
Water electrolysis for hydrogen production: from hybrid systems to self-powered/catalyzed devices
Jin-Tao
Ren
a,
Lei
Chen
a,
Hao-Yu
Wang
a,
Wen-Wen
Tian
a and
Zhong-Yong
Yuan
 *ab
*ab
aNational Institute for Advanced Materials, School of Materials Science and Engineering, Smart Sensing Interdisciplinary Science Center, Nankai University, Tianjin 300350, China
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China. E-mail: zyyuan@nankai.edu.cn
First published on 7th November 2023
Abstract
The electrocatalytic splitting of water holds great promise as a sustainable and environmentally friendly technology for hydrogen production. However, the sluggish kinetics of the oxygen evolution reaction (OER) at the anode significantly hampers the efficiency of this process. In this comprehensive perspective, we outline recent advancements in innovative strategies aimed at improving the energy and economic efficiency of conventional water electrolysis, thereby facilitating efficient hydrogen generation. These novel strategies mainly include: (i) sacrificial-agent-assisted water electrolysis, which integrates thermodynamically favorable small molecules to replace the OER while simultaneously degrading pollutants; (ii) organic upgrading-assisted water electrolysis, wherein thermodynamically and kinetically favorable organic oxidation reactions replace the OER, leading to the production of high-value chemicals alongside hydrogen; (iii) self-powered electrolysis systems, achieved by coupling water splitting with metal-based batteries or fuel cells, enabling hydrogen production without the need for additional electricity input; and (iv) self-catalyzed electrolysis systems driven by the spontaneous metal oxidation at the anode, which provides electrons for hydrogen evolution at the cathode. In particular, we emphasize the design of electrocatalysts using non-noble metal elements, elucidate the underlying reaction mechanisms, and explore the construction of efficient electrolyzers. Additionally, we discuss the prevailing challenges and future prospects, aiming to foster the development of electrocatalytic systems for highly efficient hydrogen production from water in the future.
 Jin-Tao Ren | Jin-Tao Ren received his PhD degree from Nankai University in 2020 under the supervision of Prof. Zhong-Yong Yuan. He is currently a postdoctoral fellow at Nankai University. His research interests focus on advanced nanomaterials for application in electrocatalysis, metal–air batteries, fuel cells, etc. |
 Lei Chen | Lei Chen received her BE degree from Northeast Forestry University in 2017 and obtained her ME degree from Nankai University in 2020. She is currently a PhD candidate under the supervision of Prof. Zhong-Yong Yuan at Nankai University. Her current research focuses on the fabrication of nanostructured materials for energy-related applications. |
 Hao-Yu Wang | Hao-Yu Wang received his BE degree in 2019 from Nankai University. He is currently a PhD candidate in Nankai University under the supervision of Prof. Zhong-Yong Yuan. His research interest focuses on the rational design and related applications of advanced electrocatalysts. |
 Wen-Wen Tian | Wen-Wen Tian is currently pursuing her PhD under the supervision of Prof. Zhong-Yong Yuan at Nankai University. She received her BE degree from Northeast Forestry University in 2018. Her current research focuses on the design, fabrication, and application of advanced electrocatalysts for energy conversion reactions. |
 Zhong-Yong Yuan | Zhong-Yong Yuan received his PhD degree in Physical Chemistry from Nankai University in 1999. He worked as a postdoctoral fellow at the Institute of Physics, Chinese Academy of Sciences from 1999 to 2001. He then moved to Belgium, working as a research fellow at the University of Namur from 2001 to 2005, prior to joining Nankai University as a full professor. In 2016, he was elected as a fellow of the Royal Society of Chemistry (FRSC). His research interests are mainly focused on the self-assembly of hierarchically nanoporous and nanostructured materials for energy and environmental applications. |
Broader contextExploring cutting-edge technologies for the efficient production of green hydrogen energy is imperative to alleviate the energy crisis and environmental pollution. Conventional overall water electrolysis is largely restricted by the sluggish kinetics of the anodic oxygen evolution reaction. In this original perspective, we present a comprehensive summary and discussion of the recent notable advancements in energy-efficient hydrogen production through electrolysis, employing four distinct strategies categorized based on the electrode reactions or products of the entire system. These strategies encompass (i) sacrificial-agent-assisted water electrolysis, (ii) organic upgrading-assisted water electrolysis, (iii) self-powered electrolysis systems, and (iv) self-catalyzed electrolysis systems. We will mainly focus on the development of various systems as well as the corresponding electrocatalysts and reaction mechanisms. Additionally, we thoroughly discuss and compare the advantages, differences, and critical issues of each system. The current challenges and related prospects will also be discussed, hopefully to benefit further development of energy-saving hydrogen production from renewable resources and waste products. |
1. Introduction
Because of the escalating concerns surrounding environmental pollution and the energy crisis, there is a pressing need to supplant conventional fossil fuels with sustainable energy sources.1,2 Among the potential alternatives, molecular hydrogen (H2) has garnered significant attention as a promising energy carrier due to its remarkable energy density and environmentally benign characteristics.3 Electrochemical water splitting, specifically through the utilization of a water electrolyzer, has emerged as a prominent avenue for hydrogen production owing to its affordability and zero-carbon emissions when coupled with intermittent renewable energy sources.4,5 In a conventional overall water splitting (OWS) system, the cathode drives the hydrogen evolution reaction (HER), while the anode drives the oxygen evolution reaction (OER). Thermodynamically, the HER (2H+/H2) and OER (2H2O/O2) possess equilibrium potentials of 0 and 1.23 V (vs. the reversible hydrogen electrode (RHE)), respectively.6 Thus, a theoretical minimum voltage of 1.23 V is necessary to initiate electrochemical water splitting. As such, high-performance electrocatalysts, such as Pt for the HER and IrO2/RuO2 for the OER, are highly essential to accelerate the apparent sluggish reaction kinetics, consequently, resulting in decreased overpotentials and enabling efficient electrochemical processes.Due to its potential to simplify electrolyzer configurations and reduce overall costs, overall water electrolysis has entered a prosperous and critical period in the twenty-first century. Nevertheless, despite these advantages, several inherent challenges continue to impede progress in electrocatalytic water splitting for hydrogen production.7 (i) The OER is a multi-electron coupled proton transfer process that demands significantly higher overpotentials compared to the HER to achieve equivalent current densities. (ii) Currently, with the simplest configuration of water electrolyzers, both the HER and OER take place simultaneously within a single-compartment setup. This design raises concerns regarding the formation of explosive H2/O2 gas mixtures. (iii) The coexistence of gas mixtures and electrocatalysts may generate reactive oxygen species, leading to membrane degradation and premature device failure. (iv) Gas crossover not only presents safety hazards but also reduces energy efficiency, as O2 may undergo reduction back to water at the cathode side. (v) Efficient water electrolysis necessitates a substantial power supply to drive both the HER and OER processes, resulting in a considerable energy consumption burden. While harnessing or converting energy from renewable sources like wind, thermal, solar, tidal, and self-powered energy holds promise, these approaches are still in the early stages of technological development.
Previously, a considerable emphasis has been placed on the development of advanced strategies to design high-performance electrocatalysts, including nanostructure construction, defect introduction, atomic dispersion, and heterointerface engineering.8–10 These approaches have successfully reduced the input voltage required for OWS, consequently lowering the energy cost. However, the economic viability of water splitting for commercial applications remains a considerable challenge. The primary objective of water oxidation is to extract electrons and transport them to the cathode for the production of hydrogen, which serves as the desired product of water splitting. Therefore, one potential strategy to overcome the aforementioned obstacles in electrolytic hydrogen production is to replace the OER with organic oxidation reactions that are thermodynamically more favorable.11 For instance, it has been demonstrated that electrooxidation of renewable alcohols can achieve a remarkable 67% energy saving compared to traditional water electrolysis while generating equivalent amounts of hydrogen.12
These small molecule oxidation reactions typically encompass sacrificial-agent oxidation reactions (such as urea oxidation, hydrazine oxidation, polysulfide oxidation, etc.) and electrochemical synthesis reactions (such as alcohol oxidation, aldehyde oxidation, carbohydrate oxidation, and primary amine oxidation, etc.),13–15 which possess lower theoretical equilibrium potentials (<1.23 V vs. RHE), as depicted in Fig. 1a. The former reactions are often employed for the treatment of environmental pollutants due to the toxic nature of substances like urea and hydrazine, which find widespread use as industrial raw materials and can cause significant harm to the environment. Specifically, the electrocatalytic oxidation of these small molecules offers the potential to remove toxic materials from industrial wastewater without the need for additional oxidants or complex separation methods, while also significantly reduces the overall voltage required during hydrogen production via water electrolysis. The latter reactions provide a sustainable pathway that utilizes electricity from renewable energy sources for chemical upgrading, obviating the use of organic solvents, homogeneous catalysts, and hazardous/poisonous strong oxidants (e.g., hydrogen peroxide, chloroperbenzoic acid, peroxy acids, oxone, or iodine), as well as the elevated temperatures and pressures typically associated with traditional organic oxidation reactions.16–19 Consequently, such hybrid water electrolysis offers distinct advantages when compared to overall water electrolysis (Fig. 1b). (i) By harnessing the flexible thermodynamic oxidation reactions of organic reactants, this method significantly reduces the electric energy consumption required for hydrogen production, leading to the enhanced efficiency of energy conversion. (ii) Through sacrificial-agent-assisted water electrolysis, it becomes feasible to efficiently decompose undesirable components present in wastewater, such as urea from feed/fertilizer wastewater, hydrazine from pharmaceutical/petrochemical wastewater, and polysulfide from industrial wastewater, which will contribute to the mitigation of environmental pollution. (iii) Organic upgrading-assisted water electrolysis allows for the concurrent production of value-added compounds at the anode. These compounds can find applications in various domains, including chemical synthesis, polymer production, and pharmaceutical manufacturing. (iv) The absence of oxygen production at the anode eliminates the risk associated with H2/O2 explosion and the generation of harmful reactive oxygen species. (v) Given that the anodic products in organic upgrading-assisted water electrolysis are typically non-gaseous, this method can be executed without the need for a membrane, simplifying the overall electrolysis setup. The past decade has witnessed significant advancements in the coupling of organic oxidation reactions with the HER in hybrid water electrolyzers, particularly in recent years. Nevertheless, the catalytic activities and cell configurations are still far from reaching the level required for practical applications.
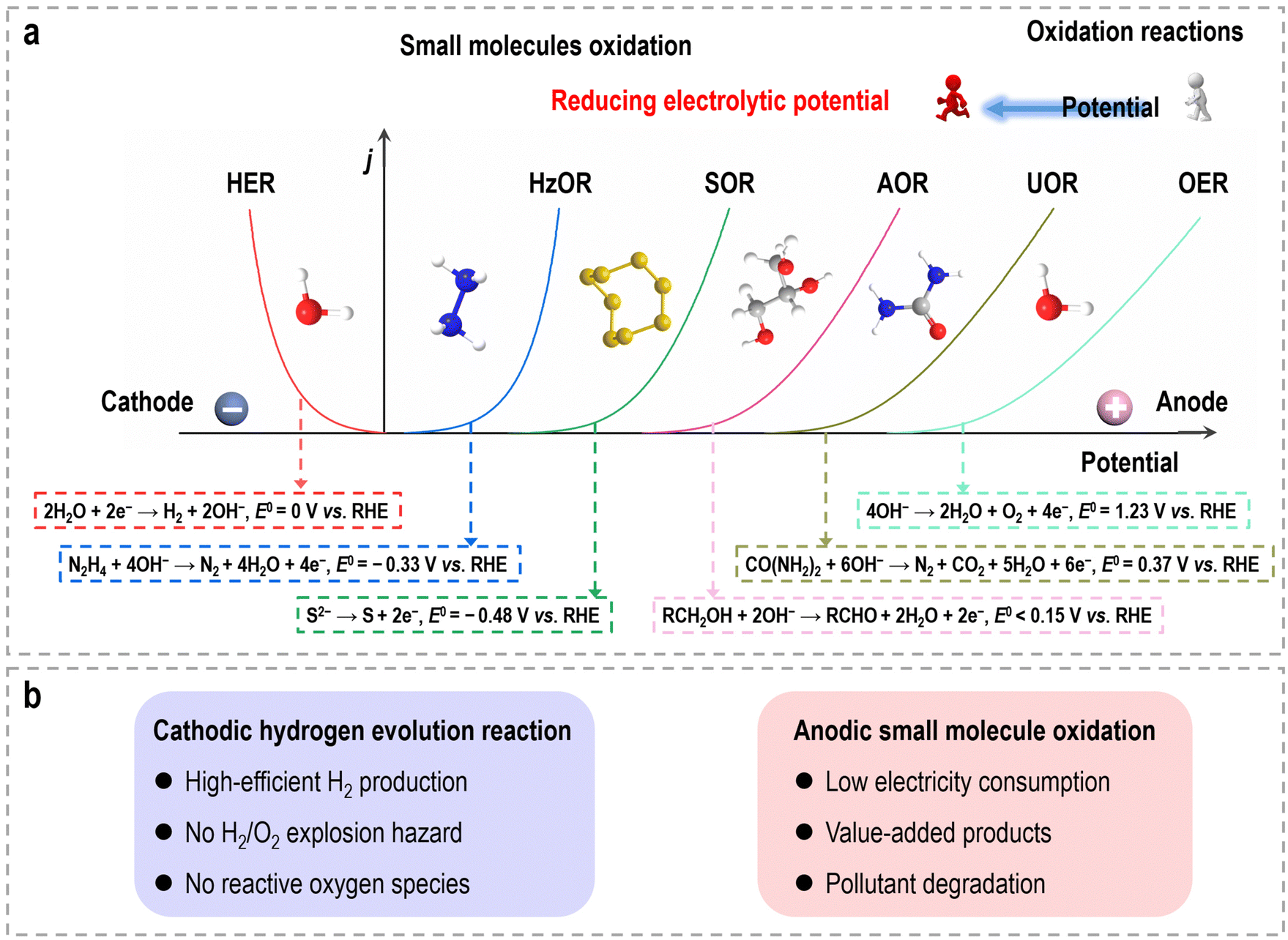 | ||
| Fig. 1 (a) Schematic diagram of polarization curves for the HER and other available/potential oxidation reactions with low-onset potential, such as the HzOR (hydrazine oxidation reaction), the SOR (polysulfide oxidation reaction), the AOR (alcohol oxidation reaction), and the UOR (urea oxidation reaction). (b) The merits of small molecule oxidation-assisted H2 production. | ||
Electrocatalytic systems conventionally rely on electricity derived from fossil fuels, incurring substantial costs and wastage of electrons during operation. Additionally, the treatment of discharged water, gases, and other low or no-value chemicals results in additional and often unforeseen expenses. In reality, the overall profitability and cost of an electrocatalytic system, whether apparent or hidden, can be primarily attributed to the efficient utilization of electrons and atoms in value-added conversions.20,21 Enhancing the atom and electron utilization, which translates to increased product profitability and decreased energy costs, lies at the core of advancing electrocatalytic systems. Consequently, the invention and development of innovative electrocatalytic systems are essential for achieving this goal. To enhance the electron economy of electrolysis, self-powered electrocatalytic systems have been devised, utilizing low-value chemical energy sourced from metal-based batteries, fuel cells, and similar technologies.22,23 These systems inherently generate electricity inside the electrocatalytic systems to drive the electrocatalytic reactions at adjacent or opposite electrodes. In contrast to traditional water electrolyzers or organic oxidation reaction-based electrolyzers, self-powered electrocatalytic systems can operate without an external energy supply. Notably, in these self-powered electrocatalytic systems, if sustainable energy derived from sources like wind and solar power is exclusively used to charge rechargeable metal-based batteries (such as zinc–air batteries), the overall economic effectiveness of these systems can be significantly enhanced.24 Similarly, due to the low equilibrium potential of metal oxidation, the electrochemical hydrogen evolution can be harnessed as the cathodic reaction in metal-based batteries, as observed in zinc–H2O batteries.25,26 In these batteries, hydrogen is generated at the cathode during the discharge process, simultaneously providing the necessary electricity. These systems are referred to as self-catalyzed electrolysis systems, as they can occur spontaneously upon the connection of the anode and the cathode. The aforementioned systems exhibit vastly improved economy throughout the reaction system, benefitting from their unique system configurations. Regrettably, comprehensive and in-depth discussions concerning approaches to maximize energy utilization in electrocatalytic systems remain notably absent in current literature.
This perspective aims to provide a concise overview of the latest advancements and notable achievements in the innovative strategies employed to enhance the energy efficiency of hydrogen generation through electrochemical approaches. From an economic standpoint, we will commence with a brief introduction elucidating the fundamentals of selecting appropriate anodic reactions and designing efficient electrocatalysts. Subsequently, we will provide a systematic summary of the current progress in different small molecule oxidation reactions at the anode side, categorizing them into sacrificial-agent reactions and organic upgrading reactions. Furthermore, we will explore self-powered hydrogen production systems and self-catalyzed hydrogen production systems, which exhibit enhanced energy utilization efficiency. Fig. 2 schematically illustrates these novel electrolyzers, facilitating clear visualization and comparison. Table 1 summarizes the relationship and distinct advantages of these system configurations. Each section will delve into key aspects of modulated catalyst strategies, catalytic activity, structure–performance relationships, related catalytic reaction mechanisms, and electrolyzer configurations. Additionally, we will discuss the remaining challenges and offer perspectives for the future development of these electrocatalytic systems. Our ultimate objective in this perspective is to provide an up-to-date account of the advancements in efficient electrocatalytic systems and underscore the potential of these captivating technologies for energy-saving H2 production in the future.
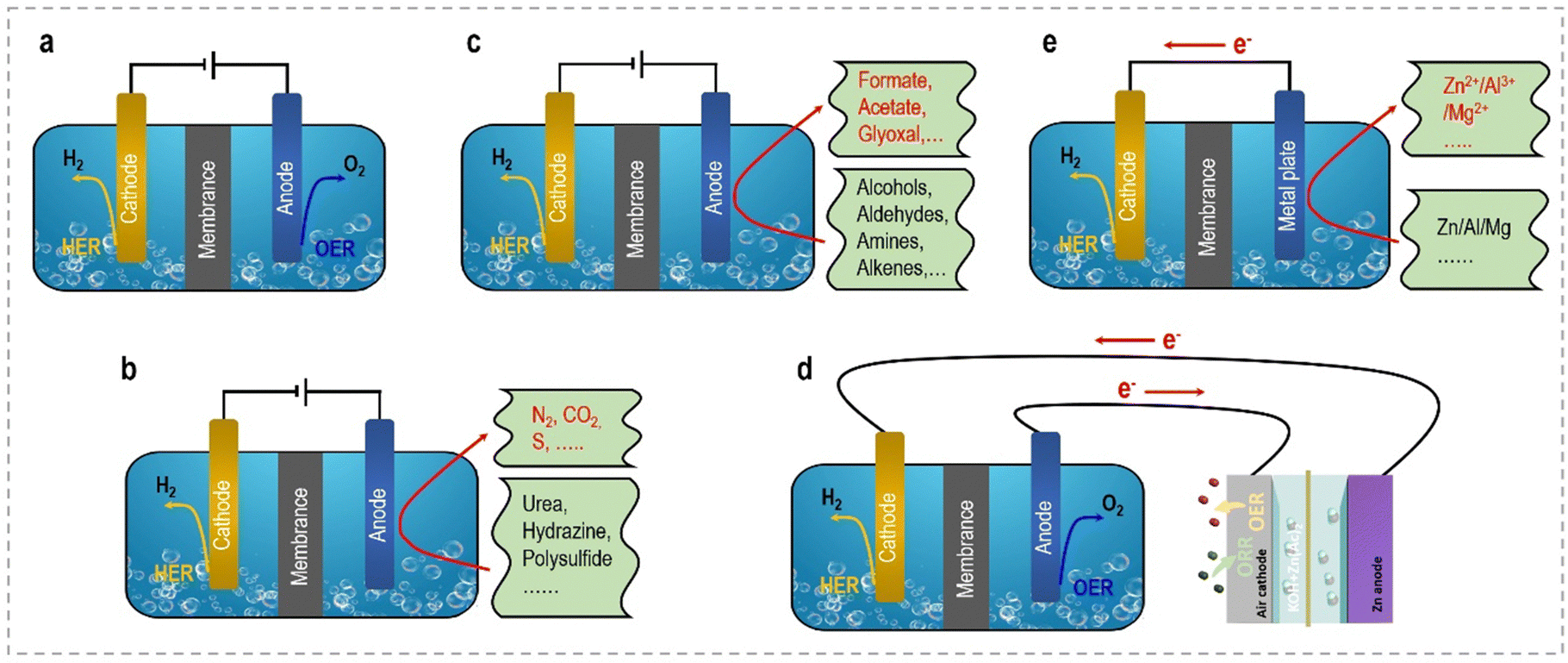 | ||
| Fig. 2 Schematic representation showing the electrolyzer/devices for energy-saving H2 production, (a) the conventional OWS system, (b) sacrificial-agent-assisted water electrolysis, (c) organic upgrading-assisted water electrolysis, (d) self-powered electrocatalytic system, and (e) self-catalyzed electrolysis system. | ||
| Types | Products | Electricity input | Electricity output | Economy efficiency | Range of current density (A cm−2) | Hydrogen production rateb (mmol cm−2 h−1) | Electron utilization | Recyclability |
|---|---|---|---|---|---|---|---|---|
| a The applied cell voltage at 2.0 V on the basis of reported literature (measured in a H-type cell). b The hydrogen production rate calculated at the largest current density. | ||||||||
| Alkaline water electrolyzer | H2 and O2 | Yes | No | Average | 0–0.4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27,28 27,28 |
7.46 | 50% | No |
| Sacrificial-agent-assisted water electrolysis | H2 and N2(CO2) | Yes | No | High, if the organics are obtained from wastewater | 0–1.0a29–31 |
18.66 | 50% | No |
| Organic upgrading-assisted water electrolysis | H2 and valued-added compounds | Yes | No | High | 0–0.6a32–34 |
11.19 | 100% | No |
| Self-powered electrolysis system | H2 + O2 or H2 + N2 | No | No | High | <0.135–37 | 1.87 | 50% | Yes |
| Self-catalyzed electrolysis system | H2 | No | Yes | Low | <0.238–40 | 3.73 | 100% | Yes |
2. Electrochemical fundamentals and electrocatalyst/device design
2.1 Fundamentals of the anodic oxidation reaction
Indeed, anodic small molecule oxidation reactions offer promising advantages for expediting hydrogen production and the generation of value-added products. The integration of these reactions into water splitting systems allows for lower cell voltages compared to the OER, thereby improving energy efficiency. Additionally, the byproducts generated during these organic oxidation reactions, such as carboxylic acids, esters, and other valuable compounds, have widespread applications in various industries, including biofuel, fine chemical, and pharmaceutical. To maximize the energy return on investment and optimize the small molecule oxidation assisted electrocatalytic water splitting process, the careful selection of suitable small molecules is crucial. Here are some considerations for this aspect: (i) Appropriate redox potentials: the chosen small molecules should have theoretical potentials lower than that of the OER. This ensures that they do not interfere or compete with the OER, thus maintaining the efficiency advantages of the electrocatalytic process. (ii) High selectivity and conversion efficiency: it is crucial to maximize the selectivity and conversion efficiency of the oxidation reactions. This enables simultaneous cathodic hydrogen production and anodic degradation of pollutants or generation of value-added products, leading to a more sustainable and efficient water splitting process. (iii) Interference with the HER: neither the chosen small molecules nor their resulting products should significantly interfere with the HER at the cathode. This ensures that hydrogen production is not compromised and maintains the overall efficiency of the water splitting system. (iv) Availability and natural resources: the selected small molecules should be readily obtainable or derived from abundant natural resources or wastewater sources. This ensures their availability and scalability for large-scale implementation. Utilizing abundant resources also contributes to the economic viability and sustainability of the process. By considering these factors, small molecules that fulfill the necessary requirements for efficient small molecule oxidation assisted electrocatalytic water splitting can be identified, allowing for simultaneous hydrogen production, pollutant degradation, and generation of value-added products. In recent years, numerous small molecules, including urea, hydrazine, polysulfide, amines, glucose, alcohols, and aldehydes, have garnered increasing attention and have been successfully employed in electrochemical hydrogen production.Various small molecule oxidation reactions increasingly studied can be broadly categorized into sacrificial-agent oxidation reactions and electrochemical upgrading reactions. To gain a deeper understanding of the reaction pathways and mechanisms involved in these small molecule oxidation reactions, researchers have extensively employed a combination of experimental techniques and theoretical calculations. By employing methods such as in situ Raman spectroscopy, in situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS), in situ X-ray photoelectron spectroscopy (XPS), in situ X-ray absorption spectroscopy (XAS), in situ X-ray diffraction (XRD), operando transmission electron microscopy (TEM), in situ differential electrochemical mass spectrometry (DEMS), high-performance liquid chromatography (HPLC), inductively coupled plasma (ICP) spectroscopy, and nuclear magnetic resonance (NMR) spectroscopy, researchers have been able to elucidate the structure, morphology, content, and dynamic changes of catalysts and reaction products during the reaction process.41,42 To illustrate the distinctions among the characterization techniques discussed in this perspective, we have compiled a summary in Table 2. This table provides insights into the capabilities, advantages, limitations, probing regions, and typical references of these techniques for a better understanding. Computational methods, including density functional theory (DFT) calculations and molecular dynamics simulations, play a crucial role in elucidating reaction mechanisms and understanding the underlying principles governing small molecule oxidation. These calculations provide insights into the electronic structure, reaction energetics, and kinetics, aiding in the interpretation and prediction of experimental observations. By combining these experimental and theoretical approaches, researchers can unravel the complex reaction pathways and mechanisms involved in small molecule oxidation reactions, which will contribute to the development of efficient catalysts, reaction optimization, and the design of sustainable processes for hydrogen production, pollutant degradation, and value-added product generation.
| Technique | Capability | Advantage | Limitation | Probing region | Typical references |
|---|---|---|---|---|---|
| In situ Raman | Analysing interfacial water structures | Non-invasive for samples | Insensitive for pure metals and unable to detect reactions occurring deeper within the electrode | Bulk | 43 and 44 |
| Detecting surface active species | High spatial resolution | Low Raman peak intensity | Interface | ||
| Probing phase reconstruction | Used in a liquid environment | Limited spectral range | |||
| In situ ATR-SEIRAS | Comparing the bonding strength of reaction intermediate adsorption | Non-destructive towards samples | Sensitivity to water | Interface | 45 and 46 |
| Identifying reaction intermediate species | Applied to various electrochemical reactions | Poor signal intensity | |||
| Relatively cheap and easy to implement | Limited depth profiling | ||||
| In situ XPS | Providing information about chemical composition, oxidation states, and electronic structure changes on the electrode surface | High sensitivity | Only surface sensitivity | Bulk | 47 and 48 |
| Non-destructive technique | Ultrahigh vacuum | Interface | |||
| Identify and quantify the chemical elements | |||||
| In situ XAS | Analysing local atomic structure | High energy resolution for precise determination of bond distances and coordination environments | Ultrahigh vacuum | Bulk | 49–52 |
| Monitoring oxidation states | No requirement for the crystallinity | Possible radiation damage to samples | Interface | ||
| Used in liquid phase to hard XAS | Complexity about operation | ||||
| In situ XRD | Probing lattice parameter and the kinetics of phase transitions | Precisely probe the crystal phase structure and lattice parameters in bulk | High requirement for crystallization | Bulk | 53–55 |
| Characterization of phase transformations | Quantitatively analyzed to determine phase compositions, and crystallite sizes | Limited spatial resolution | |||
| Relatively cheap and easy to implement | Unable to reflect local defects in samples | ||||
| Operando TEM | Insights into structural changes, growth mechanisms, and electrochemical behavior of samples | Directly observe structural reconstruction on the nanoscale | Low image resolution in liquid electrolyte | Bulk | 56 and 57 |
| Provide crystallographic information, including lattice fringes, crystal orientations, and defects | Identification of elemental distribution during reactions | Limited field of view | |||
| High spatial and temporal resolution | Damaged by electron beam | ||||
| Difficult to maintain a stable environment under radiation | |||||
| In situ DEMS | Insights into the activity and selectivity during reactions | High temporal resolution | Complex instrumentation | Interface | 58 and 59 |
| Suitable for gas phase analysis | High sensitivity | Limited mass range | |||
| Molecular information | |||||
| Obtaining quantitative information | |||||
| HPLC | Insights into the products, intermediates, and kinetics | High resolution | Time-consuming and requiring additional steps | − | 60 and 61 |
| Wide applicability | Lack of temporal resolution | ||||
| Separation of mixtures | Sensitivity to mobile phase | ||||
| High sensitivity for trace species | |||||
| ICP | Simultaneously detecting multiple elements | High sensitivity | Limited molecular information | − | 62 |
| Low interference | Complex instrumentation | ||||
| Wide applicability | Reduced sensitivity in complex samples | ||||
| Isotopic analysis | |||||
| NMR | Providing detailed structural information about molecules | Molecular-level insights | Long acquisition times | − | 63 and 64 |
| Versatility | High purity for samples | ||||
| Isotopic labelling | Complex instrumentation and high cost |
2.2 Rational design of electrocatalysts
Small molecule oxidation reactions often involve the transfer of multiple electrons, making them kinetically sluggish and complex. To address this issue and improve reaction rates while reducing overpotential, the development of efficient catalysts is crucial. Initially, catalysts based on precious metals such as Pt, Pd, Ru, and Au were commonly used for these reactions.65,66 However, the limited availability and high cost of these metals hinder their widespread application. As a result, researchers have been motivated to explore alternative, cost-effective electrocatalysts derived from nonprecious-metal elements that offer both high performance and stability.67 These nonprecious-metal catalysts encompass a wide range of materials, including transition-metal oxides/oxyhydroxides, nitrides, phosphides, sulfides, and alloy systems. Each material exhibits unique properties and catalytic mechanisms that can be tailored to specific reaction requirements. By carefully designing the composition, structure, and morphology of these catalysts, researchers aim to enhance their electrocatalytic activity, selectivity, and durability. In recent years, various design strategies have been investigated to enhance the electrocatalytic activities of catalysts based on nonprecious metals for small molecule oxidation reactions. Fig. 3 depicts recent progress in the rational design of these electrocatalysts, categorized into three principal dimensions: electronic structure engineering, surface/interface design, and morphology regulation.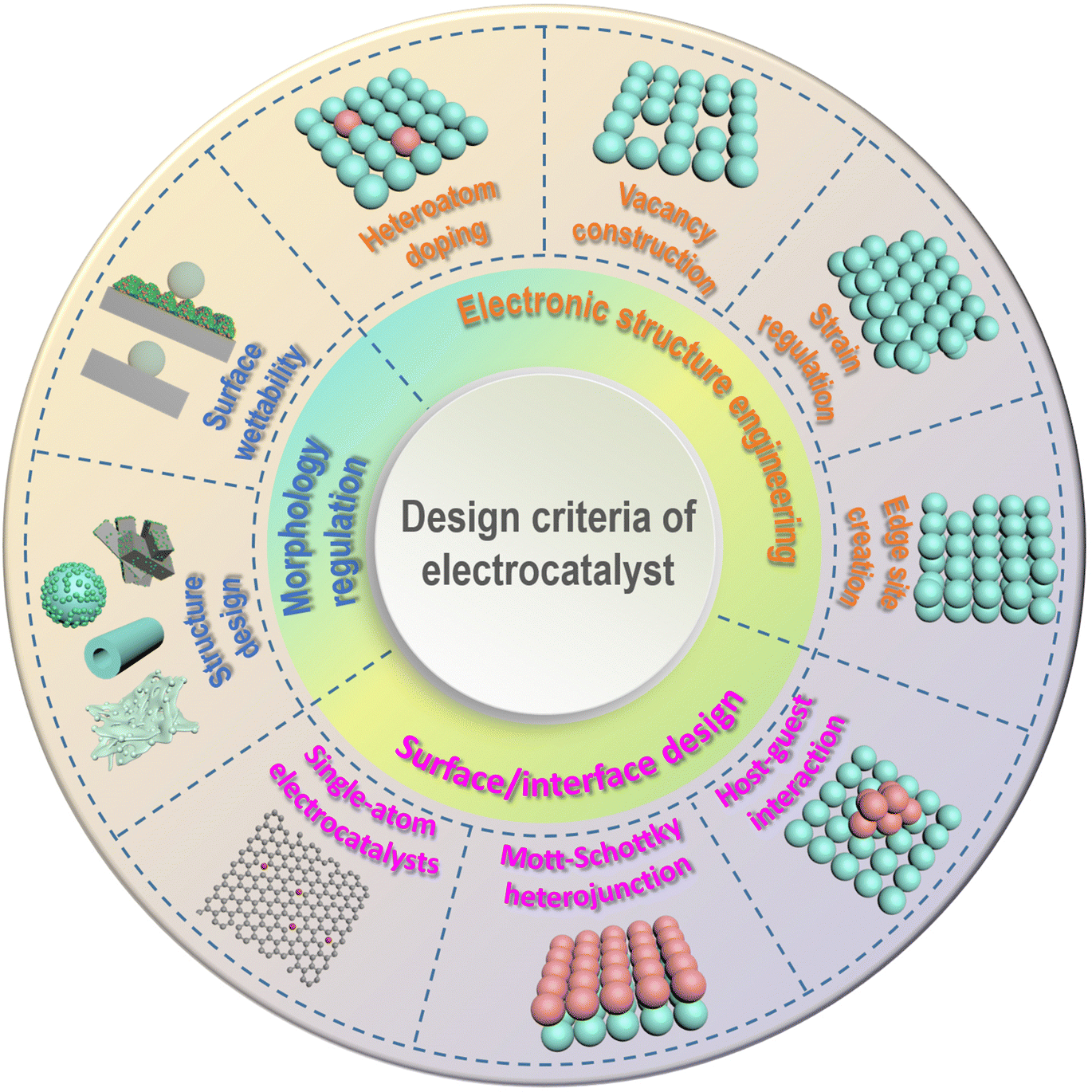 | ||
| Fig. 3 Schematic illustration of the general strategies for the rational design of electrocatalysts. | ||
Indeed, over the last decade, significant advancements have been made in the field of the HER. Through a comprehensive understanding of the fundamental mechanisms driving the HER and strategic engineering active sites, a variety of electrocatalysts with remarkable performance have been developed, rivaling that of benchmark Pt/C catalysts. Numerous insightful reviews have been published, covering various aspects of electrocatalyst design for the HER as well as their potential applicability in practical water electrolyzers.68–72 Hence, in this section, our primary focus is to summarize and discuss the design strategies employed for electrocatalysts targeting anodic oxidation reactions.
The introduction of vacancies into catalysts can have a significant impact on their electronic structure and catalytic performance. Vacancies refer to the absence of atoms at specific lattice sites within the catalysts. These vacancies can lead to localized fluctuations in electron density and atomic relaxation at the defect boundaries, influencing the catalyst's properties.79 It is worth noting that the specific effects of vacancies on catalytic performance depend on the materials, the type of reaction, and the specific conditions. The concentration, distribution, and interaction of vacancies within the catalyst are critical factors that determine their influence on catalytic activity.80 Specifically regarding the UOR, the amino group (–NH2) in the initial urea molecule possesses lone pair electrons and acts as an electron-donating moiety during the UOR, making it prone to adsorb on anion vacancies or cation sites. Fu et al. recently reported a novel and efficient electrocatalyst comprising N vacancies-enriched, Ce-doped Ni3N hierarchical nanosheets supported on carbon cloth (Ce–Ni3N@CC), aiming to optimize the UOR kinetics, particularly the rate-determining CO2 desorption step.81 DFT calculations revealed that the incorporation of Ce into Ni3N reduces the formation energy of N vacancies (Fig. 4a), resulting in a high density of N vacancies in Ce–Ni3N@CC. Furthermore, the N vacancies, in combination with Ce doping, optimize the local charge distribution around the Ni sites, thereby balancing the adsorption energy of CO2 during the rate-determining step and influencing the initial adsorption structure of urea. These factors contribute to the superior catalytic performance of Ce–Ni3N@CC for the UOR.
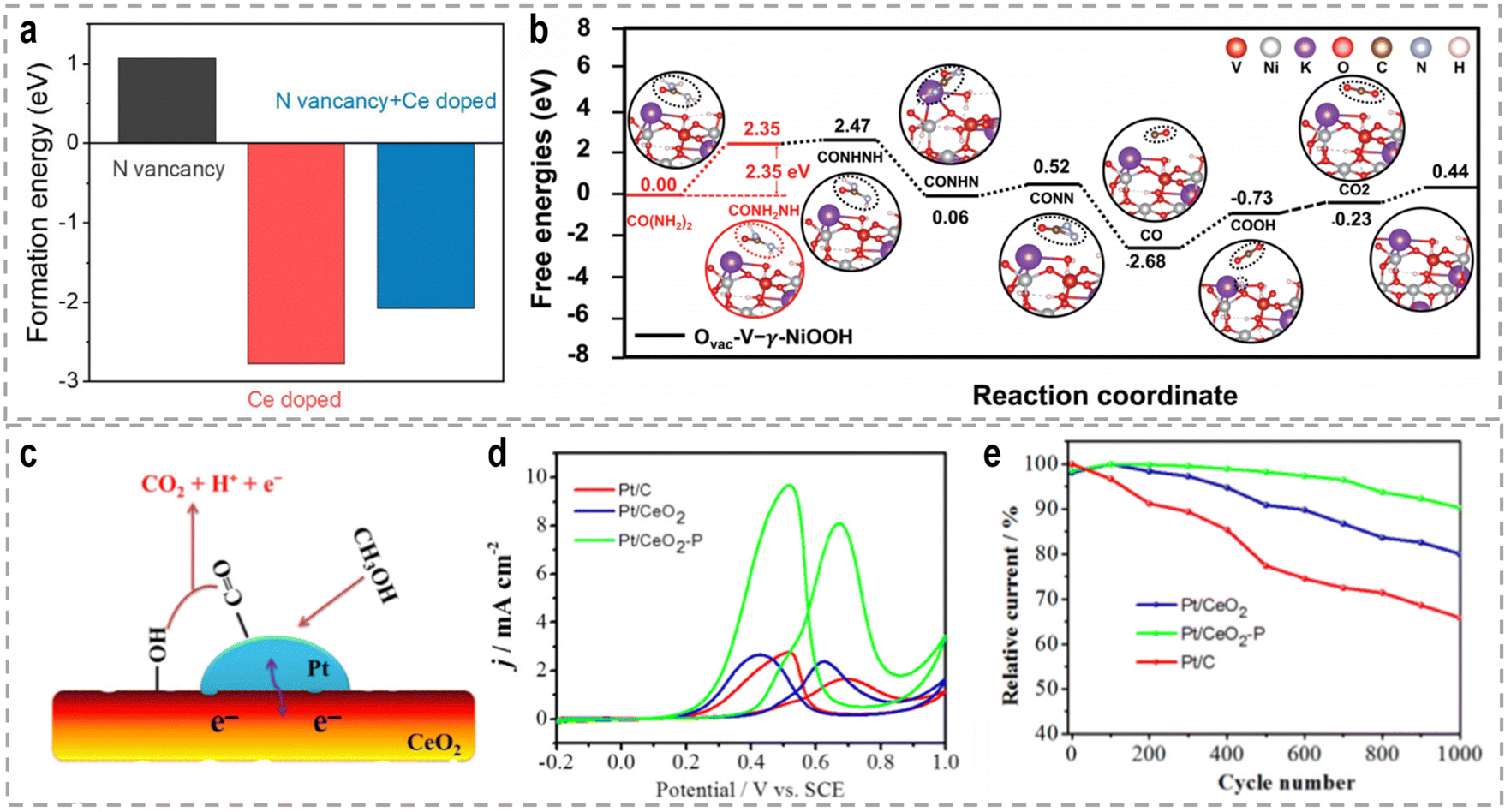 | ||
| Fig. 4 (a) Different kinds of formation energies.81 Reproduced with permission. Copyright 2022, Elsevier. (b) The Gibbs free energy profiles of the proposed UOR pathway at Ovac-V-γ-NiOOH.98 Reproduced with permission. Copyright 2022, Wiley-VCH. (c) The illustration graph of Pt on VO rich CeO2. (d) CVs of methanol oxidation. (e) Stability testing.108 Reproduced with permission. Copyright 2018, Elsevier. | ||
Strain has garnered significant attention in recent years and has been extensively investigated and reported.82 In the context of electrocatalysts, strain primarily manifests as changes in bond lengths.83,84 The strain imposed on the lattice of an electrocatalyst can alter the coordination numbers and electron densities of surface atoms, thereby modifying the surface energy and enhancing specific adsorption of target molecules. Strain defects can be intentionally introduced through various fabrication methods, such as core–shell structures, morphology modulation, alloying, and particle size regulation.85,86 By tuning the width of the surface d-band, the average energy can be adjusted, thus controlling chemical properties such as the dissociative adsorption energy of hydrogen. Lattice strain, whether compressive or tensile, can significantly modify the surface electronic structure by regulating the distances between surface atoms.87–89 For instance, previous studies have shown that a mere 1% lattice strain in Pt can shift the d-band center by approximately 0.1 eV, resulting in more than a 300% improvement in oxygen reduction reaction (ORR) activity through optimized adsorption energy.90–92 However, the conventional approach of using a metal overlayer can introduce both ligand and synergistic effects, complicating the correlation between reactivity and lattice strain. In a recent study, porous rhodium nanosheets (Rh-NSs) with tunable lattice strain were successfully synthesized by direct annealing of metastable trigonal rhodium oxide (Tri-RhO2) nanosheets in a hydrogen atmosphere.93 The magnitude of compressive strain in the Rh-NS electrocatalysts could be controlled by adjusting the annealing temperature. In stark contrast, metallic rhodium obtained by annealing rutile-RhO2 under the same conditions exhibited almost no compressive strain. Electrochemical results demonstrated that the optimized Rh-NSs-300 exhibited an ultralow working potential of −103 mV (vs. RHE) at a current density of 10 mA cm−2, with a Tafel slope of 15.4 mV dec−1 for the HzOR. DFT calculations indicated that Rh-NSs with tunable compressive strain could reduce the Gibbs free energy of the potential-determining step in the HzOR.
Recent investigations have provided compelling evidence that atoms located at the edges of 2D materials exhibit distinct properties and higher catalytic activity compared to their basal planes.94–96 Nickel hydroxide (Ni(OH)2), a well-known 3d transition metal-based material with a layered structure, has been extensively studied and considered a promising electrocatalyst for the UOR due to its favorable activity and impressive stability in alkaline environments. Luo et al. reported the synthesis of ultrathin Ni(OH)2 nanosheets with small lateral sizes through a simple liquid-phase reaction.97 The combination of theoretical calculations and experimental investigations revealed that the abundant active edges in Ni(OH)2 nanoflakes not only facilitate an accelerated phase transformation from Ni(OH)2 to the electroactive NiOOH but also enhance the kinetics of urea adsorption during the electrochemical process, resulting in significantly improved electrocatalytic activity for the UOR. Similarly, Jiao et al. reported the synthesis of O-vacancy-rich Ni(OH)2 through facile V doping, leading to the formation of Ovac–V–Ni(OH)2.98 V doping not only generates more exposed edge active sites but also introduces oxygen vacancies, shifting the rate-determining step of the UOR from *COOH deprotonation to the N–H bond cleavage process. This modification lowers the thermodynamic barrier by approximately 1.13 eV compared to pristine γ-NiOOH (Fig. 4b), thereby significantly improving the reaction performance.
The catalytic performance of the methanol oxidation reaction (MOR) is highly surface and interface sensitive. Therefore, the presence of interfaces can have a profound impact on the catalytic performance in the MOR, as it influences the optimization of electronic configurations through interfacial electron transfer in heterostructures. The construction of Pt-based heterogeneous catalysts is particularly important for optimizing the binding energies of reaction intermediates during the MOR, thereby influencing the electrocatalytic performance and stability.107 Wang et al. used the CeO2 nanorods with rich oxygen vacancies (VO) and a rough surface as the Pt support to engineer the metal-oxide interface.108 This unique property of the CeO2 substrate allows for effective modification of the particle size and dispersion of Pt nanoparticles, as well as the regulation of the Pt–ceria interaction (Fig. 4c). Notably, the abundant VO on the surface of CeO2 provides surplus electrons that transfer to the sub-surface, leading to a decrease in the Ce valence in the sub-surface of CeO2, as well as alters the electron transfer between Pt and ceria, resulting in an increased electron density in Pt. Due to the distinctive electron structure of Pt and CeO2, the catalytic activity and durability of the electrocatalyst for the MOR are significantly enhanced (Fig. 4d and e).
Mott–Schottky heterojunctions, characterized by the presence of semiconducting phases, exhibit unique properties arising from the differences in Fermi energy and charge density between the two sides of the interface. This leads to the transfer of electrons across the interface, creating a built-in electric field, a space charge region, and local charge accumulation or depletion. The band bending at the heterointerfaces induces a charge distribution, which facilitates the activation of small molecules such as urea.109 In the context of bifunctional electrocatalysis for both the HER and UOR, a bimetal heterostructure consisting of CoMn and CoMn2O4 was designed as a bifunctional electrocatalyst.110 The CoMn/CoMn2O4 Schottky synergistic effects were proposed as the basis for the UOR catalytic mechanism, in which a self-driven charge transfer occurs at the interface (Fig. 5a). This charge transfer process facilitates the adsorption of reactants and the breaking of chemical bonds, resulting in optimal adsorption energy (Fig. 5b). Consequently, both the HER and UOR are triggered. Remarkably, ultralow cell voltages of −0.069, 1.32, and 1.51 V were required to achieve a current density of 10 mA cm−2 for the HER, the OER, and urea-assisted hybrid water electrolysis, respectively.
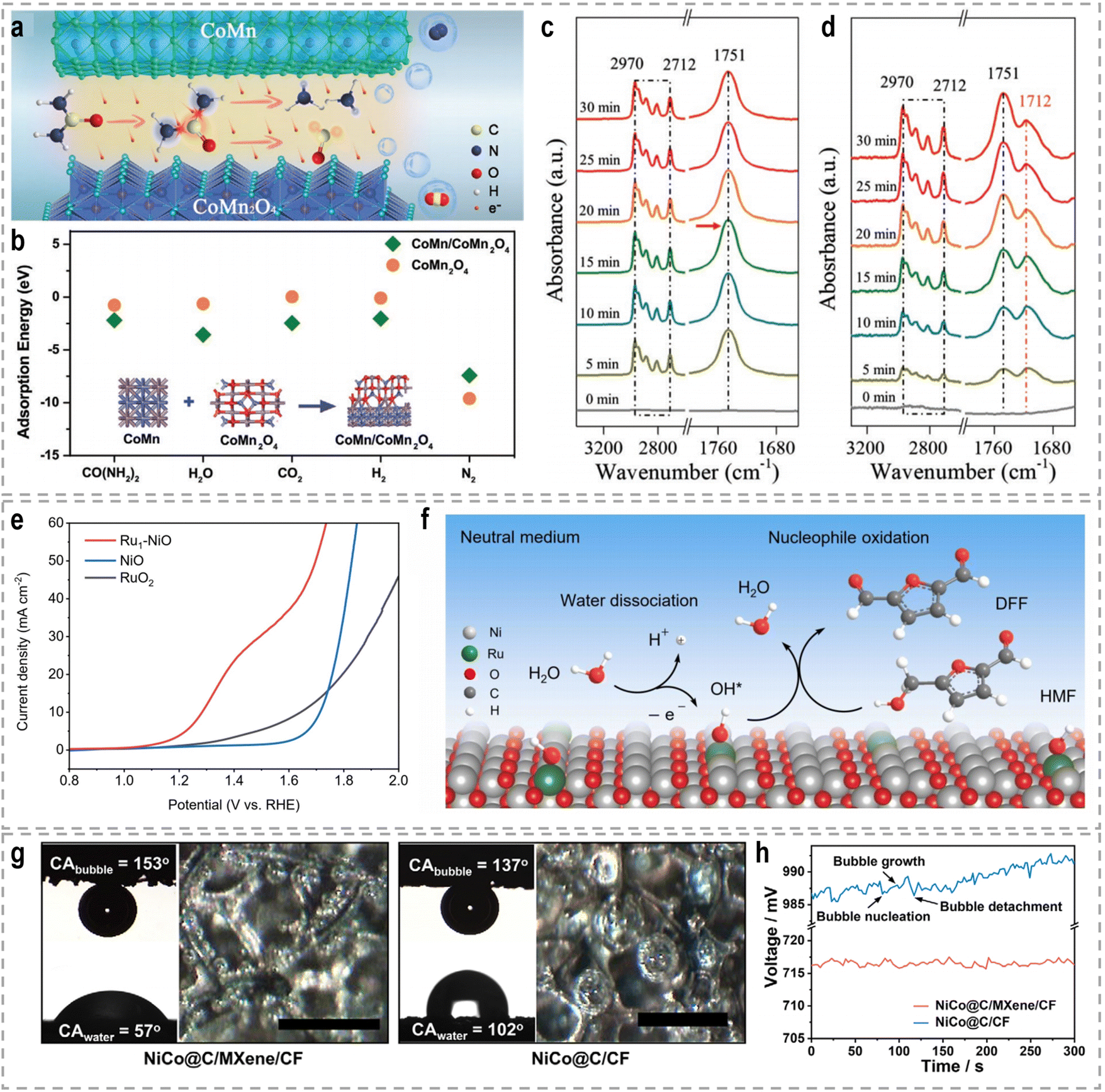 | ||
| Fig. 5 (a) The schematic UOR catalytic mechanism based on CoMn/CoMn2O4 Schottky synergistic effects. (b) Calculated adsorption energies of CO(NH2)2, H2O, CO2, H2, and N2 on CoMn2O4 and CoMn/CoMn2O4 surfaces.110 Reproduced with permission. Copyright 2020, Wiley-VCH. In situ FTIR spectra of n-valeraldehyde adsorption on (c) NiMn2O4 and (d) Au0.3 SACs-NiMn2O4.116 Reproduced with permission. Copyright 2022, Wiley-VCH. (e) Linear sweep voltammetry (LSV) curves of NiO, Ru1–NiO, and RuO2 in 1.0 M PBS with 50 mM HMF. (f) Proposed HMF oxidation reaction mechanism over Ru1–NiO in the neutral medium.117 Reproduced with permission. Copyright 2022, Wiley-VCH. (g) The contact angels of water (CAwater) and bubbles (CAbubble) on NiCo@C/MXene/CF or NiCo@C/CF, and the optical images of gas bubbles released from both electrodes during the HER. Scale bars: 500 μm. (h) Chronopotentiometric curves of NiCo@C/MXene/CF and NiCo@C/CF for hybrid seawater electrolysis.129 Reproduced with permission. Copyright 2021, Nature Publishing Group. | ||
In recent years, there has been a lot of interest in the field of single-atom electrocatalysts (SACs). SACs are characterized by discrete metal atoms that are anchored onto a suitable substrate, offering unique advantages such as near-unity atomic utilization and enhanced catalytic performance.111,112 By preventing agglomeration and dissolution phenomena, the anchored metal atoms can maintain their isolated and well-defined structures, leading to enhanced catalytic performance. The interplay between the individual metal atoms and their surrounding counterparts derived from the support materials or other metal atoms, plays a pivotal role in governing the catalytic activity of SACs. Therefore, selecting a suitable support system involves careful consideration of various factors. The support materials should possess a suitable structure, high surface area, and appropriate surface chemistry to promote strong interactions with the metal atoms. It should also exhibit good chemical stability under the harsh reaction conditions to ensure the durability of the SACs. To date, various classes of SACs with different supports have been successfully synthesized.113–115 Carbon-based SACs, including graphene, nitrogen-doped graphene, and other porous carbon materials, have been extensively explored. SACs supported by two-dimensional materials such as transition metal dichalcogenides, MXene, black phosphorus, and layered double hydroxides (LDH), have been investigated. Furthermore, support-confined SACs, such as metal alloys and carbides, have also been explored. The inherent high reactivity and stability of SACs make them promising candidates for various catalytic reactions.
Each support material offers unique properties and interactions that can be tailored to specific catalytic applications. For example, Li et al. developed Au SACs–NiMn2O4 spinel synergetic composites for the electrocatalytic upgrading of n-valeraldehyde to octane.116 Experimental and theoretical studies revealed that the presence of Au SACs on the spinel surface lowers the adsorption energy of the initial n-valeraldehyde molecule, thereby accelerating the dimerization process of alkyl radicals originating from Mn–C4H9 and Au–C4H9, leading to the formation of octane. In situ Fourier transform infrared spectroscopy (FTIR) further confirmed the enhanced adsorption performance of n-valeraldehyde on Au SACs–NiMn2O4. As shown in Fig. 5c and d, the characteristic peaks of n-valeraldehyde are observed at 1751, 2712, and 2970 cm−1, which are associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H stretching vibrations. For Au0.3 SACs–NiMn2O4, the intensity of n-valeraldehyde peaks gradually increases with prolonged reaction time. Additionally, new species are formed on the surface over time. The appearance of a peak at 1712 cm−1 indicates the vibrational mode of the carbonyl group in fatty acids, specifically n-valeric acid formation. The intensity of the n-valeric acid peak initially increases and subsequently decreases as time goes on, indicating its continuous involvement in the reaction on the surface of Au0.3 SACs–NiMn2O4. These in situ FTIR results provide evidence that the presence of Au single atoms on NiMn2O4 enhances the adsorption of n-valeraldehyde, thereby facilitating its conversion. In another example, Duan et al. reported the utilization of single-atom Ru on nickel oxide (Ru1–NiO) as a catalyst for the electrooxidation of alcohols derived from biomass into aldehyde products.117 For the oxidation reaction of 5-hydroxymethyl furfural (HMF) in a 1.0 M phosphate buffer solution (PBS), Ru1–NiO shows a low potential of 1.283 V at 10 mA cm−2 (Fig. 5e). Through a combination of cyclic voltammetry (CV), Raman spectroscopy, and operando electrochemical impedance spectroscopy analyses, the authors revealed that the remarkable activity of Ru1–NiO ascribed to the significantly improved water dissociation ability occurring at the single-atom Ru sites. As depicted in Fig. 5f, water molecules adsorbed on Ru atoms dissociate into OH* species, accompanied by simultaneous electron and proton transfers. And the resulting OH* species react with the nucleophile, HMF, yielding 2,5-diformylfuran and water.
O and C–H stretching vibrations. For Au0.3 SACs–NiMn2O4, the intensity of n-valeraldehyde peaks gradually increases with prolonged reaction time. Additionally, new species are formed on the surface over time. The appearance of a peak at 1712 cm−1 indicates the vibrational mode of the carbonyl group in fatty acids, specifically n-valeric acid formation. The intensity of the n-valeric acid peak initially increases and subsequently decreases as time goes on, indicating its continuous involvement in the reaction on the surface of Au0.3 SACs–NiMn2O4. These in situ FTIR results provide evidence that the presence of Au single atoms on NiMn2O4 enhances the adsorption of n-valeraldehyde, thereby facilitating its conversion. In another example, Duan et al. reported the utilization of single-atom Ru on nickel oxide (Ru1–NiO) as a catalyst for the electrooxidation of alcohols derived from biomass into aldehyde products.117 For the oxidation reaction of 5-hydroxymethyl furfural (HMF) in a 1.0 M phosphate buffer solution (PBS), Ru1–NiO shows a low potential of 1.283 V at 10 mA cm−2 (Fig. 5e). Through a combination of cyclic voltammetry (CV), Raman spectroscopy, and operando electrochemical impedance spectroscopy analyses, the authors revealed that the remarkable activity of Ru1–NiO ascribed to the significantly improved water dissociation ability occurring at the single-atom Ru sites. As depicted in Fig. 5f, water molecules adsorbed on Ru atoms dissociate into OH* species, accompanied by simultaneous electron and proton transfers. And the resulting OH* species react with the nucleophile, HMF, yielding 2,5-diformylfuran and water.
In contrast to SACs, dual single-atom catalysts (DSACs) possess multiple active centers with distinct electronic states within a localized domain.118 This characteristic enables DSACs to fulfill the requirements for intermediate absorption in multi-reaction steps, thereby exhibiting enhanced activity and selectivity as atomic dispersion catalysts.119,120 Pt-based catalysts have been recognized for their efficiency in the HER. The synergistic effect between metal atoms in DSACs facilitates the maintenance of a near-zero hydrogen adsorption energy, promoting efficient hydrogen production. For instance, Sun et al. employed an atomic layer deposition process to fabricate a Pt–Ru dimer catalyst for the HER.121 To assess the HER activity under varying hydrogen adsorption scenarios, the Gibbs free energy for hydrogen adsorption (ΔGH) of the Pt–Ru dimer was calculated. When one hydrogen atom is adsorbed on both sides of the Ru and Pt atoms in the Pt–Ru dimer (designated as Pt(1H)Ru(1H)), ΔGH is approximately −1 eV, indicating that the hydrogen atoms are preferentially adsorbed on the Ru atoms. Upon further adsorption of a second hydrogen atom on the dimer, ΔGH for Pt(2H)Ru(1H) reaches a high value of 0.6 eV. Remarkably, ΔGH of the Ru atom exhibits an exceptionally low value of 0.01 eV when considering the pathway from Pt(3H)Ru(3H) to Pt(3H)Ru(2H), even lower than that of a single Pt atom. This clearly demonstrates that hydrogen can easily dissociate from the Ru atom when the coverage reaches its maximum. Furthermore, the presence of Pt atoms strongly influences the electronic structure of the Ru atoms. DFT calculations reveal that the Pt–Ru dimer can be readily converted between metallic and semiconducting states upon adsorption, leaving vacant orbitals. The synergistic effect induced by Pt modulates the interaction between H and Ru, thereby enhancing the HER activity of the Pt–Ru dimer catalyst.
To facilitate efficient water splitting at high current densities, it is crucial to ensure the rapid desorption of generated gas bubbles. This is essential because the adhesion and accumulation of gas bubbles can introduce significant ionic diffusion resistance and hinder access to active sites within the system.128 Furthermore, the growth, collapse, and detachment of bubbles from the electrode surface can impose substantial mechanical strain and stretching forces, potentially leading to catalyst detachment and an overall decline in cell performance. It is evident that a hydrophilic surface plays a pivotal role in enhancing the adsorption of water while repelling the generated gas bubbles. Such characteristics are highly favorable for water splitting, especially under conditions of high current density. To achieve this, the engineering of super-wettability surfaces with special interactions at the solid–liquid–gas interfaces holds great promise. This approach can expedite the mass transfer process and reduce the ohmic resistance at the catalyst surface. Importantly, the wettability of a catalyst can be effectively adjusted by tuning its geometric structures. For instance, Sun et al. have developed a novel NiCo/MXene-based electrode by assembling NiCo-metal organic framework (MOF) nanosheets on MXene-wrapped Cu foam (MXene/CF), followed by annealing in NH3.129 The resulting NiCo@C/MXene/CF electrode exhibits a mesoporous array of NiCo-decorated carbon nanosheets (NiCo@C) with an average size of 400–800 nm and a thickness below 50 nm on the MXene/CF substrate. Importantly, this electrode possesses a remarkably high contact angle for gas bubbles (CAbubble = 153°), which effectively facilitates the rapid release of small gas bubbles (<60–80 μm) during the electrolysis process (Fig. 5g). This improvement is critical for maintaining a sufficient triple-phase interface between the electrode, electrolyte, and gas phase, thereby ensuring stable electrolysis operation under conditions of vigorous gas evolution. In contrast, when MXene is absent, the surface of NiCo@C/CF exhibits irregular microstructures due to poor chemical coupling between NiCo@C and CF. Such structural degradation not only diminishes the availability of active sites but also compromises the electrode's resistance to gas accumulation. Consequently, the NiCo@C/CF electrode experiences significant voltage fluctuations caused by the vigorous accumulation and detachment of large gas bubbles (Fig. 5h), leading to a reduction in hydrogen production efficiency. Therefore, the incorporation of macroporous gas transport channels and a nanoarray-based superaerophobic surface into the NiCo@C/MXene/CF electrode represents a substantial advancement towards optimizing the gas evolution process, particularly at higher current densities.
On the basis of the current research's achievements, the key steps and considerations for designing active catalysts towards small molecule oxidation are outlined in Fig. 6. (i) Identify the target reaction: different reactions have different requirements and mechanisms, so understanding the fundamentals of the reaction is crucial. (ii) Choose suitable catalyst materials: according to the reaction requirements, the appropriate catalyst materials are selected. (iii) Optimization of catalyst composition: adjust the catalyst composition by alloying or mixing with other materials to improve catalytic activity. Bimetallic catalysts, for example, can exhibit enhanced activity due to synergistic effects between different metal components. (iv) Surface modification: modify the surface of the chosen catalyst material to enhance its catalytic activity. (v) Electrocatalyst characterization: utilize various characterization techniques to thoroughly analyze the catalysts’ structure and properties, gaining insights into their suitability for the target reaction. (vi) Electrocatalyst testing: conduct electrochemical tests to evaluate the catalyst's performance. Compare the catalyst's activity and selectivity to benchmarks and evaluate its performance under realistic conditions. (vii) Understanding reaction mechanisms: investigate the detailed reaction mechanisms through experimental techniques and computational modeling. A deep understanding of the reaction pathways and intermediates is critical for catalyst design. (viii) Electrocatalyst stability: assess the stability and durability of the catalyst under electrochemical conditions. Catalyst degradation and poisoning should be minimized for long-term performance. (ix) Iterative design: employ the knowledge gained from experiments and modeling to refine the catalyst's design. Iterative testing and modification is necessary to optimize its performance continually. (x) Scale-up and integration: if the catalyst exhibits promise in laboratory settings, scale up its production and seamlessly integrate it into practical electrochemical devices. (xi) Environmental and economic considerations: consider the environmental and economic aspects of the catalyst design, including the use of cost-effective and sustainable materials.
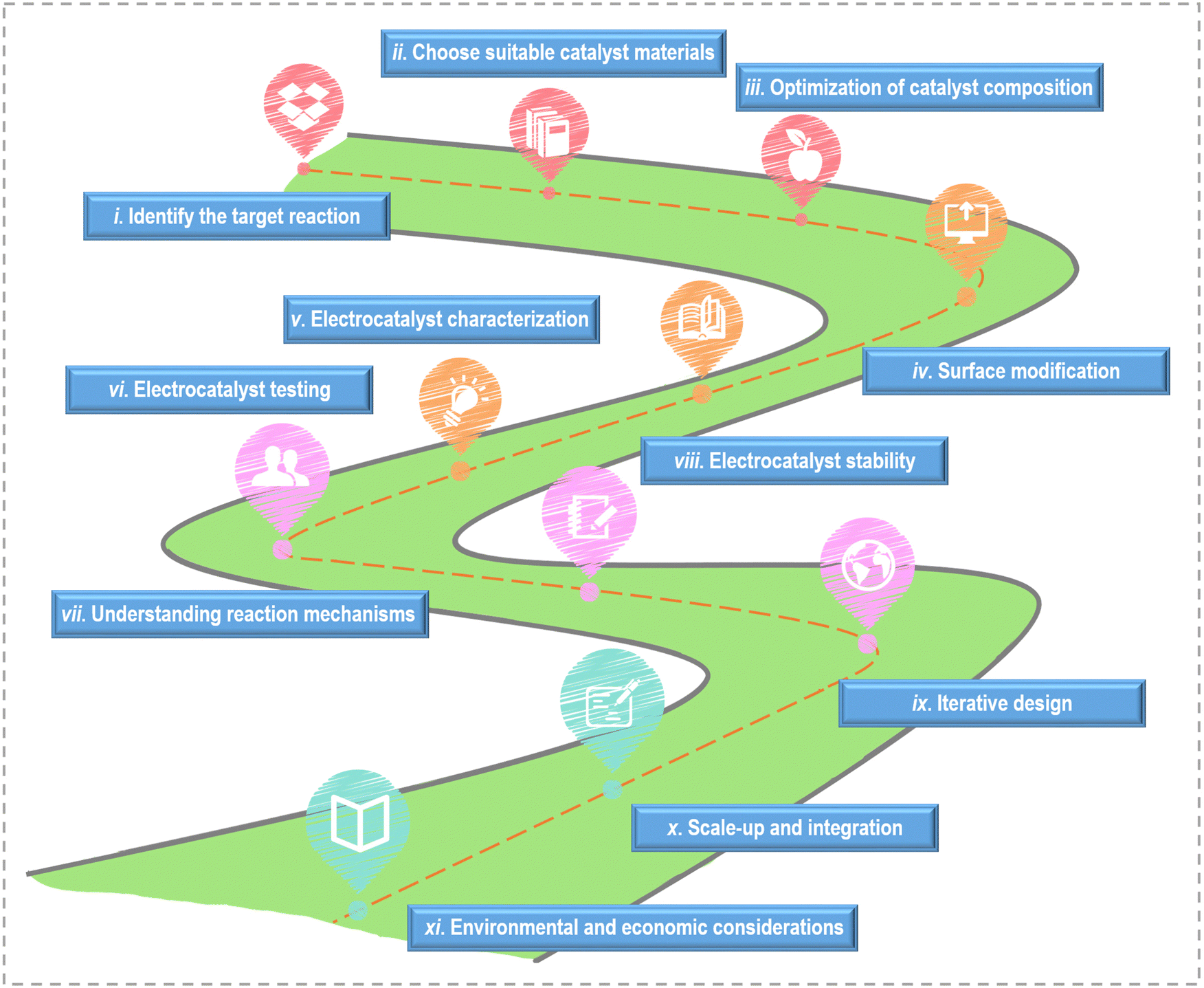 | ||
| Fig. 6 The design flow diagram towards efficient electrocatalysts. | ||
Designing active electrocatalysts for small molecule oxidation reactions is a multidisciplinary process that requires a combination of material synthesis, electrochemical characterization, and a deep understanding of the underlying reaction mechanisms. Collaboration between chemists, material scientists, and electrochemists is often crucial to succeed in this field.
2.3 Reaction devices
Besides catalyst design, the development of efficient devices plays another crucial role in enhancing the efficiency and performance of chemical reactions.130,131 For water splitting, the configuration of the electrolysis cell can significantly impact the overall performance and product selectivity. The conventional single-pool cell configuration is commonly used in lab-scale experiments to screen catalysts due to its simplicity in assembly.132,133 In this configuration, the working electrode, usually the anode for the OER and the cathode for the HER, is placed in the same electrolyte pool (Fig. 7a). However, one drawback of this configuration is the occurrence of undesired reactions at the counter electrode, where the liquid-phase products generated at the working electrode can undergo electrochemical reconversion. To address this issue, H-type cells, also referred to as two-compartment or divided cells, have been widely employed in various electrochemical reactions by incorporating an ion-exchange membrane (Fig. 7b).134 The H-type cell consists of two compartments, typically separated by a membrane, such as an anion exchange membrane (AEM), a proton exchange membrane (PEM), and a bipolar membrane (BPM), which allows for the selective transport of ions into distinct chambers while preventing direct contact between the working and counter electrodes.135–137 AEMs, acting as ionic conductors and separators, ensure the operation of the cell in mildly alkaline electrolytes. The use of a PEM as a solid polymer electrolyte enables the conduction of H+ ions from the anode to the cathode. The BPM combines the advantages of both the AEM and PEM by facilitating the anodic reaction in the basic electrode and promoting the preferential occurrence of the cathodic reaction in an acidic solution, thus reducing the cell's overpotential.138 Given that the HRE is kinetically favorable under acidic conditions and the OER under alkaline conditions, it would be preferable to realize these half reactions simultaneously in different electrolytes with a BPM.139,140 For instance, with the catalysis of bifunctional cobalt–nickel phosphide nanowire (Co–Ni–P/NF) catalysts, a water splitting electrolyzer with a BPM delivers 10 mA cm−2 at 1.567 V, while an electrolyzer with an AEM requires a voltage of 1.623 V (Fig. 7c).141 Moreover, the FEs for both the HER and OER in this BPM-assembled electrolyzer also reach 100% (Fig. 7d). Benefiting from these advantages of the BPM compared to conventional AEM and PEM, the BPM has been widely used in various systems for hydrogen production, such as photoelectrochemical water splitting,142,143 electrochemical water splitting,144,145 hybrid water splitting,146,147 alkali–acid hydrazine fuel cell,148–150 and zinc–H2 batteries.151 Nonetheless, it is worth noting that the electrochemical resistance is directly influenced by the distance between two electrodes. Due to the increased separation between the two electrodes and the presence of an ion-exchange membrane, a H-type cell exhibits inherently higher electrochemical resistance compared to a single-pool cell configuration. Consequently, the productivity of H-type cells is significantly reduced, leading to increased energy costs.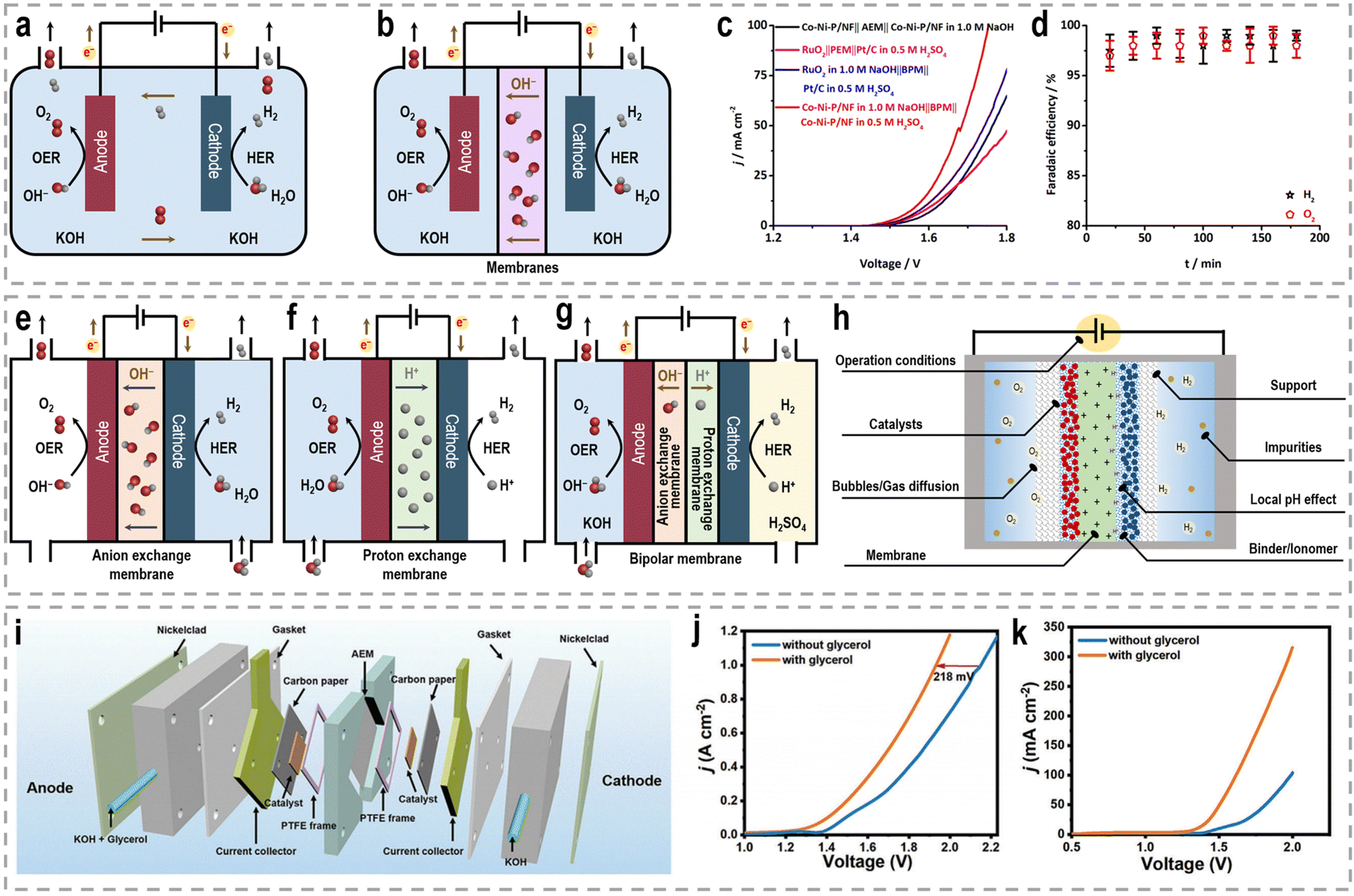 | ||
| Fig. 7 Schematic of (a) the single-pool cell configuration, and (b) H-type cell configuration for performance evaluation. Water electrolysis tests performed in a two-compartment Teflon cell separated by a polymer membrane, (c) BPM-based asymmetric water electrolysis and AEM/PEM-based water electrolysis, (d) FE of the BPM-based asymmetric water electrolysis.141 Reproduced with permission. Copyright 2020, Wiley-VCH. Schematic of (e) AEM electrolyzer, (f) PEM electrolyzer, and (g) BPM electrolyzer for performance evaluation. (h) Crucial factors to be considered for designing electrolyzers with superior activity and long-term stability. (i) Schematic illustration of the flow cell configuration. (j) LSV curves for the water electrolysis with and without glycerol addition using the flow cell. (k) LSV curves for the water electrolysis using a single-chamber electrolyzer.33 Reproduced with permission. Copyright 2023, Wiley-VCH. | ||
To address these challenges, innovative flow cells, for instance membrane reactors, have been developed to establish direct contact between the electrodes and ion-exchange membrane through the assembly of membrane electrode assemblies (MEAs). According to the type of the used membrane, these membrane reactors include an AEM electrolyzer (Fig. 7e), a PEM electrolyzer (Fig. 7f), and a BPM electrolyzer (Fig. 7g). MEAs typically consist of a five-layer structure, wherein a catalyst-coated membrane (CCM) is sandwiched between two gas diffusion layers (GDLs) or an ion-exchange membrane is placed between two gas diffusion electrodes (GDEs).152,153 Notably, the continuous circulation of the electrolyte over the electrode surface in MEAs helps overcome the mass-transport limitations encountered in conventional H-type cells or single cells.154–156 Furthermore, the zero-gap configuration achieved by pressing the GDL, catalyst layer, and ion-exchange membrane together in an MEA greatly reduces system impedance, thereby improving the reaction rate and overall energy efficiency. As an example, in a GDE-type electrolyzer operating with 1.0 M KOH electrolyte and a thickness of 3 mm in the cathode chamber, the protons/ions traverse the electrolyte with an internal resistance of approximately 1.875 Ω cm2, resulting in a voltage loss of approximately 800–900 mV at an operating current density of 500 mA cm−2.157 Recently, a series of novel flow cells have been employed for large-scale H2 production at high current densities. For instance, by utilizing highly efficient NiVRu-LDHs NAs/NF as both the cathode and anode for the HER and the glycerol oxidation reaction (GOR), respectively, a flow electrolyzer with an AEM (Fig. 7i) achieves a low cell voltage of 1.933 V to deliver a current density of 1 A cm−2 (Fig. 7j), whereas a single-chamber electrolyzer can only achieve 273.3 mA cm−2 (Fig. 7k).33 Furthermore, it demonstrated considerable formate and H2 productivities of 12.5 and 17.9 mmol cm−2 h−1, respectively, at an industry-level current density, with high faradaic efficiency (FE) of nearly 80%.
The latest advancements in electrochemical reactor and system design underscore the need for unconventional approaches to water electrolysis, as well as the optimization of more efficient and durable system designs. In Table 3, we have presented the characteristics, advantages, and limitations of various electrolytic reactors to facilitate a straightforward comparison between them. However, it is important to note that apart from the exploration of high-performance electrocatalysts and devices, the activity, long-term stability, and product yields of membrane reactors are also influenced by several critical factors. These factors encompass considerations such as the choice of membrane, support materials, binder/ionomer, gas diffusion characteristics, operational conditions, local pH values, and the presence of impurities, as depicted in Fig. 7h.
| Reactor | Separator | Electrolyte | Advantages | Drawbacks |
|---|---|---|---|---|
| Single-pool cell | None | Acidic/neutral/alkaline | Low costs | Inevitable reconversion of electrode products |
| Easy to assemble and test | ||||
| H-Type cell | Various ion exchange membrane | Depending on the selected membrane | Mature technology | Inherent high ohmic loss |
| Easy to achieve large-scale applications | Limited current density | |||
| AEM-based electrolyzer | Anion exchange membrane | Typically KOH | Fast response, long life and low price | Low OH− conductivity in polymeric membranes |
| Nonprecious metal-based catalysts | ||||
| High current density | ||||
| PEM-based electrolyzer | Proton exchange membrane (e.g., Nafion 117) | Typically pure water | Small floor space | Noble metal-based catalysts |
| Fast response | Poor durability | |||
| High current density | High capital cost | |||
| High-purity hydrogen production | ||||
| BPM-based electrolyzer | Bipolar membrane | Typically an extreme pH gradient | Without restriction of operation to electrolytes | Ion leakage, resulting in pH changes and energy loss |
| Less pH gradient | High cost and short service life |
3. Sacrificial-agent-assisted H2 production
Sacrificial-agent-assisted hydrogen production systems have indeed shown great potential for highly efficient hydrogen generation, thanks to their ultralow theoretical voltages. These systems involve the consumption of sacrificial agents such as urea and hydrazine, which provide a source of electrons and protons for the HER. However, a drawback of these systems is the generation of low-value-added byproducts during the sacrificial agent consumption process. Interestingly, these sacrificial-agent-assisted systems also hold promise for water treatment applications, as the sacrificial agents themselves, such as urea and hydrazine, are recognized as typical pollutants in wastewater (Fig. 8). Thus, the use of these sacrificial agents not only enables efficient hydrogen production but also provides a means of removing these pollutants from wastewater streams. In recent years, significant advancements have been made in sacrificial-agent-assisted hydrogen production systems, and there is ongoing research to elucidate the underlying catalytic mechanisms involved. In this section, we aim to present a comprehensive overview of the recent advancements in sacrificial-agent-assisted hydrogen production systems, for instance, urea oxidation reaction, hydrazine oxidation reaction, and polysulfide oxidation reaction, and elucidate the underlying catalytic mechanisms involved. | ||
| Fig. 8 Schematic illustration of cost-effective and sustainable H2 production from a renewable-powered and sacrificial-agent-assisted electrolyzer with cost-free seawater and industrial sewage as the feeds. | ||
3.1 Urea oxidation reaction
A significant amount of wastewater, both from industrial and domestic sources, contains urea, and approximately 80% of this wastewater is discharged directly into the environment each year.30,158 If left untreated, urea naturally decomposes, leading to the release of ammonia and nitrate. This decomposition process will result in severe environmental pollution and water resource eutrophication. To address this issue, the urea oxidation reaction (UOR, CO(NH2)2 + 6OH− → CO2 + N2 + 5H2O + 6e−, 0.37 V vs. RHE) can be employed to utilize urea from wastewater, making it an attractive approach for wastewater treatment due to its low theoretical voltage.159 By coupling the UOR with the HER, it becomes possible to simultaneously achieve wastewater treatment and highly efficient hydrogen generation. This approach offers the advantage of utilizing urea as a renewable resource for hydrogen production while addressing the environmental issue of urea-containing wastewater. However, the UOR is a complex process involving the transfer of 6e− and includes intricate adsorption and desorption steps for reactants and products. As a result, the UOR suffers from inherently sluggish kinetics, which hinders its efficiency. Thus, the development of highly efficient and active catalysts for the UOR remains a challenging task.Extensive research has been conducted on Ni-based catalysts for the UOR due to their outstanding performance. Huang's group synthesized coupled NiSe2 nanowrinkles with a Ni5P4 nanorod heterogeneous structure onto nickel foam (NiSe2@Ni5P4/NF) via the successive phosphorization and selenization processes.160 Theoretical simulations confirmed that the closely contacted interface led to fast charge transfer from Ni5P4 to NiSe2, thus constructing high-flux electron transfer pathways, enhancing carrier concentration and facilitating electron transfer. This significant electron interaction optimizes the adsorption energy of urea molecules and results in the decreased reaction energy barriers during the UOR. Experimental results revealed that this designed NiSe2@Ni5P4/NF demonstrated a higher current density of 500 mA cm−2 at a low potential of 1.402 V vs. RHE, with a small Tafel slope of 27.6 mV dec−1, as well as the excellent stability of 950 h at 100 mA cm−2. Zhang's group prepared Ce-doped Ni2P nanosheets through a simple hydrothermal and vapor-phase phosphorization method, which demonstrated excellent catalytic activities for both the HER and the UOR.161 A two-electrode urea-assisted electrolyzer utilizing the Ce-doped Ni2P nanosheets achieved a current density of 10 mA cm−2 at a cell voltage of 1.51 V, which is lower than the voltage required for overall water electrolysis (∼1.62 V). Other Ni-based catalysts, such as r-NiMoO4,162 Ni2P/CC,163 Ni–Mo nanotubes,164 Ni3Se2/MoO2@Ni12P5,165 NiO-CrOx,166 NiF3/Ni2P,167 Ni2P/NiMoP heterostructure,168 FeP4 nanotube@Ni–Co–P nanocage,169 and Ni3N/Ni0.2Mo0.8N,170 have also demonstrated compelling performance in the UOR.
The high-valence Ni3+ is commonly believed to show higher intrinsic activity towards the UOR. With respect to the UOR, the engineering of high-valence active sites can be achieved by hetero-element doping, alloying, and heterostructures. For example, the recently reported catalyst systems, such as Rh-doped NiFe-LDH,171 Ru-Co2P/N-C/NF,172 Fe, V doped NiS arrays,173 V doped FeNi3N/Ni3N,174 NiMoV LDH/NF with Mo and V dopants,175 V-Ni3N/NF,176 S-incorporated CoNiFe(oxy)hydroxides nanosheets,177 and F-doped Ni(OH)2,178 are all exhibit improved UOR performance. Wang et al. developed a W-doped NiO (Ni-WOx) catalyst, which demonstrated remarkably fast reaction kinetics and a high conversion frequency in urea oxidation, surpassing the performance of the catalysts without W elements.179 XPS and in situ X-ray absorption near edge spectra (XANES) analysis revealed that the presence of W dopants induced changes in the charge distribution around Ni atoms (Fig. 9a–c), facilitating the formation of Ni3+ active centers with excellent electrocatalytic activity during the reaction. Additionally, the electron transfer from Ni to W atoms resulted in the formation of electron-deficient and electron-rich regions, as depicted in Fig. 9d. These localized regions were beneficial for the adsorption of –NH2 and CO groups, respectively, thereby contributing to the intriguing UOR performance and faster reaction kinetics. Feng et al. synthesized NiS2–MoS2 hetero-nanorods through the sulfidation of NiMoO4 nanorods, aiming to achieve a high-valence Ni/Mo synergism for urea-assisted water electrolysis.180 The presence of Mo6+ induced the formation of Ni3+ species and facilitated electronic synergism between Ni and Mo. The resulting NiS2–MoS2 hetero-nanorods exhibited exceptional catalytic activity, stability, charge transfer ability, and catalytic kinetics compared to pure NiS2 and MoS2 phases. Through XPS analysis (Fig. 9e–g) after a stability test, it was observed that the aged sample showed an increased amount of Mo6+ and Ni3+, indicating the involvement of high-valence Mo6+ and Ni3+ species during the catalytic reaction. Besides, Ce-incorporated Ni2P nanosheets,161 N-doped carbon nanorod supported Ni2P,181 core–shell Ni(OH)2/NiO-C/WO3 hierarchical arrays,182 and Fe-doped Ni-based MOF nanosheet arrays183 were also recently reported to promote the Ni2+/Ni3+ conversion, thus accelerating the UOR kinetics. High-valence Co species are also favorable for the electrooxidation of urea.184 Jiao et al. synthesized Mo-doped cobalt carbonate hydroxide nanoarrays (CoxMoyCH) as catalysts for the UOR.185 Post-UOR XPS characterization and DFT calculations provided compelling evidence for the formation of high-valence Co3+ species within CoOOH because Co electrons transfer to Mo with the higher valence state, enhancing the conversion of Co2+ to Co3+. DFT calculations further confirmed that the incorporation of Mo sites promoted the activation and adsorption of urea on Co sites, owing to the changed electron density.
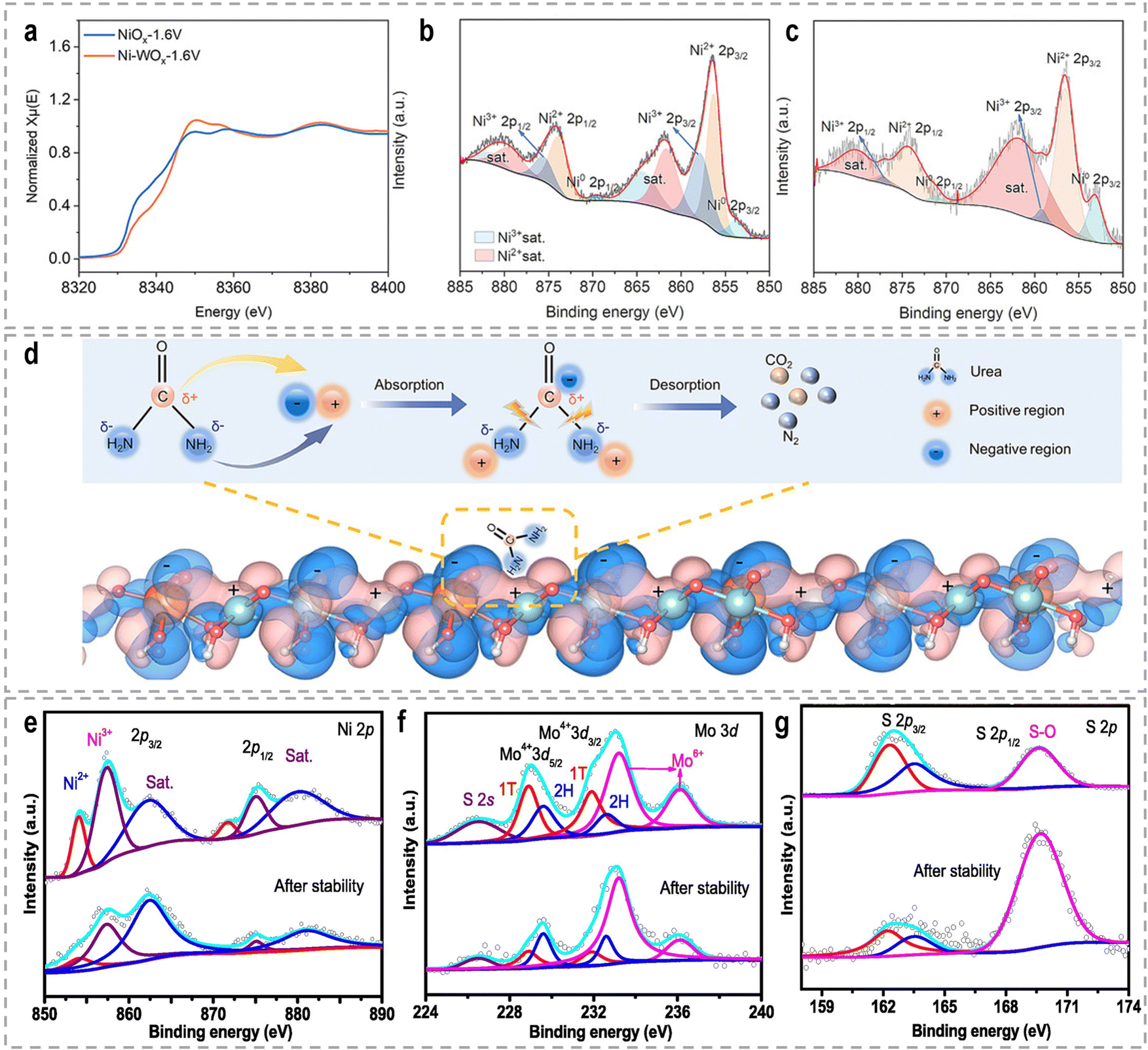 | ||
| Fig. 9 (a) Ni K-edge XANES of Ni-WOx-1.6 V and NiOx-1.6 V, respectively. (b) and (c) High-resolution Ni 2p XPS spectra of Ni–WOx and NiOx after the reaction, respectively. (d) Charge density difference for Ni–WOx from DFT calculations.179 Reproduced with permission. Copyright 2021, Wiley-VCH. XPS spectra of (e) Ni 2p, (f) Mo 3d, and (g) S 2p for the NiS2–MoS2 catalyst before and after the stability test.180 Reproduced with permission. Copyright 2022, Elsevier. | ||
The intricate nature of urea, encompassing both electron-donating amino (–NH2) and electron-withdrawing carbonyl (CO) groups, necessitates a comprehensive understanding of the interplay between these groups and the adsorption strength of urea. Such an understanding serves as a valuable foundation for designing and developing highly efficient UOR catalysts. To address this, a CoS2/MoS2 Schottky heterojunction was fabricated as a model catalyst to modulate urea molecule adsorption strength and activate its chemical bonds.109Fig. 10a–c show the experimental and theoretical findings, which reveal that the spontaneous electron transfer from CoS2 to MoS2 induces the formation of electrophilic and nucleophilic regions at the heterointerfaces. This intricate interplay facilitates the adsorption of electron-donating and electron-withdrawing groups, triggering the decomposition of urea and consequently enhancing UOR activity. Likewise, bimetallic CoMn/CoMn2O4 with a significant Mott–Schottky heterostructure exhibits analogous reaction mechanisms.110 In a study by Liu et al., a Co and V co-doped NiS2 (NCVS) catalyst was prepared and evaluated for UOR performance.186 The XPS results demonstrated a synergistic effect among the Ni, Co, and V elements. In situ electrochemistry Raman spectra (Fig. 10d) provided insights that the introduction of heteroatoms does not entirely alter the UOR pathway. Furthermore, in situ electrochemistry mass spectrometry isotope tracing experiments (Fig. 10e and f) confirmed the formation of N2 through urea intermolecular N–N coupling under the catalysis of metal sulfides. Anti-CO poisoning experiments highlighted the role of Co in expediting the oxidation of carbonaceous intermediate products and enhancing catalyst stability. Combining these findings with DFT investigations, the authors established that Ni, Co, and V elements perform as active sites, stabilizers, and catalytic promoters, respectively.
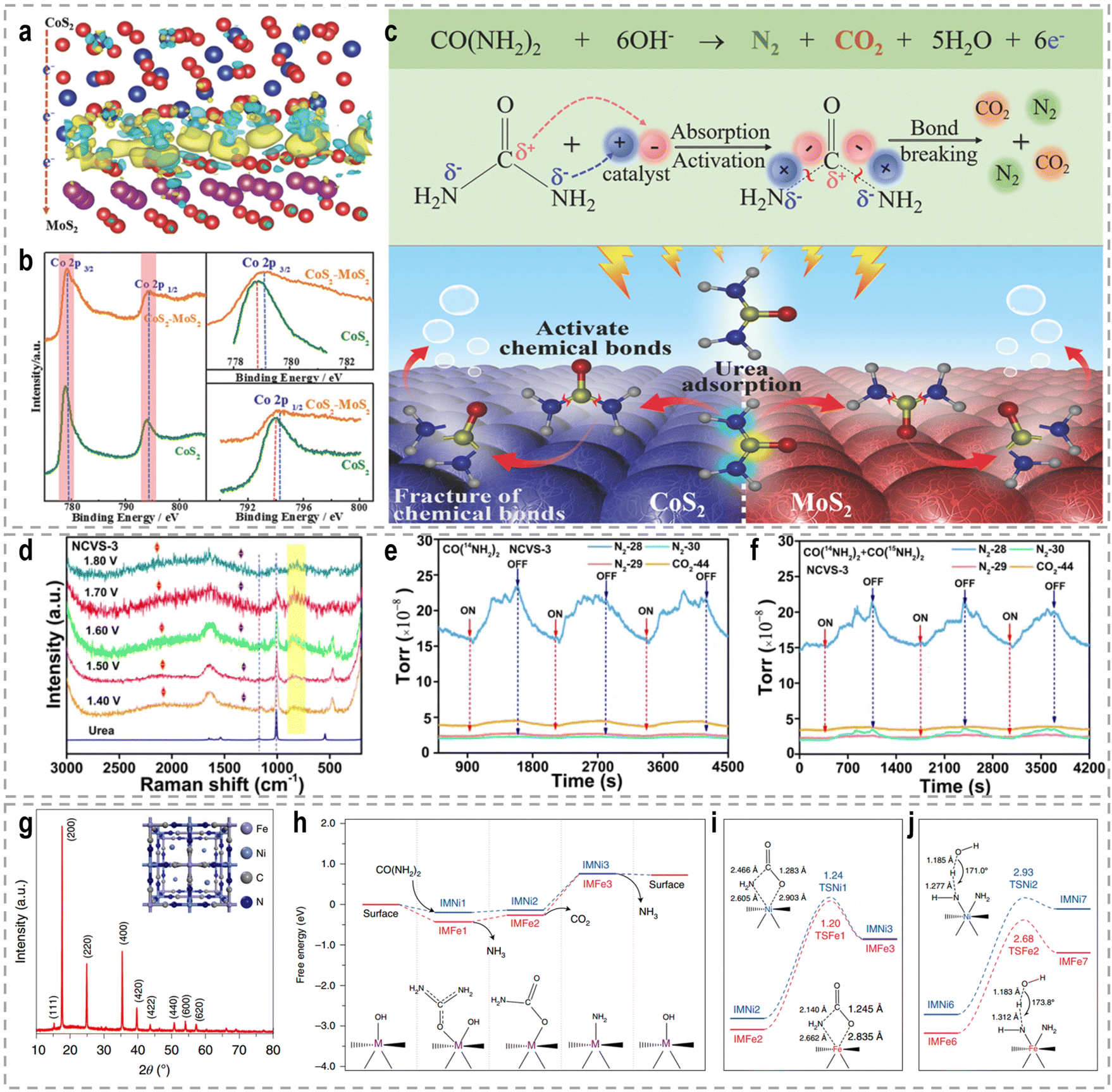 | ||
| Fig. 10 (a) The charge density difference in the heterostructure of CoS2 and MoS2. (b) XPS spectral comparison of CoS2–MoS2 and CoS2. (c) The schematic UOR catalytic mechanism using the CoS2–MoS2 Schottky catalyst.109 Reproduced with permission. Copyright 2018, Wiley-VCH. (d) In situ electrochemistry Raman spectra of the UOR by the NCVS-3 catalyst at various potentials from 1.40 to 1.80 V (vs. RHE). The in situ electrochemistry mass spectrometry isotope tracing experiment results for periodic measurement of the UOR in 1.0 M KOH with (e) 0.33 M urea [CO(14NH2)2] and (f) 0.33 M urea [CO(14NH2)2/CO(15NH2)2 = 4:1] under the catalysis of NCVS-3.186 Reproduced with permission. Copyright 2021, American Chemical Society. (g) XRD pattern with indexed peaks of the synthesized Ni2Fe(CN)6 catalyst. Inset: Schematic illustration of the catalyst. (h) The Gibbs free energy diagrams of the reaction from urea to NH3. (i) Comparison of both thermodynamically limiting and reaction determining step (RDS) of the first stage (reaction from urea to NH3) on the Ni site and on the Fe site. Inset: Bond lengths of the transition states for the CO2 dissociation reaction. (j) The comparison of both thermodynamically limiting and RDS of the second stage (from NH3 to N2) on the Ni site and on the Fe site.189 Reproduced with permission. Copyright 2021, Nature Publishing Group. | ||
The electrocatalytic oxidation of urea poses a complex mechanism involving a 6e−-transfer process, as well as multiple intermediate transfers and gas-desorption steps. Accurately understanding this reaction mechanism remains a prominent research area. Peng's group proposed a lattice-oxygen-involved pathway for the UOR by combining DFT, 18O isotope labeling mass spectrometry, and in situ FTIR.187 Their findings demonstrated the involvement of lattice oxygen in the conversion of *CO to CO2, thereby enhancing the reaction rate of the UOR. DFT calculations further revealed that lattice oxygen exhibits a preference for the Ni4+ active site in the UOR, exhibiting significantly faster reaction kinetics compared to the conventional pathway on the Ni3+ active site. Furthermore, Wang et al. investigated the electrooxidation behavior of urea using β-Ni(OH)2 as a model catalyst, unraveling the intramolecular coupling of the N–N bond during the UOR.188 Through isotope-labeled in situ DEMS, they provided compelling evidence that each N2 molecule originates from one urea molecule, indicating the intramolecular coupling process. And Co-doping was found to modulate the charge density distribution at the Ni atoms, lowering the energy barrier for the formation of the β-Ni(OH)O intermediate. This intermediate can undergo spontaneous nucleophilic dehydrogenative oxidation with a urea molecule, thereby accelerating the UOR reaction rate. Impressively, Qiao's group identified a two-stage reaction pathway for the UOR using a Ni2Fe(CN)6 catalyst (Fig. 10g), distinguishing it from conventional Ni-based materials that generate NiOOH as the active phase.189 The urea molecule is oxidized at the Ni site, forming intermediate CO2 and NH3 species, with NH3 subsequently decomposing to N2 at the Fe site. They employed an ion ammonia-selective electrode to confirm the generation of the NH3 intermediate during the UOR, and found that the concentration of NH3 in the electrolyte reached up to 0.30 ppm as the reaction time increased at an oxidation potential of 1.34 V. DFT calculations (Fig. 10h–j) further demonstrated the cooperative action of Ni and Fe sites enhanced the UOR rate. As a result, benefiting from the distinct rate-determining steps with more favorable thermal/kinetic energetics, Ni2Fe(CN)6 achieved an anodic current density of 100 mA cm−2 at a potential of 1.35 V.
3.2 Hydrazine oxidation reaction
Hydrazine (N2H4) is an important industrial raw material widely used in various applications. The hydrazine oxidation reaction (HzOR, N2H4 + 4OH− → N2 + 4H2O + 4e−, −0.33 V vs. RHE) plays a significant role in hybrid water electrolysis due to its lower theoretical voltage compared to many other anodic oxidation reactions., associated with the production of environmentally friendly products, such as H2O and N2, without producing toxic byproducts, catalyst-poisoning species, or greenhouse gases.190,191 Furthermore, the HzOR can be employed as an effective electrochemical approach to remove hydrazine from wastewater, addressing the environmental concern associated with hydrazine pollution. These advantageous features have stimulated extensive research on the HzOR for highly efficient hydrogen production in hybrid water electrolysis systems and wastewater treatment. However, the HzOR suffers from inherently sluggish kinetics due to intricate adsorption and desorption steps for intermediates. The design and optimization of catalysts aiming to improve the kinetics and efficiency of the HzOR will lead to more efficient and sustainable hydrogen production.In the field of hydrazine-assisted hydrogen production, various electrocatalysts have been developed, including both precious metals (such as Ru, Pd, and Pt) and non-precious metals (such as Fe, Co, Ni, and Cu), demonstrating their promising performance. Zhang et al. reported the use of partially exposed RuP2 nanoparticle-decorated N,P dual-doped carbon porous microsheets for both the HzOR and the HER in an alkaline environment.192 Remarkably, this catalyst achieved an ultrasmall working potential of 70 mV to reach 10 mA cm−2 for the HzOR (compared to 131 mV for 20 wt% Pt/C) in 1.0 M KOH + 0.3 M N2H4, and an extremely low overpotential of 24 mV at 10 mA cm−2 for the HER (compared to 35 mV for 20 wt% Pt/C) in 1.0 M KOH. Furthermore, the catalyst demonstrated a record-low cell voltage of 23 mV to achieve 10 mA cm−2 in a two-electrode system for overall hydrazine splitting (OHzS) (compared to 166 mV for 20 wt% Pt/C). It also exhibited an ultrahigh current density of 522 mA cm−2 at a low cell voltage of 1.0 V, significantly lower than that of conventional water splitting. Xia's group proposed a tubular CoSe2 nanosheet electrode as a bifunctional catalyst for efficient hydrogen production through hydrazine-assisted oxidation.193 The study revealed that the activity of the HzOR on CoSe2 nanosheets increased with increasing N2H4 concentration. Specifically, the as-prepared CoSe2 nanosheets demonstrated appreciable catalytic activity and strong durability in the HER and HzOR, with a current density of 10 mA cm−2 at −84 mV for the HER and −17 mV for the HzOR. Consequently, using the two-electrode hydrazine-assisted water electrolyzer utilizing CoSe2 nanosheets as a bifunctional catalyst, a current density of 10 mA cm−2 can be achieved at an ultralow cell voltage of 164 mV, much lower than that required for OWS. Moreover, the FE for H2 production reached 98.3%, demonstrating the complete utilization of electrons in the hydrazine-assisted HER process.
The strategies employed to enhance the electrochemical HzOR performance of metal-based electrocatalysts are similar to those used for the UOR. These efficient strategies including the utilization of single-atom configurations, doping with heteroatoms, constructing heterostructures, forming bifunctional active sites, and engineering hierarchical porous 3D structures have been widely reported for catalyst systems, for instance, Ru SAs on tungsten disulphide (WS2),194 Ru SAs on WO3,195 B-doped Co(OH)2 nanosheets on a Cu dendrite surface,196 CoP and N-doped Ni5P4,197 Mo doped Ni3N and Ni heterostructures,198 Cu4N/Ni3N heterostructures,199 N doped Ni1Co3Mn0.4O hybrids,200 porous Fe-doped Ni2P nanosheets,201 Ni0.6Co0.4Se/NF,202 P/Fe-NiSe2,203 and porous Ni2P hollow nanotubes.204
In the pursuit of efficient hydrazine-assisted water electrocatalysis, the development of bifunctional electrocatalysts capable of facilitating both the HzOR and HER has garnered significant attention. Zhang et al. demonstrated the construction of a P,W dual-doped Co3N nanowire array electrode, where DFT calculations revealed that P and W co-doping optimized the adsorption/desorption of hydrogen intermediates (H*) for the HER and the dehydrogenation kinetics for the HzOR.205 This work highlighted the importance of metal/nonmetal co-doping and the design of hierarchical structures in achieving enhanced bifunctional activity. Inspired by these groundbreaking findings, Wang's group developed a unique 2D/3D hierarchical structure comprising Fe, F co-doped Ni2P encapsulated by a N-doped carbon shell (Fe/F-Ni2P@NC).78 DFT calculations demonstrated that Fe and F co-doping optimized the electronic distribution and charge transfer behavior within the catalyst. Consequently, the Fe/F-Ni2P@NC catalyst exhibited remarkable bifunctional activity, achieving working potentials of 122 mV and 323 mV to attain a current density of 1000 mA cm−2 for the HzOR and HER, respectively, in an alkaline electrolyte. The OHzS test showed that Fe/F-Ni2P@NC requires a cell voltage of only 568 mV to reach 1000 mA cm−2, with excellent overall stability for 100 h above 100 mA cm−2 at the constant potential. This OHzS system saved 3.35 kW h for generating 1.0 m3 of H2 in comparison with a N2H4-free system. Additionally, Wang's group has successfully fabricated other doped catalyst systems such as Cu-doped CoFe/Co encapsulated by N-doped carbon,206 Fe/Co dual-doped Ni2P and MIL-FeCoNi heterostructure arrays,207 self-supporting Ru-doped FeP4 nanosheets,208 and Fe-doped Ni2P/CoP encapsulated by nitrogen-doped carbon layers.209 These catalysts have demonstrated promising HzOR and HER activity, further showing the potential of doped catalyst systems for efficient hydrazine-assisted water electrocatalysis.
At present, the production of H2 through water electrolysis is predominantly carried out using freshwater, which poses limitations in arid regions due to the scarcity of freshwater resources.210 To address this, electrolysis of seawater for H2 production has emerged as a promising alternative. However, the complex ionic environment of seawater presents challenges such as interference from anion side reactions and the corrosion of electrolyzers and catalysts. The competition between the chlorine electrooxidation reaction (ClOR) and the OER is a significant problem, as ClOR can lead to the release of toxic chlorine gas (Cl2) and corrosive hypochlorite (ClO−), reducing electrolysis efficiency and the lifespan of the electrolyzer. To resolve this tricky challenge, some unique tactics, such as designing selective OER catalysts and constructing Cl− blocking layers, have been proposed.211,212 Meanwhile the thermodynamically favorable oxidation reaction provides another approach to realize seawater electrolysis for hydrogen production. In the context of seawater electrolysis, the oxidation potential of the HzOR is much lower than that of the ClOR by 2.05 V (Fig. 11a), providing an advantage in avoiding chlorine-related issues without compromising the electrolysis current and H2 yield efficiency. Sun et al. explored a chlorine-free H2 production by employing hybrid seawater electrolysis (HSE) coupled with N2H4 degradation (Fig. 11b).129 They developed a self-supported NiCo/MXene-based catalyst (NiCo@C/MXene/CF) and investigated its performance in hydrazine-assisted seawater splitting in seawater electrolytes. The electrolyzer using the NiCo@C/MXene/CF catalyst achieved high activity, requiring ultralow voltages of 1.05 V to attain a current density of 500 mA cm−2 (Fig. 11c) in neutral seawater, which is 48% lower in terms of the energy input compared to commercial alkaline water electrolyzers. Importantly, this system avoided the generation of Cl2/ClO−, effectively mitigating the influence of the ClOR and enabling industrial-scale H2 production through seawater electrolysis. Furthermore, the HSE system can be integrated with photovoltaic cells powered by clean and readily available solar energy. The hybrid seawater electrolyzer, equipped with the NiCo@C/MXene/CF catalyst, could operate at a current density of ∼310 mA cm−2 and an average photovoltage of ∼0.876 V when connected to a single commercial solar cell (1.0 W). Under simulated solar illumination (AM 1.5G) with a power density of 100 mW cm−2, the system yielded hydrogen at a decent rate of 6.0 mol h−1 gcat−1 from seawater.
 | ||
| Fig. 11 (a) The Pourbaix diagram of the HzOR, HER, OER, and ClOR in artificial seawater with 0.5 M Cl− in pH 7–14. (b) The merits of HSE over ASE for energy-saving and chlorine-free hydrogen production. (c) The LSV curves of HSE using neutral or alkaline seawater as the catholyte, compared with ASE.129 Reproduced with permission. Copyright 2021, Nature Publishing Group. (d) Chronoamperometry curves of FeSA/CNT for HzOR under 0.77 V at 0.1 M N2H4 + 1.0 M KOH. DEMS analysis of N2 (m/z = 28) and NH3 (m/z = 15) (e). (f) The ratio of NH3 charge, Q(NH3) to N2 charge, Q(N2) against the electron transfer number of MSA/CNT for the HzOR. (g) Scheme for the two pathways of the HzOR by MSA/CNT.59 Reproduced with permission. Copyright 2021, Nature Publishing Group. (h) CVs on the Ni(Cu) CNP electrode during successive addition of different concentrations of hydrazine at a scan rate of 10 mV s−1 in a 1.0 M KOH solution; the inset shows the calibration curve of peak current versus the concentration of hydrazine. (i) CVs of the HzOR obtained with 10 mM hydrazine in 1.0 M KOH solution at different CV scan rates over Ni(Cu) CNPs. (j) Plot of HzOR peak potential Epversus log(v).217 Reproduced with permission. Copyright 2020, Royal Society of Chemistry. (k) CV curves of the HzOR obtained over G(CN)–Co (1.2 wt%), G-(CN)–Co(1.5 wt%), G(CN)–Co (3.4 wt%), and G(CN) using 50 mmol L−1 hydrazine in PBS (pH 7.4) at a scan rate of 10 mV s−1 and (l) the corresponding current density normalized to the total mass of Co atoms.218 Reproduced with permission. Copyright 2021, Wiley-VCH. | ||
Generally, in the oxidation of N2H4 in alkaline solution, several typical pathways (reactions (1)-(5)) can be followed to produce H2 and NH3.213
| N2H4 + 4OH− → N2 + 4H2O + 4e− | (1) |
| N2H4 + xOH− → N2 + (4 − x)/2H2 + xH2O + xe− 1 ≤ x ≤ 3 | (2) |
| N2H4 + OH− → 1/2N2 + NH3 + H2O + e− | (3) |
| N2H4 → N2 + 2H2 | (4) |
| 3N2H4 → N2 + 4NH3 | (5) |
For some polycrystalline metal catalysts like a ZrNi alloy,214 N2H4 can undergo complete electrochemical oxidation to N2 and H2O through a 4e− reaction (reaction 1), or it can be incompletely oxidized to N2 and H2 (reaction 2). Alternatively, the combination of N2 and H2 can be obtained via a fully chemical decomposition pathway (reaction 4), as observed with Pt catalysts.215,216 In addition, minor amounts of NH3 can be generated through incomplete dehydrogenation of N2H4via a 1e− electrochemical oxidation (reaction 3) or dissociative reaction (reaction 5). Wang et al. used on-line DEMS to analyze the gaseous products during HzOR catalyzed by transition metal SACs including FeSA/CNT, CoSA/CNT, and NiSA/CNT.59 Under open circuit voltage (OCV), the absence of ionic current density indicated the absence of gaseous products from the HzOR. However, upon applying an oxidation potential of 0.77 V, the ionic current density associated with N2 (m/z = 28) increased significantly, indicating the generation of N2 (Fig. 11d). Only trace amounts of NH3 (approximately 0.5 at%) were detected by DEMS during the HzOR. The content of N2 and NH3 increased with applied potential or reaction time under a fixed potential. Conversely, hydrogen was not detected under both OCV and applied potentials. Moreover, FeSA/CNT showed no activity for the electrochemical oxidation of NH3 and H2. Based on these findings, it can be concluded that the predominant formation of N2 and minor amounts of NH3 occurs during the electrochemical oxidation of N2H4, rather than through chemical decomposition reactions under ambient conditions. Similar reaction pathways were observed for CoSA/CNT and NiSA/CNT, along with dominant N2 and trace amounts of NH3 during the HzOR under applied potential, indicating similar behavior to FeSA/CNT. The calculated charge ratios of NH3 to N2 [Q(NH3)/Q(N2)] (Fig. 10e) were 0.126%, 0.141%, and 0.131% for FeSA/CNT, CoSA/CNT, and NiSA/CNT, respectively, showing a reverse correlation with the electron transfer number for the HzOR. In other words, a higher charge ratio of NH3 to N2 corresponds to a lower electron transfer number for the catalyst. The absence of mass peaks corresponding to intermediate oxygen species (m/z = 30, 44, and 46) in the DEMS detection (Fig. 11f) indicates the absence of these species during the HzOR. The absence of H2 during the HzOR rules out reactions (2) and (4), as the MSA/CNT catalysts showed no activity for hydrogen oxidation. Additionally, the increasing ammonia content with applied potential and reaction time under a fixed applied potential excludes the possibility of chemical decomposition of N2H4 to NH3via reaction (5). Therefore, the results suggest that the electrochemical oxidation of N2H4 on MSA/CNT primarily follows pathways involving reactions (1) with minor extent through reaction (3), as illustrated in Fig. 11g.
In addition to the previously mentioned pathways, evidence has also been found supporting continuous 2e− + 2e− reaction pathways in the HzOR.203 For example, Zhao et al. developed a Cu-doped Ni cubic nanopore (Ni(Cu) CNP) catalyst for the HzOR through an electrodeposition process and in situ electrochemical etching.217 They investigated the 2e− transfer mechanism of the HzOR using a double potential step chronopotentiometry test (Fig. 11h–j). The CV curves showed only oxidation peaks and no cathodic peaks during the reverse scans, confirming the irreversibility of the oxidation (or HzOR) on the Ni(Cu) CNP catalyst. Furthermore, kinetic studies revealed that increasing the electrochemical scan rates caused a slight shift towards positive peak potential in the catalytic HzOR. The number of electrons involved in the rate-determining step was estimated to be approximately 2.2, suggesting that the HzOR on the Ni(Cu) CNP electrode proceeds through a slow 2e− transfer process in the initial rate-determining step to form the diazene intermediate, followed by a fast 2e− process to generate the final product of N2. Similarly, Gawande et al. performed CV measurements (Fig. 11k and l) and determined the total number of electrons involved in the HzOR for tested single-atom Co-based catalysts to be close to 4.218 This indicates that the reaction proceeds through a 4e− process to fully oxidize hydrazine. These findings highlight the existence of both 2e− and 4e− transfer pathways in the HzOR, suggesting the involvement of different mechanisms and catalytic active sites in the electrochemical oxidation of hydrazine.
The hydrazine splitting process is typically considered a stepwise dehydrogenation process from N2H4 to N2, where the rate-determining step is often the dehydrogenation from N2H3* to N2H2* on transition metal-based catalysts.219 Impressively, Shi's group discovered a new N–N single bond breakage pathway during the HzOR using NiCoP–CoP heterostructures (Ni–Co–P/NF) as catalysts.220 They also found that the presence of hydrazine enables the instantaneous recovery of the metal phosphide active sites, ensuring robust HzOR activity during long-term operation. CV measurements on Ni–Co–P/NF in both KOH and 1.0 M KOH + 0.1 M N2H4 (Fig. 12a) showed that the in situ electrochemically oxidized Ni–Co–P/NF in the anodic potential range can be recovered back to active metal phosphide (MP) species again by N2H4. In situ Raman measurements were conducted to investigate the compositional and structural changes of the catalyst during the HzOR. The addition of 0.1 M N2H4 in the electrolyte led to the appearance of three strong N2H4-related peaks at 677, 1116, and 1598 cm−1 (Fig. 12b). These peaks correspond to N–H, N–N stretching modes of adsorbed *NH2NH2, and N–H bending modes of intermediate *NH2 on the Ni–Co–P/NF surface. Varying the applied voltages (Fig. 12c) resulted in a gradual weakening of the M-P peak density (400 cm−1) and the appearance of a weak peak near 1020 cm−1 attributed to the stretching bond of P–O in MPOx. This indicates that some MP species were gradually oxidized to MPOx during the HzOR. However, at a constant potential of 0.2 V and varying reaction time (Fig. 12d), the intensity of the M–P bond peak (400 cm−1) increased again, while the peak of the P–O bond (broad band at about 1020 cm−1) disappeared. This confirmed that the slightly oxidized species (MPOx) could be reduced and recovered back to active MP species in the presence of N2H4. Importantly, the peak of *NH2 became significantly stronger, indicating the accumulation of reaction intermediate *NH2. This suggests that the adsorbed *NH2NH2 breaks the N–N bond to form two *NH2 groups, which then gradually dehydrogenate to generate N2. DFT calculations (Fig. 12e) also supported this finding, showing that the N–N bond breaking in hydrazine molecules on the NiCoP–CoP heterostructures has a favorable reaction energy compared to the traditional stepwise dehydrogenation process.
 | ||
| Fig. 12 (a) CV curves in 1.0 M KOH (blue curves) or 1.0 M KOH + 0.005 M N2H4 obtained under different cycles of scanning of the Ni–Co–P/NF catalyst (red: first cycle, yellow: second cycle and green: third cycle). (b) Raman spectra of Ni–Co–P/NF in 1 M KOH or 1.0 M KOH + 0.1 M N2H4. In situ electrochemical Raman spectra of Ni–Co–P/NF in 1.0 M KOH + 0.1 M N2H4 (c) at varied applied potentials and (d) at different reaction time intervals under constant 0.2 V. (e) Free energy changes of the HzOR at the NiCoP(111)/CoP(011) interface with the most stable adsorption configuration of the intermediate.220 Reproduced with permission. Copyright 2023, Nature Publishing Group. | ||
3.3 Polysulfide oxidation reaction
Hydrogen sulfide (H2S) is a highly toxic pollutant released in large quantities from industries such as oil refineries and natural gas extraction.221 Electrochemical methods offer the potential for simultaneous H2 production and sulfur recovery at the cathode and anode, respectively, through the removal and utilization of this hazardous waste. Since H2S is a diprotic acid, a two-step ionization process of H2S to HS− and S2− should occur in an aqueous electrolyte. By replacing the OER with the sulfide oxidation reaction (SOR, Sx2− + S2−–2e− → Sx+12−), the theoretical voltage required for hydrogen production can be significantly reduced from 1.23 V to 0.17 V vs. RHE, and hence 86% electricity energy could be saved.222 However, the development of efficient hydrogen production systems based on the SOR is limited, primarily due to the strong poisoning effect of sulfur species on metallic electrocatalysts.223 Sulfur chemisorption and etching on the catalyst surface result in decreased activity and stability, posing challenges for industrial-scale applications. Therefore, it is crucial to develop low-cost, highly efficient, and robust electrocatalysts to enable sustainable electrocatalytic H2S decomposition and effective H2 production.Deng's group has developed a template-assisted method to fabricate an efficient electrocatalyst for simultaneous H2 and S production from H2S by coupling the HER and the SOR.223 The catalyst of CoNi@NGs consists of a non-precious CoNi nanoalloy enclosed in N-doped graphene. The developed CoNi@NGs exhibit an anode reaction onset potential of 0.25 V vs. RHE, which is 1.24 V lower than the onset potential required for the OER (Fig. 13a). This significant reduction in the required voltage demonstrates the effectiveness of the catalyst in catalysis of the SOR. Moreover, after a durability test lasting 500 h, the structure of the CoNi nanoparticles encapsulated in graphene shells remains well-maintained, indicating the catalyst's robustness in harsh environments. In situ electrochemical ultraviolet and visible (UV-Vis) spectrophotometry provides evidence for the formation of short-chain polysulfides (S22−–S42−) in the electrolyte during galvanostatic testing (Fig. 13b). Furthermore, after subjecting the electrolyte to acid treatment following long-term SOR testing, solid elemental sulfur powder can be obtained (Fig. 13c). This result confirms the successful conversion of H2S into sulfur through the electrocatalytic process. DFT calculations suggest that the high activity for the SOR can be attributed to the modulation of the graphene's electronic structure by the encapsulated metal alloy and nitrogen doping. This modulation facilitates the adsorption of S* species and the formation of polysulfide intermediates on the graphene's surface, leading to enhanced catalytic performance.
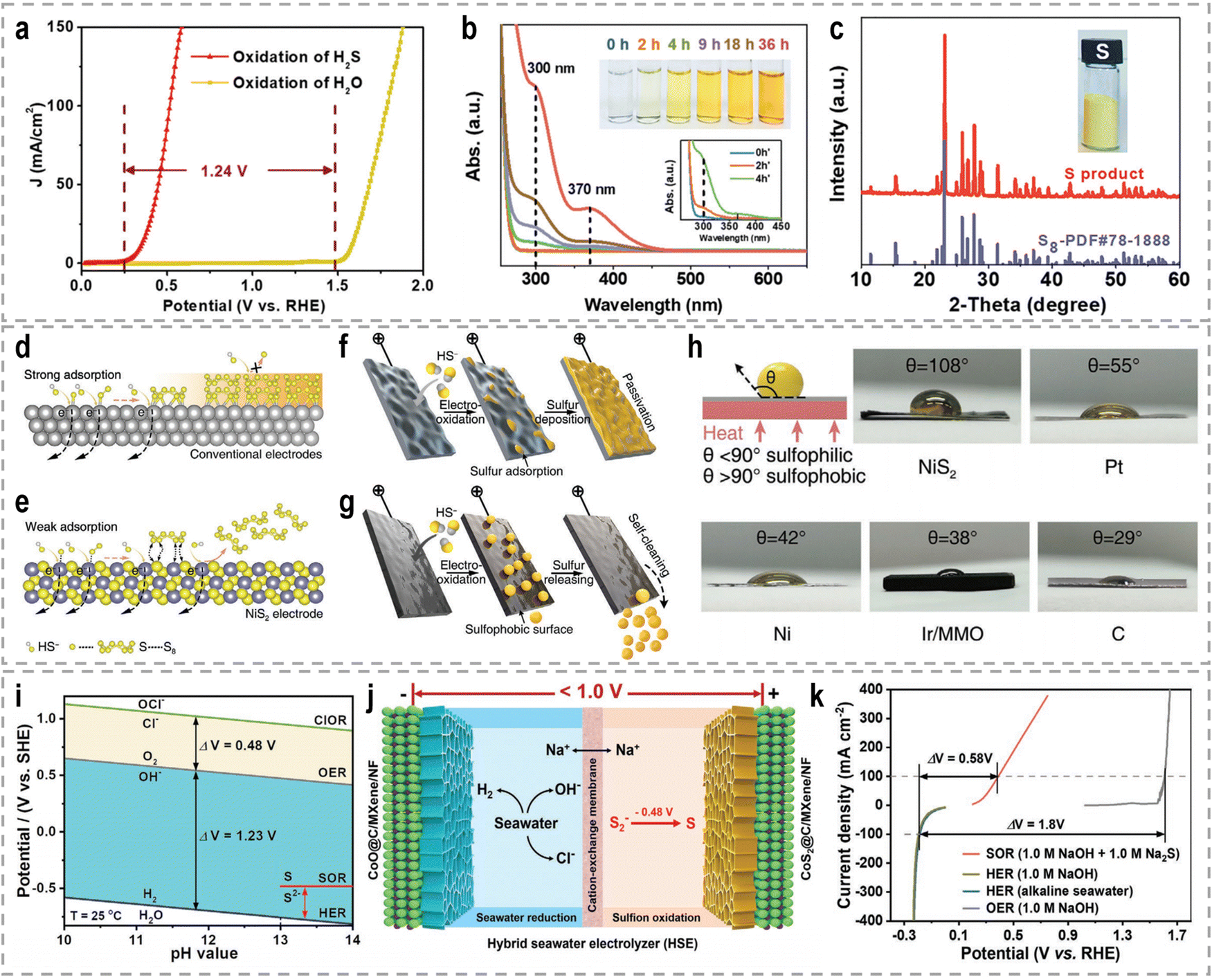 | ||
| Fig. 13 (a) Comparison of SOR and OER polarization curves for CoNi@NGs. (b) UV-Vis spectra of 250 times diluted electrolyte samples corresponding to the electrolytes in the inset photograph with the reaction proceeding in a galvanostatic test at 100 mA cm−2. (c) XRD characterization of product S. The inset photograph is the collected product of S powder.223 Reproduced with permission. Copyright 2020, Royal Society of Chemistry. Schematic illustration of sulfophobic design for desulfurization and hydrogen evolution. (d) Strong interaction between sulfides and electrodes leads to sulfur passivation whereas (e) weak interaction enables self-cleaning electrolysis. (f) Electrolytic sulfur passivates electrodes during long-term operation. (g) Sulfophobic NiS2 repels electrolytic sulfur, leading to a self-cleaning process for sulfur removal and hydrogen evolution. (h) Contact angle measurements of sulfur droplets (120 °C) on various substrates.226 Reproduced with permission. Copyright 2021, Wiley-VCH. (i) Pourbaix diagram of the SOR, HER, OER, and ClOR under alkaline conditions. (j) Schematic illustration of hydrogen production at cell voltages below 1.0 V by coupling seawater reduction with the SOR on Co-based electrocatalysts. (k) The voltage differences (ΔV) between the HER on CoO@C/MXene/NF and the SOR or the OER on CoS2@C/MXene/NF in different electrolytes.29 Reproduced with permission. Copyright 2022, Wiley-VCH. | ||
In accordance with the principles of the hard and soft acid and base theory, it is postulated that the lattice Cu(I) within the Cu2S electrode can be classified as a soft Lewis acid site.224,225 Consequently, it is expected to exhibit a higher affinity for binding with the soft-base species of HS− in the electrolyte rather than other hard or borderline acid sites. Inspired by this idea, Zhong et al. introduced a straightforward technique for the synthesis of Cu2S micro-flakes supported on nickel foam (Cu2S/NF).222 Benefiting from the synergistic effect of porous morphology, high electronic conductivity, and the enhanced sulfur adsorption on Cu(I), an anodic potential as low as 0.44 V vs. RHE was obtained to deliver 100 mA cm−2 for the SOR with high FE of exceeding 97%. Furthermore, a simple two-electrode system by utilizing Cu2S/NF as both the cathode and anode can enable a cell voltage of merely 0.64 V at 100 mA cm−2, which also exhibits a remarkable 74% reduction in energy consumption compared to conventional water splitting while generating the same quantity of hydrogen. This energy-saving efficiency is in close proximity to the theoretical energy-saving limit of 86%. The DFT calculations demonstrated that the strong adsorption of S on Cu2S can stabilize S* for chain growth. The impressive performance of Cu2S/NF can be attributed to the collective influence of several factors: (i) the inherent preference of Cu(I) for the HS− species in the electrolyte, (ii) the excellent electrical conductivity of the Cu2S/NF electrode, and (iii) the compatibility of Cu2S with sulfide electrolytes.
During the process of SOR, the formation of elemental sulfur as an electro-oxidized product leads to the passivation of the electrode surface, resulting in increased energy consumption and rendering continuous operation impractical. Moreover, the unresolved issues pertaining to interfacial chemistry contribute to the accumulation of sulfur particles within the system, inevitably diminishing the long-term activity of the electrodes. Zhang et al. recently made a significant discovery by observing a sulfophobic phenomenon (low affinity to elemental sulfur) having a weak interaction between transition metal disulfides and elemental sulfur (Fig. 13d–g).226 Utilizing sulfophobic properties, they designed a self-cleaning NiS2 electrode for highly efficient electrochemical desulfurization. Experimental analyses confirmed that NiS2 exhibits considerably lower chemical affinity to sulfur compared to conventional desulfurization electrodes such as Pt, Ni, Ir/MMO, and carbon. This was intuitively demonstrated through contact angle measurements, where NiS2 displayed a significantly higher contact angle (108°) compared to the other electrodes (Fig. 13h). Theoretical studies further revealed that the presence of more reducing Ni–S bonds facilitates electron transfer from sulfur species to catalysts, thereby enabling self-cleaning electrolysis and preventing voltage fluctuations during desulfurization. Thanks to the de-wetting properties of NiS2, passivation caused by solid sulfur deposition is circumvented, allowing for long-term self-cleaning desulfurization. When coupled with sulfur vacancy engineered NiS2 (v-NiS2) as the HER catalyst, the hybrid electrolyzer exhibited a low cell voltage of only 0.65 V to achieve 20 mA cm−2, showing efficient desulfurization and simultaneous hydrogen production. Impressively, the system demonstrated excellent voltage stability over 100 h.
The utilization of redox polysulfides (Sx+12−/Sx2−) as effective mediators in certain devices enables the maintenance of excellent redox cyclability to storage/release electrons through appropriate electrode catalysis. Taking advantage of this phenomenon, Zhang et al. developed a high-performance device by employing polysulfides as mediators and utilizing graphene-encapsulated CoNi nanoparticles (CoNi@NGs) as catalysts, achieving highly efficient conversion of surplus electricity into hydrogen energy.227 The operation mechanism of this decoupled electrolyzer involves two distinct steps. During Step 1, referred to as the “valley time” typically occurring at night, a cathodic polysulfide reduction reaction (SRR, Sx+12− + 2e− → Sx2− + S2−) takes place, accompanied by the anodic oxidation of OH− for the OER leading to O2 production. In the subsequent Step 2, known as the “peak time” typically occurring during the daytime, the HER occurs at the cathode through the reduction of H2O, while simultaneously, the SOR occurred simultaneously at the anode. Consequently, this device provides a promising strategy for peak load regulation in electricity. By utilizing the efficient catalysis of CoNi@NGs, the developed device demonstrated significant hydrogen production with low energy consumption. Furthermore, the device exhibited remarkable cyclability in the extensive recycle tests in 15 day without any performance decay.
Specifically, compared to the ClOR, the oxidation potential of the SOR can be dramatically reduced by 1.3–1.4 V (Fig. 13i). This reduction in potential offers the opportunity to completely circumvent the hazardous chlorine chemistry in seawater electrolysis, while simultaneously greatly reducing energy expenses. Hence, integrating the SOR with seawater electrolysis holds the potential for hydrogen production with added economic and environmental benefits, as shown in Fig. 13j. To address this, Wang's group developed CoS2 nanoparticles in a carbon matrix on MXene/NF (CoS2@C/MXene/NF) for the SOR(Fig. 13k).29 The utilization of an asymmetric hybrid seawater electrolyzer employing CoS2@C/MXene/NF catalysts enables operation at a low cell voltage ranging from 0.68 to 0.83 V to achieve the current densities of 200–400 mA cm−2, cutting by 56–64% compared to alkaline seawater electrolyzers (1.85–1.9 V). Importantly, the reduced oxidation potential eliminates the occurrence of the ClOR and the subsequent release of ClO−, fully avoiding the anode corrosion regardless of Cl− crossover to the anode side. Consequently, stable electrolysis can be sustained for over 180 h below 0.97 V (without iR correction) at 300 mA cm−2, in stark contrast to the rapid failure of overall water splitting observed within 30 h with a huge cell voltage of 2.45 V. DFT calculations revealed that the CoS2 (100) facet exhibits the lowest energy barrier for the rate-limiting step in the SOR, compared to the (111) and (110) facets. Furthermore, the presence of MXene coating contributes to a large contact angle for sulfur molecules (110.6°) and high conductivity (∼5600 S cm−1), effectively accelerating the reaction kinetics in the SOR.
The oxidation of polysulfides offers a promising approach for both the reutilization of toxic sulfion waste and the highly efficient production of hydrogen. However, the catalytic performance for the low-potential electrooxidation of polysulfides still faces challenges and remains inadequate. Despite the potential benefits of polysulfide oxidation, research on its practical application is currently limited. Further investigations are necessary to overcome the reaction barriers and develop efficient and practical strategies for polysulfide oxidation. One of the key challenges in polysulfide oxidation is the lack of efficient electrocatalysts, particularly bifunctional electrocatalysts that can simultaneously promote the oxidation of polysulfides and the HER. In addition to catalyst development, a comprehensive understanding of the underlying reaction mechanisms is essential for optimizing the performance of polysulfide oxidation processes. Elucidating the complex reaction pathways and identifying the key intermediates and reaction kinetics will enable the rational design and optimization of catalysts and reaction conditions. By addressing these challenges, it will be possible to enhance the catalytic performance, improve energy efficiency, and enable the practical application of polysulfide oxidation for various energy-related processes, including the reutilization of sulfion waste and the production of hydrogen.
In summary, benefitting from the ultralow thermodynamic equilibrium potential and the production of harmless by-products, as reported in the available literature shown in Table 4, the urea/hydrazine/polysulfide-assisted hybrid water electrolysis holds promise for replacing conventional water electrolysis. Remarkably, due to their notably lower reaction potentials, these hybrid electrolysis processes can also be carried out using seawater as the electrolyte, offering an attractive avenue for low-cost hydrogen production. Although the oxidation products of the UOR, HzOR, and SOR may have limited commercial value, they offer an alternative to alleviate the significant environmental pollution and eutrophication of water resources. For instance, Wang's group demonstrated that direct electrolysis of human urine yielded only slightly lower performance compared to when urea molecules were added to an alkaline electrolyte.171 Qiu's group discovered that rapid hydrazine degradation to around 3 ppb can be achieved through the HzOR.129 Zhang et al. achieved the electrocatalytic selective removal of H2S from simulated industrial syngas (49% CO, 49% H2 and 2% H2S).223 These typical examples illustrate that the UOR or the HzOR or the SOR provides viable approaches for treating urine and hydrazine-containing wastewater, even at trace amounts, resulting in both high-efficiency hydrogen production and waste degradation.
| Catalyst | Coupling reactions | Electrolyte | Overpotential at 10 mA cm−2 for cathode (mV) | Tafel slope for cathode (mV dec−1) | Potential at 10 mA cm−2 for anode (vs. RHE) | Tafel slope for anode (mV dec−1) | Cell voltage at 10 mA cm−2 | FE | Stability | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| O-NiMoP/NF | HER + UOR | 1.0 M KOH + 0.5 M urea | 54 | 49 | 1.41 V at 100 mA cm−2 | 34 | 1.55 V at 50 mA cm−2 | Cathode: 96.8% | 10 h at 20 mA cm−2 | 30 |
| 20% Pt/C(−)//Ce–Ni3N@CC(+) | HER + UOR | 1.0 M KOH + 0.5 M urea | — | — | 1.31 V | 31.1 | 1.34 V | Cathode: 100% | 15 h at 40 mA cm−2 | 81 |
| CoS2–MoS2 | HER + UOR | 1.0 M KOH + 0.5 M urea | — | — | 1.29 V | 32 | 1.29 V | — | 60 h at 10 mA cm−2 | 109 |
| CoMn/CoMn2O4 | HER + UOR | 1.0 M KOH + 0.5 M urea | 69 | 90 | 1.32 V | 38 | 1.51 V | — | 60000 s at 100 mA cm−2 |
110 |
| P-CoNi2S4 | HER + UOR | 1.0 M KOH + 0.5 M urea | 135 | 65 | 1.306 V | — | 1.402 V | — | 100 h | 159 |
| Ni–Mo nanotube | HER + UOR | 1.0 M KOH + 0.1 M urea | 44 | 55 | 1.36 V | 22 | 1.43 V | Cathode: 98.7% | 24 h at 10 mA cm−2 | 164 |
| NiF3/Ni2P@CC | HER + UOR | 1.0 M KOH + 0.33 M urea | 121 | 75 | 1.36 V | 33 | 1.83 V at 50 mA cm−2 | — | 10 h at 10 mA cm−2 | 167 |
| Ni3N/Ni0.2Mo0.8N/NF | HER + UOR | 1.0 M KOH + 0.5 M urea | 55 | 54 | 1.328 V | 17 | 1.348 V | Cathode: 100%; anode: 100% | 500 h at 10 mA cm−2 | 170 |
| NiFeRh-LDH | HER + UOR | 1.0 M KOH + 0.33 M urea | 24 | 27 | 1.346 V | 35 | 1.344 V | Cathode: 100%; | 62 h at 1.47 V | 171 |
| Ru–Co2P/N–C/NF | HER + UOR | 1.0 M KOH + 0.5 M urea | 65 | 65 | 1.351 V at 50 mA cm−2 | 60 | 1.58 V at 100 mA cm−2 | — | 20 h at 1.60 V | 172 |
| V-FeNi3N/Ni3N | HER + UOR | 1.0 M KOH + 0.33 M urea | 62 | 108.2 | 1.382 V | 29.6 | 1.46 V | — | 25 h at 50 mA cm−2 | 174 |
| NiMoV LDH/NF | HER + UOR | — | — | — | 1.40 V at 100 mA cm−2 | 24.29 | ∼1.63 V at 100 mA cm−2 | — | — | 175 |
| V-Ni3N/NF | HER + UOR | 1.0 M KOH + 0.5 M urea | 83 | 45 | 1.361 V | — | 1.416 V | — | 200 h at 10 mA cm−2 | 176 |
| Ni(OH)2/NiO-C/WO3 HAs | HER + UOR | 1.0 M KOH + 0.5 M urea | 53 | 92 | 1.340 V | 19 | 1.370 V | — | 60 h at 20 mA cm−2 | 182 |
| FeNi-MOF NSs | HER + UOR | 1.0 M KOH + 0.33 M urea | 288 mV at 100 mA cm−2 | 96.9 | 1.361 V | 28.0 | 1.431 V | — | 10 h at 10 mA cm−2 | 183 |
| Co2Mo0.2CH | HER + UOR | 1.0 M KOH + 0.33 M urea | 82 | 133 | 1.33 V | 32 | 1.40 V | — | 40 h at 10 mA cm−2 | 185 |
| Ni3N–Co3N | HER + HzOR | 1.0 M KOH + 0.1 M N2H4 | 43 | 35.1 | −88 mV | 21.6 | 71 mV | Cathode: 100% | 20 h at 0.14 V | 37 |
| Fe/F–Ni2P@NC | HER + HzOR | 1.0 M KOH + 0.5 M N2H4 | 212 mV at 100 mA cm−2 | 105 | 100 mA cm−2 at 12 mV | 64.4 | 0.568 V at 1000 mA cm−2 | Cathode: 100%; anode: 100% | 100 h at about 100 mA cm−2 | 78 |
| CoSe2 nanosheets | HER + HzOR | 1.0 M KOH + 0.5 M N2H4 | 84 | 84 | −17 mV | — | 164 mV | Cathode: 98.3% | 14 h at 10 mA cm−2 | 193 |
| WS2/Ru SAs | HER + HzOR | 1.0 M KOH + 0.5 M N2H4 | 32.1 | 53.2 | −74 mV | 42.2 | 15.4 mV | Cathode: 100% | 100 h at 10 mA cm−2 | 194 |
| N-Ni5P4@CoP/CFP | HER + HzOR | 1.0 M KOH + 0.1 M N2H4 | 56 | 63 | −32 mV | 24.9 | 0.037 V | — | 10 h at 10 mA cm−2 | 197 |
| Mo–Ni3N/Ni/NF | HER + HzOR | 1.0 M KOH + 0.1 M N2H4 | 45 | 45 | −0.3 mV | 48 | 55 mV | Cathode: 100%; anode: 100% | 10 h at 50 mA cm−2 | 198 |
| Cu1Ni2–N | HER + HzOR | 1.0 M KOH + 0.5 M N2H4 | 71.4 | 106.5 | 0.5 mV | 44.1 | 0.24 V | Cathode: 95% | 75 h at 10 mA cm−2 | 199 |
| PW–Co3N NWA/NF | HER + HzOR | 1.0 M KOH + 0.1 M N2H4 | 41 | 40 | −55 mV | 14 | 28 mV | Cathode: 96%; anode: 96% | 20 h at 0.098 V | 205 |
| Ru–FeP4/IF | HER + HzOR | 1.0 M KOH + 0.5 M N2H4 | 110 mV at 100 mA cm−2 | 51.58 | 27.0 mV at 100 mA cm−2 | 115.94 | 0.90 V at 1000 mA cm−2 | Cathode: 100%; anode: 100% | 80 h at 100 mA cm−2 | 208 |
| Ru–Cu2O/CF | HER + HzOR | — | 31 | 50 | −41 mV | 34 | 17.4 mV | — | 18 h at 0.03 V | 228 |
| Ni–C HNSA | HER + HzOR | 1.0 M KOH + 0.1 M N2H4 | 37 | 28.7 | −20 mV | 16.2 | 0.14 V at 50 mA cm−2 | Cathode: 100% | 30 h at 0.14 V | 229 |
| Ni2P/NF | HER + HzOR | 1.0 M KOH + 0.5 M N2H4 | 290 mV at 200 mA cm−2 | — | −25 mV at 50 mA cm−2 | 55 | 1.0 V at 500 mA cm−2 | Cathode: 100% | 10 h at 100 mA cm−2 | 230 |
| Ni-SN@C | HER + HzOR | 1.0 M KOH + 0.1 M N2H4 | 28 | 39 | 16.8 mV | 21 | 0.366 V | Cathode: 100% | 24 h | 231 |
| CoO@C/MXene/NF(−)//CoS2@C/MXene/NF(+) | HER + SOR | 1.0 M NaOH + 1.0 M Na2S | 232 mV at 500 mA cm−2 | 64.5 | 389 mV at 100 mA cm−2 | 60.9 | 0.97 V at 300 mA cm−2 | Cathode: 96%; anode: 80% | 180 h at 300 mA cm−2 | 29 |
| Cu2S/NF | HER + SOR | 1.0 M NaOH + 1.0 M Na2S | 180 | — | 260 mV | 68 | 0.64 V at 100 mA cm−2 | Cathode: 97% | 140 h at 10 mA cm−2 | 222 |
| v-NiS2//NiS2 | HER + SOR | 0.1 M NaOH + 50 mM Na2S | 150 mV at 145.4 mA cm−2 | 83 | 410 mV | 104 | 0.64 V at 20 mA cm−2 | Cathode: 96.5% | 100 h at 20 mA cm−2 | 226 |
| CoNi@NGs | HER + SOR | 1.0 M NaOH + 0.5 M Na2S2 | — | — | — | — | 0.82 V at 100 mA cm−2 | Cathode: >98% | 15 h at 100 mA cm−2 | 227 |
| WS2 NSs | HER + SOR | 1.0 M NaOH + 1.0 M Na2S | 214 | 64.9 | 480 mV | — | — | Cathode: 99.22% | 192 h at 1.3 V | 232 |
| Co3S4 | HER + SOR | 1.0 M NaOH + 1.0 M Na2S | 193 mV at 100 mA cm−2 | 86.4 | 262 mV at 100 mA cm−2 | 47.9 | 0.496 V at 100 mA cm−2 | Cathode: 95% | 25 h at 100 mA cm−2 | 233 |
While sacrificial-agent-assisted water electrolysis presents a promising avenue for hydrogen production, several challenges still need to be addressed in this field. (i) Maintaining a constant supply of sacrificial agents at an industrial scale can be challenging, potentially limiting the scalability of the process, especially for large-scale hydrogen production applications. (ii) The anodic products of certain sacrificial agents, such as urea and hydrazine, are gases, which can affect the purity of the hydrogen gas produced. This may necessitate the use of membrane-contained electrolyzers, increasing the overall cost of hydrogen production. (iii) Some reaction intermediates, such as CO, may be generated during these anodic reactions, potentially leading to catalyst deactivation to varying degrees. (iv) Harsh testing conditions (strong acid, strong base or high concentration salt) may result in the shortened lifetime of equipment and an increased cost for the entire system. (v) The diversity of possible intermediate products during these anodic reactions can make it challenging to fully elucidate the reaction mechanisms. Unwanted side reactions or incomplete conversion of sacrificial agents can reduce the overall efficiency of the process. To further advance the field, it is essential to investigate the structure–activity relationships of designed electrocatalysts, with a specific focus on understanding the catalytic mechanisms of the UOR/HzOR/SOR in relation to transition metal-based catalyst orientations of high-valence metal species. Such investigations will contribute to the ongoing development of improved electrocatalytic materials and enhance our understanding of the underlying processes involved in sacrificial-agent-assisted water electrolysis.
4. Organic upgrading-assisted H2 production
Currently, various small molecule compounds, such as alcohols, glucose/xylose, aldehydes, carbohydrates, and primary amines, have been extensively investigated as anodic substrates for electrochemical synthesis reactions. These compounds can undergo oxidation at the anode, where hydroxyl or aldehyde groups are converted to carboxyl groups, without generating molecular oxygen. Simultaneously, the donated electrons participate in water reduction at the cathode, producing molecular hydrogen. The oxidation reactions of these small organic molecules offer several advantages.234–236 Firstly, they exhibit significantly reduced anodic potentials compared to the OER, making them more favorable for practical electrochemical applications with lower required electrolysis voltage. This reduction in the required voltage enables more energy-efficient electrochemical processes and contributes to overall system efficiency. Secondly, the absence of molecular oxygen generation eliminates the risk of potential explosions, improving the safety of the electrochemical system. Thirdly, electrooxidation reactions at the anode can generate valuable intermediates that serve as building blocks for further chemical transformations or synthesis routes (Fig. 14). Additionally, anode electrooxidation reactions can offer high selectivity for specific products, allowing for tailored synthesis and controlled transformations of organic molecules. Hence, these electrooxidation reactions hold great promise for practical applications and have attracted considerable interest among researchers. In this section, we primarily focus on elucidating the anode electrooxidation reactions, encompassing reaction types, representative non-noble metal catalysts, catalytic performance evaluations, reaction mechanisms, and the design and construction of electrolyzers. | ||
| Fig. 14 The blueprint of hydrogen production coupled with the electrochemical upgrading of organics. | ||
4.1 Alcohol oxidation reaction
The exploration of alcohol oxidation reactions has gained significant attention due to their potential applications in direct fuel cells and synthetic chemistry. In recent years, several studies have reported various alcohol oxidation reactions as alternatives to the anodic OER in water splitting.237,238 These alternative reactions not only lower the electrolysis voltage required for hydrogen production but also enable the simultaneous generation of value-added chemicals at the anode. In the past few years, the coupling of these alcohol oxidation reactions with the HER has demonstrated successful conversions, including the generation of formate from methanol,239 acetate240/ethyl acetate241 from ethanol, benzaldehyde242/benzoic acid243,244 from benzyl alcohol, glycerate/oxalate from glycerol,245 acetone from 2-propanol,246 acrylate from 1,3-propanediol,247 glycolic acid from ethylene glycol,248 gluconic acid/gluconolactone from glucose,249 and cyclohexanone from cyclohexanol.250 These studies have achieved notable efficiency, showing the potential of alcohol oxidation reactions for practical and sustainable applications in direct fuel cells, energy storage, and synthetic chemistry.Non-Pt-based catalysts offer the potential for low-cost alternatives and improved stability and resistance to CO poisoning. The typical transition-metal-based electrocatalysts for the MOR are Co/Ni-based materials, for instance, CoxP@NiCo-LDH,256 Mo-modified Co4N nanoarrsys,257 NiCo/N-CNFs nanofibers,258 Co3O4–Ni3S4–rGO,259 CNFs@NiSe,260 h-NiSe/CNTs,261 NR-Ni(OH)2,262 defects-rich Ni3S2-CNFs,263 Ni97Bi3 aerogel,264 bimetallic NiSn nanoparticles,265 Fe–Ni nanoparticles,266 Ni(OH)2 nanosheets,267 VO-rich NiO nanosheets,268 NiCu@Cu,269 Pd/Ni(OH)2/N-rGO,270 Ni–Cu alloys,271 and Cu/NiCu NWs.272 Xiang et al. prepared cobalt hydroxide@hydroxy sulfide nanosheets on carbon paper (Co(OH)2@HOS/CP) as one of the promising catalysts for the electrooxidation of CH3OH into formate.273 Benefiting from the optimization of the composition, surface properties, electronic structure, and chemistry of Co(OH)2, Co(OH)2@HOS/CP exhibited impressive performance, delivering a current density of 10 mA cm−2 at potentials of 1.461, 1.397, 1.385, and 1.361 V vs. RHE with increasing methanol concentrations from 0.4 to 6.0 M (Fig. 15a). Notably, when Co(OH)2@HOS/CP was employed as both the anode and cathode catalysts for the concurrent production of hydrogen and formate, an ultralow cell voltage of 1.497 V was achieved to reach 10 mA cm−2 (Fig. 15b), which is 134 mV lower than that required for OWS. And this hybrid system demonstrated a remarkable FE of 100% and remarkable durability for continuous 20 h of operation without obvious decay. In another study, Wang et al. fabricated NiIr-based MOF nanosheet arrays on Ni foam (NiIr-MOF/NF) with abundant active sites for the methanol-assisted production of hydrogen.274 The synergistic effects of electronic modulation and unique morphology characteristics endow NiIr-MOF/NF with exceptional electrochemical activity. Remarkably, the two-electrode system employing NiIr-MOF/NF as a bifunctional catalyst achieved a remarkably low cell voltage of 1.39 V to obtain 10 mA cm−2, which is 170 mV lower than that required for the traditional OWS, as well as a high FE approaching 100% at the cathode for formate conversion and at the anode for H2 production in 1.0 M KOH + 4.0 M methanol.
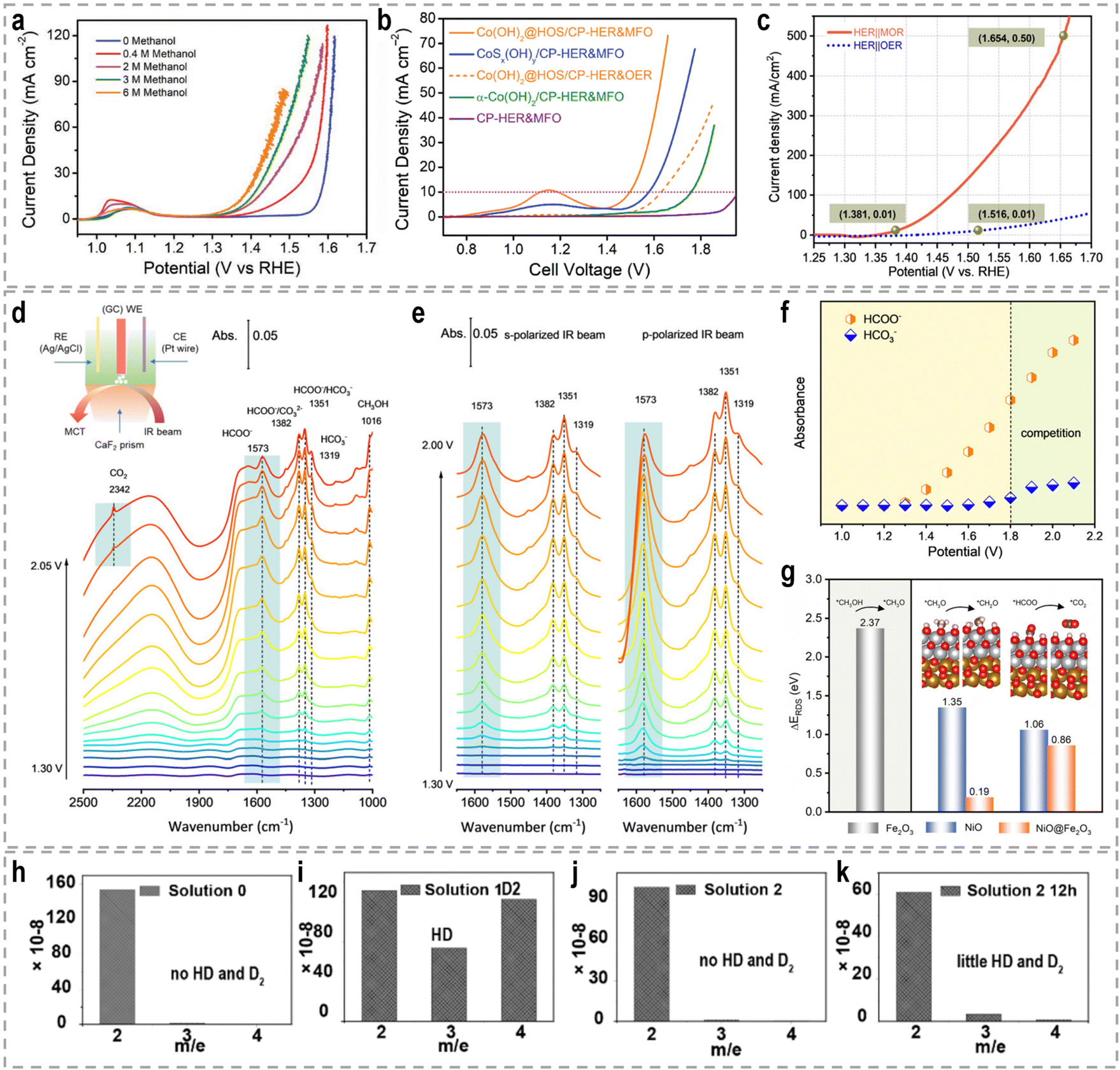 | ||
| Fig. 15 (a) The polarization curves of Co(OH)2@HOS/CP with different concentrations of methanol in 1.0 M KOH. (b) LSV curves of the as-obtained electrocatalysts in 1.0 M KOH with and without methanol in a two-electrode system.273 Reproduced with permission. Copyright 2020, Wiley-VCH. (c) LSV curves of Fe2O3/NiO/NF and MoNi4 catalysts in 1.0 M KOH with and without 1.0 M methanol electrolyte. (d) In situ infrared reflection absorption spectroscopy (IRRAS) of the MOR on the catalyst surface in the electrolyte of 1.0 M KOH with 1.0 M methanol at different potentials. (e) In situ IRRAS under a p-polarized and s-polarized IR beam in the wave number range from 1250 to 1650 cm−1. (f) The detailed variations of HCO3−1 and HCOO−1 with potential. (g) The pivotal potential gaps of the RDS during the MOR process for NiO, Fe2O3, and Fe2O3/NiO.32 Reproduced with permission. Copyright 2023, Royal Society of Chemistry. MS analyses of the produced gases from (h) solution 0 (H2O solution of 1.0 M KOH and 3.0 M Me–OH), (i) solution 1 (1.0 M KOH and 3.0 M Me–OH dissolved in D2O), (j) solution 2 (1.0 M KOH and 3.0 M D3COD dissolved in H2O) in the first hour, and (k) solution 2 in 12 h of chronoamperometry measurement at 1.4 V vs. RHE.283 Reproduced with permission. Copyright 2018, American Chemical Society. | ||
Among the non-precious electrocatalysts employed for the MOR, Ni-based materials have attracted significant attention. However, their performance in terms of current density at moderate potentials is still a challenge, primarily due to the difficulties in the dissociation of hydrogen protons.271 Furthermore, the excessive adsorption of reaction intermediates hinders the availability of catalytic active sites, thus limiting the activity and long-term stability of catalysts.275 Consequently, the selective conversion of methanol to formate at higher voltages becomes difficult, impeding the generation of hydrogen at large current densities. Previous investigations have primarily focused on the role of higher valent Ni3+ species as critical active sites in the selective oxidation of methanol to formate.276,277 Various strategies, including surface engineering, structural modification, alloying with other metals, and elemental doping, have been explored to control the density of Ni3+ active sites, aiming to enhance the performance of the MOR at larger current densities.278,279 Recently, Peng et al. proposed a novel approach by developing highly dispersed heterojunctions of FeNi oxide, wherein high valence state Ni3+ species and abundant Ni–O–Fe interfaces were anchored onto nickel foam (Fe2O3/NiO/NF).32 This innovative catalyst exhibited exceptional performance in the MOR, manifesting an onset potential of 1.328 V vs. RHE. Impressively, Fe2O3/NiO/NF achieved an absolute current density of 500 mA cm−2 at 1.654 V (Fig. 15c) with a remarkable FE > 98% in a practical dual-electrode, membrane-free electrolyzer. In-depth investigation through in situ infrared spectroscopy (Fig. 15d–f) and theoretical calculations (Fig. 15g) unveiled that the heterostructure of Fe2O3/NiO/NF effectively modulates the electronic state of NiO via robust electronic interactions, thus creating synergistic active sites that facilitate the desirable dynamic conversion of methanol to formate while impeding further oxidation. Moreover, the interface confinement effect plays a crucial role in stabilizing the metastable nickel active site, ensuring the structural stability of the catalyst during reversible redox cycling. As a result, a consistent and dynamically enhanced catalytic process is achieved, exemplifying the superior performance of this catalyst.
The selective electrooxidation of methanol to formate intermediates remains a challenging aspect of the MOR, as the typical oxidative products are CO2 and H2O.280–282 Thus, a thorough investigation into the MOR reaction mechanism is imperative. To clarify the MOR reaction mechanism, Shi's group used mass spectroscopy (MS) to analyze the produced gas.283 As depicted in Fig. 15h–k, D2 and HD can only be observed in solution 1 containing 3 M CH3OH dissolved in deuterated water (D2O), while no D2 and HD are produced in solution 2 containing methanol-4D (D3COD) in non-deuterated H2O. This observation provides evidence supporting the methanol-assisted water splitting mechanism, rather than methanol reformation, for hydrogen production. More recently, Wang et al. explored the impact of different oxyanions (TOx: T = P, S, and Se) on the coordination environments of Ni sites, aiming to optimize the electrocatalytic performance of the MOR.284 The Ni metalloids (NiTx, T = P, S, and Se) were firstly prepared through surface anionization of nickel foams. Subsequently, active amorphous NiOOH coordinated with residual oxyanions (NiOOH-TOx) were constructed via in situ anodic electrochemical oxidation, resulting in the formation of distinct coordination environments of Ni sites. Based on various in situ and ex situ experiments, they confirmed that the optimized local coordination environment of NiOOH with oxyanions effectively modulates the adsorption of OH* intermediates and methanol molecules, thus favoring the formation of CH3O* intermediates. Among the various samples, NiOOH-POx exhibited the most favorable local coordination environment and significantly enhanced the electrocatalytic activity of Ni sites towards the selective oxidation of methanol to formate.
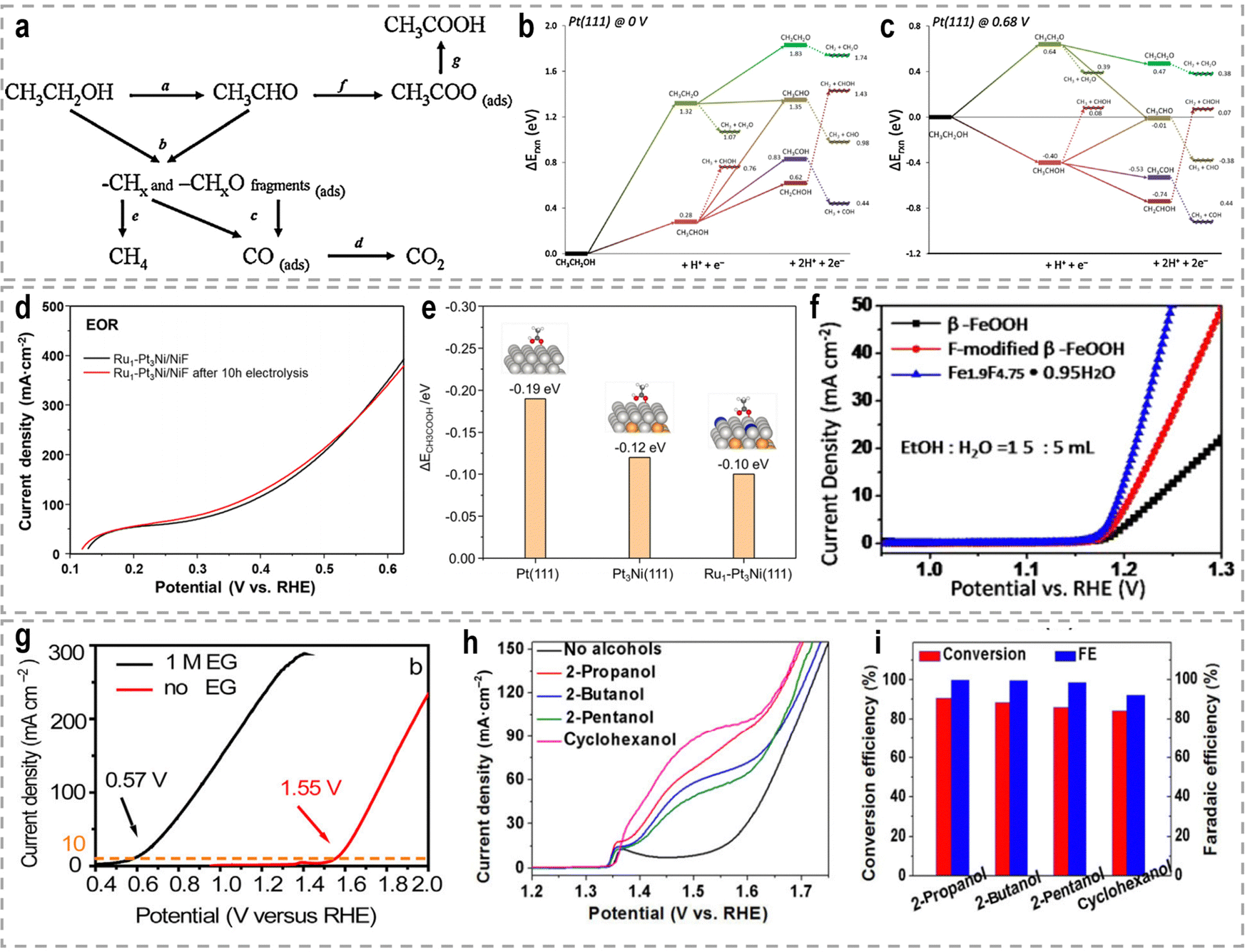 | ||
| Fig. 16 (a) Proposed reaction pathways for the electrooxidation of ethanol on a platinum surface in acidic electrolytes. Reaction energies of elementary steps (referenced to adsorbed ethanol) for dehydrogenation and C–C cleavage reactions (dashed lines) for (b) Pt(111) at 0 V and (c) 0.68 V vs. RHE.285 Reproduced with permission. Copyright 2013, Elsevier. (d) EOR polarization curves of pristine and aged Ru1–Pt3Ni/NiF. (e) Adsorption energies of CH3COOH on different catalysts facets.289 Reproduced with permission. Copyright 2023, Elsevier. (f) Polarization curves for ethanol oxidation of F-modified β-FeOOH.290 Reproduced with permission. Copyright 2018, American Chemical Society. (g) LSV curves of PdAg/NF in 0.5 M KOH with and without addition of 1.0 M ethylene glycol.248 Reproduced with permission. Copyright 2021, Elsevier. (h) LSV curves for NiS2/CFC in the absence and presence of alcohol precursors. (i) The conversions and FEs of NiS2/CFC for the electrocatalytic conversion of selected alcohols to the corresponding ketones.324 Reproduced with permission. Copyright 2017, Nature Publishing Group. | ||
During the EOR, the cleavage of C–C bonds and the prevention of CO poisoning play pivotal roles in promoting reaction kinetics. Prior investigations have focused on incorporating non-noble metals such as Co, Ni, and In into Pt/Pd nanostructures to modulate the electronic structure and weaken CO adsorption, thereby enhancing EOR performance.286–288 Huang et al. successfully developed subnanometer-sized, single-atom In-doped Pt nanowires (SA In–Pt NWs) as electrocatalysts with remarkable performance for both the HER and EOR.287 The integration of the HER and the EOR using SA In–Pt NWs/C allows circumvention of the significant overpotential associated with the sluggish OER, enabling achievement of a lower voltage of 0.62 V (compared to 2.07 V for water splitting) to attain a current density of 10 mA cm−2 for H2 production. Moreover, the anodic cell utilizing SA In–Pt NWs exhibited a high FE exceeding 93% in the conversion of ethanol to valuable acetate. Mechanistic investigations revealed that the ultrathin 1D morphology of the nanowires, combined with the presence of single-atom In species, provides a maximized number of active sites and effectively activates Pt atoms for catalytic purposes. DFT calculations ascertained that the incorporation of single-atom In efficiently reduces the limiting potential for the HER, facilitating hydrogen release. Simultaneously, it also decreased the energy barrier associated with acetate conversion and desorption, leading to the impressive selectivity and activity of the EOR in the anodic cells.
Noble metals usually show high activity for the EOR, however, an unfortunate drawback is their propensity to induce C–C bond cleavage, leading to the generation of commercially less valuable CO2 (or CO32− under alkaline conditions) during the EOR. In contrast, the partial oxidation products of ethanol, such as acetaldehyde and acetic acid, possess higher economic benefits as value-added fine chemical products. To overcome this challenge and achieve simultaneous high selectivity, activity, and stability during the EOR, Zhou et al. synthesized a unique electrocatalyst comprised of single dispersed Ru-anchored porous Pt3Ni alloy on nickel foam (Ru1–Pt3Ni/NF).289 The resulting Ru1–Pt3Ni/NF exhibited remarkable activity and selectivity for the EOR in alkaline media (Fig. 16d), with acetate being the sole detected product. DFT calculations revealed that the incorporation of Ru significantly reduced the Gibbs formation energy of adsorbed hydroxyl species while weakening the adsorption of acetic acid on the catalyst surface (Fig. 16e). These effects collectively enhanced the activity of ethanol oxidation and the selectivity for acetic acid production. When utilized as an anodic electrocatalyst for the EOR in an ethanol oxidation membrane-free cell (consisting of 2.0 M KOH + 2.0 M ethanol), Pt3Ni/NiF(−)//Ru1–Pt3Ni/NF(+) required only 0.7 V of electrolysis voltage to achieve a current density of 125 mA cm−2 for cathodic hydrogen generation.
The substitution of noble-metal catalysts with earth-abundant transition metal elements for the EOR provides several advantages such as low cost, abundant redox reactions, improved corrosion resistance, tunable properties, and environmental sustainability. For instance, by introducing electronegative F dopants into FeOOH, the binding energies between active Fe sites and reactants such as C2H5O− and OH− can be effectively moderated, leading to increased positive charge densities on the Fe sites and enhanced performance for the EOR.290 Notably, F-modified β-FeOOH showed remarkable activity, achieving a current density of 10 mA cm−2 at a potential of 1.207 V vs. RHE (Fig. 16f), with a Tafel slope of 30.1 mV dec−1. DFT calculations revealed that F dopants can weaken the adsorption of intermediates or CH3COO− on β-FeOOH, facilitating the desorption of products and promoting the generation of acetic acid as the main oxidation product. Consequently, a low cell voltage of 1.43 V was sufficient to achieve a current density of 10 mA cm−2 in a two-electrode electrolyzer. Another notable example involves the utilization of a NiOOH–CuO nano-heterostructure on Cu foam (NiOOH–CuO/CF), which exhibited abundant heterointerfaces and changed electronic structure.291 Remarkably, an ultralow potential of 1.347 V vs. RHE was attained at 200 mA cm−2, accompanied by an acetate FE of 79.1% at a high current density of 200 mA cm−2. DFT calculations indicated that the coupling between CuO and NiOOH by charge redistribution decreases the energy barrier at the rate-limiting step during the EOR. Finally, in a hybrid electrolyzer with NiOOH–CuO/CF as the anode and Pt/C/NF as the cathode, it only required a cell voltage of 1.430 V at 50 mA cm−2, 181 mV lower than that required for traditional OWS. And the acetic acid is the only liquid product, while the FE of H2 at the cathode reaches 100%. Non-noble metal-based catalysts, such as NiSn/C,292 Co–MnO/NCNTs,293 NiFeOOH,294 Cu–Ni–Fe2O3,295 NiFe-LDH,296 NiAl-LDH,297 Cu1Ni2-S/G,298 have also been developed for the EOR.
O), leading to the formation of various oxidation products.301 The specific products obtained depend on the reaction conditions, catalysts used, and applied potential. Noble metals such as Pt, Pd, Au, and their alloys always exhibit high activity for the EGOR, such as PtRh0.02@Rh NWs,302 Cu1Pd1/Ir0.03 NSs/NPG,303 Pd–Bi2Te3,304 Pd–PdSe,305 PtMo/C,306 RhCu nanoboxes,307 2D AuCu triangular nanoprisms,308 and 3D PdCu nanoflowers.309 Shi et al. developed a PdAg electrocatalyst grown in situ on nickel foam (PdAg/NF) for the EGOR in alkaline solution.248 PdAg/NF enables efficient EGOR with a low potential of 0.57 V vs. RHE at 10 mA cm−2 (Fig. 16g), which is 980 mV lower than that required for the OER in the same electrolyte without ethylene glycol. And a high FE of up to 92% can be achieved for the conversion from ethylene glycol to glycolic acid. Furthermore, glycolic acid can be electrocatalytically produced with high efficiency even at an extremely high current density of 300 mA cm−2 without the occurrence of oxygen evolution. When coupled with a Pt cathodic catalyst, the overall electrochemical cell requires a potential of 1.02 V to achieve 20 mA cm−2, for cathodic hydrogen production with a FE of 100%.
Notably, Ni and Co-based electrocatalysts have shown high activity for the EGOR. For instance, Wang et al. fabricated ultra-thin CoNi0.2P nanosheets on nickel foam (CoNi0.2P-uNS/NF).310 CoNi0.2P-uNS/NF revealed prominent electroactivity for the EGOR due to the surface electrochemical reconstruction in an alkaline environment. The assembled hybrid electrolyzer with the CoNi0.2P-uNS/NF catalyst only required 1.35 V to achieve 10 mA cm−2 for coproduction of formate and hydrogen. Chen's group replaced the OER with the EGOR over Ni3N–Ni0.2Mo0.8N nanowires, which generated formate selectively at the anode.311 Specially, ys-ZIF@CoFe-LDH NCs,312 and CoNi-MOF,313 also exhibit high performance for the EGOR.
Although noble metal catalysts demonstrate high catalytic efficiency for propanol oxidation, their scarcity and high cost pose limitations for practical applications.320,321 Therefore, there is growing interest in developing non-noble metal catalysts or earth-abundant alternatives to mitigate these challenges. Non-noble metal catalysts, including transition metal oxides, carbides, nitrides, and sulfides, have shown promising electrocatalytic performance.277,322 In noticeable example, a series of LaFe1−xCoxO3 perovskite catalysts were developed to enable electrocatalytic OER, isopropanol oxidation reaction, and glycerol oxidation reaction.323 The investigation focused on establishing a structure–composition–activity relationship, revealing distinct trends for anodic oxidation reactions arising from variations in active sites and involved reaction intermediates. Interestingly, no correlation was found between the electrochemical surface areas and the activities of electrochemical oxidation reactions. However, the phase/metal composition of the LaFe1−xCoxO3 catalysts influenced the selectivity towards different oxidation products, highlighting the trade-off between achieving high current densities and obtaining high-value chemical products. In another study, a facile vapor-phase hydrothermal method was employed to prepare a carbon fiber cloth supported single-crystalline NiS2 nanostructure (NiS2/CFC).324 This catalyst exhibited remarkable activity, selectivity, and durability in the HER, OER, and the oxidation of various alcohols including 2-propanol, 2-butanol, 2-pentanol, and cyclohexanol. In the presence of 0.45 M 2-propanol, the NiS2/CFC catalyst demonstrated a 1.2 times higher H2 production rate and a 280 mV lower cell voltage at a current density of 20 mA cm−2 compared to that required for conventional water electrolysis, accompanied by high-value-added acetone production with near-unity FEs for H2 (100%) and acetone (98%) (Fig. 16h and i).
Fortunately, nonprecious metal catalysts, particularly Ni- and Co-based materials, for example, NiOOH,329 NC/Ni–Mo–N/NF,330 NixBi1−x,331 NiOx/MWCNTs,332 Co3O4 nanosheets,333 amorphous CoOx,334 Bi-doped Co3O4,335 ZnFexCo2−xO4,336 CoOxHy,337 CuCo-oxide,338 CuCo2O4,339 CuO,340 and Cu-CuS/BM,341 have also exhibited promising glycerol oxidation activities. Li et al. fabricated a cost-effective MnO2 electrode and employed it in the glycerol-assisted hydrogen production under acidic conditions.342 Remarkably, this MnO2 catalyst exhibited exceptional activity with an anodic potential of 1.36 V vs. RHE at 10 mA cm−2 (Fig. 17a) and extremely high stability over 865 h for the GOR, which was significantly longer than the 10 h stability observed for the OER. In situ Raman spectroscopy (Fig. 17b) and DFT calculations (Fig. 17c) provided valuable insights into the underlying mechanism. The results demonstrated that glycerol molecules possess a robust electron-donating ability towards the positively charged MnO2 catalyst during the electrolysis process. The adsorption of glycerol molecules on the surface of MnO2 not only hindered the oxidation of the catalyst, preventing the formation of dissolved MnO4−, but also impeded the formation of chemisorbed oxygen species, contributing to the impressive stability observed. Recently, Wen's group developed a novel high-entropy alloy (CoNiCuMnMo HEA) self-supported electrode for the GOR and utilized self-developed machine learning-based Monte Carlo simulation to investigate its surface atomic configuration.343 To demonstrate the potential industrial prospects of this high-entropy alloy, a hybrid alkali/acid flow electrolytic cell was designed with the HEA as the anode for the alkaline GOR and the commercial RhIr/Ti as the anode for acidic HER. This system required only an applied voltage of 0.55 V to achieve a current density of 10 mA cm−2 and exhibited exceptional long-term stability over 12 days continuous running at 50 mA cm−2.
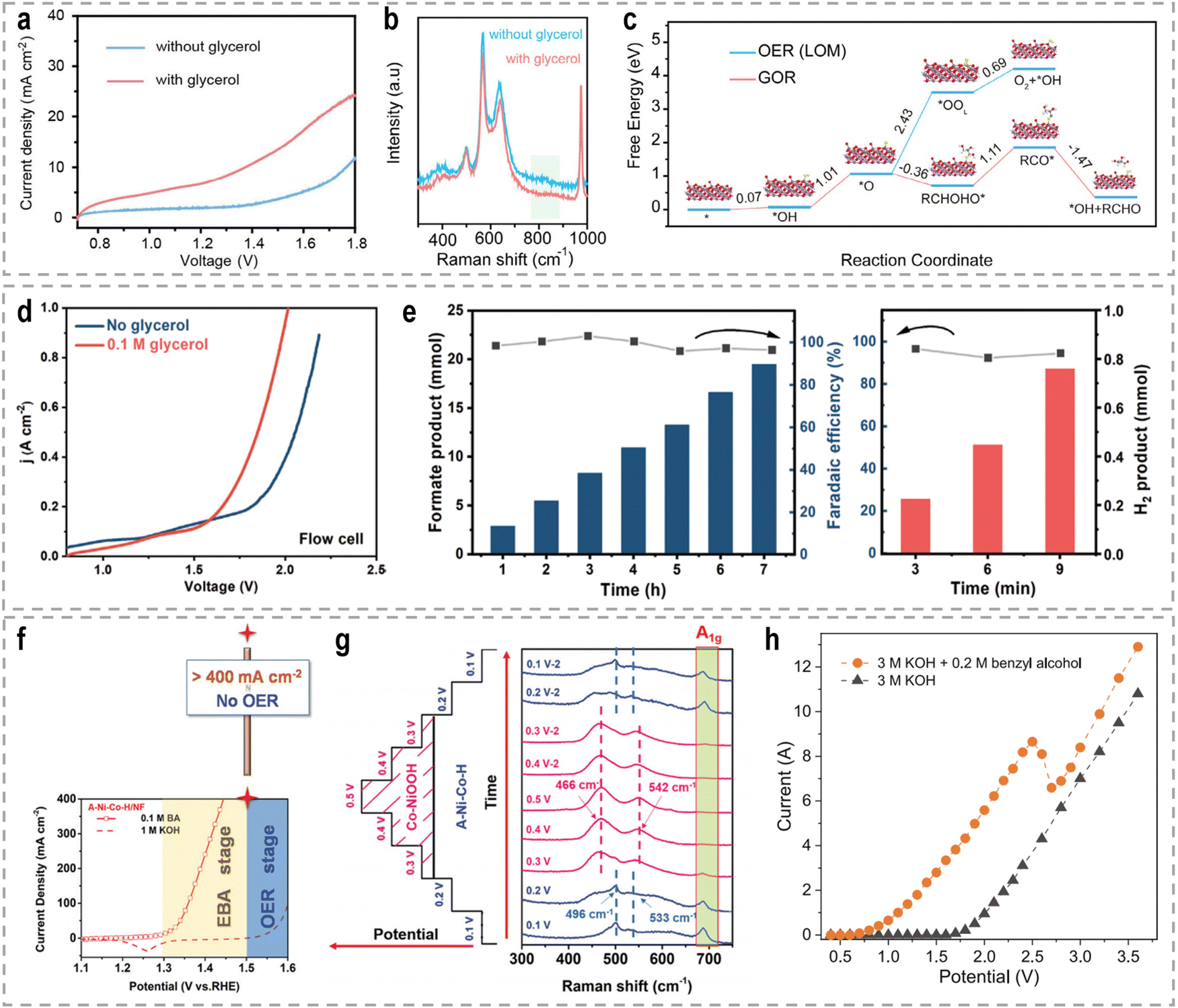 | ||
| Fig. 17 (a) Polarization curves over a MnO2/CP//Pt/C/CP electrolyzer with and without the addition of 0.2 M glycerol addition. (b) In situ Raman spectra of MnO2/CP collected with and without glycerol at an applied potential of 1.58 V. (c) Reaction free-energy diagram of the OER (LOM) and glycerol oxidation to glyceraldehyde.342 Reproduced with permission. Copyright 2021, Wiley-VCH. (d) LSV curves for the water electrolysis with and without glycerol addition using the flow cell. (e) The amount of generated formate and H2 with corresponding FEs.34 Reproduced with permission. Copyright 2023, Wiley-VCH. (f) Electrocatalytic H2 generation combined with the production of high-purity Ph-COOH at large current densities with no interference of the OER. (g) The in situ Raman spectra of the A-Ni–Co-H/NF electrode and the corresponding structural conversion under the changing potentials vs. Ag/AgCl.349 Reproduced with permission. Copyright 2020, Royal Society of Chemistry. (h) The LSV curves of Au/CoOOH in 3.0 M KOH with or without 0.2 M Ph-CH2OH at 70 °C in the membrane-free flow electrolyzer.238 Reproduced with permission. Copyright 2022, Nature Publishing Group. | ||
Despite recent advancements in biomass electrooxidation, there is still a lack of research focusing on investigating energy conversion efficiency at industrial-level current densities (∼1 A cm−2) and developing subsequent separation methods, which are crucial for practical applications. Recently, Zhang et al. reported a biphasic transition metal nitride electrode by in situ growing Ni3N/Co3N heterostructure nanowires with abundant heterointerfaces on Ni foam.34 This electrode exhibited remarkable activity for nucleophilic reaction electrocatalysis, achieving an ultrasmall work potential of 1.26 V to reach a current density of 50 mA cm−2 for the GOR and an excellent FE of 94.6% for formate production. Importantly, under a two-electrode configuration, the electrode enabled the concurrent production of formate and H2, delivering current densities of 50 and 400 mA cm−2 at 1.47 and 2.04 V, respectively, which surpass those required forthe OWS for O2 and H2 generation (1.72 and 2.22 V at 50 and 400 mA cm−2, respectively). As a proof-of-concept demonstration for practicality, a glycerol hybrid electrolysis based on a flow electrolyzer achieved an industry-level current density of 1 A cm−2 at 2.01 V (Fig. 17d) with impressive stability over 200 h for continuous operation, realizing efficient energy-saving efficiency of 9.7% compared to OWS, and outstanding productivities of 11 and 21.4 mmol cm−2 h−1 for formate and H2 at a current density of 1 A cm−2, respectively (Fig. 17e).
Li et al. fabricated N-doped nickel–molybdenum oxide (N–Mo–Ni/NF) loaded on Ni foam for the selective electrooxidation of Ph-CH2OH to benzoic acid in alkaline electrolytes.347 The N–Mo–Ni/NF electrode shows excellent electrocatalytic activity and stability at the anode and only requires a low potential of 1.338 V vs. RHE to afford 100 mA cm−2 in a 0.1 M Ph-CH2OH solution, which is 252 mV lower than that required for the OER. In addition, a yield of 98.2% of benzoic acid has been obtained with a high FE of 98.7%. Yu's group prepared 2D nickel-based nanoarrays directly grown on a carbon cloth substrate (CC@NiO/Ni3S2) using a facile one-step electrodeposition technique, enabling selective catalysis of Ph-CH2OH to Ph-COOH under alkaline conditions.348 CC@NiO/Ni3S2 demonstrated exceptional catalytic properties for the oxidation of Ph-CH2OH, exhibiting a low working potential of 1.38 V vs. RHE at 10 mA cm−2 and a remarkable selectivity of 98% towards Ph-COOH. Moreover, in the presence of 0.2 M Ph-CH2OH + 1.0 M KOH, a current density of 10 mA cm−2 was achieved at 1.458 V in the hybrid water electrolysis, 0.151 V lower than that for conventional water electrolysis. Qiu et al. fabricated amorphous nanosheets of Ni, Co hydroxide supported on Ni foam (A-Ni-Co-H/NF), enabling operation at high current densities (>400 mA cm−2) for the upgrading of Ph-CH2OH to Ph-COOH without interference from the OER (Fig. 17f).349 Notably, compared to the OER, the oxidation of Ph-CH2OH using A-Ni–Co-H/NF yielded a Ph-COOH conversion approaching 100% while significantly reducing the electric energy consumption by 0.024 W h after working for 1 h at a current density of 100 mA cm−2. The reversible structural evolution and recovery of the catalyst were observed through in situ Raman spectroscopy (Fig. 17g), confirming the formation of Co-containing nickel oxyhydroxide (Co–NiOOH) as the real active species. Co/Ni-based catalysts are commonly used for Ph-CH2OH oxidation, such as Mo–Ni alloy nanoparticle-modified MoO2,243 h-Ni(OH)2,344 Ni2P/NF,350 NiCo2O4 nanosheets,351 β-NiOOH,352 2D Ni-based MOFs,353 plasma modified nickel foam,354 Ni(OH)2 nanosheet/Ni foam,355 NiCo(OOH)x nanosheets,356 Ni–Fe thin films,357 Co–Ni LDH,358 and Co3O4 NPs/Ti.359
The electrochemical synthesis reactions discussed earlier exclusively yield hydrogen at the cathode, while no gas is generated at the anode. As a result, the implementation of a membrane-less electrolyzer becomes viable, simplifying operation under practical conditions and reducing reactor costs associated with membranes and maintenance. Duan et al. successfully demonstrated the application of a membrane-free flow electrolyzer operating under industrially relevant conditions.238 They developed cobalt oxyhydroxide nanosheet-supported gold nanoparticles (Au/CoOOH) for Ph-CH2OH-assisted hydrogen production. An impressive current density of 540 mA cm−2 was achieved at 1.5 V vs. RHE for the electrooxidation process. In a more realistic two-electrode membrane-free flow electrolyzer setup (Fig. 17h), the absolute current could be further increased to 4.8 A at 2.0 V. Experimental and theoretical findings indicate that Ph-CH2OH can be selectively enriched at the interface of Au/CoOOH and oxidized by the electrophilic oxygen species (OH*) generated on CoOOH, leading to higher activity compared to pure Au catalysts. Notably, the catalyst exhibits reversible oxidation/reduction behavior under anodic potential/open circuit conditions. Building on this observation, an intermittent potential strategy was designed for long-term alcohol electrooxidation, achieving a high current density (>250 mA cm−2) over 24 h with enhanced productivity and reduced energy consumption.
4.2 Aldehyde oxidation reactions
Aldehydes, such as 5-hydroxymethyl furfural (HMF) and furfural (FUR), are important raw materials in the synthesis of fine chemicals, biofuels, and medicines. Especially, the conversion of biomass-derived compounds containing –CHO groups, like HMF and FUR, to higher-value chemicals has attracted significant attention in recent decades.360 Conventional thermochemical oxidation methods for HMF and FUR typically require the use of precious-metal catalysts and harsh reaction conditions, including high temperatures, strong basicity, high oxygen pressures, and toxic or expensive reagents. These conditions often pose challenges in terms of cost, energy consumption, and environmental impact. In contrast, electrooxidative conversion provides a more environmentally friendly pathway for upgrading HMF and FUR. This approach operates in aqueous solution, without the need for organic solvents or oxidizing agents, and can be conducted at ambient temperature and pressure, offering several advantages, including high selectivity, mild reaction conditions, and the potential to use non-noble metal electrocatalysts.361–363 Moreover, by coupling the electrooxidation of HMF or FUR with the HER in hybrid water electrolysis, it becomes possible to simultaneously produce hydrogen and valuable chemicals, which provides a promising strategy for achieving efficient and sustainable energy conversion processes.
 | ||
| Fig. 18 (a) LSV curves for a Ni2P NPA/NF catalyst couple in 1.0 M KOH with and without 10 mM HMF.363 Reproduced with permission. Copyright 2016, Wiley-VCH. (b) Two possible pathways of HMF oxidation to FDCA.6 Reproduced with permission. Copyright 2018, American Chemical Society. Adsorption evaluation for Ir–Co3O4 and Co3O4. TPD spectra of Ir–Co3O4 and Co3O4 at HMF/He (c), ethylene (d), and CO (e) atmospheres.407 Reproduced with permission. Copyright 2021, Wiley-VCH. LSV curves of a Cu-modified glass carbon electrode in 1.0 M KOH with and without 50 mM HMF (f) and in 1.0 M KOH with and without 50 mM furfural (g). (h) The bipolar hydrogen production system of furfural oxidation in in the electrolyser using Cu foam as the anode and Pt/C as the cathode.417 Reproduced with permission. Copyright 2021, Nature Publishing Group. | ||
The catalytic conversion of HMF to FDCA requires the oxidation of both the alcohol and aldehyde groups in HMF. Fig. 18b illustrates two potential routes (route I and route II) for the synthesis of FDCA.6 In route I, the oxidation begins with the alcohol group and proceeds via DFF as the initial intermediate. Subsequent transformations lead to the conversion of DFF into 5-formyl-2-furancarboxylic acid (FFCA), which further undergoes oxidation to form FDCA. In route II, the aldehyde group in HMF is oxidized, resulting in the formation of 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) as the primary intermediate. Then HMFCA undergoes further conversion into FFCA and subsequently transforms into FDCA.397,398 Experimental techniques such as in situ SFG vibrational spectroscopy,399operando electrochemistry coupled with attenuated total reflection infrared spectroscopy,391,400 and operando surface-enhanced Raman spectroscopy401–403 have provided insights into the electrooxidation process of HMF. These studies have supported that route II is the most plausible pathway for the electrooxidation of HMF under strongly alkaline conditions. In addition to experimental techniques, DFT calculations have been employed to simulate the reaction pathway and elucidate the reaction mechanism.404 These computational studies help in understanding the transition states, solvation effects, and other factors influencing the electrooxidation process. Despite these efforts, the precise reaction mechanism and pathway for the electrooxidation of HMF to FDCA are still not fully understood. Further exploration using in situ/operando techniques and comprehensive theoretical investigations is needed to gain a detailed understanding of the intricacies of the electrooxidation process.
Among various candidates, transition metal Co-based materials have emerged as prominent catalysts for HMF oxidation. During the electrooxidation process, these Co-based catalysts generate high-valence Co species, including Co3+ and Co4+, which play specific roles in the oxidation of different functional groups in HMF. Deng et al. revealed that Co3+ primarily facilitates the oxidation of the formyl group to a carboxylate, while Co4+ exerts a pivotal effect on the initial oxidation of the hydroxyl group and significantly influences the overall reaction rate.405 On the basis of this understanding, they achieved selective synthesis of HMFCA and FDCA by applying distinct oxidation potentials. Wang's group conducted a comprehensive investigation by synthesizing a series of cobalt spinel oxides (Co3O4, ZnCo2O4, CoAl2O4) to occupy different geometric sites.406 They found that the tetrahedral (Co2+Td) site in Co3O4 enhances the chemical adsorption of HMF, while the octahedral (Co3+Oh) site facilitates HMF oxidation. Thereafter, Cu2+ was introduced into Co3O4 to improve the exposure degree of Co3+ and to boost adsorption and thus enhancing the performance for HMF oxidation. In addition to spinel oxides, this group also found that the introduction of atomically dispersed Ir sites (Ir-Co3O4) also enhances HMF adsorption by interacting with the CC group, thereby promoting HMF oxidation activity, which could be clearly reflected by the temperature-programmed desorption (TPD) results (Fig. 18c–e).407 Furthermore, Co3O4 nanosheets with oxygen vacancies (VO-Co3O4) have also been investigated by this group to understand the adsorption behavior between HMF and OH− during HMF oxidation.408In situ XAS and DFT calculations demonstrated that oxygen vacancies in Co3O4 preferentially adsorb OH−, enabling efficient coupling with HMF and enhancing the rate of HMF oxidation. These studies highlight the importance of understanding the intricate interplay between the catalyst composition, adsorption behavior, and specific reaction pathways in advancing the field of HMF oxidation. By elucidating the roles of different Co species and optimizing the catalyst composition and structure, we expediently enhance the electrocatalytic performance and selectivity for the oxidation of HMF, enabling the efficient conversion of this biomass-derived compound into valuable chemicals.
In the development of bifunctional catalysts with exceptional activities for both HER and HMF oxidation, various transition-metal-based catalysts have been synthesized and investigated for their performance in these reactions. Strategies such as surface modification, alloying, and heteroatom doping have been employed to further enhance the catalytic performance of these materials. For instance, Fu's group prepared a carbon-encapsulated MoO2–FeP heterostructure (MoO2–FeP@C) that exhibited excellent HER and HMF oxidation activity due to its unique interfacial electronic structure.372 The overpotential for the HER at 10 mA cm−2 was measured at 103 mV. In the presence of 1.0 M KOH with 10 mM HMF, MoO2–FeP@C demonstrated a potential of 1.359 V vs. RHE at 10 mA cm−2, which is lower than that required for the OER (1.474 V). Notably, when employed in an electrolyzer for cathodic H2 production and anodic FDCA production, MoO2–FeP@C achieved a low voltage of 1.486 V at 10 mA cm−2 and exhibited high selectivity for FDCA (98.6%). Moreover, MoO2–FeP@C displayed excellent catalytic performance in the electrooxidation of various biomass substrates, including benzyl alcohol, furfural, furfuryl alcohol, 4-nitrobenzyl alcohol, and 4-methoxybenzyl alcohol, coupled with the cathodic HER. Zhang et al. reported a Ni3N/carbon nanosheet (Ni3N@C) catalyst for HER and HMF oxidation.399 The strong interaction between Ni3N and carbon effectively modulates the electronic structure of Ni+, which is a crucial factor for optimizing electrochemical activity. In the presence of HMF in 1.0 M KOH, the overpotential required to achieve a current density of 50 mA cm−2 was reduced to 1.38 V vs. RHE, which is 0.22 V lower than the overpotential required for the OER. Moreover, the HER overpotential of Ni3N@C was measured at 113 mV to reach 50 mA cm−2, and the LSV curves exhibited negligible differences with or without HMF. Utilizing HMF oxidation instead of the OER as the anode reaction in a two-electrode configuration, the required potential to achieve a current density of 50 mA cm−2 was only 1.55 V, which is lower than 1.79 V required for the traditional OWS. Similarly, other bifunctional electrocatalysts, such as, Ni2P,363 and CoP,374 Ni3S2,376 CoNW/NF,409 NiCo-LDH NiCoNSs/CuNWs,410 NF@Mo–Ni0.85Se,411 CuxS@NiCo-LDHs,412 and NiSe@NiOx NWs,413 were also reported, opening up possibilities for efficient hybrid electrocatalysis in the conversion of biomass substrates.
In 2021, Wang et al. successfully realized the low-potential electrooxidation of HMF and furfural at approximately 0.1 V vs. RHE using a metallic Cu catalyst.417 Unlike conventional aldehyde electrooxidation, wherein the hydrogen atom in the –CHO group is oxidized to form H2O at high potentials (>1.0 V vs. RHE, 3OH− + R − CHO ⇌ 2H2O + R–COO− + 2e−), low-potential aldehyde oxidation involves the combination of hydrogen atoms to generate H2 (2OH− + R–CHO ⇌ 1/2H2 + R–COO− + H2O + e−). In the presence of both HMF and furfural in an electrolyte, a distinct oxidation current is observed (Fig. 18f and g), and the onset potential reaches an exceptionally low value of 0.05 V vs. RHE. Conversely, in the presence of furfuryl alcohol, which lacks an aldehyde group and only possesses a hydroxyl group, no discernible difference from pure KOH solution is observed, emphasizing the significance of the aldehyde group in this design. Consequently, in the novel bipolar H2 production system, which incorporates cathodic HER and low-potential anodic aldehyde oxidation on the developed metallic Cu catalysts, an incredibly low voltage of 0.1 V is sufficient to drive H2 generation (Fig. 18h). Notably, the assembled electrolyzer produces H2 at both the cathode and the anode, while 2-furoic acid or HMFCA is generated at the anode, with an apparent FE of about 200%. The energy input for 1 m3 H2 production is approximately 0.35 kW h, significantly lower than 5 kW h required for 1 m3 H2 generation through conventional water electrolysis. This approach presents a promising pathway for efficient hydrogen production with reduced electricity consumption. However, the catalytic performance of the low-potential electrooxidation of furfural and HMF still requires further exploration of advanced catalysts to achieve enhanced efficiency and effectiveness. Recently, this group investigated the correlation between the valence state and the adsorption behavior of the Cu-based electrocatalyst in furfural oxidation.418 Combined with the characterization of the valence state evolution and the absorption behavior on the designed mixed-valence Cu-based electrocatalyst, they found that Cu0, in its metallic form, acted as an adsorption site with low intrinsic activity for furfural oxidation. In addition, Cu+, existing in the form of Cu(OH)ads in an alkaline electrolyte, had no adsorption ability but played a crucial role in improving the performance of Cu0 by promoting the adsorption of furfural. Based on these findings, they proposed that the design principle for stable Cu-based catalysts for furfural oxidation is maintaining the stability of the valence state or the adsorption behavior of copper species.
Although H2 production through the partial FOR has been demonstrated on a limited number of metallic electrodes, the coupling of the FOR with the HER for simultaneous H2 production at both the anode and cathode remains relatively unexplored. In a recent study, Sun et al. reported a novel and cost-effective electrocatalytic system utilizing Cu3Ag7 as the anode catalyst and Ni3N/Ni as the cathode catalyst to drive the FOR and HER, respectively, under alkaline conditions.419 This system achieved H2 production with an apparent FE of 200% and demonstrated industrially relevant current densities of 100 and 500 mA cm−2 at remarkably low cell voltages of only 0.22 V and 0.60 V, respectively (Fig. 19a). Importantly, the energy consumption of this two-electrode electrolyzer for H2 production is merely 0.30 and 0.70 kW h m−3 H2 at current densities of 100 and 500 mA cm−2, respectively (Fig. 19b), which are much lower than the theoretical energy demand for OWS (4.10 and 4.70 kW h m−3 H2). To date, the reported FOR catalysts have been concentrated on Cu-based materials, such as Cu2O catalyst,420 ZrO2–CuO/Au,421 Ni doped Cu,422 Cu2O,423 hollow PdCu alloy,424 and Cu nanosheet arrays.425 Some other transition metal-based catalysts, such as Ni nanowires,426 S-Ni@Ni(OH)2/NF,427 NiCo–NiCoP@PCT,428 NiMn phosphates,429 S-doped MnO2,430 and Co–Nx–C@Co,431 have also been used for the FOR.
 | ||
| Fig. 19 (a) The two-electrode CV curves of HER/FOR (red) and HER/OER (blue) in which Cu3Ag7/CF and Ni3N/Ni/NF were employed as the anode and cathode for the former while Ni/NF and Ni3N/Ni/NF for the latter. (b) Comparative analysis of the calculated electricity consumption for H2 production between the formaldehyde (red) and paraformaldehyde (black) oxidation-integrated strategy using the Ni3N/Ni/NF(−)//Cu3Ag7/CF(+) electrode couple and traditional water electrolysis (HER/OER) using the Ni3N/Ni/NF(−)//Ni/NF(+) electrode couple.419 Reproduced with permission. Copyright 2023, Nature Publishing Group. (c) Schematic diagram of an asymmetric electrolyte electrolyzer. (d) Polarization curve for the GOR. (e) LSV curves for the asymmetric-electrolyte electrolyzer.453 Reproduced with permission. Copyright 2020, Elsevier. (f) Electrocatalytic conversion of primary amines into nitriles integrated with H2 production in water. (g) LSV curves of the NiSe anode in 1.0 M KOH with and without 1 mmol BA. (h) LSV curves of a CoP(−)//NiSe(+) electrolyzer with and without 1 mmol BA.456 Reproduced with permission. Copyright 2018, Wiley-VCH. | ||
The origin of catalytic activity and the underlying reaction mechanism for the FOR remains elusive. Wang et al. employed a novel approach by utilizing partially reduced CuO on Cu foam (CuxO@CF) catalysts to investigate the FOR mechanism.432 The CuxO@CF electrocatalyst, comprising a mixture of Cu and Cu2O phases, was synthesized through electrodeposition followed by an electrochemical activation process. To gain insights into the catalytic processes, in situ characterization studies such as in situ XAS, in situ ATR-SEIRAS, in situ DEMS, and DFT calculations were performed to identify and explore the presence of Cu0 and Cu+ species, as well as the synergistic effects governing the FOR, thereby validating the proposed reaction pathway. The obtained results unveiled that Cu0 species play a crucial role in lowering the reaction energy barrier for the HOCH2O* + HO* process, while Cu+ species are found to be more favorable for the C–H bond cleavage. Additionally, comprehensive analysis confirmed that the generated H2 molecules solely originate from HCHO decomposition.
In situ formed high-valence metal species, such as CoOx/CoOOH and NiOOH, under electrochemical reaction conditions, are always pointed out as the catalytic active centers during the GOR. Recently, Duan et al. identified two types of reducible Co3+–oxo active sites, namely adsorbed hydroxyl on Co3+ ions (μ1-OH-Co3+) and di-Co3+-bridged lattice oxygen (μ2-O-Co3+), which play crucial roles in the GOR.440 By employing operando Raman, operando/ex situ XAS and isotope-labeling experiments, they found that the μ1-OH-Co3+ and μ2-O-Co3+ species participate in different steps of the GOR. The μ1-OH-Co3+ species catalyze the oxygenation process, while the μ2-O-Co3+ species primarily catalyze the dehydrogenation step. It was also observed that the μ2-O-Co3+ species exist in a protonated form (μ2-OH-Co2+/3+) at an equilibrium state during the GOR, suggesting the fast kinetics of these species in the dehydrogenation of glucose. Additionally, DFT calculations confirmed the pivotal roles of the Co3+-oxo species, with μ1-OH-Co3+ facilitating oxygenation and μ2-O-Co3+ predominantly driving the dehydrogenation process. These oxidative steps are essential for the electrooxidation of glucose to formate.
In recent years, there has been growing interest in the coupling of the HER and the GOR on non-noble metal electrocatalysts, particularly Fe, Co, Ni, and Cu-based catalysts, such as Co@CoO heterojunctions,441 CoNi foam,442 NiCoSex,443 Ru@NiB,444 amorphous NiFe-LDH,445 NiFeOx,446 Co/N-codoped carbon,447 Co/CoP cluster,448 2D NiCo phosphate nanosheets,449 Cu/Cu2O,450 and Fe2P/SSM.451 Yu et al. fabricated NiV LDH by electrochemical and N2/H2 plasma regulations for boosting the activity of the GOR and HER.452 Specially, the electrochemically regulated NiV LDH with highly active Ni3+ exhibited a low potential of 1.23 V at 10 mA cm−2 and a FE of 94% during the GOR. While the plasma-regulated NiV LDH with a lower valence state of Ni species exhibited high HER activity, requiring an overpotential of 45 mV to deliver 10 mA cm−2. When integrated into a glucose electrolytic cell, the assembled electrolyzer just required 1.25 V to achieve 10 mA cm−2 for both production of formate and hydrogen, which is 320 mV lower than that required for water electrolysis. Additionally, Wen's group constructed a triprofitable alkaline-acidic asymmetric electrolytic cell (3A-EC), coupling the GOR under alkaline conditions with the HER under acidic conditions (Fig. 19c).453 They designed and fabricated a bifunctional electrode comprising iron-doped cobalt diselenide nanowires on conductive carbon cloth (Fe0.1-CoSe2/CC), which exhibited highly attractive electrocatalytic activity and stability for the GOR under alkaline conditions (Fig. 19d), as well as for the HER in acidic media. The assembled 3A-EC achieved an electrolytic current density of 10 mA cm−2 at an applied voltage of only 0.72 V (Fig. 19e) for H2 and gluconate generation. This remarkable performance was attributed to the successful harnessing of electrochemical energy from both glucose oxidation and neutralization processes.
4.3 Electrooxidation of amines
The electrocatalytic oxidation of primary amines provides an attractive approach to produce a variety of valuable compounds, such as imines, nitriles, amides, amine oxides, and azo compounds, which have diverse applications in pharmaceutical and agrochemical syntheses.454 Compared to the oxidation reactions of alcohols and aldehydes, the amine oxidation reaction exhibits a significantly lower thermodynamic equilibrium potential of −0.77 V vs. RHE, demonstrating a substantial thermodynamic advantage even before considering the specific reaction kinetics.455 In addition, coupling the electrooxidation of amines with the HER in a hybrid water electrolysis system not only enables the simultaneous production of H2 and valuable chemicals but also presents the potential for wastewater remediation due to amine-associated water pollution derived from the feed and fertilizer wastewater.In 2018, Zhang et al. presented a pioneering study on the coupling of electrooxidation of primary amines (–CH2–NH2) with the HER in 1.0 M KOH solution (Fig. 19f).456 They successfully electrooxidized various aromatic and aliphatic primary amines on a NiSe nanorod array anode, yielding the corresponding nitriles with excellent yields (>93%) and selectivity (>94%). Taking the conversion of benzylamine (BA) as an example, the electrooxidation of BA to benzonitrile (BN) occurred at approximately 1.34 V vs. RHE, which was significantly lower than the onset potential for the OER (∼1.55 V vs. RHE) (Fig. 19g). By utilizing NiSe as the anode for BA conversion and CoP as the cathode for the HER, a two-electrode electrolyzer was assembled, which achieved a remarkable current density of 20 mA cm−2 at only 1.59 V, much lower than 1.70 V required for the OWS (Fig. 19h). At 1.5 V, the FE for BN and H2 reached as high as 98% and ∼100%, respectively. In situ Raman spectroscopy revealed that the generated NiII/NiIII sites on the NiSe acted as redox-active species, facilitating the conversion of primary amines to nitriles. Fu et al. constructed a biphasic Mo0.8Ni0.2N–Ni3N heterojunction for enhanced BA electrooxidation by tailoring the d-band centers.457 The Mo0.8Ni0.2N–Ni3N heterojunction requires a low potential of 1.54 V to achieve 240 mA cm−2 in 0.1 M KOH/0.5 M Na2SO4/25 mM BA, along with excellent BA conversion, high BN yield, and FE > 99%. Quasi-in situ XPS characterization revealed the Ni–OOH species generate at a lower potential (1.54 V) for Mo0.8Ni0.2N–Ni3N during BA oxidation and the Ni3+/Ni2+ ratio is higher than that of Ni3N under the same conditions. Theoretical calculations indicate that charge transfer in the Mo0.8Ni0.2N–Ni3N heterojunction causes an upshift of the d-band centers, which facilitates the production and adsorption of OH* from water, leading to the easy formation of NiOOH on Ni3N and the optimized adsorption energy for BA, thus further enhancing the catalytic efficiency of BA oxidation. Propylamine electrooxidation was investigated by Zhai et al. by using vacancy-rich Ni(OH)2 atomic layers (VR-Ni(OH)2) as the catalyst.458 DFT calculations indicated that defect-induced local lean electron of the VR-Ni(OH)2 surface facilitated the conversion of amino C–N bonds to nitrile C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds. In situ FTIR spectroscopy confirmed the transformation of amino C–N bonds into nitrile CN bonds. Consequently, the VR-Ni(OH)2 catalyst demonstrated a low operating potential of 1.36 V vs. RHE to achieve a current density of 10 mA cm−2 and simultaneously exhibited an impressive FE of 96.5% at a potential of 1.38 V for the electrooxidation of propylamine to propionitrile.
N bonds. In situ FTIR spectroscopy confirmed the transformation of amino C–N bonds into nitrile CN bonds. Consequently, the VR-Ni(OH)2 catalyst demonstrated a low operating potential of 1.36 V vs. RHE to achieve a current density of 10 mA cm−2 and simultaneously exhibited an impressive FE of 96.5% at a potential of 1.38 V for the electrooxidation of propylamine to propionitrile.
The bifunctional electrocatalysts used for anodic amine oxidation and cathodic HER are also widely investigated. For instance, Guo et al. reported an atomically thin CoSe2 subnanometer belt (SB) with anion vacancies and cation substitutions, demonstrating their efficacy in enhancing the electrooxidation of butylamine to produce high-value-added butyronitrile while simultaneously generating hydrogen.459 The synthesized CoSe2/Ni-SVs SBs, specifically engineered with Se vacancies and Ni substitutions, displayed an exceptionally low onset potential of 1.3 V vs. RHE and achieved an impressive FE of about 98.5% for butyronitrile production, surpassing the performance of all previously reported Co- and Ni-based catalysts. In-depth investigations using in situ electrochemical FTIR spectroscopy and electrochemical impedance spectroscopy uncovered the underlying mechanisms behind the significantly enhanced electrooxidation performance. It was found that the optimized adsorption behavior and accelerated dehydrogenation kinetics were key contributors to the improved performance. Theoretical studies further elucidated that the Se vacancies served as robust Lewis acid sites, promoting effective N atom adsorption, while Ni substitutions played a crucial role in enhancing dehydrogenation thermodynamics by optimizing the sequence of dehydrogenation steps. Importantly, the CoSe2/Ni-SVs SBs exhibited high versatility as efficient catalysts for the electrosynthesis of propylamine, BA, and cyclohexane methylamine into nitriles, coupled with hydrogen generation. Notably, a two-electrode electrolyzer based on CoSe2/Ni-SVs SBs achieved a voltage of 1.37 V at a current density of 10 mA cm−2, which led to a remarkable reduction in cell voltage by up to 320 mV compared to that required for the conventional OWS. Other transition metal-based catalysts, including Ni–Ni3N heterostructures,460 Ni/Co MOF derivative,461 ordered Ni2Si NPs,462 V-doped Ni(OH)2,463 cobalt cyclotetraphosphate (Co2P4O12) nanorods,464 have also been reported for amine upgrading and hydrogen production.
4.4 Other electrochemical synthesis reactions
In addition to the above small organic molecules, there have been some reports on the electrooxidation of other molecules for energy-saving H2 production and organic upgrading, such as cyclohexanol,250 sterol intermediates,465 nitroalkanes,466 tetrahydroisoquinolines (THIQs),467 styrene,468 alkene,469 and C–C coupling reactions.470 For instance, Wang et al. reported Co2(OH)3Cl/FeOOH nanosheets on nickel foam synthesized by the impregnation method.250 These nanosheets exhibited dual functionality as both the cathode and anode under alkaline conditions, facilitating the H2 production and high value-added cyclohexanone production from cyclohexanol. The remarkable synergy between Co2(OH)3Cl and FeOOH obtained a substantial reduction in the anodic potential required for cyclohexanol oxidation by 120 mV, compared to the OER at a current density of 10 mA cm−2. Notably, the integration of bifunctional Co2(OH)3Cl/FeOOH catalysts into a two-electrode electrolyzer demonstrated a voltage of 1.46 V at 10 mA cm−2 to drive cathodic hydrogen generation and anodic cyclohexanone production. Upon scaling up the configuration to a 10 × 10 cm2 membrane electrode assembly reactor, an impressive cyclohexanone production rate of 3.44 g h−1 and energy savings of approximately 0.24 kW h m−3 (H2) were achieved. Zhang et al. realized the synthesis of E-nitroethene through the electrooxidation of α-nitrotoluene on NiSe nanorod arrays in an alkaline electrolyte.471 They assembled a hybrid electrolyzer with NiSe nanorod arrays serving as both the anode and cathode, which enabled simultaneous production of E-nitroethene and H2. At 1.8 V, this setup achieved a FE of 100% for H2 and a selectivity of 88% for E-nitroethene. In situ and ex situ experiments revealed that the in situ-formed NiOOH species played a crucial role in the electrocatalytic process. Furthermore, Zhang et al. reported the electrocatalytic semi-dehydrogenation of THIQs to synthesize dihydroisoquinolines (DHIQs) using a Ni2P anode in alkaline solution.472 In 1.0 M KOH, the electrooxidation of THIQs exhibited faster kinetics than the OER. In a hybrid electrolyzer employing Ni2P as both the anode and cathode, DHIQs and H2 were simultaneously produced with high activity and stability. The in situ formation of NiII/NiIII redox-active species at the anode played a vital role in the semi-dehydrogenation of THIQs to DHIQs.Table 5 provides a comprehensive summary of the electrochemical performances exhibited by anodic catalysts employed in small organic-assisted hybrid water electrolysis. Notably, the reduction in applied voltages was observed, indicating enhanced electrooxidation capabilities of various organic compounds. In addition, these reactions yielded diverse value-added products, achieving simultaneous organic upgrading and hydrogen production. However, there are several challenges that need to be addressed. (i) Although selective electrooxidation of organic compounds replaces the anodic OER in theory, practical systems often operate at voltages exceeding the theoretical voltage input for water electrolysis due to inherent limitations. Hence, developing efficient and stable catalysts, optimizing reaction conditions, and improving system designs are urgently required to reduce the operational voltages and enhance overall system efficiency. (ii) There remain diverse oxidation reactions and value-added products to select, necessitating the identification of the reaction mechanism and optimal strategies considering industrial demand, technological feasibility, and environmental implications. (iii) The corrosiveness of the organic molecules used and the radicals generated in situ can lead to a shortened lifetime of equipment, complicate the operation procedure, and increase the overall cost of the system. The aggressive nature of these chemicals can affect the durability of materials and components. (iv) In cases where carbonaceous chemicals are employed as the organic substrate, conditions such as elevated reaction temperatures, higher substrate concentrations, and the presence of aprotic electrolytes at the electrocatalyst surface can promote oxidative polymerization. This phenomenon can lead to catalyst deactivation to varying degrees, further complicating the efficiency and stability of the process. (v) Exploring alternative oxidation reactions within a hybrid water electrolysis system can enhance its versatility and potential for industrial-scale implementation. Furthermore, to realize the three-fold benefits of high-efficiency H2 production, value-added chemical synthesis, and biowaste treatment, it is crucial to explore the utilization of diversified biowaste in this electrochemical strategy. Addressing these challenges will promote the advancement of efficient and sustainable strategies in small organic-assisted hybrid water electrolysis, unlocking the potential for simultaneous hydrogen production, valuable chemical synthesis, and biowaste utilization.
| Catalysts (cathode//anode) | Electrolyte of anode | Overpotential for cathode | Tafel slope for cathode (mV dec−1) | Potential for anode | Tafel slope for anode (mV dec−1) | Cell voltage at 10 mA cm−2 | FE (H2) | FE (oxidation product) | Products | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| MoNi4//Fe2O3/NiO | 1.0 M KOH + 1.0 M methanol | − | — | 1.328 V at 10 mA cm−2 | — | 1.381 V | ∼100% | 98% | H2 + formate | 32 |
| CoxP@NiCo-LDH/NF//CoxP@NiCo-LDH/NF | 1.0 M KOH + 0.5 M methanol | 100 mV at 10 mA cm−2 | — | 1.13 V at 10 mA cm−2 | 58 | 1.43 V | ∼100% | ∼100% | H2 + formate | 256 |
| Mo–Co4N//Mo–Co4N | 1.0 M KOH + 3.0 M methanol | 45 mV at 10 mA cm−2 | 42 | 1.356 V at 10 mA cm−2 | — | 1.427 V | ∼100% | ∼100% | H2 + formate | 257 |
| CNFs@NiSe//CNFs@NiSe | 1.0 M KOH + 1.0 M methanol | — | — | 1.32 V at 100 mA cm−2 | 24 | — | 98.17% | 97.90% | H2 + formate | 260 |
| Co(OH)2@HOS/CP//Co(OH)2@HOS/CP | 1.0 M KOH + 3.0 M methanol | 148 mV at 10 mA cm−2 | — | 1.385 V at 10 mA cm−2 | 71 | 1.497 V | ∼100% | ∼100% | H2 + formate | 273 |
| Ni–Mo nanopowders/NF//Co3O4 nanosheets/CP | 1.0 M KOH + 1.0 M ethanol | — | — | 1.489 V at 10 mA cm−2 | 138 | 1.5 V | — | 98% | H2 + ethyl acetate | 241 |
| Pt/C//F-modified β-FeOOH | 15:5 volume ratio of ethanol to water |
∼0.3 V at 10 mA cm−2 | — | 1.207 V at 10 mA cm−2 | 30.1 | 1.43 V | 91.66% | 72.28% | H2 + acetate | 290 |
| Cu1Ni2-N//Cu1Ni2-S/G | 1.0 M KOH + 1.0 M ethanol | — | — | 1.37 V at 10 mA cm−2 | 15.2 | ∼1.495 V at 10 mA cm−2 | — | 96% | H2 + ethyl acetate | 298 |
| CoS2–MoS2//CoNi-PHNs | 1.0 M KOH + 1.0 M ethanol | — | — | 1.39 V at 10 mA cm−2 | 47 | 1.80 V at ∼50 mA cm−2 | 90.50% | 94.10% | H2 + acetate | 473 |
| NiS2/CFC//NiS2/CFC | 1.0 M KOH + 0.45 M 2-Propanol | 67 mV at 10 mA cm−2 | 114 | 1.348 V at 10 mA cm−2 | — | 1.41 V at 20 mA cm−2 | 100% | 98% | H2 + acetone | 324 |
| Ni3N/Co3N-NWs//Ni3N/Co3N-NWs | 1.0 M KOH + 0.1 M glycerol | 69 mV at 10 mA cm−2 | 91 | 1.18 V at 20 mA cm−2 | 131.4 | 1.47 V at 50 mA cm−2 | ∼100% | 94.6% | H2 + formate | 34 |
| CNs@CoPt//CNs@CoPt | 1.0 M KOH + 10 mM glycerol | 19.1 mV at 10 mA cm−2 | 88.7 | 1.52 V at 100 mA cm−2 | 85.4 | 1.71 V at 100 mA cm−2 | 97% | 79% | H2 + formate | 328 |
| Pt/C/CP//MnO2/CP | 0.005 M | — | — | 1.36 V at 10 mA cm−2 | 242.1 | 1.38 V | 100% | 100% | H2 + formic acid | 342 |
| H2SO4 + 0.2 M glycerol | ||||||||||
| RhIr/Ti//HEA-CoNiCuMnMo | 1.0 M KOH + 0.1 M glycerol | — | — | 1.25 V at 10 mA cm−2 | 53.4 | 0.55 V | ∼100% | ∼99% | H2 + formate | 343 |
| Ni–Mo–N/CFC//Ni–Mo–N/CFC | 1.0 M KOH + 0.1 M glycerol | 40 mV at 10 mA cm−2 | 70 | 1.30 V at 10 mA cm−2 | 87 | 1.36 V | 99.7% | 95.0% | H2 + formate | 474 |
| NF//Au/CoOOH | 3.0 M KOH + 0.2 M benzyl alcohol at 70 °C | — | — | 1.30 V at 340 mA cm−2 | — | 2.0 V at 4.8 A | 99.99% | — | H2 + benzoic acid | 238 |
| Mo–Ni alloy//Mo–Ni alloy | 1.0 M KOH + 10 mM benzyl alcohol | 48 mV at 50 mA cm−2 | 53.4 | 1.35 V at 15 mA cm−2 | 23.2 | 1.40 V at 20 mA cm−2 | ∼100% | — | H2 + benzoic acid | 243 |
| hp-Ni//hp-Ni | 1.0 M KOH + 10 mM benzyl alcohol | 219 mV at 50 mA cm−2 | — | — | — | 1.50 V at 50 mA cm−2 | ∼100% | ∼97% | H2 + benzoic acid | 244 |
| PtO2/h-Ni(OH)2//Ni(OH)2 | 1.0 M KOH + 40 × 10−3 M benzyl alcohol | 61 mV at 10 mA cm−2 | 67 | — | — | 1.48 V | — | — | H2 + benzoic acid | 344 |
| CC@NiO/Ni3S2//CC@NiO/Ni3S2 | 1.0 M KOH + 0.2 M benzyl alcohol | 91 mV at 10 mA cm−2 | 117.4 | 1.391 V at 50 mA cm−2 | 60.1 | 1.458 V | — | — | H2 + benzoic acid | 348 |
| NF//A-Ni–Co-H/NF//A-Ni–Co-H/NF | 1.0 M KOH + 0.1 M benzyl alcohol | — | — | 1.35 V at 100 mA cm−2 | — | 1.30 V at ∼20 mA cm−2 | — | ∼90–95% | H2 + benzoic acid | 349 |
| Ni2P NPA/NF//Ni2P NPA/NF | 1.0 M KOH + 10 mM HMF | 9 mV at 10 mA cm−2 | 86 | 1.40 V at ∼200 mA cm−2 | — | 1.44 V | 100% | 98% | H2 + FDCA | 363 |
| MoO2–FeP@C//MoO2–FeP@C | 1.0 M KOH + 10 mM HMF | 103 mV at 10 mA cm−2 | 48 | 1.359 V at 10 mA cm−2 | 38 | 1.486 V | — | — | H2 + FDCA | 372 |
| Ni3N@C//Ni3N@C | 1.0 M KOH + 10 mM HMF | 113 mV at 50 mA cm−2 | — | 1.38 V at 50 mA cm−2 | 48.9 | 1.55 V at 50 mA cm−2 | — | — | H2 + FDCA | 399 |
| NiCoNSs/CuNWs//NiCoNSs/CuNWs | 1.0 M KOH + 10 mM HMF | 50 mV at 10 mA cm−2 | 97.2 | 1.45 V at 50 mA cm−2 | — | 1.44 V | — | — | H2 + FDCA | 410 |
| NF@Mo–Ni0.85Se//NF@Mo–Ni0.85Se | 1.0 M KOH + 10 mM HMF | 130 mV at 10 mA cm−2 | 98.98 | — | — | 1.50 V at 50 mA cm−2 | 100% | — | H2 + FDCA | 411 |
| CuxS@NiCo-LDH//CuxS@NiCo-LDH | 1.0 M KOH + 10 mM HMF | 107 mV at 10 mA cm−2 | 35 | 1.30 V at 87 mA cm−2 | — | 1.34 V | ∼100% | ∼100% | H2 + FDCA | 412 |
| Ni2P/Ni/NF//Ni2P/Ni/NF | 1.0 M KOH + 30 mM furfural | — | — | ∼1.41 V at 200 mA cm−2 | — | 1.48 V | — | — | H2 + 2-furoicacid | 416 |
| Pt/C//metallic Cu | 1.0 M KOH + 100 mM HMF | — | — | 0.4 V at ∼1.0 mA cm−2 | — | 0.27 V at 100 mA cm−2 | 100% | 100% | H2 + FDCA | 417 |
| Pt/C//metallic Cu | 1.0 M KOH + 200 mM furfural | — | — | 0.45 V at ∼3.5 mA cm−2 | — | 0.31 V at 100 mA cm−2 | 100% | 100% | H2 + 2-furoicacid | 417 |
| Ni3N/Ni//Cu3Ag7/CF | 1.0 M KOH + 0.6 M HCHO | — | — | 0.10 V at 100 mA cm−2 | — | 0.60 V at 500 mA cm−2 | 100% | 100% | H2 + formate | 419 |
| NiFeNx//NiFeOx | 1.0 M KOH + 0.5 M glucose | 40.6 mV at 10 mA cm−2 | 39 | 1.30 at 87.6 mA cm−2 | 19 | 1.39 V at 100 mA cm−2 | — | 80% | H2 + glucaric acid | 446 |
| Pt/C//Fe2P/SSM | 1.0 M KOH + 0.5 M glucose | — | — | 1.35 V ∼10 mA cm−2 | 71 | 1.22 V | 100% | — | — | 451 |
| Fe0.1–CoSe2/CC//Fe0.1–CoSe2/CC | 1.0 M KOH + 0.5 M glucose | 270 mV at 100 mA cm−2 | 40.0 | ∼1.10 V at 10 mA cm−2 | — | 0.72 V | ∼99% | — | H2 + gluconate | 453 |
| CoS2–MoS2//VR-Ni(OH)2 | 1.0 M KOH + 10 mM propylamine | — | — | 1.36 V at 10 mA cm−2 | 31.5 | 1.36 V | — | 96.5% | H2 + propionitrile | 458 |
| CoP//CoSe2/Ni-SVs SBs | 1.0 M KOH + 20 mM butylamine | — | — | 1.37 V at 10 mA cm−2 | 54.8 | 1.37 V | 98.9% | 96.7% | H2 + butyronitrile | 459 |
5. Self-powered H2 production
One significant limitation of current water splitting technology is its reliance on an external power supply to drive the electrochemical process. This constraint renders it impractical for use in mobile devices, vehicles, and field activities where access to a continuous power source may be limited or unavailable. Therefore, there is a pressing need for the development of self-powered electrocatalytic hydrogen generation systems that can operate autonomously without the need for an external power supply. Such systems would greatly expand the applicability of hydrogen production technology and enable its use in a wider range of real-world scenarios. The concept of self-powered systems was pioneered by Wang's group in 2013 with the development of a cell-electrolyzer coupling system.475 This system combined a hybrid energy cell capable of harnessing mechanical, thermal, and solar energies to generate electricity with an electrolyzer for water splitting and chemical conversions. These self-powered electrolysis approaches offer potential advantages in terms of energy efficiency, sustainability, and decentralized hydrogen production. They can enable hydrogen generation in remote areas or off-grid locations where a constant power supply may not be readily available.476In self-powered electrolysis systems, the internally generated electricity is crucial for driving electrocatalytic reactions. These systems rely on the thermodynamic feasibility of internal redox reactions to provide the necessary electrical potential for driving the desired electrochemical reactions.477 To ensure efficient operation, the equilibrium potential difference between the redox reactions at the two electrodes must be sufficiently large to overcome the internal resistance of the system and the energy barriers associated with the catalytic reactions. The incorporation of highly active electrocatalysts is essential for promoting the internal reactions and enhancing the overall efficiency of a self-powered system.
In self-powered systems, zinc–air batteries (ZAB) and direct hydrazine fuel cells (DHzFCs) are commonly used as power suppliers. These energy sources provide the necessary electricity for driving the electrocatalytic reactions in a water splitting electrolyzer for hydrogen production. Current research in the field of self-powered systems is focused on the development of multifunctional catalysts that can enhance the kinetics of internal reactions in both chemical cells and electrolyzers. These catalysts play a crucial role in improving the overall performance and efficiency of self-powered electrolysis systems.
5.1 Aqueous zinc–air battery assisted OWS
An aqueous zinc–air battery is a type of rechargeable battery that utilizes the oxidation of zinc and the reduction of oxygen from the air to generate electricity.478 During the discharge process, zinc metal at the anode undergoes oxidation, releasing Zn2+ ions into the electrolyte solution. Meanwhile, at the air cathode, oxygen from the air combines with water and electrons from the external circuit to form hydroxide ions through the ORR process. The flow of electrons through the external circuit from the zinc anode to the air cathode provides electrical energy that can be utilized to power devices or systems. In the charging process, when an external power source is connected to the battery, the zinc oxide formed during discharge is converted back into metallic zinc, and oxygen is released back into the air through the OER at the cathode. The ORR and OER, typically governed by a 4e− process, exhibit relatively sluggish kinetics compared to the HER. Consequently, the development of efficient bifunctional catalysts for the ORR and OER at the air cathode is of paramount importance in enhancing energy efficiency.479 To realize a self-powered system based on ZABs, it is imperative to design a multifunctional electrocatalyst that can simultaneously enhance the kinetics of the ORR, OER, and HER, as depicted in Fig. 20a. Generally, due to the overpotential associated with the HER and OER, the voltage required for water splitting is at 1.5–2.3 V, exceeding the thermodynamic potential of 1.23 V. In contrast, aqueous zinc–air batteries typically possess an OCV of approximately 1.4 V. Consequently, two zinc–air batteries connected in series are necessary to drive the overall process of water splitting. | ||
| Fig. 20 (a) Illustration of the self-driven water splitting powered by zinc–air batteries. (b) Photovoltage charging and following discharging curves of the fabricated FeNiP/NPCS-based zinc–air battery, and the inset is the schematic diagram of the photovoltage charging and following discharge process.503 Reproduced with permission. Copyright 2020, Elsevier. (c) Digital images of the uninterrupted H2 production system by water splitting powered by solar photovoltaic and aqueous zinc–air batteries. (d) Current density versus time curves of this hydrogen production system powered by solar energy in day and aqueous zinc–air batteries in night.504 Reproduced with permission. Copyright 2023, Royal Society of Chemistry. | ||
In 2016, Dai's group synthesized a series of metal-free bifunctional catalysts (N and P co-doped carbon networks480 and N, P, and F tri-doped graphene481) for the ORR and the HER in a ZAB coupled OWS system. To further elevate the power densities of ZABs and decrease the applied voltage of OWS, metal-based electrocatalysts, particularly Co-based catalysts, garnered significant attention. Several catalysts were investigated, including CoP@P,N co-doped carbon (CoP@PNC),482 CoMn2−xCrxO4,483 FeCo/Co2P nanoparticles,484 hierarchical CuCoNC nanowire arrays,485 Co–N-carbon monolithic electrode,486 Co,O-doped carbon,487 Co2P/CoNPC,488 CoFeN-NCNTs//CCM,489 CoOx/CoNy@CNz,490 Co4N@NC,491 Co@NCL,492 Co3W3C/CoP/NPC,493 Co/MoC@PC,494 Co@NC-CNTs@NiFe-LDH,495 FeCo/NPC,496 Co/CoFeNC@N-CNF,497 CoFe-N-CNTs/CNFs,498 and CoFe2O4/CoFe.499 For instance, Yoo et al. fabricated a hierarchical FeCoMoS nanoflower encapsulated in nitrogen doped graphene (FeCoMoS@NG) for zinc–air batteries and overall water splitting.500 This FeCoMoS@NG catalyst exhibited excellent trifunctional activity, with a half-water potential (E1/2) for the ORR of 0.83 V vs. RHE, an overpotential at 10 mA cm−2 (η10) for the OER of 238 mV, and η10 of 137 mV for the HER. When employed as the cathode catalyst in a ZAB, FeCoMoS@NG demonstrated a significantly low charging–discharging voltage gap of 0.77 V at 2 mA cm−2, allowing for a stable working period over 70 h. Moreover, when utilized as both the cathode and anode for overall water splitting, this catalyst only required a low cell voltage of 1.58 V to achieve a current density of 10 mA cm−2 and with remarkable stability. Notably, a series connection of three zinc–air batteries effectively facilitated the operation of the OWS system for self-powered H2 production with a H2 evolution rate of 0.7 mL min−1.
To enhance the kinetics of the ORR and improve the ionic conductivity of the electrolyte, current research on ZABs primarily focuses on alkaline electrolytes. However, it is widely acknowledged that alkaline electrolytes suffer from corrosion and carbonation issues. To ensure safety and durability, there is growing interest in utilizing neutral electrolytes as an alternative to alkaline electrolytes, which would help mitigate these concerns. In the context of neutral electrolytes, the development of high-performance electrocatalysts becomes crucial to meet the demands of ZABs. Zhu et al. fabricated a single-atom Ir-embedded N-doped hierarchically porous carbon catalyst (SA-Ir/NC).501 This catalyst demonstrated exceptional performance when applied to ZABs in neutral media (0.1 M PBS with 0.02 M zinc acetate), delivering a high OCV of 1.42 V and an impressive power density of 90.4 mW cm−2. DFT calculations revealed that the chelation of single-atom Ir sites with N atoms enables the export of a stronger positive charge compared to other transition metal sites, thus facilitating O2 absorption. In addition, the long-term operation of ZABs is essential for the development of self-powered systems based on ZABs, which necessitates the use of robust electrocatalysts. Our group fabricated the FeNiP nanoparticles dispersed on heteroatom-doped graphene as an efficient electrocatalyst for ZABs.502 This catalyst exhibited high electrocatalytic activity with an overpotential of 290 mV at 10 mA cm−2 for the OER, as well as a E1/2 of 0.83 V for the ORR. When used for ZABs as air cathode, the assembled battery demonstrated a peak power density of 118 mW cm−2 and excellent cycling stability over 600 h at 10 mA cm−2. The favorable surface reconstruction properties and the robust graphene substrate are considered primary factors contributing to the outstanding stability of the ZAB system.
In recent years, significant progress has been made in electrocatalytic water splitting driven directly by electricity or solar cells. However, these approaches still present certain drawbacks. For instance, the use of electricity from a power grid is energy-consuming and economically inefficient. The intermittence of sunlight usually requires connection of energy storage devices, which suffer from complicated structures and external energy loss. zinc–air batteries offer a unique advantage since they can serve as both energy storage devices and high-discharge voltage conversion technologies. This makes them ideal for storing solar energy during light reactions and releasing it as electricity during dark reactions to drive water splitting, thereby maximizing the utilization of intermittent sunlight and eliminating its restrictions. Consequently, a sustainable and uninterrupted hydrogen production system can be realized. To achieve this goal, our group developed FeNiP nanoparticles loaded onto N,P-modified carbon nanosheets (FeNiP/NPCS) as trifunctional electrocatalysts for a zinc–air battery-powered water electrolyzer for hydrogen production.503 Solar cells were used as the energy source to charge the zinc–air battery under sunlight, effectively storing transient solar energy. Upon charging with a commercial silicon photovoltaic system for 10 minutes (as illustrated in Fig. 20b), the discharge platform with a current density of 10 mA cm−2 of the FeNiP/NPCS-catalyzed battery operated stably for 1300 minutes without any significant variations, demonstrating the significant potential for assembling a self-driven energy conversion and storage system for green H2 production. Expanding on this concept, a self-powered system based on FeNiP-loaded carbon nanofibers as trifunctional electrocatalysts achieved uninterrupted 24 h H2 production from the two-electrode water splitting electrolyzer.504 This system was powered by a silicon photovoltaic cell during the day and an aqueous zinc–air battery during the night, as depicted in Fig. 20c. The corresponding current density versus time curves are presented in Fig. 20d, and such an integrated hydrogen production device demonstrated continuous operation throughout a 15 day test period. In another example, Wang et al. proposed and assembled a self-powered energy system based on a Ni–P/Fe–P collaborative electrocatalyst for the HER, OER, and ORR.505 This system achieved a solar-to-hydrogen efficiency of 4.6% and a solar-to-water splitting device efficiency of 5.9%. Additionally, various catalysts, including core–shell Co2P@NC electrocatalysts,506 N,S-codoped 3D carbon matrix with Co9S8/CoO heterojunction (Co–S–O/NSCN),507 Co0.85Se/NC,508 and CoNi alloys coupled N-doped carbon nanotube arrays (CoNi@NCNTs/CC),509 have demonstrated the feasibility of self-powered energy systems utilizing solar energy. In summary, these solar energy-assisted systems fully utilize solar energy to generate renewable electricity and produce clean H2 without relying on external nonsolar energy sources or causing environmental pollution. They realize a closed-loop energy and substance cycle, offering a novel approach to energy conversion, storage, and the production of clean and renewable energy.
5.2 Hydrazine fuel cell-assisted OHzS
Although significant achievements have been made in the hydrogen production by hydrazine-assisted water electrolysis, most of the current research requires the utilization of external electrical energy, which poses challenges for portable devices and outdoor operations. To overcome this limitation, the concept of self-power supply has emerged, leading to innovative breakthroughs. Direct hydrazine fuel cells (DHzFCs) have garnered significant attention because they utilize hydrazine as the fuel and an oxidant, typically oxygen or air, to generate electricity through an electrochemical reaction.66,510 These fuel cells offer high theoretical power density and produce carbon-free by-products (N2 and H2O). Given the high reactivity of N2H4 with H2O and H+ to form N2H5+, alkaline solutions are commonly employed as the electrolyte in DHzFCs, and therefore transition metal-based catalysts can be used for DHzFCs.511 Additionally, hydrazine is a liquid at room temperature, which simplifies the storage and transportation compared to other gaseous or cryogenic fuels, ensuring convenience and safety in the development of DHzFCs. Generally, the OCV of DHzFCs is approximately 0.8 to 1.0 V.512 The favorable kinetics of the HzOR in the OHzS unit necessitates a voltage of about 0.3–0.5 V to achieve H2 production. As a result, one DHzFC can effectively drive the OHzS for H2 production, offering a significant advantage compared to zinc–air battery assisted overall water splitting.In 2018, inspired by the previous advancements in the fields of the HER and fuel cells, Ding's group developed a highly efficient and stable bifunctional catalyst composed of thin Fe-doped CoS2 (Fe–CoS2) nanosheets for both the HER and the HzOR.513 The Fe–CoS2 nanosheets demonstrated Pt-like activity for the HER, exhibiting low overpotentials at 10 mA cm−2 (40 mV in 1.0 M KOH, 31 mV in 0.5 M H2SO4, and 49 mV in 1.0 M PBS), high stability and 93–98% FE at all pH values. Furthermore, the Fe–CoS2 nanosheets exhibited a working potential of only 129 mV to achieve a current density of 100 mA cm−2 for the HzOR in 1.0 M KOH. Considering these excellent properties, the Fe–CoS2 nanosheets were utilized as the anode material to construct a DHzFC using H2O2 or O2 as an oxidizing agent. The assembled DHzFCs demonstrated remarkable maximum power density values of 246 mW cm−2 (H2O2) and 125 mW cm−2 (O2) (Fig. 21a), among the best reported values for DHzFCs employing Co-based electrocatalysts. To further explore the ability of the Fe–CoS2 nanosheets, they integrated a DHzFC with an electrolyzer for OHzS, forming a self-powered system for H2 production. In this configuration, hydrazine served both as the fuel for the DHzFC and as the splitting target (Fig. 21b). Notably, the Fe–CoS2 nanosheets were employed as both the anode and cathode catalysts for OHzS. The self-powered system exhibited good stability at 0.7 V for 20 h and achieved a hydrogen evolution rate of 9.95 mmol h−1 (Fig. 21c). Theoretical calculations demonstrated that the remarkable performance of the Fe–CoS2 nanosheets was attributed to the introduction of Fe dopants, which decreased the free-energy changes of H adsorption and the dehydrogenation of adsorbed NH2NH2 on CoS2.
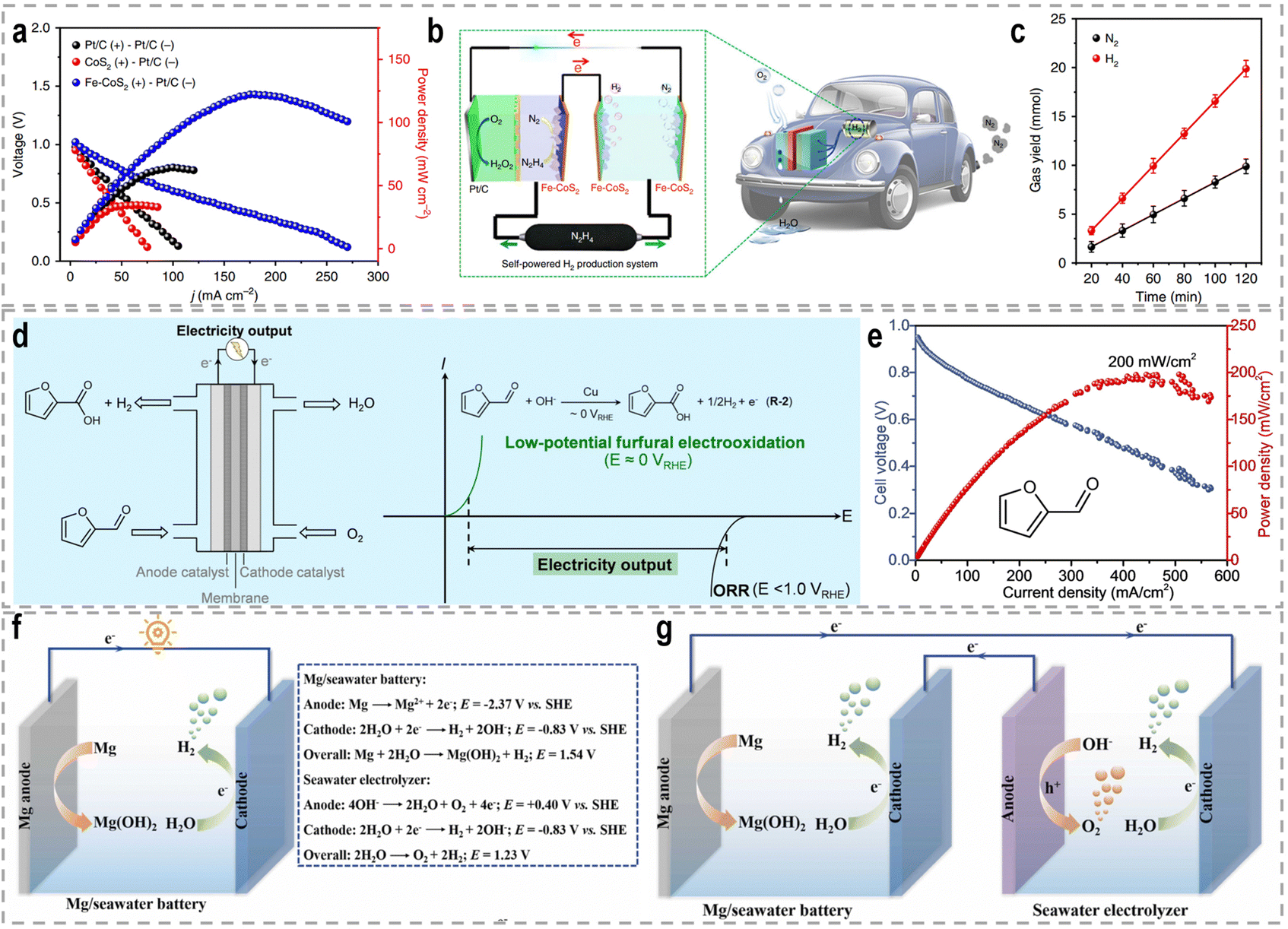 | ||
| Fig. 21 (a) Current density–voltage and current density–power density plots for the DHzFC with the oxidizing agent of O2. (b) Schematic illustration of a self-powered H2 production system integrating a DHzFC and an OHzS unit. (c) Generated amounts of H2 and N2 in the system with the hydrazine concentration of 5.3 M at 0.7 V and room temperature.513 Reproduced with permission. Copyright 2018, Nature Publishing Group. (d) Electrocatalytic coproduction of furoic acid and hydrogen in the electricity output mode. Left: Configuration of a single cell; right: polarization curves of the corresponding half-reactions. (e) Polarization and power-density curves for the assembled device using 0.1 M furfural as the substrate undergoing oxidation.517 Reproduced with permission. Copyright 2022, Wiley-VCH. (f) Illustration of the structure and working principle of the Mg/seawater battery. (g) Illustration of the Mg/seawater battery driven self-powered seawater electrolysis system.40 Reproduced with permission. Copyright 2022, Elsevier. | ||
Zhang et al. fabricated a Ru2P catalyst with a partially exposed surface (RP-CPM).192 The DHzFC utilizing the RP-CPM catalyst achieved a high power density of 64.77 mW cm−2 at an output potential of 0.36 V. Additionally, the RP-CPM catalyst exhibited excellent HER performance, delivering 10 mA cm−2 at a low overpotential of 24 mV, surpassing that of commercial Pt/C catalysts (35 mV). By integrating the DHzFC in series with a water splitting electrolyzer, a self-powered system was assembled, which is capable of supplying a high output voltage of 1.0 V and generating hydrogen with a productivity of 0.72 mmol h−1. To address the cost of electrocatalysts, they also prepared a hierarchical porous nanosheet arrays with rich Ni3N–Co3N heterointerfaces on nickel foam (Ni3N–Co3N/NF).37 This Ni3N–Co3N/NF exhibited excellent HER and HzOR performance, achieving 10 mA cm−2 at working potentials of −43 mV and −88 mV, respectively. In a constructed OHzS with Ni3N–Co3N/NF, low cell voltages of 0.071 and 0.76 V were sufficient to reach 10 and 400 mA cm−2, respectively. Additionally, to demonstrate the potential feasibility in mobile devices, a self-powered hydrogen production system was fabricated by utilizing Ni3N–Co3N/NF, which exhibited an impressive H2 production rate of 0.65 mmol h−1. Through experimental characterization and DFT calculations, they confirmed that interfacial engineering effectively regulated the electronic structure and optimized the adsorption strength of active species, thereby enhancing the kinetics of the catalytic reactions. Furthermore, the authors also explored other electrocatalysts for DHzFC and hydrazine-assisted H2 production, including Mo-doped Ni3N and Ni heterostructures,198 P, W codoped Co3N nanowires,205 and dual nanoislands on a Ni/C hybrid nanosheet array,229 all of which showed an extraordinary performance. However, it is important to note that the anodic catalysts in these DHzFCs still relied on commercial Pt/C catalysts to catalyze the ORR.514 Therefore, the development of trifunctional electrocatalysts for the HzOR, HER, and ORR remains a prominent research direction in DHzFC-powered OHzS.
Simultaneously, the utilization of hydrazine-assisted self-powered hydrogen generation under seawater conditions presents a promising avenue for promoting chlorine-free hydrogen production. In 2021, Sun et al. integrated a hybrid seawater electrolyzer with a DHzFC, with NiCo@C/MXene/CF as the anode and 20% Pt/C as the cathode in a DHzFC.129 This self-powered system demonstrated the capability to produce hydrogen from seawater at a remarkable rate of 1.6 mol h−1 gcat−1, solely relying on hydrazine as the consumable energy. Subsequently, Wang's group fabricated FeP/FeNi2P encapsulated in N, P co-doped hierarchical carbon (FeNiP-NPHC) in situ grown on nickel foam via a hydrothermal-pyrolysis-phosphidation procedure.515 Benefiting from the strong coupling effect among FeP, FeNi2P, and N, P co-doped carbon at the three-phase heterojunction interface, as well as the unique 1D/3D hierarchical structure, the prepared FeNiP-NPHC showed excellent ORR (E1/2 = 0.83 V), HzOR (E100 = 7 mV), and HER (η100 = 180 mV) performance in alkaline seawater. DFT calculations indicated that the construction of this unique interface in FeNiP-NPHC effectively modulates the d-band center and electronic structure, facilitating the fine-tuning and optimization of its trifunctional electrocatalytic behavior. As a result, a DHzFC assembled using FeNiP-NPHC displayed an OCV of 0.98 V and a peak power density of 31 mW cm−2 at ambient temperature. Furthermore, a two-electrode OHzS system using FeNiP-NPHC demonstrated the intuitive activity improvement compared to OWS in a seawater system. Finally, a self-powered system by integrating DHzFC using FeNiP-NPHC as both the anode and cathode to drive OHzS in alkaline seawater for H2 production was constructed, exhibiting nearly 100% FE for H2 production in OHzS. This group also fabricated Ru, Fe dual-doped Ni2P nanosheets as trifunctional catalysts for DHzFC powered OHzS to realize the self-powered H2 production in seawater.516 This integrated self-powered system achieved an impressive hydrogen production rate of 10.8 mmol h−1 without any other power supplies.
5.3 Others
Wang et al. found that the aldehyde groups of HMF and furfural can be selectively oxidized to their corresponding carboxylates together with hydrogen production at remarkably low potentials (onset potential as low as 0.05 V vs. RHE).417 Therefore, the combination of the HER and low-potential aldehyde oxidation can enable hydrogen production at both the anode and cathode. In contrast, the conventional hydrogen fuel cells operate through the coupling of the hydrogen oxidation reaction (HOR) and the ORR to generate electrical energy. Notably, the potential for low-potential aldehyde oxidation is comparable to that of the HOR, as depicted in Fig. 21d. Hence, it is possible to construct new types of fuel cells by combining low-potential aldehyde oxidation with the ORR. By adopting this innovative approach, simultaneous hydrogen production, biomass upgrading to carboxylates, and electricity generation can be achieved simultaneously. Based on this strategy, they coupled anodic low-potential furfural oxidation and the cathodic ORR to construct a novel electrochemical cell that generates electricity together with H2 production and furfural oxidation.517 Employing Cu as anodic and Pt/C as cathodic catalysts, an OCV of 0. 95 V was achieved, and the maximum power density reached about 200 mW cm−2 at a current density of approximately 450 mA cm−2 (Fig. 2e). Furthermore, this design concept exhibits versatility and suitability for other substrates containing similar types of aldehyde groups, including HMF, 5-methyl furfural, and 4-carboxybenzaldehyde, indicating its broad applicability.The selective electrooxidation of formaldehyde to formic acid presents an alternative pathway for hydrogen production. Considering the lower onset oxidation potential of formaldehyde (∼0.1 V vs. RHE), Fu et al. developed a direct formaldehyde fuel cell, which enabled the generation of electricity while simultaneously producing H2 and valuable formate at the anode.425 They fabricated a highly active and selective electrocatalytic anode of Cu nanosheets arrays on Cu foam, which ensures the efficient conversion of formaldehyde to formic acid and hydrogen at the anode. Upon assembling a direct formaldehyde fuel cell, the Cu nanosheet array electrode demonstrated an impressive OCV of up to 1 V and a peak power density of 350 mW cm−2. This distinctive configuration enabled the production of 1 kW h of electricity, 0.62 N m3 of hydrogen, and 53 mol of formate consuming 53 mol of formaldehyde fuel.
Mg/seawater batteries, which utilize seawater as both the electrolyte and the cathodic reactant, offer a dual functionality of electricity generation and hydrogen production at the cathode, as depicted in Fig. 21f. These batteries hold significant promise as power sources for long-term operation in deep-sea apparatuses due to their open-structured design and the use of seawater as the electrolyte. By integrating a seawater battery pack with a seawater electrolyzer, as illustrated in Fig. 21g, a self-powered seawater electrolysis system driven by Mg/seawater batteries can be realized. Notably, since hydrogen can be generated from both the cathodes of the seawater electrolyzer and the Mg/seawater batteries, the total hydrogen production rate is expected to be substantially high in this self-powered seawater electrolysis system. In 2022, Jiang's group demonstrated a self-powered direct seawater electrolysis system driven by Mg/seawater batteries, which realized continuous hydrogen production and held potential for mobile and undersea apparatus applications.40 To achieve this goal, they employed a MoNi/NiMoO4 heterostructure as the catalyst for the HER occurring at the cathodes of both the Mg/seawater batteries and the seawater electrolysis unit. Notably, the self-powered seawater electrolysis system with a MoNi/NiMoO4 catalyst achieved a high hydrogen production rate of 12.11 mL cm−2 h−1, and the conversion efficiency of Mg-to-hydrogen reaches up to 83.97%. These results surpass the performance of most of the state-of-the-art self-powered hydrogen production systems.
In order to harness solar energy for continuous self-powered hydrogen production, various energy storage systems have been integrated with photovoltaic devices. For instance, Sun et al. utilized a Li-ion battery to power a conventional water electrolyzer, which can be conveniently charged by commercial solar cells.518 Zhang et al. developed aqueous Ni–Zn batteries with an output voltage of 1.75 V to connect solar cells and water splitting devices.519 Sun et al. employed a photovoltaic cell to drive a micro zinc-ion battery array, providing a stable voltage to continuously power the seawater electrolyzer.520 These hybrid systems demonstrated the ability to sustain uninterrupted water splitting for 24 h. Besides, the supercapacitor powered OWS was also reported.521,522 Despite these innovative approaches, several key challenges persist in this field. These challenges include addressing safety concerns associated with the energy storage component, enhancing the stability and durability of the water electrolyzer, and overcoming the complexity and bulkiness of the overall system configuration.
Significant progress has been achieved in the self-powered systems for the enhanced electron utilization and H2 production without the need for an external power supply, as summarized in Table 6. However, it is important to note that these systems still have certain limitations. Zinc–air batteries, for example, are not ideal for high-current applications due to their relatively high internal resistance, which restricts their ability to deliver high power outputs. This characteristic makes them less suitable for H2 production at large current densities. Moreover, the high cost of hydrazine presents a significant constraint for the widespread adoption of hydrazine-contained energy systems for large-scale H2 production. As a result, it is crucial to continue exploring and investing in novel research and development efforts to overcome the limitations associated with current self-powered H2 production systems, ultimately facilitating the widespread adoption of self-powered H2 production technologies for a sustainable energy future.
| Catalysts | Reaction types | η 10 (mV) for HER | E 1/2 (V) for ORR | η 10 (mV) for OER or Potential (mV) for HzOR | Peak power density (mW cm−2) | Stability for zinc–air battery/fuel cells | Potential at 10 mA cm−2 for electrolyzer | Stability for electrolyzer | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| NiS2/CoS2–O NWs | ZAB assisted OWS | 174 | 0.70 | 235 | — | 30 h at 5 mA cm−2 | 1.768 | 20 h at 1.70 V | 35 |
| GO-PANi31-FP | ZAB assisted OWS | 520 | — | — | 38 | 50 h | — | — | 481 |
| CoP@PNC-DoS | ZAB assisted OWS | 173 | 0.803 | 316 | 138.57 | 150 h at 30 mA cm−2 | 1.740 | 30 h at 10 mA cm−2 | 482 |
| CoMn2−xCrxO4 | ZAB assisted OWS | 180 | 0.82 | 390 | 140.26 | 43 h at 10 mA cm−2 | — | 20 h at 10 mA cm−2 | 483 |
| FeCo/Co2P@NPCF | ZAB assisted OWS | ∼260 | 0.79 | 330 | 154 | 107 h at 10 mA cm−2 | 1.68 | 43200 s at 1.75 V |
484 |
| Cu-Foam@CuCoNC-500 | ZAB assisted OWS | 59.2 | 0.84 | 245 | 140 | 360 h at 10 mA cm−2 | 1.52 | 20 h at 1.6 V | 485 |
| CoNC | ZAB assisted OWS | 36 | 0.858 | 229 | 181.3 | 110 h at 5 mA cm−2 | 1.51 | 300 h at 30 mA cm−2 | 486 |
| CoFeN-NCNTs//CCM | ZAB assisted OWS | 151 | 0.84 | 325 | 145 | 445 h at 10 mA cm−2 | 1.63 | 55 h retaining 85.2% | 489 |
| FeZn4Co@CNFs | ZAB assisted OWS | 200 | 0.83 | 360 | 107.6 | 118 h at 10 mA cm−2 | — | — | 499 |
| FeNiP/NPCS | ZAB assisted OWS | 181 | 0.84 | 318 | 163 | 110 h at 10 mA cm−2 | 1.71 | 20 h | 503 |
| FeNiP@p-NPCF/CC | ZAB assisted OWS | 89 | 0.82 | 317 | 117 | 500 h at 10 mA cm−2 | 1.67 V at 20 mA cm−2 | 20 h | 504 |
| NiFeP | ZAB assisted OWS | — | — | 370 mV at 50 mA cm−2 | 138 | >600 h at 10 mA cm−2 | 1.9 V at 20 mA cm−2 | — | 505 |
| Fe–Co2P@Fe-N-C | ZAB assisted OWS | 77 | 0.88 | 300 | 81.3 | ∼280 h at 10 mA cm−2 | 1.58 | 10 h | 506 |
| Co0.85Se/NC | ZAB assisted OWS | 216 | 0.797 | 275 | 108 | 160 h at 20 mA cm−2 | 1.70 | 20000 s |
508 |
| Fe2P/Co@NPC | ZAB assisted OWS | 235 | 0.876 | 331 | 233.56 | 180 h | 1.73 | 60 h | 523 |
| PPy/FeTCPP/Co | ZAB assisted OWS | 240 mV (0.1 M KOH) | 0.86 | 380 mV (0.1 M KOH) | — | 24 h | — | 12 h at 10 mA cm−2 | 524 |
| NiCoOS hollow polyhedron | ZAB assisted OWS | 300 | 0.79 | 470 mV | 90 | 170 h at 5 mA cm−2 | 1.52 | 15 h | 525 |
| Fe0.5Ni0.5@N-GR | ZAB assisted OWS | 350 | 0.83 | 210 | 85 | 40 h at 20 mA cm−2 | — | — | 526 |
| Pt@CoS2-NrGO | ZAB assisted OWS | 39 | 0.85 | 235 | 114 | 55 h at 10 mA cm−2 | 1.48 | 50 h at 200 mA cm−2 | 527 |
| Ni3N–Co3N PNAs/NF | DHzFC-assisted OHzS | 43 | — | −88 mV at 10 mA cm−2 | 60.3 mW cm−2 with commercial Pt/C cathode | — | 71 mV at 10 mA cm−2 | 20 h at 0.14 V | 37 |
| NiCo@C/MXene/CF | DHzFC-assisted OHzS | 49 | — | −25 mV at 100 mA cm−2 | 53.5 mW cm−2 with commercial Pt/C cathode | — | 0.31 V at 500 mA cm−2 | 150 h at 500 mA cm−2 | 129 |
| RP-CPM | DHzFC-assisted OHzS | 24 | — | −27 mV at 10 mA cm−2 | 64.77 mW cm−2 with commercial Pt/C cathode | — | 23 mV at 10 mA cm−2 | 20 h at 10 mA cm−2 | 192 |
| Mo-Ni3N/Ni/NF | DHzFC-assisted OHzS | 45 | — | −0.3 mV at 10 mA cm−2 | 37.8 mW cm−2 with commercial Pt/C cathode | — | 55 mV at 10 mA cm−2 | 10 h at 50 mA cm−2 | 198 |
| PW-Co3N NWA/NF | DHzFC-assisted OHzS | 41 | — | −55 mV at 10 mA cm−2 | 46.3 mW cm−2 with commercial Pt/C cathode | — | 28 mV at 10 mA cm−2 | 20 h at 50 mA cm−2 | 205 |
| Fe–CoS2 | DHzFC-assisted OHzS | 40 | — | 129 mV at 100 mA cm−2 | 125 mW cm−2 with commercial Pt/C cathode | — | 0.95 V at 500 mA cm−2 | 40 h at 100 mA cm−2 | 513 |
| FeNiP-NPHC | DHzFC-assisted OHzS | 180 mV at 100 mA cm−2 | 0.83 | 7 mV at 100 mA cm−2 | 31 | — | 0.25 V at 100 mA cm−2 | 150 h at 1.35 V | 515 |
| RuFe-Ni2P@NF | DHzFC-assisted OHzS | 54 | — | 140 mV at 100 mA cm−2 | 60.1 mW cm−2 with commercial Pt/C cathode | — | 0.41 V at 200 mA cm−2 | 100 h at 100 mA cm−2 | 516 |
| Cu/Cu foam (anode) | — | — | — | — | 200 mW cm−2 with commercial Pt/C cathode | — | — | — | 517 |
| CF@Cu-NS (anode) | Direct formaldehyde fuel cell | — | — | — | 350 mW cm−2 with commercial Pt/C cathode | — | — | — | 425 |
| MoNi/NiMoO4 | Mg/seawater batteries powered seawater electrolysis | 29 | — | — | 21.08 | 100 h at 3 mA cm−2 | — | — | 40 |
| Ni/V2O3 | Mg/seawater batteries powered seawater electrolysis | 34 | — | — | 17.81 | 24 h at 3 mA cm−2 | — | — | 528 |
6. Self-catalyzed H2 production
Self-catalyzed H2 electrolysis involves driving electrocatalytic processes using self-generated electricity for hydrogen production, eliminating the need for external power sources and making the electrolysis system more efficient and sustainable.529 In self-catalyzed hydrogen production systems, metallic anodes are commonly used to obtain self-generated electricity through chemical reactions at the anode. This self-generated electricity, which can flow through an external circuit, is then utilized to drive the electrocatalytic reactions at the cathode, where hydrogen gas is evolved. By integrating the electrochemical reactions at the anode and cathode, self-catalyzed hydrogen production systems allow for simultaneous H2 production and electricity generation, making them more efficient and self-sustained. This approach reduces the dependence on external power sources, making the hydrogen production process more energy-efficient and potentially more economically viable. Exploiting this phenomenon offers tremendous potential for the development of self-catalyzed metal–H2O batteries. It is worth noting that various materials have been explored as anode materials in self-catalyzed batteries, including metals such as Zn, Al, and Mg, which exhibit spontaneous reactivity with water and thus forms the corresponding metal oxides during the discharge process.530 During the charging process, the coordinated metal ions in the electrolyte are reduced back to their original metal electrode, allowing for the regeneration of the metal anode. This cyclic process of oxidation and reduction enables the self-sustainability of the metal–H2O battery and provides a continuous source of hydrogen and electricity. Of course, each metal anode has its own advantages and limitations in terms of electrochemical reactivity, cost, and stability. Given the challenges associated with the large-scale deployment of hydrogen, including risks of explosion and high storage and transportation costs, the self-powered, functional, and portable metal–H2O batteries hold promise for on-demand H2 generation and energy supply.6.1 Metal–H2O battery
Lithium (Li), being the most prevalent anode metal, exhibits a sufficiently low equilibrium potential (−3.040 V vs. standard hydrogen electrode (SHE)), providing ample driving force for water splitting reactions. However, a direct reaction between the pure Li metal and water is hindered due to the explosive nature of Li in an aqueous electrolyte. To circumvent this limitation, Guo et al. designed an asymmetric dual cell employing an organic/aqueous electrolyte with a Li1.35T1.75Al0.25P2.7Si0.3O12 (LTAP) film serving as the separator, wherein the oxidation of the Li anode provides electrons for the cathodic hydrogen production, as depicted in Fig. 22a.531 Benefiting from the specific operational conditions, this Li-based battery can be adapted as either a Li–O2 battery (O2-rich environments) or a Li–H2O fuel cell (O2-free environments). Although this Li-based fuel cell demonstrates remarkable reactivity and a wide working voltage range, it still exhibits some drawbacks such as low hydrogen production efficiency, limited availability of lithium resources, safety concerns associated with the organic electrolyte, and high cost of the separator, and a suboptimal electrical energy output persists. To address these challenges, alternative alkaline-earth metals have been explored for constructing functional batteries. In 2018, Wen et al. reported a Zn–H2O battery operating in an alkaline-acid hybrid system, enabling cathodic H2 generation and electricity production.532 This battery configuration involved a Zn anode and a commercial Pt/C cathode, as illustrated in Fig. 22b. To enhance the kinetics of Zn oxidation and the HER at the cathode, NaOH (4.0 M) and H2SO4 (2.0 M) were employed as dual electrolytes in the anode and cathode, respectively, separated by a bipolar membrane to prevent direct neutralization of the anolyte and catholyte with the release of heat energy. The hybrid alkaline–acid Zn–H2O fuel cell theoretically exhibited an OCV of 1.32 V and an energy density of 1082.4 W h kg−1 with hydrogen production at the cathode according to the Nernst equation. Experimentally, the cell demonstrated impressive performance, with an OCV of 1.249 V, an energy density of up to 934 W h kg−1, and a power density of 80 mW cm−2 (Fig. 22c). Furthermore, it exhibited a hydrogen production rate of 0.166 mL s−1 at a voltage of 0.6 V, along with excellent long-term stability at a current density of 10 mA cm−2 with an output voltage of 1.16 V. | ||
| Fig. 22 (a) Schematic illustration of a Li–water fuel cell which consists of a Li anode in the organic electrolyte and a cathode electrode in the alkaline electrolyte, separated by a LTAP film.531 Reproduced with permission. Copyright 2017, American Chemical Society. (b) Schematic illustration of the as-proposed Zn–H2O fuel cell, separated by a bipolar membrane. (c) Polarization curves (left-hand y axis) and power density (right-hand y axis) for Zn–H2O fuel cells using Pt/CNTs, Pt/C, and CNTs.532 Reproduced with permission. Copyright 2018, Wiley-VCH. (d) Schematic illustration of aqueous Zn– or Al–CO2 systems and their reaction mechanism.535 Reproduced with permission. Copyright 2019, Wiley-VCH. (e) Self-co-electrolysis system assembled using a Zn plate as the anode, N,Cu-CoP/CC as the cathode, and neutral 1.0 M PBS as the electrolyte that can generate H2 gas, NaZnPO4, and electric energy concurrently.546 Reproduced with permission. Copyright 2022, Wiley-VCH. (f) Concept of the coupled systems of electrolytic hydrogen evolution and sulfur production with the assistance of the Fe2+/Fe3+ redox cycle and H2S absorption reaction.547 Reproduced with permission. Copyright 2018, Wiley-VCH. (g) The Fe(II) electrooxidation coupled with HER, and the Fe(III) can be chemically reduced to Fe(II) by SO2. (h) Schematic diagram of the cycle of iron ion redox by adding SO2. (i) LSV curves using the flow cell electrolyzer in 0.1 M FeCl3 and 0.5 M H2SO4 electrolyte with continuous inflow SO2.550 Reproduced with permission. Copyright 2023, Wiley-VCH. | ||
Although Zn–H2O batteries exhibit significant potential for future applications without the use of organic electrolytes, there are concerns regarding their high cost of bipolar membranes and potential risks associated with dual-electrolyte systems employing strong acid and strong alkaline solutions. To address these challenges, Hou et al. presented an alkaline Zn–H2O battery utilizing 1.0 M KOH as the anolyte and catholyte.533 The HER performance in this battery was enhanced by the strong coupling effect of the constructed interface between Ni2P nanoparticles and Ni-MOF nanorod arrays, leading to a reduced overpotential of 132 mV at 10 mA cm−2. Although the alkaline Zn–H2O battery achieved a notable power density of 4.1 mW cm−2, its current density remained constrained by the relatively low ionic conductivity resulting from the implementation of the ion exchange membrane. In contrast to alkaline–acid Zn–H2O batteries, this alkaline device demonstrated stable discharge–charge cycling behavior at 5 mA cm−2 over 90 cycles, highlighting its promising potential for long-term operation. Nadeema et al. developed an Al–H2O battery in a single-pool electrolyzer, employing a low concentration of alkaline solution (0.1 M KOH) as the electrolyte.534 To enhance the kinetics of the HER, they engineered a novel Co@CoAl-LDH/N-doped graphene catalyst. This innovative catalyst exhibited excellent performance for the HER in the Al–H2O battery. The assembled Al–H2O battery demonstrated a relatively high cell voltage of approximately 1.0 V at a current density of 5 mA cm−2. The specific capacity of the battery was estimated to be approximately 538 mA h g−1. Notably, this configuration eliminated the risks associated with a dual-electrolyte system and minimized the additional costs associated with the use of an ion exchange membrane.
Kim et al. have designed a noteworthy aqueous Zn- or Al-based CO2 system for both hydrogen production and electrical energy generation.535 In contrast to the aforementioned metal–H2O batteries, these Zn– or Al–CO2 devices do not utilize strong alkaline or acid solutions in the cathode pool. Instead, CO2 introduced into the electrolyte would react with water to form H2CO3. Upon the oxidation of the metal at the anode, the H+ ions dissociated from H2CO3 undergo reduction to yield H2, as depicted in Fig. 22d. Simultaneously, K+ ions migrate from the anode pool to the cathode pool through a glass membrane to maintain charge balance. The Al–CO2 system exhibited a high OCV of 1.3 V and achieved a maximum power density of 125 mW cm−2. Furthermore, the actual H2 generation rate was determined to be 0.681 mL min−1 at a current of 100 mA, which closely aligns with the theoretically predicted generation rate of 0.696 mL min−1. This measurement highlights a remarkable FE of 97.9% for H2 production, emphasizing the efficiency of the system in converting electrical energy to hydrogen gas.
The Mg/seawater battery represents another type of metal–H2O battery that not only enables electricity generation but also facilitates the production of clean hydrogen using abundant seawater as a resource.536–538 In comparison to the traditional Mg/dissolved oxygen battery,539,540 which relies on the oxygen concentration present in seawater, the Mg/seawater battery exhibits significant advantages. Notably, the cathodic reaction of the Mg/seawater battery involves the HER directly from seawater, eliminating the dependency on dissolved oxygen and thereby enhancing the potential for improved discharging performance.540–543 The performance of the Mg/seawater battery is primarily governed by the cathode, and the sluggish kinetics of the HER in neutral seawater necessitates the development of ideal cathode catalysts to minimize the polarization of the cathodic HER. Early works on the Mg/seawater batteries employed Pt-based cathodes.536 To address this challenge, Jiang et al. developed a unique porous CoP/Co2P heterostructure electrode for the pH-universal HER.544 The optimized porous CoP/Co2P heterostructure demonstrates remarkable HER performance, with low overpotentials of 87, 133, and 454 mV at 10 mA cm−2 in acidic, alkaline, and seawater media, respectively. The assembled Mg/seawater battery with the porous CoP/Co2P heterostructure cathode exhibited promising performance, achieving a peak power density of 6.28 mW cm−2, maintaining satisfactory stability over 24 h. Subsequently, this group also developed heterostructured MoNi/NiMoO4 catalysts for Mg/seawater batteries, which yielded a remarkable peak power density of up to 21.08 mW cm−2.40
The metal–H2O batteries discussed herein fully meet the principles of green chemistry, demonstrating both non-toxicity and high efficiency. However, the generation of electrical energy and value-added chemicals in these systems relies on the consumption of pure metals, which increases the cost of these systems. Kékedy-Nagy et al. found that MgNH4PO3 could be synthesised by the spontaneous combination of Mg2+, NH4+ and PO43−.545 Notably, Mg as an active alkaline–earth metal element possesses a low equilibrium potential (−2.372 V vs. SHE) and readily undergoes oxidation to form Mg2+ ions. By employing NH4H2PO4 solution as the electrolyte, the Mg–H2O battery can simultaneously produce MgNH4PO3 at the anode and hydrogen gas at the cathode.
Due to the high cost and oxidation activity, Mg is not an ideal material for application in aqueous solutions for safety and cost concerns. As an alternative, Zn has emerged as a promising anode material for metal–H2O batteries due to its moderate oxidation kinetics. While alkaline electrolytes have been commonly used in Zn–H2O batteries to enhance reaction kinetics, the utilization of neutral electrolytes has been rarely reported. Shi's group firstly reported a neutral Zn–H2O battery by utilizing PBS as the electrolyte, as depicted in Fig. 22e.546 To accelerate the reaction kinetics of the Zn–H2O battery, a N and Cu dual-doped CoP (N,Cu–CoP) catalyst was synthesized as the cathode material, which showed excellent activity for the HER with a low overpotential of 68 V to reach 10 mA cm−2 in 1.0 M PBS electrolyte. The N,Cu–CoP-catalyzed Zn–H2O battery exhibited an OCV of 0.79 V, a maximum power density of 1.83 mW cm−2 and simultaneously generated a hydrogen production rate of 13.7 mL cm−2 h−1 without requiring an external energy supply. Furthermore, by adjusting the pH values (6, 7, and 8) and concentrations (0.01, 0.1, and 1.0 M) of the PBS electrolyte, various valuable solid-state chemicals, such as NaZnPO4 (an important raw material for heat-reflective materials, Ni–Zn battery anode materials, and light-emitting diodes) and Zn3(PO4)2 can be controllably synthesized at the anode during the discharge process.
Besides the electrooxidation of pure metal, the oxidation of multivalent metals with the low potential (e.g., 0.69 V vs. RHE for Cu+/Cu2+, and 0.75 V vs. RHE for Fe2+/Fe3+) has been considered as a suitable substitution reaction of the OER for energy-saving H2 production. For instance, with the electrodes of CC@N-CoP (cathode)//CC(anode) in 0.5 M H2SO4 (catholyte)//0.5 M H2SO4 with 0.96 M FeSO4/0.74 M Fe3(SO4)2 (anolyte), the fabricated electrolyzer only requires 0.89 V to drive a current density of 10 mA cm−2 due to the lower redox potential of Fe2+/Fe3+, saving 53% energy consumption compared to the traditional OWS.547 Wang et al. used a Cu+/Cu2+ redox cycle to replace the OER process, and the assembled hybrid system could deliver 100 mA cm−2 at 0.94 V (2.02 V in the case of OWS), correspondingly electricity consumption is 2.23 kW h N m−3 H2 at 100 mA cm−2 (4.80 kW h N m−3 H2 for OWS).548 However, the anodic reaction of metal ions also faces a challenging obstacle of how to improve the continuity of the electrooxidation for metal ions. To address this issue, researchers have proposed the introduction of a reducing agent, which facilitates the spontaneous conversion of high-valent cations into low-valence components. Fig. 22f provides an illustrative example, wherein a coupled H2S absorption and Fe2+/Fe3+ redox cycle is employed. This approach ensures the uninterrupted electrooxidation of metal ions while simultaneously enabling low-potential H2 production when coupled with the HER. Notably, glucose47 and ascorbate548 were introduced into the Cu+/Cu2+ redox cycle, glucose/starch/cellulose549 and H2S547 were coupled with the Fe2+/Fe3+ redox cycle. However, the utilization of these reducing agents in redox systems may not be economically or environmentally sustainable due to the consumption of organic resources or chemicals. In a recent study inspired by the reducing ability of SO2, Wang's group proposed a continuous Fe2+/Fe3+ redox assisted by the SO2 waste gas under ambient conditions for hydrogen production (Fig. 22g).550 In this system, SO2 chemically reduces Fe3+ to Fe2+, and the resulting Fe2+ is subsequently electrochemically oxidized to Fe3+, providing electrons for cathodic H2 production. Meanwhile, SO2 was converted to sulfuric acid (Fig. 22h). Due to the low oxidation potential of Fe2+, the assembled electrolyzer achieved a significantly lower electrolytic voltage of 0.97 V at 10 mA cm−2 compared to traditional water electrolysis (1.85 V) (Fig. 22i).
6.2 Others
In contrast to water, sulfide ions (S2−) exhibit a greater propensity to donate electrons through the SOR (for instance, S2− → S + 2e−, E0 = −0.48 V vs. SHE), as the SOR is thermodynamically more favorable than the OER. Weng et al. developed WS2 nanosheets (NSs) using a low-temperature molten-salt-assisted method, which demonstrated remarkable electrocatalytic properties for the alkaline SOR and the acidic HER.232 The researchers assembled a hybrid alkali–acid electrochemical cell, with WS2 NSs serving as the alkaline anode and the acidic cathode. The anolyte and catholyte were separated by a cation exchange membrane (CEM) and an anion exchange membrane (AEM), respectively, with deionized water flowing through the middle chamber, as depicted in Fig. 23a. In this cell, the SOR occurs at the alkaline anode, releasing electrons that flow through the external circuit and are consumed at the cathode through the HER. Na+ and SO42− ions are simultaneously transported into the middle chamber via the CEM and AEM, respectively, completing the circuit. For comparison, a conventional cell was also established by employing 1.0 M NaOH as the anolyte and catholyte for reference purposes. The LSV curves of the hybrid alkali–acid SOR/HER cell, alkali–alkali SOR/HER cell, and alkali–alkali OER/HER cell are presented in Fig. 23b. The hybrid alkali–acid cell demonstrated the ability to achieve oxidation conversion of S2− and hydrogen generation under bias-free voltage conditions. In contrast, the alkali–alkali SOR/HER cell required a higher voltage of 0.73 V to drive the electrolysis and an applied voltage of 1.17 V to achieve a current density of 10 mA cm−2. Notably, the alkali–alkali OER/HER cell demanded a significantly higher voltage (>2.0 V) to initiate water splitting due to the high overpotential of WS2 NSs for the OER. The alkali–acid electrochemical cell operated as a self-powered cell exhibits an OCV of about 0.45 V, slightly lower than the theoretical output voltage (0.53 V), and can deliver a current density of ∼2.93 mA cm−2 without an applied voltage. These results highlight that the asymmetric alkali-acid electrolyte substantially reduces the electricity required for electrochemical H2 generation, attributable to the pH gradient between the anode and cathode chambers. Moreover, these results demonstrate that the optimized SOR/HER electrolysis system (acid: 2.5 M H2SO4, alkaline: 1.0 M NaOH/2.0 M Na2S) can provide a maximum current of 8.54 mA cm−2 at a bias-free voltage while simultaneously producing hydrogen and degrading sulfide. | ||
| Fig. 23 (a) Illustration of the home made H2 production flow-cell system coupled with the SOR (a peristaltic pump is used to flow deionized water through the intermediate transition chamber, so that the ion concentration can be maintained relatively stable in the middle chamber). (b) Comparison of LSV curves for the alkali–alkali OER/HER cell, alkali–alkali SOR/HER cell, and alkali–acid SOR/HER cell.232 Reproduced with permission. Copyright 2021, Wiley-VCH. Electrochemical performance of the Zn–Hz battery. (c) Schematic illustration of the Zn–Hz battery. (d) Discharge and charge voltage profiles. (e) Galvanostatic discharge–charge cycling curves at 5 mA cm−2.38 Reproduced with permission. Copyright 2022, Wiley-VCH. | ||
Inspired by the simultaneous resource utilization and energy storage characterization of Zn–CO2 batteries, Wang's group has developed a rechargeable alkaline Zn–hydrazine (Zn–Hz) battery that enables efficient and separate hydrogen generation through decoupled hydrazine splitting.38 Unlike typical decoupled electrolysis approaches, the decoupled hydrazine splitting in the Zn–Hz battery is achieved using bifunctional electrocatalysts in a two-electrode system, enabling hydrogen generation without the need for purification through temporal separation. The proposed Zn–Hz battery consists of bifunctional electrocatalysts as the cathode and Zn foil as the anode, aiming to the separate electrochemical reactions of the HER during discharge process and the HzOR during the charging process (Fig. 23c). During the discharge process, hydrogen is generated at the cathode through the electrochemical HER from H2O, while hydrazine oxidation occurs at the cathode during the charging process, enabling separate hydrogen generation through temporally decoupled electrochemical hydrazine splitting. To drive this device, a 3D hierarchical Mo2C/Ni@C/CS catalyst is fabricated, wherein Mo2C and Ni nanoparticles are encapsulated in porous carbon and uniformly decorated on a 3D carbon sphere. This catalyst demonstrates bifunctional activity for both the HER and the HzOR, achieving small potentials of −76 and 42 mV to achieve a current density of 10 10 mA cm−2, respectively. When employed as the cathode catalyst in the Zn–Hz battery, an OCV of 0.366 V is obtained. Notably, the discharge voltage is 0.364 V at 0.4 mA cm−2, while the charge voltage is 0.379 V at 0.4 mA cm−2 (Fig. 23d), indicating that the Zn–Hz battery can achieve an ultrahigh energy efficiency of over 96%. Moreover, this novel battery has the ability to simultaneously produce hydrogen and generate electric energy, eliminating the continuous energy consumption associated with conventional decoupled electrolysis and achieving efficient hydrogen evolution. As revealed in Fig. 23e, the battery exhibits excellent durability for 600 cycles (200 h) at 5 mA cm−2 with a slight voltage change.
Significant achievements have been made in the development of self-catalyzed systems for much enhanced electron utilization and without the need for an external power supply (Table 7). Even though these systems can reduce the consumption of electrical energy through additional reactions, they often require large quantities of sacrificial anode metal, leading to a significant reduction in the overall economic effectiveness. Therefore, it is imperative to develop electrocatalytic systems that fully utilize atoms and electrons, achieving an internal power supply while minimizing the waste of electrode materials. To accomplish those objectives, two strategies are proposed. (i) Assembling rechargeable functional batteries for electrode material recycling and high-value chemical production: by designing and implementing rechargeable batteries with functional electrodes, the electrode materials can be effectively recycled and utilized for the production of high-value chemicals. This approach not only promotes sustainability by reducing waste but also enables the recovery of valuable resources from the electrode materials. (ii) Converting electrode materials into high-value-added chemicals during the battery discharge process: rather than considering the electrode materials as consumables, this strategy aims to transform them into valuable chemicals during the discharge process of the battery. By harnessing the electrochemical reactions within the battery, the electrode materials can undergo controlled transformations, leading to the production of high-value-added chemicals. This approach maximizes the utilization of electrode materials, minimizing waste and enhancing the overall efficiency of the system. These strategies provide promising approaches for the development of atom- and electron-optimized electrocatalytic systems, which not only achieve internal power supply but also enable the conversion of electrode materials into valuable products. By adopting these approaches, researchers can advance the self-catalyzed systems towards more sustainable and efficient electrochemical processes.
| Catalysts | Reaction types | Electrolyte | Anode | Open circuit voltage | Peak power density (mW cm−2) | Stability | Recyclability | Ref. |
|---|---|---|---|---|---|---|---|---|
| Pt/CNTs | Zn–H2O battery | 4.0 M NaOH (anode)/2.0 M H2SO4 (cathode) | Zn | 1.25 V | 80 | 18 h at 5, 10, and 20 mA cm−2 | — | 532 |
| Ni-MOF/Ni2P@EG | Zn–H2O battery | 1.0 M KOH (anode + cathode) | Zn | — | 4.1 | — | 90 cycles (∼35 h) at 5 mA cm−2 | 533 |
| N,Cu-CoP/CC | Zn–H2O battery | 1.0 M PBS (anode + cathode) | Zn | 0.79 V | 1.83 | — | — | 546 |
| Mo-WC@NCS | Zn–H2O battery | 1.0 M KOH (anode)/0.5 M H2SO4 (cathode) | Zn | 1.08 V | 41.4 | 10 h at 10 mA cm−2 | — | 551 |
| Mo–Co0.85SeVSe/NC | Zn–H2O battery | 1.0 M KOH (anode + cathode) | Zn | — | 3.9 | 12 h at 10 mA cm−2 | — | 552 |
| Co@CoAl/NG | Al–H2O battery | 0.1 M KOH (anode + cathode) | Al | ∼1.24 V | — | — | — | 534 |
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ | Al–CO2 battery | 4.0 M NaOH (anode + cathode) | Al | 1.3 V | 125.4 | — | — | 535 |
| MoNi/NiMoO4 heterostructure | Mg/seawater battery | Simulated seawater (0.5 M NaCl) (anode + cathode) | Mg | 1.18–1.56 V | 21.08 | 100 h at 3 mA cm−2 | — | 40 |
| Ni/V2O3 | Mg/seawater battery | Simulated seawater (3.5% NaCl) (anode + cathode) | Mg | 1.16–1.39 V | 17.81 | 24 h at 3 mA cm−2 | — | 528 |
| CoP/Co2P heterostructure | Mg/seawater battery | Simulated seawater (3.5% NaCl) (anode + cathode) | Mg | 1.2–1.4 V | 6.28 | 24 h at 3 mA cm−2 | — | 544 |
| Ni-doped MoO3 | Mg/seawater battery | Simulated seawater (3.5% NaCl solution) (anode + cathode) | Mg | 1.23 V | 6.54 | 24 h at 3 mA cm−2 | — | 553 |
| Mo2C/Ni@C/CS | Zn–Hz battery | 1.0 M KOH + 0.2 M N2H4 (cathode) | Zn | 0.366 V | — | — | 240 cycles (200 h) at 5 mA cm−2 | 38 |
| NiCoP/NF | Zn–Hz battery | 1.0 M KOH + 0.2 M N2H4 (cathode) | Zn | 0.315 V | — | — | 240 cycles (80 h) at 5 mA cm−2 | 554 |
7. Conclusion and perspectives
This perspective highlights recent advancements in four innovative strategies for energy-saving hydrogen production, aiming to focus on the discussion of the strategies to synthesize the catalysts, select suitable organic molecules, discuss reaction mechanisms, overcome challenges in traditional water electrolysis, and show various advantages in hybrid electrolysis. Water splitting assisted by the oxidation of sacrificial-agent species (e.g., urea, hydrazine, and polysulfide) provides an elegant approach for highly efficient electrochemical hydrogen production at lower electrolysis voltages, simultaneously enabling wastewater treatment at the anode. The typical electrooxidation reactions of small organic molecules, including alcohols, aldehydes, amines, and biomass, open a sustainable strategy for the upgrading of organic compounds at the anode while achieving energy-saving hydrogen production at the cathode, which also mitigates the production of explosive H2/O2 mixtures and reactive oxygen species. Self-powered electrocatalytic systems provide a promising approach for uninterrupted hydrogen production without the need for an external power supply, making them suitable for mobile devices, vehicles, and field activities. Self-catalyzed electrolysis systems can achieve 100% electron and atom utilization for hydrogen production in the absence of external energy input. These coupling processes significantly reduce the cell voltage for energy-saving hydrogen production and enhance the atom/electron utilization of electrolyzers. Despite the superior advantages and significant achievement in these coupling approaches, these technologies are still in the early stages of development, and the transformation efficiency remains unsatisfactory. However, through further advancements in electrode development and electrolyzer design, it is anticipated that these appealing technologies will find practical applications in chemical manufacturing. Continued research and innovation in this field hold promise for achieving efficient and sustainable hydrogen production.Theoretically, there are several important issues that are needed to be addressed for further development and enhanced energy utilization for the energy-saving hydrogen production. These aspects include the rational design of high-performance electrodes, identification of the reaction mechanism, exploration of novel reactions, design of electrolyzer systems, and comprehensive economic cost analysis, which are crucial for a sustainable and low-carbon energy future. Regarding the future direction in this burgeoning field, we provide some perspectives as follows (Fig. 24).
 | ||
| Fig. 24 Prospects for developing energy-saving H2 production. | ||
7.1 Catalyst preparation
Electrode materials play a crucial role in determining the electrochemical reaction kinetics. Exploring effective electrode catalysts through morphology engineering and active-site design is essential for improving reaction kinetics and facilitating the desired reactions. At present, the catalyst design strategies primarily originate from traditional water electrolysis, and there is still a need for targeted catalyst design specifically tailored for small molecule oxidation reactions. It is necessary to gain a deep understanding of the catalytic reaction mechanisms involved and realize targeted designs for each small molecule oxidation reaction to fully exploit the potential of catalysts. Additionally, since small molecules often contain diverse functional groups, it is important to establish a fundamental understanding of the relationships between these groups and catalysts. This understanding can greatly benefit the design of highly efficient catalysts by optimizing the specific interactions and reactivity of different functional groups. Furthermore, with respect to biomass-assisted hybrid water electrolysis systems, especially for alcohols and aldehydes, these are opportunities to lower the thermodynamic equilibrium potentials, produce value-added chemicals, and utilize a wide range of raw materials. However, the complete oxidation of these compounds often leads to the generation of worthless CO2 and H2O. It is crucial to conduct in-depth research to facilitate specific reaction kinetics and selectively control the production of particular valuable products, which will enable the fabrication of efficient and highly applicable electrocatalysts for the hybrid water electrolysis. Moreover, the utilization of bifunctional catalysts based on earth-abundant elements for hybrid water electrolyzers is highly desirable in terms of cost reduction and practical applications, making the technology more economically viable and scalable.7.2 Mechanism investigation
Currently, the mechanisms underlying small molecule oxidation reactions remain a huge debate. A comprehensive understanding of the principal electrocatalytic and interfacial processes holds great potential for precisely controlling the reaction pathway and fine-tuning the selectivity of desirable products. A reasonable investigation of the intricate reaction mechanism is of paramount importance for exploring efficient catalysts. It is worth noting that many non-precious metal-based electrocatalysts often undergo a reconstruction process during anodic oxidation reactions, during which high-valence metal sites are formed and subsequently serve as the true active sites. Consequently, it is imperative to develop advanced in situ characterization techniques to analyze pivotal intermediates and catalytic species. To attain this objective, in situ/operando technologies must be employed in conjunction with existing techniques, such as in situ XAS, in situ XRD, in situ Raman spectroscopy, and in situ FTIR, to monitor the behavior of key intermediates and identify the actual active sites at the atomic scale. The profound identification of active sites and a comprehensive understating of structure–property–activity relationships will undoubtedly provide valuable insights for the intelligent design of effective materials. Furthermore, simultaneous theoretical investigation using DFT and machine learning provides another powerful approach to identify the reaction mechanism and facilitate the design of advanced catalyst systems. By combining the presently available operando techniques with advanced DFT simulations, a more reasonable understanding of structural evolution and the reaction mechanism during catalysis can be achieved. The new mechanistic insights thus obtained will serve as invaluable feedback for knowledge-driven reaction engineering and catalyst optimization endeavors.7.3 Reaction exploration
The development of novel electrooxidation reactions holds significant importance for a range of applications, including biomass upgrading, industrial organic pollutant removal, and fine chemical production. In particular, the efficient electrooxidation of organic pollutants by coupling the HER provides a sustainable approach for simultaneously producing valuable chemicals and purifying water contaminated by industrial wastes. In contrast to the electrolysis of pure water, the electrolysis of seawater for hydrogen production presents a more cost-effective alternative that obviates the need for desalination or purification units. However, the practical implementation of seawater electrolysis is severely hindered by the occurrence of the ClOR at the anode. This competing reaction interferes with the desired OER, resulting in the generation of undesirable chlorine and/or hypochlorite species that gradually corrode catalysts, membranes, and other crucial components during long-term tests. It is noteworthy that the oxidation of small molecules occurs at lower potentials compared to both the OER and ClOR. Consequently, coupling the electrooxidation of small molecules with the HER holds potential to enable high-current-density seawater electrolysis without the detrimental interference of the ClOR. This advancement offers a promising avenue for large-scale, environmentally friendly hydrogen production from seawater.7.4 Cell configuration design
The successful presentation of the feasibility of using the prepared electrocatalysts under industrial-scale current densities, reaching up to 1 A cm−2, represents a critical milestone for their potential applications. However, achieving both high selectivity and efficiency at such high current densities poses a significant challenge. Overcoming this challenge necessitates close collaboration to simultaneously improve catalytic performance and optimize electrolyzer design. To address these requirements, the utilization of a flow electrolyzer assembly proves advantageous. Such an assembly facilitates the efficient transport of reactants and ensures the effective removal of products from the catalyst surface. This design strategy maximizes the contact between reactants and active sites, allowing for the attainment of a substantial current density at low voltage. Importantly, this configuration also impedes the occurrence of the competitive OER that tends to arise at high working voltages. The advantages of the flow electrolyzer assembly extend beyond improved performance, as it also offers scalability to larger modules and holds significant commercial potential.7.5 Product separation
The downstream separation and purification of valuable products generated at the anode play a crucial role in industrial applications, necessitating further considerations and improvements. Many electrocatalytic systems yield gases or liquids as products, which require subsequent separation and purification steps. While the separation of gas-phase products can be conveniently achieved using dual-pool reactors, the cost associated with separating soluble products from electrolytes, particularly at low concentrations, is often high. Therefore, the production of insoluble or easily separable chemicals in or from electrolytes is highly desirable for the advancement of electrocatalytic systems. Urgent efforts are required to develop efficient methods for the separation of target products. Furthermore, it is essential to explore novel molecules that can be oxidized to yield hydrophobic, rather than hydrophilic, valuable products. This approach would facilitate easier separation and purification processes. Additionally, to maximize the utilization of the product/precursor mixed electrolyte obtained after the electrocatalytic reaction, other chemicals can be introduced into the electrolyte mixture to generate novel high-value compounds through subsequent reactions such as precipitation and phase separation. Simultaneously, improving the selectivity of ion-exchange membranes is of significance to prevent the crossover of small molecules during real-world applications. Enhancing the membrane's selectivity will enable more efficient separation and purification processes, contributing to the overall performance and practicality of electrocatalytic systems.7.6 Economic evaluation
In evaluating the feasibility and profitability of any coupled process, careful consideration must be given to the economics, particularly energy costs. The profitability of a reaction system is determined by the balance between consumption costs and the value of products generated. When considering consumption costs, both visible and invisible factors should be taken into account. Visible consumption encompasses the costs of raw materials, electrocatalysts, and all components of devices, which are typically considered in economic analyses. However, it is equally important to consider the often overlooked invisible consumption, which includes electricity, treatment costs for products or pollutants, and electrolytes. The cost of electricity plays a pivotal role in the economic analysis, and exploring strategies to minimize energy consumption is crucial. Renewable energy sources such as solar or wind can be harnessed to reduce the overall energy cost and enhance the economic feasibility of the electrocatalytic systems. In this context, self-powered electrocatalytic systems, particularly those stored chemical energy derived from sustainable energy sources, hold promise for future development. Therefore, a combination of a clean power source, moderate operating conditions, aqueous electrolytes, high selectivity/faradaic efficiency, and nonprecious metal-based electrocatalysts emerges as a key research field for maximizing the profitability of a whole electrocatalytic system. By addressing these factors, the overall energy costs can be minimized, making the hydrogen production more economically viable.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22105108 and 22179065) and China Postdoctoral Science Foundation (2020M680860).Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- G. Chen, H. Zhong and X. Feng, Chem. Sci., 2021, 12, 15802–15820 RSC.
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- X. Wang, W. Li, D. Xiong, D. Y. Petrovykh and L. Liu, Adv. Funct. Mater., 2016, 26, 4067–4077 CrossRef CAS.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- A. Ali, F. Long and P. K. Shen, Electrochem. Energy Rev., 2022, 5, 1 CrossRef CAS.
- L. Sun, Q. Luo, Z. Dai and F. Ma, Coord. Chem. Rev., 2021, 444, 214049 CrossRef CAS.
- H. Xu, H. Shang, C. Wang and Y. Du, Coord. Chem. Rev., 2020, 418, 213374 CrossRef CAS.
- G. Chen, X. Li and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202209014 CrossRef CAS.
- V. Bambagioni, M. Bevilacqua, C. Bianchini, J. Filippi, A. Lavacchi, A. Marchionni, F. Vizza and P. K. Shen, ChemSusChem, 2010, 3, 851–855 CrossRef CAS PubMed.
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- J. L. Zhang, Q. X. Zhao, M. Y. Cheng, W. M. Xuan and Y. Liu, Tungsten, 2023, 5, 261–269 CrossRef.
- H. Y. Wang, M. L. Sun, J. T. Ren and Z. Y. Yuan, Adv. Energy Mater., 2022, 13, 2203568 CrossRef.
- Y. Li, X. Wei, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 19550–19571 CrossRef CAS PubMed.
- W. J. Cui, S. M. Zhang, Z. Y. Tian, C. Li, Y. M. Wang, B. R. Yu, Y. Y. Ma and Z. G. Han, Tungsten, 2022, 4, 109–120 CrossRef.
- C. Deng, C. Y. Toe, X. Li, J. Tan, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2022, 12, 2201047 CrossRef CAS.
- W. Xue, H. Liu, B. Zhao, C. Tang, B. Y. Xia and B. You, ChemSusChem, 2022, 16, e202201512 CrossRef PubMed.
- H. Xu, L. Chen and J. Shi, Energy Environ. Sci., 2023, 16, 1334–1363 RSC.
- J. Li, K. Ji, B. Li, M. Xu, Y. Wang, H. Zhou, Q. Shi and H. Duan, Angew. Chem., Int. Ed., 2023, 62, e202304852 CrossRef PubMed.
- T. W. Chen, G. Anushya, S. M. Chen, P. Kalimuthu, V. Mariyappan, P. Gajendran and R. Ramachandran, Materials, 2022, 15, 458 CrossRef CAS.
- N. Kornienko, Joule, 2018, 2, 207–209 CrossRef CAS.
- F. Zhang, M. Gao, S. Huang, H. Zhang, X. Wang, L. Liu, M. Han and Q. Wang, Adv. Mater., 2022, 34, 2104562 CrossRef CAS.
- F. Wang, Y. Li, X. Xia, W. Cai, Q. Chen and M. Chen, Adv. Energy Mater., 2021, 11, 2100667 CrossRef CAS.
- R. Li, K. Xiang, Z. Peng, Y. Zou and S. Wang, Adv. Energy Mater., 2021, 11, 2102292 CrossRef CAS.
- Z. Yu, Y. Li, V. Martin-Diaconescu, L. Simonelli, J. Ruiz Esquius, I. Amorim, A. Araujo, L. Meng, J. L. Faria and L. Liu, Adv. Funct. Mater., 2022, 32, 2206138 CrossRef CAS.
- S. Zhang, C. Tan, R. Yan, X. Zou, F. L. Hu, Y. Mi, C. Yan and S. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202302795 CrossRef CAS.
- L. Zhang, Z. Wang and J. Qiu, Adv. Mater., 2022, 34, 2109321 CrossRef CAS.
- H. Jiang, M. Sun, S. Wu, B. Huang, C. S. Lee and W. Zhang, Adv. Funct. Mater., 2021, 31, 2104951 CrossRef CAS.
- J. Chi, L. Guo, J. Mao, T. Cui, J. Zhu, Y. Xia, J. Lai and L. Wang, Adv. Funct. Mater., 2023, 33, 2300625 CrossRef CAS.
- Y. Hao, D. Yu, S. Zhu, C. H. Kuo, Y. M. Chang, L. Wang, H. Y. Chen, M. Shao and S. Peng, Energy Environ. Sci., 2023, 16, 1100–1110 RSC.
- Q. Qian, X. He, Z. Li, Y. Chen, Y. Feng, M. Cheng, H. Zhang, W. Wang, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2023, 35, 2300935 CrossRef CAS.
- Y. Zhu, Q. Qian, Y. Chen, X. He, X. Shi, W. Wang, Z. Li, Y. Feng, G. Zhang and F. Cheng, Adv. Funct. Mater., 2023, 33, 2300547 CrossRef CAS.
- J. Yin, Y. Li, F. Lv, M. Lu, K. Sun, W. Wang, L. Wang, F. Cheng, Y. Li, P. Xi and S. Guo, Adv. Mater., 2017, 29, 1704681 CrossRef.
- K. Ding, J. Hu, W. Jin, L. Zhao, Y. Liu, Z. Wu, B. Weng, H. Hou and X. Ji, Adv. Funct. Mater., 2022, 32, 2201944 CrossRef CAS.
- Q. Qian, J. Zhang, J. Li, Y. Li, X. Jin, Y. Zhu, Y. Liu, Z. Li, A. El-Harairy, C. Xiao, G. Zhang and Y. Xie, Angew. Chem., Int. Ed., 2021, 60, 5984–5993 CrossRef CAS.
- Y. Feng, Q. Shi, J. Lin, E. Chai, X. Zhang, Z. Liu, L. Jiao and Y. Wang, Adv. Mater., 2022, 34, 2207747 CrossRef CAS.
- R. Tang, Y. Yang, Y. Zhou and X. Y. Yu, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202301925.
- Y. Xu, H. Lv, H. Lu, Q. Quan, W. Li, X. Cui, G. Liu and L. Jiang, Nano Energy, 2022, 98, 107295 CrossRef CAS.
- Y. Xie, Y. Sun, H. Tao, X. Wang, J. Wu, K. Ma, L. Wang, Z. Kang and Y. Zhang, Adv. Funct. Mater., 2022, 32, 2111777 CrossRef CAS.
- T. Zhao, Y. Wang, S. Karuturi, K. Catchpole, Q. Zhang and C. Zhao, Carbon Energy, 2020, 2, 582–613 CrossRef CAS.
- Y. H. Wang, S. Zheng, W. M. Yang, R. Y. Zhou, Q. F. He, P. Radjenovic, J. C. Dong, S. Li, J. Zheng, Z. L. Yang, G. Attard, F. Pan, Z. Q. Tian and J. F. Li, Nature, 2021, 600, 81–85 CrossRef CAS.
- J. C. Dong, X. G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z. L. Yang, D. Y. Wu, J. M. Feliu, C. T. Williams, Z. Q. Tian and J. F. Li, Nat. Energy, 2018, 4, 60–67 CrossRef.
- R. L. Wei, Y. Liu, Z. Chen, W. S. Jia, Y. Y. Yang and W. B. Cai, J. Electroanal. Chem., 2021, 896, 115254 CrossRef CAS.
- J. E. Avilés Acosta, J. C. Lin, D. Un Lee, T. F. Jaramillo and C. Hahn, ChemCatChem, 2023, 15, e202300520 CrossRef.
- Y. Zhang, B. Zhou, Z. Wei, W. Zhou, D. Wang, J. Tian, T. Wang, S. Zhao, J. Liu, L. Tao and S. Wang, Adv. Mater., 2021, 33, 2104791 CrossRef CAS.
- A. Sivanantham, P. Ganesan, A. Vinu and S. Shanmugam, ACS Catal., 2019, 10, 463–493 CrossRef.
- Y. Kim, S. Park, S. J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13, 4301–4311 RSC.
- J. Wang, H. Y. Tan, T. R. Kuo, S. C. Lin, C. S. Hsu, Y. Zhu, Y. C. Chu, T. L. Chen, J. F. Lee and H. M. Chen, Small, 2021, 17, 2005713 CrossRef CAS PubMed.
- V. Celorrio, A. S. Leach, H. Huang, S. Hayama, A. Freeman, D. W. Inwood, D. J. Fermin and A. E. Russell, ACS Catal., 2021, 11, 6431–6439 CrossRef CAS.
- P. Tang, H. J. Lee, K. Hurlbutt, P. Y. Huang, S. Narayanan, C. Wang, D. Gianolio, R. Arrigo, J. Chen, J. H. Warner and M. Pasta, ACS Catal., 2022, 12, 3173–3180 CrossRef CAS.
- F. Gong, M. Liu, S. Ye, L. Gong, G. Zeng, L. Xu, X. Zhang, Y. Zhang, L. Zhou, S. Fang and J. Liu, Adv. Funct. Mater., 2021, 31, 2101715 CrossRef CAS.
- S. J. Blair, M. Doucet, V. A. Niemann, K. H. Stone, M. E. Kreider, J. F. Browning, C. E. Halbert, H. Wang, P. Benedek, E. J. McShane, A. C. Nielander, A. Gallo and T. F. Jaramillo, Energy Environ. Sci., 2023, 16, 3391–3406 RSC.
- H. Zhu, Z. Zhu, J. Hao, S. Sun, S. Lu, C. Wang, P. Ma, W. Dong and M. Du, Chem. Eng. J., 2022, 431, 133251 CrossRef CAS.
- Y. Nomura, K. Yamamoto, M. Fujii, T. Hirayama, E. Igaki and K. Saitoh, Nat. Commun., 2020, 11, 2824 CrossRef CAS.
- B. Song, Y. Yang, M. Rabbani, T. T. Yang, K. He, X. Hu, Y. Yuan, P. Ghildiyal, V. P. Dravid, M. R. Zachariah, W. A. Saidi, Y. Liu and R. Shahbazian-Yassar, ACS Nano, 2020, 14, 15131–15143 CrossRef CAS PubMed.
- B. Qin, Y. Li, H. Wang, G. Yang, Y. Cao, H. Yu, Q. Zhang, H. Liang and F. Peng, Nano Energy, 2019, 60, 43–51 CrossRef CAS.
- J. Zhang, Y. Wang, C. Yang, S. Chen, Z. Li, Y. Cheng, H. Wang, Y. Xiang, S. Lu and S. Wang, Nano Res., 2021, 14, 4650–4657 CrossRef CAS.
- M. K. Kim, H. Lee, J. H. Won, W. Sim, S. J. Kang, H. Choi, M. Sharma, H. S. Oh, S. Ringe, Y. Kwon and H. M. Jeong, Adv. Funct. Mater., 2021, 32, 2107349 CrossRef.
- H. Liu, T. H. Lee, Y. Chen, E. W. Cochran and W. Li, Green Chem., 2021, 23, 5056–5063 RSC.
- M. Scohy, S. Abbou, V. Martin, B. Gilles, E. Sibert, L. Dubau and F. Maillard, ACS Catal., 2019, 9, 9859–9869 CrossRef CAS.
- R. Jay, A. L. Jadhav, L. W. Gordon and R. J. Messinger, Chem. Mater., 2022, 34, 4486–4495 CrossRef CAS.
- M. He, J. Chen, A. Hu, Z. Yan, L. Cao and J. Long, Energy Storage Mater., 2023, 62, 102941 CrossRef.
- Y. Xu and B. Zhang, ChemElectroChem, 2019, 6, 3214–3226 CrossRef CAS.
- A. Serov and C. Kwak, Appl. Catal., B, 2010, 98, 1–9 CrossRef CAS.
- J. Ren, Z. Hu, C. Chen, Y. Liu and Z. Yuan, J. Energy Chem., 2017, 26, 1196–1202 CrossRef.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- Y. Lei, Y. Wang, Y. Liu, C. Song, Q. Li, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 20794–20812 CrossRef CAS PubMed.
- C. Li and J. B. Baek, ACS Omega, 2020, 5, 31–40 CrossRef CAS PubMed.
- J. T. Ren, C. Y. Wan, T. Y. Pei, X. W. Lv and Z. Y. Yuan, Appl. Catal., B, 2020, 266, 118633 CrossRef CAS.
- C. Huang, J. Zhou, D. Duan, Q. Zhou, J. Wang, B. Peng, L. Yu and Y. Yu, Chin. J. Catal., 2022, 43, 2091–2110 CrossRef CAS.
- A. H. Al-Naggar, N. M. Shinde, J. S. Kim and R. S. Mane, Coord. Chem. Rev., 2023, 474, 214864 CrossRef CAS.
- J. Qian, X. Liu, C. Zhong, G. Xu, H. Li, W. Zhou, B. You, F. Wang, D. Gao and D. Chao, Adv. Funct. Mater., 2022, 33, 2212021 CrossRef.
- R. Zhang, X. Du, S. Li, J. Guan, Y. Fang, X. Li, Y. Dai and M. Zhang, J. Electroanal. Chem., 2022, 921, 116679 CrossRef CAS.
- W. Zhang, X. Liu, Q. Yu, X. Wang, H. Mao, J. Chi, B. Li, J. Wan and L. Wang, Chem. Eng. J., 2023, 454, 140210 CrossRef CAS.
- Z. Wu, Y. Zhao, W. Jin, B. Jia, J. Wang and T. Ma, Adv. Funct. Mater., 2020, 31, 2009070 CrossRef.
- R. Li, B. Hu, T. Yu, H. Chen, Y. Wang and S. Song, Adv. Sci., 2020, 7, 1902830 CrossRef CAS PubMed.
- M. Li, X. Wu, K. Liu, Y. Zhang, X. Jiang, D. Sun, Y. Tang, K. Huang and G. Fu, J. Energy Chem., 2022, 69, 506–515 CrossRef CAS.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- X. Wang, Y. Tuo, Y. Zhou, D. Wang, S. Wang and J. Zhang, Chem. Eng. J., 2021, 403, 126297 CrossRef CAS.
- L. Wang, Z. Zeng, W. Gao, T. Maxson, D. Raciti, M. Giroux, X. Pan, C. Wang and J. Greeley, Science, 2019, 363, 870–874 CrossRef CAS PubMed.
- H. Wang, S. Xu, C. Tsai, Y. Li, C. Liu, J. Zhao, Y. Liu, H. Yuan, F. Abild-Pedersen, F. B. Prinz, J. K. Nørskov and Y. Cui, Science, 2016, 354, 1031–1036 CrossRef CAS PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- Q. Mao, S. Jiao, K. Ren, S. Wang, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Eng. J., 2021, 426, 131227 CrossRef CAS.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Norskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- X. Tian, X. Zhao, Y. Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- M. Escudero-Escribano, A. Verdaguer-Casadevall, P. Malacrida, U. Grønbjerg, B. P. Knudsen, A. K. Jepsen, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, J. Am. Chem. Soc., 2012, 134, 16476–16479 CrossRef CAS PubMed.
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velázquez-Palenzuela, V. Tripkovic, J. Schiøtz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS PubMed.
- J. Shi, Q. Sun, W. Zhu, T. Cheng, F. Liao, M. Ma, J. Yang, H. Yang, Z. Fan and M. Shao, Chem. Eng. J., 2023, 463, 142385 CrossRef CAS.
- B. Ni and X. Wang, Chem. Sci., 2015, 6, 3572–3576 RSC.
- J. Hu, B. Huang, C. Zhang, Z. Wang, Y. An, D. Zhou, H. Lin, M. K. H. Leung and S. Yang, Energy Environ. Sci., 2017, 10, 593–603 RSC.
- X. Wang, Y. Ouyang, L. Jiao, H. Wang, L. Xie, J. Wu, J. Guo and H. Dai, Nat. Nanotechnol., 2011, 6, 563–567 CrossRef CAS PubMed.
- W. Yang, X. Yang, C. Hou, B. Li, H. Gao, J. Lin and X. Luo, Appl. Catal., B, 2019, 259, 118020 CrossRef CAS.
- H. Qin, Y. Ye, J. Li, W. Jia, S. Zheng, X. Cao, G. Lin and L. Jiao, Adv. Funct. Mater., 2023, 33, 2209698 CrossRef CAS.
- M. Luo, W. Sun, B. B. Xu, H. Pan and Y. Jiang, Adv. Energy Mater., 2020, 11, 2002762 CrossRef.
- N. C. S. Selvam, L. Du, B. Y. Xia, P. J. Yoo and B. You, Adv. Funct. Mater., 2021, 31, 2008190 CrossRef CAS.
- J. T. Ren, G. G. Yuan, C. C. Weng and Z. Y. Yuan, ACS Sustainable Chem. Eng., 2017, 6, 707–718 CrossRef.
- L. Chen, J. T. Ren and Z. Y. Yuan, Adv. Energy Mater., 2023, 13, 2203720 CrossRef CAS.
- X. Liu, P. Wang, X. Liang, Q. Zhang, Z. Wang, Y. Liu, Z. Zheng, Y. Dai and B. Huang, Mater. Today Energy, 2020, 18, 100524 CrossRef CAS.
- Z. Zhu, Y. Zhang, H. Wang, B. Y. Xia and B. You, ChemCatChem, 2022, 14, e202201100 CrossRef CAS.
- Y. Yang, M. Luo, W. Zhang, Y. Sun, X. Chen and S. Guo, Chem, 2018, 4, 2054–2083 CAS.
- Z. Zhu, X. Chen, J. Liu, S. Fan and B. You, Energy Fuels, 2023 DOI:10.1021/acs.energyfuels.3c02121.
- N. S. Porter, H. Wu, Z. Quan and J. Fang, Acc. Chem. Res., 2013, 46, 1867–1877 CrossRef CAS PubMed.
- L. Tao, Y. Shi, Y.-C. Huang, R. Chen, Y. Zhang, J. Huo, Y. Zou, G. Yu, J. Luo, C.-L. Dong and S. Wang, Nano Energy, 2018, 53, 604–612 CrossRef CAS.
- C. Li, Y. Liu, Z. Zhuo, H. Ju, D. Li, Y. Guo, X. Wu, H. Li and T. Zhai, Adv. Energy Mater., 2018, 8, 1801775 CrossRef.
- C. Wang, H. Lu, Z. Mao, C. Yan, G. Shen and X. Wang, Adv. Funct. Mater., 2020, 30, 2000556 CrossRef CAS.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Perez-Ramirez and J. Lu, Adv. Mater., 2021, 33, 2003075 CrossRef CAS PubMed.
- Y. Wang, D. Wang and Y. Li, Adv. Mater., 2021, 33, 2008151 CrossRef CAS PubMed.
- X. Zhao, D. He, B. Y. Xia, Y. Sun and B. You, Adv. Mater., 2023, 35, 2210703 CrossRef CAS PubMed.
- M. Qin, S. Fan, X. Li, Z. Niu, C. Bai and G. Chen, Small, 2022, 18, 2201359 CrossRef CAS PubMed.
- R. Ge, Y. Wang, Z. Li, M. Xu, S.-M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200211 CrossRef CAS PubMed.
- S. Chen, M. Cui, Z. Yin, J. Xiong, L. Mi and Y. Li, ChemSusChem, 2021, 14, 73–93 CrossRef CAS PubMed.
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Adv. Energy Mater., 2021, 12, 2102715 CrossRef.
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T. K. Sham, M. Gu, L. M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2019, 10, 4936 CrossRef PubMed.
- C. Zhan, Y. Xu, L. Bu, H. Zhu, Y. Feng, T. Yang, Y. Zhang, Z. Yang, B. Huang, Q. Shao and X. Huang, Nat. Commun., 2021, 12, 6261 CrossRef CAS PubMed.
- J. T. Ren, Y. Yao and Z. Y. Yuan, Green Energy Environ., 2021, 6, 620–643 CrossRef CAS.
- J. T. Ren, G. G. Yuan, C. C. Weng, L. Chen and Z. Y. Yuan, Nanoscale, 2018, 10, 10620–10628 RSC.
- Y. Niu, Y. Yuan, Q. Zhang, F. Chang, L. Yang, Z. Chen and Z. Bai, Nano Energy, 2021, 82, 105699 CrossRef CAS.
- Y. Li, M. Lu, Y. Wu, Q. Ji, H. Xu, J. Gao, G. Qian and Q. Zhang, J. Mater. Chem. A, 2020, 8, 18215–18219 RSC.
- H. F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- C. C. Weng, X. W. Lv, J. T. Ren, T. Y. Ma and Z. Y. Yuan, Electrochem. Energy Rev., 2022, 5, 19 CrossRef CAS.
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Nat. Commun., 2021, 12, 4182 CrossRef CAS PubMed.
- Y. Pei, H. Zhong and F. Jin, Energy Sci. Eng., 2021, 9, 1012–1032 CrossRef CAS.
- Y. Ido, A. Fukazawa, Y. Furutani, Y. Sato, N. Shida and M. Atobe, ChemSusChem, 2021, 14, 5405–5409 CrossRef CAS PubMed.
- Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193 RSC.
- Q. Xu, L. Zhang, J. Zhang, J. Wang, Y. Hu, H. Jiang and C. Li, EnergyChem, 2022, 4, 100087 CrossRef CAS.
- W. Zhu, X. Fu, A. Wang, M. Ren, Z. Wei, C. Tang, X. Sun and J. Wang, Appl. Catal., B, 2022, 317, 121726 CrossRef CAS.
- Z. Yan and T. E. Mallouk, Acc. Mater. Res., 2021, 2, 1156–1166 CrossRef CAS.
- B. Mayerhöfer, D. McLaughlin, T. Böhm, M. Hegelheimer, D. Seeberger and S. Thiele, ACS Appl. Energy Mater., 2020, 3, 9635–9644 CrossRef.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
- F. Sun, D. He, K. Yang, J. Qiu and Z. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203929 CrossRef CAS PubMed.
- I. Amorim, J. Xu, N. Zhang, Z. Yu, A. Araújo, F. Bento and L. Liu, Chem. Eng. J., 2021, 420, 130454 CrossRef CAS.
- B. S. Kim, S. C. Park, D. H. Kim, G. H. Moon, J. G. Oh, J. Jang, M. S. Kang, K. B. Yoon and Y. S. Kang, Small, 2020, 16, 2002641 CrossRef CAS PubMed.
- J. Xu, I. Amorim, Y. Li, J. Li, Z. Yu, B. Zhang, A. Araujo, N. Zhang and L. Liu, Carbon Energy, 2020, 2, 646–655 CrossRef CAS.
- K. Sun, R. Liu, Y. Chen, E. Verlage, N. S. Lewis and C. Xiang, Adv. Energy Mater., 2016, 6, 1600379 CrossRef.
- J. Luo, D. A. Vermaas, D. Bi, A. Hagfeldt, W. A. Smith and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600100 CrossRef.
- Z. Wang, P. Cai, Q. Chen, X. Yin, K. Chen, Z. Lu and Z. Wen, J. Colloid Interface Sci., 2023, 636, 610–617 CrossRef CAS PubMed.
- Y. Li, J. Chen, P. Cai and Z. Wen, J. Mater. Chem. A, 2018, 6, 4948–4954 RSC.
- L. Fan, Y. Ji, G. Wang, Z. Zhang, L. Yi, K. Chen, X. Liu and Z. Wen, J. Energy Chem., 2022, 72, 424–431 CrossRef CAS.
- B. Liu, G. Wang, X. Feng, L. Dai, Z. Wen and S. Ci, Nanoscale, 2022, 14, 12841–12848 RSC.
- X. Liu, W. Sun, X. Hu, J. Chen and Z. Wen, Chem. Eng. J., 2023, 474, 145355 CrossRef CAS.
- M. Zhang, H. Li, J. Chen, F. X. Ma, L. Zhen, Z. Wen and C. Y. Xu, Adv. Funct. Mater., 2023, 33, 2303189 CrossRef CAS.
- G. Wang, J. Chen, P. Cai, J. Jia and Z. Wen, J. Mater. Chem. A, 2018, 6, 17763–17770 RSC.
- H. Li, M. Zhang, L. Yi, Y. Liu, K. Chen, P. Shao and Z. Wen, Appl. Catal., B, 2021, 280, 119412 CrossRef CAS.
- R. He, N. Xu, I. M. U. Hasan, L. Peng, L. Li, H. Huang and J. Qiao, EcoMat, 2023, 5, e12346 CrossRef CAS.
- M. K. Cho, A. Lim, S. Y. Lee, H. J. Kim, S. J. Yoo, Y. E. Sung, H. S. Park and J. H. Jang, J. Electrochem. Sci. Technol., 2017, 8, 183–196 CrossRef CAS.
- W. Yang, K. Dastafkan, C. Jia and C. Zhao, Adv. Mater. Technol., 2018, 3, 1700377 CrossRef.
- J. E. Park, J. Lim, M. S. Lim, S. Kim, O. H. Kim, D. W. Lee, J. H. Lee, Y. H. Cho and Y. E. Sung, Electrochim. Acta, 2019, 323, 134808 CrossRef CAS.
- J. Hyun, W. Jo, S. H. Yang, S. H. Shin, G. Doo, S. Choi, D. H. Lee, D. W. Lee, E. Oh, J. Y. Lee and H. T. Kim, J. Power Sources, 2022, 543, 231835 CrossRef CAS.
- L. Yuan, S. Zeng, X. Zhang, X. Ji and S. Zhang, Mater. Rep.: Energy, 2023, 3, 100177 CAS.
- J. Li, J. Li, T. Liu, L. Chen, Y. Li, H. Wang, X. Chen, M. Gong, Z. P. Liu and X. Yang, Angew. Chem., Int. Ed., 2021, 60, 26656–26662 CrossRef CAS PubMed.
- X. F. Lu, S. L. Zhang, W. L. Sim, S. Gao and X. W. D. Lou, Angew. Chem., Int. Ed., 2021, 60, 22885–22891 CrossRef CAS PubMed.
- J. Li, X. Xu, X. Hou, S. Zhang, G. Su, W. Tian, H. Wang, M. Huang and A. Toghan, Nano Res., 2023, 16, 8853–8862 CrossRef CAS.
- K. Xiong, L. Yu, Y. Xiang, H. Zhang, J. Chen and Y. Gao, J. Alloys Compd., 2022, 912, 165234 CrossRef CAS.
- Y. Tong, P. Chen, M. Zhang, T. Zhou, L. Zhang, W. Chu, C. Wu and Y. Xie, ACS Catal., 2017, 8, 1–7 CrossRef.
- D. Liu, T. Liu, L. Zhang, F. Qu, G. Du, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2017, 5, 3208–3213 RSC.
- J. Y. Zhang, T. He, M. Wang, R. Qi, Y. Yan, Z. Dong, H. Liu, H. Wang and B. Y. Xia, Nano Energy, 2019, 60, 894–902 CrossRef CAS.
- S. Feng, J. Luo, J. Li, Y. Yu, Z. Kang, W. Huang, Q. Chen, P. Deng, Y. Shen and X. Tian, Mater. Today Phys., 2022, 23, 100646 CrossRef CAS.
- X. Cao, T. Wang, H. Qin, G. Lin, L. Zhao and L. Jiao, Nano Res., 2022, 16, 3665–3671 CrossRef.
- K. Wang, W. Huang, Q. Cao, Y. Zhao, X. Sun, R. Ding, W. Lin, E. Liu and P. Gao, Chem. Eng. J., 2022, 427, 130865 CrossRef CAS.
- T. Wang, X. Cao and L. Jiao, eScience, 2021, 1, 69–74 CrossRef.
- J. Zhang, S. Huang, P. Ning, P. Xin, Z. Chen, Q. Wang, K. Uvdal and Z. Hu, Nano Res., 2021, 15, 1916–1925 CrossRef.
- R. Q. Li, X. Y. Wan, B. L. Chen, R. Y. Cao, Q. H. Ji, J. Deng, K. G. Qu, X. B. Wang and Y. C. Zhu, Chem. Eng. J., 2021, 409, 128240 CrossRef CAS.
- H. Sun, W. Zhang, J. G. Li, Z. Li, X. Ao, K. H. Xue, K. K. Ostrikov, J. Tang and C. Wang, Appl. Catal., B, 2021, 284, 119740 CrossRef CAS.
- Y. Xu, T. Ren, K. Ren, S. Yu, M. Liu, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Eng. J., 2021, 408, 127308 CrossRef CAS.
- X. Feng, Y. Shi, Y. Chen, Z. Xu and H. Guan, J. Ind. Eng. Chem., 2022, 113, 170–180 CrossRef CAS.
- J. Wang, Y. Sun, Y. Qi and C. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 57392–57402 CrossRef CAS PubMed.
- Z. Wang, W. Liu, J. Bao, Y. Song, X. She, Y. Hua, G. Lv, J. Yuan, H. Li and H. Xu, Chem. Eng. J., 2022, 430, 133100 CrossRef CAS.
- R. Q. Li, Q. Liu, Y. Zhou, M. Lu, J. Hou, K. Qu, Y. Zhu and O. Fontaine, J. Mater. Chem. A, 2021, 9, 4159–4166 RSC.
- C. Fang, D. Zhang, X. Wang and R. Li, Inorg. Chem. Front., 2022, 9, 3643–3653 RSC.
- S. J. Patil, N. R. Chodankar, S. K. Hwang, G. S. Rama Raju, Y. S. Huh and Y. K. Han, Small, 2022, 18, 2103326 CrossRef CAS PubMed.
- L. Wang, Y. Zhu, Y. Wen, S. Li, C. Cui, F. Ni, Y. Liu, H. Lin, Y. Li, H. Peng and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10577–10582 CrossRef CAS PubMed.
- S. Wang, L. Zhao, J. Li, X. Tian, X. Wu and L. Feng, J. Energy Chem., 2022, 66, 483–492 CrossRef CAS.
- X. Zhang, G. Ma, L. Shui, G. Zhou and X. Wang, J. Energy Chem., 2022, 72, 88–96 CrossRef CAS.
- J. Zhao, Y. Zhang, H. Guo, J. Ren, H. Zhang, Y. Wu and R. Song, Chem. Eng. J., 2022, 433, 134497 CrossRef CAS.
- X. Zhang, X. Fang, K. Zhu, W. Yuan, T. Jiang, H. Xue and J. Tian, J. Power Sources, 2022, 520, 230882 CrossRef CAS.
- Y. Jiang, S. Gao, G. Xu and X. Song, J. Mater. Chem. A, 2021, 9, 5664–5674 RSC.
- S. Zheng, H. Qin, X. Cao, T. Wang, W. Lu and L. Jiao, J. Energy Chem., 2022, 70, 258–265 CrossRef CAS.
- Z. Ji, Y. Song, S. Zhao, Y. Li, J. Liu and W. Hu, ACS Catal., 2021, 12, 569–579 CrossRef.
- L. Zhang, L. Wang, H. Lin, Y. Liu, J. Ye, Y. Wen, A. Chen, L. Wang, F. Ni, Z. Zhou, S. Sun, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2019, 58, 16820–16825 CrossRef CAS PubMed.
- W. Chen, L. Xu, X. Zhu, Y. C. Huang, W. Zhou, D. Wang, Y. Zhou, S. Du, Q. Li, C. Xie, L. Tao, C. L. Dong, J. Liu, Y. Wang, R. Chen, H. Su, C. Chen, Y. Zou, Y. Li, Q. Liu and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 7297–7307 CrossRef CAS PubMed.
- S. K. Geng, Y. Zheng, S. Q. Li, H. Su, X. Zhao, J. Hu, H. B. Shu, M. Jaroniec, P. Chen, Q. H. Liu and S. Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- T. Wang, Q. Wang, Y. Wang, Y. Da, W. Zhou, Y. Shao, D. Li, S. Zhan, J. Yuan and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 13466–13471 CrossRef CAS PubMed.
- L. Zhou, M. Shao, C. Zhang, J. Zhao, S. He, D. Rao, M. Wei, D. G. Evans and X. Duan, Adv. Mater., 2017, 29, 1604080 CrossRef PubMed.
- Y. Li, J. Zhang, Y. Liu, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Sci. Adv., 2020, 6, eabb4197 CrossRef CAS PubMed.
- J. Y. Zhang, H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 7649–7653 CrossRef CAS PubMed.
- J. Li, Y. Li, J. Wang, C. Zhang, H. Ma, C. Zhu, D. Fan, Z. Guo, M. Xu, Y. Wang and H. Ma, Adv. Funct. Mater., 2022, 32, 2109439 CrossRef CAS.
- J. Li, C. Zhang, C. Zhang, H. Ma, Y. Yang, Z. Guo, Y. Wang and H. Ma, Chem. Eng. J., 2022, 430, 132953 CrossRef CAS.
- S. Zhuang, Y. Tang, X. Tai, Q. Huang, P. Wan, Y. Chen, Y. Sun, J. Pan and X. J. Yang, Appl. Catal., B, 2022, 306, 121132 CrossRef CAS.
- S. Zhang, C. Zhang, X. Zheng, G. Su, H. Wang and M. Huang, Appl. Catal., B, 2023, 324, 122207 CrossRef CAS.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li and G. Zhang, Adv. Funct. Mater., 2021, 31, 2103673 CrossRef CAS.
- Z. Wang, L. Xu, F. Huang, L. Qu, J. Li, K. A. Owusu, Z. Liu, Z. Lin, B. Xiang, X. Liu, K. Zhao, X. Liao, W. Yang, Y. B. Cheng and L. Mai, Adv. Energy Mater., 2019, 9, 1900390 CrossRef.
- T. Wang, Y. Cao, H. Wu, C. Feng, Y. Ding and H. Mei, Int. J. Hydrogen Energy, 2022, 47, 5766–5778 CrossRef CAS.
- H. Wang and S. Tao, Nanoscale Adv., 2021, 3, 2280–2286 RSC.
- Z. Feng, E. Wang, S. Huang and J. Liu, Nanoscale, 2020, 12, 4426–4434 RSC.
- H. Y. Wang, L. Wang, J. T. Ren, W. W. Tian, M. L. Sun and Z. Y. Yuan, Nano-Micro Lett., 2023, 15, 155 CrossRef CAS PubMed.
- T. J. Wang, G. R. Xu, H. Y. Sun, H. Huang, F. M. Li, P. Chen and Y. Chen, Nanoscale, 2020, 12, 11526–11535 RSC.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Nat. Commun., 2020, 11, 1853 CrossRef CAS PubMed.
- X. Liu, H. Mao, G. Liu, Q. Yu, S. Wu, B. Li, G. Zhou, Z. Li and L. Wang, Chem. Eng. J., 2023, 451, 138699 CrossRef CAS.
- Q. Yu, J. Chi, G. Liu, X. Wang, X. Liu, Z. Li, Y. Deng, X. Wang and L. Wang, Sci. China: Mater., 2022, 65, 1539–1549 CAS.
- T. Cui, J. Chi, J. Zhu, X. Sun, J. Lai, Z. Li and L. Wang, Appl. Catal., B, 2022, 319, 121950 CrossRef CAS.
- X. Wang, W. Zhang, Q. Yu, X. Liu, Q. Liang, X. Meng, X. Wang and L. Wang, Chem. Eng. J., 2022, 446, 136987 CrossRef CAS.
- L. Quan, X. Chen, J. Liu, S. Fan, B. Y. Xia and B. You, Adv. Funct. Mater., 2023, 33, 2307643 CrossRef CAS.
- H. Y. Wang, C. C. Weng, J. T. Ren and Z. Y. Yuan, Front. Chem. Sci. Eng., 2021, 15, 1408–1426 CrossRef.
- J. Liu, S. Duan, H. Shi, T. Wang, X. Yang, Y. Huang, G. Wu and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202210753 CrossRef CAS PubMed.
- K. Asazawa, T. Sakamoto, S. Yamaguchi, K. Yamada, H. Fujikawa, H. Tanaka and K. Oguro, J. Electrochem. Soc., 2009, 156, B509 CrossRef CAS.
- W. X. Yin, Z. P. Li, J. K. Zhu and H. Y. Qin, J. Power Sources, 2008, 182, 520–523 CrossRef CAS.
- K. Yamada, K. Yasuda, H. Tanaka, Y. Miyazaki and T. Kobayashi, J. Power Sources, 2003, 122, 132–137 CrossRef CAS.
- T. Kodera, M. Honda and H. Kita, Electrochim. Acta, 1985, 30, 669–675 CrossRef CAS.
- Q. Sun, Y. Li, J. Wang, B. Cao, Y. Yu, C. Zhou, G. Zhang, Z. Wang and C. Zhao, J. Mater. Chem. A, 2020, 8, 21084–21093 RSC.
- R. G. Kadam, T. Zhang, D. Zaoralova, M. Medved, A. Bakandritsos, O. Tomanec, M. Petr, J. Zhu Chen, J. T. Miller, M. Otyepka, R. Zboril, T. Asefa and M. B. Gawande, Small, 2021, 17, 2006477 CrossRef CAS PubMed.
- J. Wang, X. Guan, H. Li, S. Zeng, R. Li, Q. Yao, H. Chen, Y. Zheng and K. Qu, Nano Energy, 2022, 100, 107467 CrossRef CAS.
- L. Zhu, J. Huang, G. Meng, T. Wu, C. Chen, H. Tian, Y. Chen, F. Kong, Z. Chang, X. Cui and J. Shi, Nat. Commun., 2023, 14, 1997 CrossRef CAS PubMed.
- S. K. Apte, S. N. Garaje, S. D. Naik, R. P. Waichal and B. B. Kale, Green Chem., 2013, 15, 3459 RSC.
- Y. Pei, J. Cheng, H. Zhong, Z. Pi, Y. Zhao and F. Jin, Green Chem., 2021, 23, 6975–6983 RSC.
- M. Zhang, J. Guan, Y. Tu, S. Chen, Y. Wang, S. Wang, L. Yu, C. Ma, D. Deng and X. Bao, Energy Environ. Sci., 2020, 13, 119–126 RSC.
- L. Zhang, D. Liu, Z. Muhammad, F. Wan, W. Xie, Y. Wang, L. Song, Z. Niu and J. Chen, Adv. Mater., 2019, 31, 1903955 CrossRef CAS PubMed.
- X. Wu, A. Markir, L. Ma, Y. Xu, H. Jiang, D. P. Leonard, W. Shin, T. Wu, J. Lu and X. Ji, Angew. Chem., Int. Ed., 2019, 58, 12640–12645 CrossRef CAS PubMed.
- S. Zhang, Q. Zhou, Z. Shen, X. Jin, Y. Zhang, M. Shi, J. Zhou, J. Liu, Z. Lu, Y. N. Zhou and H. Zhang, Adv. Funct. Mater., 2021, 31, 2101922 CrossRef CAS.
- M. Zhang, J. Guan, Y. Tu, S. Wang and D. Deng, Innovation, 2021, 2, 100144 CAS.
- P. Shen, B. Zhou, Z. Chen, W. Xiao, Y. Fu, J. Wan, Z. Wu and L. Wang, Appl. Catal., B, 2023, 325, 122305 CrossRef CAS.
- Y. Zhu, J. Zhang, Q. Qian, Y. Li, Z. Li, Y. Liu, C. Xiao, G. Zhang and Y. Xie, Angew. Chem., Int. Ed., 2022, 61, e202113082 CrossRef CAS PubMed.
- C. Tang, R. Zhang, W. Lu, Z. Wang, D. Liu, S. Hao, G. Du, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2017, 56, 842–846 CrossRef CAS PubMed.
- H. Jin, X. Wang, C. Tang, A. Vasileff, L. Li, A. Slattery and S. Z. Qiao, Adv. Mater., 2021, 33, 2007508 CrossRef CAS PubMed.
- L. Yi, Y. Ji, P. Shao, J. Chen, J. Li, H. Li, K. Chen, X. Peng and Z. Wen, Angew. Chem., Int. Ed., 2021, 60, 21550–21557 CrossRef CAS PubMed.
- Z. Xiao, C. Lu, J. Wang, Y. Qian, B. Wang, Q. Zhang, A. Tang and H. Yang, Adv. Funct. Mater., 2023, 33, 2212183 CrossRef CAS.
- L. Du, Y. Sun and B. You, Mater. Rep.: Energy, 2021, 1, 100004 CAS.
- H. Zhao, D. Lu, J. Wang, W. Tu, D. Wu, S. W. Koh, P. Gao, Z. J. Xu, S. Deng, Y. Zhou, B. You and H. Li, Nat. Commun., 2021, 12, 2008 CrossRef CAS PubMed.
- Y. Yan, H. Zhou, S. M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- F. Arshad, T. u Haq, I. Hussain and F. Sher, ACS Appl. Energy Mater., 2021, 4, 8685–8701 CrossRef CAS.
- Z. Li, Y. Yan, S. M. Xu, H. Zhou, M. Xu, L. Ma, M. Shao, X. Kong, B. Wang, L. Zheng and H. Duan, Nat. Commun., 2022, 13, 147 CrossRef CAS PubMed.
- K. Yin, Y. Chao, F. Lv, L. Tao, W. Zhang, S. Lu, M. Li, Q. Zhang, L. Gu, H. Li and S. Guo, J. Am. Chem. Soc., 2021, 143, 10822–10827 CrossRef CAS PubMed.
- Y. X. Chen, A. Lavacchi, H. A. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 4036 CrossRef CAS PubMed.
- L. Dai, Q. Qin, X. Zhao, C. Xu, C. Hu, S. Mo, Y. O. Wang, S. Lin, Z. Tang and N. Zheng, ACS Cent. Sci., 2016, 2, 538–544 CrossRef CAS PubMed.
- J. Zheng, X. Chen, X. Zhong, S. Li, T. Liu, G. Zhuang, X. Li, S. Deng, D. Mei and J. G. Wang, Adv. Funct. Mater., 2017, 27, 1704169 CrossRef.
- X. Cui, M. Chen, R. Xiong, J. Sun, X. Liu and B. Geng, J. Mater. Chem. A, 2019, 7, 16501–16507 RSC.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- X. Yu, E. C. dos Santos, J. White, G. Salazar-Alvarez, L. G. M. Pettersson, A. Cornell and M. Johnsson, Small, 2021, 17, 2104288 CrossRef CAS PubMed.
- Y. Zhao, S. Xing, X. Meng, J. Zeng, S. Yin, X. Li and Y. Chen, Nanoscale, 2019, 11, 9319–9326 RSC.
- J. Mahmoudian, M. Bellini, M. V. Pagliaro, W. Oberhauser, M. Innocenti, F. Vizza and H. A. Miller, ACS Sustainable Chem. Eng., 2017, 5, 6090–6098 CrossRef CAS.
- D. Si, B. Xiong, L. Chen and J. Shi, Chem Catal., 2021, 1, 941–955 CrossRef CAS.
- C. Lin, P. Zhang, S. Wang, Q. Zhou, B. Na, H. Li, J. Tian, Y. Zhang, C. Deng, L. Meng, J. Wu, C. Liu, J. Hu and L. Zhang, J. Alloys Compd., 2020, 823, 153784 CrossRef CAS.
- M. Qin, R. Fan, J. Chen, H. Wang, X. Zheng, S. Mao, R. Du and Y. Wang, Chem. Eng. J., 2022, 442, 136264 CrossRef CAS.
- Z. Xia, X. Zhang, H. Sun, S. Wang and G. Sun, Nano Energy, 2019, 65, 104048 CrossRef CAS.
- J. Wang, B. Zhang, W. Guo, L. Wang, J. Chen, H. Pan and W. Sun, Adv. Mater., 2023, 35, 2211099 CrossRef CAS PubMed.
- G. Chen, H. Wan, W. Ma, N. Zhang, Y. Cao, X. Liu, J. Wang and R. Ma, Adv. Energy Mater., 2019, 10, 1902535 CrossRef.
- W. Chen, S. Luo, M. Sun, M. Tang, X. Fan, Y. Cheng, X. Wu, Y. Liao, B. Huang and Z. Quan, Small, 2022, 18, 2107803 CrossRef CAS PubMed.
- P. Wang, H. Cui and C. Wang, Chem. Eng. J., 2022, 429, 132435 CrossRef CAS.
- M. Li, X. Deng, Y. Liang, K. Xiang, D. Wu, B. Zhao, H. Yang, J. L. Luo and X. Z. Fu, J. Energy Chem., 2020, 50, 314–323 CrossRef.
- T. Wang, X. Cao, H. Qin, X. Chen, J. Li and L. Jiao, J. Mater. Chem. A, 2021, 9, 21094–21100 RSC.
- F. Chen, S. Guo, S. Yu, C. Zhang, M. Guo and C. Li, J. Colloid Interface Sci., 2023, 646, 43–53 CrossRef CAS PubMed.
- M. B. Askari and S. M. Rozati, J. Alloys Compd., 2022, 900, 163408 CrossRef CAS.
- B. Zhao, J. W. Liu, Y. R. Yin, D. Wu, J. L. Luo and X. Z. Fu, J. Mater. Chem. A, 2019, 7, 25878–25886 RSC.
- B. Zhao, J. Liu, C. Xu, R. Feng, P. Sui, L. Wang, J. Zhang, J. L. Luo and X. Z. Fu, Adv. Funct. Mater., 2020, 31, 2008812 CrossRef.
- X. Wang, S. Xi, W. S. V. Lee, P. Huang, P. Cui, L. Zhao, W. Hao, X. Zhao, Z. Wang, H. Wu, H. Wang, C. Diao, A. Borgna, Y. Du, Z. G. Yu, S. Pennycook and J. Xue, Nat. Commun., 2020, 11, 4647 CrossRef CAS PubMed.
- B. Zhao, J. Liu, X. Wang, C. Xu, P. Sui, R. Feng, L. Wang, J. Zhang, J. L. Luo and X. Z. Fu, Nano Energy, 2021, 80, 105530 CrossRef CAS.
- A. A. Dubale, Y. Zheng, H. Wang, R. Hubner, Y. Li, J. Yang, J. Zhang, N. K. Sethi, L. He, Z. Zheng and W. Liu, Angew. Chem., Int. Ed., 2020, 59, 13891–13899 CrossRef CAS PubMed.
- J. Li, Z. Luo, Y. Zuo, J. Liu, T. Zhang, P. Tang, J. Arbiol, J. Llorca and A. Cabot, Appl. Catal., B, 2018, 234, 10–18 CrossRef CAS.
- S. L. Candelaria, N. M. Bedford, T. J. Woehl, N. S. Rentz, A. R. Showalter, S. Pylypenko, B. A. Bunker, S. Lee, B. Reinhart, Y. Ren, S. P. Ertem, E. B. Coughlin, N. A. Sather, J. L. Horan, A. M. Herring and L. F. Greenlee, ACS Catal., 2016, 7, 365–379 CrossRef.
- J. Hao, J. Liu, D. Wu, M. Chen, Y. Liang, Q. Wang, L. Wang, X. Z. Fu and J. L. Luo, Appl. Catal., B, 2021, 281, 119510 CrossRef CAS.
- W. Yang, X. Yang, J. Jia, C. Hou, H. Gao, Y. Mao, C. Wang, J. Lin and X. Luo, Appl. Catal., B, 2019, 244, 1096–1102 CrossRef CAS.
- F. Arshad, A. Tahir, T. U. Haq, H. Duran, I. Hussain and F. Sher, Int. J. Hydrogen Energy, 2022, 47, 36118–36128 CrossRef CAS.
- N. Moazzami, S. Khadempir, H. Karimi-Maleh, F. Karimi and C. Karaman, Int. J. Hydrogen Energy, 2023, 48, 6680–6690 CrossRef CAS.
- X. Cui, P. Xiao, J. Wang, M. Zhou, W. Guo, Y. Yang, Y. He, Z. Wang, Y. Yang, Y. Zhang and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 4488–4493 CrossRef CAS PubMed.
- D. Wu, W. Zhang and D. Cheng, ACS Appl. Mater. Interfaces, 2017, 9, 19843–19851 CrossRef CAS PubMed.
- K. Xiang, D. Wu, X. Deng, M. Li, S. Chen, P. Hao, X. Guo, J. L. Luo and X. Z. Fu, Adv. Funct. Mater., 2020, 30, 1909610 CrossRef CAS.
- Y. Xu, M. Liu, M. Wang, T. Ren, K. Ren, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 300, 120753 CrossRef CAS.
- Q. Wang, L. Chen, S. Guan, X. Zhang, B. Wang, X. Cao, Z. Yu, Y. He, D. G. Evans, J. Feng and D. Li, ACS Catal., 2018, 8, 3104–3115 CrossRef CAS.
- C. Cao, D. D. Ma, J. Jia, Q. Xu, X. T. Wu and Q. L. Zhu, Adv. Mater., 2021, 33, 2008631 CrossRef CAS PubMed.
- M. Zhang, J. Zhu, R. Wan, B. Liu, D. Zhang, C. Zhang, J. Wang and J. Niu, Chem. Mater., 2022, 34, 959–969 CrossRef CAS.
- C. Liu, W. Zhou, J. Zhang, Z. Chen, S. Liu, Y. Zhang, J. Yang, L. Xu, W. Hu, Y. Chen and Y. Deng, Adv. Energy Mater., 2020, 10, 2001397 CrossRef CAS.
- B. Dong, W. Li, X. Huang, Z. Ali, T. Zhang, Z. Yang and Y. Hou, Nano Energy, 2019, 55, 37–41 CrossRef CAS.
- J. Klein, F. Argast, A. K. Engstfeld, S. Brimaud and R. J. Behm, Electrochim. Acta, 2019, 311, 244–254 CrossRef CAS.
- J. Park, H. J. Kim, A. Oh, T. Kwon, H. Baik, S. I. Choi and K. Lee, Nanoscale, 2018, 10, 21178–21185 RSC.
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed.
- X. Wei, S. Wang, Z. Hua, L. Chen and J. Shi, ACS Appl. Mater. Interfaces, 2018, 10, 25422–25428 CrossRef CAS PubMed.
- S. Li, R. Ma, J. Hu, Z. Li, L. Liu, X. Wang, Y. Lu, G. E. Sterbinsky, S. Liu, L. Zheng, J. Liu, D. Liu and J. Wang, Nat. Commun., 2022, 13, 2916 CrossRef CAS PubMed.
- B. Braunchweig, D. Hibbitts, M. Neurock and A. Wieckowski, Catal. Today, 2013, 202, 197–209 CrossRef CAS.
- M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li, H. Fei, Y. Zhu, R. Yu, J. Luo, K. Zang, Z. Lin, M. Ding, J. Huang, H. Sun, J. Guo, X. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef CAS.
- Y. Zhu, X. Zhu, L. Bu, Q. Shao, Y. Li, Z. Hu, C. T. Chen, C. W. Pao, S. Yang and X. Huang, Adv. Funct. Mater., 2020, 30, 2004310 CrossRef CAS.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS PubMed.
- C. A. Zhou, S. Wang, K. Ma, L. Song, L. Zheng and H. Yue, Appl. Catal., B, 2023, 321, 122065 CrossRef CAS.
- G. F. Chen, Y. Luo, L. X. Ding and H. Wang, ACS Catal., 2017, 8, 526–530 CrossRef.
- H. Sun, L. Li, Y. Chen, H. Kim, X. Xu, D. Guan, Z. Hu, L. Zhang, Z. Shao and W. Jung, Appl. Catal., B, 2023, 325, 122388 CrossRef CAS.
- L. R. Vidales-Gallardo, E. N. Armendáriz-Mireles, G. G. Suarez-Velázquez, E. Rocha-Rangel and W. J. Pech-Rodríguez, J. Mater. Res., 2021, 36, 4207–4215 CrossRef CAS.
- M. Zhang, Z. Song, Z. Wang, A. Wang, G. Zhu and S. Shao, J. Colloid Interface Sci., 2021, 590, 164–174 CrossRef CAS PubMed.
- J. Xu, B. X. Wang, D. Lyu, T. Wang and Z. Wang, Int. J. Hydrogen Energy, 2023, 48, 10724–10736 CrossRef CAS.
- M. Ghalkhani, R. Abdullah Mirzaie, A. Banimostafa, E. Sohouli and E. Hashemi, Int. J. Hydrogen Energy, 2023, 48, 21214–21223 CrossRef CAS.
- J. Shi, H. He, Y. Guo, F. Ji, J. Li, Y. Zhang, C. Deng, L. Fan and W. Cai, J. Energy Chem., 2023, 85, 76–82 CrossRef CAS.
- L. Xu, Z. Wang, X. Chen, Z. Qu, F. Li and W. Yang, Electrochim. Acta, 2018, 260, 898–904 CrossRef CAS PubMed.
- Z. Wang, X. Liao, M. Zhou, F. Huang, K. A. Owusu, J. Li, Z. Lin, Q. Sun, X. Hong, C. Sun, Y. B. Cheng, Y. Zhao and L. Mai, Energy Environ. Mater., 2022, 6, e12409 CrossRef.
- Y. Zhao, Z. H. Yuan, J. T. Huang, M. Y. Wang, B. He, Y. Ding, P. J. Jin and Y. Chen, Nanoscale, 2023, 15, 1947–1952 RSC.
- Y. Wang, J. Liu, H. Yuan, F. Liu, T. Hu and B. Yang, Adv. Funct. Mater., 2023, 33, 2211909 CrossRef CAS.
- H. Pu, K. Dong, T. Zhang, H. Dai, Y. Wang and Y. Deng, J. Mater. Chem. A, 2022, 10, 10614–10624 RSC.
- X. Jiang, Z. Dong, Q. Zhang, G. R. Xu, J. Lai, Z. Li and L. Wang, J. Mater. Chem. A, 2022, 10, 20571–20579 RSC.
- L. Luo, C. Fu, F. Yang, X. Li, F. Jiang, Y. Guo, F. Zhu, L. Yang, S. Shen and J. Zhang, ACS Catal., 2019, 10, 1171–1184 CrossRef.
- H. Xu, B. Huang, Y. Zhao, G. He and H. Chen, Inorg. Chem., 2022, 61, 4533–4540 CrossRef CAS PubMed.
- Y. Qin, W. Zhang, F. Wang, J. Li, J. Ye, X. Sheng, C. Li, X. Liang, P. Liu, X. Wang, X. Zheng, Y. Ren, C. Xu and Z. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200899 CrossRef CAS PubMed.
- W. J. Pech-Rodríguez, C. Calles-Arriaga, D. González-Quijano, G. Vargas-Gutiérrez, C. Morais, T. W. Napporn and F. J. Rodríguez-Varela, J. Power Sources, 2018, 375, 335–344 CrossRef.
- B. Qiao, T. Yang, S. Shi, N. Jia, Y. Chen, X. Chen, Z. An and P. Chen, Small, 2021, 17, 2006534 CrossRef CAS PubMed.
- H. Xu, B. Yan, J. Wang, K. Zhang, S. Li, Z. Xiong, C. Wang, Y. Shiraishi, Y. Du and P. Yang, J. Mater. Chem. A, 2017, 5, 15932–15939 RSC.
- X. Guo, H. Shang, J. Guo, H. Xu and Y. Du, Appl. Surf. Sci., 2019, 481, 1532–1537 CrossRef CAS.
- X. H. Wang, Z. N. Zhang, Z. Wang, Y. Ding, Q. G. Zhai, Y. C. Jiang, S. N. Li and Y. Chen, Chem. Eng. J., 2023, 465, 142938 CrossRef CAS.
- X. Liu, Z. Fang, X. Teng, Y. Niu, S. Gong, W. Chen, T. J. Meyer and Z. Chen, J. Energy Chem., 2022, 72, 432–441 CrossRef CAS.
- M. Huang, C. Cao, L. Liu, W. Wei, Q.-L. Zhu and Z. Huang, eScience, 2023, 3, 100118 CrossRef.
- L. Jiao, W. Wei, X. Li, C. B. Hong, S. G. Han, M. I. Khan and Q. L. Zhu, Rare Metals, 2022, 41, 3654–3661 CrossRef CAS.
- D. Si, M. Wang, X. Yang, C. Wang, K. Shi, B. Huang, L. Chen and J. Shi, Appl. Catal., B, 2023, 331, 122664 CrossRef CAS.
- I. Mangoufis-Giasin, O. Piqué, P. Khanipour, K. J. J. Mayrhofer, F. Calle-Vallejo and I. Katsounaros, J. Catal., 2021, 400, 166–172 CrossRef CAS.
- X. Fu, C. Wan, Y. Huang and X. Duan, Adv. Funct. Mater., 2022, 32, 2106401 CrossRef CAS.
- M. L. Chelaghmia, M. Nacef, H. Fisli, A. M. Affoune, M. Pontie, A. Makhlouf, T. Derabla, O. Khelifi and F. Aissat, RSC Adv., 2020, 10, 36941–36948 RSC.
- J. Ye, J. Liu, C. Xu, S. P. Jiang and Y. Tong, Electrochem. Commun., 2007, 9, 2760–2763 CrossRef CAS.
- C. Poochai, Appl. Surf. Sci., 2017, 396, 1793–1801 CrossRef CAS.
- Y.-Q. Kang, Q. Xue, Y. Zhao, X. F. Li, P. J. Jin and Y. Chen, Small, 2018, 14, 1801239 CrossRef PubMed.
- B. Wang, L. Tao, Y. Cheng, F. Yang, Y. Jin, C. Zhou, H. Yu and Y. Yang, Catalysts, 2019, 9, 387 CrossRef CAS.
- M. Yang, S. Fan, J. Chen, Y. Chen, C. Li, J. Meng, H. Qing, Y. Liu and Z. Xiao, Chem. Eng. Sci., 2023, 278, 118923 CrossRef CAS.
- A. C. Brix, M. Dreyer, A. Koul, M. Krebs, A. Rabe, U. Hagemann, S. Varhade, C. Andronescu, M. Behrens, W. Schuhmann and D. M. Morales, ChemElectroChem, 2022, 9, e202200092 CrossRef CAS.
- T. Wu, X. Zhu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Nano Res., 2017, 11, 1004–1017 CrossRef.
- L. Fan, B. Liu, X. Liu, N. Senthilkumar, G. Wang and Z. Wen, Energy Technol., 2021, 9, 2000804 CrossRef CAS.
- M. Guschakowski and U. Schroder, ChemSusChem, 2021, 14, 5216–5225 CrossRef CAS PubMed.
- Y. Kwon, K. J. P. Schouten and M. T. M. Koper, ChemCatChem, 2011, 3, 1176–1185 CrossRef CAS.
- S. Li, W. Xie, Y. Song, Y. Li, Y. Song, J. Li and M. Shao, Chem. Eng. J., 2022, 437, 135473 CrossRef CAS.
- M. K. Goetz, M. T. Bender and K. S. Choi, Nat. Commun., 2022, 13, 5848 CrossRef CAS PubMed.
- Y. Xu, M. Liu, S. Wang, K. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2021, 298, 120493 CrossRef CAS.
- M. S. E. Houache, K. Hughes, R. Safari, G. A. Botton and E. A. Baranova, ACS Appl. Mater. Interfaces, 2020, 12, 15095–15107 CrossRef CAS PubMed.
- D. M. Morales, D. Jambrec, M. A. Kazakova, M. Braun, N. Sikdar, A. Koul, A. C. Brix, S. Seisel, C. Andronescu and W. Schuhmann, ACS Catal., 2022, 12, 982–992 CrossRef CAS.
- X. Feng, K. X. Guo, L. F. Fan, G. X. Wang, S. Q. Ci and Z. H. Wen, J. Electrochem., 2023, 29, 2215005 Search PubMed.
- T. G. Vo, P. Y. Ho and C. Y. Chiang, Appl. Catal., B, 2022, 300, 120723 CrossRef CAS.
- Y. Wang, Y. Q. Zhu, Z. Xie, S. M. Xu, M. Xu, Z. Li, L. Ma, R. Ge, H. Zhou, Z. Li, X. Kong, L. Zheng, J. Zhou and H. Duan, ACS Catal., 2022, 12, 12432–12443 CrossRef CAS.
- H. Wan, C. Dai, L. Jin, S. Luo, F. Meng, G. Chen, Y. Duan, C. Liu, Q. Xu, J. Lu and Z. J. Xu, ACS Appl. Mater. Interfaces, 2022, 14, 14293–14301 CrossRef CAS PubMed.
- X. Huang, Y. Guo, Y. Zou and J. Jiang, Appl. Catal., B, 2022, 309, 121247 CrossRef CAS.
- L. S. Oh, M. Park, Y. S. Park, Y. Kim, W. Yoon, J. Hwang, E. Lim, J. H. Park, S. M. Choi, M. H. Seo, W. B. Kim and H. J. Kim, Adv. Mater., 2023, 35, 2203285 CrossRef CAS PubMed.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- C. Liu, M. Hirohara, T. Maekawa, R. Chang, T. Hayashi and C. Y. Chiang, Appl. Catal., B, 2020, 265, 118543 CrossRef CAS.
- J. Du, Y. Qin, T. Dou, J. Ge, Y. Wang, X. Zhao, F. Zhang and X. Lei, ACS Appl. Nano Mater., 2022, 5, 10174–10182 CrossRef CAS.
- Y. Li, X. Wei, S. Han, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 21464–21472 CrossRef CAS PubMed.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- X. Chen, X. Zhong, B. Yuan, S. Li, Y. Gu, Q. Zhang, G. Zhuang, X. Li, S. Deng and J. G. Wang, Green Chem., 2019, 21, 578–588 RSC.
- Z. Zhao, M. M. Flores Espinosa, J. Zhou, W. Xue, X. Duan, J. Miao and Y. Huang, Nano Res., 2019, 12, 1467–1472 CrossRef CAS.
- Y. Han, C. Yu, H. Huang, Q. Wei, J. Dong, L. Chen and J. Qiu, SmartMat, 2023 DOI:10.1002/smm2.1206.
- J. Wan, X. Mu, Y. Jin, J. Zhu, Y. Xiong, T. Li and R. Li, Green Chem., 2022, 24, 4870–4876 RSC.
- R. Li, P. Kuang, L. Wang, H. Tang and J. Yu, Chem. Eng. J., 2022, 431, 134137 CrossRef CAS.
- H. Huang, C. Yu, X. Han, H. Huang, Q. Wei, W. Guo, Z. Wang and J. Qiu, Energy Environ. Sci., 2020, 13, 4990–4999 RSC.
- F. Li, C. Liu, H. Lin, Y. Sun, H. Yu, S. Xue, J. Cao, X. Jia and S. Chen, J. Colloid Interface Sci., 2023, 640, 329–337 CrossRef CAS PubMed.
- M. Xu, J. Geng, H. Xu, S. Zhang and H. Zhang, Inorg. Chem. Front., 2023, 10, 2053–2059 RSC.
- Q. Xue, Z. Xia, W. Gou, J. Bu, J. Li, H. Xiao and Y. Qu, ACS Catal., 2022, 13, 400–406 CrossRef.
- Z. Liang, D. Jiang, X. Wang, M. Shakouri, T. Zhang, Z. Li, P. Tang, J. Llorca, L. Liu, Y. Yuan, M. Heggen, R. E. Dunin-Borkowski, J. R. Morante, A. Cabot and J. Arbiol, Adv. Funct. Mater., 2021, 31, 2106349 CrossRef CAS.
- J. N. Hausmann, P. V. Menezes, G. Vijaykumar, K. Laun, T. Diemant, I. Zebger, T. Jacob, M. Driess and P. W. Menezes, Adv. Energy Mater., 2022, 12, 2202098 CrossRef CAS.
- L. Ming, X. Y. Wu, S. S. Wang, W. Wu and C. Z. Lu, Green Chem., 2021, 23, 7825–7830 RSC.
- M. Zhang, Z. Xu, B. Liu, Y. Duan, Z. Zheng, L. Li, Q. Zhou, V. G. Matveeva, Z. Hu, J. Yu and K. Yan, AIChE J., 2023, 69, e18077 CrossRef CAS.
- L. Wei, M. D. Hossain, M. J. Boyd, J. Aviles-Acosta, M. E. Kreider, A. C. Nielander, M. B. Stevens, T. F. Jaramillo, M. Bajdich and C. Hahn, ACS Catal., 2023, 13, 4272–4282 CrossRef CAS.
- N. Shilpa, A. Pandikassala, P. Krishnaraj, P. S. Walko, R. N. Devi and S. Kurungot, ACS Appl. Mater. Interfaces, 2022, 14, 16222–16232 CrossRef CAS PubMed.
- Z. Yin, Y. Zheng, H. Wang, J. Li, Q. Zhu, Y. Wang, N. Ma, G. Hu, B. He, A. Knop-Gericke, R. Schlogl and D. Ma, ACS Nano, 2017, 11, 12365–12377 CrossRef CAS PubMed.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- Y. Xie, Z. Zhou, N. Yang and G. Zhao, Adv. Funct. Mater., 2021, 31, 2102886 CrossRef CAS.
- P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren, B. Zhang, B. J. J. Timmer, M. S. G. Ahlquist and L. Sun, Angew. Chem., Int. Ed., 2019, 58, 9155–9159 CrossRef CAS PubMed.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- Y. Zhao, M. Cai, J. Xian, Y. Sun and G. Li, J. Mater. Chem. A, 2021, 9, 20164–20183 RSC.
- H. Zhou, Y. Ren, B. Yao, Z. Li, M. Xu, L. Ma, X. Kong, L. Zheng, M. Shao and H. Duan, Nat. Commun., 2023, 14, 5621 CrossRef CAS PubMed.
- M. Yang, Z. Yuan, R. Peng, S. Wang and Y. Zou, Energy Environ. Mater., 2022, 5, 1117–1138 CrossRef CAS.
- M. Li, L. Chen, S. Ye, G. Fan, L. Yang, X. Zhang and F. Li, J. Mater. Chem. A, 2019, 7, 13695–13704 RSC.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Green Chem., 2014, 16, 3778–3786 RSC.
- M. Sahoo, S. Mansingh, S. Subudhi, P. Mohapatra and K. Parida, Catal. Sci. Technol., 2019, 9, 4678–4692 RSC.
- Y. Li, Y. Hu and X. F. Wu, Chem. Soc. Rev., 2018, 47, 172–194 RSC.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed.
- X. Huang, J. Song, M. Hua, Z. Xie, S. Liu, T. Wu, G. Yang and B. Han, Green Chem., 2020, 22, 843–849 RSC.
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, ACS Energy Lett., 2016, 1, 386–390 CrossRef CAS.
- D. H. Nam, B. J. Taitt and K. S. Choi, ACS Catal., 2018, 8, 1197–1206 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- B. J. Taitt, D. H. Nam and K. S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- F. J. Holzhäuser, T. Janke, F. Öztas, C. Broicher and R. Palkovits, Adv. Sustainable Syst., 2020, 4, 1900151 CrossRef.
- X. Lu, K. H. Wu, B. Zhang, J. Chen, F. Li, B. J. Su, P. Yan, J. M. Chen and W. Qi, Angew. Chem., Int. Ed., 2021, 60, 14528–14535 CrossRef CAS PubMed.
- W. J. Liu, L. Dang, Z. Xu, H. Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- M. Zhang, Y. Liu, B. Liu, Z. Chen, H. Xu and K. Yan, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS.
- B. Liu, S. Xu, M. Zhang, X. Li, D. Decarolis, Y. Liu, Y. Wang, E. K. Gibson, C. R. A. Catlow and K. Yan, Green Chem., 2021, 23, 4034–4043 RSC.
- M. Zhou, J. Chen and Y. Li, Catal. Sci. Technol., 2022, 12, 4288–4297 RSC.
- J. Chen, Y. Wang, M. Zhou and Y. Li, Chem. Sci., 2022, 13, 4647–4653 RSC.
- R. Zhang, S. Jiang, Y. Rao, S. Chen, Q. Yue and Y. Kang, Green Chem., 2021, 23, 2525–2530 RSC.
- S. Liang, L. Pan, T. Thomas, B. Zhu, C. Chen, J. Zhang, H. Shen, J. Liu and M. Yang, Chem. Eng. J., 2021, 415, 128864 CrossRef CAS.
- F. Liu, N. Lin, D. Xin, X. Li, L. Cong, F. Han and H. Lin, Catal. Sci. Technol., 2023, 13, 3182–3191 RSC.
- J. Wu, Z. Zhai, T. Yu, X. Wu, S. Huang, W. Cao, Y. Jiang, J. Pei and S. Yin, J. Energy Chem., 2023, 86, 480–489 CrossRef CAS.
- B. Xia, G. Wang, S. Cui, J. Guo, H. Xu, Z. Liu and S. Q. Zang, Chin. Chem. Lett., 2023, 34, 107810 CrossRef CAS.
- A. R. Poerwoprajitno, L. Gloag, J. Watt, S. Cychy, S. Cheong, P. V. Kumar, T. M. Benedetti, C. Deng, K.-H. Wu, C. E. Marjo, D. L. Huber, M. Muhler, J. J. Gooding, W. Schuhmann, D. W. Wang and R. D. Tilley, Angew. Chem., Int. Ed., 2020, 59, 15487–15491 CrossRef CAS PubMed.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- B. Zhang, H. Fu and T. Mu, Green Chem., 2022, 24, 877–884 RSC.
- L. Gao, Y. Bao, S. Gan, Z. Sun, Z. Song, D. Han, F. Li and L. Niu, ChemSusChem, 2018, 11, 2547–2553 CrossRef CAS PubMed.
- J. Wu, J. Chen, T. Yu, Z. Zhai, Y. Zhu, X. Wu and S. Yin, ACS Catal., 2023, 13, 13257–13266 CrossRef CAS.
- H. Chen, J. Wang, Y. Yao, Z. Zhang, Z. Yang, J. Li, K. Chen, X. Lu, P. Ouyang and J. Fu, ChemElectroChem, 2019, 6, 5797–5801 CrossRef CAS.
- K. Gu, D. Wang, C. Xie, T. Wang, G. Huang, Y. Liu, Y. Zou, L. Tao and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 20253–20258 CrossRef CAS PubMed.
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. Shah, I. E. L. Stephens and M. M. Titirici, Adv. Energy Mater., 2021, 11, 2101180 CrossRef CAS.
- G. Grabowski, J. Lewkowski and R. Skowroński, Electrochim. Acta, 1991, 36, 1995 CrossRef CAS.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- A. M. Román, J. C. Hasse, J. W. Medlin and A. Holewinski, ACS Catal., 2019, 9, 10305–10316 CrossRef.
- N. Heidary and N. Kornienko, Chem. Commun., 2020, 56, 8726–8734 RSC.
- N. Heidary and N. Kornienko, Chem. Commun., 2019, 55, 11996–11999 RSC.
- N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806 RSC.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1904780 CrossRef CAS.
- X. Deng, G. Y. Xu, Y. J. Zhang, L. Wang, J. Zhang, J. F. Li, X. Z. Fu and J. L. Luo, Angew. Chem., Int. Ed., 2021, 60, 20535–20542 CrossRef CAS PubMed.
- Y. Lu, C. L. Dong, Y. C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- Y. Lu, T. Liu, C. L. Dong, Y. C. Huang, Y. Li, J. Chen, Y. Zou and S. Wang, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed.
- Y. Lu, T. Liu, C. L. Dong, C. Yang, L. Zhou, Y. C. Huang, Y. Li, B. Zhou, Y. Zou and S. Wang, Adv. Mater., 2022, 34, 2107185 CrossRef CAS PubMed.
- Z. Zhou, C. Chen, M. Gao, B. Xia and J. Zhang, Green Chem., 2019, 21, 6699–6706 RSC.
- R. Zheng, C. Zhao, J. Xiong, X. Teng, W. Chen, Z. Hu and Z. Chen, Sustainable Energy Fuels, 2021, 5, 4023–4031 RSC.
- C. Yang, C. Wang, L. Zhou, W. Duan, Y. Song, F. Zhang, Y. Zhen, J. Zhang, W. Bao, Y. Lu, D. Wang and F. Fu, Chem. Eng. J., 2021, 422, 130125 CrossRef CAS.
- X. Deng, X. Kang, M. Li, K. Xiang, C. Wang, Z. Guo, J. Zhang, X. Z. Fu and J. L. Luo, J. Mater. Chem. A, 2020, 8, 1138–1146 RSC.
- L. Gao, Z. Liu, J. Ma, L. Zhong, Z. Song, J. Xu, S. Gan, D. Han and L. Niu, Appl. Catal., B, 2020, 261, 118235 CrossRef.
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS.
- P. Nilges and U. Schröder, Energy Environ. Sci., 2013, 6, 2925 RSC.
- N. Jiang, X. Liu, J. Dong, B. You, X. Liu and Y. Sun, ChemNanoMat, 2017, 3, 491–495 CrossRef CAS.
- T. Wang, L. Tao, X. Zhu, C. Chen, W. Chen, S. Du, Y. Zhou, B. Zhou, D. Wang, C. Xie, P. Long, W. Li, Y. Wang, R. Chen, Y. Zou, X.-Z. Fu, Y. Li, X. Duan and S. Wang, Nat. Catal., 2021, 5, 66–73 CrossRef.
- M. Yang, Y. Li, C. L. Dong, S. Li, L. Xu, W. Chen, J. Wu, Y. Lu, Y. Pan, Y. Wu, Y. Luo, Y. C. Huang, S. Wang and Y. Zou, Adv. Mater., 2023, 35, 2304203 CrossRef CAS PubMed.
- G. Li, G. Han, L. Wang, X. Cui, N. K. Moehring, P. R. Kidambi, D. E. Jiang and Y. Sun, Nat. Commun., 2023, 14, 525 CrossRef CAS PubMed.
- L. Xiao, W. Dai, S. Mou, X. Wang, Q. Cheng and F. Dong, Energy Environ. Sci., 2023, 16, 2696–2704 RSC.
- K. V. Özdokur, Ç. C. Koçak, Ç. Eden, Z. Demir, Ç. Çirak, E. Yavuz and B. Çağlar, ChemistrySelect, 2022, 7, e202201411 CrossRef.
- W. Liao, Y. W. Chen, Y. C. Liao, X. Y. Lin, S. Yau, J. J. Shyue, S. Y. Wu and H. T. Chen, Electrochim. Acta, 2020, 333, 135542 CrossRef CAS.
- M. Li, T. Wang, W. Zhao, S. Wang and Y. Zou, Nano-Micro Lett., 2022, 14, 211 CrossRef CAS PubMed.
- X. Zhang, T. Y. Liu, Y. Zhou, L. Zhang, X. C. Zhou, J. J. Feng and A. J. Wang, Appl. Catal., B, 2023, 328, 122530 CrossRef CAS.
- Y. Yang, X. Wu, M. Ahmad, F. Si, S. Chen, C. Liu, Y. Zhang, L. Wang, J. Zhang, J. L. Luo and X. Z. Fu, Angew. Chem., Int. Ed., 2023, 62, e202302950 CrossRef CAS PubMed.
- Š. Trafela, J. Zavašnik, S. Šturm and K. Žužek Rožman, Electrochim. Acta, 2020, 362, 137180 CrossRef.
- W. Huang, Y. Guo, X. Cen, Y. Tong, L. Chen and P. Chen, ACS Appl. Energy Mater., 2023, 6, 7221–7229 CrossRef CAS.
- Z. Zhou, L. Zeng, G. Xiong, L. Yang, H. Yuan, J. Yu, S. Xu, D. Wang, X. Zhang, H. Liu and W. Zhou, Chem. Eng. J., 2021, 426, 129214 CrossRef CAS.
- I. Elghamry, S. A. Al-Jendan, M. M. Saleh and M. E. Abdelsalam, RSC Adv., 2022, 12, 20656–20671 RSC.
- W. Ruan, S. Shi, Q. Wang, X. Zhang, W. Hao, C. Yuan, B. Ma, G. Cheng and F. Teng, J. Alloys Compd., 2022, 925, 166748 CrossRef CAS.
- M. Qin, S. Fan, X. Li, Z. Yin, L. Wang and A. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 38256–38265 CrossRef CAS PubMed.
- Y. Pan, Y. Li, C. L. Dong, Y. C. Huang, J. Wu, J. Shi, Y. Lu, M. Yang, S. Wang and Y. Zou, Chem, 2023, 9, 963–977 CAS.
- P. Zhang, Y. J. Guo, J. Chen, Y. R. Zhao, J. Chang, H. Junge, M. Beller and Y. Li, Nat. Catal., 2018, 1, 332–338 CrossRef CAS.
- M. van der Ham, E. van Keulen, M. T. M. Koper, A. A. Tashvigh and J. H. Bitter, Angew. Chem., Int. Ed., 2023, 62, e202306701 CrossRef CAS PubMed.
- W. Tang, L. Zhang, T. Qiu, H. Tan, Y. Wang, W. Liu and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202305843 CrossRef CAS PubMed.
- N. Fujiwara, S. I. Yamazaki, Z. Siroma, T. Ioroi, H. Senoh and K. Yasuda, Electrochem. Commun., 2009, 11, 390–392 CrossRef CAS.
- X. Xu, Z. Ma, D. Li, Z. Su, X. Dong, H. Huang and M. Qi, ACS Appl. Nano Mater., 2022, 5, 4983–4990 CrossRef CAS.
- Y. Guo, J. Liu, Y. T. Xu, B. Zhao, X. Wang, X. Z. Fu, R. Sun and C. P. Wong, Sci. Bull., 2019, 64, 764–773 CrossRef CAS PubMed.
- G. Moggia, T. Kenis, N. Daems and T. Breugelmans, ChemElectroChem, 2020, 7, 86–95 CrossRef CAS.
- Y. Q. Zhu, H. Zhou, J. Dong, S. M. Xu, M. Xu, L. Zheng, Q. Xu, L. Ma, Z. Li, M. Shao and H. Duan, Angew. Chem., Int. Ed., 2023, 62, e202219048 CrossRef CAS PubMed.
- M. Wu, J. Zhao, C. Li and R. Liu, J. Mater. Chem. A, 2022, 10, 4791–4799 RSC.
- Y. Wang, W. Yan, M. Ni, C. Zhu and H. Du, Chem. Commun., 2023, 59, 2485–2488 RSC.
- X. Lin, H. Zhong, W. Hu and J. Du, Inorg. Chem., 2023, 62, 10513–10521 CrossRef CAS PubMed.
- H. Liu, R. Zhang, L. Chen, L. Wang, Y. Guo and Y. Yang, Adv. Sustainable Syst., 2021, 5, 2000184 CrossRef CAS.
- Y. Song, X. Wan, Y. Miao, J. Li, Z. Ren, B. Jin, H. Zhou, Z. Li and M. Shao, Appl. Catal., B, 2023, 333, 122808 CrossRef CAS.
- W. J. Liu, Z. Xu, D. Zhao, X. Q. Pan, H. C. Li, X. Hu, Z. Y. Fan, W. K. Wang, G. H. Zhao, S. Jin, G. W. Huber and H. Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- Y. Xin, F. Wang, L. Chen, Y. Li and K. Shen, Green Chem., 2022, 24, 6544–6555 RSC.
- Y. Zhang, Y. Qiu, Z. Ma, Y. Wang, Y. Zhang, Y. Ying, Y. Jiang, Y. Zhu and S. Liu, J. Mater. Chem. A, 2021, 9, 10893–10908 RSC.
- Y. Shu, B. Li, J. Chen, Q. Xu, H. Pang and X. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 2360–2367 CrossRef CAS PubMed.
- G. Yuan, S. Yu, J. Jie, C. Wang, Q. Li and H. Pang, Chin. Chem. Lett., 2020, 31, 1941–1945 CrossRef CAS.
- P. Du, J. Zhang, Y. Liu and M. Huang, Electrochem. Commun., 2017, 83, 11–15 CrossRef CAS.
- L. Dong, G. R. Chang, Y. Feng, X. Z. Yao and X. Y. Yu, Rare Met., 2022, 41, 1583–1594 CrossRef CAS.
- D. Zheng, J. Li, S. Ci, P. Cai, Y. Ding, M. Zhang and Z. Wen, Appl. Catal., B, 2020, 277, 119178 CrossRef CAS.
- M. T. Schümperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108–1117 CrossRef.
- B. You, G. Han and Y. Sun, Chem. Commun., 2018, 54, 5943–5955 RSC.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS PubMed.
- Y. Li, Y. Jiao, H. Yan, G. Yang, Y. Liu, C. Tian, A. Wu and H. Fu, Angew. Chem., Int. Ed., 2023, 62, e202306640 CrossRef CAS PubMed.
- W. Wang, Y. Wang, R. Yang, Q. Wen, Y. Liu, Z. Jiang, H. Li and T. Zhai, Angew. Chem., Int. Ed., 2020, 59, 16974–16981 CrossRef CAS PubMed.
- L. Zeng, W. Chen, Q. Zhang, S. Xu, W. Zhang, F. Lv, Q. Huang, S. Wang, K. Yin, M. Li, Y. Yang, L. Gu and S. Guo, ACS Catal., 2022, 12, 11391–11401 CrossRef CAS.
- F. Ma, S. Wang, L. Han, Y. Guo, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, Z. Zheng and B. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 56140–56150 CrossRef CAS PubMed.
- M. Xiang, Z. Xu, Q. Wu, Y. Wang and Z. Yan, J. Power Sources, 2022, 535, 231461 CrossRef CAS.
- I. Mondal, J. N. Hausmann, G. Vijaykumar, S. Mebs, H. Dau, M. Driess and P. W. Menezes, Adv. Energy Mater., 2022, 12, 2200269 CrossRef CAS.
- S. Bai, L. Chen, J. Bai, C. Lv, S. Xu, D. Zhang, H. Meng, C. Guo, H. Yang and C. Shang, Inorg. Chem. Front., 2023, 10, 4695–4701 RSC.
- K. Chen, W. Zhang, Y. Bai, W. Gong, N. Zhang, R. Long and Y. Xiong, Chin. Chem. Lett., 2023, 34, 107319 CrossRef CAS.
- J. He, S. Li, C. Li, K. Li, Y. Xu, M. Wang, S. Zhao, J. Zhang, X. Zhong, X. Li, Z. Zhang and J. Wang, AIChE J., 2023, 69, e18153 CrossRef CAS.
- G. Shukla, D. Yadav, S. Singh and M. Shankar Singh, Adv. Synth. Catal., 2022, 364, 1982–1988 CrossRef CAS.
- M. Xiang, Z. Xu, J. Wang, X. Yang and Z. Yan, Chem. – Eur. J., 2021, 27, 7502–7506 CrossRef CAS PubMed.
- K. Dang, H. Dong, L. Wang, M. Jiang, S. Jiang, W. Sun, D. Wang and Y. Tian, Adv. Mater., 2022, 34, 2200302 CrossRef CAS PubMed.
- Y. Lum, J. E. Huang, Z. Wang, M. Luo, D. H. Nam, W. R. Leow, B. Chen, J. Wicks, Y. C. Li, Y. Wang, C. T. Dinh, J. Li, T. T. Zhuang, F. Li, T. K. Sham, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 14–22 CrossRef CAS.
- M. Jin, L. Ma, L. Zhou, K. Ji, X. Xue, B. J. Li and H. Duan, Sci. China: Chem., 2022, 65, 2307–2317 CrossRef CAS.
- X. Chong, C. Liu, C. Wang, R. Yang and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22010–22016 CrossRef CAS PubMed.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12014–12017 CrossRef CAS PubMed.
- W. Wang, Y. B. Zhu, Q. Wen, Y. Wang, J. Xia, C. Li, M. W. Chen, Y. Liu, H. Li, H. A. Wu and T. Zhai, Adv. Mater., 2019, 31, 1900528 CrossRef PubMed.
- Y. Li, X. Wei, L. Chen, J. Shi and M. He, Nat. Commun., 2019, 10, 5335 CrossRef PubMed.
- Y. Yang, H. Zhang, Z.-H. Lin, Y. Liu, J. Chen, Z. Lin, Y. S. Zhou, C. P. Wong and Z. L. Wang, Energy Environ. Sci., 2013, 6, 2429 RSC.
- B. Zhang, C. Zhang, O. Yang, W. Yuan, Y. Liu, L. He, Y. Hu, Z. Zhao, L. Zhou, J. Wang and Z. L. Wang, ACS Nano, 2022, 16, 15286–15296 CrossRef CAS PubMed.
- N. Zhai, Z. Wen, X. Chen, A. Wei, M. Sha, J. Fu, Y. Liu, J. Zhong and X. Sun, Adv. Energy Mater., 2020, 10, 2001041 CrossRef CAS.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- J. Fu, R. Liang, G. Liu, A. Yu, Z. Bai, L. Yang and Z. Chen, Adv. Mater., 2019, 31, 1805230 CrossRef PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 2230–2234 CrossRef CAS PubMed.
- J. Zhang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 13296–13300 CrossRef CAS PubMed.
- Y. Li, Y. Liu, Q. Qian, G. Wang and G. Zhang, Energy Storage Mater., 2020, 28, 27–36 CrossRef.
- W. Liu, J. Bao, L. Xu, M. Guan and Y. Lei, J. Power Sources, 2020, 479, 229099 CrossRef CAS.
- Q. Shi, Q. Liu, Y. Ma, Z. Fang, Z. Liang, G. Shao, B. Tang, W. Yang, L. Qin and X. Fang, Adv. Energy Mater., 2020, 10, 1903854 CrossRef CAS.
- H. Sun, Q. Li, Y. Lian, C. Zhang, P. Qi, Q. Mu, H. Jin, B. Zhang, M. Chen, Z. Deng and Y. Peng, Appl. Catal., B, 2020, 263, 118139 CrossRef CAS.
- H. J. Son, M. J. Kim and S. H. Ahn, Chem. Eng. J., 2021, 414, 128739 CrossRef CAS.
- X. Duan, N. Pan, C. Sun, K. Zhang, X. Zhu, M. Zhang, L. Song and H. Zheng, J. Energy Chem., 2021, 56, 290–298 CrossRef CAS.
- H. Liu, J. Guan, S. Yang, Y. Yu, R. Shao, Z. Zhang, M. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, 2003649 CrossRef CAS PubMed.
- G. Zhou, G. Liu, X. Liu, Q. Yu, H. Mao, Z. Xiao and L. Wang, Adv. Funct. Mater., 2021, 32, 2107608 CrossRef.
- J. Liu, C. Wang, H. Sun, H. Wang, F. Rong, L. He, Y. Lou, S. Zhang, Z. Zhang and M. Du, Appl. Catal., B, 2020, 279, 119407 CrossRef CAS.
- H. Ge, G. Li, J. Shen, W. Ma, X. Meng and L. Xu, Appl. Catal., B, 2020, 275, 119104 CrossRef CAS.
- Q. Liu, Q. Shi, Y. Ma, Z. Fang, Z. Zhou, G. Shao, H. Liu and W. Yang, Chem. Eng. J., 2021, 423, 130313 CrossRef CAS.
- Y. Zhang, W. Shi, L. Bo, Y. Shen, X. Ji, L. Xia, X. Guan, Y. Wang and J. Tong, Chem. Eng. J., 2022, 431, 134188 CrossRef CAS.
- L. Zhang, Y. Zhu, Z. Nie, Z. Li, Y. Ye, L. Li, J. Hong, Z. Bi, Y. Zhou and G. Hu, ACS Nano, 2021, 15, 13399–13414 CrossRef CAS PubMed.
- L. Yan, Z. Xu, X. Liu, S. Mahmood, J. Shen, J. Ning, S. Li, Y. Zhong and Y. Hu, Chem. Eng. J., 2022, 446, 137049 CrossRef CAS.
- H. X. Zhong, J. Wang, Q. Zhang, F. Meng, D. Bao, T. Liu, X. Y. Yang, Z. W. Chang, J. M. Yan and X. B. Zhang, Adv. Sustainable Syst., 2017, 1, 1700020 CrossRef.
- J. Li, T. Tan, Y. Xie, J. Chu, L. Li, B. Ouyang, E. Kan and W. Zhang, J. Colloid Interface Sci., 2023, 640, 78–90 CrossRef CAS PubMed.
- S. Wang, J. Wang, X. Wang, L. Li, J. Qin and M. Cao, J. Energy Chem., 2021, 53, 422–432 CrossRef CAS.
- F. Wang, Z. Xiao, X. Liu, J. Ren, T. Xing, Z. Li, X. Li and Y. Chen, J. Power Sources, 2022, 521, 230925 CrossRef CAS.
- S. Ramakrishnan, J. Balamurugan, M. Vinothkannan, A. R. Kim, S. Sengodan and D. J. Yoo, Appl. Catal., B, 2020, 279, 119381 CrossRef CAS.
- X. Luo, M. Yang, W. Song, Q. Fang, X. Wei, L. Jiao, W. Xu, Y. Kang, H. Wang, N. Wu, W. Gu, L. Zheng, L. Hu and C. Zhu, Adv. Funct. Mater., 2021, 31, 2101193 CrossRef CAS.
- J. T. Ren, Y. D. Ying, Y. P. Liu, W. Li and Z. Y. Yuan, J. Energy Chem., 2022, 71, 619–630 CrossRef CAS.
- J. T. Ren, Y. S. Wang, L. Chen, L. J. Gao, W. W. Tian and Z. Y. Yuan, Chem. Eng. J., 2020, 389, 124408 CrossRef CAS.
- J. T. Ren, L. Chen, L. Wang, X. L. Song, Q. H. Kong and Z. Y. Yuan, J. Mater. Chem. A, 2023, 11, 2899–2909 RSC.
- R. Yang, X. Zheng, M. Qin, B. Lin, X. Shi and Y. Wang, Adv. Sci., 2022, 9, 2201594 CrossRef CAS PubMed.
- X. W. Lv, W. S. Xu, W. W. Tian, H. Y. Wang and Z. Y. Yuan, Small, 2021, 17, 2101856 CrossRef CAS PubMed.
- W. W. Tian, J. T. Ren, X. W. Lv, L. J. Gao and Z. Y. Yuan, ACS Sustainable Chem. Eng., 2020, 9, 510–520 CrossRef.
- W. W. Tian, Y. D. Ying, J. T. Ren and Z. Y. Yuan, J. Mater. Chem. A, 2023, 11, 8024–8037 RSC.
- W. W. Tian, J. T. Ren and Z. Y. Yuan, Appl. Catal., B, 2022, 317, 121764 CrossRef CAS.
- D. Khalafallah, M. Zhi and Z. Hong, ChemCatChem, 2020, 13, 81–110 CrossRef.
- A. Serov, M. Padilla, A. J. Roy, P. Atanassov, T. Sakamoto, K. Asazawa and H. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 10336–10339 CrossRef CAS PubMed.
- H. Sun, L. Gao, A. Kumar, Z. Cao, Z. Chang, W. Liu and X. Sun, ACS Appl. Energy Mater., 2022, 5, 9455–9462 CrossRef CAS.
- X. Liu, J. He, S. Zhao, Y. Liu, Z. Zhao, J. Luo, G. Hu, X. Sun and Y. Ding, Nat. Commun., 2018, 9, 4365 CrossRef PubMed.
- Z. Li, W. Wang, Q. Qian, Y. Zhu, Y. Feng, Y. Zhang, H. Zhang, M. Cheng and G. Zhang, eScience, 2022, 2, 416–427 CrossRef.
- Q. Yu, X. Liu, G. Liu, X. Wang, Z. Li, B. Li, Z. Wu and L. Wang, Adv. Funct. Mater., 2022, 32, 2205767 CrossRef CAS.
- X. Zhai, Q. Yu, J. Chi, X. Wang, B. Li, B. Yang, Z. Li, J. Lai and L. Wang, Nano Energy, 2023, 105, 108008 CrossRef CAS.
- T. Wang, Z. Huang, T. Liu, L. Tao, J. Tian, K. Gu, X. Wei, P. Zhou, L. Gan, S. Du, Y. Zou, R. Chen, Y. Li, X. Z. Fu and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202115636 CrossRef CAS PubMed.
- Z. Sun, G. Wang, S. W. Koh, J. Ge, H. Zhao, W. Hong, J. Fei, Y. Zhao, P. Gao, H. Miao and H. Li, Adv. Funct. Mater., 2020, 30, 2002138 CrossRef CAS.
- Q. Zhang, B. He, L. Tang, Z. Zhou, L. Kang, J. Sun, T. Zhang, Q. Li, C. Li, J. Zhao, Z. Zhang, L. Wei and Y. Yao, Adv. Funct. Mater., 2019, 29, 1808889 CrossRef.
- Y. Wu, Z. Tian, S. Yuan, Z. Qi, Y. Feng, Y. Wang, R. Huang, Y. Zhao, J. Sun, W. Zhao, W. Guo, J. Feng and J. Sun, Chem. Eng. J., 2021, 411, 128538 CrossRef CAS.
- R. Xiao, P. Huang, T. Xiong, J. Wei, F. Wang, J. Deng, Z. Wang and M. S. Balogun, J. Mater. Chem. A, 2023, 11, 374–384 RSC.
- R. Xiao, F. Wang, L. Luo, X. Yao, Y. Huang, Z. Wang and M. S. Balogun, Small Methods, 2023, 7, 2201659 CrossRef CAS PubMed.
- Y. Bai, Y. Wang, Z. Qiao, Y. Yang, L. Deng, C. Li, X. Chen, S. Wang, Y. Huang, X. Zhang and D. Cao, J. Mater. Chem. A, 2022, 10, 16037–16045 RSC.
- J. Yang, X. Wang, B. Li, L. Ma, L. Shi, Y. Xiong and H. Xu, Adv. Funct. Mater., 2017, 27, 1606497 CrossRef.
- Z. Bai, S. Li, J. Fu, Q. Zhang, F. Chang, L. Yang, J. Lu and Z. Chen, Nano Energy, 2019, 58, 680–686 CrossRef CAS.
- P. Liu, D. Gao, W. Xiao, L. Ma, K. Sun, P. Xi, D. Xue and J. Wang, Adv. Funct. Mater., 2018, 28, 1706928 CrossRef.
- N. Logeshwaran, S. Ramakrishnan, S. S. Chandrasekaran, M. Vinothkannan, A. R. Kim, S. Sengodan, D. B. Velusamy, P. Varadhan, J.-H. He and D. J. Yoo, Appl. Catal., B, 2021, 297, 120405 CrossRef CAS.
- G. Liu, H. Lv, Q. Quan, X. Li, H. Lu, W. Li, X. Cui and L. Jiang, Chem. Eng. J., 2022, 450, 138079 CrossRef CAS.
- Y. Feng, S. Yan, X. Zhang and Y. Wang, Acc. Chem. Res., 2023, 56, 1645–1655 CrossRef CAS PubMed.
- S. Yan, Y. Feng, J. Lin and Y. Wang, Adv. Mater., 2023, 35, 2212078 CrossRef CAS PubMed.
- Z. Guo, Y. Wang, Y. Song, C. Li, X. Su, Y. Wang, W. B. Cai and Y. Xia, ACS Energy Lett., 2017, 2, 36–44 CrossRef CAS.
- P. Cai, Y. Li, G. Wang and Z. Wen, Angew. Chem., Int. Ed., 2018, 57, 3910–3915 CrossRef CAS PubMed.
- F. Cheng, L. Wang, H. Wang, C. Lei, B. Yang, Z. Li, Q. Zhang, L. Lei, S. Wang and Y. Hou, Nano Energy, 2020, 71, 104621 CrossRef CAS.
- A. Nadeema, G. Pandurang Kharabe, D. Prakash Biswal and S. Kurungot, ChemElectroChem, 2020, 7, 2582–2591 CrossRef CAS.
- C. Kim, J. Kim, S. Joo, Y. Yang, J. Shin, M. Liu, J. Cho and G. Kim, Angew. Chem., Int. Ed., 2019, 58, 9506–9511 CrossRef CAS PubMed.
- Q. Liu, Z. Yan, E. Wang, S. Wang and G. Sun, Int. J. Hydrogen Energy, 2017, 42, 23045–23053 CrossRef CAS.
- P. K. Shen, A. C. C. Tseung and C. Kuo, J. Power Sources, 1994, 47, 119–127 CrossRef CAS.
- Ø. Hasvold, H. Henriksen, E. Melvir, G. Citi, B. Ø. Johansen, T. Kjønigsen and R. Galetti, J. Power Sources, 1997, 65, 253–261 CrossRef.
- M. Shinohara, E. Araki, M. Mochizuki, T. Kanazawa and K. Suyehiro, J. Power Sources, 2009, 187, 253–260 CrossRef CAS.
- R. Hahn, J. Mainert, F. Glaw and K. D. Lang, J. Power Sources, 2015, 288, 26–35 CrossRef CAS.
- W. S. D. Wilcock and P. C. Kauffman, J. Power Sources, 1997, 66, 71–75 CrossRef CAS.
- J. M. Bergthorson, Y. Yavor, J. Palecka, W. Georges, M. Soo, J. Vickery, S. Goroshin, D. L. Frost and A. J. Higgins, Appl. Energy, 2017, 186, 13–27 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- G. Liu, M. Wang, Y. Xu, X. Wang, X. Li, J. Liu, X. Cui and L. Jiang, J. Power Sources, 2021, 486, 229351 CrossRef CAS.
- L. Kekedy-Nagy, M. Abolhassani, S. I. Perez Bakovic, Z. Anari, J. P. Moore Ii, B. G. Pollet and L. F. Greenlee, J. Am. Chem. Soc., 2020, 142, 18844–18858 CrossRef CAS PubMed.
- H. Xu, G. Xu, L. Chen and J. Shi, Adv. Mater., 2022, 34, 2200058 CrossRef CAS PubMed.
- Q. Zhou, Z. Shen, C. Zhu, J. Li, Z. Ding, P. Wang, F. Pan, Z. Zhang, H. Ma, S. Wang and H. Zhang, Adv. Mater., 2018, 30, 1800140 CrossRef PubMed.
- B. Zhou, Y. Zhang, T. Wang, W. Zhou, J. Liu, Y. Zou, L. Tao, Y. Lu and S. Wang, Chem Catal., 2021, 1, 1493–1504 CrossRef CAS.
- L. Yang, W. Liu, Z. Zhang, X. Du, J. Gong, L. Dong and Y. Deng, Electrochim. Acta, 2017, 246, 1163–1173 CrossRef CAS.
- Z. Wu, Y. Zhang, L. Zhang, B. Zhou, Z. Wei, D. Wang, W. Lu, J. Jia, L. Tao, T. Wang and S. Wang, Adv. Funct. Mater., 2023, 33, 2212479 CrossRef CAS.
- L. Wang, Z. Li, K. Wang, Q. Dai, C. Lei, B. Yang, Q. Zhang, L. Lei, M. K. H. Leung and Y. Hou, Nano Energy, 2020, 74, 104850 CrossRef CAS.
- Q. Dai, L. Wang, K. Wang, X. Sang, Z. Li, B. Yang, J. Chen, L. Lei, L. Dai and Y. Hou, Adv. Funct. Mater., 2021, 32, 2109556 CrossRef.
- T. Yang, Y. Xu, H. Lv, M. Wang, X. Cui, G. Liu and L. Jiang, ACS Sustainable Chem. Eng., 2021, 9, 13106–13113 CrossRef CAS.
- H. Y. Wang, L. Wang, J. T. Ren, W. Tian, M. Sun, Y. Feng and Z. Y. Yuan, ACS Nano, 2023, 17, 10965–10975 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |