
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of aging conditions at equal OH exposure in an oxidation flow reactor on the composition of toluene-derived secondary organic aerosols†
Hendryk
Czech
 *abc,
Pasi
Yli-Pirilä
b,
Petri
Tiitta‡
b,
Mika
Ihalainen
b,
Anni
Hartikainen
b,
Eric
Schneider
ad,
Patrick
Martens§
a,
Andreas
Paul
ae,
Thorsten
Hohaus
e,
Christopher P.
Rüger
ad,
Jorma
Jokiniemi
b,
Ralf
Zimmermann
acd and
Olli
Sippula
bf
*abc,
Pasi
Yli-Pirilä
b,
Petri
Tiitta‡
b,
Mika
Ihalainen
b,
Anni
Hartikainen
b,
Eric
Schneider
ad,
Patrick
Martens§
a,
Andreas
Paul
ae,
Thorsten
Hohaus
e,
Christopher P.
Rüger
ad,
Jorma
Jokiniemi
b,
Ralf
Zimmermann
acd and
Olli
Sippula
bf
aChair of Analytical Chemistry, University of Rostock, 18059, Rostock, Germany. E-mail: hendryk.czech@uni-rostock.de
bFine Particle and Aerosol Technology Laboratory, Department of Environmental and Biological Science, University of Eastern Finland, 70211, Kuopio, Finland
cCooperation Group “Comprehensive Molecular Analytics” (CMA), Helmholtz Zentrum München, 81379, München, Germany
dDepartment “Life, Light and Matter”, University of Rostock, 18059, Rostock, Germany
eInstitute of Energy and Climate Research: Troposphere (IEK-8), Forschungszentrum Jülich, 52425, Jülich, Germany
fDepartment of Chemistry, University of Eastern Finland, 80101, Joensuu, Finland
First published on 7th June 2024
Abstract
Oxidation flow reactors (OFRs) have been increasingly used to conduct research on secondary aerosol formation potential and composition in laboratory and field studies by exposing aerosols to high levels of oxidants in short time periods. In order to assess the atmospheric relevance of the triggered chemical reactions, kinetic models have been developed to reveal the production of atmospheric oxidants and the fate of volatile organic compounds (VOCs). However, it is unknown how different OFR conditions generating the same OH exposure affect the chemical and physical properties of the secondary aerosol particle phase because the model is based on gas phase chemistry. Toluene as a well-investigated precursor of secondary organic aerosols (SOAs) was aged in the high-volume OFR “PEAR” at three different external OH reactivities (OHRext) and in an environmental chamber at the same OH exposure of (1.09 ± 0.09) × 1011 s cm−3. These specific OFR conditions altered the majority of the investigated chemical and physical properties of the toluene-derived SOA (tol-SOA). However, OFR-aging at low OHRext associated with atmospherically relevant conditions did not lead to physical and chemical SOA properties most similar to chamber-generated SOAs at the same low OHRext. Particularly, for the most detailed chemical analysis by electrospray (ESI) high-resolution Orbitrap mass spectrometry (HRMS), tol-SOA from the PEAR at high OHRext, deviating from atmospherically relevant conditions, and the chamber were most different to tol-SOA from the PEAR with low and medium OHRext. Our study challenges the concept of atmospherically relevant OFR aerosol aging and motivates further exploration of OFR conditions on secondary aerosol composition.
Environmental significanceOxidation flow reactors (OFRs) became a widely used device for laboratory atmospheric aging studies, especially for bridging the gap between the maximum age achievable in an environmental chamber and aerosol particle lifetime in the atmosphere. In OFRs, aerosols are exposed to high concentrations of oxidants within a short time in order to cover exposures to OH radicals equivalent to up to two weeks. This approach has been criticized to induce irrelevant chemical reactions, so kinetic models have been developed to define constraints for atmospherically relevant OFR operation. Experimental results from toluene photooxidation challenge the concept of atmospherically relevant OFR conditions and explore which physical-chemical properties of secondary organic aerosols from toluene photooxidation may be reproduced from complementary experiments in an environmental chamber. |
1. Introduction
Secondary organic aerosols (SOAs) from the oxidation of volatile organic compounds (VOCs) largely contribute to the ambient organic aerosol burden from both anthropogenic and biogenic sources.1,2 Depending on the detailed chemical composition and optical properties, SOAs may affect the climate3–5 and human health.6–9 The oxidative gas-to-particle conversion of VOCs to SOAs is initiated by reactions with hydroxyl (OH) radicals, ozone (O3), and nitrate radicals (NO3) or via photolysis, with OH being the dominant atmospheric oxidant during daytime.10Laboratory atmospheric aging of VOCs, VOC mixtures including plant emissions, and combustion aerosols is conducted to understand atmospheric processes and composition as a bottom-up approach.11 Environmental chambers are well-established and regarded as the most realistic model for laboratory atmospheric aging. Both aerosol and atmospheric oxidants are injected or generated at near-ambient levels, so the aerosol residence in the environmental chamber appears close to the atmospheric residence time of an aerosol under equivalent conditions.12 Losses to the inner chamber walls continuously decrease aerosol concentrations, which can be corrected by inert tracers or experiments with model aerosols without aging. Thus, the uncertainty of aerosol measurement increases with longer residence in environmental chambers. Consequently, atmospheric aging in environmental chambers of common size has an inherent upper limit of about two equivalent days in the atmosphere. Therefore, some environmental chambers with large volumes were built to minimize wall effects for studies on atmospheric aerosol concentrations at remote sites (SAPHIR with 270 m3)13 to the upper level like in dense wildfire plumes (PHOTO-LAC with 1800 m3).14 Nevertheless, residence times of aerosol particles in the troposphere of up to two weeks cannot be reached.
Oxidation flow reactors (OFRs) expose aerosols to high levels of OH radicals on the time scale of a few minutes.15 In OFR254 mode, OH radicals are generated from the photolysis of O3 at 254 nm to singlet oxygen O(1D), which subsequently reacts with water vapor to give two OH radical equivalents. Alternatively, ozone may be generated in situ by first photolysis of molecular oxygen (O2) to triplet oxygen atoms O(3P) (OFR185 mode) and subsequent reaction with O2. Moreover, OH radicals may be formed by the photolysis of water vapor.15 In these ways, high OH exposures (time × OH concentration) of 1 × 1012 s cm−3 equivalent to up to 14 days and beyond may be reached, and were used to study OH-initiated SOA formation from VOCs,16,17 combustion aerosols,18,19 ambient air20,21 and also heterogeneous oxidation of carbonaceous particulate matter.22,23 However, concerns have been raised that OFR chemistry may be generally irrelevant to tropospheric chemistry.15
Peng et al. (2016, 2017, and 2019)24–27 developed a model for the potential aerosol mass (PAM) OFR28,29 to define the space of atmospherically relevant reactions and beyond, for example by the classes “safer”, “transition” and “riskier” concerning the contribution of non-OH oxidation reactions under low-NOx conditions, or the fate of peroxyalkyl radicals (RO2). This model is based on gas phase chemistry and assesses the atmospherically relevant space using the ratios of atmospheric oxidants and established reaction pathways. The crucial parameters of OFR aging are the external OH reactivity (OHRext), which is the sum of the product of reactive species concentrations with their rate constants with OH radicals, the photon flux, ozone addition or in situ generation, and the level of humidity as well as the level of NOx. Combined with the OFR residence time, the OFR fate of individual reactive species, such as by reactions with OH radicals, ozone or photolysis, may be calculated and compared to the fate under real atmospheric conditions. However, it is unknown how OFR conditions of different atmospheric relevance affect the physical and chemical properties of the formed secondary aerosol particles, which has consequences for toxicological studies with SOAs and aged aerosols,30–33 because the atmospheric representativity of the formed aerosol particle characteristics presumably plays a critical role.
In this study, the SOA precursor toluene was photochemically aged in the high-volume OFR “PEAR”,34 operated in OFR254-7 mode (external feed of 7 ppm ozone into the OFR and photolysis at 254 nm) without NOx addition.15 Toluene concentrations and photon flux were adjusted in order to obtain “safer”, “transition” and “riskier” OFR conditions at an equal OH exposure, i.e. an equal photochemical equivalent age.24 Furthermore, photochemical aging of toluene was conducted in an environmental chamber as a reference for the most realistic laboratory model for atmospheric processing. The physical and chemical properties of a toluene-derived SOA (tol-SOA) under all conditions were analyzed by several online and offline analytical techniques and discussed concerning the effect of OFR conditions and the similarity of OFR-generated tol-SOA to chamber-generated tol-SOA.
2. Materials and methods
2.1. Generation of tol-SOA
Different concentrations of toluene vapor (“low”, “medium”, and “high”) were generated by gentle heating of a reservoir filled with toluene. The reservoir was connected by stainless steel tubing, so toluene vapor entered the main flow of 100 L min−1 of purified air (737-15 zero air generator; Aadco Inc., VT, USA) to the “Photochemical Emission Aging flowtube Reactor” (PEAR), so the mode of the residence time distribution appeared at ∼70 s. The PEAR is a high-volume oxidation flow reactor made of stainless steel with a volume of 139 L and an inner surface to volume ratio of 16.4 m−1. It consists of a conical inlet diffuser, a cylindrical center part and a conical outlet. Inside the cylinder center part, four UV lamps are mounted, which may be turned on and off individually. More information on the PEAR reactor may be found elsewhere.34Prior to entering the PEAR, water vapor, ozone, and toluene vapor were mixed (OFR254-7 mode) to obtain 50% relative humidity, 7 ppm of ozone and three different toluene concentrations. The three different settings for the PEAR were chosen to achieve “safer”, “transition” and “riskier” according to the “OFR Exposures Estimator v3.1”24,27 by varying toluene concentrations (and consequently OHRext) and UV lamp intensity but keeping the same OH exposure of (1.09 ± 0.09) × 1011 s cm−3 (Table 1). OH exposure was determined online using the ratio of introduced toluene into the PEAR and toluene at the PEAR outlet, according to the concept of “photochemical clock”,35 which was measured by single-photon ionization time-of-flight mass spectrometry (SPI-TOFMS, see Section 2.2.1.). Particle measurements and sampling after the PEAR were corrected for wall losses using the loss function for silver particles at 100 L min−1 as provided in Ihalainen et al. (2019).34 Due to the assumed relatively high volatility of tol-SOA, true losses are likely higher, so the wall loss-corrected results denote a lower limit of particle concentrations.
| Experiment | [tol]in [ppb] | OHRext [s−1] | OHexp [s cm−3] | Photo flux @ 254 nm [cm−2 s−1] | OFR conditions | Y SOA [%] |
|---|---|---|---|---|---|---|
| low1 | 316 | 37 | 9.94 × 1010 | 8.71 × 1014 | Safer | 4.4 |
| low2 | 335 | 39 | 1.05 × 1011 | 8.71 × 1014 | Safer | 4.3 |
| low3 | 298 | 35 | 1.16 × 1011 | 8.71 × 1014 | Safer | 3.1 |
| med1 | 680 | 79 | 1.07 × 1011 | 1.76 × 1015 | Transition | 5.0 |
| med2 | 673 | 78 | 1.10 × 1011 | 1.76 × 1015 | Transition | 4.5 |
| med3 | 663 | 77 | 1.17 × 1011 | 1.76 × 1015 | Transition | 3.8 |
| high1 | 3840 | 445 | 1.08 × 1011 | 7.06 × 1016 | Riskier | 6.2 |
| high2 | 3850 | 446 | 1.09 × 1011 | 7.06 × 1016 | Riskier | 5.8 |
| high3 | 4010 | 465 | 1.14 × 1011 | 7.06 × 1016 | Riskier | 4.8 |
| cham1 | 309 | 36 | 9.99 × 1010 | 0 | — | 39.7 |
| cham2 | 303 | 35 | 1.07 × 1011 | 0 | — | 40.5 |
| cham3 | 313 | 36 | 1.07 × 1011 | 0 | — | 39.9 |
Complementary to OFR-aging, the experiment was repeated under similar conditions in the ILMARI smog chamber of 27 m3 volume36 with a target concentration as for “low” OFR conditions (Table 1). As an OH source without NOx generation, H2O2 was added to the chamber with an air flow over one hour by vaporizing 1 mL of an aqueous H2O2 solution in a flask. The resulting concentration of H2O2 was estimated to be <2.7 ppm, which is similar to other chamber experiments.37 Subsequently, toluene was injected into the chamber to reach the same concentration as used in “safer” OFR aging, which required about 5 h of residence. Finally, 340 nm blacklights were turned on with full power for approximately five hours until the same OH exposure as in OFR experiments was reached. The background concentrations of PM, NO2 and O3 in the chamber were below 0.3 μg m−3, 4 ppb and 2.4 ppb, respectively. All chamber experiments were conducted at a relative humidity of 50% and a temperature of approximately 20 °C. Particle measurements and sampling from the chamber were corrected for wall losses using the loss function for polydisperse ammonium sulfate particles as provided in Leskinen et al. (2015).36 Due to the assumed relatively high volatility of tol-SOA, true losses are also likely higher in the chamber, so the wall loss-corrected results denote a lower limit of particle concentrations. Online data on chamber aging were obtained by data acquisition for 30 min after UV lamps had been switched off.
A summary of the experimental conditions is listed in Table 1.
The assessment of non-OH chemistry24,27 revealed that the reactions of methane, ethane, and benzene with O(1D) and O(3P) would be comparable to ambient air conditions, while the reactions of terpenes α-pinene and γ-terpenene as well as isoprene would be underestimated in all three OFR conditions. The photolysis of toluene at 254 nm becomes slightly overestimated in “high” with 10% atmospheric fate, while acetylacetone photolysis accounts for 55%, which appears clearly outside the considered atmospherically relevant range. However, the model proposed by Peng et al. (2016)24 assumes photodissociation quantum yields of unity, so the model prediction denotes an upper limit for the fate of photolysis. Therefore, the fates of toluene in the PEAR may be closer to the fates in the atmosphere and other processes determine potential deviations from atmospheric SOA formation. Experiments with toluene exposed to different intensities of 254 nm UV light without the addition of ozone may reduce this bias in the model result.
Organic peroxy radicals (RO2) may be formed from the reaction of O2 with carbon-centered radicals. The subsequent atmospheric fate of RO2 involves possible reactions with hydroperoxyl radicals (HO2), NO or previous RO2 isomerization. In the case of primary RO2 and acyl RO2, reactions between different RO2 (RO2 + RO2) may be significant in the atmosphere but are often underrepresented in OFR aging. In OFR aging with high levels of OH and high OHRext, the RO2 fate is dominated by OH (RO2 + OH), deviating from atmospheric processing.26 Furthermore, sufficiently long RO2 lifetimes for a relevant contribution of isomerization to the RO2 fate may be achieved by conditions with high UV intensities, but then photolysis at 254 nm becomes a relevant fate of toluene, which is not anymore in the atmospherically relevant range either. Thus OFR254-7 conditions cannot lead to overall good, i.e., atmospherically relevant aging.26 Even in “low” belonging to “safer” OFR conditions (Table 1), RO2 + OH accounted for 56.3%, which increased in “high”” to 87.9%, with accordingly minor contributions from RO2 + HO2 and negligible contributions from RO2 + RO2 and RO2 isomerization (Table S1†). However, the fates of individual RO2 radicals depend on the inherent RO2 structure and may significantly deviate from the model result.
2.2. Online measurements
The background concentrations of reactive gases O3, NO and NO2 were analyzed using a trace-level chemiluminescence NO–NO2–NOx analyzer (Thermo 42i-TL; Thermo Fisher Scientific Inc., USA) and a UV photometric ozone analyzer (Thermo 49i; Thermo Fisher Scientific Inc., USA).
 | (1) |
The optical properties of OFR-derived tol-SOA were analyzed using a seven-wavelength aethalometer (AE33; Magee Scientific, USA). Wavelength pairs 370 and 520 nm were used to derive the Ångström Absorption Exponent (AAE),
 | (2) |
The chemical composition was analyzed with a soot-particle time-of-flight aerosol mass spectrometer (SP-AMS, Aerodyne Research Inc., USA), operated in tungsten vaporizer mode only because of the absence of soot particles in the experiments. The vaporizer temperature was set to 600 °C and volatiliz non-refractory particle components such as organic aerosols (OAs), nitrate, ammonium, sulfate and chloride. Intensities at m/z 44 were corrected for interfering carbon dioxide with a trace gas analyzer (GMP 343; Vaisala Oy, Finland). Elemental ratios O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C and H:C of OAs were determined by the “improved ambient method”,42 which allows the calculation of the average carbon oxidation state (OSC) according to
:C and H:C of OAs were determined by the “improved ambient method”,42 which allows the calculation of the average carbon oxidation state (OSC) according to
| OSC ≈ 2·O:C − H:C | (3) |
Further details of the SP-AMS operation can be found in Hartikainen et al. (2020).18
2.3. PM filter sampling
Samples of PM were collected after the PEAR on quartz fiber filters of 47 mm in diameter (Munktell, Filtrak; Sweden) through 6 mm stainless steel tubing at a flow rate of 20 L min. In order to reduce differences in filter loads, PM was collected for 3.5 h, 2 h and 0.5 h under “low”, “medium” and “high” OFR conditions with total carbon filter loads from 20 to 90 μg cm−2. At the end of each chamber experiment, PM samples were collected for 1 h at a flow rate of 60 L min−1 to obtain the same filter loading as for “low”. All filter samples were immediately stored after collection at −20 °C.2.4. Offline PM analysis
Filter samples were analyzed by multi-wavelength thermal optical carbon analysis (MW-TOCA) hyphenated to SPI-TOFMS. The MW-TOCA consisted of a DRI Model 2001 A carbon analyzer, which was retrofitted with seven laser diodes of wavelengths (λ) in visible UV and near-infrared ranges, replacing the He–Ne laser for optical correction of charring.43 This modification enables the measurement of laser transmittance over a spectral range and connects the optical properties of PM to their carbon content. Due to the low filter load and inherent absence of elemental carbon in tol-SOA, no correction for the shadow effects was applied44 and ATN was related to the carbon content of tol-SOA to obtain the mass absorption efficiency MAE for each λ: | (4) |
The thermal refractiveness of the tol-SOA and its carbon content were determined according to the Improve_A temperature protocol, formally defining four fractions of organic carbon (OC1–OC4) and three fractions of elemental carbon (EC1–EC3).46 In all samples of this study, the laser transmittance returned to its baseline value not before EC3, and thus no true EC was measured but only pyrolytic OC. Nevertheless, the fractionation of EC was kept in the discussion because it might reveal tol-SOA properties.
Evolved gas analysis was conducted at a modified quartz inlet of the TOCA, so an aliquot of the total flow was taken from the oven and analyzed by SPI-TOFMS47 as described in Section 2.2.1. SPI mass spectra were acquired at 1 s time resolution and summed to one spectrum per carbon fraction. In principle, SPI with 118 nm photons may ionize all compounds with an ionization energy below 10.49 eV, mainly generating molecular ions. However, ion yields may vary between different compound classes up to one order of magnitude.48
For tol-SOA analysis by high-resolution Orbitrap mass spectrometry, half of each PM filter sample was extracted by adding 5 mL of methanol (LC-MS grade) with gentle shaking for 60 min, and then filtered with a 0.2 μm pore size PTFE membrane (Sartorius, Goettingen, Germany) in a stainless-steel filter using a glass syringe. Mass spectrometric analysis was performed on a Velos Orbitrap (Thermo Scientific, Bremen, Germany) with electrospray ionization in both positive and negative ionization modes (ESI+/ESI−). ESI was operated with a direct-infusion ion source electrospray setup using a spray voltage of 4.2/3.4 kV and a syringe flow rate of 5 μL min. High-resolution data in the frequency domain were collected in the equivalent range of 50 to 1000 Da, resulting in a resolving power of >100000 at m/z 400 and a mass accuracy below 3 ppm. The exported mass spectra were processed using in-house MATLAB (R2020; The MathWorks, USA) functions driven by the graphical user interface “CERES Processing”. For elemental composition and sum formula assignments, the following restrictions were deployed in the range of 70 to 650 Da for the sum formula CcHhOo with 3 ≤ c ≤ 35, 3 ≤ h ≤ 60, o ≤ 24, and a maximum error of 3 ppm. Additional restrictions of 0.4–2.4 for H:C, 0–1.4 for O:C and 0–20 double bond equivalents (DBEs) were applied.
2.5. Statistical data analysis
Statistical testing was conducted with the Statistic Toolbox in MATLAB (R2020; The MathWorks, USA). Analysis of variance (ANVOA) was performed with Bonferroni correction in multiple testing at a significance level of 0.05 using the commands “anova1” and “multcompare”. Principal component analysis was performed on the covariance matrix of L1-normalized SPI mass spectra using the command “pca”. Correlations were either calculated as unadjusted correlation (or congruence, rc) for pattern comparison or as the Pearson correlation coefficient (r).3. Results
3.1. Particle concentration, size distribution and SOA yields
The particle sizes generated under three different OFR conditions showed a shift toward larger particle size modes from “low” to “high” (Fig. S1†). While “low” and “medium” generated particles with geometric mean diameters (GMD) from 32 to 38 nm and 35 to 42 nm, respectively, “high” OFR conditions increased the GMD to 60 to 63 nm. However, all particle sizes of OFR-generated tol-SOA were significantly smaller than those of chamber-generated tol-SOA, which peaked between 150 and 200 nm. In a mass-based size distribution, the particle size distributions of all OFR conditions interfere more with GMDs of 90 to 97 nm, 99 to 114 nm, and 120 to 126 nm, but still it is evident that the concentration of tol-SOA originates from larger particles generated in chamber experiments with GMDs from 180 to 128 nm (Fig. 1). Furthermore, with a YSOA of only 3.1 to 6.2%, OFR-aging generated significantly less SOA than chamber-aging with a YSOA of ∼40%, which agrees well with the conclusion of Lambe et al. (2015)49 on generally higher YSOA in chamber- than in PAM OFR-aging. YSOA typically increases exponentially with the aerosol concentration in chamber experiments;40 however, when comparing OFR-aging in “low” and “high”, the more than one order of magnitude higher concentration of toluene and the resulting SOA caused only an increase in YSOA from (3.9 ± 0.7) % to (5.6 ± 0.7) %.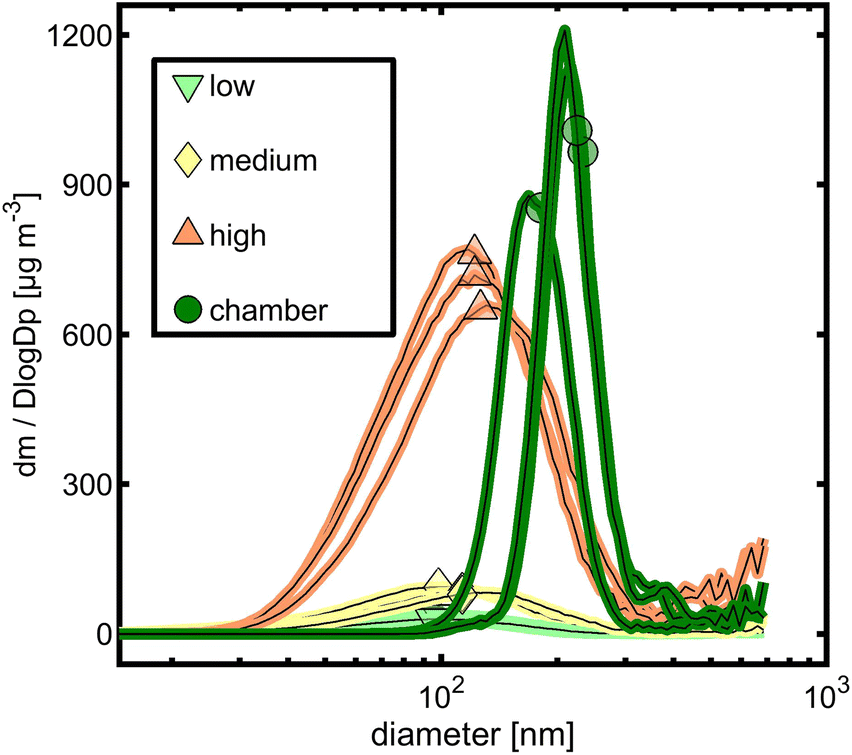 | ||
| Fig. 1 Size distributions derived from the SMPS based on particle mass concentration assuming spherical particles with a density of 1.5 g cm−3. Symbols represent the geometric mean particle diameter. | ||
In SOA formation, organic vapors are oxidized in the gas phase and form low-volatile organic compounds (LVOCs), condensing on existing particles or forming new particles by nucleation. The lifetime of LVOCs is determined by their conversion into the particle phase (τaer), further oxidation to products of higher volatility (τOH) and losses to the walls (τwall).18 The vast majority of LVOCs condense on particles under all OFR conditions when using the particle size distribution after OFR processing (Table S2†). However, it is unknown how fast surface area particles are formed, and hence the results give an upper limit. When using only half of the particle number concentration measured after the PEAR, 10 to 19% of the LVOCs exit the OFR in the gas phase under “low” conditions (Table S3†). This tendency is in line with the observation of decreasing YSOA from “high” to “low”, but it would require an even lower condensation sink than half of the measured particle size distribution to explain quantitative differences in YSOA. In the chamber, the same OH exposure is received by toluene, but with a longer residence time and lower ratio of surface to volume compared to the PEAR. A quantity to compare the fate of LVOCs in the OFR and chamber provides the ratio condensation sink concerning condensation on particles CSpar related to losses on the inner wall CSwall. With 409 ± 50, “high” OFR conditions are closer to 350 ± 65 for chamber-aging, while “low” and “medium” OFR conditions have a CSpar/CSpar of 30 ± 6 and 65 ± 14, respectively. Hence, “high” conditions suppress LVOC wall losses on a relative scale, similar to the conditions in the chamber.
Lambe et al. (2015)49 modeled the time required for a gas phase species to condense on pre-existing particles, and thus when seed particles were used, they provided an initial condensation sink in contrast to our study. With the assumption of a 10–100 μm2 cm−3 particle surface, 150 g mol−1 average SOA molar mass and an accommodation coefficient of 1, a substantial fraction of oxidized vapors exit the reactor in the gas phase, this not increasing YSOA. In our study, the longer residence time of 5 h compared to the PEAR residence time of 70 s cancels out the effect from the 10 times higher OHRext (and 3 to 4 times higher particle surface area) under “high” OFR conditions compared to “cham” and provides an explanation for the significant differences in YSOA between the OFR and chamber.
3.2. Optical properties
In the absence of NOx, toluene is less susceptible to the formation of light-absorbing organic carbon (“brown carbon”, BrC) due to the decomposition of aromatic rings as chromophores and the absence of the formation of strongly absorbing nitroaromatics by addition of NO2.50 The MAEλ is a measure of light absorption related to the amount of particulate carbon. Because of small attenuation in laser transmittance after filter sample penetration, MAEλ could be determined only for 405 and 450 nm in the MW-TOCA. The mean MAEλ values for all OFR and chamber experiments appeared closely together, and the associated experimental and measurement uncertainties do not allow us to conclude an effect from the aging conditions (Fig. 2a).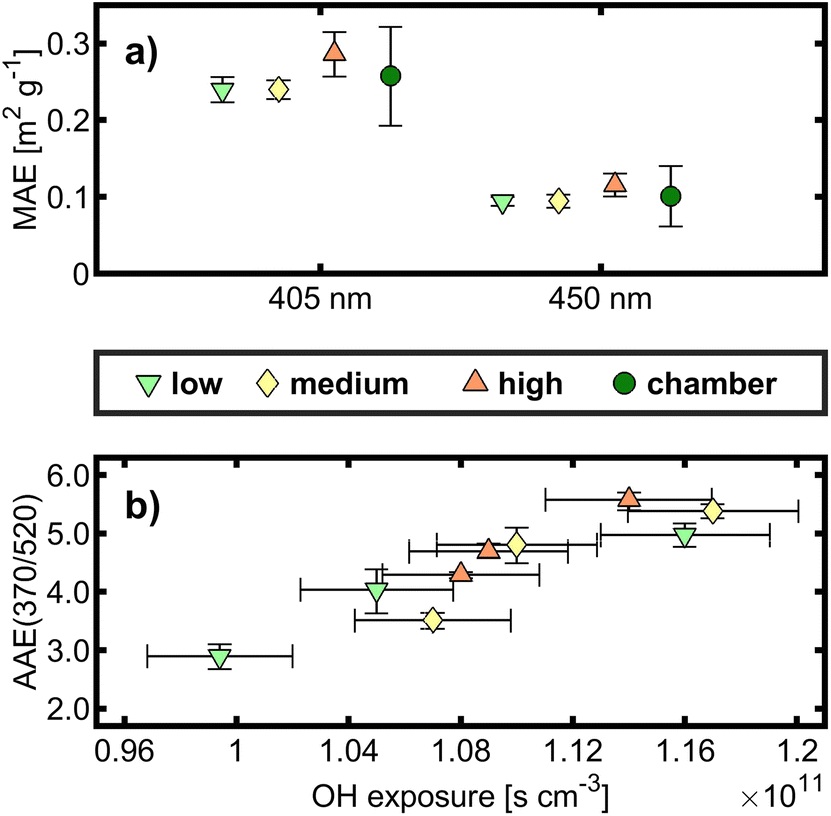 | ||
| Fig. 2 (a) Mean Mass Absorption Efficiency (MAE) related to total carbon (TC) at 405 and 450 nm derived from MW-TOCA with standard deviation and (b) correlation of the Angström Absorption Exponent (AAE) between 370 and 520 nm, derived from an aethalometer, to OH exposure with a 95% confidence interval for the means of the individual experiments. | ||
A widely used approach to calculate the AAE is an exponential fit of the aerosol absorption coefficient vs. all seven wavelengths of the aethalometer, and hence the absolute AAE from other studies may differ compared to the AAE obtained from the wavelength pair 370 nm/532 nm. In contrast to the quantities discussed in Section 3.1., the AAE did not cluster according to OFR conditions, spanning the AAE range from 3 to 5.5. Among all experiments with the same targeted OH exposure, small differences in the exact OH exposure in the order of 5 × 109 cm3 s were found to be determining for the AAE, having a Pearson correlation coefficient of 0.91 (Fig. 2b). Therefore, the OFR conditions of this study do not seem to affect the BrC formed from toluene photooxidation.
3.3. Thermal refractiveness
3.4. Chemical composition
3.4.1.1. Aerosol mass spectrometry (AMS). The bulk elemental composition was determined by Aerosol Mass Spectrometry (AMS). The elemental ratios O
:C and H:C (Fig. 3a) as well as the molar ratio of organic matter to organic carbon (OM:OC) were significantly different between all individual tol-SOA experiments (Table S4†). The largest O:C of 0.94 was obtained for “low” OFR conditions and the lowest for “high” (O:C of 0.8), with chamber-generated tol-SOA appearing in between (O:C of 0.85), which is in line with a previous study on m-xylene and α-pinene photooxidation attributing the higher O:C in the OFR to a possibly faster gas phase oxidation than nucleation.52 OM:OC ranged from 2.4 (“low”) to 2.2 (“high”) also with chamber-generated tol-SOA in between (OM:OC of 2.3). However, despite significance, the differences have low physical meaning because they originate from the high repeatability of the experiments and low uncertainties. In contrast to O:C, H:C only exhibited significant differences between all OFR- (H:C of 1.64 to 1.65) and chamber-generated tol-SOA (H:C of 1.47). Consequently, the average carbon oxidation state (OSC) is affected, showing similarly high OSC for “low” (OSC = 0.25) and chamber-generated tol-SOA (OSC = 0.24) but lower OSC of 0.18 and −0.06 for “medium” and “high” OFR-generated tol-SOA.
 | ||
| Fig. 3 (a) Van Krevelen diagram with the average carbon oxidation state indicated by dashed lines and (b) triangular space according to Ng et al. (2010)53 with magnification of the narrow space of tol-SOA. Symbols without edges refer to individual data points, and symbols with black edges denote the mean values of one aging condition. | ||
In AMS data analysis, the relative intensities of organic ions at m/z 43 and m/z 44 have been used to delineate the space of ambient organic aerosols and classify them into hydrocarbon-like, biomass burning-line and oxidized organic aerosols (HOA, BBOA and OOA) in the triangular space.53 Ions at m/z 43 may belong to C3H7+ or C2H3O+, associated with primary aerosol sources spanning the range of the x-axis. Upon atmospheric aging, oxygenated organic species such as carboxylic acids are formed, which increase the intensity of CO2+ at m/z 44, thus moving organic aerosols towards the vertex of the triangular space. As expected from previous experiments,34 the data points of tol-SOA were distributed along the hypotenuse of the triangle (Fig. 3b). However, despite lower O:C than “low” and “medium” OFR-generated tol-SOA, chamber-generated tol-SOA had the largest contribution of f44 and hence was classified as the most intensely aged organic aerosol. Furthermore, OFR-generated tol-SOA lined up on the hypotenuse from “high” to “low” OFR conditions with increasing f44, although all tol-SOA received the same OH exposure.
3.4.2.1. Single-photon ionization time-of-flight mass spectrometry. Mass spectrometry with soft ionization for evolved gas analysis in analytical pyrolysis provides insights into the molecular composition beyond bulk properties. During MW-TOCA, a small subsample of the evolving tol-SOA is analyzed by single-photon ionization time-of-flight mass spectrometry (SPI-TOFMS), thus giving molecular ions of thermally desorbed tol-SOA constituents or thermal fragments for individual fractions of OCs. Highly oxygenated organic aerosols already start to decompose at temperatures >80 °C in thermal desorption,54 so detecting compounds in evolved gas analysis likely underwent loss of H2O, CO2 or other small molecules in addition to larger thermal fragments detected by SPI-TOFMS. Therefore, the fractions OC2 and OC3 were combined to form OC23 for further analysis. OC1 and OC4 were discarded because of sampling artifacts and unspecific pyrolysis products, respectively.
The SPI mass spectra of OC23 revealed multiplets with dispersions of ±2 u and a distance of 14 u, indicating homologous series with structural increments of –CH2–, –OCH3 or –OH (Fig. 4a–d). The mostly absence of odd m/z emphasizes the low yield of fragment ions, but the large yield of molecular ions in SPI. However, the mass spectra were very similar having an uncentered correlation coefficient larger than 0.935. Therefore, principal component analysis (PCA) was conducted on the centered SPI mass spectra after L1-normalization. Eigenvalue analysis by the broken stick method55 (Fig. S3†) supports the use of only two principal components (PCs), which account for 77% of the total variance. It is evident that the experimental intra-class to total variance was sufficiently low to separate all four types of tol-SOA with some interference between the 50% confidence ellipsoids of “low” and “medium” (Fig. 4e). Based on the PCA scores, “high” and chamber-generated tol-SOA were most different in chemical composition, while “low” and “medium” appeared in between. Nevertheless, with an explained total variance of 31%, the second PC still has a large contribution, so tol-SOA generated in “high” and in the chamber seems to contain mutual molecular features.
 | ||
| Fig. 4 Average and normalized OC23 SPI mass spectra of tol-SOA generated under (a) “low”, (b) medium, and (c) “high” OFR conditions and in (d) the chamber. Colored stems in (a–d) indicate m/z highly correlated (rc > 0.95, angle of ±18.2°) with the center (“+” in biplot) of the four tol-SOA classes in (e) principal component (PC) biplot space. For simplicity, only variables with the highest loadings in either PC1 and/or PC2 are labelled. | ||
Overall, the first two PC loadings did not give a clear picture of the chemical composition, so most correlating m/z values were determined by extracting m/z within ±18.2° of the line between the origin and the center of the scores, equal to rc > 0.95. It becomes apparent that those highly correlated thermal fragments increase in mass from “low” to “high” with intensity-weighted m/z means of 88 (“low”), 104 (“medium”), 142 (“high”) and 88 (“cham”) rounded to the nearest integer. Hence, tol-SOA from “low” and chamber-aging were likely more oxidized than tol-SOA from “medium” and “high” with more intense decomposition. However, larger m/z values mainly appear in the negative values of the first two PC loadings, so chamber-generated tol-SOA seems to cover the entire mass spectral range spanned by the three OFR conditions.
3.4.2.2. High-resolution mass spectrometry ESI(±). Chemical analysis of methanolic tol-SOA extracts by ESI(±)-Orbitrap mass spectrometry revealed only minor differences in the Van Krevelen space of O
:C vs. H:C but noticeable differences in the intensity-weighted means of elemental ratios (Table 2). O:C, H:C and OSC were decreasing from “low” to “high” for both ionization modes, while chamber-generated tol-SOA had elemental ratios and OSc slightly below those of “high”. Interestingly, among all multiple comparisons only the pairs “low” and “medium” as well as “high” and “cham” gave an insignificant difference at the 5% significance level. Based on the sum formula, the modified aromaticity index (AImod) may be derived, providing classifications into saturated aliphatic (AImod = 0), unsaturated aliphatic (0 < AImod < 0.5), monoaromatic (0.5 < AImod < 0.67) or polyaromatic (AImod > 0.5) compounds for individual sum formulae.56 Generally, the AImod values for all types of tol-SOA were low and between 0.04 and 0.11 for ESI+ and between 0.03 and 0.07 for ESI−. However, chamber-generated tol-SOA had significantly higher AImod compared to “low” in ESI+ and generally significantly higher AImod compared to all other tol-SOA in ESI−. The mean values of the relative number of sodium adducts [M + Na+] to total ions indicate that [M + Na+] are formed in the order low > medium > high > chamber but without significant differences among the tol-SOA types.
:C, H:C, and OSC), modified aromaticity index (AImod) and rel. intensity of sodium adducts of tol-SOA analysis ESI(+)- and ESI(−)-Orbitrap mass spectrometry
| Experiment | (+)O:C |
(+)H:C |
(+)OSc | (+)AImod | (+)rel Na | (−)O:C |
(−)H:C |
(−)OSc | (−)AImod |
|---|---|---|---|---|---|---|---|---|---|
| low1 | 0.75 | 1.43 | 0.06 | 0.06 | 0.63 | 0.86 | 1.13 | 0.59 | 0.04 |
| low2 | 0.76 | 1.43 | 0.09 | 0.04 | 0.71 | 0.85 | 1.14 | 0.56 | 0.04 |
| low3 | 0.78 | 1.44 | 0.11 | 0.04 | 0.76 | 0.88 | 1.16 | 0.60 | 0.03 |
| med1 | 0.71 | 1.37 | 0.05 | 0.07 | 0.61 | 0.79 | 1.13 | 0.46 | 0.05 |
| med2 | 0.74 | 1.41 | 0.07 | 0.05 | 0.72 | 0.81 | 1.14 | 0.49 | 0.04 |
| med3 | 0.73 | 1.39 | 0.07 | 0.05 | 0.69 | 0.83 | 1.14 | 0.51 | 0.04 |
| high1 | 0.60 | 1.27 | −0.06 | 0.11 | 0.51 | 0.72 | 1.14 | 0.31 | 0.04 |
| high2 | 0.64 | 1.31 | −0.03 | 0.07 | 0.69 | 0.72 | 1.13 | 0.30 | 0.05 |
| high3 | 0.63 | 1.30 | −0.05 | 0.09 | 0.63 | 0.73 | 1.14 | 0.31 | 0.04 |
| cham1 | 0.57 | 1.28 | −0.14 | 0.11 | 0.47 | 0.71 | 1.09 | 0.33 | 0.07 |
| cham2 | 0.62 | 1.26 | −0.02 | 0.10 | 0.55 | 0.70 | 1.09 | 0.31 | 0.07 |
| cham3 | 0.65 | 1.28 | 0.01 | 0.07 | 0.69 | 0.70 | 1.09 | 0.32 | 0.07 |
To explore further the differences in intensity-weighted mean elemental ratios and derived quantities, all twelve individual mass spectra were L1-normalized, set to the same number of variables, i.e. m/z, and merged to one mean mass spectrum for each type of tol-SOA. In both ionization modes, the same main peaks could be observed in all tol-SOA; however, differences appeared in the mass spectrometric pattern (Fig. S4†). In ESI(+), three regions of interest (ROIs) may be defined between m/z 100 and 200 (A), (b) m/z 200 and 300 (B), and m/z 300 and 400 (C). ROIs A and C are present in all tol-SOA, but there is a decrease in the ratio A/C from “low” over “medium” and “high” to “cham”. Simultaneously, peaks in ROI B start to vanish in the same order, but with a single distinct peak at m/z 213.038 with the sum formula C7H10O6, detected as [M + Na]+. With lower intensities, the same ion was detected in all OFR-generated tol-SOA, and hence an impurity or artifact of the chamber can be ruled out. Elemental analysis of the main peaks of all ROIs revealed that ROIs A and C contained monomers and dimers of toluene with 7 and 14 carbon atoms and O:C between 0.55 and 0.75. In contrast, peaks in ROI B belong to sum formulae with carbon numbers slightly larger than seven, but distinctly lower than 14, such as [C8H14O7+Na]+ (m/z 245.064) or [C9H16O8+Na]+ (m/z 275.074). Hence, ROIs A and C contain monomers and dimers of toluene-SOA, whereas ROI B includes products from the reaction of C7 units with units of a lower carbon number or possibly further processing of dimers by fragmentation. A more detailed view of dimer formation is provided by the analysis of the carbon number (#C) vs. the average carbon oxidation state (OSC) together with the relative peak intensities for each #C (Fig. 5). Among the tol-SOA types, “low” contains the largest proportion of species with a #C of 8 and 9, while in all other tol-SOA a local minimum occurs in the #C vs. peak intensity space. Furthermore, two local maxima appear at a #C of 12 and 14, which can be assigned to dimers. However, the ratio of C12 to C14 species decreases in the order chamber > low > medium > high, thus having the highest similarity between “cham” and “low”. Nevertheless, the strongest overall correlation between the #C vs. peak intensity patterns of the three OFR-generated SOAs to “cham” was obtained for “medium” with 0.97 (0.95 for “low” and 0.89 for “high”).
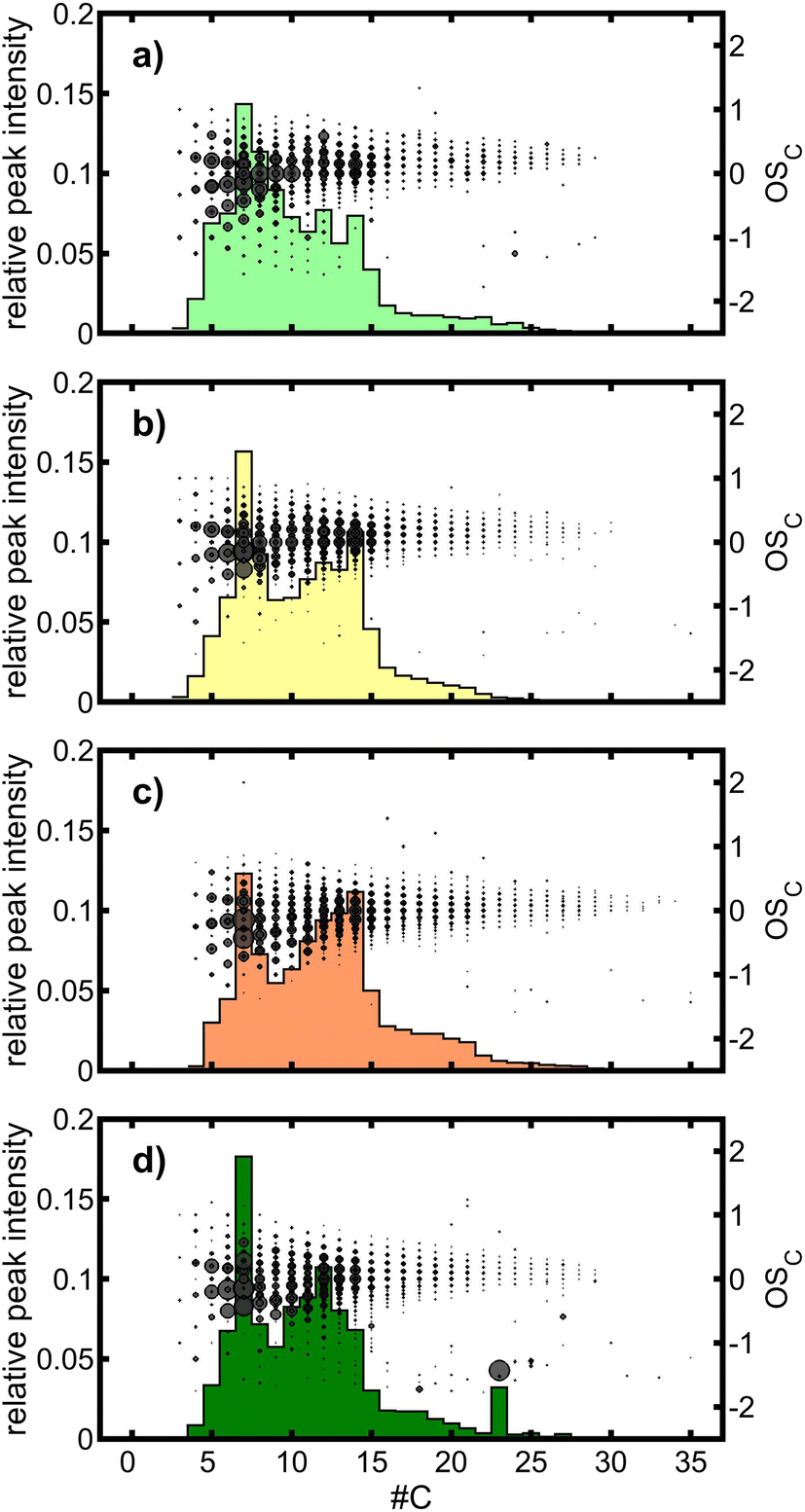 | ||
| Fig. 5 #C vs. OSC with summed relative intensities for each #C for ESI(+) of (a) “low”, (b) “medium”, (c) “high” and (d) “cham”. The contribution to #C = 23 in “cham” mainly belongs to [C23H40O4 + H]+, which could be a common plasticizer from the family of cyclohexanedicarboxylic acid esters. | ||
Due to its high abundance in the atmosphere, several secondary products of toluene from atmospheric aging were identified and partly used as markers, such as 2,3-dihydroxy-4-oxopentanoic acid (DHOPA) with the sum formula C5H8O5.57 Such a compound was detected as a sodium adduct [C5H8O5 + Na]+ in all tol-SOA with relative intensities in the order: “low” (0.61%) > “medium” (0.58%) > “cham” (0.11%) ≈ “high” (0.10%). Hence, the yield of DHOPA was the largest under “low” OFR conditions, but those from “high” were most comparable to the chamber-generated tol-SOA. However, the peak of [C5H8O5 + Na]+ may also belong to isomers of DHOPA and does not provide an appropriate marker isolation from the complex mixture of toluene oxidation products.
In ESI(−) mass spectra, larger m/z values increase in relative intensity from “low” to “high” conditions and show qualitatively the same main peaks as in ESI(+). Some of the most intense peaks detected in all tol-SOA appear at m/z 191.0198 ([C6H8O7–H]−), m/z 173.0092 ([C6H6O6–H]−) and m/z 161.009 ([C5H6O6–H]−), indicating highly oxidized or sugar-like components with O:C ≥ 1. ROIs derived from the pattern in ESI− mass spectra were not as apparent as in ESI+, but matched the possible structures of monomers and dimers derived from elemental analysis. From “low” to “high” OFR conditions, the intensities of m/z < 250 decreased while m/z > 250 increased. Moreover, most intense peaks of m/z > 250 in “high” shifted to larger m/z, agreeing with a shift of the #C vs. peak intensity pattern of “high” to larger #C. Generally, monomers and dimers appeared at #C of 6 and 13 in OFR-generated tol-SOA with favoring monomers in “low”, dimers in “high” and none in “medium” OFR experiments (Fig. S5†). In contrast, chamber-generated tol-SOA exhibits a maximum in the #C vs. peak intensity pattern at #C = 11 and a local minimum at #C = 7, which equals the carbon number of toluene. Therefore, acidic tol-SOA components of monomers and dimers underwent fragmentation involving carbon loss. The #C vs. OSC and peak intensity derived from ESI(−) mass spectra were less similar between chamber- and OFR-generated tol-SOA than derived from ESI(+). The largest similarity to “cham” was found for “medium” (rc = 0.94), the lowest for “low” (rc = 0.87) and “high” in between (rc = 0.90).
Peak intensities in direct-infusion ESI mass spectra may suffer from matrix effects, so an alternative approach for data analysis is based on binary treatment of m/z being present or not present in samples, regardless of their intensity. In a Venn diagram, peaks are grouped according to their occurrence in the different types of tol-SOA. To limit the influence of different concentrations associated with a larger number of peaks above a signal-to-noise of six, particularly, in the higher m/z range, the input data for the Venn diagram were restricted to a maximum #C of 15. Of the 798 detected peaks in ESI+, 356 appeared in all tol-SOA, accounting for 57%, 61%, 62%, and 58% of the peaks detected in “low”, “medium”, “high” and “cham”, respectively. Moreover, 75%, 83%, and 77% of the peaks in “low”, “medium”, and “high” also appeared in “cham”. In ESI−, 265 out of 479 peaks were detected in all tol-SOA, accounting for 78%, 76%, 67%, and 70% of the peaks detected in “low”, “medium”, “riskier” and “cham”, respectively. Moreover, 79%, 82%, and 80% of the peaks in “low”, “medium”, and “high” also appeared in “cham”. Overall, the alternative binary peak analysis demonstrates fairly low differences among the tol-SOA types and the high similarity of OFR-generated to chamber-generated tol-SOA.
4. Discussion
OFRs are widely used to study the fate and impact of primary aerosols released into the atmosphere. Their main advantage lies in the potential coverage of the entire range of atmospheric particle lifetime, but at the cost of different mechanisms of secondary aerosol formation and subsequent multiphase chemistry. Recent work has modelled the range of OFR conditions for the OFR “potential aerosol mass” (PAM), triggering similar gas phase chemistry to that in the troposphere.15 However, the effect of OFR conditions, regarded as atmospherically relevant or irrelevant, on the physical and chemical properties of SOAs has not been systematically investigated yet, which motivated our case study on toluene photooxidation products. OFR experiments with the PEAR were conducted with settings of OH reactivity and photon fluxes (“low”, “medium”, and “high”), appearing in the range of “safer”, “transition” and “riskier” OFR conditions according to the PAM model. Chamber experiments were conducted as a laboratory reference for atmospherically relevant tol-SOA.Our results reveal that “low” conditions, representing “safer” OFR conditions, do not stringently prove a higher similarity to atmospherically relevant SOAs when compared to complementary environmental chamber experiments conducted at comparable concentrations to the literature.58–60 For the aerosol properties O:C from AMS, elemental quantities and the molecular composition derived from ESI±, as well as the SOA yield, “low” even showed the least comparability to “cham”. Moreover, insignificant and fairly small differences were found between both OFR- (all conditions) and chamber-derived tol-SOA for the optical properties AAE and MAEλ as well as the distribution of carbonaceous species among the thermal fractions of carbon.
On a quantitative scale, tol-SOA from OFRs was generally produced in lower yields than from chamber aging in agreement with Lambe et al. (2015).49 Here, the approach of constant OH exposure with high OH concentrations and short time shows its disadvantage as oxidized vapor may require more time to condense and form SOA than the residence time typically permitted in most OFRs, so the majority of low-volatile compounds exit the OFR in the gas phase. This may partially be counteracted by using a seed aerosol, which provides an additional condensation sink and consequently enhances YSOA. In the same manner, the shift of the particle size distribution for “low” over “medium” to “high” would be avoided and could appear closer to the chamber-generated tol-SOA.
In order to cover the complex composition of SOAs, bulk properties and parametrizations are used. OFR conditions did not show any effect on H:C, but led to decreasing O:C from “low” to “high” OFR conditions, so OSC decreased as well. Although “low” and “cham” agree well, similar OSC originates from different H:C and O:C, pointing towards a different chemical composition. This is further supported by the parametrization in the triangular space f43vs. f44,53 where chamber-generated tol-SOA revealed the largest contribution of f44, thus the most advanced aging.
The most substantial differences between “low” and “cham” were observed in the more detailed chemical composition, particularly in ESI high-resolution Orbitrap mass spectrometry. Surprisingly, in pattern analysis “high” and in peak number-based comparison “medium” showed the largest correlation with “cham”, which could be attributed to differences in dimer formation. However, thermodesorption SPI mass spectra suggested that chamber-generated tol-SOA contains molecular features of all three OFR conditions.
Overall, our study on tol-SOA challenges the concept of “safer”, i.e., atmospherically relevant OFR conditions. However, due to the wide space of parameters, an experimental approach is required to elucidate the effect of OFR conditions on SOA composition and gas phase species, which have not been addressed in this study. This would include aging under high-NOx conditions,25 SOA precursors with a stronger tendency towards oligomer formation,31,61 the application of seed aerosols, different initial O3 concentrations in OFR254 mode, and the comparison to the OFR185 mode with in situ ozone generation. Furthermore, recent process-level modelling enabled the connection between environmental chamber and OFR results,62 which might be incorporated in OFR condition assessment.
As an extension of physical and chemical aerosol properties, the toxicity of aged aerosols has been studied using an OFR. Fine particulate matter, including specifically SOAs, is harmful to human health,6,63 so OFRs enable toxicological studies with in vitro exposure to investigate specific biological endpoints and SOAs from individual precursors. Cytotoxicity and inflammatory responses start with oxidative stress, which has been linked to the concentrations of airborne reactive oxygen species (ROS).8,30,64 In the OFR, high levels of ROS may be substantially formed over the full length of the reactor. Although the majority of ROS is short-lived,65 a significant fraction of the ROS is leaving the OFR because of its short residence time. In environmental chambers, the residence time is orders of magnitude higher, so those short-living ROS would already react during the longer residence in environmental chambers and lower concentrations of short-living ROS are present at the end of the aging experiment. Ozone as part of the ROS class is removed using denuders,8 which likely reduce other ROS as well, but systematic research on different ROS would be beneficial to rule out if cell cultures exposed in the air–liquid interface possibly receive a significantly higher dose of ROS from OFRs than chamber-generated to aged aerosols.
Besides ROS, polycyclic aromatic hydrocarbons (PAHs) are health-relevant ambient aerosol constituents, which become potent after metabolization in the human body. Recently, Hrdina et al. (2022)9 pointed out the similarity of PAH metabolism in the human body and (short) atmospheric aging, leading both to oxygenated PAHs. Here, a deeper analysis and understanding of OFR chemistry products is required since the detailed chemical structure of aerosol constituents, i.e., the position of a functional group in the carbon backbone, may be crucial to their toxicity.
The most carcinogenic PAHs, including benzo(a)pyrene, are predominantly located in the particle phase, so direct oxidation by OH is slower compared to homogeneous gas phase oxidation of more volatile PAHs like naphthalene.66 Additionally, PAHs may be shielded from atmospheric oxidants by condensing SOAs as a coating, further decelerating their degradation.67 Hence, the underestimation of heterogeneous oxidation would be another task for process-level modelling in order to harmonize PAH degradation in OFRs and environmental chambers and determine the highest carcinogenic/mutagenic potential of PAHs during their atmospheric lifetime, which would be beneficial for OFR studies on aged combustion aerosols.
5. Conclusion
OFR-aging conditions by means of different ratios of atmospheric oxidants and photon fluxes but with a similar level of OH exposure affect the physical-chemical properties of tol-SOA. While the relative effect towards less atmospherically relevant conditions was substantial for YSOA and particle size distribution, only small differences were observed for chemical bulk properties, such as elemental ratios, but there were larger discrepancies in the more detailed chemical composition. Most surprisingly, for many physical-chemical properties of OFR-generated tol-SOA less atmospherically relevant conditions exhibit the largest agreement with chamber-generated tol-SOA, which resembles photochemical aging more closely than an OFR at the same OH exposure. OFRs may bridge the gap between the upper limit of the equivalent photochemical aerosol age achievable in chambers and the maximum atmospheric particle residence time, but the value from their application depends on the research objective and considered aerosol properties.Author contributions
HC: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, visualization, supervision, and project administration; PYP: investigation and data curation; AH: validation, formal analysis, and writing – review & editing; MI: investigation and data curation; PT: formal analysis and investigation; PM: investigation, formal analysis and writing – review & editing; ES: investigation, formal analysis, and writing – review & editing; AP: formal analysis; TH: writing – review & editing and supervision; JJ: resources and funding acquisition; CR: methodology, software, writing – review & editing, and supervision; RZ: resources and funding acquisition; OS: resources, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Gert Jakobi (Helmholtz Centre München) for aethalometer data processing and evaluation. This research was supported by the Academy of Finland projects ASTRO (grant no. 304459) and BBrCAC (grant no. 341597), the Helmholtz International Laboratory aeroHEALTH (Interlabs-0005), and the German Science Foundation (DFG; grant no. ZI 764/24-1).References
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang and D. R. Worsnop, Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 2007, 34(13), L13801 CrossRef.
- J. M. Kelly, R. M. Doherty, F. M. O'Connor and G. W. Mann, The impact of biogenic, anthropogenic, and biomass burning volatile organic compound emissions on regional and seasonal variations in secondary organic aerosol, Atmos. Chem. Phys., 2018, 18, 7393–7422 CrossRef.
- Z. Fang, C. Li, Q. He, H. Czech, T. Gröger, J. Zeng, H. Fang, S. Xiao, M. Pardo, E. Hartner, D. Meidan, X. Wang, R. Zimmermann, A. Laskin and Y. Rudich, Secondary organic aerosols produced from photochemical oxidation of secondarily evaporated biomass burning organic gases: chemical composition, toxicity, optical properties, and climate effect, Environ. Int., 2021, 157, 106801 CrossRef CAS PubMed.
- T. F. Mentel, E. Kleist, S. Andres, M. Dal Maso, T. Hohaus, A. Kiendler-Scharr, Y. Rudich, M. Springer, R. Tillmann, R. Uerlings, A. Wahner and J. Wildt, Secondary aerosol formation from stress-induced biogenic emissions and possible climate feedbacks, Atmos. Chem. Phys., 2013, 13, 8755–8770 CrossRef.
- M. Shrivastava, R. C. Easter, X. Liu, A. Zelenyuk, B. Singh, K. Zhang, P.-L. Ma, D. Chand, S. Ghan, J. L. Jimenez, Q. Zhang, J. Fast, P. J. Rasch and P. Tiitta, Global transformation and fate of SOA, J. Geophys. Res.: Atmos., 2015, 120, 4169–4195 CrossRef CAS.
- H. O. T. Pye, C. K. Ward-Caviness, B. N. Murphy, K. W. Appel and K. M. Seltzer, Secondary organic aerosol association with cardiorespiratory disease mortality in the United States, Nat. Commun., 2021, 12, 7215 CrossRef CAS PubMed.
- K. von Stackelberg, J. Buonocore, P. V. Bhave and J. A. Schwartz, Public health impacts of secondary particulate formation from aromatic hydrocarbons in gasoline, Environ. Health, 2013, 12, 19 CrossRef PubMed.
- S. Offer, E. Hartner, S. Di Bucchianico, C. Bisig, S. Bauer, J. Pantzke, E. J. Zimmermann, X. Cao, S. Binder, E. Kuhn, A. Huber, S. Jeong, U. Käfer, P. Martens, A. Mesceriakovas, J. Bendl, R. Brejcha, A. Buchholz, D. Gat, T. Hohaus, N. Rastak, G. Jakobi, M. Kalberer, T. Kanashova, Y. Hu, C. Ogris, A. Marsico, F. Theis, M. Pardo, T. Gröger, S. Oeder, J. Orasche, A. Paul, T. Ziehm, Z.-H. Zhang, T. Adam, O. Sippula, M. Sklorz, J. Schnelle-Kreis, H. Czech, A. Kiendler-Scharr, Y. Rudich and R. Zimmermann, Effect of Atmospheric Aging on Soot Particle Toxicity in Lung Cell Models at the Air-Liquid Interface: Differential Toxicological Impacts of Biogenic and Anthropogenic Secondary Organic Aerosols (SOAs), Environ. Health Perspect., 2022, 130, 27003–27021 CrossRef CAS PubMed.
- A. I. H. Hrdina, I. N. Kohale, S. Kaushal, J. Kelly, N. E. Selin, B. P. Engelward and J. H. Kroll, The Parallel Transformations of Polycyclic Aromatic Hydrocarbons in the Body and in the Atmosphere, Environ. Health Perspect., 2022, 130, 25004 CrossRef CAS PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- J. B. Burkholder, J. P. D. Abbatt, I. Barnes, J. M. Roberts, M. L. Melamed, M. Ammann, A. K. Bertram, C. D. Cappa, A. G. Carlton, L. J. Carpenter, J. N. Crowley, Y. Dubowski, C. George, D. E. Heard, H. Herrmann, F. N. Keutsch, J. H. Kroll, V. F. McNeill, N. L. Ng, S. A. Nizkorodov, J. J. Orlando, C. J. Percival, B. Picquet-Varrault, Y. Rudich, P. W. Seakins, J. D. Surratt, H. Tanimoto, J. A. Thornton, Z. Tong, G. S. Tyndall, A. Wahner, C. J. Weschler, K. R. Wilson and P. J. Ziemann, The Essential Role for Laboratory Studies in Atmospheric Chemistry, Environ. Sci. Technol., 2017, 51, 2519–2528 CrossRef CAS PubMed.
- B. Chu, T. Chen, T. Liu, Q. Ma, Y. Mu, Y. Wang, J. Ma, P. Zhang, J. Liu, C. Liu, H. Gui, R. Hu, B. Hu, X. Wang, Y. Wang, P. Xie, J. Chen, Q. Liu, J. Jiang, J. Li, K. He, W. Liu, G. Jiang, J. Hao and H. He, Application of smog chambers in atmospheric process studies, Natl. Sci. Rev., 2022, 9(2), nwab103 CrossRef CAS PubMed.
- T. F. Mentel and A. Wahner, A Large Reaction Chamber for Nighttime Atmospheric Chemistry: Design and Characteristics of the Chamber, Forschungszentrum Jülich, Berichte des Forschungszentrums Jülich 3196, Jülich, Germany, 1996, https://books.google.fi/books/about/A_Large_Reaction_Chamber_for_Nighttime_A.html?id=3WBpPwAACAAJ%26redir_esc=y Search PubMed.
- H. Czech, O. Popovicheva, D. G. Chernov, A. Kozlov, E. Schneider, V. P. Shmargunov, M. Sueur, C. P. Rüger, C. Afonso, V. Uzhegov, V. S. Kozlov, M. V. Panchenko and R. Zimmermann, Wildfire plume ageing in the Photochemical Large Aerosol Chamber (PHOTO-LAC), Environ. Sci.: Processes Impacts, 2024, 26, 35–55 RSC.
- Z. Peng and J. L. Jimenez, Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 2020, 49, 2570–2616 RSC.
- E. Ahlberg, J. Falk, A. Eriksson, T. Holst, W. H. Brune, A. Kristensson, P. Roldin and B. Svenningsson, Secondary organic aerosol from VOC mixtures in an oxidation flow reactor, Atmos. Environ., 2017, 161, 210–220 CrossRef CAS.
- P. H. Chowdhury, Q. He, R. Carmieli, C. Li, Y. Rudich and M. Pardo, Connecting the Oxidative Potential of Secondary Organic Aerosols with Reactive Oxygen Species in Exposed Lung Cells, Environ. Sci. Technol., 2019, 53, 13949–13958 CrossRef CAS PubMed.
- A. Hartikainen, P. Tiitta, M. Ihalainen, P. Yli-Pirilä, J. Orasche, H. Czech, M. Kortelainen, H. Lamberg, H. Suhonen, H. Koponen, L. Hao, R. Zimmermann, J. Jokiniemi, J. Tissari and O. Sippula, Photochemical transformation of residential wood combustion emissions: dependence of organic aerosol composition on OH exposure, Atmos. Chem. Phys., 2020, 20, 6357–6378 CrossRef CAS.
- P. Simonen, J. Kalliokoski, P. Karjalainen, T. Rönkkö, H. Timonen, S. Saarikoski, M. Aurela, M. Bloss, G. Triantafyllopoulos, A. Kontses, S. Amanatidis, A. Dimaratos, Z. Samaras, J. Keskinen, M. Dal Maso and L. Ntziachristos, Characterization of laboratory and real driving emissions of individual Euro 6 light-duty vehicles - Fresh particles and secondary aerosol formation, Environ. Pollut., 2019, 255, 113175 CrossRef CAS PubMed.
- B. B. Palm, P. Campuzano-Jost, A. M. Ortega, D. A. Day, L. Kaser, W. Jud, T. Karl, A. Hansel, J. F. Hunter, E. S. Cross, J. H. Kroll, Z. Peng, W. H. Brune and J. L. Jimenez, In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor, Atmos. Chem. Phys., 2016, 16, 2943–2970 CrossRef CAS.
- W. Hu, H. Zhou, W. Chen, Y. Ye, T. Pan, Y. Wang, W. Song, H. Zhang, W. Deng, M. Zhu, C. Wang, C. Wu, C. Ye, Z. Wang, B. Yuan, S. Huang, M. Shao, Z. Peng, D. A. Day, P. Campuzano-Jost, A. T. Lambe, D. R. Worsnop, J. L. Jimenez and X. Wang, Oxidation Flow Reactor Results in a Chinese Megacity Emphasize the Important Contribution of S/IVOCs to Ambient SOA Formation, Environ. Sci. Technol., 2022, 56, 6880–6893 CrossRef CAS PubMed.
- J. H. Kroll, C. Y. Lim, S. H. Kessler and K. R. Wilson, Heterogeneous Oxidation of Atmospheric Organic Aerosol: Kinetics of Changes to the Amount and Oxidation State of Particle-Phase Organic Carbon, J. Phys. Chem. A, 2015, 119, 10767–10783 CrossRef CAS PubMed.
- M. Pardo, H. Czech, S. Offer, M. Sklorz, S. Di Bucchianico, E. Hartner, J. Pantzke, E. Kuhn, A. Paul, T. Ziehm, Z.-H. Zhang, G. Jakobi, S. Bauer, A. Huber, E. J. Zimmermann, N. Rastak, S. Binder, R. Brejcha, E. Schneider, J. Orasche, C. P. Rüger, T. Gröger, S. Oeder, J. Schnelle-Kreis, T. Hohaus, M. Kalberer, O. Sippula, A. Kiendler-Scharr, R. Zimmermann and Y. Rudich, Atmospheric aging increases the cytotoxicity of bare soot particles in BEAS-2B lung cells, Aerosol Sci. Technol., 2023, 57, 367–383 CrossRef CAS.
- Z. Peng, D. A. Day, A. M. Ortega, B. B. Palm, W. Hu, H. Stark, R. Li, K. Tsigaridis, W. H. Brune and J. L. Jimenez, Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 2016, 16, 4283–4305 CrossRef CAS.
- Z. Peng and J. L. Jimenez, Modeling of the chemistry in oxidation flow reactors with high initial NO, Atmos. Chem. Phys., 2017, 17, 11991–12010 CrossRef CAS.
- Z. Peng, J. Lee-Taylor, J. J. Orlando, G. S. Tyndall and J. L. Jimenez, Organic peroxy radical chemistry in oxidation flow reactors and environmental chambers and their atmospheric relevance, Atmos. Chem. Phys., 2019, 19, 813–834 CrossRef CAS.
- Z. Peng, D. A. Day, H. Stark, R. Li, J. Lee-Taylor, B. B. Palm, W. H. Brune and J. L. Jimenez, HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 2015, 8, 4863–4890 CrossRef CAS.
- E. Kang, M. J. Root and W. H. Brune, Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 2007, 7, 9925–9972 CrossRef.
- E. A. Bruns, I. El Haddad, A. Keller, F. Klein, N. K. Kumar, S. M. Pieber, J. C. Corbin, J. G. Slowik, W. H. Brune, U. Baltensperger and A. S. H. Prévôt, Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition, Atmos. Meas. Tech., 2015, 8, 2315–2332 CrossRef CAS.
- F. Khan, K. Kwapiszewska, Y. Zhang, Y. Chen, A. T. Lambe, A. Kołodziejczyk, N. Jalal, K. Rudzinski, A. Martínez-Romero, R. C. Fry, J. D. Surratt and R. Szmigielski, Toxicological Responses of α-Pinene-Derived Secondary Organic Aerosol and Its Molecular Tracers in Human Lung Cell Lines, Chem. Res. Toxicol., 2021, 34, 817–832 Search PubMed.
- M. Pardo, S. Offer, E. Hartner, S. Di Bucchianico, C. Bisig, S. Bauer, J. Pantzke, E. J. Zimmermann, X. Cao, S. Binder, E. Kuhn, A. Huber, S. Jeong, U. Käfer, E. Schneider, A. Mesceriakovas, J. Bendl, R. Brejcha, A. Buchholz, D. Gat, T. Hohaus, N. Rastak, E. Karg, G. Jakobi, M. Kalberer, T. Kanashova, Y. Hu, C. Ogris, A. Marsico, F. Theis, T. Shalit, T. Gröger, C. P. Rüger, S. Oeder, J. Orasche, A. Paul, T. Ziehm, Z.-H. Zhang, T. Adam, O. Sippula, M. Sklorz, J. Schnelle-Kreis, H. Czech, A. Kiendler-Scharr, R. Zimmermann and Y. Rudich, Exposure to naphthalene and β-pinene-derived secondary organic aerosol induced divergent changes in transcript levels of BEAS-2B cells, Environ. Int., 2022, 166, 107366 CrossRef CAS PubMed.
- Z. Leni, M. N. Ess, A. Keller, J. D. Allan, H. Hellén, K. Saarnio, K. R. Williams, A. S. Brown, M. Salathe, N. Baumlin, K. Vasilatou and M. Geiser, Role of Secondary Organic Matter on Soot Particle Toxicity in Reconstituted Human Bronchial Epithelia Exposed at the Air-Liquid Interface, Environ. Sci. Technol., 2022, 56, 17007–17017 CrossRef CAS PubMed.
- Y.-S. Lau, H.-Y. Poon, B. Organ, H.-C. Chuang, M.-N. Chan, H. Guo, S. S. H. Ho and K.-F. Ho, Toxicological effects of fresh and aged gasoline exhaust particles in Hong Kong, J. Hazard. Mater., 2023, 441, 129846 CrossRef CAS PubMed.
- M. Ihalainen, P. Tiitta, H. Czech, P. Yli-Pirilä, A. Hartikainen, M. Kortelainen, J. Tissari, B. Stengel, M. Sklorz, H. Suhonen, H. Lamberg, A. Leskinen, A. Kiendler-Scharr, H. Harndorf, R. Zimmermann, J. Jokiniemi and O. Sippula, A novel high-volume Photochemical Emission Aging flow tube Reactor (PEAR), Aerosol Sci. Technol., 2019, 53, 276–294 CrossRef CAS.
- P. Barmet, J. Dommen, P. F. DeCarlo, T. Tritscher, A. P. Praplan, S. M. Platt, A. S. H. Prévôt, N. M. Donahue and U. Baltensperger, OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 2012, 5, 647–656 CrossRef CAS.
- A. Leskinen, P. Yli-Pirilä, K. Kuuspalo, O. Sippula, P. Jalava, M.-R. Hirvonen, J. Jokiniemi, A. Virtanen, M. Komppula and K. E. J. Lehtinen, Characterization and testing of a new environmental chamber, Atmos. Meas. Tech., 2015, 8, 2267–2278 CrossRef.
- N. L. Ng, J. H. Kroll, A. W. H. Chan, P. S. Chhabra, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 2007, 7, 4085–4126 Search PubMed.
- H. Czech, O. Sippula, M. Kortelainen, J. Tissari, C. Radischat, J. Passig, T. Streibel, J. Jokiniemi and R. Zimmermann, On-line analysis of organic emissions from residential wood combustion with single-photon ionisation time-of-flight mass spectrometry (SPI-TOFMS), Fuel, 2016, 177, 334–342 CrossRef CAS.
- D. Butcher, Vacuum Ultraviolet Radiation for Single-Photoionization Mass Spectrometry: A Review, Microchem. J., 1999, 62, 354–362 CrossRef CAS.
- J. R. Odum, T. Hoffmann, F. Bowman, D. Collins, R. C. Flagan and J. H. Seinfeld, Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 1996, 30, 2580–2585 CrossRef CAS.
- V. Bernardoni, L. Ferrero, E. Bolzacchini, A. C. Forello, A. Gregorič, D. Massabò, G. Močnik, P. Prati, M. Rigler, L. Santagostini, F. Soldan, S. Valentini, G. Valli and R. Vecchi, Determination of Aethalometer multiple-scattering enhancement parameters and impact on source apportionment during the winter 2017/18 EMEP/ACTRIS/COLOSSAL campaign in Milan, Atmos. Meas. Tech., 2021, 14(4), 2919–2940 CrossRef CAS.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Elemental ratio measurements of organic compounds using aerosol mass spectrometry, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- L.-W. A. Chen, J. C. Chow, X. L. Wang, J. A. Robles, B. J. Sumlin, D. H. Lowenthal, R. Zimmermann and J. G. Watson, Multi-wavelength optical measurement to enhance thermal/optical analysis for carbonaceous aerosol, Atmos. Meas. Tech., 2015, 8, 451–461 CrossRef CAS.
- J. C. Chow, L.-W. A. Chen, X. Wang, M. C. Green and J. G. Watson, Improved estimation of PM2.5 brown carbon contributions to filter light attenuation, Particuology, 2021, 56, 1–9 CrossRef CAS.
- V. Bernardoni, L. Ferrero, E. Bolzacchini, A. C. Forello, A. Gregorič, D. Massabò, G. Močnik, P. Prati, M. Rigler, L. Santagostini, F. Soldan, S. Valentini, G. Valli and R. Vecchi, Determination of Aethalometer multiple-scattering enhancement parameters and impact on source apportionment during the winter 2017/18 EMEP/ACTRIS/COLOSSAL campaign in Milan, Atmos. Meas. Tech., 2021, 14, 2919–2940 CrossRef CAS.
- J. C. Chow, J. G. Watson, L.-W. A. Chen, M. O. Chang, N. F. Robinson, D. Trimble and S. Kohl, The IMPROVE_A Temperature Protocol for Thermal/Optical Carbon Analysis, J. Air Waste Manage. Assoc., 2007, 57, 1014–1023 CrossRef CAS PubMed.
- J. Grabowsky, T. Streibel, M. Sklorz, J. C. Chow, J. G. Watson, A. Mamakos and R. Zimmermann, Hyphenation of a carbon analyzer to photo-ionization mass spectrometry to unravel the organic composition of particulate matter on a molecular level, Anal. Bioanal. Chem., 2011, 401, 3153–3164 CrossRef CAS PubMed.
- T. Adam and R. Zimmermann, Determination of single photon ionization cross sections for quantitative analysis of complex organic mixtures, Anal. Bioanal. Chem., 2007, 389, 1941–1951 CrossRef CAS PubMed.
- A. T. Lambe, P. S. Chhabra, T. B. Onasch, W. H. Brune, J. F. Hunter, J. H. Kroll, M. J. Cummings, J. F. Brogan, Y. Parmar, D. R. Worsnop, C. E. Kolb and P. Davidovits, Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 2015, 15, 3063–3075 CrossRef CAS.
- P. Lin, J. Liu, J. E. Shilling, S. M. Kathmann, J. Laskin and A. Laskin, Molecular characterization of brown carbon (BrC) chromophores in secondary organic aerosol generated from photo-oxidation of toluene, Phys. Chem. Chem. Phys., 2015, 17, 23312–23325 RSC.
- J. Ma, X. Li, P. Gu, T. R. Dallmann, A. A. Presto and N. M. Donahue, Estimating ambient particulate organic carbon concentrations and partitioning using thermal optical measurements and the volatility basis set, Aerosol Sci. Technol., 2016, 50, 638–651 CrossRef CAS.
- A. T. Lambe, A. T. Ahern, L. R. Williams, J. G. Slowik, J. P. S. Wong, J. P. D. Abbatt, W. H. Brune, N. L. Ng, J. P. Wright, D. R. Croasdale, D. R. Worsnop, P. Davidovits and T. B. Onasch, Characterization of aerosol photooxidation flow reactors, Atmos. Meas. Tech., 2011, 4, 445–461 CrossRef CAS.
- N. L. Ng, M. R. Canagaratna, Q. Zhang, J. L. Jimenez, J. Tian, I. M. Ulbrich, J. H. Kroll, K. S. Docherty, P. S. Chhabra, R. Bahreini, S. M. Murphy, J. H. Seinfeld, L. Hildebrandt, N. M. Donahue, P. F. DeCarlo, V. A. Lanz, A. S. H. Prévôt, E. Dinar, Y. Rudich and D. R. Worsnop, Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 2010, 10, 4625–4641 CrossRef CAS.
- Z. Zhao, X. Yang, J. Lee, R. Tolentino, R. Mayorga, W. Zhang and H. Zhang, Diverse Reactions in Highly Functionalized Organic Aerosols during Thermal Desorption, ACS Earth Space Chem., 2020, 4, 283–296 CrossRef CAS.
- D. A. Jackson, Stopping Rules in Principal Components Analysis: A Comparison of Heuristical and Statistical Approaches, Ecology, 1993, 74, 2204–2214 CrossRef.
- B. P. Koch and T. Dittmar, Erratum: from mass to structure: an aromaticity index for high-resolution mass data of natural organic matter, Rapid Commun. Mass Spectrom., 2016, 30, 250 CrossRef CAS.
- T. E. Kleindienst, T. S. Conver, C. D. McIver and E. O. Edney, Determination of Secondary Organic Aerosol Products from the Photooxidation of Toluene and their Implications in Ambient PM2.5, J. Atmos. Chem., 2004, 47, 79–100 CrossRef CAS.
- L. Hildebrandt, N. M. Donahue and S. N. Pandis, High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 2009, 9, 693–733 CrossRef.
- S. V. Dhulipala, S. Bhandari and L. Hildebrandt Ruiz, Formation of oxidized organic compounds from Cl-initiated oxidation of toluene, Atmos. Environ., 2019, 199, 265–273 CrossRef CAS.
- L. Li, P. Tang, S. Nakao and D. R. Cocker III, Impact of molecular structure on secondary organic aerosol formation from aromatic hydrocarbon photooxidation under low-NOx conditions, Atmos. Chem. Phys., 2016, 16, 10793–10808 CrossRef CAS.
- S. Wang, J. Ye, R. Soong, B. Wu, L. Yu, A. J. Simpson and A. W. H. Chan, Relationship between chemical composition and oxidative potential of secondary organic aerosol from polycyclic aromatic hydrocarbons, Atmos. Chem. Phys., 2018, 18, 3987–4003 CrossRef CAS.
- Y. He, A. T. Lambe, J. H. Seinfeld, C. D. Cappa, J. R. Pierce and S. H. Jathar, Process-Level Modeling Can Simultaneously Explain Secondary Organic Aerosol Evolution in Chambers and Flow Reactors, Environ. Sci. Technol., 2022, 56, 6262–6273 CrossRef CAS PubMed.
- P. Tarín-Carrasco, U. Im, C. Geels, L. Palacios-Peña and P. Jiménez-Guerrero, Contribution of fine particulate matter to present and future premature mortality over Europe: a non-linear response, Environ. Int., 2021, 153, 106517 CrossRef PubMed.
- J. Li, J. Li, G. Wang, K. F. Ho, W. Dai, T. Zhang, Q. Wang, C. Wu, L. Li, L. Li and Q. Zhang, Effects of atmospheric aging processes on in vitro induced oxidative stress and chemical composition of biomass burning aerosols, J. Hazard. Mater., 2021, 401, 123750 CrossRef CAS PubMed.
- S. J. Fuller, F. Wragg, J. Nutter and M. Kalberer, Comparison of on-line and off-line methods to quantify reactive oxygen species (ROS) in atmospheric aerosols, Atmos. Environ., 2014, 92, 97–103 CrossRef CAS.
- I. J. Keyte, R. M. Harrison and G. Lammel, Chemical reactivity and long-range transport potential of polycyclic aromatic hydrocarbons--a review, Chem. Soc. Rev., 2013, 42, 9333–9391 RSC.
- M. Shrivastava, S. Lou, A. Zelenyuk, R. C. Easter, R. A. Corley, B. D. Thrall, P. J. Rasch, J. D. Fast, S. L. Massey Simonich, H. Shen and S. Tao, Global long-range transport and lung cancer risk from polycyclic aromatic hydrocarbons shielded by coatings of organic aerosol, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 1246–1251 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00027g |
| ‡ Now at: Envineer Oy, Kuopio, Finland. |
| § Now at: Desert Research Institute (DRI), Reno, NV, USA. |
| This journal is © The Royal Society of Chemistry 2024 |