
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of relative humidity on the molecular composition of secondary organic aerosols from α-pinene ozonolysis†
Hao
Luo‡
 abc,
Yindong
Guo‡
ab,
Hongru
Shen
ab,
Dan Dan
Huang
d,
Yijun
Zhang
abcef and
Defeng
Zhao
*abcegh
abc,
Yindong
Guo‡
ab,
Hongru
Shen
ab,
Dan Dan
Huang
d,
Yijun
Zhang
abcef and
Defeng
Zhao
*abcegh
aDepartment of Atmospheric and Oceanic Sciences & Institute of Atmospheric Sciences, Fudan University, Shanghai, 200438, China. E-mail: dfzhao@fudan.edu.cn
bShanghai Frontiers Science Center of Atmosphere-Ocean Interaction, Fudan University, Shanghai, 200438, China
cShanghai Key Laboratory of Ocean-land-atmosphere Boundary Dynamics and Climate Change, Fudan University, Shanghai, 200438, China
dState Environmental Protection Key Laboratory of Formation and Prevention of Urban Air Pollution Complex, Shanghai Academy of Environment Sciences, Shanghai, 200233, China
eNational Observations and Research Station for Wetland Ecosystems of the Yangtze Estuary, Fudan University, Shanghai, 200438, China
fShanghai Institute of Pollution Control and Ecological Security, Shanghai, 200092, China
gInstitute of Eco-Chongming (IEC), 20 Cuiniao Rd., Chongming, Shanghai, 202162, China
hKey Laboratory of Polar Atmosphere-ocean-ice System for Weather and Climate, Ministry of Education, Fudan University, Shanghai, 200438, China
First published on 9th April 2024
Abstract
Secondary organic aerosols (SOAs) originating from the oxidation of biogenic volatile organic compounds such as monoterpenes by atmospheric oxidants (e.g. OH, ozone, and NO3), constitute a widespread source of organic aerosols in the atmosphere. Among monoterpenes, α-pinene has the highest emission rates and its ozonolysis is often used as a canonical SOA system. However, the molecular composition of SOAs obtained from monoterpene ozonolysis as a function of relative humidity (RH) remains unclear. Herein, we investigated the real-time molecular composition of SOAs obtained from the ozonolysis of α-pinene using extractive electrospray ionization coupled with long time-of-flight mass spectrometry (EESI-LTOF-MS). We investigated the dependence of the molecular composition on RH in the presence and absence of seed aerosols. We characterized a large number of organic compounds, including less oxygenated and highly oxygenated organic molecules (HOMs). In the presence of a ammonium sulfate (AS) seed aerosol, the fractions of both monomers and dimers in the SOAs from α-pinene ozonolysis remained largely unchanged as RH increased from 3% to 84%, which can be attributed to a similar extent of increase in the absolute abundance of both dimers and monomers with increasing RH. The increase of the absolute abundance of monomers is likely due to the enhanced partitioning of less oxygenated semi-volatile monomer products (such as C10H16Ox≤6) at higher RH. The increase in the absolute abundance of dimers may be attributed to acid-catalyzed reactions, which is corroborated by a marked change in the distribution pattern of dimers. The average O/C of the most abundant product families in the SOAs, such as C10H16Ox, decreased with increasing RH due to the decreasing fractions of more oxygenated products (C10H16Ox>6). However, the elemental composition (O/C and H/C) of the total SOA remained stable with increasing RH. In contrast, in the absence of the seed aerosol, an increase in the monomer fraction and a decrease in the dimer fraction were observed with increasing RH. These changes were attributed to a combination of different extents of condensation enhancement of monomer and dimer vapors by increasing RH and different vapor wall losses of monomers and dimers. Our results provide new insights into the RH-dependent molecular chemical composition of α-pinene SOAs. We also highlight the necessity to characterize the composition of SOAs at the molecular level.
Environmental significanceSecondary organic aerosol (SOAs) formed from monoterpene oxidation has an important effect in climate change and human health. Relative humidity (RH) in the atmosphere is an important environmental factor that can affect SOA composition, which further affects volatility and SOA yield. However, the RH-dependence of the chemical composition of SOAs from monoterpene ozonolysis is still unclear. We show how the molecular composition of SOAs formed in α-pinene ozonolysis, a canonical biogenic oxidation system, depends on RH and how the elemental composition (O/C, H/C) remains largely unaffected. This work emphasizes the necessity to characterize the composition at the molecular level. RH-dependent SOA composition highlights the necessity to consider the influence of RH in atmospheric numerical models to improve the prediction of SOA concentration and composition. |
1 Introduction
Secondary organic aerosols (SOAs) play a critical role in climate change and human health. Biogenic SOAs formed through the atmospheric oxidation of biogenic volatile organic compounds (BVOCs), particularly isoprene and monoterpenes, contribute largely to SOAs.1–3 Among monoterpenes, mostly from terrestrial vegetation, α-pinene ranks the highest in global emission rates, which can be oxidized by OH, NO3, and O3. These oxidation processes lead to the formation of various products that partition between the gas and particle phase,4–9 ultimately resulting in SOA formation.α-Pinene ozonolysis is a canonical SOA system.5,10,11 Our understanding of SOA formation in α-pinene ozonolysis has improved substantially in the past two decades.4,10,12–19 In particular, the role of highly oxygenated organic molecules (HOMs) with low or extremely low volatile organic compounds (LVOCs or ELVOCs) in the formation of SOAs has been recognized,20–22 which was first observed in the α-pinene ozonolysis system and later in various oxidation systems.7–9,23,24 Previous studies have measured the elemental composition25,26 and identified multifunctional particle-phase products, including monomers with carboxylic acid groups and high-molecular-weight compounds.5,10,11,27–29 However, the molecular composition of SOAs formed in α-pinene ozonolysis and its dependence on environmental factors remain elusive.
Particularly, the effect of the relative humidity (RH) on the molecular composition of SOA from α-pinene ozonolysis is still unclear. Although RH theoretically affects gas-particle partitioning and leads to different gas- and particle-phase chemistry, as shown in other reaction systems and modeled by a number of previous studies,30–38 previous studies have reported different and even contradictory findings regarding whether RH influences the composition of SOAs from α-pinene ozonolysis. For example, Qin et al.30 found that neither the ion families in the mass spectra nor the atomic ratios of various elements of SOAs formed in α-pinene ozonolysis changes substantially with increasing RH. In contrast, Zhang et al.10 observed that the average molecular weight of α-pinene ozonolysis SOA is lower under higher RH (55% versus <5% RH). Kidd et al.39 also reported that the viscosity of SOA decreases with increasing RH, and carboxylic acids increase with increasing RH, suggesting that the composition of SOA is influenced by RH. Caudillo et al.40 found that the distribution of particle phase products in the α-pinene ozonolysis at 223 K at high RH is quite different from that at low RH. Recently, Surdu et al.33 observed an increase in some less oxygenated compounds, such as C10H16O2–4, with increasing RH in the absence of seed aerosol at both 243 and 263 K. This increase results in an increase of the SOA mass at higher RH, and is attributed to the enhanced partitioning of these less oxygenated semi-volatile organic compounds (SVOCs) into the particle phase. In fact, even if RH does not alter the bulk elemental composition and SOA mass concentration, RH may still affect the specific composition of SOA.10,27 This discrepancy in the literature highlights the need for further investigation into the effects of RH on both the detailed molecular composition and into the underlying mechanism.
In the present study, we investigated the RH dependence of the SOA molecular composition from α-pinene ozonolysis using online mass spectrometry, and discussed the underlying mechanisms and roles of particle-phase chemistry. We also compared the influence of the presence and absence of ammonium sulfate (AS) seed aerosol on the RH dependence of the SOA molecular composition.
2 Materials and methods
2.1 Flow tube setup and experiments
SOA in α-pinene ozonolysis was formed in a homemade 1.8 m Pyrex glass flow tube (Fig. 1). The temperature of the flow tube was maintained at 298 K by water bath (RH25-12A, Labtech). The flow tube has an inner diameter of 20 cm and a volume of 62.8 L. The typical flow rates used were 2.85–3.00 LPM. The residence time in the flow tube for the experiments in this study was ∼14.5 min, which could be adjusted by a movable sampling inlet. The detailed experimental conditions are presented in Table S1.† α-Pinene (∼160 ppb, ≥99%, Sigma-Aldrich) was oxidized by O3 (∼480 ppb) in the absence and presence of ammonium sulfate (AS) seed aerosol (∼3 × 104 μm2 cm−3). O3 was generated by passing zero air through an ultraviolet ozone generator (UVP, 97-0067-02), and subsequently measured using an ozone analyzer (Model 49i, Thermo Scientific) in each experiment. The VOC vapor was generated consistently using a diffusion technique,41 and its concentration was measured through a weighing method. In brief, the volatile organic compounds were placed in a cylindrical diffusion vial with a volume of 4 ml, covered with a headspace cap and penetrated with a PEEK tube. The diffusion vial was placed in a glass bottle, which was temperature-controlled with a water bath. Zero air entered the diffusion bottle from the bottom through the air inlet, flowed upward through the diffusion vial and exited at the top of the glass bottle, finally into the flow tube. Such a device has been used in previous studies.42,43 AS seed aerosols were generated using a constant-rate atomizer (3076, TSI) by atomizing AS solution, and subsequently dried by a Nafion dryer (Perma Pure). The RH was gradually increased ranging from (3 ± 1)% to (84 ± 3)% in the presence of AS seed, and from <1% to (91 ± 2)% in the absence of seed. The RH was controlled by adjusting the flow ratio of dry air from a zero air generator (747-30, AADCO) with <1% RH and humid zero air humidified using a Nafion tube with ultrapure water (18.2 MΩ cm, MilliQ). The RH and temperature were continuously monitored by a temperature and humidity sensor (HMT333, Vaisala). All experiments in this study were performed in the presence of an excessive amount of 2-propanol, which served as an OH scavenger so that >90% of OH was scavenged by 2-propanol. Before each experiment, the flow tube was flushed with dry zero air. Then, the flows with humid air, with O3, and with AS seed aerosol (if added) entered the dark flow tube. VOC vapors mixed with dry zero air were subsequently added into the flow tube. Particle concentrations and the RH in the flow tube both reached a steady state after ∼60 min once all conditions were set.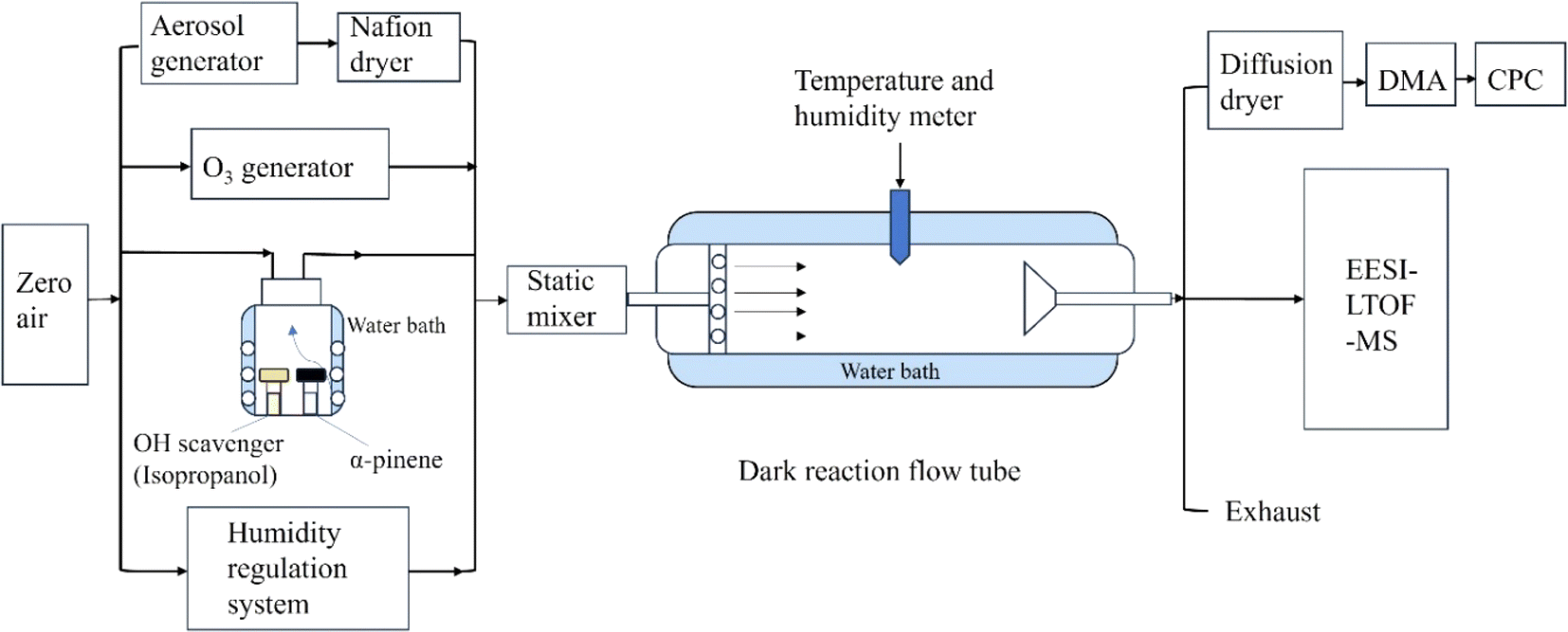 | ||
| Fig. 1 The schematic diagram of the flow tube system used in this study. | ||
2.2 Instrumentation and data processing
The chemical components of α-pinene ozonolysis SOA were characterized online by an Extractive Electrospray Ionization inlet coupled with a Long Time-of-Flight Mass Spectrometer (EESI-LTOF-MS, Aerodyne) with a mass resolution of ∼8500. The details of the instrument have been previously described by Lopez-Hilfiker et al.44 In brief, particles and gases are sampled continuously through a charcoal denuder, which can remove most volatile gas species efficiently. After the denuder, the particle flow intersects a charged droplet spray, and then collides with the electrosprayed droplets. Soluble components in particles are extracted by solvents from charged droplets, and then the droplets evaporate through a heated stainless-steel capillary, finally yielding charged aerosol ions that are detected by a time-of-flight mass spectrometer. The electrospray working solution was 100 ppm NaI in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water (MilliQ):acetonitrile (UHPLC-MS grade, Sigma-Aldrich) mixture, enabling the organics detected as [M + Na+]. Particles were sampled at a flow rate of ∼0.9 L min−1. An auto-valve was used to enable automatic alternates between direct sampling particles and sampling particle-free air through a HEPA filter (AQ, Parker). The durations of the direct sampling particles and filter sampling are 8 min and 7 min, respectively. The difference of the spectra between direct sampling and sampling through the filter was used to derive the particle signal devoid of any instrument background signal (including residue gases passing through the denuder and impurities from the spraying solution). Mass spectra were recorded at a frequency of 0.2 Hz. The chemical composition of SOA at each RH was measured and averaged for two 8 min periods after the particle concentrations became stable for each experimental condition. Mass spectral data were analyzed using Tofware v3.2.3. A total of 145 compounds were identified, including less oxygenated and highly oxygenated organic molecules (Table S2†). The paired sample T-test was used to examine whether the dimer distribution at two different RH is significantly different. A Scanning Mobility Particle Sizer (SMPS 3938, TSI) was used to monitor the particle size distribution.
:1 water (MilliQ):acetonitrile (UHPLC-MS grade, Sigma-Aldrich) mixture, enabling the organics detected as [M + Na+]. Particles were sampled at a flow rate of ∼0.9 L min−1. An auto-valve was used to enable automatic alternates between direct sampling particles and sampling particle-free air through a HEPA filter (AQ, Parker). The durations of the direct sampling particles and filter sampling are 8 min and 7 min, respectively. The difference of the spectra between direct sampling and sampling through the filter was used to derive the particle signal devoid of any instrument background signal (including residue gases passing through the denuder and impurities from the spraying solution). Mass spectra were recorded at a frequency of 0.2 Hz. The chemical composition of SOA at each RH was measured and averaged for two 8 min periods after the particle concentrations became stable for each experimental condition. Mass spectral data were analyzed using Tofware v3.2.3. A total of 145 compounds were identified, including less oxygenated and highly oxygenated organic molecules (Table S2†). The paired sample T-test was used to examine whether the dimer distribution at two different RH is significantly different. A Scanning Mobility Particle Sizer (SMPS 3938, TSI) was used to monitor the particle size distribution.
3 Results and discussion
3.1 RH dependence of the SOA composition in the presence of the AS seed aerosol
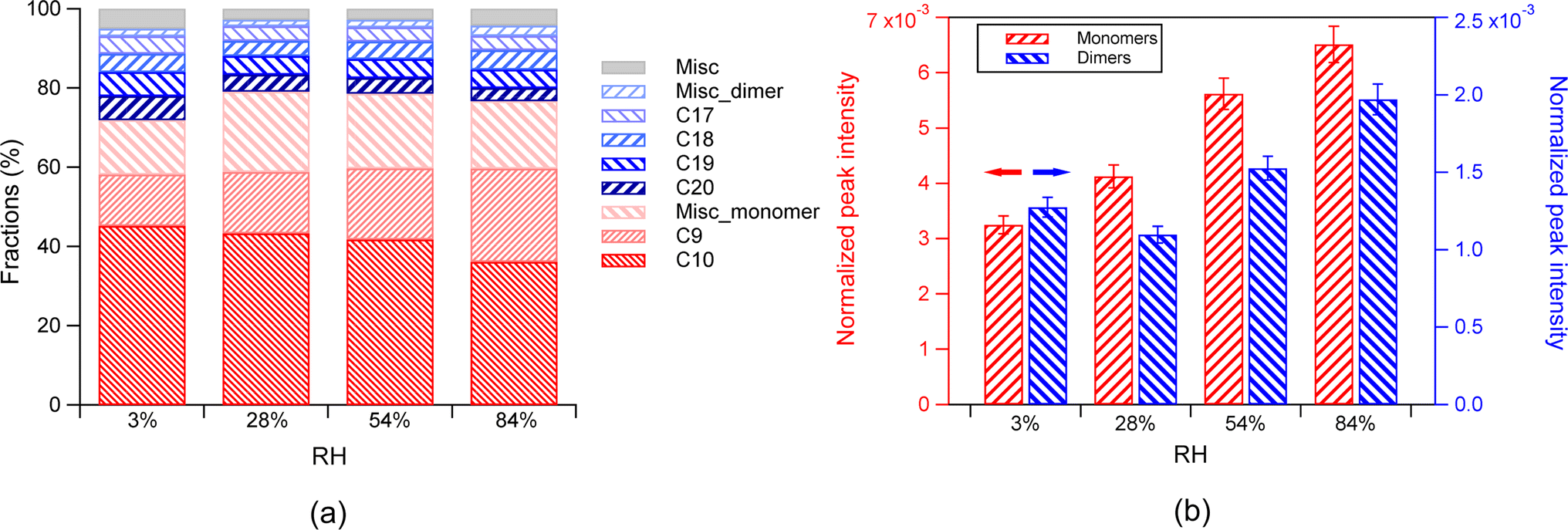 | ||
| Fig. 2 (a) The fractions of monomers and dimers in SOAs in the presence of the AS seed aerosol at different RH ((3 ± 1)%, (28 ± 2)%, (54 ± 2)%, and (84 ± 3)%). The fraction of each class is normalized to the total products. Misc_monomer denotes other monomers, except for the C9 and C10 monomers. Misc_dimer denotes other dimers, except for the C17-20 dimers. Misc denotes the other SOA components, which are not identified. (b) The absolute intensity of total monomers (in red, left y-axis) and total dimers (in blue, right y-axis) in the presence of the AS seed aerosol under different RH. The error bar represents 1σ. Note that the peak intensity in mass spectra was normalized to the peak intensity of NaINa+ (m/z 173), which is the most abundant peak from ion source. | ||
Despite the general invariant fractions of monomers and dimers with increasing RH, the absolute abundance of total monomers increased with increasing RH. The absolute abundance of total dimers also increased with increasing RH in general, despite a slight decrease from 3% to 28% RH (Fig. 2b). The difference of the absolute abundance between different RH was statistically significant (p values between any two adjacent RH conditions from the T-test are below 0.01). The dependence of the fractions of monomers and dimers on RH is determined by the dependence of their absolute abundance on RH. For a change of RH from 3% to 84%, the absolute abundance of monomers increased by a factor of 2.9 (Fig. S2(b and d)†), and the dimers increased with a comparable factor (3.3). These changes finally led to a largely stable fraction with increasing RH both in the total monomers and dimers (Fig. 2b). We would like to note that while the normalized dimer abundance is not completely monotonic as a function of RH, it generally shows an increasing trend with RH. We compare the abundance at the lowest and highest RH in order to quantitatively describe the extent of changes of abundance as a function of RH.
The influence of RH on the chemical composition of SOA from α-pinene ozonolysis can be attributed to either the influence on gas-phase reactions or multi-phase processes. Although RH can affect the gas-phase products of α-pinene ozonolysis by the reaction of water with the Criegee radical,19,45 Li et al.23 reported that the concentrations of all main HOM (monomers and dimers) did not change in the RH ranging from 3% to 92% at 293 K. Caudillo et al.40 also reported that the RH range from 20% to 60% does not have a significant influence on the distribution of HOM products at 223 K. Therefore, HOM in the gas phase is unlikely to be affected by RH. As HOM contributes to most of the SOA from α-pinene ozonolysis,22 the influence of RH on the SOA composition here is likely attributed to the influence on multi-phase processes, such as particle-phase reactions and gas–particle partitioning of organics.46,47
The increase in the absolute abundance of particle-phase monomers is likely attributed to their decreasing activity and increasing bulk-phase diffusivity with increasing RH, as reported by Surdu et al.,33 who found a similar RH dependence of the absolute abundance of C10H16O2–8 in the particle phase at 243 K.33 Specifically, increasing RH can promote the particle water content. A higher particle water content leads to higher water activity in the water–organic mixture. This can reduce the mole fraction and thereby the activity of these organic compounds in the particle phase, and thus ultimately reduce the equilibrated gas-phase concentration of condensable vapors. Therefore, the gas–particle equilibrium undergoes a shift towards condensation at higher RH. Increasing RH can also enhance the bulk diffusivity and further enhance the partitioning of organics. In addition, heterogeneous reactive uptake may play a role in the enhancement of monomers and dimers by RH. Previous studies have reported that water-soluble organics, such as glyoxal and methylglyoxal, undergo reactive uptake and subsequently contribute to the increase in the SOA mass at high RH.48,49 In this study, the significant increase in monomers with increasing RH may also be contributed by the enhanced reactive uptake and subsequent reactions of hydrophilic compounds.10
In contrast to monomers, the increase in the absolute abundance of particle-phase dimers with increasing RH is unlikely to be explained to the decrease in their activity and increase in bulk-phase diffusivity with increasing RH. Dimers generally have much lower volatility than monomers. Thus, most of them are expected to reside in the particle-phase, and be less sensitive to partitioning. In this study, the enhancement of dimers with increasing RH may be instead contributed by particle-phase reactions. Such a speculation is substantiated by the observation that the distribution of the dimer species exhibited distinct patterns between the dry condition (3% RH) and the other three RH conditions (Fig. 3, Table S3†). Specifically, the fractions of C20 generally decrease and the fractions of C16–19 with low oxygen number generally increase, indicating a change in the dimer composition despite the largely invariant total fractions of dimers and monomers. This observation suggests that there were particle-phase reactions forming dimers, which was influenced by RH. Previous studies have shown that the presence of the AS seed aerosol can enhance the aerosol acidity, and the acid-catalyzed reactions may facilitate the formation of dimers.50–55 Acid-catalyzed reactions have been shown to play a crucial role in the formation of SOA,50 particularly in the case of oligomers in SOA from α-pinene ozonolysis.51–53,56 For example, Du et al. found that neutralization of the acidity of H2SO4 by NH3 resulted in the suppression of oligomer formation in the SOA by β-caryophyllene ozonolysis, indicating that the oligomer formation is acid-catalyzed.54 Such an acid-catalyzed process may result in the increase in dimer abundance with increasing RH in this study. In addition, multiphase reactions may play a role in the increase of dimers with increasing RH.
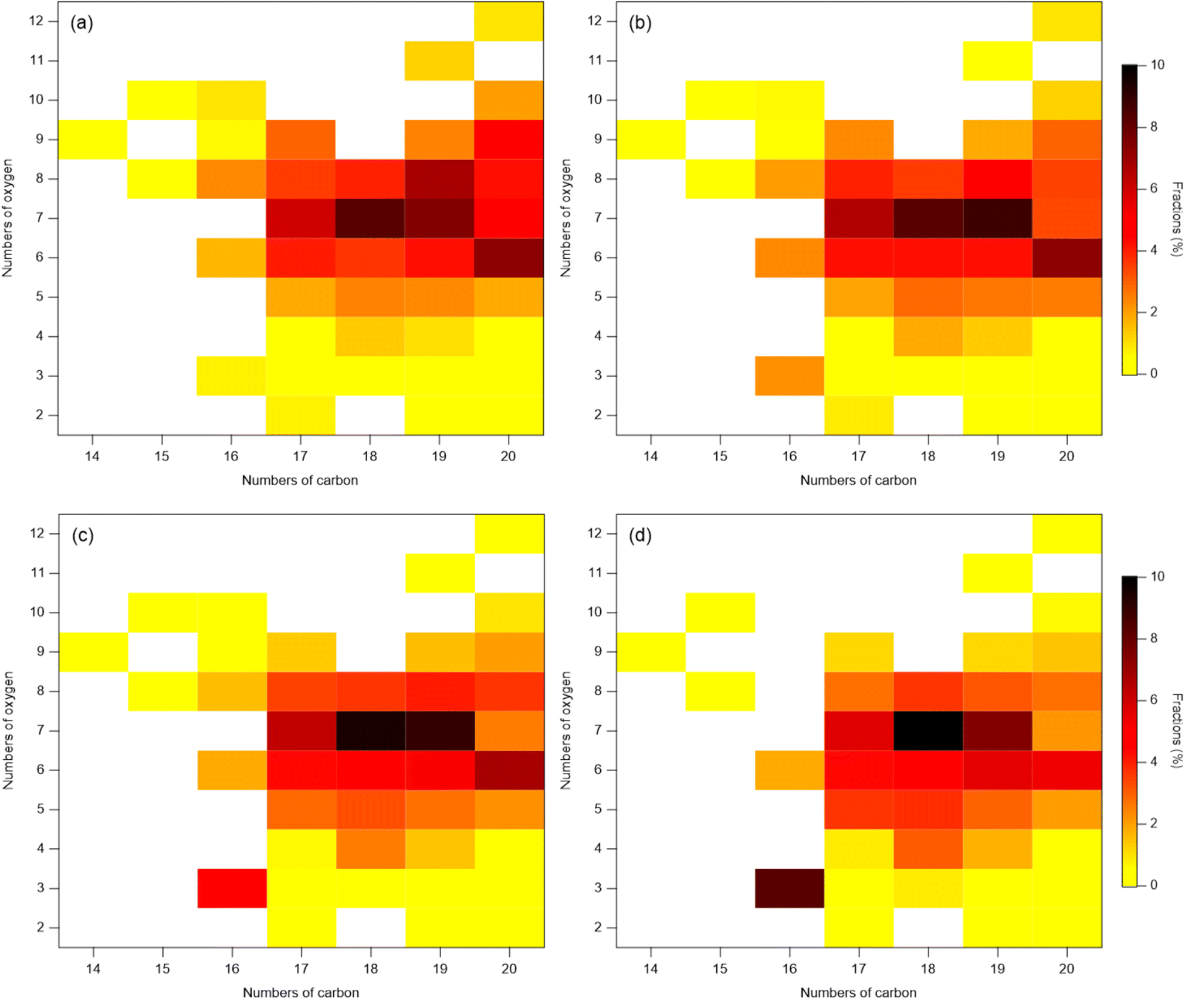 | ||
| Fig. 3 Distribution of dimers in SOA formed in the presence of AS seeds during α-pinene ozonolysis under different RH: (a) (3 ± 1)%, (b) (28 ± 2)%, (c) (54 ± 2)%, and (d) (84 ± 3)%. Color bar shows the fractions of each class of compounds in total dimers presented as a function of the number of oxygen and carbon atoms in the molecules. Each data point is the sum over ions with a different number of hydrogen atoms. | ||
The dependence of the dimer abundance on RH is in contrast with the study by Surdu et al.,33 who reported that the absolute abundance of dimers in SOA formed in α-pinene ozonolysis at 243 K remains unchanged with increasing RH. The discrepancy between our study and that of Surdu et al. may be attributed to the disparity in the temperature employed. While 298 K was used in this study, a much lower temperature (243 K/263 K) was used by Surdu et al.33 At low temperature, particle-phase reactions between monomers forming dimers occur at a slower rate due to reduced rate constants and particle-phase diffusivity. Therefore, the enhancement of dimer formation by RH at low temperature is expected to be less significant compared to that at high temperature. Moreover, the HOM formed at low temperature are less oxygenated,57 which may result in reduced reactivity for dimer formation in the particle-phase due to less functional groups in these HOM. Consequently, these HOM are not enhanced by RH at low temperature.
Among monomers, both the absolute abundance of total C10 species and C9 species increased with increasing RH (Fig. S3b†). As RH increased from 3% to 84%, the increase in the total C9 monomers (7 times) is much higher than the increase in the total products (0.9 times), including both monomers and dimers, while the increase of the C10 species (0.5 times) was lower than that of the total products. This phenomenon resulted in a general increase in the C9 fraction and a general decrease in the C10 fraction. The increase of the C9 species with increasing RH can be attributed to the enhanced partitioning at higher RH, as discussed above. The smaller increase of the absolute abundance of C10 than the total products may be attributed to their larger reactive loss from forming dimers than C9. The amount of C20 dimers, which are likely formed from the C10 compounds, increased more than that of the C18 dimer, which is likely formed from the C9 compounds. Additionally, we still cannot rule out that the increase in monomers can be attributed in part to the decomposition of some dimers, even if the absolute abundance of total dimers increased with increasing RH. For example, the increase of C9 can be caused by the decomposition of dimers.29 Yao et al. found that the decomposition rates of most dimer peroxides increases with increasing RH, while the fraction of dimers in products decreased with increasing RH, suggesting that water acts as a plasticizer.15 We would like to note that the reaction time in this study is significantly shorter (<20 min) compared to the studies by Yao et al.15 (one to several hours) and Pospisilova et al.29 (∼120 min). Thus, the decomposition of dimers is expected to be much less significant than that in those studies.
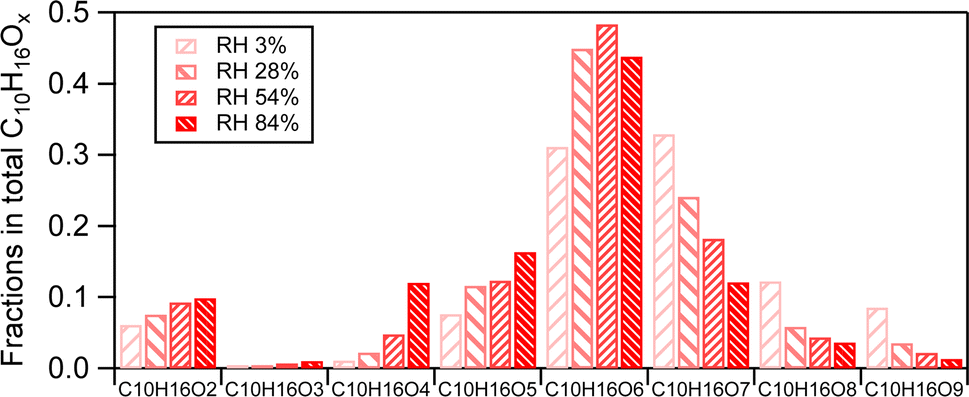 | ||
| Fig. 4 The fractions of individual C10H16Ox(x=2–9) compounds in the family under different RH in the presence of the AS seed aerosol. The fraction of each C10H16Ox is normalized to the total intensity of the C10H16Ox family. The gradual color gradient from light to deep represents the RH range from (3 ± 1)%, (28 ± 2)%, (54 ± 2)%, and (84 ± 3)%. | ||
The changes in fractions of individual C10H16Ox(x=2–9) compounds within their family can be attributed to changes in their absolute abundance and in the total compounds in SOA. The increase of the absolute abundance of C10H16Ox≤6 with increasing RH was more pronounced than that of C10H16Ox>6 (Fig. S2†). This is attributed to the higher volatility of C10H16Ox≤6 than C10H16Ox>6. As discussed above, RH can enhance the partitioning of more volatile products than less volatile products. In this study, the C10H16Ox≤6 products are classified as a semi-volatile compound class (logC*(300K) (C10H16Ox≤6) ranges from 0.13 to 2.3), and the C10H16Ox>6 products are classified as LVOCs (logC*(300K) ranges from −3.2 to −0.8) based on the method to estimate volatility by previous studies.33,58 Less oxygenated C10H16Ox are more volatile than more oxygenated C10H16Ox. Therefore, the enhancement of C10H16Ox≤6 due to gas-particle partitioning by increasing RH was higher than that of C10H16Ox>6. This is similar to the rationale of the larger enhancement of monomers compared to dimers by RH, which is further discussed in Section 3.2.1.
We would like to note that in our study, the influence of vapor wall losses, i.e., vapor deposited on the reactor walls, on the particle formation for C10H16Ox≤6 products is insignificant as RH increases due to the much larger timescale of the vapor–wall equilibration (τv,w) compared to that of vapor directly condensing on particles (vapor–particle, (τv,p)) (Fig. S5†). These time scales were calculated according to the method used by Huang et al. (2016) and references therein, and more details of the timescale estimates are provided in the ESI†). Our finding is similar to the finding of Huang et al.,59 who found that the condensation of SVOCs onto suspended particles remains unaffected by the initial seed surface area. Similarly, with regard to the C10H16Ox>6 products, which belong to LVOCs, the condensation onto the suspended particle from vapor wall losses can also be ignored (Fig. S5†).
Our finding of the increasing abundance of C10H16Ox≤6 with rising RH agrees with a recent study by Surdu et al., who reported that C10H16Ox=2–3 in the particle phase increased by a factor of 1.5 to 2 when RH rises from 20% RH to 60% RH at 263 K.33 However, C10H16Ox>6 remained constant in the study of Surdu et al.,33 which is different from our finding. This disparity between their study and this study may be attributed to the difference in temperature, as we discussed in Section 3.1.1. As C10H16Ox>6 are lower volatility organic compounds at low temperature (263 K) and mostly reside in the particle-phase similar to dimers, the influence of RH on their gas–particle partitioning is very limited.60
The stable O/C ratio of total SOA products with increasing RH is consistent with a number of previous studies. For example, Qin et al. observed a stable O/C ratio of 0.52 ± 0.02 in α-pinene ozonolysis SOA, regardless of the RH changes.30 Zhang et al. found that the O/C and H/C of SOA formed in α-pinene ozonolysis, measured by aerosol mass spectrometer (AMS), is independent of RH,10 and the O/C of α-pinene SOA remains stable in the range of 0.45 to 0.50 despite a RH increase from 5% to 50%. Moreover, the range of O/C in our study is similar to the previous studies.10,30,33 However, our study is in contrast with the study by Surdu et al. that reported the O/C of SOA formed in α-pinene ozonolysis at 263 K decreased from 0.55 to 0.40 with RH increasing from 10% to 80%.33 The discrepancy may also be attributed to the difference in the reaction temperature. As mentioned above, the product composition of various families varied with RH, which compensated with each other in our study. In the study of Surdu et al.,33 which was conducted at 243 K, C10H16Ox is the most dominant product. The absolute abundance of other products (such as dimers) does not change significantly with increasing RH. Therefore, the change O/C of SOA with increasing RH is largely determined by the dependence of individual C10H16Ox on RH. At lower temperature, high RH makes more semi-volatile or intermediate volatile organic compounds (S/IVOC) condense to particles. Thus, C10H16O2–3 increases with increasing RH. In contrast, the C10H16Ox=6–8 are classified as the low volatility organic compounds, and their absolute abundance remains almost constant. Overall, these trends finally lead to a decrease in the O/C ratio of total SOA with increasing RH in the study of Surdu et al.33
3.2 RH dependence of the SOA composition in the absence of seed aerosol
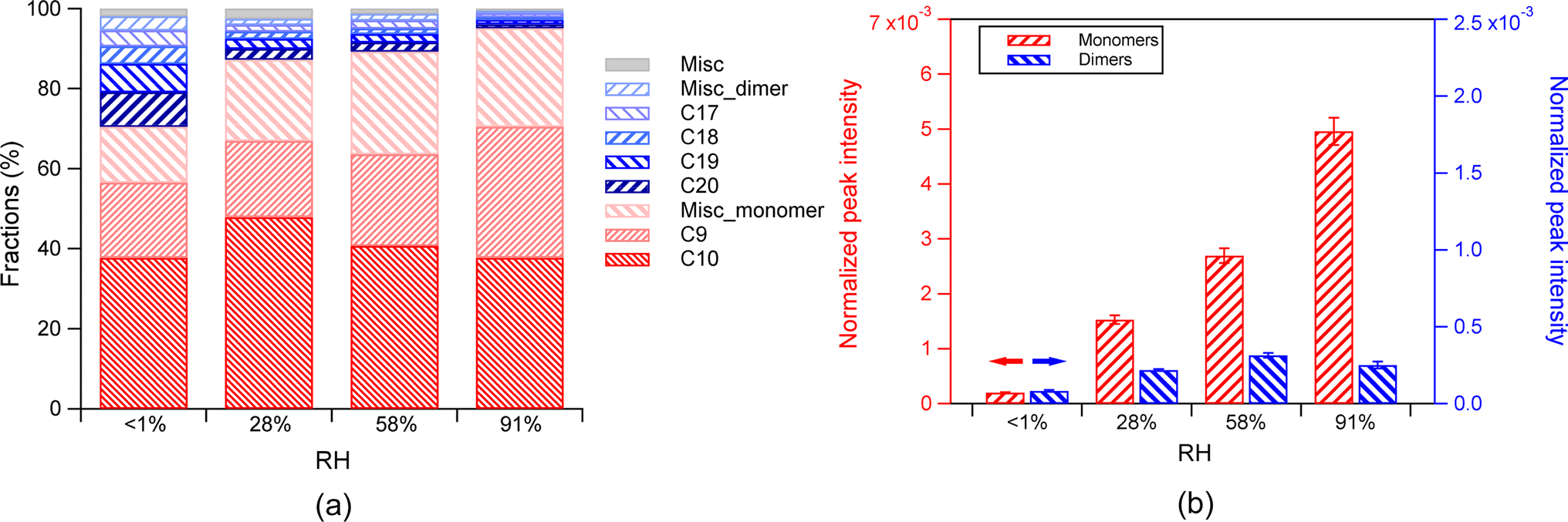 | ||
| Fig. 5 (a) The fractions of monomers and dimers in the SOA in the absence of the AS seed aerosol at different RH (<1%, (28 ± 1)%, (58 ± 2)%, and (91 ± 2)%). The fraction of each class is normalized to the total products. Misc_monomer denotes other monomers, except for the C9 and C10 monomers. Misc_dimer denotes other dimers, except for the C17–20 dimers. Misc denotes the other SOA components, which are not identified. (b) The absolute intensity of the total monomers (in red, left y-axis) and total dimers (in blue, right y axis) in the absence of the AS seed aerosol under different RH. The error bar represents 1σ. Note that the peak intensity in the mass spectra was normalized to the peak intensity of NaINa+ (m/z 173), which is the most abundant peak from the ion source. | ||
The dependence of the fractions of monomers and dimers on RH was attributed to the fact that the absolute abundance of monomers increased by 24 times, and that of dimers increased by 2 times from <1% RH to 91% RH. The enhancement of monomers can be attributed to the enhanced partitioning by RH, as discussed above in Section 3.1.1. The larger extent of increase in monomers compared to dimers with increasing RH can be attributed to their different volatility and thus their different sensitivity to gas–particle partitioning, as discussed above.
However, the increase of monomer abundance with increasing RH was much stronger in the absence of seed aerosol than that in the presence of the AS seed aerosol. This difference can be attributed to vapor wall loss, i.e., the equilibration shift between vapor-particle and vapor-wall competition, as reported by Huang et al.59 The particle loading was generally lower in the absence of seed aerosol than that in the presence of the AS seed aerosol. The estimated timescale of the vapor–particle (τv,p) equilibration is longer or similar to that of vapor–wall equilibration (Fig. S5†). The surface area concentrations increased with increasing RH in the absence of seed aerosol (Fig. S1d†), which can lead to enhanced condensation of organic vapors onto particles at higher RH. Therefore, the influence of the competition between the vapor–particle partitioning and vapor–wall partitioning on the monomer abundance in particles could not be neglected. Furthermore, the increase in the absolute abundance of monomers was partly contributed by favored condensation of vapors onto particles at higher RH that would otherwise deposit onto the reactor walls, in addition to the enhanced condensation by RH, particularly for low RH conditions in the experiments with the absence of seed particles.
Overall, the vapor wall loss and the enhanced condensation of organic vapors at higher RH resulted in an increase of monomer fraction and a decrease in dimer fraction in the absence of seed aerosol.
The increase of C10H16Ox(x<6) with increasing RH can be attributed to the enhanced condensation by RH and/or vapor wall loss, as discussed above (Sections 3.1.2 and 3.2.1). Comparing the estimated timescales for vapor–particle and vapor–wall equilibration, specifically for C10H16Ox≤6 (represented by C10H16O3, Fig. S5†), we see that the vapor wall loss is likely to be most significant within the RH range of <1% to 28%. This is also true for C10H16Ox>6 and dimers. However, the vapor wall loss should only contribute minimally to the increase of C10H16Ox≤6, as the RH increased from 28% to 91% because the timescale of vapor-particle equilibration is much shorter than that of the vapor–wall equilibration in that RH range (Fig. S5†). Therefore, the increase in C10H16Ox≤6 at the RH range from 28% to 91% is likely contributed mostly by RH via enhancing the gas–particle partitioning, as discussed in Section 3.1.1. Regarding C10H16Ox>6, their absolute abundance increased and reached a plateau (Fig. S2a†) at >58% RH. We cannot clearly distinguish whether the vapor wall loss or the enhanced partitioning by RH is more important in the increase of C10H16Ox>6 with increasing RH. This is because the timescales of vapor–particle equilibrium and vapor-wall equilibrium of LVOCs represented by C10H16O9 were comparable. The plateau above >58% RH might be because the vapor wall losses at 58% RH and 91% RH were similar as the particle surface area concentration and thus timescales of vapor–particle equilibrium versus vapor–wall equilibrium of C10H16Ox>6 were similar. Overall, the fact that less oxygenated C10H16Ox increased more sharply with increasing RH than more oxygenated C10H16Ox is more likely attributed to the lower sensitivity of the lower volatile compounds to the enhanced condensation by RH rather than the vapor wall loss. Otherwise, more oxygenated compounds would increase more with increasing RH than less oxygenated compounds.
For low volatility compounds, such as C19H28O7–11, the abundance also increased largely as a function of RH (Fig. S7†). This dependence on RH could be attributed to the vapor wall loss or the enhanced condensation by RH. If the dependence on RH resulted from the enhanced condensation by RH, it is expected to be more pronounced for compounds with higher volatility. In contrast, if the dependence resulted from the vapor wall loss, it is expected to be more pronounced with lower volatility. We found that the increment of the absolute abundance of C19H28Ox=7–11 with increasing RH (from 28% RH to 91% RH) is higher than that of C10H16Ox=7–9 (Fig. S8†). As C10H16Ox=7–9 has higher volatility than C19H28Ox=7–11, this finding suggests that for these low volatility compounds (LVOCs, represented by C10H16O9 or ELVOCs, represented by C19H28O9), the condensation from vapor deposited onto the wall may be more important than RH enhancement for promoting the abundance in the absence of seed aerosol.
4 Conclusion
In this study, we investigated the RH dependence of the molecular composition of SOA formed in α-pinene ozonolysis in the absence and presence of ammonium sulfate seed aerosol. The fractions of both monomers and dimers in SOA formed in the presence of AS seed aerosol remained largely unchanged with increasing RH, which is due to their similar extent of increase in their absolute abundance with increasing RH. The increase in the absolute abundance of monomers is likely attributed to the increase in bulk diffusivity and enhanced partitioning of less oxygenated semi-volatile monomer products (such as C10H16Ox≤6) with increasing RH. The increase of dimers with increasing RH is likely due to acid-catalyzed reactions, which is supported by the fact that distribution of dimers changed markedly with increasing RH. The RH dependence of SOA composition resulted in a slight decrease in the overall molecular weight of SOA with increasing RH. The average O/C of the most abundant product family, C10H16Ox, decreased with increasing RH. This is attributed to the more pronounced increase in less oxygenated compounds (C10H16Ox≤6) compared to more oxygenated compounds (C10H16Ox>6). Despite the changes in the O/C within the most abundant families, the O/C of the total SOA remained largely invariant with increasing RH, which is attributed to the counteracting trend of various families with increasing RH in altering the overall O/C of SOA.In the absence of seed aerosol, we observed an increase in the monomer fraction and a decrease in the dimer fraction from <1% RH to 91% RH as a result of the more pronounced increase in the absolute abundance of monomers with increasing RH than dimers. Moreover, a significant decrease in the overall molecular weight of SOA with increasing RH was observed. The dependence of SOA components on RH is attributed to enhanced condensation by higher RH (for C10H16Ox≤6) and/or to vapor wall loss (for C10H16Ox>6 and dimers), i.e., the equilibration shift between vapor–particle and vapor–wall competition for the compounds with lower volatility. This finding is based on the comparison of the timescale of the condensation from the vapor–wall to the suspended particles with that from the direct vapor condensation onto the particle, and different responses of compounds with different volatility to increasing RH.
Admittedly, there are still some limitations in this study. The detailed dimer formation mechanism of particle-phase reactions between C10 compounds are still unclear, and the reaction mechanism need further study. The reason for the RH dependence of C10H14Ox compounds lacks direct evidence. Furthermore, the relative importance of the vapor wall loss and the enhanced partitioning by RH in the increase of C10H16Ox>6 with increasing RH in the absence of seed aerosol cannot be clearly distinguished. In addition, although the RH dependence of the SOA composition was likely attributed to multiphase processes rather than the influence of RH on the gas-phase HOM, the gas-phase HOM was not directly measured due to limited instrumentation. Previous studies reported that HOM from α-pinene is independent of RH.23,40 Yet, in the ozonolysis of other monoterpenes, e.g., limonene compared with Δ3-carene, RH-dependent product distribution in the gas-phase has also been reported to play a vital role in the composition and yield of SOA.62 Therefore, more mechanistic studies are warranted in the future.
As a canonical biogenic oxidation system, α-pinene ozonolysis in the atmosphere takes place under varying RH. The RH-dependent chemical composition of SOA can further affect its physico-chemical properties. For example, the viscosity39 and volatility63 of α-pinene ozonolysis SOA have been found to be influenced by RH. The RH-dependent chemical composition may also play a role in SOA mass concentration, as the molecular level composition affects volatility and SOA yield. However, the effect of RH on SOA yields remains inconclusive.10,17,18,62,64,65 The RH-dependent chemical composition can also influence the CCN activity of SOA. The decrease of the average molecular weight with increasing RH may enhance the hygroscopic parameter κ, which is inversely proportional to the molar volume of solute, and thus to the molecular weight (provided that the density of different organics is largely similar).66–68 Moreover, the RH-dependent SOA composition reported here highlights the necessity to consider the influence of RH on the SOA formation in atmospheric numerical models.69,70 Additionally, this work emphasizes the necessity to characterize the composition at the molecular level, as the RH-dependent composition is not manifested in the elemental composition (O/C, H/C), which remains largely unchanged with increasing RH.10
Author contributions
D. Z. conceptualized the idea. Y. G. conducted the data collection with the aid of H. S. and H. L. H. L. and Y. G. analyzed the experimental data. H. L. and D. Z. wrote and edited the manuscript with the input from all authors. All co-authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 92044301), Shanghai Pilot Program for Basic Research – Fudan University 21TQ1400100 (22TQ010), and Shanghai International Science and Technology Partnership Project (No. 20230711400, 21230780200). Dan Dan Huang acknowledges the financial support from the Science and Technology Commission of Shanghai Municipality (21230711000).References
- X. Ding, Q.-F. He, R.-Q. Shen, Q.-Q. Yu and X.-M. Wang, Spatial distributions of secondary organic aerosols from isoprene, monoterpenes, β-caryophyllene, and aromatics over China during summer, J. Geophys. Res.: Atmos., 2014, 119(20), 877–891, DOI:10.1002/2014jd021748.
- H. Zhang, L. D. Yee, B. H. Lee, M. P. Curtis, D. R. Worton, G. Isaacman-VanWertz, J. H. Offenberg, M. Lewandowski, T. E. Kleindienst and M. R. Beaver, et al., Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(9), 2038–2043, DOI:10.1073/pnas.1717513115.
- G. McFiggans, T. F. Mentel, J. Wildt, I. Pullinen, S. Kang, E. Kleist, S. Schmitt, M. Springer, R. Tillmann and C. Wu, et al., Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 2019, 565(7741), 587–593, DOI:10.1038/s41586-018-0871-y.
- A. Lee, A. H. Goldstein, M. D. Keywood, S. Gao, V. Varutbangkul, R. Bahreini, N. L. Ng, R. C. Flagan and J. H. Seinfeld, Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes, J. Geophys. Res., 2006, 111(D7), D07302, DOI:10.1029/2005jd006437.
- M. Claeys, Y. Iinuma, R. Szmigielski, J. D. Surratt, F. Blockhuys, C. Van Alsenoy, O. Boge, B. Sierau, Y. Gomez-Gonzalez and R. Vermeylen, et al., Terpenylic acid and related compounds from the oxidation of alpha-pinene: implications for new particle formation and growth above forests, Environ. Sci. Technol., 2009, 43(18), 6976–6982, DOI:10.1021/es9007596.
- J. L. Fry, D. C. Draper, K. C. Barsanti, J. N. Smith, J. Ortega, P. M. Winkler, M. J. Lawler, S. S. Brown, P. M. Edwards and R. C. Cohen, et al., Secondary organic aerosol formation and organic nitrate yield from NO3 oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 2014, 48(20), 11944–11953, DOI:10.1021/es502204x.
- H. Shen, L. Vereecken, S. Kang, I. Pullinen, H. Fuchs, D. Zhao and T. F. Mentel, Unexpected significance of a minor reaction pathway in daytime formation of biogenic highly oxygenated organic compounds, Sci. Adv., 2022, 8, 1–11 Search PubMed.
- Y. Guo, H. Shen, I. Pullinen, H. Luo, S. Kang, L. Vereecken, H. Fuchs, M. Hallquist, I.-H. Acir and R. Tillmann, et al., Identification of highly oxygenated organic molecules and their role in aerosol formation in the reaction of limonene with nitrate radical, Atmos. Chem. Phys., 2022, 22(17), 11323–11346, DOI:10.5194/acp-22-11323-2022.
- H. Luo, L. Vereecken, H. Shen, S. Kang, I. Pullinen, M. Hallquist, H. Fuchs, A. Wahner, A. Kiendler-Scharr and T. F. Mentel, et al., Formation of highly oxygenated organic molecules from the oxidation of limonene by OH radical: significant contribution of H-abstraction pathway, Atmos. Chem. Phys., 2023, 23(13), 7297–7319, DOI:10.5194/acp-23-7297-2023.
- X. Zhang, R. C. McVay, D. D. Huang, N. F. Dalleska, B. Aumont, R. C. Flagan and J. H. Seinfeld, Formation and evolution of molecular products in alpha-pinene secondary organic aerosol, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(46), 14168–14173, DOI:10.1073/pnas.1517742112.
- M. E. Jenkin, D. E. Shallcross and J. N. Harvey, Development and application of a possible mechanism for the generation of cis-pinic acid from the ozonolysis of α- and β-pinene, Atmos. Environ., 2000, 34, 2837–2850 CrossRef CAS.
- D. F. Zhao, M. Kaminski, P. Schlag, H. Fuchs, I. H. Acir, B. Bohn, R. Häseler, A. Kiendler-Scharr, F. Rohrer and R. Tillmann, et al., Secondary organic aerosol formation from hydroxyl radical oxidation and ozonolysis of monoterpenes, Atmos. Chem. Phys., 2015, 15(2), 991–1012, DOI:10.5194/acp-15-991-2015.
- T. Kurtén, K. H. Møller, T. B. Nguyen, R. H. Schwantes, P. K. Misztal, L. Su, P. O. Wennberg, J. L. Fry and H. G. Kjaergaard, Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields From α-Pinene + NO3, J. Phys. Chem. Lett., 2017, 8(13), 2826–2834, DOI:10.1021/acs.jpclett.7b01038.
- Q. Ye, E. S. Robinson, X. Ding, P. Ye, R. C. Sullivan and N. M. Donahue, Mixing of secondary organic aerosols versus relative humidity, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(45), 12649–12654, DOI:10.1073/pnas.1604536113.
- M. Yao, Z. Li, C. Li, H. Xiao, S. Wang, A. W. H. Chan and Y. Zhao, Isomer-Resolved Reactivity of Organic Peroxides in Monoterpene-Derived Secondary Organic Aerosol, Environ. Sci. Technol., 2022, 56(8), 4882–4893, DOI:10.1021/acs.est.2c01297.
- Z. Zhao, C. Le, Q. Xu, W. Peng, H. Jiang, Y.-H. Lin, D. R. Cocker and H. Zhang, Compositional Evolution of Secondary Organic Aerosol as Temperature and Relative Humidity Cycle in Atmospherically Relevant Ranges, ACS Earth Space Chem., 2019, 3(11), 2549–2558, DOI:10.1021/acsearthspacechem.9b00232.
- N. L. Prisle, G. J. Engelhart, M. Bilde and N. M. Donahue, Humidity influence on gas-particle phase partitioning of α-pinene + O3 secondary organic aerosol, Geophys. Res. Lett., 2010, 37(1), L01802, DOI:10.1029/2009gl041402.
- A. M. Jonsson, M. Hallquist and E. Ljungstrom, Impact of humidity on the ozone initiated oxidation of limonene, Δ3-carene, and α-pinene, Environ. Sci. Technol., 2006, 40(1), 188–194, DOI:10.1021/es051163w.
- T. Berndt and O. B. Stratmann, F. Gas-phase ozonolysis of α-pinene: gaseous products and particle formation, Atmos. Environ., 2003, 37, 3933–3945, DOI:10.1016/S1352-2310(03)00501-6.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg and T. F. Mentel, et al., Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119(6), 3472–3509, DOI:10.1021/acs.chemrev.8b00395.
- M. Ehn, E. Kleist, H. Junninen, T. Petäjä, G. Lönn, S. Schobesberger, M. Dal Maso, A. Trimborn, M. Kulmala and D. R. Worsnop, et al., Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 2012, 12(11), 5113–5127, DOI:10.5194/acp-12-5113-2012.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann and B. Lee, et al., A large source of low-volatility secondary organic aerosol, Nature, 2014, 506(7489), 476–479, DOI:10.1038/nature13032.
- X. Li, S. Chee, J. Hao, J. P. D. Abbatt, J. Jiang and J. N. Smith, Relative humidity effect on the formation of highly oxidized molecules and new particles during monoterpene oxidation, Atmos. Chem. Phys., 2019, 19(3), 1555–1570, DOI:10.5194/acp-19-1555-2019.
- Y. Zhao, J. A. Thornton and H. O. T. Pye, Quantitative constraints on autoxidation and dimer formation from direct probing of monoterpene-derived peroxy radical chemistry, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(48), 12142–12147, DOI:10.1073/pnas.1812147115.
- J. E. Shilling, Q. Chen, S. M. King, T. Rosenoern, J. H. Kroll, D. R. Worsnop, P. F. DeCarlo, A. C. Aiken, D. Sueper and J. L. Jimenez, et al., Loading-dependent elemental composition of α-pinene SOA particles, Atmos. Chem. Phys., 2009, 9(3), 771–782, DOI:10.5194/acp-9-771-2009.
- W. A. Hall and M. V. Johnston, Oligomer Content of α-Pinene Secondary Organic Aerosol, Aerosol Sci. Technol., 2011, 45(1), 37–45, DOI:10.1080/02786826.2010.517580.
- K. Kristensen, T. Cui, H. Zhang, A. Gold, M. Glasius and J. D. Surratt, Dimers in α-pinene secondary organic aerosol: effect of hydroxyl radical, ozone, relative humidity and aerosol acidity, Atmos. Chem. Phys., 2014, 14(8), 4201–4218, DOI:10.5194/acp-14-4201-2014.
- L. Müller, M. C. Reinnig, J. Warnke and T. Hoffmann, Unambiguous identification of esters as oligomers in secondary organic aerosol formed from cyclohexene and cyclohexene/α-pinene ozonolysis, Atmos. Chem. Phys., 2008, 8, 1423–1433 CrossRef.
- V. Pospisilova, F. D. Lopez-Hilfiker, D. M. Bell, I. El Haddad, C. Mohr, W. Huang, L. Heikkinen, M. Xiao, J. Dommen and A. S. H. Prevot, et al., On the fate of oxygenated organic molecules in atmospheric aerosol particles, Sci. Adv., 2020, 6(11), 1–11, DOI:10.1126/sciadv.aax8922.
- Y. Qin, J. Ye, P. Ohno, J. Zhai, Y. Han, P. Liu, J. Wang, R. A. Zaveri and S. T. Martin, Humidity Dependence of the Condensational Growth of α-Pinene Secondary Organic Aerosol Particles, Environ. Sci. Technol., 2021, 55, 14360–14369, DOI:10.1021/acs.est.1c01738.
- J. F. Pankow, An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol, Atmos. Environ., 1994, 28, 185–188 CrossRef CAS.
- N. M. Donahue, A. L. Robinson, C. O. Stanier and S. N. Pandis, Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 2006, 40(8), 2635–2643, DOI:10.1021/es052297c.
- M. Surdu, H. Lamkaddam, D. S. Wang, D. M. Bell, M. Xiao, C. P. Lee, D. Li, L. Caudillo, G. Marie and W. Scholz, et al., Molecular Understanding of the Enhancement in Organic Aerosol Mass at High Relative Humidity, Environ. Sci. Technol., 2023, 57, 2297–2309, DOI:10.1021/acs.est.2c04587.
- Y. Li and M. Shiraiwa, Timescales of secondary organic aerosols to reach equilibrium at various temperatures and relative humidities, Atmos. Chem. Phys., 2019, 19(9), 5959–5971, DOI:10.5194/acp-19-5959-2019.
- K. Gorkowski, T. C. Preston and A. Zuend, Relative-humidity-dependent organic aerosol thermodynamics via an efficient reduced-complexity model, Atmos. Chem. Phys., 2019, 19(21), 13383–13407, DOI:10.5194/acp-19-13383-2019.
- M. L. Hinks, J. Montoya-Aguilera, L. Ellison, P. Lin, A. Laskin, J. Laskin, M. Shiraiwa, D. Dabdub and S. A. Nizkorodov, Effect of relative humidity on the composition of secondary organic aerosol from the oxidation of toluene, Atmos. Chem. Phys., 2018, 18(3), 1643–1652, DOI:10.5194/acp-18-1643-2018.
- T. B. Nguyen, P. J. Roach, J. Laskin, A. Laskin and S. A. Nizkorodov, Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 2011, 11(14), 6931–6944, DOI:10.5194/acp-11-6931-2011.
- A. P. Bateman, S. A. Nizkorodov, J. Laskinb and A. Laskinc, Physical chemistry of aerosols, Phys. Chem. Chem. Phys., 2009, 11(36), 7931–7942, 10.1039/b916865f.
- C. Kidd, V. Perraud, L. M. Wingen and B. J. Finlayson-Pitts, Integrating phase and composition of secondary organic aerosol from the ozonolysis of α-pinene, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(21), 7552–7557, DOI:10.1073/pnas.1322558111.
- L. Caudillo, B. Rörup, M. Heinritzi, G. Marie, M. Simon, A. C. Wagner, T. Müller, M. Granzin, A. Amorim and F. Ataei, et al., Chemical composition of nanoparticles from α-pinene nucleation and the influence of isoprene and relative humidity at low temperature, Atmos. Chem. Phys., 2021, 21(22), 17099–17114, DOI:10.5194/acp-21-17099-2021.
- M. Gautrois and R. Koppmann, Diffusion technique for the production of gas standards for atmospheric measurements, J. Chromatogr. A, 1999, 848, 239–249 CrossRef CAS.
- M. Gautrois and R. Koppmann, Diffusion technique for the production of gas standards for atmospheric measurements, J. Chromatogr. A, 1999, 848(1–2), 239–249, DOI:10.1016/S0021-9673(99)00424-0.
- T. F. Mentel, J. Wildt, A. Kiendler-Scharr, E. Kleist, R. Tillmann, M. Dal Maso, R. Fisseha, T. Hohaus, H. Spahn and R. Uerlings, et al., Photochemical production of aerosols from real plant emissions, Atmos. Chem. Phys., 2009, 9(13), 4387–4406, DOI:10.5194/acp-9-4387-2009.
- F. D. Lopez-Hilfiker, V. Pospisilova, W. Huang, M. Kalberer, C. Mohr, G. Stefenelli, J. A. Thornton, U. Baltensperger, A. S. H. Prevot and J. G. Slowik, An extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF) for online measurement of atmospheric aerosol particles, Atmos. Meas. Tech., 2019, 12(9), 4867–4886, DOI:10.5194/amt-12-4867-2019.
- J. Fick, L. Pommer, C. Nilsson and B. Andersson, Effect of OH radicals, relative humidity, and time on the composition of the products formed in the ozonolysis of α-pinene, Atmos. Environ., 2003, 37(29), 4087–4096, DOI:10.1016/s1352-2310(03)00522-3.
- R. A. Zaveri, J. E. Shilling, A. Zelenyuk, M. A. Zawadowicz, K. Suski, S. China, D. M. Bell, D. Veghte and A. Laskin, Particle-Phase Diffusion Modulates Partitioning of Semivolatile Organic Compounds to Aged Secondary Organic Aerosol, Environ. Sci. Technol., 2020, 54, 2595–2605, DOI:10.1021/acs.est.9b05514.
- M. Shiraiwa, A. Zuend, A. K. Bertram and J. H. Seinfeld, Gas–particle partitioning of atmospheric aerosols: interplay of physical state, non-ideal mixing and morphology, Phys. Chem. Chem. Phys., 2013, 15(27), 11441–11453, 10.1039/c3cp51595h.
- R. M. Kamens, H. Zhang, E. H. Chen, Y. Zhou, H. M. Parikh, R. L. Wilson, K. E. Galloway and E. P. Rosen, Secondary organic aerosol formation from toluene in an atmospheric hydrocarbon mixture: Water and particle seed effects, Atmos. Environ., 2011, 45(13), 2324–2334, DOI:10.1016/j.atmosenv.2010.11.007.
- R. Volkamer, P. J. Ziemann and M. J. Molina, Secondary Organic Aerosol Formation from Acetylene (C2H2): seed effect on SOA yields due to organic photochemistry in the aerosol aqueous phase, Atmos. Chem. Phys., 2009, 9(6), 1907–1928, DOI:10.5194/acp-9-1907-2009.
- M. Jang, N. M. Czoschke, S. Lee and R. M. Kamens, Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 2002, 298(5594), 814–817, DOI:10.1126/science.1075798.
- S. Gao, N. L. Ng, M. Keywood, V. Varutbangkul, R. Bahreini, A. Nenes, J. He, K. Y. Yoo, J. L. Beauchamp and R. P. Hodyss, et al., Particle phase acidity and oligomer formation in secondary organic aerosol, Environ. Sci. Technol., 2004, 38(24), 6582–6589, DOI:10.1021/es049125k.
- N. M. Czoschke and M. Jang, Acidity effects on the formation of α-pinene ozone SOA in the presence of inorganic seed, Atmos. Environ., 2006, 40(23), 4370–4380, DOI:10.1016/j.atmosenv.2006.03.030.
- N. Czoschke, Effect of acidic seed on biogenic secondary organic aerosol growth, Atmos. Environ., 2003, 37(30), 4287–4299, DOI:10.1016/s1352-2310(03)00511-9.
- L. Du, L. Xu, K. Li, C. George and M. Ge, NH3 Weakens the Enhancing Effect of SO2 on Biogenic Secondary Organic Aerosol Formation, Environ. Sci. Technol. Lett., 2023, 10(2), 145–151, DOI:10.1021/acs.estlett.2c00959.
- J. A. Faust, J. P. Wong, A. K. Lee and J. P. Abbatt, Role of Aerosol Liquid Water in Secondary Organic Aerosol Formation from Volatile Organic Compounds, Environ. Sci. Technol., 2017, 51(3), 1405–1413, DOI:10.1021/acs.est.6b04700.
- C. Wong, S. Liu and S. A. Nizkorodov, Highly Acidic Conditions Drastically Alter the Chemical Composition and Absorption Coefficient of α-Pinene Secondary Organic Aerosol, ACS Earth Space Chem., 2022, 6(12), 2983–2994, DOI:10.1021/acsearthspacechem.2c00249.
- M. Simon, L. Dada, M. Heinritzi, W. Scholz, D. Stolzenburg, L. Fischer, A. C. Wagner, A. Kürten, B. Rörup and X.-C. He, et al., Molecular understanding of new-particle formation from α-pinene between −50 and +25 °C, Atmos. Chem. Phys., 2020, 20(15), 9183–9207, DOI:10.5194/acp-20-9183-2020.
- N. M. Donahue, S. A. Epstein, S. N. Pandis and A. L. Robinson, A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 2011, 11(7), 3303–3318, DOI:10.5194/acp-11-3303-2011.
- D. D. Huang, X. Zhang, N. F. Dalleska, H. Lignell, M. M. Coggon, C.-M. Chan, R. C. Flagan, J. H. Seinfeld and C. K. Chan, A note on the effects of inorganic seed aerosol on the oxidation state of secondary organic aerosol-α-Pinene ozonolysis, J. Geophys. Res.: Atmos., 2016, 121(20), 476–483, DOI:10.1002/2016jd025999.
- Q. Ye, M. Wang, V. Hofbauer, D. Stolzenburg, D. Chen, M. Schervish, A. Vogel, R. L. Mauldin, R. Baalbaki and S. Brilke, et al., Molecular Composition and Volatility of Nucleated Particles from α-Pinene Oxidation between −50 °C and +25 °C, Environ. Sci. Technol., 2019, 53(21), 12357–12365, DOI:10.1021/acs.est.9b03265.
- L. Poulain, A. Tilgner, M. Brüggemann, P. Mettke, L. He, J. Anders, O. Böge, A. Mutzel and H. Herrmann, Particle-Phase Uptake and Chemistry of Highly Oxygenated Organic Molecules (HOMs) From α-Pinene OH Oxidation, J. Geophys. Res.: Atmos., 2022, 127(16), 1–22, DOI:10.1029/2021jd036414.
- S. Zhang, L. Du, Z. Yang, N. T. Tchinda, J. Li and K. Li, Contrasting impacts of humidity on the ozonolysis of monoterpenes: insights into the multi-generation chemical mechanism, Atmos. Chem. Phys., 2023, 23(18), 10809–10822, DOI:10.5194/acp-23-10809-2023.
- B.-H. Lee, J. R. Pierce, G. J. Engelhart and S. N. Pandis, Volatility of secondary organic aerosol from the ozonolysis of monoterpenes, Atmos. Environ., 2011, 45(14), 2443–2452, DOI:10.1016/j.atmosenv.2011.02.004.
- L. Stirnweis, C. Marcolli, J. Dommen, P. Barmet, C. Frege, S. M. Platt, E. A. Bruns, M. Krapf, J. G. Slowik and R. Wolf, et al., Assessing the influence of NOx concentrations and relative humidity on secondary organic aerosol yields from α-pinene photo-oxidation through smog chamber experiments and modelling calculations, Atmos. Chem. Phys., 2017, 17(8), 5035–5061, DOI:10.5194/acp-17-5035-2017.
- I. I. I. D. R. Cocker, S. L. Cleggb, R. C. Flagana and J. H. Seinfeld, The effect of water on gas–particle partitioning of secondary, Atmos. Environ., 2001, 35, 6049–6072 CrossRef.
- M. D. Petters and S. M. Kreidenweis, A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 2007, 7, 1961–1971 CrossRef CAS.
- M. D. Petters, S. M. Kreidenweis, A. J. Prenni, R. C. Sullivan, C. M. Carrico, K. A. Koehler and P. J. Ziemann, Role of molecular size in cloud droplet activation, Geophys. Res. Lett., 2009, 36(22), L22801, DOI:10.1029/2009gl040131.
- C. Zhang, Y. Guo, H. Shen, H. Luo, I. Pullinen, S. H. Schmitt, M. Wang, H. Fuchs, A. Kiendler-Scharr and A. Wahner, et al., Contrasting Influence of Nitrogen Oxides on the Cloud Condensation Nuclei Activity of Monoterpene-Derived Secondary Organic Aerosol in Daytime and Nighttime Oxidation, Geophys. Res. Lett., 2023, 50(4), 1–11, DOI:10.1029/2022gl102110.
- H. O. T. Pye, B. N. Murphy, L. Xu, N. L. Ng, A. G. Carlton, H. Guo, R. Weber, P. Vasilakos, K. W. Appel and S. H. Budisulistiorini, et al., On the implications of aerosol liquid water and phase separation for organic aerosol mass, Atmos. Chem. Phys., 2017, 17(1), 343–369, DOI:10.5194/acp-17-343-2017.
- S. H. Jathar, A. Mahmud, K. C. Barsanti, W. E. Asher, J. F. Pankow and M. J. Kleeman, Water uptake by organic aerosol and its influence on gas/particle partitioning of secondary organic aerosol in the United States, Atmos. Environ., 2016, 129, 142–154, DOI:10.1016/j.atmosenv.2016.01.001.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the experimental conditions; the molecular formula of the SOA products; the absolute abundance of monomer (C10H16Ox) and dimers (C17–20); average molecule weight, O/C, and H/C; the estimated vapor–particle (τv,p) and vapor–wall equilibration timescale (τv,w); the absolute abundance of C9, C10, and total products; the fraction of C9 and C10 monomers with different O/C ratios; average oxygen number of monomer families under different RH; the abundance ratio of C10H16Ox and C19H28Ox at 91% RH and 28% RH in the absence of AS seed aerosol; the absolute abundance of C19H28Ox in the absence of seed aerosol. See DOI: https://doi.org/10.1039/d3ea00149k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |