
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Does gas-phase sulfur dioxide remove films of atmosphere-extracted organic material from the aqueous aerosol air–water interface?†
Edward J.
Stuckey
 ab,
Rebecca J. L.
Welbourn
b,
Stephanie H.
Jones
c,
Alexander J.
Armstrong
b,
Matthew
Wilkinson
d,
James I. L.
Morison
d and
Martin D.
King
*a
ab,
Rebecca J. L.
Welbourn
b,
Stephanie H.
Jones
c,
Alexander J.
Armstrong
b,
Matthew
Wilkinson
d,
James I. L.
Morison
d and
Martin D.
King
*a
aCentre of Climate, Ocean and Atmospheres, Department of Earth Sciences, Royal Holloway University of London, Queens Building, Egham, Surrey, TW20 0EX, UK. E-mail: m.king@rhul.ac.uk
bISIS Pulsed Neutron and Muon Source, Rutherford Appleton Laboratory, Oxford, OX11 0QX, UK
cInstitute of Meteorology and Climate Research, Atmospheric Aerosol Research (IMK-AAF), Karlsruhe Institute of Technology (KIT), 76021 Karlsruhe, Germany
dForest Research, Alice Holt Lodge, Farnham, Surrey, GU10 4LH, UK
First published on 16th October 2024
Abstract
The reaction of gas-phase SO2 with unsaturated carbon–carbon double bonds forms organosulfates at the surface of the aerosol. Previous studies have focused on the reaction products and not the fate of organic films in the atmosphere. Neutron reflectometry was used to study the interaction of gas-phase SO2 at the air–water interface with organic material extracted from atmospheric particulate matter and pure proxy chemicals to determine whether the reaction of organic films with SO2 removes the film and if a product film is formed. Films formed from atmospheric aerosol collected in urban and woodland environments typically produced a layer of approximately 0.6 nm thickness, whereas a thick (>40 nm) film was formed by the woodsmoke sample. Fitting of this thicker woodsmoke film suggested a three-layered structure at the interface that has been interpreted to be consistent with a surfactant-rich layer next to the air–water interface, a mid-layer rich in polyaromatic hydrocarbons (PAH), and topped with a more aliphatic region. The multilayer structure of atmospheric extracted material at the air–water interface is potentially an exciting result that requires further study. Gas-phase SO2 was confirmed to react with pure insoluble surfactant molecules at the air–water interface that contained carbon–carbon double bonds (oleic acid) and did not react with a similar saturated surfactant (stearic acid). No reaction was observed during the interaction of SO2 and atmospheric material extracted from urban and woodland environments, and no material appeared to be removed from the interface; however, films made from woodsmoke-extracted material did appear to be altered by SO2 but there was no significant loss of material. In addition, the gas-phase ozone mixing ratios in the neutron blockhouse, which have historically been of some concern for reactions with organics, were found to be of the order 15 ppb, with no evidence of additional production in the neutron beam-path. Owing to a lack of substantial removal of material from real atmospheric extracted films, SO2 is not considered atmospherically significant for the removal of organic films from the air–water interface.
Environmental significanceThe environmental significance of this work is that gas-phase sulfur dioxide, with concentrations in the order of extreme conditions on our atmosphere, may alter but does not remove films formed of atmosphere-extracted material from the air–water interface. An additional finding of this work is that atmosphere-extracted material from woodsmoke may form multi-layered structures at the air–water interface. One portion of the multi-layered structure can be attributed to PAHs, and this layer appears to be altered by exposure to SO2. |
1 Introduction
The Earth's climate is influenced by atmospheric aerosols directly, through their ability to scatter and absorb light, and indirectly by their role as cloud condensation nuclei.1–3 The potential for aerosol to influence the climate is dictated in part by their chemical, optical and physical properties1–11 which are varied by factors such as source, environment, diurnal and seasonal variations,6,12–23 and chemical reactions e.g., oxidation.2,6,7,9,24–33 Owing to their importance in the atmosphere, aerosols have long remained a critical topic of atmospheric research; however, despite developments in understanding of the influence of atmospheric aerosol particles on the climate, uncertainties in their direct and indirect contributions to the radiative balance of the climate remain significant.2,34 Uncertainties in aerosol effects on the atmosphere come in part from variation of physical and chemical properties of aerosols between sources, environments and with time, as well as their highly complex chemical composition which makes definitive characterization difficult.The capability of an aerosol particle to affect the atmosphere can be altered by the presence of organic molecules on the particulate surface, which can result in the formation of a thin film of between a few Angstroms and up to hundreds of nanometers in thickness.26–32,35–42 These film-coated particulates may be considered to have core–shell morphology.43–45 The organic film may alter the light scattering and absorption of the aerosol particle,7,9,31,36 and also its potential to act as a cloud condensation nucleus.30,35,40,46 These films are typically formed from a combination of lipids,32,47 fatty acids28,30,31,41,48–56 and biomass burning products5,27,45,57 and are subject to chemical oxidation and cloud processing.26,27,29,30,32,35,53,58–62 The accumulation of organic films at the surface of an atmospheric particle, and their subsequent oxidation by atmospheric oxidants such as OH and NO3 radicals, O3, and SO2, will result in dynamic changes to the hygroscopic and optical properties of a particle throughout its lifetime.6,7,26,29,31,32,35–37,39,40,59,63–65 By considering the lifetime of aerosol organic films or film components, relative to the physical lifetime of the host particulate in the atmosphere, it is possible to determine the significance of film oxidation. If organic film lifetimes from oxidation are significantly shorter or longer than the physical lifetime of aerosol particulates then the aerosol may be considered to be constantly uncoated or coated within atmospheric models. If the organic film lifetime is comparable to that of the aerosol, or some material remains at the interface post-oxidation, then both the kinetics and light scattering influence of the product film will need to be considered in atmospheric models.
The reaction of gaseous sulfur dioxide with organics has been proposed to be atmospherically significant owing to its ability to react with atmospherically relevant organics to form organosulfates66–70. Previous studies on organic-SO2 reactions are limited, however, they include: measuring the formation of organosulfates through the reaction of SO2 and unsaturated fatty acids and alkenes in a flow tube with diffuse reflectance infrared Fourier transform spectroscopy; Ultra-High-Performance Liquid Chromatography coupled with High-Resolution Mass Spectrometry and a Switchable Reagent Ion Time-of-Flight Mass Spectrometer;66,67 the reaction of SO2 and intermediates of monoterpene ozonolysis with varying humidity observed using electrospray mass spectrometry;68 and the production of organosulfates from the reaction of fatty acids in ambient particulate matter (PM2.5) extracted from southern China.71 These previous studies focused on the characterization of the products of organosulfate formation but did not consider whether the organic material remains surface active throughout the reaction with gas-phase SO2. The objective of the work herein is to determine whether organic films consisting of proxy (pure chemical) and atmospheric extracted material remain at the air–water interface of a particle is when it is exposed to gas-phase SO2.
Organic films have previously been measured at the air–water interface as a proxy for atmospheric processes by a variety of techniques, e.g. surface tension,72–74 Fourier Transform Infrared spectroscopy (FTIR),72–75 mass spectrometry,69,76 Brewster angle microscopy,77 and X-ray26,42,78 and neutron scattering techniques.26–31,37,61,62,79–83 The current investigation implemented specular neutron reflectometry owing to its ability to make real-time, non-destructive measurements of the structure of thin films at the air water interface with Ångström resolution normal to the plane of the interface. Neutron reflectometry was used to determine the thickness of a film and the amount of material at the interface throughout its reaction as performed previously by the authors herein.26–31
Films of organic material spread on a water surface can be used to mimic aerosol organic films.26–31,41,55,61,62,84 Organic films have been shown to partially decay when exposed to varying atmospherically relevant oxidants, leaving some or no material at the interface.26–31 The changes of the organic film in the presence of an oxidant allows the lifetime of the organic film to be determined,26,27,29,31–33,85 which is a measurable analogue for atmospheric reactions. Investigations are typically performed using atmospheric proxies such as oleic acid and stearic acid, the reactions of which have been observed with O3,26,31,41,54,55,75,79,86–92 OH27,64,70,85,93–99 and nitrate radicals.28,57,61,62,64,88,100–104 Oleic acid is commonly used as an atmospheric proxy material owing to its prevalence in the atmosphere as an organic molecule with a single unsaturated carbon–carbon double bond, while stearic acid provides a similar saturated counterpart.31 However, in order to provide a more realistic representation of atmospheric processes, water-insoluble organic matter extracted from the atmosphere of characteristic regions of urban, woodland, and woodsmoke environments have been used within this work, analogous to prior work by some of the authors.6,13,26,27
The current study investigated the stability of organic films at the air water interface when exposed to SO2 using neutron reflectometry. The significance of SO2 as an oxidant in the atmosphere can be inferred by determining if there is a change in the morphology and presence of organic films after their reaction with SO2. Films were also exposed to other atmospherically relevant gases for comparison. Films of extracted atmospheric organic matter were studied as well as the commonly studied atmospheric proxies: oleic and stearic acid. By comparing real and proxy films exposed to SO2, conclusions may be drawn with greater confidence as to the relevance of any observed reactions with atmospheric proxies, and hence regarding the significance of SO2 in the atmosphere.
2 Methods
2.1 Sample extraction and preparation
Atmospheric material was extracted from two different environments defined as ‘urban’ and ‘woodland’, and one direct source which is defined as ‘woodsmoke’. Urban aerosol was collected in sampling periods of ∼30 days over the months of September, October and November on a rooftop at a height of ∼15 m at the campus of Royal Holloway, University of London, which is in close proximity to three major motorways, an international airport, and the city of London. Owing to the proximity of the sampling site to these major sources of pollutants, this sample was deemed ‘urban’. Woodland aerosol was collected over sampling periods of ∼30 days in the months of April and May. The sampling site was at a height of ∼24 m above the canopy of the Straits Enclosure of the Alice Holt Forest, Hampshire, UK, which is a predominantly oak woodland. Woodsmoke aerosol was collected from the chimney of a domestic woodburner in which seasoned Weymouth pine (Pinus strobus) was burned over a period of 6 hours and aerosol from the smoke was collected during the main burn.Urban and woodland atmospheric particulate material was collected using an air pump at a flow rate of 30 L min−1. Air was pulled through a stainless-steel pipe (10 cm × 1/4” OD) into a filter holder under ambient temperature and pressure. Quartz filters (47 mm, Whatman) were pre-combusted in a furnace at 550 °C for 4 hours and were encased in Perfluoroalkoxy (PFA) Savillex filter holders for the urban samples and aluminum filter holders for the woodland and woodsmoke samples. The aluminum filter holders were fabricated at Royal Holloway, University of London. The glassware used in atmospheric material extraction was cleaned multiple times using a combination of ultrapure water (>18 MΩ cm) and chloroform (Sigma-Aldrich, 0.5–1% ethanol as stabilizer) and constructed and deconstructed in a clean glove bag or laminar air flow hood. The filter samples were stored in the dark at −18 °C in glass Petri dishes wrapped in aluminum foil and a sealed plastic bag until extraction.
In order to isolate the water-insoluble portion of the sample, the filter was shaken vigorously in a conical flask in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of water:chloroform. The solution was then filtered through a further quartz filter to remove remnants of the original filter. The glassware and filter remnants were rinsed three times further with water and chloroform. The chloroform portion contained water-insoluble surfactant material which was used to form a thin film at the air–water interface. Glassware was rinsed with ultrapure water and chloroform. A separating funnel was used to separate the chloroform solution, which was then evaporated under nitrogen and blown down, producing a waxy or oily residue (sample origin dependent) and this was dissolved in 2 ml of chloroform. These solutions were stored in the dark in amber glass bottles at −18 °C until their measurement using neutron reflectometry. The atmospheric proxies were used in a deuterated form to provide additional neutron contrast and increased reflectivity. The samples extracted from the atmosphere are a very complex mixture of chemicals and are likely to be more representative of typical organic material in the atmospheric aerosol than a single proxy chemical, however, these atmospheric samples could not possibly span all possible organic molecules in the atmosphere and are merely indicative of the environments they were extracted from.
:1 solution of water:chloroform. The solution was then filtered through a further quartz filter to remove remnants of the original filter. The glassware and filter remnants were rinsed three times further with water and chloroform. The chloroform portion contained water-insoluble surfactant material which was used to form a thin film at the air–water interface. Glassware was rinsed with ultrapure water and chloroform. A separating funnel was used to separate the chloroform solution, which was then evaporated under nitrogen and blown down, producing a waxy or oily residue (sample origin dependent) and this was dissolved in 2 ml of chloroform. These solutions were stored in the dark in amber glass bottles at −18 °C until their measurement using neutron reflectometry. The atmospheric proxies were used in a deuterated form to provide additional neutron contrast and increased reflectivity. The samples extracted from the atmosphere are a very complex mixture of chemicals and are likely to be more representative of typical organic material in the atmospheric aerosol than a single proxy chemical, however, these atmospheric samples could not possibly span all possible organic molecules in the atmosphere and are merely indicative of the environments they were extracted from.
The deuterated atmospheric proxy materials d17-oleic and d35-stearic acids were purchased from Sigma Aldrich and were used without further purification after being dissolved in ultrapure chloroform.
2.2 Neutron reflectometry & data analysis
Neutron reflectometry is a non-destructive technique that can provide structural information normal to the plane of an interface.37,105,106 The penetrative capabilities and Ångström resolution of neutron reflection techniques enable it to be used to measure changes to the thickness and scattering length density of an interfacial organic film throughout its oxidation as a function of time.Neutron reflectometry measures the specular reflected intensity, I, of a collimated beam of neutrons, which is then normalized by the incident neutron beam, I0, to give reflectivity, 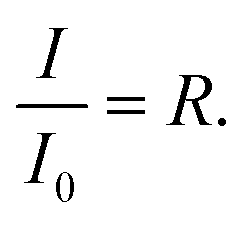 Typically, neutron reflectivity, R, is presented as a function of momentum transfer, Q,105 known henceforth as the reflectivity profile, where
Typically, neutron reflectivity, R, is presented as a function of momentum transfer, Q,105 known henceforth as the reflectivity profile, where 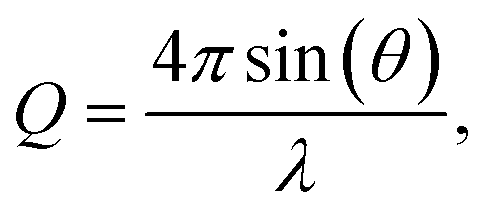 λ is the neutron wavelength and θ is the angle of incidence. The applications and foundational theory of neutron reflectometry are described elsewhere.37,79,82,84,106,107 The instrument used in this experiment, INTER, is a multi-wavelength instrument with a wavelength range of 1.7–17 Å, which can elucidate detailed structural information in real time.79,107–109 Incident angles, θ, of 0.8° and 2.3° were used for the initial measurement of the highly scattering samples providing a total Q-range of 0.01–0.3 Å−1 and an angle of 0.8° was maintained for monitoring the samples throughout their exposure to gases.
λ is the neutron wavelength and θ is the angle of incidence. The applications and foundational theory of neutron reflectometry are described elsewhere.37,79,82,84,106,107 The instrument used in this experiment, INTER, is a multi-wavelength instrument with a wavelength range of 1.7–17 Å, which can elucidate detailed structural information in real time.79,107–109 Incident angles, θ, of 0.8° and 2.3° were used for the initial measurement of the highly scattering samples providing a total Q-range of 0.01–0.3 Å−1 and an angle of 0.8° was maintained for monitoring the samples throughout their exposure to gases.
Changes to the intensity of specularly reflected neutrons arise from the variation in the scattering length density, ρ, the scattering length per unit volume, between matter in the direction perpendicular to the interface (material specific property). The reflectivity profile is also affected by the thickness, δ, of each medium the neutrons pass through. Owing to the differences in scattering length between hydrogen and deuterium, substitutions between these isotopes can be implemented to alter the scattering length density of a sample or bulk material in order to hide or highlight information from part of the sample in the reflectivity profile – this is a method known as contrast matching.82,106,110 Contrast matching was applied in this work to remove the scattering contributions from water by matching the scattering length density of the water below the film to that of air (zero scattering length density) above it.
To obtain structural information, a model must be fitted to the reflectivity profile. A model of scattering length density versus distance through the film, normal to the interface, (scattering length density profile) is optimized within constraints to reproduce the measured reflectivity profile. These models are typically composed of a layered structure between two infinitely thick bulk materials, with each layer being described by a series of properties: thickness, scattering length density and inter-layer roughness, all of which contribute to the sample's reflectivity.37,111 In the work herein the infinitely thick layers are air and air-contrast-matched water either side of the film. The reflectometry fitting package refnx,111 was used in this work which uses the Abelès method to reproduce reflectivity.36 The model was described by a single layer between two infinitely thick layers except for woodsmoke, where a single layer was found to be insufficient to reproduce the reflectivity profile and a multi-layered structure was used. The initial parameterization of the samples was predominantly determined from previous literature,13,31,112,113 except for the woodland sample which was estimated based on values of film thickness and scattering length density for atmospheric extracted material observed by Shepherd et al.27 owing to a lack of previous measurements. Interfacial and film roughness, neutron background, film thickness and scattering length densities of the films were varied using a combination of Markov Chain Monte Carlo analysis (MCMC)114,115 and nested sampling116 within refnx for each sample, within pre-defined ranges based on reasonable physical limitations.
During the oxidation reaction, the precise atomic composition of the layer cannot be known so the scattering length density cannot be directly calculated. However, for these systems the scattering length density and thickness parameters are correlated so the product of scattering length density and thickness, ρδ, may be followed as a relative measure of the amount of material at the air–water interface. The product, ρδ, is the scattering length per unit area, and is taken as a proxy for surface excess, Γ, or the amount of scattering material at the interface, which has previously been used as a kinetic variable i.e.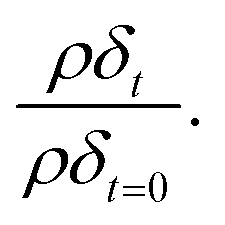 31 For a thin film between two bulk media of scattering length density zero, changes in reflectivity, and therefore in the intensity of the reflected beam, can be used as a measure of the change in the amount of material at the interface owing to the proportional relationship between the square root of neutron reflectivity intensity,
31 For a thin film between two bulk media of scattering length density zero, changes in reflectivity, and therefore in the intensity of the reflected beam, can be used as a measure of the change in the amount of material at the interface owing to the proportional relationship between the square root of neutron reflectivity intensity, 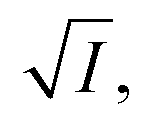 and the surface excess at a given point in momentum transfer. The quantity
and the surface excess at a given point in momentum transfer. The quantity  is used as the kinetic variable as this is the most direct, relevant data we are able to extract from reflectivity measurements. Henceforth this method is referred to as the ‘total counts method’. The ESI† has further details, including a comparison of this relationship to more usual data-fitting methods. The work herein utilizes this approach to determine changes in the film on exposure to different atmospheric gases, including oxidants, with time, using the neutron reflectometry instrument INTER, at the ISIS neutron and muon source, UK.
is used as the kinetic variable as this is the most direct, relevant data we are able to extract from reflectivity measurements. Henceforth this method is referred to as the ‘total counts method’. The ESI† has further details, including a comparison of this relationship to more usual data-fitting methods. The work herein utilizes this approach to determine changes in the film on exposure to different atmospheric gases, including oxidants, with time, using the neutron reflectometry instrument INTER, at the ISIS neutron and muon source, UK.
2.3 Experimental set up
A polytetrafluoroethylene (PTFE) Langmuir trough (240 × 75 × 40 mm) was housed within a Tedlar gas bag with a 6.4 L capacity and aluminum foil windows for the neutron beam-path. Gases were transported through quarter inch PTFE tubing with stainless steel fittings. The trough was initially cleaned using hexane, toluene, chloroform and ultrapure water, and subsequently with chloroform and ultrapure water between samples. The water-insoluble surfactants (listed in Table 1) were dissolved in chloroform in concentrations of 1 mg ml−1 for oleic and stearic acid and unknown concentrations for the atmospheric extracts. These solutions were spread onto air contrast matched water (91.9%/8.1% H2O/D2O v/v) using a microliter syringe to produce a surface pressure of ∼15 mN m−1, based on prior measurements in the same trough using a surface tensiometer. Detailed experimental and theoretical consideration of spreading insoluble surfactants at an air–water interface can be found in Davies and Rideal.117 Gases (zero air, nitrogen, ozone, and sulfur dioxide, the composition of which are summarized in Table 1) were flowed through the Tedlar bag at 0.3 L min−1 to provide an atmosphere of the desired gas with a typical mixing time in the chamber of Gases (zero air, nitrogen, oxygen and sulfur dioxide) were sourced from Air-Liquide and used without further purification. Ozone was produced using a commercial ozonizer (UVP Ltd) through the photolysis of O2 (continuous flow of 0.3 L min−1) in a quartz glass tube using a mercury pen-ray lamp. The mixing ratio of the ozone produced was measured in ppb using a ThermoScientific ozone analyzer, Model 49i. The chamber was purged with nitrogen between samples. All measurements were carried out in the dark at an ambient temperature of ∼20 °C. The reaction described here was studied in the dark. The reactions of sulfur dioxide in the presence of light may be faster owing to promotion to a triplet state and further information is contained in Passananti et al.66 Further investigation of the effect of light on the reaction presented here is beyond the scope of this work. Owing to the reaction being measured at the air water interface, there is an abundance of liquid water present in the chamber and the conditions near the interface are representative of a humid environment. The temperature of all the experiments was maintained at 20 °C. The humidity was not controlled but was kept constant. All incoming gas flows were dry and approximately 75 ml of water was added to the area surrounding the trough to minimise evaporation. The relative humidity a few nanometres over the trough was assumed to be >95%. The gas flow rate was 0.3 L min−1 and not varied. The mixing time in the gas volume above the trough was ∼1300 seconds (i.e. very small compared to the total time of the reaction experiment).
Gases (zero air, nitrogen, oxygen and sulfur dioxide) were sourced from Air-Liquide and used without further purification. Ozone was produced using a commercial ozonizer (UVP Ltd) through the photolysis of O2 (continuous flow of 0.3 L min−1) in a quartz glass tube using a mercury pen-ray lamp. The mixing ratio of the ozone produced was measured in ppb using a ThermoScientific ozone analyzer, Model 49i. The chamber was purged with nitrogen between samples. All measurements were carried out in the dark at an ambient temperature of ∼20 °C. The reaction described here was studied in the dark. The reactions of sulfur dioxide in the presence of light may be faster owing to promotion to a triplet state and further information is contained in Passananti et al.66 Further investigation of the effect of light on the reaction presented here is beyond the scope of this work. Owing to the reaction being measured at the air water interface, there is an abundance of liquid water present in the chamber and the conditions near the interface are representative of a humid environment. The temperature of all the experiments was maintained at 20 °C. The humidity was not controlled but was kept constant. All incoming gas flows were dry and approximately 75 ml of water was added to the area surrounding the trough to minimise evaporation. The relative humidity a few nanometres over the trough was assumed to be >95%. The gas flow rate was 0.3 L min−1 and not varied. The mixing time in the gas volume above the trough was ∼1300 seconds (i.e. very small compared to the total time of the reaction experiment).
| Pre gas exposure characterisation | Exposure conditions | |||||
|---|---|---|---|---|---|---|
| Material | Film thickness/Å2 | Scattering length density/10−6 Å−2 | Gas | Mixing ratio | ||
| This work | Other works | This work | Other works | |||
| a Gas suspended in air. b Produced in situ using a UV lamp. c A 3 layered structure was required to fit the woodsmoke data to a scattering length density profile, discussed at length in 3.3. Values for thickness and SLD calculated from this work are of each component of the three layered structure and shown in the order: a hydrocarbon or alkane/alkene layer, a layer of polycyclic aromatic hydrocarbons, and a layer alkyl surfactants. d Estimated using NIST scattering length density calculator *error not reported for urban and woodland samples due to small reflectivity and very high correlation between SLD & thickness for thin, low scattering films. | ||||||
| Urban | 6.4* | 3.6–10.2 (ref. 27) | 1.2* | 0.68–0.89 (ref. 27) | SO2 in air | 450 ppba |
| Urban | 6.3* | 3.6–10.2 (ref. 27) | 1.3* | 0.68–0.89 (ref. 27) | Zero air | 80/20% O2/N2 |
| Woodsmokec |
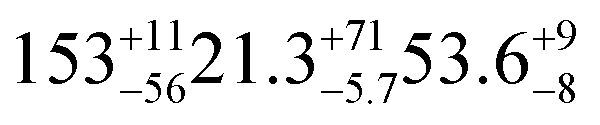
|
18.6 ± 0.5 (ref. 27) |
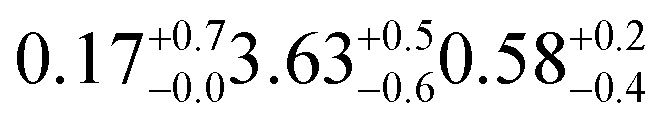
|
1.72 ± 0.05 (ref. 27) | SO2 in air | 450 ppba |
| Woodsmokec |

|
18.6 ± 0.5 (ref. 27) |

|
1.72 ± 0.05 (ref. 27) | Zero air | 80/20% O2/N2 |
| Woodland | 6.4* | — | 1.0* | — | SO2 in air | 450 ppba |
| d17-oleic | 18.3 ± 0.5 | 20 (ref. 31) | 3.30 ± 0.09 | 3.3d | SO2 in air | 450 ppba |
| d17-oleic | 18.6 ± 0.8 | 20 (ref. 31) | 3.30 ± 0.13 | 3.3d | Zero air | 80/20% O2/N2 |
| d17-oleic | 19.5 ± 0.5 | 20 (ref. 31) | 3.28 ± 0.08 | 3.3d | N2 | 99.999% |
| d17-oleic | 17.7 ± 0.6 | 20 (ref. 31) | 3.29 ± 0.11 | 3.3d | O3 in O2 | 400–450 ppba,b |
| d35-stearic | 19.3 ± 0.4 | 21 (ref. 113) | 6.0 ± 0.10 | 6.4d | SO2 in air | 450 ppba |
2.4 Background ozone monitoring
Surface active organic films with unsaturated carbon–carbon double bonds are sensitive to reactions with gas-phase ozone. Debate about background generation of ozone within the beam-path at neutron spallation sources has been a concern for experiments involving unsaturated organic films which are sensitive to oxidation for some time. Therefore measurements of ozone mixing ratios were performed using a ThermoScientific ozone analyzer, Model 49i, to monitor ozone mixing ratios within the experimental area. The ozone analyzer was calibrated to zero with N2 gas. Measurements were sampled from both outside and inside the blockhouse that shields users from neutron radiation and inside the Tedlar bag at the point of neutron measurement with N2 or O2 flowing at 0.3 L min−1 (i.e. contained volume within the beam-path). The results showed mixing ratios of 18 ppb, 12 ppb, 15 ppb and 0 ppb outside the blockhouse, inside the blockhouse and in the Tedlar bag (direct beam-path) for O2 and N2 respectively. These mixing ratios of ozone were similar to mean concentrations of ozone observed in indoor environments in the UK (∼5 ppb),53 thus while low mixing ratios of ozone were observed this is not considered significant in the reaction with organic thin films on the timescale of this work and suggests ozone production within the beam path is not a significant issue for neutron reflectometry measurements.2.5 Infrared absorption of product films
To provide additional information on any reaction products between an organic film and gas-phase SO2, infrared absorption measurements were carried out on the product film from the exposure of oleic acid to SO2 for 2 hours. All measurements were taken using a Nicolet iS50 FTIR Spectrometer. The surface-active products from this reaction were collected from the air–water interface and extracted by adding 5 ml of chloroform to the solution. The surface extract solution of chloroform and water was well-mixed and the chloroform component was separated using a separating funnel. The reaction of oleic monolayers with SO2 and extraction of product films was repeated five times and the products were combined to produce one sample. Excess chloroform was removed from the combined solutions by gently blowing nitrogen over the new solution until only 5 ml remained. A drop of this solution was added to a CaF2 window, evaporated under nitrogen, and subjected to FTIR analyses concentrating on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretch region visible at ∼3004 cm−1.
C–H stretch region visible at ∼3004 cm−1.
3 Results & discussion
The thickness and scattering length densities obtained via neutron reflectometry of organic films at the air–water interface consisting of proxy and atmospheric extracted material will be presented, discussed and compared with existing literature. Changes to the organic films when exposed to zero air, nitrogen, ozone, and sulfur dioxide over long periods of time will then be considered. Lastly, the structure and oxidation of the film formed from the woodsmoke extract will be presented and discussed separately.The thickness of films and their scattering length densities from reproduced neutron reflection profiles are summarized in Table 1, and typical pre-reaction reflectivity profiles are presented in Fig. 1. Note that all the samples show notable reflectivity compared to a null measurement of a blank interface of air-contrast matched water and air. While the signal from deuterated proxies and woodsmoke films are much higher in reflectivity, films of weakly scattering, undeuterated atmospheric extract have been shown to be observable using neutron reflectometry as demonstrated here and previously by Shepherd et al.27
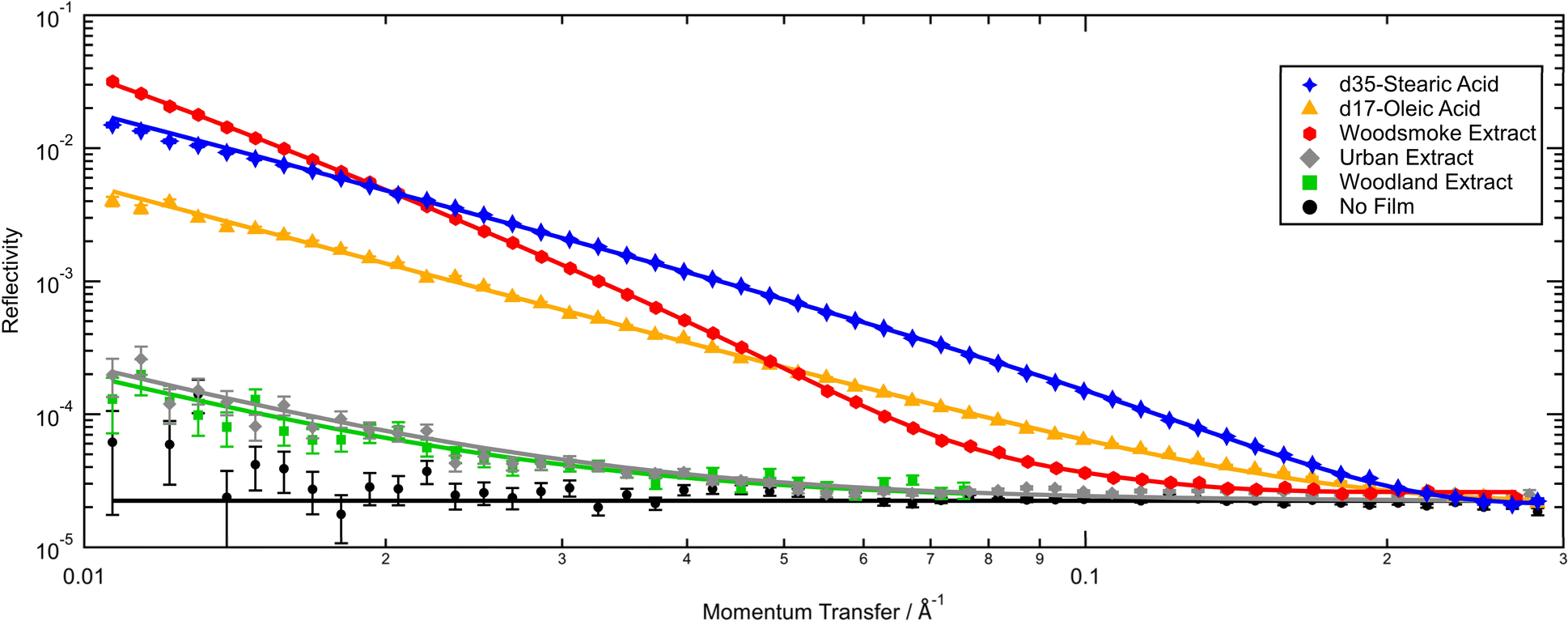 | ||
| Fig. 1 Neutron reflectivity profiles (points) and the ‘best fits’ (lines) of organic films formed from deuterated atmospheric proxy and real atmospheric material at the interface of air and air-contrast matched water. Films were measured under 0.3 L min−1 zero air flow from a cylinder in a gas-tight environment. | ||
3.1 Film properties – scattering length density and thickness
As far as the authors are aware, this work is the first instance of the measurement of a thin film at the air–water interface formed from atmospheric material extracted from woodland environments. The initial thickness and scattering length density of the film formed by atmospheric extracted material from deciduous woodland were 6.4 Å and 1.0 × 10−6 Å−2. Owing to a lack of previous comparable literature, the woodland sample can only be compared with those from other environments. The woodland sample is similar in film thickness to the urban sample presented here, however, the scattering length density is lower. Although the woodland sample was collected in a deciduous 90 hectare plantation, the sampling location is also impacted by sources of traffic and urban pollution so may be more representative of a rural sample rather than a remotely located woodland.
3.2 Film kinetics
The proportion of organic scattering material at the air–water interface throughout exposure to various atmospherically relevant gases calculated by the total counts method is presented in Fig. 2. The errors are estimated using a Poisson distribution for the raw counts and displayed as one standard deviation. The change in the amount of material at the interface with time was used to determine the significance of gas-phase SO2 in the removal of films from the air–water interface in comparison to zero air. The mixing ratio of SO2 used in this work is large with respect to what can be found in the atmosphere, and is similar to mixing ratios found in volcanic plumes118 and slightly in excess of those found in power stations with poor desulfuring technology.119 The mixing ratio of SO2 used here is typical of extreme values found in the atmosphere and was used to elicit any large observable effect with the observed organic films. While the kinetics of reactions of SO2 with organics have been shown to be mixing ratio dependent, the focus of the work presented here is on whether the film is removed or altered by SO2 and thus a large mixing ratio was used to investigate an effect, i.e. whether the film is removed or altered by SO2.67,68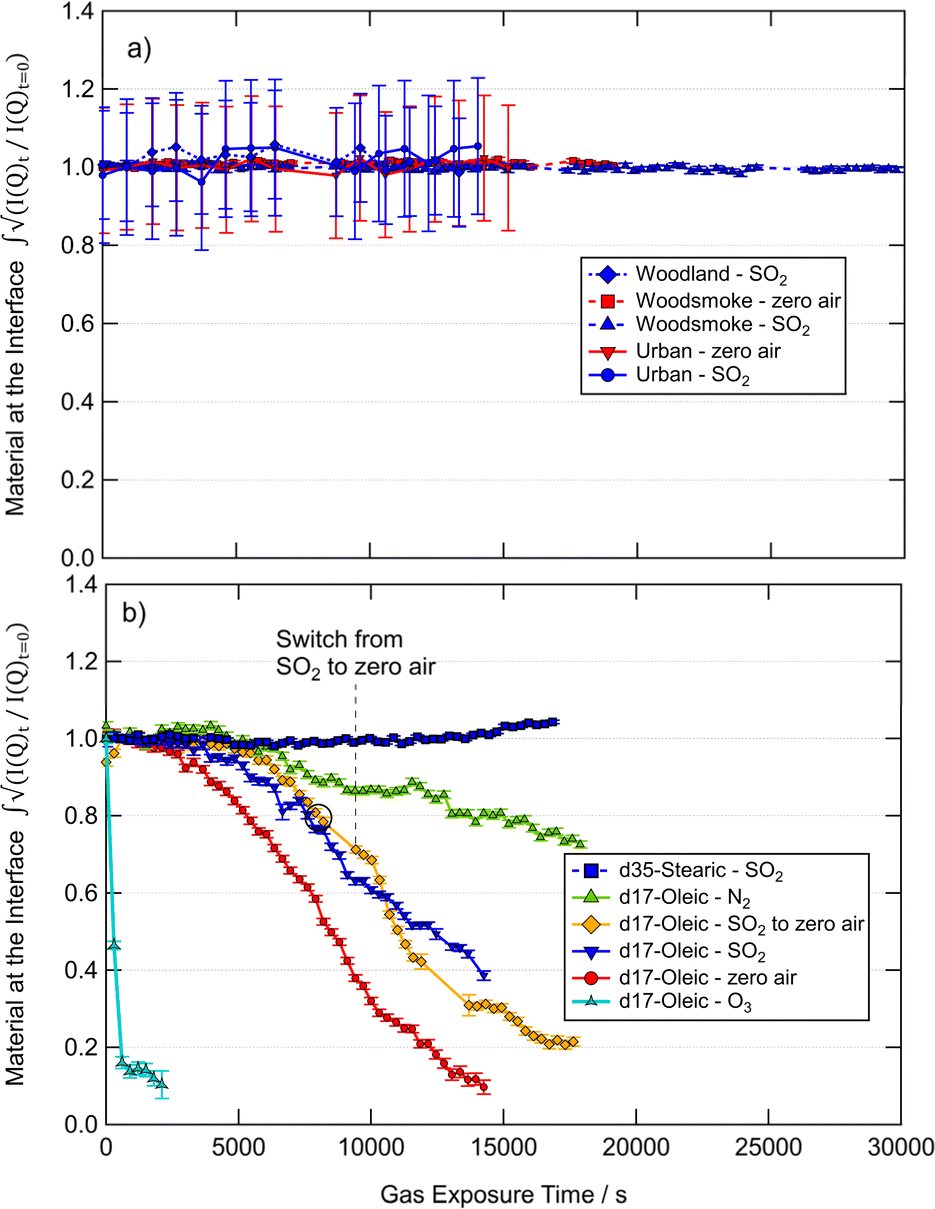 | ||
| Fig. 2 The relative proportion of material remaining at the air–water interface throughout the reaction of monolayers of deuterated atmospheric proxies (d17-oleic acid, d35-stearic acid) and real atmospheric extracts from woodland, urban and woodsmoke environments with atmospherically relevant gases. Gas flow was maintained at 0.3 L min−1. Errors show one standard deviation. | ||
The film of stearic acid showed no significant changes at the interface with increased exposure time to SO2 which indicates no reaction and no removal of material from the interface. The reaction was a control as unsaturated carbon–carbon double bonds are needed for the reaction of organics with SO2. A small increase is observed in the stearic acid film measurement at long exposure times to SO2, this may be attributed to a change in the packing of the stearic acid molecules at the air–water interface owing to a change in pH of the sub-phase which is becoming more acidic with exposure to SO2.
The oleic acid films showed a loss of scattering material throughout exposure to O3, zero air, SO2, and N2. The exposure of the oleic film to N2 demonstrated the relative stability of the film in the trough, and a comparison of oleic acid to that of woodsmoke, woodland and urban samples (Fig. 2) demonstrate relative surface instability of oleic acid in comparison to films of atmospheric extracted material. The rate of loss of oleic acid from the interface under N2 is similar to controls in previous experiments (e.g.Fig. 2 and 4 from King et al., 2020 (ref. 31)). Exposure of the oleic acid film to zero air showed a greater and more rapid loss of material from the interface in comparison to N2, indicating a reaction of oleic acid and O2 which has been previously demonstrated.31 Interestingly, the interaction of SO2 (in air) and the oleic film resulted in a slightly slower loss of material than with zero air. Additionally, in one measurement where the gas was changed from SO2 to zero air, it can be seen that the rate of change of material at the interface varies from being similar to the oleic acid decay under SO2 to being more representative of the oleic acid decay under air. Gas-phase SO2 may be reducing the rate of material loss from the interface owing to the competition between SO2 and O2 for the unsaturated carbon–carbon double bond. The reaction of SO2 and oleic acid may produce a more stable surface product than with O2 which ultimately slowed the rate of loss of material from the interface in comparison to zero air. However, this is one possible mechanism, and further spectroscopy focused experiments would be required to determine if this is the correct interpretation. What has been shown is that the rate of loss of oleic acid films at the air–water interface is comparable between air and SO2, thus, from an atmospheric perspective, the reaction of oleic acid with SO2 is not more significant than the reaction of oleic acid and O2 in respect to the removal of aerosol organic films.
FTIR analysis of the product of reaction of SO2 with oleic acid extracted from the air–water interface showed that the CC–H stretch had been removed during the exposure of oleic acid to SO2 (Fig. 3); a comparison of the post-reaction spectra and that of a standard literature example120 is shown in Fig. 3. Note the loss of the peak at ∼3004 cm−1, which has previously been assigned to CC–H stretch, indicates a reaction of SO2 with the double bond which may result in the formation of S–O bonds (organosulfates),66,121 and the loss of this stretch in the FTIR supports the idea of reaction between SO2 and unsaturated carbon–carbon double bonds.
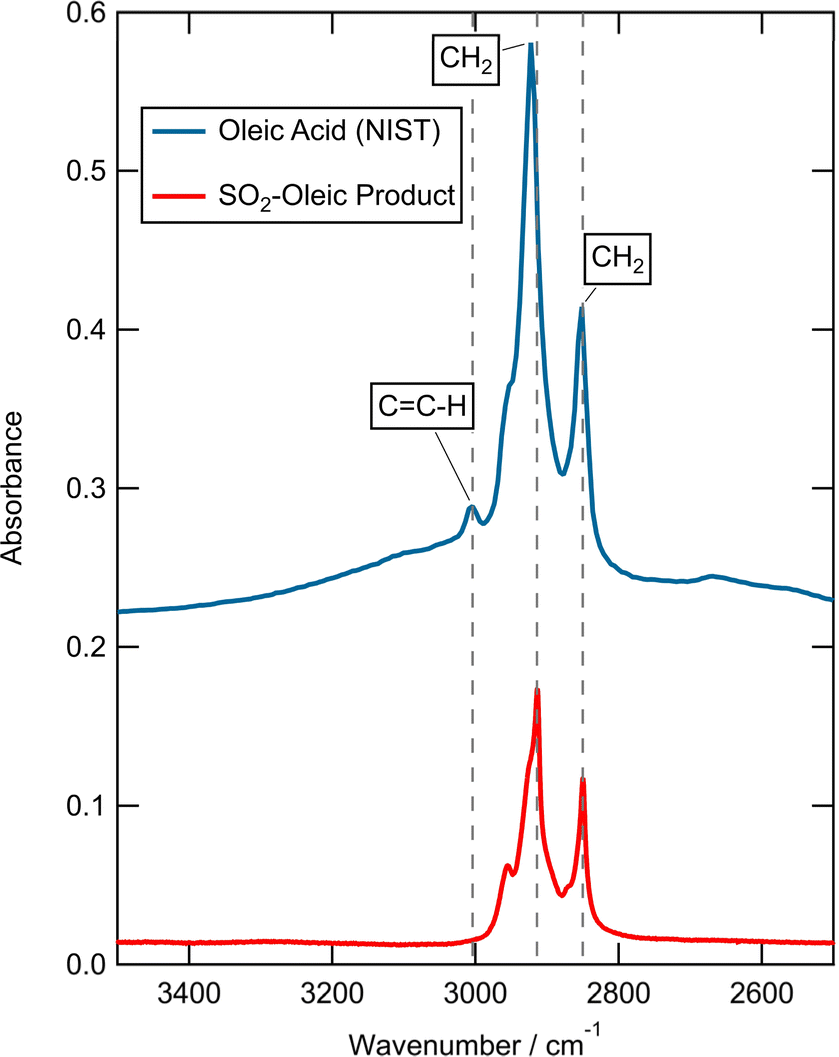 | ||
| Fig. 3 FTIR spectra of organic material extracted from the air–water interface after the reaction of SO2 and oleic (red, bottom) versus literature oleic120 (blue, top) (translated on y-axis by 0.2).51 The product following reaction of oleic acid with SO2 shows a loss of the CC–H peak indicating a reaction with SO2 has occurred. | ||
3.3 Woodsmoke film structure & reaction with SO2
The reflectivity profile of the film formed from the woodsmoke extracted film is shown in Fig. 1. A model of a single, thin film did not adequately reproduce the measured reflectivity profile for the woodsmoke samples and a more complex model was required to produce a better fit. The woodsmoke film was modelled as a multi-layer structure with the best-fit scattering length density profiles shown in Fig. 4 and 5. Note that the scattering length densities and total film thickness shown here significantly exceed those found previously by Shepherd et al.27 for woodsmoke samples. However, a much greater amount of material was deposited at the air–water interface in this investigation and the sample was produced by the burning of a different wood (Weymouth pine, Pinus strobus, cf. Wild cherry, Prunus avium). | ||
| Fig. 4 The neutron scattering length density profile of two films of woodsmoke extracted organic material at the air–water interface before and after exposure to SO2 (top) and zero-air (bottom). The exposure of the film to SO2 caused a decrease in the scattering length density of the central component, which may allude to a reaction of SO2 and PAHs, which the large scattering length density of the central component has been ascribed to. Little change in the scattering length density profile of the film before and after exposure to zero-air indicates no reaction. | ||
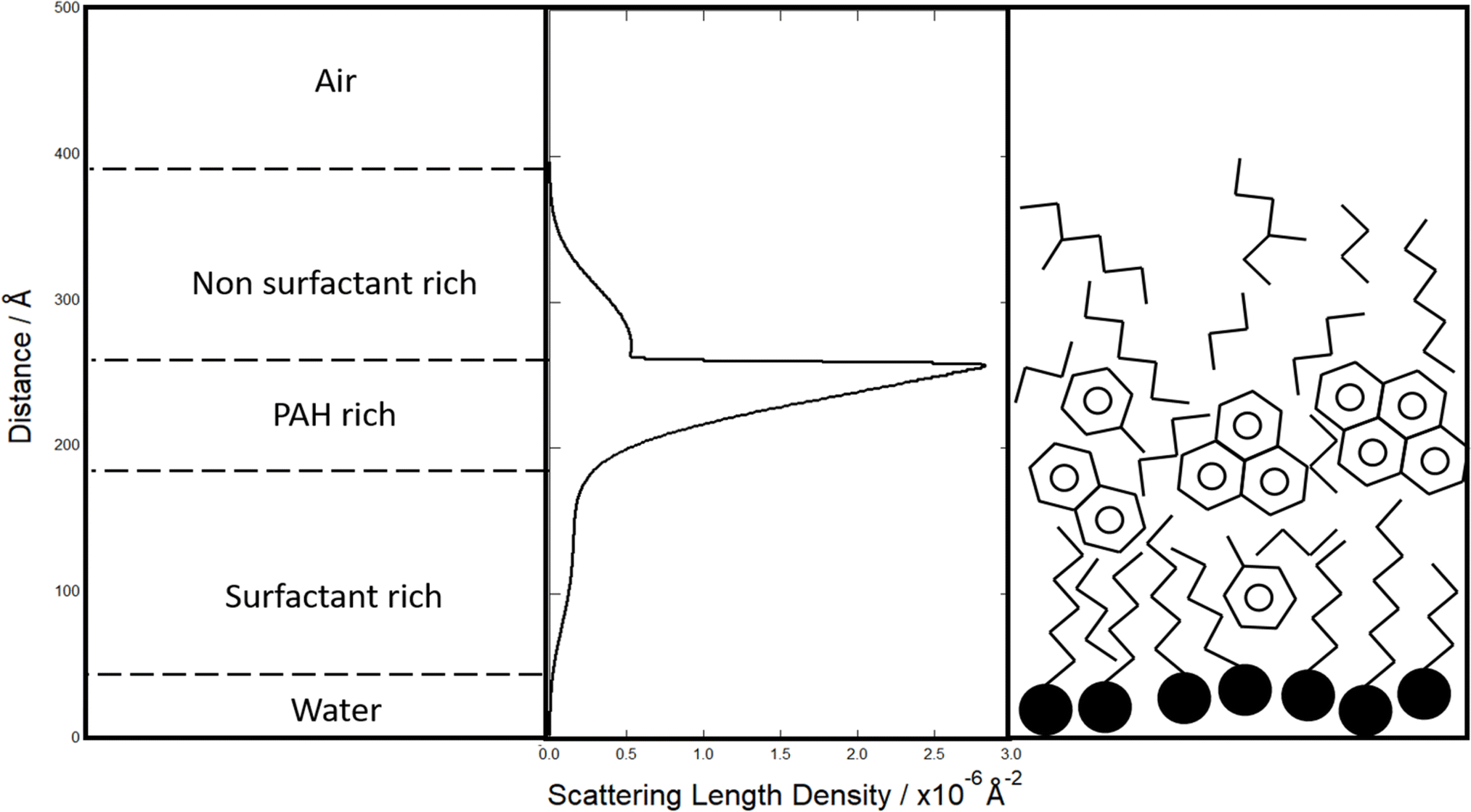 | ||
| Fig. 5 A cartoon interpretation of the modelled scattering length density of an organic film at the air–water interface formed from atmospheric extracted material from deciduous wood burning. The structure was determined based on the approximated scattering length densities and surface activity of typical deciduous burning products. The presented diagram suggests a potential three-layered structure of surfactants, PAHs, and non-surfactants on top of the film. | ||
Despite the woodsmoke sample being deposited as a homogenous solution, the best-fit scattering length density profile (Fig. 4) shows a separation of low and high scattering regions which may indicate some ordering of compounds within the film. While ordering of proxy atmospheric organic material has been observed previously,78,123,124 this is the first instance to the authors' knowledge that this ordering has been observed with the use of material extracted from the atmosphere. Furthermore, both a repeat deposition of the film and reparameterization of the scattering model resulted in similar structures.
The scattering length density profiles shown in Fig. 4 and 5 are the result of extensive searches through possible scattering length density profiles using MCMC analyses to find the best-fit of the measured reflectivity of these samples. The scattering length density profiles indicate a three component structure with a lower (<0.5 × 10−6 Å−2), medium (0.5–1.5 × 10−6 Å−2) and a higher (2.5–3.0 × 10−6 Å−2) scattering length density. A comparison of these components with Table 2 (ref. 113 and 114) shows that these three film components may be interpreted as a layer of alkyl surfactants (lower), a layer of polycyclic aromatic hydrocarbons (higher), and a hydrocarbon or alkane/alkene layer (medium). These are suggested based on the scattering length densities. The alkyl layer is likely the closest to the water and composed of organic acids to give some surface active nature, supporting the other components on top as a stable air–water film. A cartoon of this is shown in Fig. 5. Readers should note that incomplete surface coverage or hydration of the film will result in a lowering of the SLD, and the interpretation here is just one solution and additional studies are needed to confirm in full detail.
| Compounds | Formula | Density/g cm−1 | SLD/10−6 Å−2 |
|---|---|---|---|
| Fluoranthene | C16H10 | 1.25 | 2.57 |
| Pyrene | C16H10 | 1.27 | 2.61 |
| benzo [a]pyrene | C20H12 | 1.35–1.4 | 2.84–2.94 |
| Phenanthrene | C14H10 | 0.98–1.18 | 1.84–2.22 |
| Pelargonic acid | C9H18O2 | 0.91 | 0.14 |
| Lauric acid | C12H24O2 | 0.88 | 0.04 |
| Myristic acid | C14H28O2 | 0.86 | 0.00 |
| Behenic acid | C22H44O2 | 0.90 | −0.11 |
| Catechol | C6H6O2 | 1.34 | 2.13 |
| 4-Hydroxybenzoic acid | C7H6O3 | 1.46 | 2.64 |
| Vanillic acid | C8H8O4 | 1.40 | 2.33 |
| Levoglucosan | C6H10O5 | 1.70 | 1.29 |
| Dehydroabietic acid | C20H28O2 | 1.10 | 0.88 |
| β-Sitosterol | C29H50O | 1.00 | 0.17 |
The results of the exposure of the woodsmoke film to SO2 and zero air are shown in Fig. 4. There is a slight change in the structure following exposure to SO2, with virtually no change following exposure to zero air. However, the integrated area under the scattering length density profile does not substantially change indicating that there is no loss of material from the surface and results in more of a rearrangement following the reaction. This is supported by the total counts method shown in Fig. 2. Fig. 4 demonstrates that the most significant change to the scattering length density profile of the woodsmoke film after exposure to SO2 is in the central component – which is expected to be rich in polyaromatic hydrocarbons, PAHs. Thus, whilst SO2 may react with material extracted from woodsmoke it does not possess the capability to remove woodsmoke material from the air–water interface which has previously been shown to be removed by ozone and OH radicals albeit with thinner woodsmoke films.27
4 Conclusions
Real atmospheric aerosol extracted from urban and woodland environments formed thin, stable, organic films at the air–water interface and were found to be ∼0.6 nm thick. Woodsmoke extract also formed films and produced thicker films (>40 nm). There is initial evidence that these thicker films exhibit layered ordering, and these thick layered films are unusual. There was no evidence of a reaction of SO2 with films consisting of material extracted from urban an woodland environments that removed them from the air–water interface. Reactions of proxy insoluble surfactants at the air–water interface were also studied. The reaction of oleic acid with SO2 showed changes in the FTIR spectrum consistent with the formation of organosulfates, it is presumed that the urban and woodland samples lack a significant number of unsaturated carbon–carbon double bonds and have likely been aged in the atmosphere prior to extraction. Thus whilst films from wood burning aerosol did not appear to react and leave the interface this may be because SO2 reacted but did not remove films at the air–water interface. Analysis of the scattering length density profile of the wood burning film indicates the film to be multilayered and that some reaction may have occurred with the central component of the three-layered film.Monitoring of ozone mixing ratios throughout the neutron reflectometry measurements showed that ozone concentrations within the path of the neutron beam remained equivalent to background levels within the ISIS complex, indicating that there was no significant production of ozone from the spallation source and other irradiated components along the beam-path.
The work herein demonstrates that SO2 may not appreciably remove the organic material studied from the air–water interface at a rate substantially greater than in air. Note whilst the organic samples used here are of a complexity that represents atmospheric aerosol they are certainty not representative of all atmospheric aerosol. Atmospheric mixing ratios of SO2 are typically 2–20 ppb125 and at 450 ppb our experiment was in excess of typical concentrations in the atmosphere with the exception of a plume from a fossil fuel power station with poor desulfuring technology or a volcanic eruption.124 While some evidence of SO2-organic reactions was observed, the thickness of measured films remained relatively similar during exposure to SO2. Thus, for changes to the thickness of organic films at the air–water interface, the reaction of SO2 in the dark may not be atmospherically significant with respect to the removal of thin films consisting of insoluble matter.
Data availability
The data presented in this article are available as part of the ISIS open access data policy. See DOI: 10.5286/ISIS.E.RB2210230, 10.5286/ISIS.E.RB2310464.Author contributions
Contributions have been defined by the CRediT contributor role taxonomy. Edward J. Stuckey: data curation, formal analysis, investigation, methodology, software, validation, visualization, writing – original draft, writing – review & editing. Rebecca J. L. Welbourn: data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, writing – review & editing. Stephanie H. Jones: investigation, methodology, validation, writing – review & editing. Alexander J. Armstrong: methodology, software, writing – review & editing. Matthew Wilkinson: resources, writing – review & editing. James I. L. Morison: resources, writing – review & editing. Martin D. King*: conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the work by the support network at the ISIS neutron and muon source for providing the beam time and making this experiment possible by maintaining the facility, providing engineering works for our sample environment and providing constant support throughout the experiments RB2210230 and RB2310464. Particular acknowledgement is extended to Andy Church, Tom Charleston, Sarah Langham, James Taylor, Ludmilla Mee and Maura Martellini. Acknowledgements are made to NERC (NE/T00732X/1) for funding this work. Further acknowledgements are extended to the Earth Science support teams at Royal Holloway, university of London, whose engineering of the filter holders made the extraction of atmospheric particulate material possible. The authors would like to thank Forestry Research, UK, for allowing us to sample woodland particulate material from the Alice Holt plantation. SHJ is grateful to the German Research Foundation (DFG) for funding (JO 1849/1-1). Lastly, thanks is given to William Bloss of the University of Birmingham for the generous loan of the ozone monitoring equipment.References
- U. Pöschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- IPCC, Climate Change 2021: the Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2021, In Press Search PubMed.
- J. Haywood and O. Boucher, Rev. Geophys., 2000, 38, 513–543 CrossRef CAS.
- P. K. Quinn, D. B. Collins, V. H. Grassian, K. A. Prather and T. S. Bates, Chem. Rev., 2015, 115, 4383–4399 CrossRef CAS PubMed.
- Y. Feng, V. Ramanathan and V. R. Kotamarthi, Atmos. Chem. Phys., 2013, 13, 8607–8621 CrossRef.
- C. R. Barker, M. L. Poole, M. Wilkinson, J. Morison, A. Wilson, G. Little, E. J. Stuckey, R. J. L. Welbourn, A. D. Ward and M. D. King, Environ. Sci.: Atmos., 2023, 3, 1008–1024 CAS.
- T. Moise, J. M. Flores and Y. Rudich, Chem. Rev., 2015, 115, 4400–4439 CrossRef CAS PubMed.
- Y. Chen and T. C. Bond, Atmos. Chem. Phys., 2010, 10, 1773–1787 CrossRef CAS.
- A. T. Lambe, C. D. Cappa, P. Massoli, T. B. Onasch, S. D. Forestieri, A. T. Martin, M. J. Cummings, D. R. Croasdale, W. H. Brune, D. R. Worsnop and P. Davidovits, Environ. Sci. Technol., 2013, 47, 6349–6357 CrossRef CAS PubMed.
- A. Virkkula, I. K. Koponen, K. Teinilä, R. Hillamo, V.-M. Kerminen and M. Kulmala, Geophys. Res. Lett., 2006, 33, L06805 CrossRef.
- E. F. Mikhailov, G. N. Mironov, C. Pöhlker, X. Chi, M. L. Krüger, M. Shiraiwa, J.-D. Förster, U. Pöschl, S. S. Vlasenko, T. I. Ryshkevich, M. Weigand, A. L. D. Kilcoyne and M. O. Andreae, Atmos. Chem. Phys., 2015, 15, 8847–8869 CrossRef CAS.
- V. R. Després, J. A. Huffman, S. M. Burrows, C. Hoose, A. S. Safatov, G. Buryak, J. Fröhlich-Nowoisky, W. Elbert, M. O. Andreae, U. Pöschl and R. Jaenicke, Tellus B, 2012, 64, 15598 CrossRef.
- R. H. Shepherd, M. D. King, A. A. Marks, N. Brough and A. D. Ward, Atmos. Chem. Phys., 2018, 18, 5235–5252 CrossRef CAS.
- O. Dubovik, B. Holben, T. F. Eck, A. Smirnov, Y. J. Kaufman, M. D. King, D. Tanré and I. Slutsker, J. Atmos. Sci., 2002, 59, 590–608 CrossRef.
- R. Jaenicke and P. V. Hobbs, Tropospheric Aerosols, University of Washington, Seattle, 54th edn, 1993, ch. 1, pp. 1–31 Search PubMed.
- A. Massling, M. Stock and A. Wiedensohler, Atmos. Environ., 2005, 39, 3911–3922 CrossRef CAS.
- J. H. Slade, A. P. Ault, A. T. Bui, J. C. Ditto, Z. Lei, A. L. Bondy, N. E. Olson, R. D. Cook, S. J. Desrochers, R. M. Harvey, M. H. Erickson, H. W. Wallace, S. L. Alvarez, J. H. Flynn, B. E. Boor, G. A. Petrucci, D. R. Gentner, R. J. Griffin and P. B. Shepson, Environ. Sci. Technol., 2019, 53, 4977–4987 CrossRef CAS PubMed.
- R. M. Kirpes, D. Bonanno, N. W. May, M. Fraund, A. J. Barget, R. C. Moffet, A. P. Ault and K. A. Pratt, ACS Cent. Sci., 2019, 5, 1760–1767 CrossRef CAS PubMed.
- D. J. Delene and J. A. Ogren, J. Atmos. Sci., 2002, 59, 1135–1150 CrossRef.
- A. Virkkula, H. Grythe, J. Backman, T. Petäjä, M. Busetto, C. Lanconelli, A. Lupi, S. Becagli, R. Traversi, M. Severi, V. Vitale, P. Sheridan and E. Andrews, Atmos. Chem. Phys., 2022, 22, 5033–5069 CrossRef CAS.
- S. T. Martin, M. O. Andreae, P. Artaxo, D. Baumgardner, Q. Chen, A. H. Goldstein, A. Guenther, C. L. Heald, O. L. Mayol-Bracero, P. H. McMurry, T. Pauliquevis, U. Pöschl, K. A. Prather, G. C. Roberts, S. R. Saleska, M. A. Silva Dias, D. V. Spracklen, E. Swietlicki and I. Trebs, Rev. Geophys., 2010, 48, RG2002 CrossRef.
- H.-W. Xiao, H.-Y. Xiao, C.-Y. Shen, Z.-Y. Zhang and A.-M. Long, Atmosphere, 2018, 9, 298 CrossRef.
- D. C. S. Beddows, R. J. Donovan, R. M. Harrison, M. R. Heal, R. P. Kinnersley, M. D. King, D. H. Nicholson and K. C. Thompson, J. Environ. Monit., 2004, 6, 124 RSC.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- B. Ervens, B. J. Turpin and R. J. Weber, Atmos. Chem. Phys., 2011, 11, 11069–11102 CrossRef CAS.
- S. H. Jones, M. D. King, A. D. Ward, A. R. Rennie, A. C. Jones and T. Arnold, Atmos. Environ., 2017, 161, 274–287 CrossRef CAS.
- R. H. Shepherd, M. D. King, A. R. Rennie, A. D. Ward, M. M. Frey, N. Brough, J. Eveson, S. Del Vento, A. Milsom, C. Pfrang, M. W. A. Skoda and R. J. L. Welbourn, Environ. Sci.: Atmos., 2022, 2, 574–590 CAS.
- M. D. King, A. R. Rennie, C. Pfrang, A. V. Hughes and K. C. Thompson, Atmos. Environ., 2010, 44, 1822–1825 CrossRef CAS.
- S. H. Jones, M. D. King, A. R. Rennie, A. D. Ward, R. A. Campbell and A. V. Hughes, J. Phys. Chem. A, 2023, 127, 8922–8934 CrossRef CAS PubMed.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V. Hughes, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- M. D. King, S. H. Jones, C. O. M. Lucas, K. C. Thompson, A. R. Rennie, A. D. Ward, A. A. Marks, F. N. Fisher, C. Pfrang, A. V. Hughes and R. A. Campbell, Phys. Chem. Chem. Phys., 2020, 22, 28032–28044 RSC.
- E. Aumann and A. Tabazadeh, J. Geophys. Res., 2008, 113, D23205 CrossRef.
- Y. Rudich, N. M. Donahue and T. F. Mentel, Annu. Rev. Phys. Chem., 2007, 58, 321–352 CrossRef CAS PubMed.
- J. B. Burkholder, J. P. D. Abbatt, I. Barnes, J. M. Roberts, M. L. Melamed, M. Ammann, A. K. Bertram, C. D. Cappa, A. G. Carlton, L. J. Carpenter, J. N. Crowley, Y. Dubowski, C. George, D. E. Heard, H. Herrmann, F. N. Keutsch, J. H. Kroll, V. F. McNeill, N. L. Ng, S. A. Nizkorodov, J. J. Orlando, C. J. Percival, B. Picquet-Varrault, Y. Rudich, P. W. Seakins, J. D. Surratt, H. Tanimoto, J. A. Thornton, Z. Tong, G. S. Tyndall, A. Wahner, C. J. Weschler, K. R. Wilson and P. J. Ziemann, Environ. Sci. Technol., 2017, 51, 2519–2528 CrossRef CAS PubMed.
- C. N. Cruz and S. N. Pandis, J. Geophys. Res.: Atmos., 1998, 103, 13111–13123 CrossRef CAS.
- O. S. Heavens, Rep. Prog. Phys., 1960, 23, 301 CrossRef.
- B. J. Kirby, P. A. Kienzle, B. B. Maranville, N. F. Berk, J. Krycka, F. Heinrich and C. F. Majkrzak, Curr. Opin. Colloid Interface Sci., 2012, 17, 44–53 CrossRef CAS.
- J. Q. Xiong, M. Zhong, C. Fang, L. C. Chen and M. Lippmann, Environ. Sci. Technol., 1998, 32, 3536–3541 CrossRef CAS.
- D. J. Donaldson and V. Vaida, Chem. Rev., 2006, 106, 1445–1461 CrossRef CAS PubMed.
- P. S. Gill, T. E. Graedel and C. J. Weschler, Rev. Geophys., 1983, 21, 903 CrossRef CAS.
- B. Woden, M. W. A. Skoda, A. Milsom, C. Gubb, A. Maestro, J. Tellam and C. Pfrang, Atmos. Chem. Phys., 2021, 21, 1325–1340 CrossRef CAS.
- A. Milsom, A. M. Squires, I. Quant, N. J. Terrill, S. Huband, B. Woden, E. R. Cabrera-Martinez and C. Pfrang, J. Phys. Chem. A, 2022, 126, 7331–7341 CrossRef CAS PubMed.
- M. Z. Jacobson, Nature, 2001, 409, 695–697 CrossRef CAS PubMed.
- T. C. Bond, G. Habib and R. W. Bergstrom, J. Geophys. Res., 2006, 111, D20211 CrossRef.
- D. A. Lack and C. D. Cappa, Atmos. Chem. Phys., 2010, 10, 4207–4220 CrossRef CAS.
- N. Sareen, A. N. Schwier, T. L. Lathem, A. Nenes and V. F. McNeill, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2723–2728 CrossRef CAS PubMed.
- H. Tervahattu, J. Geophys. Res., 2002, 107, 4053 Search PubMed.
- L. Tinel, S. Rossignol, A. Bianco, M. Passananti, S. Perrier, X. Wang, M. Brigante, D. J. Donaldson and C. George, Environ. Sci. Technol., 2016, 50, 11041–11048 CrossRef CAS PubMed.
- H. Tervahattu, J. Geophys. Res., 2002, 107, 4319 Search PubMed.
- H. Tervahattu, J. Juhanoja, V. Vaida, A. F. Tuck, J. V. Niemi, K. Kupiainen, M. Kulmala and H. Vehkamäki, J. Geophys. Res.: Atmos., 2005, 110, n/a CrossRef.
- N. Hayeck, I. Mussa, S. Perrier and C. George, ACS Earth Space Chem., 2020, 4, 1247–1253 CrossRef CAS.
- B. A. Wellen, E. A. Lach and H. C. Allen, Phys. Chem. Chem. Phys., 2017, 19, 26551–26558 RSC.
- L. F. Voss, M. F. Bazerbashi, C. P. Beekman, C. M. Hadad and H. C. Allen, J. Geophys. Res.: Atmos., 2007 DOI:10.1029/2006jd007677.
- E. P. Rosen, E. R. Garland and T. Baer, J. Phys. Chem. A, 2008, 112, 10315–10324 CrossRef CAS PubMed.
- C. Pfrang, M. Shiraiwa and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 4537–4557 CrossRef CAS.
- J. He, H. Zhang, W. Wang, Y. Ma, M. Yang, Y. He, Z. Liu, K. Yu and J. Jiang, Environ. Res., 2022, 212, 113232 CrossRef CAS PubMed.
- M. Shiraiwa, U. Pöschl and D. A. Knopf, Environ. Sci. Technol., 2012, 46, 6630–6636 CrossRef CAS PubMed.
- T. L. Eliason, J. B. Gilman and V. Vaida, Atmos. Environ., 2004, 38, 1367–1378 CrossRef CAS.
- C. Y. Lim, E. C. Browne, R. A. Sugrue and J. H. Kroll, Geophys. Res. Lett., 2017, 44, 2949–2957 CrossRef CAS.
- J. M. Anglada, M. T. C. Martins-Costa, J. S. Francisco and M. F. Ruiz-López, J. Am. Chem. Soc., 2020, 142, 16140–16155 CrossRef CAS PubMed.
- F. Sebastiani, R. A. Campbell and C. Pfrang, Environ. Sci.: Atmos., 2022, 2, 1324–1337 CAS.
- F. Sebastiani, R. A. Campbell, K. Rastogi and C. Pfrang, Atmos. Chem. Phys., 2018, 18, 3249–3268 CrossRef CAS.
- M. D. King, K. C. Thompson and A. D. Ward, J. Am. Chem. Soc., 2004, 126, 16710–16711 CrossRef CAS PubMed.
- D. J. Donaldson and K. T. Valsaraj, Environ. Sci. Technol., 2010, 44, 865–873 CrossRef CAS PubMed.
- H. Tervahattu, J. Geophys. Res., 2002, 107, 4319 Search PubMed.
- M. Passananti, L. Kong, J. Shang, Y. Dupart, S. Perrier, J. Chen, D. J. Donaldson and C. George, Angew. Chem., Int. Ed., 2016, 55, 10336–10339 CrossRef CAS PubMed.
- J. Shang, M. Passananti, Y. Dupart, R. Ciuraru, L. Tinel, S. Rossignol, S. Perrier, T. Zhu and C. George, Environ. Sci. Technol. Lett., 2016, 3, 67–72 CrossRef CAS.
- J. Ye, J. P. D. Abbatt and A. W. H. Chan, Atmos. Chem. Phys., 2018, 18, 5549–5565 CrossRef CAS.
- M. Brüggemann, N. Hayeck, C. Bonnineau, S. Pesce, P. A. Alpert, S. Perrier, C. Zuth, T. Hoffmann, J. Chen and C. George, Faraday Discuss., 2017, 200, 59–74 RSC.
- A. D. Estillore, J. V. Trueblood and V. H. Grassian, Chem. Sci., 2016, 7, 6604–6616 RSC.
- M. Zhu, B. Jiang, S. Li, Q. Yu, X. Yu, Y. Zhang, X. Bi, J. Yu, C. George, Z. Yu and X. Wang, Environ. Sci. Technol. Lett., 2019, 6, 318–322 CrossRef CAS.
- N. C. Auvil, M. G. Vazquez de Vasquez and H. C. Allen, ACS Earth Space Chem., 2021, 5, 2947–2956 CrossRef CAS.
- M. Xu, N. T. Tsona, S. Cheng, J. Li and L. Du, Sci. Total Environ., 2021, 782, 146893 CrossRef CAS PubMed.
- S. Li, L. Du, N. T. Tsona and W. Wang, Chemosphere, 2018, 196, 323–330 CrossRef CAS PubMed.
- Y. Dubowski, J. Vieceli, D. J. Tobias, A. Gomez, A. Lin, S. A. Nizkorodov, T. M. McIntire and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2004, 108, 10473–10485 CrossRef CAS.
- F. Karagulian, A. Scott Lea, C. W. Dilbeck and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2008, 10, 528–541 RSC.
- E. Adams and H. Allen, Atmosphere, 2013, 4, 315–336 CrossRef.
- A. Milsom, A. M. Squires, M. W. A. Skoda, P. Gutfreund, E. Mason, N. J. Terrill and C. Pfrang, Environ. Sci.: Atmos., 2022, 2, 964–977 CAS.
- M. W. A. Skoda, B. Thomas, M. Hagreen, F. Sebastiani and C. Pfrang, RSC Adv., 2017, 7, 34208–34214 RSC.
- E. Staples, L. Thompson, I. Tucker, J. Penfold, R. K. Thomas and J. R. Lu, Langmuir, 1993, 9, 1651–1656 CrossRef CAS.
- R. Dalgliesh, Curr. Opin. Colloid Interface Sci., 2002, 7, 244–248 CrossRef CAS.
- F. Cousin and A. Chennevière, EPJ Web Conf., 2018, 188, 04001 CrossRef.
- K. C. Thompson, A. R. Rennie, M. D. King, S. J. O. Hardman, C. O. M. Lucas, C. Pfrang, B. R. Hughes and A. V. Hughes, Langmuir, 2010, 26, 17295–17303 CrossRef CAS PubMed.
- F. Sebastiani, R. A. Campbell and C. Pfrang, RSC Adv., 2015, 5, 107105–107111 RSC.
- P. J. Ziemann and R. Atkinson, Chem. Soc. Rev., 2012, 41, 6582 RSC.
- J. Zahardis and G. A. Petrucci, Atmos. Chem. Phys., 2007, 7, 1237–1274 CrossRef CAS.
- H.-M. Hung and C.-W. Tang, J. Phys. Chem. A, 2010, 114, 13104–13112 CrossRef CAS PubMed.
- H.-M. Hung, Y. Katrib and S. T. Martin, J. Phys. Chem. A, 2005, 109, 4517–4530 CrossRef CAS PubMed.
- Y. Katrib, G. Biskos, P. R. Buseck, P. Davidovits, J. T. Jayne, M. Mochida, M. E. Wise, D. R. Worsnop and S. T. Martin, J. Phys. Chem. A, 2005, 109, 10910–10919 CrossRef CAS PubMed.
- J. W. L. Lee, V. Carrascón, P. J. Gallimore, S. J. Fuller, A. Björkegren, D. R. Spring, F. D. Pope and M. Kalberer, Phys. Chem. Chem. Phys., 2012, 14, 8023 RSC.
- J. Vieceli, O. L. Ma and D. J. Tobias, J. Phys. Chem. A, 2004, 108, 5806–5814 CrossRef CAS.
- P. J. Gallimore, P. T. Griffiths, F. D. Pope, J. P. Reid and M. Kalberer, J. Geophys. Res.: Atmos., 2017, 122, 4364–4377 CrossRef CAS.
- T. Nah, S. H. Kessler, K. E. Daumit, J. H. Kroll, S. R. Leone and K. R. Wilson, J. Phys. Chem. A, 2014, 118, 4106–4119 CrossRef CAS PubMed.
- T. Nah, S. H. Kessler, K. E. Daumit, J. H. Kroll, S. R. Leone and K. R. Wilson, Phys. Chem. Chem. Phys., 2013, 15, 18649 RSC.
- R. C. Chapleski, Y. Zhang, D. Troya and J. R. Morris, Chem. Soc. Rev., 2016, 45, 3731–3746 RSC.
- X. Zhang, K. M. Barraza, K. T. Upton and J. L. Beauchamp, Chem. Phys. Lett., 2017, 683, 76–82 CrossRef CAS.
- I. J. George, A. Vlasenko, J. G. Slowik, K. Broekhuizen and J. P. D. Abbatt, Atmos. Chem. Phys., 2007, 7, 4187–4201 CrossRef CAS.
- J. D. Smith, J. H. Kroll, C. D. Cappa, D. L. Che, C. L. Liu, M. Ahmed, S. R. Leone, D. R. Worsnop and K. R. Wilson, Atmos. Chem. Phys., 2009, 9, 3209–3222 CrossRef CAS.
- A. T. Lambe, A. T. Ahern, L. R. Williams, J. G. Slowik, J. P. S. Wong, J. P. D. Abbatt, W. H. Brune, N. L. Ng, J. P. Wright, D. R. Croasdale, D. R. Worsnop, P. Davidovits and T. B. Onasch, Atmos. Meas. Tech., 2011, 4, 445–461 CrossRef CAS.
- L. Qi, S. Nakao and D. R. Cocker, J. Air Waste Manage. Assoc., 2012, 62, 1359–1369 CrossRef CAS PubMed.
- R. A. Zaveri, C. M. Berkowitz, F. J. Brechtel, M. K. Gilles, J. M. Hubbe, J. T. Jayne, L. I. Kleinman, A. Laskin, S. Madronich, T. B. Onasch, M. S. Pekour, S. R. Springston, J. A. Thornton, A. V. Tivanski and D. R. Worsnop, J. Geophys. Res., 2010, 115, D12304 Search PubMed.
- K. S. Docherty and P. J. Ziemann, J. Phys. Chem. A, 2006, 110, 3567–3577 CrossRef CAS PubMed.
- L. F. Gamon and U. Wille, Acc. Chem. Res., 2016, 49, 2136–2145 CrossRef CAS PubMed.
- D. K. Farmer, A. Matsunaga, K. S. Docherty, J. D. Surratt, J. H. Seinfeld, P. J. Ziemann and J. L. Jimenez, Proceedings of the National Academy of Sciences, 2010, vol. 107, pp. 6670–6675 Search PubMed.
- J. Daillant and A. Gibaud, X-Ray and Neuron Reflectivity: Principles and Applications, Springer Berlin Heidelberg, Berlin, Heidelberg, 1999, vol. 58 Search PubMed.
- J. Penfold and R. K. Thomas, J. Phys.: Condens. Matter, 1990, 2, 1369–1412 CrossRef CAS.
- T. R. Charlton, R. L. S. Coleman, R. M. Dalgliesh, C. J. Kinane, C. Neylon, S. Langridge, J. Plomp, N. G. J. Webb and J. R. P. Webster, Neutron News., 2011, 22, 15–18 CrossRef.
- J. Webster, S. Holt and R. Dalgliesh, Phys. B, 2006, 385–386, 1164–1166 CrossRef CAS.
- J. R. P. Webster, S. Langridge, R. M. Dalgliesh and T. R. Charlton, Eur. Phys. J. Plus, 2011, 126, 112 CrossRef.
- A. E. Whitten and J. Trewhella, Methods Mol. Biol, 2009, 554, 307–323 CrossRef PubMed.
- A. R. J. Nelson and S. W. Prescott, J. Appl. Crystallogr., 2019, 52, 193–200 CrossRef CAS PubMed.
- S. W. Chiu, E. Jakobsson, S. Subramaniam and H. L. Scott, Biophys. J., 1999, 77, 2462–2469 CrossRef CAS PubMed.
- C. Kong, H. Zhang, Z. Zhao and Q. Zheng, Chem. Res. Chin. Univ., 2013, 29, 545–550 CrossRef CAS.
- D. W. Hogg and D. Foreman-Mackey, Astrophys. J., Suppl. Ser., 2018, 236, 11 CrossRef.
- D. van Ravenzwaaij, P. Cassey and S. D. Brown, Psychonomic Bulletin & Review, 2018, 25, 143–154 Search PubMed.
- J. Skilling, Bayesian Anal., 2006, 1, 833–859 Search PubMed.
- J. T. Davies and E. K. Rideal, Interfacial Phenomena, AcademicPress, 1961 Search PubMed.
- P. A. Paez, M. G. Cogliati, A. T. Caselli and A. M. Monasterio, J. South Am. Earth Sci., 2021, 110, 103365 CrossRef CAS.
- R.-H. Shie, T.-H. Yuan and C.-C. Chan, J. Air Waste Manage. Assoc., 2013, 63, 702–711 CrossRef CAS PubMed.
- National Institute of Standards and Technology, NIST Chemistry WebBook - SRD 69, https://webbook.nist.gov/chemistry/, accessed 23 May 2023 Search PubMed.
- J. Shang, M. Passananti, Y. Dupart, R. Ciuraru, L. Tinel, S. Rossignol, S. Perrier, T. Zhu and C. George, Environ. Sci. Technol. Lett., 2016, 3, 67–72 CrossRef CAS.
- J. C. Acosta Navarro, S. Smolander, H. Struthers, E. Zorita, A. M. L. Ekman, J. O. Kaplan, A. Guenther, A. Arneth and I. Riipinen, J. Geophys. Res.: Atmos., 2014, 119, 6867–6885 CrossRef CAS PubMed.
- C. Pfrang, K. Rastogi, E. R. Cabrera-Martinez, A. M. Seddon, C. Dicko, A. Labrador, T. S. Plivelic, N. Cowieson and A. M. Squires, Nat. Commun., 2017, 8, 1724 CrossRef CAS PubMed.
- A. Milsom, A. M. Squires, A. D. Ward and C. Pfrang, Atmos. Chem. Phys., 2022, 22, 4895–4907 CrossRef CAS.
- J. Stedman, J. Abbott, P. Willis and J. Bower, Review of Background Air-Quality Data and Methods to Combine These with Process Contributions, Bristol, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data accessible through ISIS open access data. See DOI: https://doi.org/10.1039/d4ea00098f |
| This journal is © The Royal Society of Chemistry 2024 |