 15.0 ppm. HRMS (ES): m/z 1042.0038 (calc. 1042.0022) [M − Cl]+, 100%.Synthesis of 2ReL2
A mixture of L2 (75 mg, 0.160 mmol) and Re(CO)5Cl (115 mg, 0.32 mmol) were heated at ∼100 °C in degassed chlorobenzene (15 ml) for 3 h under nitrogen. After cooling, the yellow-orange solution was taken to dryness in vacuo and the resultant solid was triturated with pentane to give a yellow solid that was filtered in air and recrystallised from acetone by vapour diffusion of diethyl ether. Yield = 60 mg (35%). 1H (CDCl3, 500 MHz, major isomer): 9.41 (dd, 5.1, 0.9 Hz, 1H, H7), 9.27 (dd, 5.1, 1.3 Hz, 1H, H1), 9.20 (dd, 8.8, 1.0 Hz, 1H, H5), 8.38 (dd, 8.3, 1.4 Hz, 1H, H3), 7.92 (dd, 8.7, 5.1 Hz, 1H, H6), 7.78 (dd, 8.3, 5.0 Hz, 1H, H2), 7.61 (s, 1H, H4), 7.60–7.31 (m, 14H), 6.87 (m, 1H), 5.51 (d, 9.8 Hz, 1H, NH), 5.13 (t, 10.8 Hz, 1H, CH2), 4.61 (dd, 11.4, 4.3 Hz, 1H, CH2) ppm. 13C{1H} (CDCl3, 150 MHz): 196.8 (CO), 192.6 (d, 7.3 Hz, CO), 192.2 (d, 6.8 Hz, CO), 190.0 (d, 68.3 Hz, CO), 188.8 (CO), 154.1 (CH), 152.8 (CH), 148.4 (C), 146.9 (C), 144.9 (C), 137.6 (CH), 136.3 (d, 14.9 Hz, C), 134.3 (CH), 133.3 (d, 9.9 Hz, CH), 133.2 (CH), 132.5 (CH), 132.3 (CH), 132.2 (C), 131.8 (CH), 131.6 (CH), 131.2 (d, 7.4 Hz, CH), 130.0 (C), 129.5 (CH), 129.4 (CH), 129.2 (d, 10.6 Hz, CH), 128.5 (d, 40.2 Hz, C), 128.3 (d, 49.8 Hz, C), 127.8 (C), 126.4 (CH), 126.1 (CH), 123.8 (CH), 114.5 (CH), 62.2 (d, 10.8 Hz, CH2) ppm. 31P{1H} (CDCl3, 202 MHz): 11.8 ppm. HRMS (ES): m/z 1046.0201 (calc. 1046.0206) [M − Cl]+, 100%.Synthesis of ReAuL1
To a solution of ReL1 (100 mg, 0.148 mmol) in CH2Cl2 (10 ml) was added Au(THT)Cl (48 mg, 0.148 mmol). After stirring for 1 h, the volatiles were removed in vacuo and the resultant yellow solid crystallised from chloroform by the slow infusion of diethyl ether. Yield (several crops) = 131 mg (88%). 1H (CD2Cl2, 400 MHz): 9.20 (dd, 5.1, 1.4 Hz, 1H, H7), 9.17 (dd, 5.1, 1.4 Hz, 1H, H1), 9.02 (d, 1.6 Hz, 1H, NCH), 8.60 (dd, 8.3, 1.4 Hz, 1H, H3), 8.20 (ddd, 7.8, 4.3, 1.2 Hz, 1H, Ph), 7.76 (dd, 8.3, 5.2 Hz, 1H, H2), 7.72 (m, 2H, H5, Ph), 7.58–7.38 (m, 9H, H6), 7.51 (s, 1H, H4), 6.99 (ddd, 12.9, 7.8, 1.0 Hz, 1H, Ph) ppm. 13C{1H} (CD2Cl2, 150 MHz): 197.7 (CO), 197.6 (CO), 189.5 (CO), 161.3 (d, 6.6 Hz, CH), 152.9 (CH), 151.8 (CH), 147.3 (C), 146.9 (C), 145.7 (C), 138.6 (CH), 137.7 (d, 7.6 Hz, C), 135.1 (CH), 135.1 (d, 6.6 Hz, CH), 134.5 (d, 14.3 Hz, CH), 134.2 (d, 13.8 Hz, CH), 133.2 (d, 7.9 Hz, CH), 132.1 (2 × CH), 132.0 (CH), 131.0 (C), 129.7 (d, 12.5 Hz, CH), 129.6 (d, 12.3 Hz, CH), 129.4 (d, 6.6 Hz, C), 129.3 (C), 129.0 (d, 3.1 Hz, C), 127.7 (C), 126.0 (CH), 125.3 (CH), 113.3 (CH) ppm. 31P{1H} (CD2Cl2, 162 MHz): 29.7 ppm. HRMS (ES): m/z 970.0291 (calc. 970.0310) [M − Cl]+, 100%.Synthesis of ReAuL2
Prepared similarly to ReAuL1. It was crystallised by slow evaporation of a concentrated solution in CDCl3. Yield (several crops) = 111 mg (75%). 1H (CDCl3, 400 MHz): 8.89 (d, 4.8 Hz, 1H, H7), 8.74 (d, 4.9 Hz, 1H, H1), 8.37 (d, 8.3 Hz, 1H, H3), 7.94 (d, 8.4 Hz, 1H, H5), 7.52 (t, 7.1 Hz, 1H), 7.48–7.06 (m, 12H), 6.67 (dd, 13.2, 7.7 Hz, 1H, Ph), 6.48 (s, 1H, H4), 5.83 (br, 1H, NH), 4.69 (d, 16.1 Hz, 1H, CH2), 3.86 (d, 15.9 Hz, 1H, CH2) ppm. 13C{1H} (CDCl3, 100 MHz): 197.3 (CO), 197.1 (CO), 189.3 (CO), 151.6 (CH), 148.1 (CH), 147.1 (C), 141.1 (d, 15.0 Hz, C), 141.0 (C), 140.7 (C), 136.7 (CH), 134.9 (CH), 134.7 (CH), 134.3 (d, 6.7 Hz, CH), 134.0 (CH), 133.8 (CH), 132.4 (d, 9.7 Hz, CH), 132.3 (CH), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (d, 9.5 Hz, CH), 129.1 (C), 128.5 (C), 128.2 (C), 128.0 (d, 9.9 Hz, CH), 125.6 (CH), 125.3 (C), 123.9 (CH), 123.6 (C), 100.4 (CH), 45.1 (d, 12.1 Hz, CH2) ppm. 31P{1H} (CDCl3, 162 MHz): 23.7 ppm. HRMS (ES): m/z 972.0460 (calc. 972.0467) [M − Cl]+, 100%.Synthesis of RuL1
A mixture of L1·EtOH (100 mg, 0.195 mmol) and [Ru(bipy)2(MeCN)2]2BF4 (130 mg, 0.195 mmol) was heated at ∼100 °C in degassed ethoxyethanol (8 ml) for 15 h under nitrogen. After cooling, the orange-red precipitate was filtered off under N2 and dried at the pump. Yield = 65 mg (32%). The mother liquor was taken to dryness in vacuo and the residue was extracted into EtOH. After filtering, ethanol was removed in vacuo to give a second crop of the product. Yield = 98 mg (48%). Note: attempts to use the PF6- salt of the ruthenium starting material led to significant by-product formation. 1H (d6-dmso, 400 MHz): 9.14 (d, 4.3 Hz, 1H, NCH), 8.78 (d, 8.0 Hz, 2H), 8.74 (d, 8.0 Hz, 2H), 8.50 (dd, 8.4, 1.0 Hz, 1H, H7), 8.24 (d, 8.4 Hz, 1H, H1), 8.20 (ddd, 7.8, 3.6, 1.2 Hz, 1H, Ph), 8.12 (td, 8.0, 1.4 Hz, 2H), 8.04 (s, 1H, H4), 8.02 (tt, 7.9 Hz, 2H), 7.98 (dd, 5.4, 1.2 Hz, 1H, H3), 7.91 (dd, 5.3, 1.2 Hz, 1H, H5), 7.74 (m, 3H), 7.63–7.43 (m, H), 7.60 (dd, 8.4, 5.2 Hz, 1H, H2), 7.37–7.18 (m, 15H), 6.93 (ddd, 7.7, 4.5, 0.8 Hz, 1H) ppm. 31P{1H} (d6-dmso, 162 MHz): −11.2 ppm. HRMS (ES): m/z 440.5989 (calc. 440.5992) [M]2+, 100%.Synthesis of RuAuL1
To a solution of RuL1 (80 mg, 9.1 × 10−5 mol) in dry, degassed acetone (10 ml) was added, as a solid, Au(THT)Cl (29 mg, 9.1 × 10−5 mol). After stirring for 3 h at room temperature, the solution was filtered to remove the slight precipitate and taken to dryness at the pump. The orange-red residue was subsequently triturated with diethyl ether in air, filtered and air-dried. Yield = 105 mg (90%). 1H (d6-dmso, 400 MHz): 9.14 (d, 4.3 Hz, 1H, NCH), 8.78 (d, 8.0 Hz, 2H), 8.74 (d, 8.0 Hz, 2H), 8.50 (dd, 8.4, 1.0 Hz, 1H, H7), 8.24 (d, 8.4 Hz, 1H, H1), 8.20 (ddd, 7.8, 3.6, 1.2 Hz, 1H, Ph), 8.12 (td, 8.0, 1.4 Hz, 2H), 8.04 (s, 1H, H4), 8.02 (tt, 7.9 Hz, 2H), 7.98 (dd, 5.4, 1.2 Hz, 1H, H3), 7.91 (dd, 5.3, 1.2 Hz, 1H, H5), 7.74 (m, 3H), 7.63–7.43 (m, H), 7.60 (dd, 8.4, 5.2 Hz, 1H, H2), 7.37–7.18 (m, 15H), 6.93 (ddd, 7.7, 4.5, 0.8 Hz, 1H) ppm. 31P{1H} (d6-dmso, 162 MHz): −11.2 ppm. HRMS (ES): m/z 1200.1371 (calc. 1200.1353) [M + BF4]+, 10%; 556.5734 [M]2+, 100%.Synthesis of AuL1
To a solution of [Au(THT)Cl] (63 mg, 0.195 mmol) in CH2Cl2 (20 ml) was added solid L1·EtOH and the solution was stirred for 2 h. Volatiles were removed in vacuo to give a yellow solid. Yield = quant. Crystals suitable for SCXRD were obtained by vapour diffusion of Et2O into a concentrated solution in CH2Cl2. 1H (CDCl3, 400 MHz): 9.08 (d, 1.4 Hz, 1H, NCH), 9.05 (dd, 4.3, 1.6 Hz, 1H, H7), 9.03 (dd, 4.3, 1.6 Hz, 1H, H1), 8.28 (dd, 8.1, 1.6 Hz, 1H, H3), 8.24 (ddd, 7.7, 4.4, 0.9 Hz, 1H, Ph), 7.71 (dd, 8.2, 1.7 Hz, 1H, H5), 7.67 (m, 1H, Ph), 7.57–7.37 (m, 10H), 7.33 (dd, 8.3, 4.3 Hz, 1H, H6), 7.23 (s, 1H, H4), 6.93 (ddd, 12.9, 7.9, 0.9 Hz, 1H, Ph) ppm. 13C{1H} (CDCl3, 75 MHz): 158.3 (d, 8.3 Hz, CH), 150.3 (CH), 149.5 (CH), 146.1 (C), 145.3 (C), 138.3 (d, 8.5 Hz, C), 136.6 (CH), 134.5 (d, 7.2 Hz, CH), 134.4 (CH), 134.3 (CH), 132.4 (CH), 132.1 (2 × CH), 131.4 (d, 8.4 Hz, CH), 129.6 (CH), 129.5 (CH), 129.5 (C), 129.4 (C), 128.9 (d, 13.0 Hz, C), 125.4 (C), 123.4 (CH), 122.6 (CH), 112.6 (CH) ppm. 31P{1H} (CDCl3, 162 MHz): 29.0 ppm. HRMS (ES): m/z 664.1249 (calc. 664.1217) [M − Cl]+, 100%.Synthesis of AuL2
Prepared similarly to AuL1. Yield = quant. 1H (CDCl3, 400 MHz): 8.95 (dd, 4.3, 1.4 Hz, 1H, H7), 8.72 (dd, 4.3, 1.6 Hz, 1H, H1), 8.17 (d, 8.3 Hz, 1H, H5), 7.66 (dd, 8.0, 1.5 Hz, 1H, H3), 7.60 (d br, 6.8 Hz, 1H, Ph), 7.44 (t, 7.9 Hz, 1H, Ph), 7.28–7.18 (m, 8H), 7.01 (t, 7.5 Hz, 5H), 6.75 (dd, 10.4, 8.2 Hz, 1H, Ph), 6.33 (s, 1H, H4), 6.00 (s br, 1H, NH), 4.89 (d, 5.2 Hz, 2H, CH2) ppm. 13C{1H} (CDCl3, 75 MHz): 149.5 (CH), 146.3 (C), 146.1 (CH), 143.2 (d, 13.1 Hz, C), 141.7 (C), 141.0 (C), 134.6 (CH), 133.9 (CH), 133.8 (CH), 133.6 (CH), 131.8 (d, 9.7 Hz, CH), 131.7 (CH), 131.1 (CH), 131.0 (d, 7.9 Hz, CH), 130.2 (CH), 129.9 (C), 129.8 (C), 129.0 (CH), 128.9 (CH), 128.0 (d, 7.2 Hz, CH), 122.9 (CH), 122.2 (C), 121.9 (CH), 100.2 (CH), 48.5 (d, 9.8 Hz, CH2) ppm. 31P{1H} (CDCl3, 162 MHz): 29.0 ppm. HRMS (ES): m/z 666.1391 (calc. 666.1373) [M − Cl]+, 100%.Synthesis of IrAuL1
A solution of AuL1 (50 mg, 7.1 × 10−5 mol) and [Ir(Ppy)2(MeCN)2]BF4 (48 mg, 7.1 × 10−5 mol) in CH2Cl2 (30 ml) was stirred at RT for 10 days. Volatiles were removed in vacuo and the solid residue was triturated with diethyl ether to give a golden-yellow solid that was isolated by filtration and air-dried. Yield = 84 mg (92%). 1H (d6-acetone, 400 MHz): 9.21 (d, 1.4 Hz, 1H, NCH), 8.62 (dd, 8.4, 1.3 Hz, 1H, H3), 8.33 (ddd, 7.7, 4.3, 1.1 Hz, 1H, Ph), 8.23 (dd, 5.0, 1.4 Hz, 1H, H7), 8.20 (dd, 5.1, 1.4 Hz, 1H, H1), 8.09 (d, 8.2 Hz, 2H), 7.87 (m, 2H, H2/H5), 7.83–7.73 (m, 6H), 7.67 (dd, 8.3, 5.0 Hz, 1H, H6), 7.63 (tt, 7.7, 1.6, 1.4 Hz, 1H), 7.61–7.43 (m, 12H), 7.59 (s, 1H, H4), 7.05–6.80 (m, 7H), 6.31 (dm, 7.5 Hz, 2H) ppm. 13C{1H} (d6-acetone, 100 MHz): 167.8 (C), 162.3 (d, 6.3 Hz, CH), 151.4 (CH), 150.2 (CH), 149.9 (d, 17.0 Hz, C), 149.5 (CH), 149.3 (CH), 147.9 (C), 147.0 (C), 145.7 (C), 144.3 (C), 138.6 (CH), 138.5 (CH), 135.2 (CH), 135.0 (d, 6.8 Hz, CH), 134.5 (CH), 134.3 (CH), 134.2 (CH), 134.0 (d, 8.0 Hz, CH), 132.4 (obs, CH), 132.3 (CH), 131.8 (d, 3.8 Hz, CH), 131.6 (C), 130.3 (CH), 129.8 (CH), 129.7 (CH), 129.3 (d, 12.6 Hz, C), 128.8 (C), 128.2 (C), 127.2 (CH), 126.6 (CH), 124.9 (CH), 123.5 (d, 5.3 Hz, CH), 122.6 (CH), 119.8 (CH), 113.6 (CH) ppm. 31P{1H} (d6-acetone, 162 MHz): 30.1 ppm. HRMS (ES): m/z 1200.1844 (calc. 1200.1848) [M]+, 100%.Synthesis of IrAuL2
Prepared similarly to IrAuL1. Yield = 82 mg (90%). 1H (d6-acetone, 400 MHz): 8.68 (d, 8.5 Hz, 1H, H7), 8.28 (d, 8.4 Hz, 1H, H3), 8.22 (dd, 4.8, 1.0 Hz, 1H, H5), 8.09 (t, 8.4 Hz, 2H, Ph), 7.86 (dd, 5.0, 1.2 Hz, 1H, H1), 7.80 (d, 1.1 Hz, 1H), 7.79–7.71 (m, 5H, H6), 7.60 (dd, 8.3, 5.0 Hz, 1H, H2), 7.56–7.39 (m, 13H), 7.34 (t, 7.7 Hz, 1H), 6.98–6.76 (m, 9H), 6.80 (s, 1H, H4), 6.31 (dd, 4.6, 0.7 Hz, 1H), 6.29 (dd, 4.6, 0.9 Hz, 1H), 4.90 (m, 2H, CH2) ppm. 13C{1H} (d6-acetone, 125 MHz): 167.8 (d, 12.6 Hz, C), 150.8 (C), 150.7 (CH), 150.3 (C), 149.3 (d, 3.3 Hz, CH), 147.7 (C), 146.3 (CH), 144.3 (d, 9.8 Hz, C), 143.1 (C), 141.5 (C), 141.1 (d, 10.7, C), 138.5 (d, 7.8 Hz, CH), 136.1 (CH), 136.6 (CH), 134.5 (CH), 134.4 (2 × CH), 133.9 (d, 7.6 Hz, CH), 133.3 (C), 132.9 (CH), 132.5 (d, 2.6 Hz, CH), 132.4 (d, 2.5 Hz, CH), 131.8 (CH), 131.7 (CH), 130.3 (CH), 130.2 (CH), 130.0 (d, 8.9 Hz, CH), 129.8 (2 × CH), 129.7 (2 × CH), 128.6 (d, 3.8 Hz, C), 128.4 (d, 9.5 Hz, CH), 128.0 (d, 5.6 Hz, C), 127.3 (C), 126.8 (C), 126.5 (CH), 125.7 (CH), 125.0 (C), 124.9 (CH), 124.8 (CH), 123.5 (CH), 123.4 (CH), 122.5 (CH), 122.4 (CH), 119.8 (2 × CH), 100.5 (CH), 47.1 (d, 12.4 Hz, CH2) ppm. 31P{1H} (d6-acetone, 162 MHz): 25.1 ppm. HRMS (ES): m/z 1202.2000 (calc. 1202.2005) [M]+, 85%.Synthesis of PtL1
A mixture of L1·EtOH (0.1 g, 0.195 mmol) and Pt(COD)Cl2 (73 mg, 0.195 mmol) was stirred in acetone (25 ml) at RT for several hours whereupon a pale yellow solid precipitated. The solid was filtered, washed with acetone and air-dried. Yield = 122 mg (85%). The solid was poorly soluble in all solvents except for DMSO and heating was required to get a sample for NMR spectroscopy. However, NMR analysis was not useful as the 31P{1H} and 1H NMR spectra were relatively poor possibly due to competition with the solvent on dissolution. HRMS (ES): m/z 698.0889 (calc. 698.0889) [M − Cl]+, 100%.Synthesis of PdL1
Prepared similarly to the PtL1 complex and isolated as a bright yellow solid. Yield = 113 mg (90%). The solid was poorly soluble in all solvents except for DMSO and heating was required to get a sample for NMR spectroscopy. However, NMR analysis was not useful as the 31P{1H} and 1H NMR spectra were relatively poor possibly due to competition with the solvent on dissolution. HRMS (ES): m/z 572.0533 (calc. 572.0508) [M − 2Cl − H]+, 70%.Synthesis of RePtL1
A 1:1 mixture of Re(CO)5Cl (50 mg, 0.138 mmol) and PtL1 (101 mg, 0.138 mmol) in PhCl (30 ml) was heated at 100 °C for 3 h. After allowing to cool to RT, the yellow solid was isolated by filtration and air-dried. Yield = 84 mg (60%). IR (ATR) 2018, 1910, 1873 cm−1. 1H (d6-dmso, 400 MHz, 353 K, major isomer): 9.49 (s, 1H, NCH) 9.34 (d, 5.0 Hz, 2H, H1/7), 8.88 (d, 8.1 Hz, 1H, H5), 8.53 (d, 8.4 Hz, 1H, H3), 8.16–8.06 (br, 2H), 8.06 (dd, 8.2, 5.2 Hz, 1H, H6), 7.91–7.85 (m, 2H), 7.80 (dd, 8.4, 5.0, 1H, H2), 7.70–7.50 (br, 9H), 7.35–7.22 (m, 2H), 7.33 (s, 1H) ppm. 13C{1H} (d6-dmso, 150 MHz, APT): 197.9 (CO), 190.1 (CO), 170.8 (CH), 154.2 (d, 10.5 Hz, CH), 148.6 (C), 145.9 (C), 145.5 (C), 140.1 (CH), 139.6 (d, 7.9 Hz, CH), 136.4 (CH), 135.4 (CH), 134.6 (CH), 134.3 (d, 10.7 Hz, CH), 133.9 (d, 10.8 Hz, CH), 133.8 (CH), 132.8 (CH), 132.5 (CH), 129.8 (2 × CH), 129.3 (d, 12.3 Hz, CH), 129.3 (C), 128.3 (C), 128.2 (C), 127.5 (CH), 127.1 (d, 10.6 Hz, C), 126.6 (C), 126.3 (CH), 122.2 (CH) ppm. 31P{1H} (CD2Cl2, 162 MHz): 1.3 (minor, 1JP–Pt = 3620 Hz), 1.0 (major, 1JP–Pt = 3620 Hz) ppm. HRMS (ES): m/z 1003.9990 (calc. 1003.9983) [M − Cl]+, 20%; 1045.0259 [M − Cl + MeCN]+, 100%.
Synthesis of RePdL1
Prepared similarly to the Pt complex above. Yield = 84 mg (60%). IR (ATR) 2016, 1910, 1877 cm−1. 1H (CDCl3, 400 MHz, major isomer): 10.03 (s, 1H, NCH), 9.17 (m, 2H, H1/7), 8.25 (dd, 8.5, 1.4 Hz, 1H, H3), 7.97 (br, 2H), 7.93 (dd, 8.4, 5.2 Hz, 1H), 7.80–7.74 (m, 2H), 7.65–7.05 (m, 11H), 7.53 (s, 1H, H4), 6.94 (m, 1H) ppm. 31P{1H} (CDCl3, 162 MHz): 19.3 (major), 14.0 (minor) ppm. HRMS (ES): m/z 923.9659 (calc. 923.9656) [M − Cl]+, 100%.Synthesis of IrPtL1
A mixture of [Ir(Ppy)2(MeCN)2]BF4 (50 mg, 7.4 × 10−5 mol) and PtL1 (48 mg, 7.4 × 10−5 mol) were stirred in CH2Cl2 (40 ml) for seven days. The light orange solution was then taken to dryness and the residue was triturated with diethyl ether to give a bright orange solid which was filtered off and air-dried. Yield = 85 mg (87%). 1H (d6-dmso, 400 MHz): 9.47 (s, 0.5H), 9.44 (s, 0.5H), 8.92 (m, 1H), 8.45 (dd, 8.5, 1.2 Hz, 0.5H), 8.37 (dd, 8.5, 1.2 Hz, 0.5H), 8.34 (s, 0.5H), 8.33–8.18 (m, 3H), 8.10 (m, 1H), 8.05–7.50 (m, 20.5H), 7.43 (dd, 10.0, 5.7 Hz, 1H), 7.30 (m, 2H), 7.11–6.92 (m, 5H), 6.33 (d, 7.5 Hz, 1H), 6.29 (dd, 11.6, 7.7 Hz, 1H) ppm. 31P{1H} (d6-dmso, 162 MHz): 1.7 (1JP–Pt = 3622 Hz), 1.0 (1JP–Pt = 3622 Hz) ppm. HRMS (ES): m/z 1233.1541 (calc. 1233.1519) [M]+, 10%; 1244.1818 [M + HCO2]+, 45%; 599.5925 [M − Cl]2+, 100%.Synthesis of IrPdL1
A mixture of [Ir(Ppy)2(MeCN)2]BF4 (50 mg, 7.4 × 10−5 mol) and PdL1 (54 mg, 7.4 × 10−5 mol) was stirred in CH2Cl2 (40 ml) for seven days. The light orange solution was then taken to dryness and the residue was triturated with diethyl ether to give a bright orange solid which was filtered off and air-dried. Yield = 70 mg (75%). The solid was recrystallised from CH2Cl2 by slow evaporation. 1H (d6-dmso, 400 MHz, major isomer): 9.62 (s, 1H, NCH), 9.09 (br m, 1H, H1), 9.07 (dd, 5.4, 1.3 Hz, 1H, H7), 8.95 (dd, 5.4, 1.1 Hz, 1H, H5), 8.64 (dd, 8.3, 1.3 Hz, 1H, H3), 8.39 (ddd, 8.1, 4.7, 1.4 Hz, 1H), 8.15 (t, 7.5 Hz, 1H), 8.09 (td, 7.7, 1.2 Hz, 1H), 8.02 (m, 1H), 7.92 (dd, 8.3, 5.4 Hz, 1H, H2), 7.86 (m, 1H), 7.75–7.53 (m, 12H), 7.54 (s, 1H), 7.43 (m, 1H, H2), 7.33 (dd, 8.5, 5.5 Hz, 1H, H6), 7.17 (t, 7.3 Hz, 1H), 6.85–6.75 (m, 5H), 6.30 (t, 8.6 Hz, 2H), 6.20 (tt, 7.4, 7.4, 1.5 Hz, 1H), 6.06 (t, 7.5 Hz, 1H), 5.96 (m, 1H), 5.41 (ddd, 7.6, 4.5, 0.7 Hz, 1H) ppm. 31P{1H} (d6-dmso, 162 MHz): −3.1 (minor), −3.4 (major) ppm. HRMS (ES): m/z 1144.0914 (calc. 1144.0906) [M]+, 10%.Author contributions
PDN – Conceptualisation, investigation, methodology, supervision, and writing. JAP – analysis (DFT) and writing. BA – investigation. SJAP – supervision and writing. BMK – analysis (SCXRD) and writing.Data availability
The data supporting this article have been included in the ESI.†Crystallographic data for 2ReL1, 2ReL2, ReAuL2 and AuL1 have been deposited at the CCDC under the identifiers 2372883, 2372884, 2372885 and 2372886† respectively and can be obtained from https://www.ccdc.cam.ac.uk.
Conflicts of interest
There are no conflicts to declare.References
- M. A. Stevens and A. L. Colebatch, Chem. Soc. Rev., 2022, 51, 1881 RSC; D. Liu, M. Zhang, H.-H. Huang, Q. Feng, C. Su, A. Mo, J.-W. Wang, Z. Qi, X. Zhang, L. Jiang and Z. Chen, ACS Sustainable Chem. Eng., 2021, 9, 28A Search PubMed; J.-W. Wang, X. Zhang, L. Velasco, M. Karnahl, Z. Li, Z.-M. Luo, Y. Huang, J. Yu, W. Hu and X. Zhang, J. Am. Chem. Soc. Au, 2023, 3, 1984 Search PubMed; Z. Fickenscher and E. Hey-Hawkins, Molecules, 2023, 28, 4233 CrossRef CAS PubMed; V. K. Rawat, K. Higashida and M. Sawamura, ACS Catal., 2022, 12, 8325 CrossRef; M. Sato, V. K. Rawat, K. Higashida and M. Sawamura, Chem. – Eur. J., 2023, 29, e202301917 CrossRef PubMed; T. Ishizuka, A. Hosokawa, T. Kawanishi, H. Kotani, Y. Zhi and T. Kojima, 20216016059 CrossRef PubMed; S. Singh, D. Nautiyal, F. Thetiot, N. Le Poul, T. Goswami, A. Kumar and S. Kumar, Inorg. Chem., 2021, 60, 16059 CrossRef PubMed; S. Singh, D. Nautiyal, F. Thetiot, N. Le Poul, T. Goswami, A. Kumar and S. Kumar, Inorg. Chem., 2000, 35, 3523 Search PubMed.
- A. Luengo, V. Fernández-Moreira, I. Marzo and M. Concepción Gimeno, Inorg. Chem., 2017, 56, 15159 CrossRef CAS PubMed; A. Luengo, M. Redrado, I. Marzo, V. Fernández-Moreira and M. Concepción Gimeno, Inorg. Chem., 2020, 59, 8960 CrossRef PubMed; Z. Huang and J. J. Wilson, Eur. J. Inorg. Chem., 2021, 1312 CrossRef; B. T. Elie, K. Hubbard, B. Layek, W. S. Yang, S. Prabha, J. W. Ramos and M. Contel, ACS Pharmacol. Transl. Sci., 2020, 3, 644 CrossRef PubMed; A. Jain, Coord. Chem. Rev., 2019, 401, 213067 CrossRef; L. Ma, L. Li and G. Zhu, Inorg. Chem. Front., 2022, 9, 2424 RSC; L. Ma, L. Li and G. Zhu, Inorg. Chem. Front., 2000, 35, 3523 Search PubMed.
- M. Wenzel, A. de Almeida, E. Bigaeva, P. Kavanagh, M. Picquet, P. Le Gendre, E. Bodio and A. Casini, Inorg. Chem., 2016, 55, 2544 CrossRef CAS PubMed; J. A. Platts, B. M. Kariuki and P. D. Newman, Molecules, 2024, 29, 1150 CrossRef PubMed.
- V. J. Catalano, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos and D. Pascual, Dalton Trans., 2014, 43, 16468 RSC; M. Osawa, M. Hoshino and Y. Wakatsuki, Angew. Chem., Int. Ed., 2001, 40, 3472 CrossRef CAS PubMed; H. Li, D. Lupp, P. K. Das, L. Yang, T. P. Gonçalves, M.-H. Huang, M. El Hajoui, L.-C. Liang and K.-W. Huang, ACS Catal., 2021, 11, 4071 CrossRef; H. Li, Y. Wang, Z. Lai and K.-W. Huang, ACS Catal., 2017, 7, 4446 CrossRef.
- J. Zhang and M. Rueping, Chem. Soc. Rev., 2023, 52, 4099 RSC; A. Y. Chan, A. Ghosh, J. T. Yarranton, J. Twilton, J. Jin, D. M. Arias-Rotondo, H. A. Sakai, J. K. McCusker and D. W. C. MacMillan, Science, 2023, 382, 191 CrossRef CAS PubMed; Y. Gao, L. Gao, E. Zhu, Y. Yang, M. Jie, Ji. Zhang, Z. Pan and C. Xia, Nat. Commun., 2023, 14, 7917 CrossRef PubMed; M. D. Palkowitz, M. A. Emmanuel and M. S. Oderinde, Acc. Chem. Res., 2023, 56, 2851 CrossRef PubMed; S. M. Treacy and T. Rovis, Synthesis, 2024, 56, 1967 CrossRef PubMed , A–L; L. Tian, N. A. Till, B. Kudisch, D. W. C. MacMillan and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 4555 CrossRef PubMed; A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485 CrossRef PubMed.
- X. Chen, F. J. Femia, J. W. Babich and J. Zubieta, Inorg. Chim. Acta, 2001, 315, 147 CrossRef CAS PubMed.
- U. Monkowius, M. Zabel, M. Fleck and H. Yersin, Z. Naturforsch., 2009, 64b, 1513 CrossRef.
- M. Navarro, J. Miranda-Pizarro, J. J. Moreno, C. Navarro-Gilabert, I. Fernández and J. Campos, Chem. Commun., 2021, 57, 9280 RSC; S. Y. Ho and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, m549 CrossRef CAS; A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644 CrossRef; W.-C. Liu, Y. Kim and F. P. Gabbaï, Chem. – Eur. J., 2021, 27, 6701 CrossRef PubMed; K. N. Kouroulis, S. K. Hadjikakou, N. Kourkoumelis, M. Kubicki, L. Male, M. Hursthouse, S. Skoulika, A. K. Metsios, V. Y. Tyurin, A. V. Dolganov, E. R. Milaeva and N. Hadjiliadis, Dalton Trans., 2009, 10446 RSC.
- C. Khin, A. S. K. Hashmi and F. Rominger, Eur. J. Inorg. Chem., 2010, 1063 CrossRef CAS; C. L. Carpenter-Warren, M. Cunnington, M. R. J. Elsegood, A. Kenny, A. R. Hill, C. R. Miles and M. B. Smith, Inorg. Chim. Acta, 2017, 462, 289 CrossRef; T. Traut-Johnstone, S. Kanyanda, F. H. Kriel, T. Viljoena, P. D. R. Kotze, W. E. van Zyl, J. Coates, D. J. G. Rees, M. Meyer, R. Hewer and D. B. G. Williams, J. Inorg. Biochem., 2015, 145, 108 CrossRef PubMed; J. A. Platts, B. M. Kariuki and P. D. Newman, Molecules, 2024, 29, 1150 CrossRef PubMed; Y. Inoguchi, B. Milewski-Marhia and H. Schmidbauer, Chem. Ber., 1982, 115, 3085 CrossRef.
- E. T. Luis, G. E. Ball, A. Gilbert, H. Iranmanesh, C. W. Newdick and J. E. Beves, J. Coord. Chem., 2016, 69, 1686 CrossRef CAS.
- M. Koprowski, R.-M. Sebastián, V. Maraval, M. Zablocka, V. Cadierno, B. Donnadieu, A. Igau, A.-M. Caminade and J.-P. Majoral, Organometallics, 2002, 21, 4680 CrossRef CAS.
- T. Zhang, N. Bhuvanesh and J. A. Gladysz, Eur. J. Inorg. Chem., 2017, 1017 CrossRef CAS.
- A. D. Hunter, T. R. Williams, B. M. Zarzyczny, H. W. Bottesch II, S. A. Dolan, K. A. McDowell, D. N. Thomas and C. H. Mahler, Organometallics, 2016, 35, 2701 CrossRef CAS; C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef.
- D. W. Meek and T. J. Mazanec, Acc. Chem. Res., 1981, 14, 266 CrossRef CAS.
- N. Yoshikawa, S. Yamabe, S. Sakaki, N. Kanehisa, T. Inoue and H. Takashima, J. Mol. Struct., 2015, 1094, 98 CrossRef CAS.
- CrysAlisPro, Rigaku OD, Yarnton, England, 2022–24 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra, theoretical description and figures, and crystallographic data. CCDC 2372883–2372886. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02130d |
| This journal is © The Royal Society of Chemistry 2024 |