
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecondary aerosol formation from mixtures of marine volatile organic compounds in a potential aerosol mass oxidative flow reactor†
Alexia N.
Moore‡
 a,
Lucia
Cancelada‡
a,
Ke'La A.
Kimble
a and
Kimberly A.
Prather
*ab
a,
Lucia
Cancelada‡
a,
Ke'La A.
Kimble
a and
Kimberly A.
Prather
*ab
aDepartment of Chemistry and Biochemistry, University of California, La Jolla, San Diego, CA 92093, USA. E-mail: kprather@ucsd.edu
bScripps Institution of Oceanography, University of California, La Jolla, San Diego, CA 92037, USA
First published on 22nd February 2024
Abstract
Increasing recognition of the significant contributions secondary organic aerosols can make in marine environments has led to an increase in research focused on understanding the reactions controlling their formation. Most marine laboratory studies to date have focused on the oxidation of individual volatile organic compounds (VOCs), particularly dimethyl sulfide (DMS). Thus, a lack of understanding exists in how complex marine VOC mixtures affect secondary marine aerosol formation and composition. To address this gap, we conducted controlled lab experiments that compare the effects of oxidizing single common marine VOCs versus VOC mixtures on secondary marine aerosol production. We used a potential aerosol mass oxidative flow reactor to investigate marine-relevant VOCs, including DMS, dimethyl disulfide (DMDS), and isoprene. Ion chromatography, chemical ionization mass spectrometry, aerosol time-of-flight mass spectrometry, and particle sizing instruments were employed to study how these mixtures influence the overall composition of marine aerosols. Our findings reveal that mixtures significantly alter the production and composition of secondary marine aerosols. Specifically, we found that isoprene, when oxidized in the presence of DMS and DMDS, affects methanesulfonic acid (MSA) and sulfate ratios, as well as overall aerosol yields. These insights suggest further studies on realistic marine VOC mixtures will help understand and predict the dynamics of secondary marine aerosol formation, therefore improving air quality and climate models and enabling more accurate predictions of marine aerosol impacts on cloud formation and properties.
Environmental significanceSecondary marine aerosols (SMA) have been shown to significantly contribute to the marine atmosphere by affecting the formation and lifespan of clouds. Prior research has focused on single VOC studies, leading to a gap in understanding SMA. In this work we look at how mixtures of marine relevant gases impact SMA formation by analyzing the aerosol yields and composition. We found that mixtures of VOCs impacted the production of sulfate and methanesulfonic acid, two species that play a key role in aerosol formation and growth. These results can be used to improve the parametrization of SMA in climate models and their predictive power of future Earth scenarios. |
Introduction
Dimethyl sulfide (DMS) has been estimated to account for more than half of the natural gas-phase sulfur emissions.1 Oxidation of DMS has been shown to produce secondary aerosols comprised primarily of sulfate and methanesulfonic acid (MSA) formed through H abstraction and OH addition reaction pathways.2,3 These oxidation products have atmospheric significance with MSA playing a major role in particle growth,4,5 and sulfuric acid enhancing new particle formation.6,7 Sulfate aerosols can offset the warming effect of greenhouse gases by scattering solar radiation and influencing cloud albedo and lifetime.8,9 In the marine environment, secondary aerosols have significantly different cloud forming potential from primary marine aerosols.10 Knowing the factors controlling the production of secondary marine aerosols and their physicochemical properties is required to predict the formation, lifetime, and properties of marine clouds. For instance, it has been shown that the aerosol mass needed to achieve a similar cloud albedo is 5 times larger for sulfate than for MSA-formed clouds.11 In the Arctic Sea region, the highest daily concentration of MSA was found to be associated with new particle formation event days over summer.12 Meanwhile, studies along the Antarctic coast have not only estimated the branching ratio between MSA and non-sea salt sulfate from DMS oxidation to be 0.84, but also showed an enrichment of non-sea salt sulfate with respect to MSA for aged air masses with low DMS content.13 However, in areas with higher anthropogenic activity, DMS conversion to MSA was found to be inhibited,14 thus highlighting that a more complex picture of gaseous emissions will likely lead to differences in secondary marine aerosol particle composition and concentrations.Laboratory studies have examined the oxidation of reduced sulfur compounds over a range of conditions. Van Rooy et al. oxidized DMS and dimethyl disulfide (DMDS) in a smog chamber with relative humidity ranging from 35 to 40%, using hydroxyl radical, nitrate radical, and O(3P) to examine the yields and formation mechanisms of sulfuric acid and MSA.15 Using these conditions, they determined a higher humidity was necessary for the enhancement of MSA formation. Sulfate and MSA yields from DMS oxidation varied among experiments depending on the environmental conditions and overall ratio and type of gas phase species present. In the past, MSA yields have ranged from 0.5% to 45% with the highest concentrations occurring under high NOx conditions. Van Rooy et al. estimated 25% of the aerosol mass from DMS oxidation by O(3P) and NOx were explained by MSA formation, but this number was much lower only in the presence of the hydroxyl radical. Other authors have also examined DMS oxidation reactions by OH radicals over a range of NOx concentrations, as well as changes in RH.3 However, few studies have considered the impact of other non-sulfur volatile organic compounds (VOCs) on the products and aerosol yield of DMS oxidation. For example, isoprene, another relatively less studied contributor to marine gas emissions, has been reported as a secondary organic aerosol (SOA) precursor in both terrestrial and marine environments.16–18 Isoprene SOA potential increases in the presence of acidic particles.17,18 Chen and Jang conducted a study examining the photooxidation of DMS and isoprene in a chamber under various NOx and humidity conditions and reported a substantial increase in the isoprene yield in the presence of DMS at atmospherically relevant RH. This discovery has significant implications for the marine environment as the oxidation of DMS can provide acidic seed particles that promote isoprene aerosol formation. However, they found that isoprene decreased the MSA yield from 48% to 25.8%.19 Controlled laboratory experiments are needed to further understand the mechanisms and processes that control the formation of secondary aerosols in marine environments. Herein, we use simplified mixtures comprised of several marine-relevant VOCs to help elucidate secondary marine aerosol properties and yields.
In this study, a flow reactor was used to oxidize mixtures of DMS, DMDS, and isoprene with hydroxyl radical simulating periods ranging from 0.7 to 19.1 days of equivalent oxidation to determine the impact on aerosol yields and sulfate to MSA ratios. To our knowledge, this is the first reported study of reduced sulfur oxidation reactions in a Potential Aerosol Mass Oxidative Flow Reactor (PAM-OFR).20,21 Aerosol yield and composition were probed as a function of precursor gases (DMS, DMDS, isoprene, and mixtures of DMS/isoprene, DMDS/isoprene, DMS/DMS, and DMS/DMDS/isoprene) and extent of OH exposure.
Methods
Experimental setup
The experimental setup used in this study is shown in Fig. 1. Dry zero air was generated using the Model 1001 Zero Air Source (Sabio Environmental). DMS and isoprene concentrations were adjusted using a G-Cal permeation device at room temperature (VICI Metronics) to give a permeation rate of 300 ng min−1 (±5% at 25 °C) and 247 ng min−1 (±5% at 25 °C), respectively. DMDS concentrations were adjusted by flowing air through a Dynacal permeation tube (VICI Metronics) housed inside a custom-built permeation oven. Permeation rate was 150 ng min−1 (±25% at 50 °C). Mass flow controllers (Alicat Scientific) were used to adjust the flows and calibrated against a Defender 520 volumetric primary flow standard (Mesa Labs). Humidified zero air was introduced to make up a total flow of 5 L min−1. The final concentrations are shown in Table 1. The gas phase mixture was sent through a potential aerosol mass oxidation flow reactor (PAM-OFR, Aerodyne Inc.) at relative humidity ranging between 50–70%.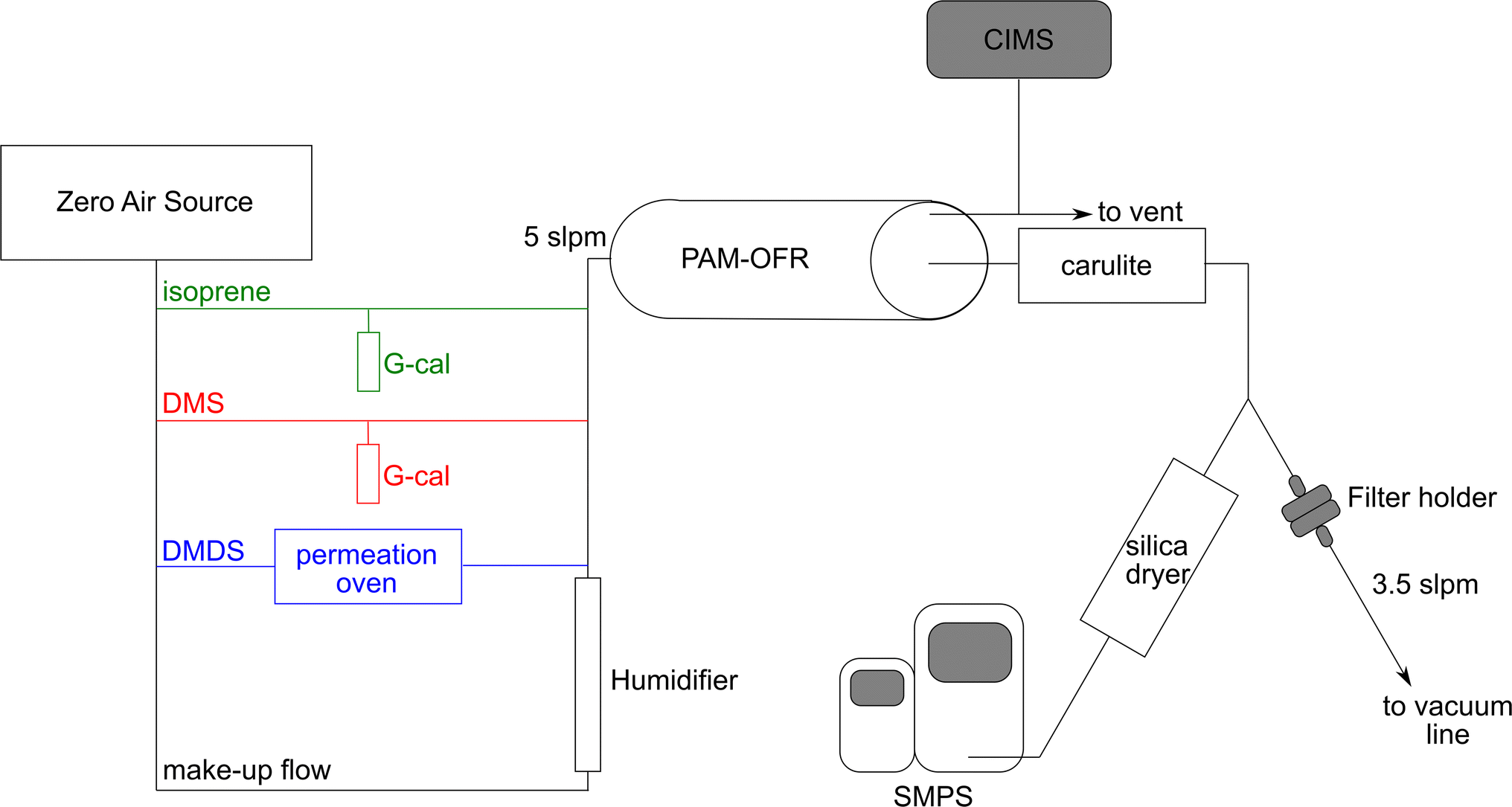 | ||
| Fig. 1 Schematic diagram of experimental setup used for secondary marine aerosol reaction studies. | ||
| Experiment | VOC source | VOC concentration (ppb) | OH exposure (mol s−1 cm−3) | OH reaction rate constant (cm3 mol−1 s−1) |
|---|---|---|---|---|
| DMS | G-cal | 16 | 9.6 × 1010 to 2.5 × 1012 | 0.44 × 10−11 (1) |
| DMDS | Permeation tube | 9 | 20 × 10−11 (2) | |
| Isoprene | G-cal | 15 | 10 × 10−11 (3) | |
| DMS + isoprene | G-cal | 15 (DMS) | ||
| 11 (isoprene) | ||||
| DMDS + isoprene | Permeation tube (DMDS) | 6 (DMDS) | ||
| G-cal (isoprene) | 15 (isoprene) | |||
| DMS + DMDS | G-cal (DMS) | 18 (DMS) | ||
| Permeation tube (DMDS) | 9 (DMDS) | |||
| DMS + DMDS + isoprene | G-cal (DMS) | 15 (DMS) | ||
| Permeation tube (DMDS) | 9 (DMDS) | |||
| G-cal (isoprene) | 12 (isoprene) |
The PAM-OFR uses UV lamps with wavelengths of λ = 185 and 254 nm (OFR185 mode) to produce a high concentration of OH radicals.20 Calibration of OH exposure in the OFR was determined by introducing carbon monoxide (initial concentration ≈ 1 ppm) to the OFR at the same airflow rate and relative humidity used during the experiments and measuring the CO decay as a function of lamp voltage using a CO analyzer (APMA-370, Horiba Ltd). The change in CO concentration yielded the OH exposure versus lamp voltage relationship using the known CO + OH rate coefficient kOH+CO, 298K = 1.5 × 10−13 cm3 mol−1 s−1.22 Calibration results are shown in Fig. S1.† The residence time of gases in the OFR was 2.7 min. A diffusion ozone denuder (Carulite-200, Ozone Solutions) was located after the OFR to remove ozone and avoid further oxidation of reaction products. A custom-built silica drier (length: 28 in, diameter: 4 in) was used to dry the aerosols under 25% RH before they were introduced into the particle sizing instrument.
Aerosol yield and characterization were conducted using two experimental approaches. In aerosol yield experiments, lamp voltage was changed every 40 minutes to produce more OH radical and increase the equivalent days of aging. Seven OH exposures were used, ranging from 9.6 × 1010 to 2.5 × 1012 mol s−1 cm−3, equivalent to 0.7 to 19.1 days of aging assuming typical tropospheric conditions ([OH] = 1.5 × 106 mol cm−3). For aerosol characterization experiments, a single OH exposure of 3.8 × 1011 mol s−1 cm−3 was used, equivalent to 2.9 days of aging. Each filter was collected for 22 hours to ensure enough aerosol mass was collected for detection of MSA and sulfate with ion chromatography (see below). More details about yield and aerosol characterization experiments are included in Tables 1 and S1.†
Quantification of VOCs
During the aerosol yield experiments, VOCs (DMS, DMDS, and isoprene) were quantified after the PAM-OFR in real time using a chemical ionization time-of-flight mass spectrometer (CI-ToF-MS), with benzene as the reagent ion.26,27 A concentration of 300 ppm benzene vapor was generated by passing 10 standard cubic centimeters per minute (sccm) of ultrahigh-purity N2 gas over a cylinder of liquid benzene further diluted to concentration with added N2. Benzene vapor was passed through a 20 mCi Po-210 α-source to generate benzene cluster cation reagent ions, and further drawn through an inline critical orifice at 1.8 standard liters per minute (slpm) into the ion–molecule region (IMR) of the CI-ToF-MS. Sample analyte was similarly drawn into the IMR at the same flow rate as the reagent. The IMR pressure was maintained at 60 Torr and voltage at 60 V for all analyses. Analyte ions generated through charge transfer and ligand switching reactions with benzene cluster cations were focused by a radio frequency ion funnel, and subsequently transferred by an RF-only quadrupole into an orthogonal extraction time-of-flight analyzer (Tofwerk). Co-summed mass spectra from 5–500 m/z were obtained at 1 Hz and analyzed using the Tofware plugin for Igor Pro 8 software. External calibrations were run for DMS, DMDS, and isoprene using the G-cal permeation tubes (DMS and isoprene) and Dynacal permeation tube (DMDS). In addition to the external calibration, the experimental setup was constructed to minimize stainless steel by using a Teflon-coated OFR and reducing the number of stainless-steel fittings to limit reactions on metal surfaces. The supplier for both the Dynacal and G-cal devices (VICI Metronics) assures these standards consist of pure compounds. To ensure this purity, we verified the absence of contaminating species using the CI-ToF-MS spectra.Quantification of sulfate and methanesulfonic acid
To quantify sulfate and MSA produced by VOC oxidation, filters were collected after the PAM-OFR at 3.5 L min−1 for 22 hours (Fig. 1). Pallflex Tissuquartz filters (Pall) were combusted for 6 hours at 500 °C to reduce background contamination signal. After collection, filters were stored at −20 °C until the time they were processed. 5 mL of Milli-Q water (Millipore, resistivity of 18.2 MΩ cm at 25 °C and total organic carbon below 5 ppb) and 30 minutes of sonication were used to extract the filters. The extracts were analyzed using a Dionex ICS-2000 ion chromatography (IC) system. The ICS-2000 was equipped with an eluent generator (23 mM KOH isocratic run), a Dionex IonPac AS18 2 × 250 mm column, an AG18 2 × 50 mm guard column, and an AERS 500 2 mm electric suppressor. The DS6 conductivity detector was held at 35 °C. The sample injection volume was 25 μL and the flow rate was 0.25 mL min−1. Under these conditions, MSA was eluted at 3.97 min and sulfate at 6.27 min. MSA (Alfa Aesar, purity > 98%) and sulfate (Inorganic Ventures, [SO42−] = 1001 ± 4 μg mL−1) standard solutions were prepared in Milli-Q water, and calibration solutions were run in the IC along with the filter extracts.Particle size distribution measurements
During all experiments, particle size distributions of the aerosol produced in the PAM-OFR were measured using a scanning mobility particle sizer (SMPS) at a RH below 25%. The SMPS consists of a differential mobility analyzer (DMA) Model 3081, an electrostatic classifier Model 3082, and a water-based condensation particle counter Model 3787 (TSI, Inc.) and was operated at a sample flow rate of 0.6 L min−1 and a sheath flow rate of 3.0 L min−1. Each SMPS scan was 5 minutes over electrical mobility diameters ranging between 14–750 nm. Electrical mobility diameters are assumed to be equal to physical diameters, and thus size and volume distributions presented in Fig. 2 are shown with physical diameters.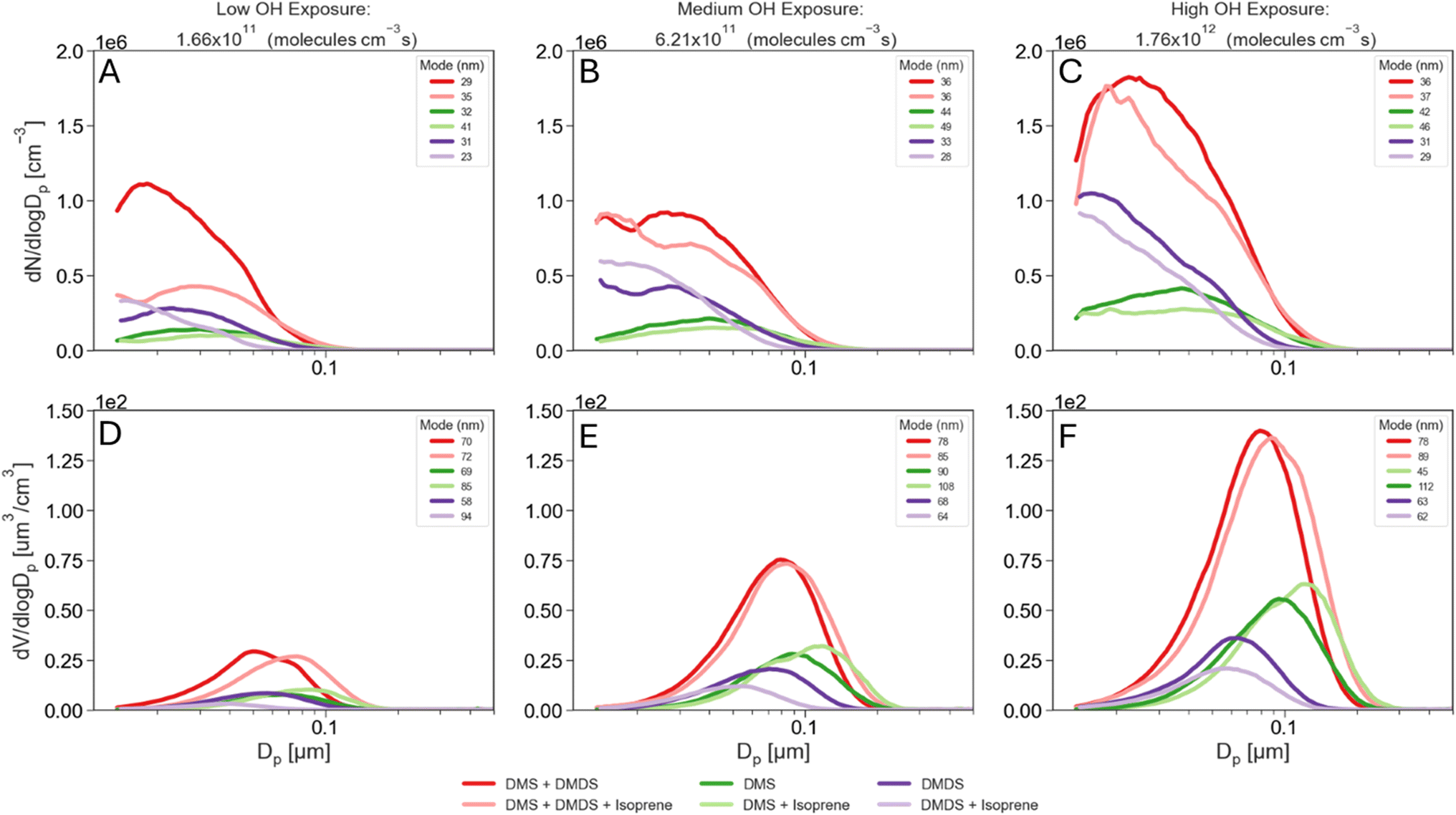 | ||
| Fig. 2 Particle number (top) and volume (bottom) distributions for low (A and D), medium (B and E) and high (C and F) OH exposure (VOCs and mixtures combined on each panel). | ||
Real-time composition of secondary aerosols
The compositions of 20 and 60 nm secondary aerosols were measured using an aerosol time-of-flight mass spectrometer (ATOFMS).28,29 Particles were size selected at 20 nm using a DMA and then passed through a sublimation-deposition growth tube at a flow of 200 sccm, where they were grown with 1,8-bis(tetramethylguanidino)-naphthalene (TMGN) at 100 °C.30 Particles were then size-selected post growth at 400 nm using a second DMA. This newly developed MALDI method was used because the standard ATOFMS cannot detect particles below 100 nm. After the second size selection, particles were sent into the ATOFMS for size-resolved chemical analysis. In the ATOFMS, the particle beam is first collimated using an aerodynamic lens before measuring the speed of each particle between two continuous wave lasers to determine their size. The signal from the continuous scattering lasers triggers a Nd:YAG desorption ionization laser at 266 nm which produces positive and negative ions from each individual particle. Since the oxidized products preferentially form negative ions, only the negative ion mass spectra are displayed here.Results and discussion
Particle size distributions for the oxidation of single VOCs
Fig. 2 presents aerosol size distributions resulting from the oxidation of DMS and DMDS at varying OH exposures. Panels A to C show particle number distributions while D to F show particle volume distributions. The total number and volume concentrations and the median number-weighed and volume-weighed diameters for these distributions are presented in Fig. S2.† At low OH exposures, equivalent of 1.66 × 1011 mol s−1 per cm−3 (as shown in Fig. 2A and S2†), oxidation of DMDS generates more aerosols than DMS, consistent with DMDS having a faster initial OH rate constant (20 × 10−11 cm3 mol−1 s−1) compared to DMS (0.44 × 10−11 cm3 mol−1 s−1).23,24 At this low OH exposure, DMDS also produced more aerosols by volume than DMS. However, at medium and high exposures, despite DMDS producing more particles than DMS, the total volume concentration is higher for DMS than for DMDS (Fig. 2D and S2†). In their DMDS oxidation chamber experiments, Van Rooy et al. observed that aerosol mass continued to increase even after complete consumption of DMDS.15 This led them to hypothesize that slower secondary reactions also contribute to aerosol mass. In our study, this hypothesis provides an explanation for the differences between DMS and DMDS. At the lowest OH exposure, where the fastest DMDS initial oxidation rate is dominant, DMS produced fewer aerosols by number and volume than DMDS. As the OH exposure increased, DMDS was quickly consumed and the products of this initial oxidation led to slower secondary reactions compared to those of DMS, therefore yielding a lower total aerosol volume. Consistent with this, the median number-weighted diameters from Fig. S2† show that DMDS produced smaller particles and higher number concentrations than DMS, which again is expected based on the faster DMDS initial oxidation rate. Typically, particles below 100 nm are dominated by nucleation processes, whereas particles above 100 nm are dominated by condensation processes. In this study, we found that reactions in the PAM-OFR were primarily dominated by nucleation (Fig. S2†). Despite the short residence times in the PAM-OFR, we found that about 5% of DMS total aerosol production occurred above 100 nm. In contrast, the DMDS experiments showed less than 1% of particles above 100 nm, suggesting that secondary oxidation reactions with DMS occur faster than those with DMDS, allowing both nucleation and condensation processes to take place. The median volume-weighed diameter for the DMS experiment is around 69 nm at the lowest OH exposure and shifts to 95 nm at the highest OH exposure, further highlighting the particle growth observed in the number size distributions. The volume size distributions for the DMDS experiment peak around 63 nm on average at all OH exposure times. Increasing the OH exposure does not result in a shift in the volume size distribution, indicating that DMDS oxidation is dominated by new particle formation.Particle size distributions for the oxidation of mixtures
The particle number size distributions for the mixture of DMS and isoprene, DMDS and isoprene, and DMS and DMDS are shown in Fig. 2A–C. An additional experiment using a mixture of all three VOCs is also shown. In Fig. 2 and S2,† the median number-weighed diameter for the DMS and isoprene mixture, particle size distribution shifts to larger sizes compared to DMS alone, showing that larger particles are being formed in the presence of isoprene. The addition of isoprene not only shifts the distribution but also impacts the production of aerosols by decreasing the number of secondary aerosols in the DMS experiment. Isoprene also seems to decrease the number of aerosols produced when mixed with DMDS at high OH exposure (Fig. S2†). In contrast to DMS, the presence of isoprene with DMDS enhances the median number-weighed diameter of the distribution in the smaller size range.The aerosol volume size distributions for the mixtures are shown in Fig. 2D–F. Larger total volumes were obtained for the DMS and isoprene mixture compared to DMS alone while the opposite occurs for the combination of DMDS and isoprene (Fig. S2†). Ahlberg et al. found in organic mixture experiments that isoprene shifted the size distribution to larger particles while also reducing the number of particles,16 a similar behavior to that is observed for DMS. Since isoprene has a faster reaction rate with OH than DMS and a slower reaction rate than DMDS (Table 1), we hypothesize isoprene had a greater impact on the initial DMS aerosol production. Because of the slower initial DMS oxidation and the lower partition coefficient of isoprene PAM-OFR oxidation products to the aerosol phase,18 fewer particles undergo condensation and growth, yielding larger total volume concentrations for the DMS and isoprene experiment (Fig. S2†). As shown in Fig. 2A–C, the mixture between DMS, DMDS, and isoprene resulted in a slight decrease in aerosol number production compared to the mixture between DMS and DMDS. This behavior reflects the trends that DMS and DMDS have shown in their individual experiments. Number concentrations decrease both for DMS and DMDS upon the addition of isoprene, therefore explaining the decrease in number concentrations for the DMS, DMDS and isoprene mixture. On the other hand, volume concentrations increase upon the addition of isoprene for DMS and decrease for DMDS. Aerosol volume showed a slight increase upon the addition of isoprene to DMS and DMDS together, which resembles DMS behavior and suggests again that condensation and growth might become the dominant process when DMS is present.
Aerosol yields for the oxidation of single VOCs versus mixtures
Fig. 3A presents the aerosol yields for DMS, DMDS, and isoprene, each measured independently. Aerosol yield, calculated from the amount of reacted gas and aerosol mass formed, was highest for DMS, followed by DMDS, and then isoprene. The aerosol yield from DMS oxidation increased consistently with OH exposure, reaching a peak yield of 90%. This is higher than the 50% yield found by Chen and Jang in a chamber study at 45% RH.19 Other chamber studies reported lower estimates for DMS and DMDS aerosol yield due to the absence of a steady state of aerosol mass with yields of 12% and 25%.15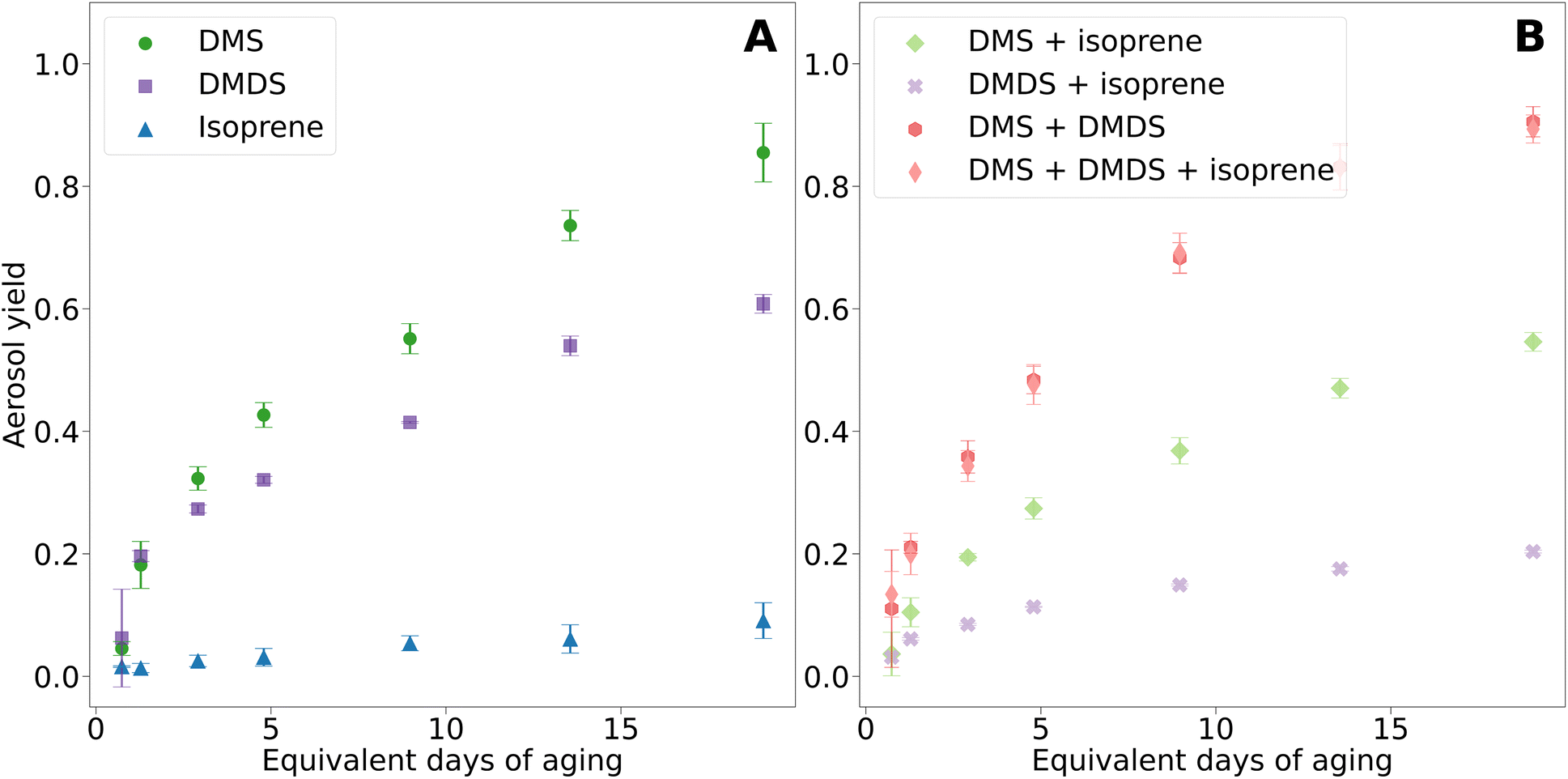 | ||
| Fig. 3 Aerosol yields for VOCs and mixtures. (A) Pure compounds. (B) Mixtures. Error bars represent the standard deviation of three trials. | ||
In this study, DMS and DMDS had similar aerosol yields at low equivalent days of aging. However, DMS aerosol yield surpassed that of DMDS for 2.9 days and longer aging times. Interestingly, the non-reacted gases produced noteworthy results (Fig. S3†). At 1.3 days of aging, less than 4% of the initial DMDS gas phase concentrations remained, and by 19.1 days of aging, less than 1% was left. Despite DMDS being almost completely reacted at the highest OH exposure, the aerosol yield was only 61%, suggesting the formation of smaller more volatile gas phase products that did not condense on the secondary aerosols.
For DMS, the non-reacted gas phase concentrations were 52% and 9% at 1.3 and 19.1 days of aging. However, at the highest OH exposure, DMS aerosol yield was 85%. As anticipated, isoprene had the lowest aerosol yield, reaching a maximum yield of 10% at the highest OH exposure. At 1.3 days of aging, 11% of isoprene remained unreacted and 8% at 19 days of aging. Despite most of isoprene being reacted, isoprene oxidation in the PAM-OFR resulted in low yields due to the absence of isoprene epoxy diols (IEPOX) aerosol formation and no enhancement of acidic seed particles.31 These results suggest that the majority of isoprene degraded to form volatile gas phase compounds.18,32
Fig. 3B shows aerosol yields from mixtures of DMS and isoprene, DMDS and isoprene, DMS and DMDS, and DMS, DMDS, and isoprene. The overall yield from the oxidation of DMS and isoprene together was lower than that of DMS oxidation only. While the formation of isoprene SOA has been shown to be enhanced in the presence of acidic particles, such as DMS oxidation products (sulfate and MSA), the mixture with isoprene decreased sulfate formation from DMS, as shown in Fig. 4 and discussed in the next section.18 We hypothesize that isoprene and DMS oxidation products are competing for atmospheric oxidants. These results are consistent with previous chamber studies that have shown lower DMS yields in the presence of isoprene.19 A similar behavior was observed in the case of the DMDS and isoprene mixture in our study; aerosol yield substantially decreased compared to DMDS alone. In this case, at the highest OH exposure, the aerosol yield experienced a 3-fold decrease, leading to a value close to the yield of isoprene alone. For DMS, this decrease was not as dramatic (1.3-fold).
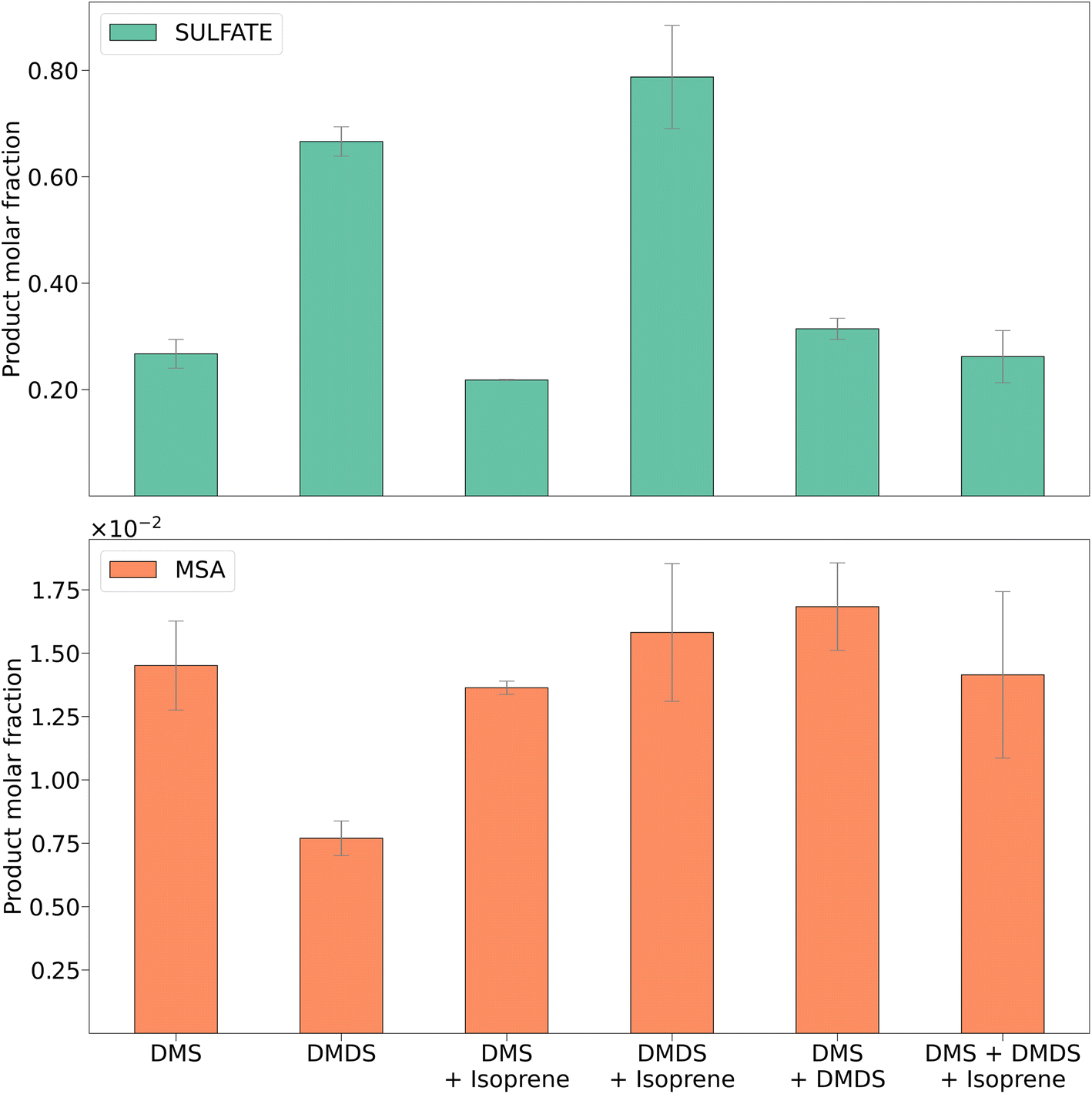 | ||
| Fig. 4 MSA and sulfate molar fractions for DMS, DMDS, and mixtures with isoprene. Error bars represent the standard deviation of four trials. | ||
Oxidation of DMS and DMDS mixtures resulted in the highest aerosol yield of any combination, yielding 94%. This result was surprising although we hypothesize that rapid oxidation of DMS to sulfate helped provide the aerosol seeds for the DMDS oxidation products to condense upon leading to a higher yield. Isoprene added to the mixture of DMS and DMDS resulted in a small decrease in the aerosol yield of 89%. The impact of the addition of isoprene on the overall yield of the oxidation of the DMS and DMDS mixture is minimal compared to the impact on DMS or DMDS oxidation alone. We hypothesize the inhibitory effect of isoprene on aerosol yield is minimized due to the fast oxidation mechanism mentioned above. This behavior also corresponds with the small shift to larger particle sizes in the volume size distribution.
Fig. S5† shows aerosol yields for VOCs and mixtures plotted against the increase in aerosol mass (ΔM, μg m−3). To gain more knowledge on the properties of the particle phase species, these data were fitted to an absorptive partitioning model.33,34 At first glance, no dramatic differences were observed between the experiments. This is not surprising given that for all of them except isoprene alone, MSA and sulfate were the main products, and therefore, the most abundant particle phase species. As seen in the top panel of Fig. S5,† DMS yields increased at a faster rate than DMDS, at least for ΔM lower than 20 μg m−3. Again, this agrees with what was suggested above; DMS oxidation products may partition to the aerosol phase at a higher rate than DMDS. Interestingly, the mixture with isoprene seemed to affect the absorption properties only if DMDS was present (Fig. S5,† bottom panel). This could also support the idea that some of the products from DMDS oxidation partition to the particle phase at a lower ratio than for DMS. No differences were observed between the DMS alone and DMS and isoprene curves.
Methanesulfonic acid and sulfate ratios in bulk secondary aerosol
The major DMS oxidation products found in the secondary aerosols were MSA and sulfate, which has been shown previously.2Fig. 4 shows a comparison of the concentrations of these products measured using ion chromatography. Product molar fractions were calculated as the ratio between moles of sulfate or MSA extracted and the initial amount of DMS and/or DMDS over the duration of the filter sampling experiments. For comparison, Fig. S7† shows aerosol concentrations of sulfate and MSA in μg m−3 for each experiment.Although dominant, the concentration of sulfate showed a significant decrease for DMS oxidation in the presence of isoprene (Fig. S7,†p < 0.05 for a paired t-test). The concentration of MSA remained unchanged with and without isoprene (Fig. S8†). This leads to the conclusion that isoprene might interfere with the DMS oxidation pathway that forms sulfate. In the DMDS experiment, we also found sulfate to be the dominant oxidation product. When isoprene was added, both sulfate and MSA increased, but the change was significant for the latter (Fig. S8,†p < 0.01 for a paired t-test). As opposed to what occurred during the DMS experiment, isoprene does not appear to inhibit either oxidation pathway for DMDS.
Formation of MSA in these experiments is consistent with previous experiments. The reactions were run between 50–70% RH and without addition of NOx. Barnes et al. found little MSA formed under similar low NOx and high RH conditions.35 Van Rooy et al. suggested that NOx and high RH were required to produce a significant amount of MSA.15 We found low concentrations of MSA due to the low NOx concentration. However, adding a non-sulfur compound increased the MSA product concentrations. In contrast to previous chamber work that suggested high concentrations of NOx (in the order of hundreds of ppb) were required for the oxidation pathway of DMS to MSA, our results suggest that in a PAM-OFR this is not the case.
The MSA and sulfate results obtained for mixtures of DMS and DMDS and DMS, DMDS, and isoprene did not show significant changes between each experiment or when compared to DMS alone. This could possibly be attributed, again, to the faster aerosol production exhibited by DMS that outcompeted slower reactions in the DMDS oxidation pathway.
MSA and sulfate ratios in individual secondary aerosols
Online single particle analysis via ATOFMS produced consistent chemical composition results to those found using ion chromatography. Fig. S9† shows the average mass spectra of 60 nm particles produced by the various VOCs and mixtures after they have been coated with matrix 1,8-bis(tetramethylguanidino)naphthalene (TMGN) for real-time analysis using ATOFMS.30 Similar to offline results, the most abundant peaks present in the 60 nm single particle spectra are sulfate and MSA. The ability of the ATOFMS to detect MSA and sulfate ions has been previously demonstrated by Gaston et al., which validates the peak assignments provided here.36 In addition, sulfuric acid (195H2SO4HSO4−) is present as well.37Fig. 5 shows the fraction of particles containing sulfate, MSA, and sulfuric acid along with their respective peak areas after subtracting the background from the matrix. Since ATOFMS cannot determine mass concentrations, the peak area of the ions provides insight on the particle composition. While DMS alone produced the greatest number of particles containing MSA (92%), the VOC mixture of DMS, DMDS, and isoprene combined produced particles with the highest peak area of MSA present. DMDS, both individually and in the presence of isoprene, produced the lowest amount of MSA out of all the mixtures, though there is a slight increase in the number of particles with MSA present once isoprene is added (4.9% to 5.6% with the addition of isoprene). DMS, on the other hand, shows a decrease in the number of particles containing MSA when DMDS or isoprene are added. When comparing the single-particle and bulk data for MSA, while both the DMS and DMDS experiments produced similar amounts of MSA in the bulk, there is significantly less MSA present at 60 nm for DMDS. It appears either MSA produced from DMDS is concentrated in a smaller number of particles than in the DMS experiment or the MSA fraction is lower for this size of particles. We hypothesize particle growth rates and different oxidation mechanisms impact the MSA fraction.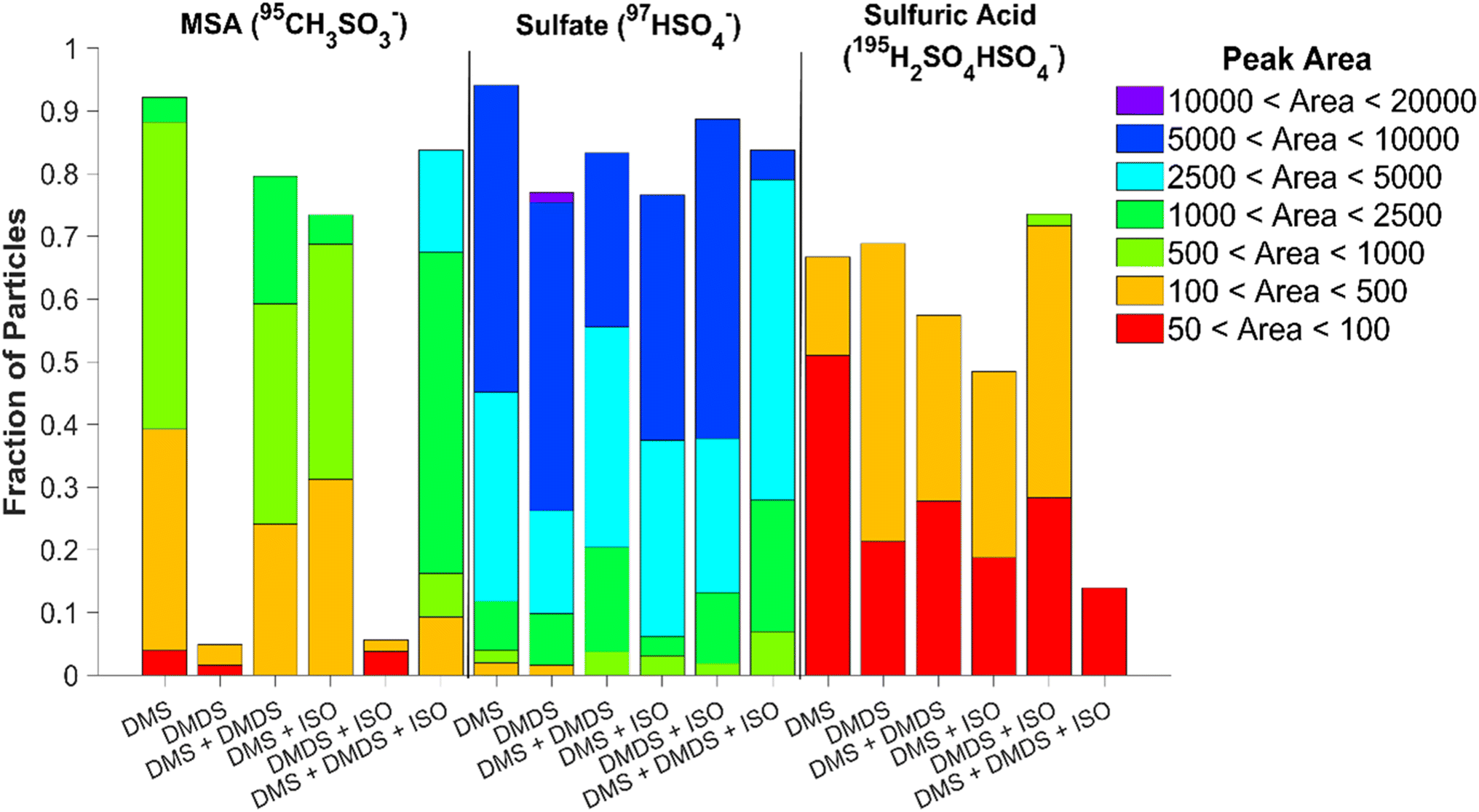 | ||
| Fig. 5 Fraction of 60 nm particles produced by the different VOCs and mixtures that contain MSA, sulfate, and sulfuric acid. Colors represent the fraction of particles that present peak areas falling in specific ranges. | ||
All single particle experiments showed that over 75% of the particles contained sulfate, with DMS producing the greatest number of particles containing sulfate. This number decreases with the addition of DMDS and/or isoprene. The highest sulfate peak area was present in the DMDS experiment, while a larger fraction of sulfate-containing particles was produced by the oxidation of DMDS and isoprene together. While the sulfuric acid peak areas were relatively low for all experiments, the highest values were found in particles produced from the oxidation of DMDS; notably these peak areas increased with the addition of isoprene. To determine if there were any differences in the composition of smaller particles, 20 nm particles were also measured (Fig. S10†). Compared to 60 nm particles, the 20 nm particles show no presence of sulfuric acid but similar trends for sulfate and MSA (Fig. S11†). Increased content of sulfuric acid in single particles has been attributed to longer particle lifetime and cloud processing.37 The higher fraction of sulfuric acid in the DMDS particles is likely due to its faster OH reaction rate compared to DMS; sulfuric acid formation has been shown experimentally in a recent flow reactor study.38 In addition, the particles with the smallest fraction of MSA, those produced from DMDS oxidation, have the largest fraction of sulfuric acid.
Conclusion
The oxidation processes of reduced marine sulfur gas phase compounds have been previously studied in both field and laboratory experiments. In marine environments, sulfur species have been shown to produce secondary marine aerosols that play a critical role in cloud formation. This represents the first laboratory experiment looking at the oxidation of DMDS in a PAM-OFR, as well as the oxidation of mixtures of DMS, DMDS, and isoprene, three major marine VOCs with a crucial role in the atmosphere. We report aerosol yields of reduced sulfur compounds in a PAM-OFR that have not been measured before. We found that DMDS forms more aerosols than DMS at equivalent aging times. DMS particle phase products grow faster than those produced by DMDS oxidation. In the case of DMS, there was enough time in the OFR for condensation to occur without preexisting aerosols added. This shows that, although particle formation processes in the PAM-OFR are dominated by nucleation, fast reactions can allow for condensation processes to also occur.In this study, we determined that oxidation of DMS results in a higher aerosol yield than DMDS and isoprene. At the highest exposure time, almost 90% of DMS is oxidized to the aerosol phase and only 60% and 10% for DMDS and isoprene, respectively. Although DMS had the highest yields, DMS loss was slower than DMDS loss resulting in 52% and 4%, respectively, of unreacted gas at the lowest OH exposure. The remaining 39% of DMDS oxidation products in the gas phase (versus only 6% from DMS) adds a substantial amount of gas phase products that have the potential to alter the composition of the atmosphere impacting further oxidation reactions and enhancing condensation on existing aerosols. This impact has been shown in our DMS and DMDS mixture experiment, which led to a higher yield likely because of gas phase DMDS oxidation products condensing on recently produced aerosols. With DMDS having a lower yield but still producing more particles in number, the mixing ratios of DMDS in marine environments could have a significant impact on the cloudiness over the oceans by increasing the number of cloud condensation nuclei present, potentially influencing marine cloud cover. Interestingly, when mixing DMDS and DMS, the fraction of particles containing MSA, sulfate, and sulfuric acid concentrations decreased compared to DMS alone, for 60 nm particles. These results suggest a potential enhancement of other oxidation products when these two sulfur species are mixed. The presence of DMDS considerably alters the composition, number, and size of secondary aerosols, supporting the need to study other marine-relevant sulfur species, including methanethiol. In addition to marine-derived sulfur compounds, anthropogenic sulfur species, such as benzothiazole, should also be studied as they have been shown to be present in marine environments.39
DMS and DMDS are known to produce similar OH oxidation products as shown in previous chamber experiments.15,40 We found that for both VOCs the main oxidation product was sulfate followed by MSA and, when mixed, the product ratio between the two was altered. Although the comprehensive characterization of all oxidation products, e.g. by untargeted methods, is outside the scope of this work, on-going experiments using electrospray ionization coupled to high-resolution Orbitrap mass spectrometry have confirmed MSA and sulfate as main oxidation products, followed by a plethora of sulfur-containing organic compounds in low concentrations. Fig. S12† shows positive and negative mode Orbitrap spectra from the mixture of DMS, DMDS, and isoprene. Our results also suggest that the production of MSA in a humid environment did not require the addition of NOx into the system. The introduction of isoprene to the mix exhibited a contrasting effect on these sulfur compounds: while it reduced sulfate concentration in DMS oxidation, it enhanced both sulfate and MSA levels in DMDS. Furthermore, the different composition of the single particle aerosols from mixtures of these VOCs will likely impact the composition of the atmosphere. The combination of the three intensified the amount of MSA and sulfate present compared to each individual gas, leading to a potential increase of acidity in the atmosphere. This emphasizes the complex interactions between trace gases in atmospheric systems. Even a single compound like isoprene can markedly alter DMS and DMDS oxidation pathways. This study underscores the importance of examining mixtures to comprehend alterations in oxidation products and relative concentrations. Future work will expand the complexity of these mixtures by sampling marine air in coastal environments through the PAM-OFR to explore the impact on aerosol production, thus complementing recent studies carried out by our group and collaborators on marine VOC emissions from laboratory mesocosms.41 Moreover, while experiments were always performed at a humidity close to 70%, the present work did not explore the influence of relative humidity and is certainly a point to be addressed in future studies. Going forward, further research is required to unravel the mechanisms that govern the impact of marine organic compounds on the oxidation of reduced sulfur compounds, as well as the presence of oxidants other than OH (e.g. NOx, Cl), thus shedding light on the behavior of secondary marine aerosols and their influence in cloud formation and lifetime.
Author contributions
ANM, LC, and KAP conceptualized the study. ANM, LC, and KAK conducted the experiments. ANM, LC, and KAK analyzed the data and composed the figures. ANM and LC led the manuscript writing. ANM, LC, KAK, and KAP contributed to the editing and reviewing of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation through the NSF Center for Aerosol Impacts on Chemistry of the Environment, an NSF Center for Chemical Innovation (CHE-1801971). Any opinions, findings, and conclusions, or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. We thank Kathryn J. Mayer, Neal Arakawa, Jonathan H. Slade and Robert S. Pomeroy for their assistance and helpful feedback.References
- K. G. S. Dani and F. Loreto, Trade-Off Between Dimethyl Sulfide and Isoprene Emissions from Marine Phytoplankton, Trends Plant Sci., 2017, 22(5), 361–372 CrossRef CAS PubMed.
- T. S. Bates, J. A. Calhoun and P. K. Quinn, Variations in the methanesulfonate to sulfate molar ratio in submicrometer marine aerosol particles over the south Pacific Ocean, J. Geophys. Res. Atmos., 1992, 97(D9), 9859–9865 CrossRef CAS.
- Q. Ye, et al., Product distribution, kinetics, and aerosol formation from the OH oxidation of dimethyl sulfide under different RO2 regimes, Atmos. Chem. Phys., 2022, 22(24), 16003–16015 CrossRef CAS.
- H. Berresheim, et al., Gas-aerosol relationships of H2SO4, MSA, and OH: Observations in the coastal marine boundary layer at Mace Head, Ireland, J. Geophys. Res. Atmos., 2002, 107(D19), 12 CrossRef.
- E. S. Saltzman, et al., Methane sulfonic acid in the marine atmosphere, J. Geophys. Res. Oceans, 1983, 88(NC15), 897–902 CrossRef.
- M. Kulmala, et al., Chemistry of Atmospheric Nucleation: On the Recent Advances on Precursor Characterization and Atmospheric Cluster Composition in Connection with Atmospheric New Particle Formation, in Annual Review of Physical Chemistry, ed. M. A. Johnson and T. J. Martinez, Annual Reviews, Palo Alto, 2014, vol. 65, pp. 21–37 Search PubMed.
- R. Y. Zhang, et al., Nucleation and Growth of Nanoparticles in the Atmosphere, Chem. Rev., 2012, 112(3), 1957–2011 CrossRef CAS PubMed.
- U. Lohmann and J. Feichter, Global indirect aerosol effects: a review, Atmos. Chem. Phys., 2005, 5, 715–737 CrossRef CAS.
- S. Twomey, Pollution and planetary albedo, Atmos. Environ., 1974, 8(12), 1251–1256 CrossRef.
- K. J. Mayer, et al., Secondary Marine Aerosol Plays a Dominant Role over Primary Sea Spray Aerosol in Cloud Formation, ACS Cent. Sci., 2020, 6(12), 2259–2266 CrossRef CAS PubMed.
- A. L. Hodshire, et al., The potential role of methanesulfonic acid (MSA) in aerosol formation and growth and the associated radiative forcings, Atmos. Chem. Phys., 2019, 19(5), 3137–3160 CrossRef CAS.
- M. Dall'Osto, et al., Arctic sea ice melt leads to atmospheric new particle formation, Sci. Rep., 2017, 7, 10 CrossRef PubMed.
- S. Becagli, et al., Factors controlling atmospheric DMS and its oxidation products (MSA and nssSO42-) in the aerosol at Terra Nova Bay, Antarctica, Atmos. Chem. Phys., 2022, 22(14), 9245–9263 CrossRef CAS.
- B. Jiang, et al., Modification of the Conversion of Dimethylsulfide to Methanesulfonic Acid by Anthropogenic Pollution as Revealed by Long-Term Observations, ACS Earth Space Chem., 2021, 5(10), 2839–2845 CrossRef CAS.
- P. Van Rooy, et al., Methanesulfonic acid and sulfuric acid Aerosol Formed through oxidation of reduced sulfur compounds in a humid environment, Atmos. Environ., 2021, 261, 7 CrossRef.
- E. Ahlberg, et al., Secondary organic aerosol from VOC mixtures in an oxidation flow reactor, Atmos. Environ., 2017, 161, 210–220 CrossRef CAS.
- M. Claeys, et al., Formation of secondary organic aerosols through photooxidation of isoprene, Science, 2004, 303(5661), 1173–1176 CrossRef CAS PubMed.
- A. Lambe, et al., Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 2015, 250, 1 Search PubMed.
- T. Y. Chen and M. Jang, Secondary organic aerosol formation from photooxidation of a mixture of dimethyl sulfide and isoprene, Atmos. Environ., 2012, 46, 271–278 CrossRef CAS.
- E. Kang, et al., Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 2007, 7(22), 5727–5744 CrossRef CAS.
- Z. Peng and J. L. Jimenez, Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 2020, 49(9), 2570–2616 RSC.
- W. C. Chen and R. A. Marcus, On the theory of the reaction rate of vibrationally excited CO molecules with OH radicals, J. Chem. Phys., 2006, 124(2), 5 Search PubMed.
- R. Atkinson, et al., Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Supplement IV. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry, J. Phys. Chem. Ref. Data, 1992, 21(6), 1125–1568 CrossRef CAS.
- R. Atkinson, et al., Evaluated kinetic and photochemical data for atmospheric chemistry: Supplement III, Int. J. Chem. Kinet., 1989, 21(2), 115–150 CrossRef CAS.
- J. B. Burkholder, et al., Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies. Evaluation Number 19, Jet Propulsion Laboratory, Pasadena, 2019 Search PubMed.
- M. J. Kim, et al., Revisiting benzene cluster cations for the chemical ionization of dimethyl sulfide and select volatile organic compounds, Atmos. Meas. Tech., 2016, 9(4), 1473–1484 CrossRef.
- A. Lavi, et al., The sensitivity of benzene cluster cation chemical ionization mass spectrometry to select biogenic terpenes, Atmos. Meas. Tech., 2018, 11(6), 3251–3262 CrossRef CAS.
- E. Gard, et al., Real-time analysis of individual atmospheric aerosol particles: Design and performance of a portable ATOFMS, Anal. Chem., 1997, 69(20), 4083–4091 CrossRef CAS.
- K. A. Pratt, et al., Development and Characterization of an Aircraft Aerosol Time-of-Flight Mass Spectrometer, Anal. Chem., 2009, 81(5), 1792–1800 CrossRef CAS PubMed.
- L. Li, et al., Nanoparticle growth in thermally diffusive sublimation-condensation systems with low vapor pressure solids, J. Aerosol Sci., 2023, 173, 13 CrossRef.
- W. W. Hu, et al., Volatility and lifetime against OH heterogeneous reaction of ambient isoprene-epoxydiols-derived secondary organic aerosol (IEPOX-SOA), Atmos. Chem. Phys., 2016, 16(18), 11563–11580 CrossRef CAS.
- J. H. Kroll, et al., Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol, Phys. Chem. Chem. Phys., 2009, 11(36), 8005–8014 RSC.
- J. R. Odum, et al., Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 1996, 30(8), 2580–2585 CrossRef CAS.
- J. H. Slade, et al., Nitrate radical oxidation of γ-terpinene: hydroxy nitrate, total organic nitrate, and secondary organic aerosol yields, Atmos. Chem. Phys., 2017, 17(14), 8635–8650 CrossRef CAS.
- I. Barnes, J. Hjorth and N. Mihalopoulos, Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere, Chem. Rev., 2006, 106(3), 940–975 CrossRef CAS PubMed.
- C. J. Gaston, et al., Real-Time Detection and Mixing State of Methanesulfonate in Single Particles at an Inland Urban Location during a Phytoplankton Bloom, Environ. Sci. Technol., 2010, 44(5), 1566–1572 CrossRef CAS PubMed.
- K. A. Pratt and K. A. Prather, Aircraft measurements of vertical profiles of aerosol mixing states, J. Geophys. Res. Atmos., 2010, 115, 10 Search PubMed.
- T. Berndt, et al., Direct sulfuric acid formation from the gas-phase oxidation of reduced-sulfur compounds, Nat. Commun., 2023, 14(1), 10 Search PubMed.
- E. B. Franklin, et al., Atmospheric Benzothiazoles in a Coastal Marine Environment, Environ. Sci. Technol., 2021, 55(23), 15705–15714 CrossRef CAS PubMed.
- P. Van Rooy, et al., Characterization of secondary products formed through oxidation of reduced sulfur compounds, Atmos. Environ., 2021, 256, 12 CrossRef.
- D. B. Kilgour, et al., Marine gas-phase sulfur emissions during an induced phytoplankton bloom, Atmos. Chem. Phys., 2022, 22(2), 1601–1613 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00169e |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |