
Unified ORR mechanism criteria via charge–spin–coordination of Fe functional units
Kexin
Song
,
Binbin
Yang
,
Xu
Zou
,
Wei
Zhang
 * and
Weitao
Zheng
*
* and
Weitao
Zheng
*
Key Laboratory of Automobile Materials MOE, and School of Materials Science & Engineering, and Jilin Provincial International Cooperation, Key Laboratory of High-Efficiency Clean Energy Materials, and Electron Microscopy Center, and International Center of Future Science, Jilin University, Changchun, 130012, Jilin, China. E-mail: weizhang@jlu.edu.cn; wtzheng@jlu.edu.cn
First published on 10th November 2023
Abstract
For oxygen reduction reaction (ORR) consisting of complex multi-electron and proton-coupled elementary steps, it has been always a core issue to address controlling the adsorption properties of oxygen-containing species (OCs) on the surface and interface of catalysts. Since the unique 3d orbital electronic configuration of Fe functional units (Fe-FUs) enables strong interactions with OCs, sufficient power is provided for the ORR. Inspired by the separation of the three powers, initialized from fingerprinting “charge–spin–coordination” of the catalytic system, we explored and summarized electronic and geometric structures via the descriptors for electronic configuration. Next, the specific catalytic mechanism of Fe-FUs in multiple forms was analyzed, perfectly interpreting the structure–activity relationship in Fe-based catalysts. Finally, the corresponding solutions were put forward by summarizing the bottleneck issues in the deactivation and degradation. This review aims to fully gain high utilization of active components, thereby achieving the win–win goal of combining activity and stability for Fe-based catalysts.
 Kexin Song | Kexin Song obtained his master's degree from Jilin University in 2022. He is currently a doctoral candidate of Jilin University under the supervision of Professor Wei Zhang. He is engaged in design, synthesis and application of carbon-based single-atom catalysts. His research focuses on the development of novel metal centers and coordination environments and the exploration of dynamic configuration phenomena of single-atom catalysts using advanced in situ techniques. |
 Binbin Yang | Binbin Yang received her master's degree from Lanzhou University of Technology in 2021. She is currently a doctoral candidate of Jilin University under the supervision of Professors Wei Zhang and Weitao Zheng. Her current research focuses on design, synthesis and application of carbon-based materials for rechargeable alkali metal ion batteries. The specific strategy is to explore the reaction mechanism of modified carbon materials in alkali metal ion batteries at the electronic and atomic levels. |
 Xu Zou | Xu Zou obtained her PhD degree from Jilin University in 2020. She is now an associate researcher at the School of Materials Science & Engineering, College of Materials, Jilin University, and has been selected for the “Dingxin Scholar from Jilin University” support program. She has been engaged in design, synthesis and application research of catalytic materials for long term, aiming to reveal the relationship between the “basic gene” and the performance of materials, develop new catalytic materials, and develop green synthesis technology of catalytic materials. |
 Wei Zhang | Wei Zhang, Director of the electron microscopy center and Tang-Auchin Scholar-Leading Professor at Jilin University. He is also a member of the Surface and Interface Physics Professional Committee of the Chinese Physical Society, Chairman of the Jilin Electron Microscopy Society and a Fellow of the Royal Society of Chemistry (FRSC). Prof. Zhang is selected as “Highly Cited Researcher 2023” in the field of “Cross-field” by Clarivate. He obtained his PhD degree from the Institute of Metal Research (IMR) Chinese Academy of Sciences in 2004. After that, he held positions at NIMS Japan, Samsung AIT South Korea, FHI-MPG Germany, DTU Denmark, and CIC Energigune. His main research interests are surface/interface chemistry of catalytic and energy materials, and electron microscopy of advanced materials. |
 Weitao Zheng | Weitao Zheng, Cheung Kong Distinguished Professor at Jilin University. He received his PhD degree from Jilin University in 1990. Then, he held positions at Linkoping University, Chiba Institute of Technology and Nanyang Technological University. He is also the Vice Chairman of the Crystallographic Society of China, and the member of the International Centre for Diffraction Data (ICDD). His research interests include energy materials, functional thin films, and catalysts. |
Broader contextThe oxygen reduction reaction (ORR) is a key cathode reaction in fuel cells and metal–air batteries. Reasonable design of catalysts is of great significance to improve their overall working efficiency. As the most potential alternative to noble-metal catalysts, Fe-FUs in different forms often undergo appropriate interactions with oxygen-containing intermediate species (OCs) due to a unique 3d electron configuration, thus providing sufficient power for the ORR. This review aims to comprehensively analyze the specific reaction and deactivation mechanisms of Fe-FUs with different forms in ORR processes from the “charge–spin–coordination” perspective, aiming to provide sufficient theoretical guidance and design principles for developing Fe-based catalysts. In-depth exploration of the structure–activity relationship of different Fe-FUs in the ORR process is expected to achieve the win–win goal of combining activity and stability, realizing the large-scale application of non-noble metals in fuel cells and metal–air batteries. |
1 Introduction
As the three essential factors regulating the oxygen reduction reaction (ORR), charge, spin and coordination appear independent of each other but are closely correlated. Their complementary ORR with sluggish kinetics (5–6 orders of magnitude lower than hydrogen oxidation) provides an opportunity for a relatively comprehensive design of catalysts.1,2 An ideal expectation of ORR catalysts aims to establish an appropriate adsorption state with oxygen-containing species (OCs), achieving fast proton and electron transfers.3–6 It is known that the unfilled 3d valence orbital (3d64s2) makes Fe a promising candidate among non-noble metals.7–11 The highly delocalized valence-shell electronic configuration enables the diversity of regulating the Fe electronic configuration, and the unique d–p hybrid also ensures efficient electron transfer with OCs.12–14 So these unique structural advantages contribute to Fe as a leader in the ORR.Attributed to the diversity of experimental design and preparation methods, the Fe functional units (Fe-FUs) in multiple existing forms in a catalytic system are indicative of the complex reaction mechanism. They can either directly or indirectly act as active sites to promote efficient and fast adsorption/desorption with OCs. Concretely speaking, all the existence forms of Fe-FUs (e.g., nanoparticles, nanoclusters and single atoms), coordination environment (types/numbers of coordination atoms) and geometric configuration (defects/edges and planar/nonplanar) affect the electronic and magnetic properties of the active sites to a variety of degrees, i.e., unfolding around “charge–spin–coordination”.15–19 However, Fe-based catalysts suffer essentially from the following disadvantages toward their large-scale commercialization: (1) the catalytic mechanism of Fe-FUs remains unambiguous attributed to the diverse structures and complex compositions; (2) there is plenty of room for improvement in the dynamic balance of catalytic activity and stability.20–25 Complex working conditions cause irreversible phenomena such as aggregation, dissolution and phase transition of Fe components, and also induce structural damage to carbon supports. These malignant reconstructions are largely limiting the practical application of Fe-based catalysts.
Thus, the activity sources and deactivation mechanisms of different Fe-FUs were comprehensively investigated (Fig. 1). Initialized from the electronic configuration, three important descriptors are abstracted to elaborate on the active source in Fe-based catalysts. Next, we explore the structure–activity relationship at the microscopy level by analyzing the catalytic behavior and mechanism of Fe-FUs present in multiple forms. Finally, the optimization strategies to balance the activity and stability are proposed according to the deactivation and degradation of the Fe-based catalyst. Our review is highly integrated with Fe-FU internal factors of “charge–spin–coordination” to provide a sound theoretical basis for designing and optimizing efficient ORR catalysts.
 | ||
| Fig. 1 Research status and bottleneck problems of Fe-based catalysts. | ||
2 Basic theory of the ORR
2.1 The reaction principles of the ORR
As a multi-electron–proton coupling reaction, the ORR usually consists of complex elementary steps. According to the difference in reaction pathways, the ORR is mainly carried out through association mechanism (AM) and dissociation mechanism (DM), accompanied by the occurrence of two-electron and four-electron reactions,26–28 as illustrated in Fig. 2. The adsorption mode of O2 molecules at the active sites is usually carried out in three ways: Griffiths mode, Pauling mode, and Yeager mode.29 Taking the AM as an example, O2 adsorbs on the active sites in Yeager mode to form M–O–O–M structures. Subsequently, accompanied by the breaking of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the addition of two protons and electrons, 2M–OH was formed, followed by the addition of two protons and electrons to form H2O (O2 + 4H+ + 4e− → 2H2O) or OH− (O2 + 2H2O + 4e− → 4OH−). The pathway composed of the 2+2 electron mentioned-above is called the indirect four-electron reaction. In the DM, O2 molecules act on the active sites in the Pauling mode to form M–OO structures. After the M–O–O–H structure is formed by adding a proton and electron in one step, further addition of a proton and electron usually forms the following two species: (1) H2O2 (M–O–O–H + H+ + e− → M + H2O2) or HO2− (M–O–O–H + H2O + e− → M + HO2−) is the strongly oxidizing species, following a two-electron reaction. (2) M–O intermediate state. Next, M–OH and the final product OH− or H2O are formed through two steps of proton and electron addition, respectively, forming a direct four-electron reaction.28,30–32 The interaction strength between abundant OCs and active sites in multiple elemental steps, as well as the difficulty of electron and proton transfer, jointly determines the reaction rate of the ORR.33 The reaction mechanism and pathway of Fe-based catalysts with various Fe-FUs in the ORR are also key issues worth further analyzing and exploring.
O bond and the addition of two protons and electrons, 2M–OH was formed, followed by the addition of two protons and electrons to form H2O (O2 + 4H+ + 4e− → 2H2O) or OH− (O2 + 2H2O + 4e− → 4OH−). The pathway composed of the 2+2 electron mentioned-above is called the indirect four-electron reaction. In the DM, O2 molecules act on the active sites in the Pauling mode to form M–OO structures. After the M–O–O–H structure is formed by adding a proton and electron in one step, further addition of a proton and electron usually forms the following two species: (1) H2O2 (M–O–O–H + H+ + e− → M + H2O2) or HO2− (M–O–O–H + H2O + e− → M + HO2−) is the strongly oxidizing species, following a two-electron reaction. (2) M–O intermediate state. Next, M–OH and the final product OH− or H2O are formed through two steps of proton and electron addition, respectively, forming a direct four-electron reaction.28,30–32 The interaction strength between abundant OCs and active sites in multiple elemental steps, as well as the difficulty of electron and proton transfer, jointly determines the reaction rate of the ORR.33 The reaction mechanism and pathway of Fe-based catalysts with various Fe-FUs in the ORR are also key issues worth further analyzing and exploring.
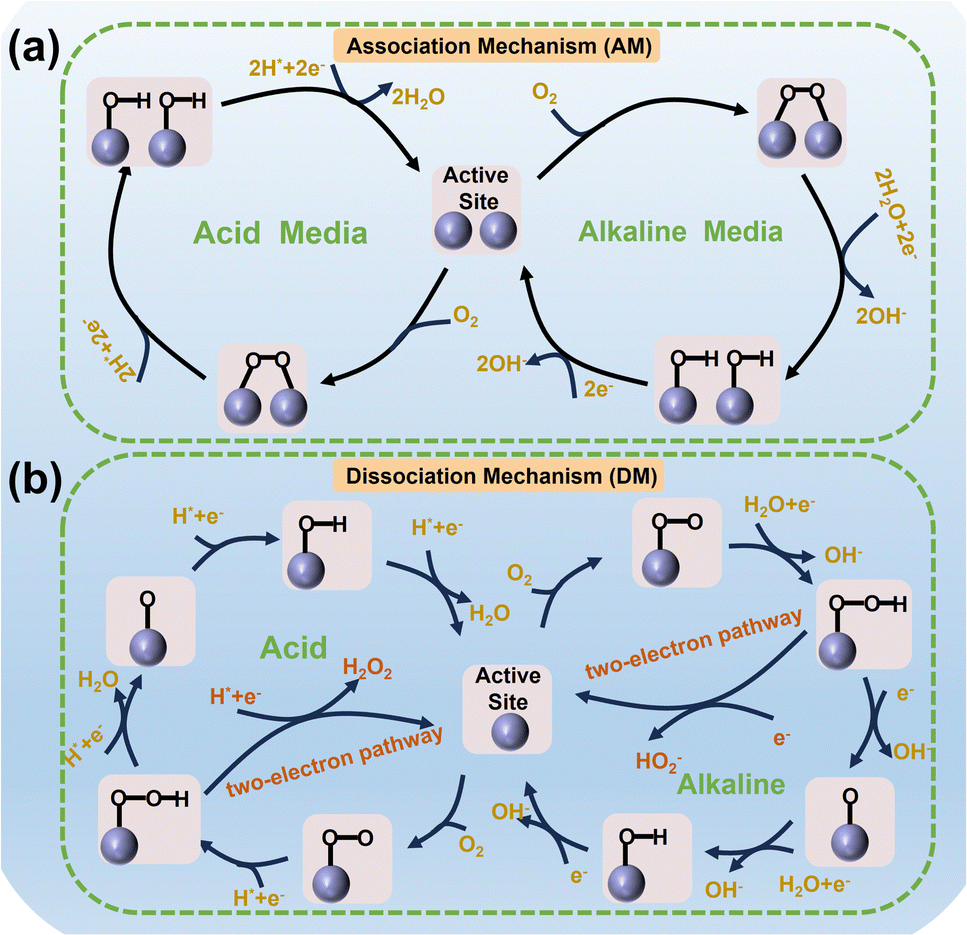 | ||
| Fig. 2 Multiple reaction pathways of the ORR with (a) AM and (b) DM in acid and alkaline media. | ||
2.2 Descriptors for the electronic configuration
Searching for descriptors that can accurately describe the electronic and geometric configuration of a catalyst is a primary prerequisite to defining “charge–spin–coordination” clearly.34–36 Advanced theoretical calculation and experimental characterization have induced a wealth of descriptors for electronic configuration (Fig. 3), providing sufficient convenience for describing the electronic structure of active components, such as electron delocalization degree, spin state and state density at the Fermi level.37 These appropriate descriptors can reasonably broadcast the source and change the trend of catalytic activity and provide inverted guidance for precisely regulating the fine structure at the atomic levels. Therefore, it is expected to establish more general, accurate and appropriate descriptors toward further revealing and exploring the catalytic mechanism of different Fe-FUs in Fe-based catalysts.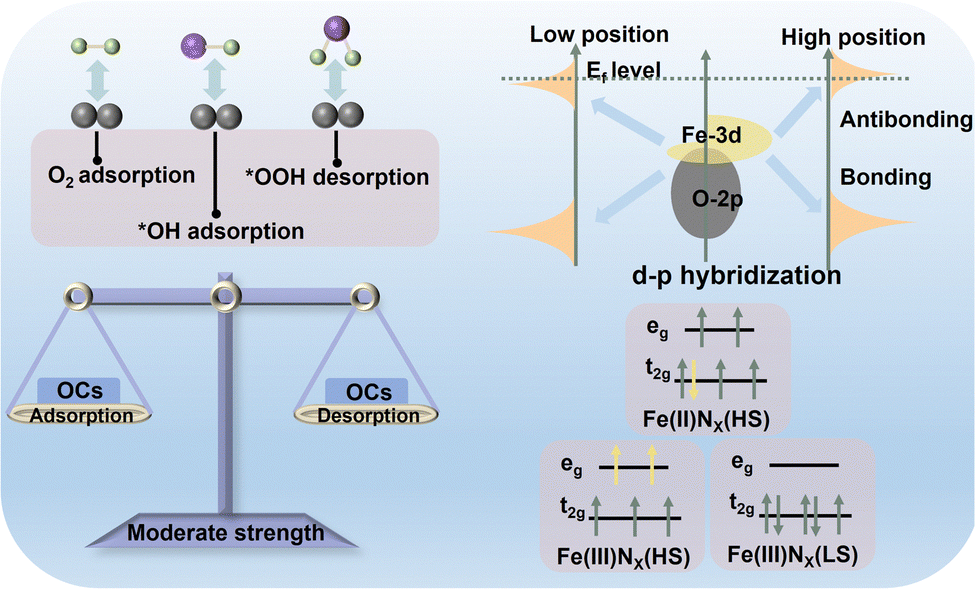 | ||
| Fig. 3 Three descriptors for the electronic configuration of Fe-based catalysts. | ||
3 Specific ORR mechanisms of Fe-FUs in multiple forms
3.1 Fe-FUs with crystalline structures
Nanomaterials exhibit many excellent physicochemical properties (specific electrical, magnetic, and optical properties) on account of their small size effect, surface and boundary effect, quantum size effect and tunnel effect, and once shone in electrocatalysis.60–62 Early studies on Fe-based catalysts were conducted with nanoscale Fe-FUs as the starting point.63–65 The unstable carbon reduction during high-temperature pyrolysis causes the Fe components in the precursor to be loaded onto the carbon supports in multiple forms of Fe-FUs with crystalline structures (Fe-NPs, FeOx and FeCx), which participates in the catalytic process by acting directly or indirectly as active sites.66 The differences in the type and presence of nano-scale Fe-FUs lead to some controversy in the source of catalytic activity. Therefore, it is imperative to reveal the complex mechanism of multiple Fe-FUs with crystal structures in catalysis.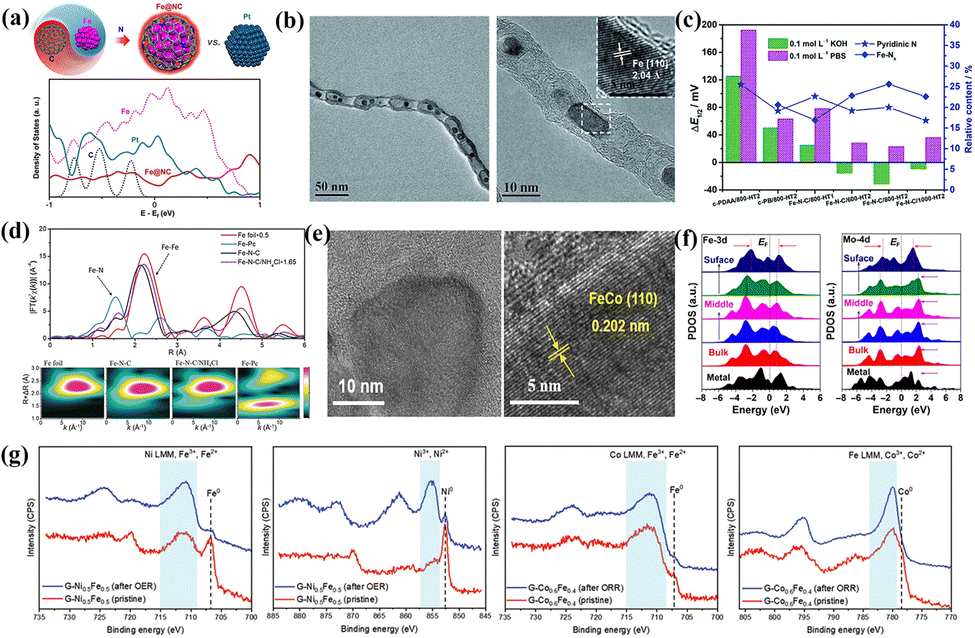 | ||
| Fig. 4 (a) Model structure and partial density of states (PDOS) of Fe@NC and Pt/C. Reproduced with permission.67 Copyright 2020, Wiley-VCH Verlag. (b) Low and high-resolution TEM images of Pod-Fe. Reproduced with permission.73 Copyright 2013, Wiley-VCH Verlag. (c) The relationship between the relative content of nitrogenous species and ORR activity. Reproduced with permission.74 Copyright 2020, The Royal Society of Chemistry. (d) Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra and corresponding wavelet transform of the target catalyst and reference sample. Reproduced with permission.75 Copyright 2020, Wiley-VCH Verlag. (e) TEM images of alloying FeCo-NPs species with different resolutions. Reproduced with permission.76 Copyright 2022, Wiley-VCH Verlag. (f) The PDOS of Fe and Mo with different forms in Fe2Mo. Reproduced with permission.77 Copyright 2022, Springer. (g) Fe/Ni/Co 2p XPS spectra of the initial condition and stability testing. Reproduced with permission.78 Copyright 2020, Wiley-VCH Verlag. | ||
Fe-NPs, the most common electron donors in chainmail catalysts, continuously transfer electrons to the external GLs to reduce the local work function, thus achieving enhanced C-2p and O-2p hybridization. Bao and coworkers prepared a peasecod-like chainmail structure of carbon nanotubes coated with Fe-NPs (Fig. 4b).73 It accelerated the adsorption/activation of O2 molecules and O–O bond breaking, effectively alleviating the poor stability caused by direct corrosion of acidic electrolytes. It is also a typical means for modulating the type and quality of GLs to optimize the catalytic activity. Introducing the more electronegative N (3.04) in the GLs forms abundant nitrogen-containing active species (pyridine nitrogen, pyrrole nitrogen and graphite nitrogen), in situ formation of g-C3N4 species also accelerates Fe-FUs anchoring on carbon carriers.79–81 Inspired by this scenario, Liu et al. prepared ORR catalysts with nitrogen-doped GLs wrapped with a TM-NP (Fe, Co, Ni) structure.80 The bidirectional regulation of internal TM-NPs and nitrogen on a carbon structure provides more active sites for the ORR. In addition, it is also an open question whether the nitrogen source is converted into high-activity FeNx species in the chainmail structures. Yang and co-workers prepared Fe–N–C/800-HT2 catalysts with Fe-NPs wrapped in graphene-like nanosheets.74Fig. 4c indicated that different nitrogen-containing species with appropriate proportions provided more active sites through synergistic effects. Unlike internal Fe-NPs, exposed Fe-NPs as non-catalytic species should avoid their formation. The common methods to remove such species are as follows: high-temperature Cl2 treatment, ball milling, high-concentration H2O2 oxidation, and chemical etching,82,83 but accompanied by irreversible destruction of active sites and carbon supports. Low-temperature NH4Cl treatment using NH3 and HCl gas generated by decomposition can produce rich pore structures for the carbon supports, and etching treatment on the exposed Fe-NPs.75 Thus, this approach achieves a directed transition of Fe-NPs from inactive to active states, corresponding to an increased ratio of Fe–N to Fe–Fe in the EXAFS spectrum (Fig. 4d).
High-temperature pyrolysis is a common strategy for preparing chainmail catalysts abundant in Fe-NPs, often producing byproducts with different structures, such as nitrogen-doped carbon (NC) and FeNx sites. In summary, the catalytic effect of internal Fe-NPs can be divided into the following three levels: (1) improving the local work function of GLs as electron donors; (2) synergistic catalysis of internal Fe-NPs and surface nitrogen; (3) FeNx configurations formed between nitrogen-containing species and Fe-NPs act as active sites. Thus, an in-depth analysis of the structural characteristics of chainmail catalysts is an essential source for balancing catalytic activity and stability.
In addition to alloying M-NPs composed of 3d transition metals, the PtFe alloy catalysts have an extensive research foundation for the ORR. The transition from disordered to ordered phases and the surface composition in PtFe alloys are essential factors determining the catalytic activity and stability. Guan's group used a two-step direct annealing method to load uniformly ordered PtFe alloys onto single-walled carbon nanohorns protected by thin nitrogen-doped GLs.90 The secondary annealing treatment under a hydrogen atmosphere perfectly transformed partially ordered fcc/fct-PtFe NPs to highly ordered fct-PtFe NPs (order degree: >80%, average size: 5.2 nm). The ordered fct-PtFe structure accelerated the electron transfer and provided strong resistance to the leaching and aggregation of alloying NPs. Besides, the enhanced ORR activity of PtFe nanowires (NMs) was mainly rooted in the exposed (111) crystal plane. Zhong and co-workers found that the PtFe-NMs with a Pt content of 24% exhibited mixed fcc/bcc structures and apparent strain lattice, thus showing the highest mass activity (3.4 A mgPt−1) and durability (activity decreased by 2% after 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 CV cycles).91 Compared with pure Pt-NMs, the (111)/(200) ratio after introducing Fe in PtnFe100−n increased significantly, which demonstrated that exposure to specific (111) crystal planes can greatly enhance ORR activity.
000 CV cycles).91 Compared with pure Pt-NMs, the (111)/(200) ratio after introducing Fe in PtnFe100−n increased significantly, which demonstrated that exposure to specific (111) crystal planes can greatly enhance ORR activity.
The promoting effect of multiple alloying is essential to enhance the ORR performance of catalysts. Although the introduction of complementary dual/multi-metal components makes full use of charge transfer and geometric effects to optimize the electronic configuration of the active site, the development of Fe-based alloy catalysts still confronted the following problems:92–94 (1) it is challenging to achieve controlled preparation of single alloying species; (2) the Fe component in the alloying system plays a “leading” or “auxiliary” role, depending on a variety of catalytic systems; (3) it is still complicated and confusing to clarify in situ reconstruction of alloying Fe-NPs in the electrochemical process. The popularization and sublimation of the alloying concept are boosters for the future development of Fe-based catalysts.
FeOx species serve more as functionalized units in the ORR. Su's group explored specific ORR behaviors of Fe3O4 and nitrogen-containing species under alkaline conditions (FeN4 > N > Fe3O4).101 The poor conductivity and spinel structure made Fe3O4 exhibit worse intrinsic activity than nitrogen-doped carbon black (NCB). In addition, the introduction of N facilitated the pathway transferred from two-electron to four-electron to elevate energy conversion efficiency. Liu et al. explored the ORR behavior of FeOx and hydroxides after loading graphite oxide (GO) (Fig. 5a).103 The presence of α-Fe2O3 and hydroxides resulted in an optimal rearrangement of the GO electronic structure, inducing an increased open potential (Eon) and limiting current density (Jk). Fe3O4 derived the ideal four-electron pathway to achieve perfect selectivity. The strong electron attraction effect of FeOx impacts the electronic structure of the adjacent carbon and Fe3N. Fe3N can overcome the low electron conductivity to improve the ORR activity.104 Fan's group verified this view by constructing a composite system of Fe3O4 and bamboo-like multi-walled carbon nanotubes (MW-CNTs),105 in which Fe components existed in Fe3O4 (002) and Fe3N (002) crystal planes (Fig. 5b). The enhanced surface wettability after Fe3O4 optimization significantly broadens the three-phase reaction interface. The heterogeneous interface composed of FeN3, Fe3O4 and carbon promoted the fast electron transfer from the GLs to Fe3O4 and the adsorption and activation of OCs. Niu et al. analyzed the specific catalytic activity of exposed crystal planes of Fe2O3 through morphology engineering (Fig. 5c).106 The high index crystal planes (206) and (119) in concave octahedral CO–Fe2O3 exhibited high intrinsic ORR activity, attributed to the high surface energy and low coordination structure at the surface atoms. The synergistic effect of strong coupling interfaces in heterostructures exceeds the prediction of mixing rules and often achieves unexpected catalytic results. Fan et al. successfully prepared ORR catalysts with controllable heterostructures (α-Fe2O3/Fe3O4/hNCNC).107 With the increased heating time and pyrolysis temperature, the FeOx presented the following trend in Fig. 5d: α-Fe2O3–α-Fe2O3/Fe3O4–Fe3O4. The interfacial coupling effect exists in sufficient reaction zones on the heterostructures (α-Fe2O3/Fe3O4), balancing the adsorption strength of the OCs at the active sites.
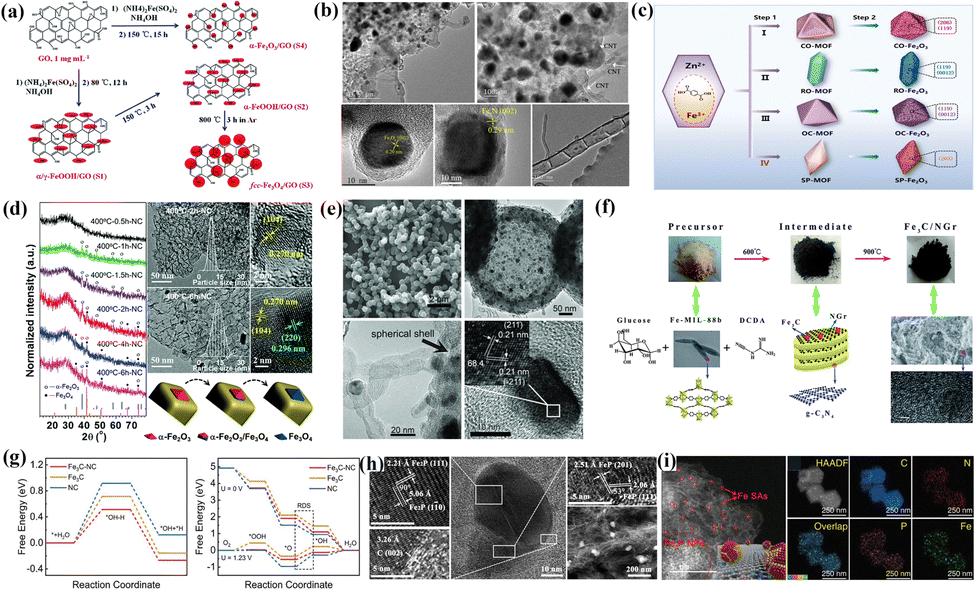 | ||
| Fig. 5 (a) Synthetic procedure and simple structure of four FeOx species. Reproduced with permission.103 Copyright 2016, Royal of Society Chemistry. (b) TEM images of Fe3O4/Fe–N–C-CNT catalysts at different scales. Reproduced with permission.105 Copyright 2020, Elsevier. (c) Fe2O3 with different micromorphologies and their corresponding exposed crystal planes. Reproduced with permission.106 Copyright 2021, Elsevier BV. (d) Characterization and analysis of the microstructure and phase structure of α-Fe2O3/Fe3O4/hNCNC catalysts. Reproduced with permission.107 Copyright 2018, Royal of Society Chemistry. (e) SEM, TEM and HRTEM images and corresponding lattice information of the Fe3C/C-700 catalysts. Reproduced with permission.108 Copyright 2014, Wiley-VCH Verlag. (f) Synthetic process and corresponding structural information for Fe3C/NGr. Reproduced with permission.109 Copyright 2015, Royal of Society Chemistry. (g) The variations of Gibbs free energy in Fe3C-NC and control samples for water dissociation and the ORR. Reproduced with permission.110 Copyright 2022,Wiley-VCH Verlag. (h) HRTEM images of different regions in Fe2P/FeP-PNC catalysts. Reproduced with permission.111 Copyright 2021,Wiley-VCH GmbH. (i) HAADF-STEM image and EDS mapping of the Fe SAs-Fe2P NPs/NPCFs catalyst. Reproduced with permission.112 Copyright 2022,Wiley-VCH GmbH. | ||
The ORR activity of FeOx is closely related to the crystallinity, crystal forms and exposed crystal planes, leading to different effects on the reactive activity and selectivity. The specific roles of Fe3O4 are reflected in the three points: (1) optimization of catalytic selectivity; (2) improved electrode wettability increases the exposed active sites; (3) excellent electronic acceptors optimize the electronic configuration of active sites. The α-crystal forms in Fe2O3 species can improve the Eon and JL for catalysts. However, FeOx falls short of the conductivity required by the ORR, which is not conducive to fast electron transfer. The low intrinsic ORR activity leaves room for further improvement. Effectively overcoming these difficulties is expected to realize the rapid development of FeOx in the ORR field.
Similar to the preparation of pure-phase Fe-NPs, FeCx species are usually present in the inner cores to form the chainmail catalysts. Li and co-workers demonstrated the positive role of FeCx species in the chainmail structure by ball milling and acid leaching.108 The internal encapsulation effect of FeCx (Fig. 5e) provided the catalyst with good electro-catalytic properties under acidic and alkaline conditions. The GLs-dominated chainmail layers can produce a more desirable synergistic effect with the Fe3C cores by introducing tiny metals and nitrogen. Huang et al. controllably embedded uniformly sized Fe3C-NPs (10 nm) into the Fe/N-rich GLs with large pore structures and high electrical conductivity (Fig. 5f).109 The enhanced surface hydrophilicity provided rich three-phase interfaces for catalytic reactions, facilitating exposure of active sites (Fe–N and C–N bonding) and catalytic mass transfer. The protons bound to the OCs under alkaline conditions mainly originate from the dissociation of water, which limits the overall kinetics in the ORR. Therefore, the Fe3C-loaded NC constructed by Cao et al. exhibited the lowest energy barrier of water dissociation and the free-energy change of protonation of *O intermediate (Fig. 5g) due to the electron delocalization effect.110 Thanks to the synergistic catalysis of the dual sites, the E1/2 of the Fe3C@NPW catalyst (0.87 V) was higher than the commercial Pt/C (0.85 V), confirming the reliability of the dual-site combination strategy. The synergistic catalysis between multi-components with heterostructures is expected to achieve unexpected catalytic effects. Zhong and co-workers established a simulated hydrothermal solid-phase system to explore the relationship between the existence form (hybridization states and graphitization degrees) of the multi-component hybrid Fe2O3/Fe5C2-NPs and the catalytic activity.116 Therefore, the synergistic catalysis and rich interfacial structure in the heterogeneous Fe2O3/Fe5C2/Fe–N–C promoted the efficient electron/charge transfer for a fast ORR process.
With the popularity of chainmail catalysts, early research on Fe3C, which is considered to contribute less to the ORR, has been pushed back due to a new understanding. The adjustive local work function of the GLs by Fe3C species and the protective effect of the GLs serve as the primary sources of the activity and stability for the chainmail catalysts,110,114,115,117–119 but there are the following problems in its development: (1) the unclear relationship between the quantity of GLs and the strength of the electron shuttle effect; (2) the non-quantitative relationship between the doping effect of the GLs and the activity; (3) whether there are active sites at the interface between the inner metal cores and carbon supports. Achieving the dynamic balance of catalytic activity and stability is expected to reach large-scale commercial applications in advanced energy storage devices.
Controlling the relative ratio of Fe and P atoms can regulate the d-band center and energy band structure to varying degrees in FePx species. Ni et al. reported that the chainmail structure composed of FeP-NPs encapsulated in P-doped GLs showed excellent ORR activity in both acidic and alkaline systems.123 The interfacial synergistic effect induced by the charge transfer between FeP-NPs and GLs reduces the surface work function of the catalyst, similar to the chainmail catalysts. In addition, the heteroatom-doped carbon support loaded with FeP/Fe2P heterostructures also exhibits excellent potential in the ORR. Li and co-workers elucidated the ORR activity origin of FeP/Fe2P heterostructures embedded with N, S, and P co-doped carbon structures:120 (1) the interfacial charge transfer and coupling effects induced by heterostructures leads to rearrangement of electronic structures at the reaction interface; (2) heteroatom doping further optimizes the charge distribution and spin states of heterostructures and carbon substrates. Xue et al. also found that the FeP/Fe2P heterostructures and P heteroatoms can effectively compensate for the loss of activity in N-doped materials under acidic conditions.111 The HRTEM image shown in Fig. 5h perfectly interpreted the existence of FeP and Fe2P heterostructures and corresponding crystal planes. The heterostructures formed between FeP and other transition metal phosphides (such as Cu3P, FeNiP) have also been reported to have certain application potential in the ORR.121,122 Precisely designing the atomic-level coupled catalysts and exploring the reaction mechanism can provide a strong basis for efficient ORR catalysts. Chen and co-workers co-loaded Fe-SACs and Fe2P-NPs onto N and P co-doped carbon support to form a Fe-SAS–Fe2PNPS/NPCFs-2.5 coupling catalyst.112 The local distribution of Fe SACs and Fe2P NPs on carbon substrate is shown in Fig. 5i. The synergistic effect of P atoms and heterostructures elevated the Fe-3d orbital, resulting in a lower oxidation state and less positive charge of the active Fe components. In situ spectra confirmed that P–Fe–O–H* intermediate species formed at the Fe–P site and N4–Fe–O–H* intermediate species formed at the FeN4 site jointly promoted ORRs in acidic media. Under alkaline conditions, only Fe–N4 sites are in service.
FePx exhibits high electrocatalytic activity due to rich active sites and electron transfer channels, making it a candidate for ORR catalysts. The strategies to improve its activity mainly focus on heteroatom doping, constructing the heterostructures, regulating the vacancy and defects, and exposing the optimal crystal planes.124 It should be clarified that the development of FePx species in the ORR still has the following problems: (1) the controllability of FePx's composition purity, particle size and micro-morphology needs to be further improved; (2) the stability and corrosion resistance still need to be further improved to adapt to complex reaction environments and long-term usage requirements; (3) there is still great room for boosting the intrinsic ORR activity. Doping, alloying, interface engineering and surface modification are expected to achieve enhanced ORR activity and stability.121,122,125 The effective combination of advanced experimental characterization and theoretical calculation is expected to promote a deep understanding of the catalytic mechanism and reaction kinetics of FeP and Fe2P in the ORR, to guide the experimental design and optimization of catalytic performance.
The application of Fe components in the ORR field is developed from traditional metal macrocyclic compounds. The excellent FeN4 coordination structure exhibits certain potential but still faces the dilemma of insufficient activity and stability.126–130 High-temperature pyrolysis can effectively convert Fe components into Fe NPs, FeOx, FeCx and FePx species and anchor them on carbon supports, effectively overcoming these challenges.15,88,131–134 Fe-FUs with crystalline structures in the catalytic system contribute to the ORR kinetics by acting as active sites directly or indirectly. The ORR activity of common Fe-FUs with multiple crystalline structures is shown in Table 1, where alloying Fe-NPs appear to exhibit a higher intrinsic catalytic activity. The efficient strategies of modulating the Fe-NPs mainly revolve around enhancing the conductivity, improving the intrinsic catalytic activity and increasing the exposed active sites. Exploring how crystal structure, chemical state and coordination structure promote different catalytic behaviors is significant for the rapid development of Fe-based nanomaterials.
| Fe-FUs with crystalline structures | Electrolyte | E on (V vs. RHE) | E 1/2 (V vs. RHE) | Ref. |
|---|---|---|---|---|
| Fe–N–C/800-HT2 | 0.1 M KOH | 1.01 | 0.881 | 74 |
| 0.1 M PBS | 0.862 | 0.743 | ||
| Fe–N–C/NH4Cl | 0.1 M HClO4 | 0.831 | 75 | |
| Fe0.25Co0.75/NC-800 | 0.1 M KOH | 0.99 | 0.86 | 76 |
| Fe2Mo/NC | 0.1 M KOH | 0.91 | 77 | |
| 0.5 M H2SO4 | 0.80 | |||
| PtFe@NC/SWCNHS | 0.1 M KOH | 1.01 | 0.938 | 90 |
| FeNC | 0.1 M KOH | 1.03 | 0.881 | 105 |
| CO-Fe2O3 | 0.1 M KOH | 0.80 | 0.70 | 106 |
| α-Fe2O3/Fe3O4/hNCNC | 0.1 M KOH | 1.03 | 0.838 | 107 |
| Fe3C/C-700 | 0.1 M HClO4 | 0.90 | 0.73 | 108 |
| Fe3C/NGr-1 | 0.1 M KOH | 1.07 | 0.90 | 109 |
| Fe3C/NPW | 0.1 M KOH | 0.87 | 110 | |
| Fe2O3/Fe5O2/Fe–N–C-3 | 0.1 M KOH | 0.87 | 116 | |
| FeP@PGL | 0.1 M KOH | 1.01 | 0.90 | 123 |
| 0.5 M H2SO4 | 0.95 | 0.81 | ||
| FeNSPC1000 | 0.1 M KOH | 0.833 | 120 | |
| Fe2P/FeP-PNC | 0.1 M HClO4 | 0.70 | 111 | |
| Fe-SAs–Fe2P NPs/NPCFs-2.5 | 0.1 M KOH | 1.03 | 0.91 | 112 |
| 0.5 M H2SO4 | 0.93 | 0.78 |
3.2 Non-crystalline iron species
When Fe components change from crystalline to non-crystalline states, the electrons near the Fermi energy level transform from quasi-continuous to highly discrete conditions, significantly changing their physicochemical properties. Unlike the surface catalysis carried out by crystalline species, Fe-based single-atom catalysts (SACs) with non-crystalline states achieve efficient electron transfer in both surface and bulk-limited domains, significantly improving the utilization efficiency of Fe components.135–138 In addition, the well-defined coordination structure of Fe-based SACs provides sufficient opportunities for achieving high catalytic activity and selectivity and serves as a suitable research model to reveal the catalytic mechanism in depth.139–142 Therefore, clarifying the key factors such as the chemical state, coordination environment and geometric configuration of Fe centers can effectively reveal the triple effect theory among “charge–coordination–spin”.In addition, the type of nitrogen-containing species is also a vital factor affecting the intrinsic activity of FeNx units.159–161 Wu's group confirmed that NH3 high-temperature treatment can successfully achieve the perfect conversion of pyrrole-type to pyridine-type FeNx (Fig. 6a).159 In pyrrole type FeNx, the Fe center in a high electron deficient state not only facilitates the adsorption and activation of O2 molecules and the desorption of *OH, but also enhances the four-electron selectivity (Fig. 6b). Han et al. believed that the pyridine nitrogen easily interacted with Fe atoms to form FeN4 active sites in Fig. 6c. Graphite nitrogen rearranged the electronic configuration of the adjacent FeN4 sites and improved its intrinsic ORR activity.162 In summary, pyridine and pyrrole nitrogen with planar sp2 hybrid structures are usually present at the edges and defects of the graphene skeleton, where high electron delocalization ensures rapid electron transfer during catalysis. Implementing Fe-based SACs with high-density FeNx units is also a hot research direction, with ZIFs shining brightly here.13,89,163,164 Li et al. used the cavity structure of ZIF-8 (dc = 11.6 Å, dp = 3.4 Å) to prepare a Fe-ISAS/CN-SACs catalyst with a single-atom Fe content of 2.16 wt%.13 The rapid electron transfer between *OH and Fe active sites stimulates excellent ORR activity (E1/2 = 0.90 V, Jk = 37.83 mV cm−2 at 0.85 V) in Fig. 6d. In addition, Wu's group used the spatial fence effect of sacrificial metals to accurately regulate the Fe doping amount in ZIFs, investigating the effects on particle size, nitrogen-containing species, and graphitization degree.165 The efficient binding between the active Fe sites (mainly D1 sites) and *OOH species accelerated the breaking of O–O bonding (Fig. 6e). Most preparations are accompanied by uncontrollable factors such as non-equilibrium high-temperature carbonization and nitrogen doping, making it difficult to elucidate the formation mechanism of Fe–N bonding. Li et al. were surprised to find that ultrafine FeOx particles were thermally activated, transformed into active FeN4 sites at 400 °C, and transformed into shorter bond lengths and symmetric coordination structures at 700 °C (Fig. 6f).166 Although excessive temperature increases the graphitization of catalysts, it is also accompanied by losing the nitrogen-containing species and destroying the symmetric coordination structures.
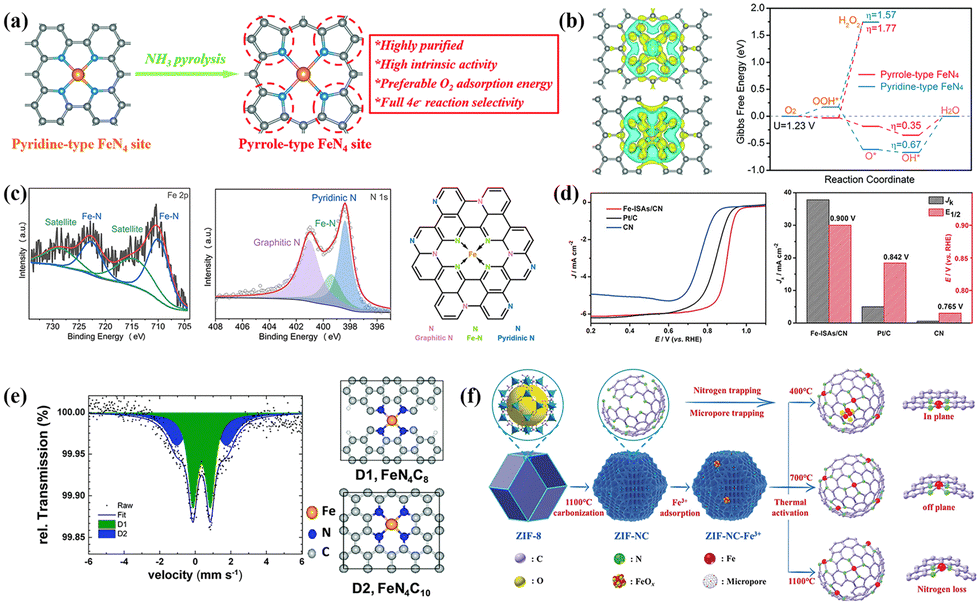 | ||
| Fig. 6 (a) Transformation process and advantages of pyridine-type to pyrrole-type FeN4 sites. Reproduced with permission.159 Copyright 2020, Royal of Society Chemistry. (b) Charge density difference and variations of Gibbs free energy for different FeN4 types. Reproduced with permission.159 Copyright 2020, Royal of Society Chemistry. (c) Nitrogen-containing species and structure model of the Fe–N–C/N-OMC catalyst. Reproduced with permission.162 Copyright 2021, Elsevier. (d) Comparison of ORR activity between Fe-ISAs/CN and control catalysts. Reproduced with permission.13 Copyright 2017,Wiley-VCH Verlag. (e) Mössbauer spectrum and structure of D1 and D2 sites of the 1.5 Fe-ZIF catalyst with optimal performance. Reproduced with permission.165 Copyright 2019, Royal of Society Chemistry. (f) Effect of thermal activation temperature on Fe–N bonding mechanism. Reproduced with permission.166 Copyright 2019, Wiley-VCH Verlag. | ||
For SACs, high specific surface energy can induce the formation of a symbiotic system containing single atoms and M-NPs. Investigating the synergistic effect between “inactive” species and nearby atomic sites has positive implications. Zhang and co-workers believed that introducing Fe3O4-NPs led to a certain degree of rearrangement of the electronic configuration at the surrounding FeN4 sites, enhancing the interaction with the O2 molecules in an alkaline environment.167 The construction of the hierarchical porous structure realized the fast mass transfer and electron transport, and also affected the nucleation of Fe3O4-NPs, nitrogen-containing species and graphitization (Fig. 7a). Chen et al. also verified that the electron transfer effect between the FeN4 and FexC species increased the charge density of the Fe active sites, thereby optimizing the adsorption strength of *OH intermediates.168 DFT calculation in Fig. 7b confirmed that the rate-determining step (RDS) of Fe–N4–Fe10 was *OH protonation (OH* + e− + H+ → H2O(l) + H*) with a low over-potential (η = 0.27 V). Yin et al. first revealed that the M-NPs and FeN4 composite structure facilitated the direct four-electron pathway (dissociation mechanism) under acid conditions, where O2 molecules broke the O–O bonding at the active sites and then protonated.169 The dissociation pathway inhibited the formation of strong oxidizing species such as hydrogen peroxide, which contributed to improving activity and stability for the ORR. In contrast, the single-atom sites tended to undergo an indirect four-electron pathway according to the association mechanism, that is, the adsorbed O2 molecules were protonated and then dissociated from *O–OH. Although the formation of Fe NPs reduces the density of single-atom sites, it will significantly improve the utilization rate of metal components.
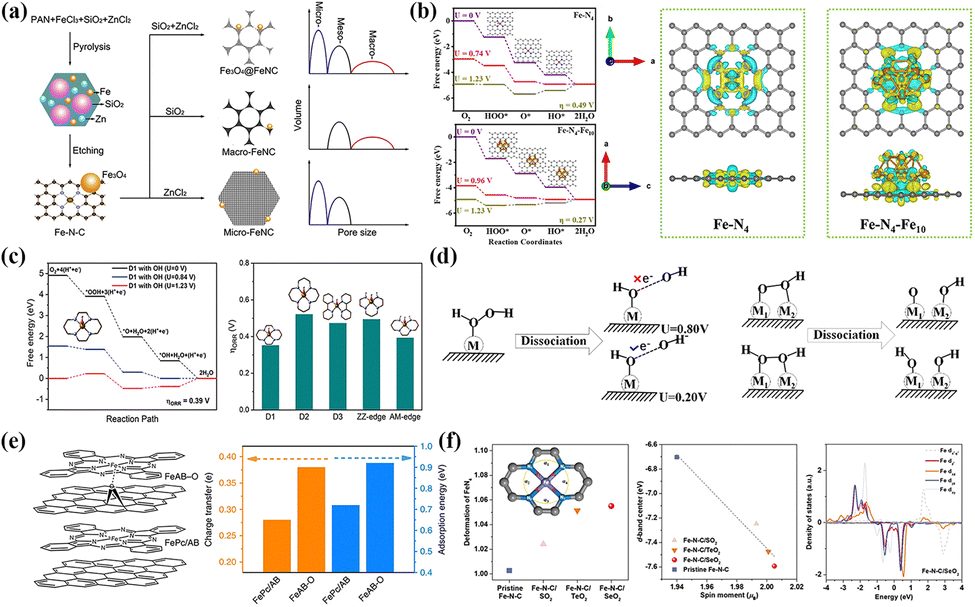 | ||
| Fig. 7 (a) Synthesis progress of Fe3O4@FeNC catalyst and influence mechanism of pore size distribution. Reproduced with permission.167 Copyright 2020, Elsevier Ltd. (b) The charge density difference and variations of Gibbs free energy of Fe–N4 and Fe–N4–Fe10 systems. Reproduced with permission.168 Copyright 2023, Elsevier. (c) The variations of Gibbs free energy at Fe–N4 sites modified by an axial OH ligand and comparison of overpotential at different sites. Reproduced with permission.175 Copyright 2020, Wiley-VCH Verlag. (d) The dissociation mechanisms of *OOH and *H2O2 species at single and dual atom sites. Reproduced with permission.176 Copyright 2020, Elsevier BV. (e) Structure diagram of FeAB-O modified by an axial O ligand and adsorption capacity for O2 molecules. Reproduced with permission.174 Copyright 2020, Springer Nature. (f) The effects of XO2 modification on geometry, d-band center and spin structure for the FeN4 site. Reproduced with permission.173 Copyright 2022, Wiley-VCH Verlag. | ||
Fe-based SACs rich in FeNx sites have an extensive research foundation in the ORR field, mainly to improve the density and intrinsic activity of FeNx sites. The current research difficulties mainly focus on exploring the optimal coordination environment and the in situ reconstruction during the reaction process. The differences in coordination environments may trigger complex chain reactions such as nitrogen-containing species, spin states and d-band electronic configurations. Before oxygen protonation and other processes, the dynamic reconstruction of FeN4 sites is also controversial. Introducing advanced in situ technology and theoretical calculations into near-real model catalysts provides a sharp edge in solving the above difficulties.
Axial OH ligands formed spontaneously on Fe-NC catalysts under electrochemical conditions serve as electronic structure regulators to optimize the electronic configuration of Fe centers. Some people believe that they exist in Fe centers dominated by D3 sites. In contrast, they spontaneously form on one side of the D1 or ZZ edge sites, enhancing the ORR activity on the opposite side.177,178 Yang et al. suggested that Fe–N4(OH)–C structures formed after modification of FeN4 sites (D1, D3 and edge sites) by axial OH ligands at specific electrode potentials may be true catalytic active sites.175 The formation of axial OH species alleviated the excessive adsorption strength with Fe centers leading to problematic desorption under alkaline conditions (Fig. 7c). Xie's group used DFT calculation to reveal that the OH ligand enhanced the Eon of the catalysts, but also greatly affected the selectivity of H2O2 generation.176 For single-atom sites, the formation of ⋅OH radical via a 2+2 electron pathway at high potential was not conducive to improved stability (Fig. 7d). Furthermore, the strong interaction between Fe sites and *OOH species made *OH desorption challenging. However, the dual-metal sites could alleviate this strong interaction well to improve the catalytic efficiency. Specifically, based on the experimental verification of H2O2 yield versus catalyst loading, the ORR process of TM-SACs catalysts proceeded mainly in the 2+2 electron pathway. It suggested an intrinsic correlation between the modification of axial ligands and catalytic selectivity.
In addition to the case described above, introducing oxygen-containing ligands/functional groups also becomes an effective means of fine-tuning the electronic configuration. Chen et al. proposed an axial ligand-induced electron redistribution strategy to form Fe–O ligands between FePc and AB–O groups (Fig. 7e).174 The strong electron localization generated on the Fe–O ligand disrupted the original symmetric distribution state, achieving higher adsorption energy (0.92 eV) and charge transfer (0.38 eV) for O2 molecules. Modulating the spin polarization and optimizing the magnetic moment can reduce the energy barrier or overcome the spin inhibition to enhance ORR activity as an effective means to modify the electronic degree of freedom. Constructing Fe centers in Fe-based SAC with high-spin states can be divided into two methods: (1) grafting of axial ligands to the active Fe sites; (2) regulation of the coordination environment in the first/second shells. Wang et al. demonstrated that the introduction of XO2 functional groups with different polar (SeO2 > TeO2 > SO2) into the FeN4 site resulted in a deviation from the plane centers and changed length of the Fe–N bond on the diagonal, achieving a perfect transition from the planar symmetric structure (D4h) to the distorted structure.173 As depicted in Fig. 7f, the geometric distortion broke the simplicity of the electronic states and caused some electrons to shift from spin-down to spin-up orbitals, thus inducing the electronic rearrangement and the successful modulation of the spin moments in the Fe centers.
The local coordination environment of the Fe centers can be micro-modified by the axial ligand modification, thus optimizing the charge distribution and spin state. Unlike the doping mechanism, the ligand structure acted on the central atoms through weak bonding to achieve precise regulation without destroying the original FeN4 coordination configuration. Plentiful axial ligand modification strategies intend to optimize the Fe d-band center, thus weakening the adsorption strength with OCs.174,179–181 Although meaningful progress has been made in optimizing atomic configuration through the axial ligands, the selective diversity of axial ligands and unclear mechanism of action still significantly limit the development of this strategy. Therefore, finding more effective general ligand-modification strategies is the key to studying the structure–activity relationship of atomic sites.
3.2.3.1 Fe nanoclusters (NCs). Lacking strong constraining forces to prevent metal atoms from aggregating leads to forming symbiotic systems of single-atoms (SAs) and nanoclusters (NCs) in Fe-SACs. With traditional conditions it was believed that the formation of NCs was not conducive to high site density (SD) and increased turnover frequency (TOF), so it was not easy to achieve high catalytic activity. Recent studies have shown that the high utilization of atomically dispersed active sites is a utopian scenario, and not all active sites serve in catalysis.182–184 Although NCs formed by multiple atoms formally reduce the atomic utilization rate, multiple atomic sites exposed on micro-crystal surfaces in catalytic reactions perform their respective duties. In addition, strong electron transfer effects are induced between NCs and SAs due to unblocked electron transfer paths and short-range interactions. Therefore, studying the controllable formation and mode action of NCs in catalysis is still essential to achieve high catalytic efficiency of Fe-based SACs.
NCs shine brightly in heterogeneous catalysis, so exploring their specific action mechanisms in various reactions is urgent. Ao et al.155 confirmed that Fe-NCs as electronic structure modifiers caused the Fe d-band center to shift downward, effectively alleviating the excessive adsorption strength with *OH species in alkaline ORRs (Fig. 8a). Furthermore, the Fe component catalyzed pyrrole and oxide nitrogen into graphitic and pyridine nitrogen pyrolytically, raising the relative content of active species. As a result, FeAC@FeSA-NC catalysts with nanocomposite structures exhibited higher E1/2 (0.912 V) and cycling stability than commercial Pt/C. The weaker metal–support interaction leads to the dissolution of the active components (NPs, NCs) in the acidic electrolyte to a certain extent. The limited regulatory effect on the SA site leads to poor electrochemical performance in acidic ORR. Shui and co-workers successfully synthesized a catalytic symbiosis system of Fe-NCs and FeN4 co-existing using the moderate coordination strength between protonated N-doped carbon supports with metals.185 Unhindered electron transfer pathways and short-range interactions between Fe-NCs and SAs promote the spontaneous formation of *OH ligands at adjacent FeN4 sites (Fig. 8b). It increased the binding strength of the opposite side of the FeN4 sites with *OH species and reduced the energy barrier to 0.35 eV, significantly improving the intrinsic catalytic activity.
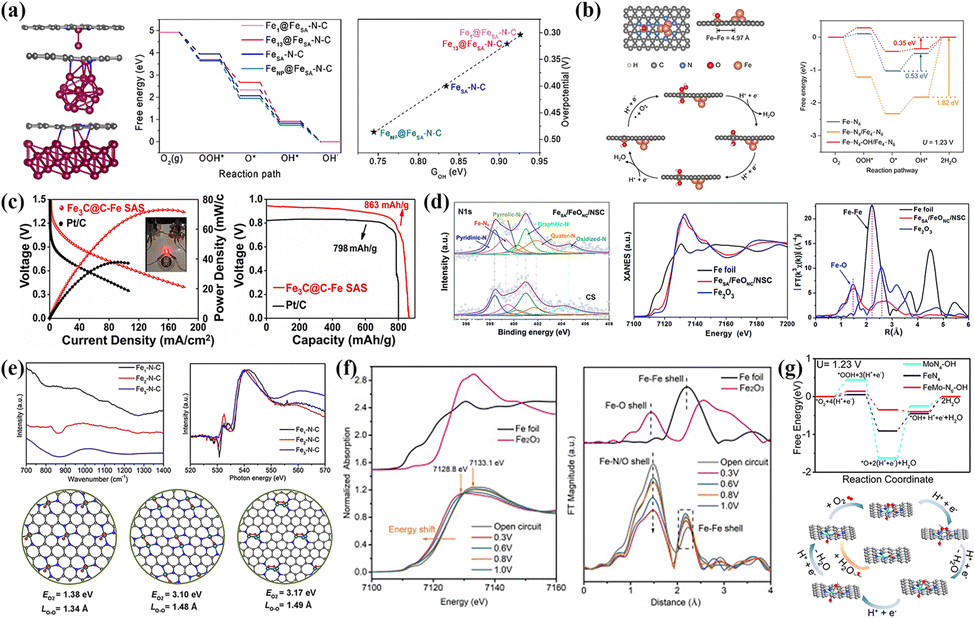 | ||
| Fig. 8 (a) Model structures, variations of Gibbs free energy and overpotential of FeSA, Fe13 and FeNP. Reproduced with permission.155 Copyright 2019, American Chemical Society. (b) Model structures, ORR mechanism and variations of Gibbs free energy of the Fe–N4/Fe4–N6 system. Reproduced with permission.185 Copyright 2022, Springer Nature. (c) The discharge polarization and voltage-capacity curves of Fe3C@C–Fe SAS and Pt/C-based zinc–air batteries. Reproduced with permission.118 Copyright 2021, Elsevier BV. (d) XANES and EXAFS spectra of FeSA/FeONC/NSC and control samples. Reproduced with permission.186 Copyright 2020, The Royal Society of Chemistry. (e) The interaction strength of O2 molecules in different adsorption modes at active sites. Reproduced with permission.187 Copyright 2019, Elsevier Inc. (f) Operando X-ray absorption near edge structure (XANES) and EXAFS spectra of planar-like Fe2N6 sites at different potentials in acidic systems. Reproduced with permission.188 Copyright 2020, Elsevier Inc. (g) The variations of Gibbs Free energy at single-atom and dual-atom sites and the ORR mechanism in acidic systems. Reproduced with permission.85 Copyright 2022, American Chemical Society. | ||
Zinc–air batteries have attracted much attention due to their low cost, high theoretical energy density and high safety, but they are also plagued by the intolerance of electrode materials under acidic and alkaline conditions. Fe-based NCs seem to have potential as high-efficiency cathode catalysts in neutral environments. Wei et al. believed that bringing in Fe3C nanocrystals reduced the adsorption energy between FeN4 sites and O2 molecules in 0.1 M PBS, facilitating the subsequent adsorption and activation progress.118 The electron transfer effect also reduced the energy barrier of *OH desorption to alleviate the over-strong adsorption strength. Based on the excellent ORR activity (E1/2 = 0.90 V) under neutral conditions, the zinc–air battery constructed with this catalyst displayed a high maximum power density (74.8 mW cm−2) and specific capacity (863 mA h g−1) in Fig. 8c, as well as excellent stability. The N/S-mediated porous carbon co-loaded with Fe2O3-NCs and Fe-SAs prepared by Lei et al. also exhibited better ORR activity and discharge performance in zinc–air batteries compared with commercial Pt/C.186 In the XPS spectrum (Fig. 8d), the higher Fe–Nx content came from the g-C3N4 mediation. The strong coupling between the two components induced an optimized electronic configuration of the Fe-SAS sites from the XANES spectrum. In addition, the N and S-mediated porous carbon provides space for active sites and a favorable three-phase interface for the catalytic process.
In the catalytic process, the Fe-NCs act as electronic structure regulators and auxiliary units to optimize the electronic configuration of Fe-SAs. To put it bluntly, the high utilization efficiency of active Fe components is realized in an absolute sense.51,189–191 However, the following controversies still exist in the study of NCs: (1) as a metastable state, it is difficult to accurately regulate the atomic numbers and particle sizes of NCs in the carbonization process; (2) different types of NCs exhibit different synergistic catalytic mechanisms with FeN4 sites due to structural differences; (3) inhibiting the dissolution of Fe-NCs under acidic conditions is the key to achieving the balance between enhanced activity and stability for the ORR. Therefore, elucidating the specific mechanism of NCs is particularly meaningful for realizing the efficient utilization of Fe-based components in catalysis.
3.2.3.2 Dual-atom catalysts (DACs). At single atomic sites, the ORR tends to proceed in a two-electron or an indirect four-electron pathway (association mechanism). The OCs generated during the reaction are prone to attack the active sites, reducing the energy conversion efficiency. In addition, the OH ligands formed spontaneously at the single-atom sites mask the high atomic rate after occupying the surface.192–196 In recent years, the emergence of dual-atom catalysts (DACs) has broken the linear connection between the d-band electronic configuration and the adsorption strength of OCs, hopeful of achieving the catalytic effect of 1 + 1 > 2.6,85,197,198 Firstly, the adsorption of O2 molecules on dual-atom sites is more inclined to the Pauling mode, which promotes the breakage and activation of O–O bonding and the direct four-electron step smoothly. Secondly, heteronuclear atoms with different electronegativities constitute a micro-internal electric field, achieving precise modulation of the electronic configuration (charge distribution, energy band and spin state) of the active sites at the atomic scale. Reasonably elucidating the enhanced ORR mechanism of dual-metal sites is significant for efficiently utilizing Fe active components.
Unlike the single-atom sites, the simultaneous adsorption of two oxygen atoms in the O2 molecule on the adjacent dual-atom sites allows more electrons to be injected into the empty O-2p orbitals in peroxo-like O2 mode. It usually greatly increases the activation efficiency of O2 molecules.13,199,200 Ye et al. explored the adsorption mode of O2 molecules on atomic sites and clusters by low-temperature Fourier-transform infrared spectroscopy (LT-FTIR), as shown in Fig. 8e.187 The 1200–1350 cm−1 and 800–1000 cm−1 peaks correspond to supero-like (Fe1–N–C) and peroxo-like (Fe2–N–C and Fe3–N–C) vibration modes, respectively. Compared to Fe1–N–C, Fe2–N–C and Fe3–N–C showed higher adsorption energy (EO2) and longer O–O bonding stretch. The peak at 532 eV in the O K-edge NEXAFS spectra corresponded to the hybridization of Fe-3d and O-2p, in which Fe2-NC exhibited the highest peak intensity. Fe2 dual-atom sites enhanced the interaction strength of O2 molecules in the peroxo-like O2 mode, resulting in the optimal ORR activity (E1/2 = 0.78 V) and stability of Fe2-NC catalysts in acidic systems. The in situ technique realizes the dynamic change of electronic configuration in Fe2 sites during operating conditions. Xie and co-workers utilized in situ synchrotron radiation to prove that the initial state Ox–Fe3+–Fe2+ changed to the final state Fe2+–Fe2+ in Fe2N6 sites.188 With the decrease of application potential, the near-edge absorption edge moved in the low-energy direction and the white line peak widened in the XANES spectra (Fig. 8f). The relative change of Fe–N/O and Fe–Fe shells in the FT-EXAFS spectra demonstrated the stretching or compression of Fe–Fe bonding caused by OCs adsorbing on active sites. The low interaction of planar Fe2N6 sites with *O2 and *OH intermediates leads to rapid electron transfer, showing faster intrinsic activity and selectivity than single sites.
The difference in atomic radius and electronegativity gives heteronuclear atom pairs more desirable catalytic power. The Fe/Co sites loaded hollow carbon prepared by Li's group exhibited higher Eon (1.06 V) and E1/2 (0.863 V) under alkaline conditions than commercial Pt/C.195 The electron transfer effect in Fe/Co sites enhanced the activation capacity of O2 molecules and the four-electron selectivity. The Fe component catalyzed the high-graphitization carbon support, and also realizes the hollow structure by promoting the decomposition of metal–imidazole–metal coordination bonding. Zhang et al. suggested introducing Co atoms in FeCo–C/N catalysts optimized the Fe d-band configuration and reduced the energy barrier for generating *OOH species.201 Li and co-workers verified that FeMoN6 sites made O2 molecules effectively activate in the bridge-cis adsorption mode, triggering the critical process: *OOH + H+ + e− → *O + H2O.85 Due to the downward movement of the Fe-3d orbital caused by introducing Mo, FeMo–N6–(OH) exhibited the lowest energy barrier in the *OH protonation in Fig. 8g, which proved excellent ORR potential. In the heteronuclear cases, the Fe atoms act as the “main force”, while the other atoms act as an “auxiliary” to rearrange the electronic configuration of the Fe atoms to some extent. The optimized adsorption method of O2 gives high catalytic activity and ideal reaction selectivity. The multiple metal atoms in the catalytic system constitute a miniature “dynamo”, providing sufficient power for rapid catalytic kinetics.
Compared to SACs, DACs usually exhibit higher intrinsic activity, which stems from three effects: (1) the modulation effect. One atom in the atomic pairs acts as the active site, and the other remotely modulates its electronic structure; (2) the collaboration effect. Adsorption and activation of O2 molecules occur by dual-atom pairs in Yeager mode simultaneously; (3) the multi-function effect. Disparate atoms are responsible for multiple catalytic processes, realizing multifunctional catalysis in the reaction process. The introduction of the heteroatoms will lead to the change of net charge distribution in DACs and the overlapping energy band structure between the metal sites and the OCs, thus affecting their interaction. However, there are still shortcomings in the precisely controlled formation of dual-atom sites and the in-depth understanding of kinetic catalytic mechanisms. The effective combination of machine learning and theoretical simulation is committed to exploring dual-site models with ORR potential, which is expected to guide the rational design of DACs with satisfied performance and stability.
3.2.3.3 Hetero-atom modification. Introducing some non-metallic atoms (B, P, S, Se) with relatively weak electronegativity into the coordination environment of FeNx sites can effectively break the symmetric charge distribution and spin state of the Fe centers. Specifically, the differences in the atomic radius, electronegativity and binding state of the doping atoms will have different degrees of influence on the hydrophilicity/hydrophobicity, the electron conductivity of the carbon support, and the electronic configuration of the metal centers, thus affecting the interaction strength with the OCs.202–204 For the N/F/O heteroatoms with strong electronegativity, the strong electron affinity leads to the neighboring C atoms exhibiting high positive charge density. The active sites' charge density or spin state is optimally regulated through the electron transfer effect with Fe atoms. The B with smaller electronegativity makes the surrounding C atoms slightly polarized by valence-forming electron pairs, increasing the local positive charge density of B.205–207 In addition, the electron-deficient B can receive electrons from the C atoms with sp2 hybridized states, transferring the Fermi energy level to the conduction band. P is the group V element along with N, and its introduction can induce an asymmetric charge distribution in the active sites and increase the degree of charge delocalization of C atoms.112,208 The introduction of S/Se, equivalent to the electronegativity of C atoms, can redistribute the spin density of the active sites to achieve a moderate state suitable for the adsorption of OCs.209–211
Doping atoms introduce different coordination environments in FeNx sites due to differences in atomic radius and electronegativity. Yuan et al. partially replaced the nitrogen-rich coordination environment with a foreign P atom, breaking the symmetric charge distribution and geometric configuration of the Fe centers.212 The rearrangement of the charge density and the relative change of Fe–N bonding length allowed for a suitable adsorption/desorption strength between the OCs and the active sites, significantly improving the reaction kinetics. The increased intrinsic activity and the large aperture structure conferred high activity (E1/2 = 0.941 V, Eon = 0.867 V) and stability for the ORR under alkaline conditions (Fig. 9a). Zhu et al. synthesized Fe-NPC catalysts with Fe–N/P double coordination using axially introduced P atoms (Fig. 9b),213 in which the quasi-octahedral O2–FeNxPy molecule was considered the active site. Adding P leads to the reconstruction of the electronic configuration of the Fe center, effectively promoting the adsorption process of *OH intermediates. Therefore, the P doping mode plays a specific role in promoting modification of the electronic configuration.
 | ||
| Fig. 9 (a) The influence mechanism of high-pyrolysis temperature on ORR activity and specific surface area for Fe–N/P–C catalysts. Reproduced with permission from ref. 212. Copyright 2020, American Chemical Society. (b) Structure optimization before and after adsorbing OCs (*O, *OH, *OOH) on the FeNPC catalyst and exploring the ORR activity source. Reproduced with permission from ref. 213. Copyright 2019, The Royal Society of Chemistry. (c) The effects of P and S modifications on charge density difference and Gibbs free energy at FeN4 sites. Reproduced with permission from ref. 18. Copyright 2018, Springer Nature. (d) Analysis of the S chemical state in Fe-ISA/SNC and molecular structure of FeN4S2. Reproduced with permission from ref. 214. Copyright 2018, Wiley-Blackwell. (e) Spin polarization state of Fe centers and relative contents of D1, D2 and D3 sites in Fe1Se1-NC and Fe1-NC. Reproduced with permission from ref. 215. Copyright 2022, Elsevier. | ||
Compared with N, S is a p-block element with a larger radius and less electronegativity. Its introduction is beneficial to producing numerous defect structures on carbon supports and adjusting the electronic configuration of Fe centers.216,217 Li's group cleverly used the Kirkendall effect to construct Fe-SAs/NPS-HC catalysts with functionalized hollow structures and S/P co-doping.18 The impact of PZS monomer can be divided into two structural and electronic modulation levels: (1) induced ZIF-8 to decompose at a lower temperature (∼400 °C); (2) the S and P elements modulated the second shell of the Fe centers. The electron transfer effect induced by S/P atoms weakens the positive charge density (Feδ+) of Fe centers, thus achieving the efficient binding to *OH species (Fig. 9c). They also accurately explored the linear relationship between S doping amount and ORR activity by controlling the ratios of polymeric monomers to achieve precise doping of S/N elements (1:1).214 The connection between the oxidation state of S and the K-edge position is shown in Fig. 9d. In the constructed FeN4S2 unit, the less electronegative S atom was positively charged by losing an electron to hold all three bonds, while its neighbor N atom was saturated and negatively charged by accepting the electron. Se element has excellent properties such as a large atomic radius, abundant d-band electrons, fast electron transfer and high polarity, exhibiting rich potential in electrocatalysis. Therefore, Chen et al. added SeO2 as a selenium source in the balling to prepare dual-metal Fe/Se and N co-doped catalysts.215 The introduction of the Se source provided abundant atomic Se sites and modulated the spin state and charge distribution of Fe centers. The introduction of Se significantly increased the relative content of D3 sites (Fig. 9e). The low filling rate of eg at D3 sites with a low spin state leads to an appropriate adsorption strength with OCs, showing excellent ORR activity under alkaline and acidic conditions.
Unlike metal atoms, non-metal atoms prefer to dope in the first or second coordination shells of the FeNx molecules. The short/long-range interactions modulate the metal centers' electronic configuration and spin state in different degrees, thus achieving excellent ORR activity. However, the study of the doping effect is still limited by the following problems: (1) different preparation strategies can hardly lead to significant differences in the number and position of doping atoms; (2) the unclear relationship between the physicochemical properties of doping atoms and catalytic activity is still being investigated; (3) the types of doping atoms are limited, leading to a lack of a broader understanding of the doping mechanism. As an effective strategy for catalyst modification, it is imperative to investigate how specific doping mechanisms act on catalytic activity and selectivity, which is also an important direction for future development. In addition, compared to crystalline Fe-FUs, the ORR activity of non-crystalline Fe species appears to be superior, as can be seen from the comparison between Tables 1 and 2. The gradual nanometerization of Fe active components have gradually become the main research direction of Fe-based catalysts, and it is possible to achieve both ORR activity and stability in acidic media. The above research ideas provide enormous opportunities for the application of Fe-based catalysts in fuel cells and metal–air batteries.
| Non-crystalline iron species | Electrolyte | E on (V vs. RHE) | E 1/2 (V vs. RHE) | Ref. |
|---|---|---|---|---|
| HP-FeN4 | 0.5 M H2SO4 | 0.95 | 0.80 | 159 |
| Fe–N–C/N-OMC | 0.5 M H2SO4 | 1.08 | 0.93 | 162 |
| Fe-ISAs/CN | 0.1 M KOH | 0.986 | 0.90 | 13 |
| Fe3O4@FeNC | 0.1 M KOH | 1.007 | 0.89 | 167 |
| Fe/FexC@Fe–N–C-900 | 0.1 M KOH | 1.01 | 0.91 | 168 |
| FeAB-O | 0.1 M KOH | 0.90 | 174 | |
| FeAC@FeSA–N–C | 0.1 M KOH | 0.912 | 155 | |
| FeSA/FeAC-2DNPC | 0.5 M H2SO4 | 0.81 | 185 | |
| Fe3C@C-Fe SAS | 0.1 M PBS | 0.99 | 0.91 | 118 |
| FeSA/FeONC/NSC | 0.1 M KOH | 0.99 | 0.86 | 186 |
| Fe2–N–C | 0.5 M H2SO4 | 0.78 | 187 | |
| Fe2N6 | 0.1 M HClO4 | 0.84 | 188 | |
| (Fe, Co)/N–C | 0.1 M HClO4 | 1.06 | 0.863 | 195 |
| FeMo–N–C | 0.1 M HClO4 | 0.98 | 0.84 | 85 |
| Fe–N/P–C-700 | 0.1 M KOH | 0.941 | 0.867 | 212 |
| FeNPC | 0.1 M KOH | 1.03 | 0.88 | 213 |
| Fe-SAs/NPS-HC | 0.1 M KOH | 0.912 | 18 | |
| Fe-ISA/SNC | 0.1 M KOH | 0.896 | 214 | |
| Fe1Se1-NC | 0.1 M KOH | 1.0 | 0.88 | 215 |
4 Bottlenecks for Fe-based catalysts
As described earlier, Fe components in multiple forms exhibit ORR activity on the same order of magnitude as the commercial catalysts. As the ultimate destination of ORR semi-reaction, fuel cells and metal–air batteries have more stringent requirements on the performance parameters of catalysts due to harsh and complex working environments (divided into external and internal environments).3,112,218–220 The external environment refers to the wide ranging fluctuation of the battery at the applied potential (0.6–1.5 V) with the moment of starting and stopping. The internal environment means that the catalyst is in a strongly oxidizing operating environment, such as corrosive electrolytes and oxidized OCs. During long-term operation, the applied electric field and the strongly oxidizing working environment make the catalyst structure undergo a vicious cycle of multiple oxidation–reduction processes. It will cause irreversible damage to the Fe active components and carbon supports, such as deactivation, gradual aging and reduced cycle life. Specifically, Fe active components will suffer from irreversible changes such as poisoning, shedding and dissolution, oxidation and phase transition and aggregation in the working environment, and the carbon support that accommodates them will also be malignantly damaged through collapse and corrosion (Fig. 10). Therefore, the catalysts should maintain high ORR activity while considering good stability and an anti-poisoning ability to achieve the purpose of “killing two birds with one stone”.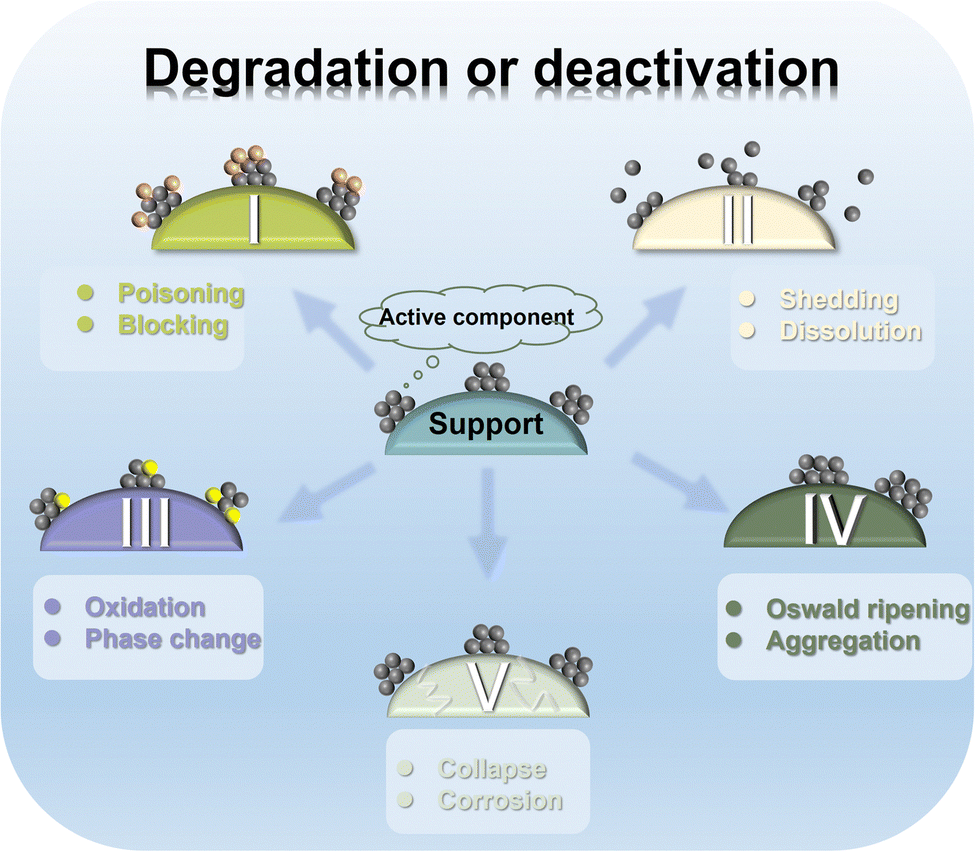 | ||
| Fig. 10 Mechanism of deactivation and degradation for Fe-based catalysts. | ||
4.1 The level of active Fe components
| Fe2+ + H2O2 → Fe3+ + OH⋅ + OH− | (4.1) |
| Fe3+ + H2O2 → Fe2+ + OOH⋅ + OH+ | (4.2) |
| OH⋅ + RH → H2O2 + R⋅ | (4.3) |
| R⋅ + Fe3+ → Fe2+ degradation product | (4.4) |
The proposed mechanism of protonation deactivation adds an innovative explanation for the degraded performance of Fe-based catalysts: N-protonation or the adsorption of anions after N-protonation.236–238 The N-protonation theory holds that protons in an acidic environment combine with lone electron pairs in nitrogen-containing species to protonate, unbeneficial in promoting the subsequent reducing oxygen adsorption. Subsequent studies further refined the theory that anion adsorption onto the nitrogen sites after protonation leads to the deactivation of catalysts. However, further research must confirm whether this deactivation mechanism is the primary source of decreased ORR activity. In addition to the malignant reconstruction, the poisoning of the active sites is also a cause of catalytic deactivation. The fuel cell utilizes air as the source of O2 molecules under experimental conditions. The impurity gases such as sulfide, nitride and CO will preferentially occupy the active sites, leading to the poisoning phenomenon.
4.2 The level of carbon support
In addition to serving as a place for loading active sites, carbon support also provides a conductive network for fast electron transfer in catalytic reactions. Therefore, the physicochemical properties of carbon support largely determine the catalytic performance. Strong metal–support interaction is critical in determining the anchoring of metal components and efficient electron transfer, so more stringent requirements are put forward for the stability and microstructure of the carbon support.239,240 In the real working environment of fuel cells, strong corrosion electrolytes and electric fields will cause varying degrees of damage to the carbon support, causing the deactivation and aging problems of catalysts. The carbon materials with strong chemical stability and high electrical conductivity make them suitable supports for metal components. However, under the attack of the strong oxidizing environment and species generated during the reaction, carbon supports usually face structural corrosion or collapse. The weakened metal–support interaction results in the loss of active metal components. The slow electro-oxidation of the carbon supports causes the catalyst flooding.235,241,242 The micropore structure of carbon supports changes from the original hydrophobic to the hydrophilic system, which makes the reduction product of H2O too late to diffuse out, thus causing micropore flooding. This phenomenon leads to the loss of catalytic kinetics and impacts the mass transfer in catalysis. The geometrical microstructure is also a crucial factor limiting the catalytic performance.The ideal hierarchically porous structure is a crucial factor in achieving fast electron/proton inter-coupling and diffusive mass transfer in the ORR.243–245 In practice, the selection diversity of precursor and the complexity under experimental conditions lead to the difficulty of achieving the synthesized carbon supports with ideal hierarchical porous structures. The undesirable utilization of active sites leads to catalysts exhibiting low-volume catalytic activity. Therefore, thicker catalyst layers in practical applications will significantly reduce the crack connection of the four channels, indirectly leading to phenomena such as deactivation and poisoning of the active sites.
5 Solution and outlook
The diversity and functionality of Fe-FUs afford Fe-based catalysts great potential in the ORR. Exploring the specific action mechanisms of Fe-FUs in multiple forms aims to maximize Fe active components' utilization, thereby narrowing the gap between commercialization and utilization. To meet the good desire for both activity and stability, the future development direction and corresponding solution strategies prospects of Fe-based catalysts are as follows:5.1 Dynamic balance between catalytic activity and stability
The trade-off between catalytic activity and the antioxidant capacity of the Fe-based catalysts is the key to guaranteeing excellent stability. Fe components must maintain a low specific surface energy to satisfy the thermodynamic stability, i.e., preventing the Ostwald ripening effect and Fenton reaction. In addition, Fe-NC catalysts should resist electrochemical oxidative solubilization and oxidation under strong oxidizing species, namely, strong electrochemical stability.5.2 Fine regulation of carbon support
The high specific surface area can accommodate abundant active sites, and their good electrical conductivity becomes an ideal carrier for charge transfer. The design of hydrophobic and hierarchical porous structures is also crucial for establishing efficient three-phase interfaces, aiming to provide fast mass transfer channels. Highly graphitized carbon supports have strong corrosion and oxidation resistance, and the local microscopic defects contribute to the breakage of O–O bonding. Therefore, it is essential to improve the relative ratio of sp2/sp3 hybridization states to balance high activity and stability.5.3 Construction of strong metal–support interactions (SMSI)
Specifically, the electronic rearrangement of heterogeneous structures induced by the SMSI effect is expected to achieve fast charge transfer. The excessive charge accumulation caused by rich interface sites enhances the adsorption and activation of the reactive species. In addition, the SMSI effect can also prevent metal components from shedding, dissolution and agglomeration in the electrochemical conditions, maintaining the efficient utilization of metal components.5.4 Precise protection of active units
Maintaining relative stability in an electric field and corrosion environment is an important prerequisite for maintaining the catalytic activity and stability of Fe-FUs. The common means of physical protection is the construction of chainmail structures, which avoids direct contact between the reaction environment and active units. From the perspective of the electronic structure, the chemical protection strategy avoids the attack of strong oxidizing species on the active units by regulating the coordination environment, including introducing axial ligands, heterogeneous atoms and multi-nanoscale components.5.5 Novel design of active sites
The gradual nanosizing of Fe-FUs seems to be the future main development trend of Fe-based catalysts. Theoretically, the atomic-scale Fe component has high atomic utilization efficiency, but the actual reaction process does not reach the ideal effect. The rapid development of dual-metal and nano-cluster configurations seems to hold the key to this puzzle, and it shines brightly in the ORR under acidic conditions. Seeking more elements to construct novel coordination structures is also one of the options to pursue.5.6 Application of advanced technology
The rapid development of in situ spectroscopy technology can effectively reveal the dynamic changes of Fe components and the interaction between OCs and active sites during the ORR process. The complexity and uncontrollability of the experimental procedure are compensated by the theoretical calculation of thermodynamic simulation, dynamic simulation and molecular dynamics simulation, and machine learning provides more convenience for the screening and optimization of Fe-FUs. Determining the actual structure–activity relationship in the Fe-based catalysts is fundamentally expected to realize a substantial commercial process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (51872115, 52101256) is greatly acknowledged.References
- C.-X. Zhao, B.-Q. Li, J.-N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4448–4463 CrossRef CAS
.
- M. Xiao, Y. Chen, J. Zhu, H. Zhang, X. Zhao, L. Gao, X. Wang, J. Zhao, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, J. Am. Chem. Soc., 2019, 141, 17763–17770 CrossRef CAS PubMed
- Z. Zhuang, Y. Li, R. Yu, L. Xia, J. Yang, Z. Lang, J. Zhu, J. Huang, J. Wang, Y. Wang, L. Fan, J. Wu, Y. Zhao, D. Wang and Y. Li, Nat. Catal., 2022, 5, 300–310 CrossRef CAS
- J. Liu, S. Liu, F. Yan, Z. Wen, W. Chen, X. Liu, Q. Liu, J. Shang, R. Yu, D. Su and J. Shui, J. Am. Chem. Soc., 2022, 144, 19106–19114 CrossRef CAS
- X. Shu, Q. Chen, M. Yang, M. Liu, J. Ma and J. Zhang, Adv. Energy Mater., 2023, 13, 2202871 CrossRef CAS
- H. Li, S. Di, P. Niu, S. Wang, J. Wang and L. Li, Energy Environ. Sci., 2022, 15, 1601–1610 RSC
- H. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song, Z. Fu and P. Huang, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS
- Y. Wang, F. Chu, J. Zeng, Q. Wang, T. Naren, Y. Li, Y. Cheng, Y. Lei and F. Wu, ACS Nano, 2021, 15, 210–239 CrossRef CAS
- Y. Kang, J. Wang, Y. Wei, Y. Wu, D. Xia and L. Gan, Nano Res., 2022, 15, 6148–6155 CrossRef CAS
- D. Takimoto, S. Toma, Y. Suda, T. Shirokura, Y. Tokura, K. Fukuda, M. Matsumoto, H. Imai and W. Sugimoto, Nat. Commun., 2023, 14, 19 CrossRef CAS
- J. Huang, L. Sementa, Z. Liu, G. Barcaro, M. Feng, E. Liu, L. Jiao, M. Xu, D. Leshchev, S.-J. Lee, M. Li, C. Wan, E. Zhu, Y. Liu, B. Peng, X. Duan, W. A. Goddard, A. Fortunelli, Q. Jia and Y. Huang, Nat. Catal., 2022, 5, 513–523 CrossRef CAS
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed
- L. Jiao, R. Zhang, G. Wan, W. Yang, X. Wan, H. Zhou, J. Shui, S.-H. Yu and H.-L. Jiang, Nat. Commun., 2020, 11, 2831 CrossRef CAS PubMed
- T. Wang, X. Sang, W. Zheng, B. Yang, S. Yao, C. Lei, Z. Li, Q. He, J. Lu, L. Lei, L. Dai and Y. Hou, Adv. Mater., 2020, 32, 2002430 CrossRef CAS PubMed
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed
- G. Chen, R. Lu, C. Li, J. Yu, X. Li, L. Ni, Q. Zhang, G. Zhu, S. Liu, J. Zhang, U. I. Kramm, Y. Zhao, G. Wu, J. Xie and X. Feng, Adv. Mater., 2023, 35, 2300907 CrossRef CAS PubMed
- Y. Li, Y. Ding, B. Zhang, Y. Huang, H. Qi, P. Das, L. Zhang, X. Wang, Z.-S. Wu and X. Bao, Energy Environ. Sci., 2023, 16, 2629–2636 RSC
- Y. Han, Q. Wei, Y. Fu, D. Zhang, P. Li, X. Shan, H. Yang, X. Zhan, X. Liu and W. Yang, Small, 2023, 19, 2300683 CrossRef CAS
- C. Qi, H. Yang, Z. Sun, H. Wang, N. Xu, G. Zhu, L. Wang, W. Jiang, X. Yu, X. Li, Q. Xiao, P. Qiu and W. Luo, Angew. Chem., Int. Ed., 2023, 62, e202308344 CrossRef CAS PubMed
- Y. Zhao, Z. Shen, J. Huo, X. Cao, P. Ou, J. Qu, X. Nie, J. Zhang, M. Wu, G. Wang and H. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308349 CrossRef CAS
- L. Zong, K. Fan, P. Li, F. Lu, B. Li and L. Wang, Adv. Energy Mater., 2023, 13, 2203611 CrossRef CAS
- X. Wei, S. Song, W. Cai, X. Luo, L. Jiao, Q. Fang, X. Wang, N. Wu, Z. Luo, H. Wang, Z. Zhu, J. Li, L. Zheng, W. Gu, W. Song, S. Guo and C. Zhu, Chem, 2023, 9, 181–197 CAS
- H. Li, S. Kelly, D. Guevarra, Z. Wang, Y. Wang, J. A. Haber, M. Anand, G. T. K. K. Gunasooriya, C. S. Abraham, S. Vijay, J. M. Gregoire and J. K. Nørskov, Nat. Catal., 2021, 4, 463–468 CrossRef CAS
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- Y. Zhao, D. P. Adiyeri Saseendran, C. Huang, C. A. Triana, W. R. Marks, H. Chen, H. Zhao and G. R. Patzke, Chem. Rev., 2023, 123, 6257–6358 CrossRef CAS
- Z. Shi, J. Zhang, Z.-S. Liu, H. Wang and D. P. Wilkinson, Electrochim. Acta, 2006, 51, 1905–1916 CrossRef CAS
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS
- J. Lan, V. V. Rybkin and M. Iannuzzi, J. Phys. Chem. Lett., 2020, 11, 3724–3730 CrossRef CAS PubMed
- N. M. Marković and P. N. Ross, Surf. Sci. Rep., 2002, 45, 117–229 CrossRef
- N. Marzari, A. Ferretti and C. Wolverton, Nat. Mater., 2021, 20, 736–749 CrossRef CAS
- S. Chen, Y. Gao, W. Wang, O. V. Prezhdo and L. Xu, ACS Nano, 2023, 17, 1522–1532 CrossRef CAS
- Z. Hou, C. Cui, Y. Li, Y. Gao, D. Zhu, Y. Gu, G. Pan, Y. Zhu and T. Zhang, Adv. Mater., 2023, 35, 2209876 CrossRef CAS PubMed
- Z.-J. Zhao, S. Liu, S. Zha, D. Cheng, F. Studt, G. Henkelman and J. Gong, Nat. Rev. Mater., 2019, 4, 792–804 CrossRef
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- B. L. Vanzetti, Scientia, 1916, 10, 149 Search PubMed
- P. Sabatier, Ber. Dtsch. Chem. Ges., 1911, 44, 1984–2001 CrossRef CAS
- M. Shao, P. Liu, J. Zhang and R. Adzic, J. Phys. Chem. B, 2007, 111, 6772–6775 CrossRef CAS PubMed
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS
- J. K. Nørskov, Prog. Surf. Sci., 1991, 38, 103–144 CrossRef
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed
- L. Gong, H. Zhang, Y. Wang, E. Luo, K. Li, L. Gao, Y. Wang, Z. Wu, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed., 2020, 59, 13923–13928 CrossRef CAS
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed
- J. Li, W. Xia, J. Tang, Y. Gao, C. Jiang, Y. Jia, T. Chen, Z. Hou, R. Qi, D. Jiang, T. Asahi, X. Xu, T. Wang, J. He and Y. Yamauchi, J. Am. Chem. Soc., 2022, 144, 9280–9291 CrossRef CAS PubMed
- H. Bai, J. Feng, D. Liu, P. Zhou, R. Wu, C. T. Kwok, W. F. Ip, W. Feng, X. Sui, H. Liu and H. Pan, Small, 2023, 19, 2205638 CrossRef CAS PubMed
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T.-S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Chem, 2020, 6, 3440–3454 CAS
- W. Zhong, Y. Qiu, H. Shen, X. Wang, J. Yuan, C. Jia, S. Bi and J. Jiang, J. Am. Chem. Soc., 2021, 143, 4405–4413 CrossRef CAS PubMed
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC
- P. Li, L. Zhou, C. Zhao, H. Ju, Q. Gao, W. Si, L. Cheng, J. Hao, M. Li, Y. Chen, C. Jia and X. Guo, Rep. Prog. Phys., 2022, 85, 086401 CrossRef
- Y. Liu, X. Liu, Z. Lv, R. Liu, L. Li, J. Wang, W. Yang, X. Jiang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2022, 61, e202117617 CrossRef CAS
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed
- J. Wang, Z. Li, Y. Wu and Y. Li, Adv. Mater., 2018, 30, 1801649 CrossRef
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS
- S. Dou, X. Wang and S. Wang, Small Methods, 2019, 3, 1800211 CrossRef
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Adv. Mater., 2019, 31, 1806545 CrossRef PubMed
- C.-X. Zhao, J.-N. Liu, J. Wang, D. Ren, B.-Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC
- T. Asset and P. Atanassov, Joule, 2020, 4, 33–44 CrossRef CAS
- W. Wang, Q. Jia, S. Mukerjee and S. Chen, ACS Catal., 2019, 9, 10126–10141 CrossRef CAS
- L. Yu, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 15294–15297 CrossRef CAS PubMed
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC
- Y. Tu, P. Ren, D. Deng and X. Bao, Nano Energy, 2018, 52, 494–500 CrossRef CAS
- Y. Feng, K. Song, W. Zhang, X. Zhou, S. J. Yoo, J.-G. Kim, S. Qiao, Y. Qi, X. Zou, Z. Chen, T. Qin, N. Yue, Z. Wang, D. Li and W. Zheng, J. Energy Chem., 2022, 70, 211–218 CrossRef CAS
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed
- G. Zhang, L. Li, M. Chen and F. Yang, J. Mater. Chem. A, 2020, 8, 9256–9267 RSC
- M.-X. Chen, M. Zhu, M. Zuo, S.-Q. Chu, J. Zhang, Y. Wu, H.-W. Liang and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 1627–1633 CrossRef CAS PubMed
- G. Zhu, H. Yang, Y. Jiang, Z. Sun, X. Li, J. Yang, H. Wang, R. Zou, W. Jiang, P. Qiu and W. Luo, Adv. Sci., 2022, 9, 2200394 CrossRef CAS PubMed
- J. Hu, C. Zhang, M. Sun, Q. Qi, S. Luo, H. Song, J. Xiao, B. Huang, M. K. H. Leung and Y. Zhang, Nano Res., 2022, 15, 4950–4957 CrossRef CAS
- H. Khani, N. S. Grundish, D. O. Wipf and J. B. Goodenough, Adv. Energy Mater., 2020, 10, 1903215 CrossRef CAS
- N. Yang, L. Li, J. Li, W. Ding and Z. Wei, Chem. Sci., 2018, 9, 5795–5804 RSC
- Y. Liu, H. Jiang, Y. Zhu, X. Yang and C. Li, J. Mater. Chem. A, 2016, 4, 1694–1701 RSC
- J. A. Varnell, J. S. Sotiropoulos, T. M. Brown, K. Subedi, R. T. Haasch, C. E. Schulz and A. A. Gewirth, ACS Energy Lett., 2018, 3, 823–828 CrossRef CAS
- J. A. Varnell, E. C. M. Tse, C. E. Schulz, T. T. Fister, R. T. Haasch, J. Timoshenko, A. I. Frenkel and A. A. Gewirth, Nat. Commun., 2016, 7, 12582 CrossRef CAS PubMed
- B. Jeong, D. Shin, H. Jeon, J. D. Ocon, B. S. Mun, J. Baik, H.-J. Shin and J. Lee, ChemSusChem, 2014, 7, 1289–1294 CrossRef CAS PubMed
- Y. Nakaya and S. Furukawa, Chem. Rev., 2022, 123, 5859–5947 CrossRef PubMed
- P. Zhu, X. Xiong, X. Wang, C. Ye, J. Li, W. Sun, X. Sun, J. Jiang, Z. Zhuang, D. Wang and Y. Li, Nano Lett., 2022, 22, 9507–9515 CrossRef CAS PubMed
- J. Guan, S. Yang, T. Liu, Y. Yu, J. Niu, Z. Zhang and F. Wang, Angew. Chem., Int. Ed., 2021, 60, 21899–21904 CrossRef CAS PubMed
- M. Jahan, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2012, 134, 6707–6713 CrossRef CAS PubMed
- Q. Liu, X. Liu, L. Zheng and J. Shui, Angew. Chem., Int. Ed., 2018, 57, 1204–1208 CrossRef CAS PubMed
- J. Han, X. Meng, L. Lu, J. Bian, Z. Li and C. Sun, Adv. Funct. Mater., 2019, 29, 1808872 CrossRef CAS
- S. He, Y. Liu, H. Zhan and L. Guan, ACS Catal., 2021, 11, 9355–9365 CrossRef CAS
- Z. Kong, Y. Maswadeh, J. A. Vargas, S. Shan, Z.-P. Wu, H. Kareem, A. C. Leff, D. T. Tran, F. Chang, S. Yan, S. Nam, X. Zhao, J. M. Lee, J. Luo, S. Shastri, G. Yu, V. Petkov and C.-J. Zhong, J. Am. Chem. Soc., 2020, 142, 1287–1299 CrossRef CAS PubMed
- F. Xiao, Q. Wang, G.-L. Xu, X. Qin, I. Hwang, C.-J. Sun, M. Liu, W. Hua, H.-W. Wu, S. Zhu, J.-C. Li, J.-G. Wang, Y. Zhu, D. Wu, Z. Wei, M. Gu, K. Amine and M. Shao, Nat. Catal., 2022, 5, 503–512 CrossRef CAS
- W. Zhu, Y. Pei, J. C. Douglin, J. Zhang, H. Zhao, J. Xue, Q. Wang, R. Li, Y. Qin, Y. Yin, D. R. Dekel and M. D. Guiver, Appl. Catal., B, 2021, 299, 120656 CrossRef CAS
- Y.-R. Hao, H. Xue, L. Lv, J. Sun, N. Guo, T. Song, H. Dong, J. Zhang and Q. Wang, Appl. Catal., B, 2021, 295, 120314 CrossRef CAS
- R. F. Ziolo, E. P. Giannelis, B. A. Weinstein, M. P. O'Horo, B. N. Ganguly, V. Mehrotra, M. W. Russell and D. R. Huffman, Science, 1992, 257, 219–223 CrossRef CAS PubMed
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed
- W. A. Challener, C. Peng, A. V. Itagi, D. Karns, W. Peng, Y. Peng, X. Yang, X. Zhu, N. J. Gokemeijer, Y. T. Hsia, G. Ju, R. E. Rottmayer, M. A. Seigler and E. C. Gage, Nat. Photonics, 2009, 3, 220–224 CrossRef CAS
- T. P. Almeida, T. Kasama, A. R. Muxworthy, W. Williams, L. Nagy, T. W. Hansen, P. D. Brown and R. E. Dunin-Borkowski, Nat. Commun., 2014, 5, 5154 CrossRef CAS PubMed
- R. Karunagaran, C. Coghlan, T. T. Tung, S. Kabiri, D. N. H. Tran, C. J. Doonan and D. Losic, New J. Chem., 2017, 41, 15180–15186 RSC
- Y. Ma, H. Wang, J. Key, V. Linkov, S. Ji, X. Mao, Q. Wang and R. Wang, Int. J. Hydrogen Energy, 2014, 39, 14777–14782 CrossRef CAS
- Y. Zhu, B. Zhang, D.-W. Wang and D. S. Su, ChemSusChem, 2015, 8, 4016–4021 CrossRef CAS PubMed
- Z. Fan, J. Li, W. Yang, Q. Fu, K. Sun, Y.-C. Song, Z. Wei, Q. Liao and X. Zhu, Chem. Eng. J., 2020, 385, 123393 CrossRef CAS
- X. Liu and W. Hu, RSC Adv., 2016, 6, 29848–29854 RSC
- W. Feng, M. Liu, J. Liu, Y. Song and F. Wang, Catal. Sci. Technol., 2018, 8, 4900–4906 RSC
- K. Xu, H. Bao, C. Tang, K. Maliutina, F. Li and L. Fan, Mater. Today Energy, 2020, 18, 100500 CrossRef CAS
- Y. Niu, Y. Yuan, Q. Zhang, F. Chang, L. Yang, Z. Chen and Z. Bai, Nano Energy, 2021, 82, 105699 CrossRef CAS
- H. Fan, K. Mao, M. Liu, O. Zhuo, J. Zhao, T. Sun, Y. Jiang, X. Du, X. Zhang, Q. Wu, R. Che, L. Yang, Q. Wu, X. Wang and Z. Hu, J. Mater. Chem. A, 2018, 6, 21313–21319 RSC
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed
- H. Huang, X. Feng, C. Du, S. Wu and W. Song, J. Mater. Chem. A, 2015, 3, 4976–4982 RSC
- M. Cao, Y. Liu, K. Sun, H. Li, X. Lin, P. Zhang, L. Zhou, A. Wang, S. Mehdi, X. Wu, J. Jiang and B. Li, Small, 2022, 18, 2202014 CrossRef CAS PubMed
- D. Xue, F. Yu, Q. Ying, Y. Wu, W.-J. Lee, S.-H. Kwon and Y. Yang, ChemCatChem, 2021, 13, 4431–4441 CrossRef CAS
- Y. Pan, X. Ma, M. Wang, X. Yang, S. Liu, H.-C. Chen, Z. Zhuang, Y. Zhang, W.-C. Cheong, C. Zhang, X. Cao, R. Shen, Q. Xu, W. Zhu, Y. Liu, X. Wang, X. Zhang, W. Yan, J. Li, H. M. Chen, C. Chen and Y. Li, Adv. Mater., 2022, 34, 2203621 CrossRef CAS PubMed
- Y. Pan, M. Li, W. Mi, M. Wang, J. Li, Y. Zhao, X. Ma, B. Wang, W. Zhu, Z. Cui, H. Yin and Y. Liu, Nano Res., 2022, 15, 7976–7985 CrossRef CAS
- X. Cui, L. Gao, S. Lei, S. Liang, J. Zhang, C. D. Sewell, W. Xue, Q. Liu, Z. Lin and Y. Yang, Adv. Funct. Mater., 2021, 31, 2009197 CrossRef CAS
- Q. Zhou, Y. Yang, Q. Ye, T. Xue, M. Tu, Y. Liu, H. Li, X.-H. Yan, Z.-L. Zou, B.-P. Wang, Y.-J. Lu and F.-L. Han, Mater. Today Energy, 2023, 31, 101194 CrossRef CAS
- X. Guo, S. Liu, X. Wan, J. Zhang, Y. Liu, X. Zheng, Q. Kong and Z. Jin, Nano Lett., 2022, 22, 4879–4887 CrossRef CAS PubMed
- H. Tan, Y. Li, J. Kim, T. Takei, Z. Wang, X. Xu, J. Wang, Y. Bando, Y.-M. Kang, J. Tang and Y. Yamauchi, Adv. Sci., 2018, 5, 1800120 CrossRef PubMed
- X. Wei, S. Song, N. Wu, X. Luo, L. Zheng, L. Jiao, H. Wang, Q. Fang, L. Hu, W. Gu, W. Song and C. Zhu, Nano Energy, 2021, 84, 105840 CrossRef CAS
- F. Zhou, P. Yu, F. Sun, G. Zhang, X. Liu and L. Wang, J. Mater. Chem. A, 2021, 9, 6831–6840 RSC
- J. Zou, M. Zheng, H. Wu, Z. Pan, J. Xu, X. Zeng, T. Lan, Y. Yao and Z. Li, J. Energy Storage, 2023, 70, 107950 CrossRef
- X. Yang, F. Wang, Z. Jing, M. Chen, B. Wang, L. Wang, G. Qu, Y. Kong and L. Xu, Small, 2023, 19, 2301985 CrossRef CAS PubMed
- Q. Yu, X. Liu, G. Liu, X. Wang, Z. Li, B. Li, Z. Wu and L. Wang, Adv. Funct. Mater., 2022, 32, 2205767 CrossRef CAS
- B. Ni, R. Chen, L. Wu, P. Sun and T. Chen, Sci. China Mater., 2021, 64, 1159–1172 CrossRef CAS
- M. Du, D. Li, S. Liu and J. Yan, Chin. Chem. Lett., 2023, 34, 108156 CrossRef CAS
- T. Sun, S. Li, T. Li, X. Chen, D. Xu and H.-G. Wang, Energy Technol., 2022, 10, 2100877 CrossRef CAS
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS
- S.-i Yamazaki, Coord. Chem. Rev., 2018, 373, 148–166 CrossRef CAS
- J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254, 2755–2791 CrossRef CAS
- J. H. Zagal and M. T. M. Koper, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS PubMed
- Y. Wen, B. Wang, C. Huang, L. Wang and D. Hulicova-Jurcakova, Chem. – Eur. J., 2015, 21, 80–85 CrossRef CAS PubMed
- M. Xiao, J. Zhu, L. Ma, Z. Jin, J. Ge, X. Deng, Y. Hou, Q. He, J. Li, Q. Jia, S. Mukerjee, R. Yang, Z. Jiang, D. Su, C. Liu and W. Xing, ACS Catal., 2018, 8, 2824–2832 CrossRef CAS
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed
- X. Yan, Y. Jia, K. Wang, Z. Jin, C.-L. Dong, Y.-C. Huang, J. Chen and X. Yao, Carbon Energy, 2020, 2, 452–460 CrossRef CAS
- Z. Yang, Y. Wang, M. Zhu, Z. Li, W. Chen, W. Wei, T. Yuan, Y. Qu, Q. Xu, C. Zhao, X. Wang, P. Li, Y. Li, Y. Wu and Y. Li, ACS Catal., 2019, 9, 2158–2163 CrossRef CAS
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef CAS PubMed
- Z. Li, Y. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C.-T. He, C. Peng, W.-C. Cheong, Q. Li, M. Zhang, Z. Chen, N. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X.-M. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS PubMed
- K. Liu, J. Fu, Y. Lin, T. Luo, G. Ni, H. Li, Z. Lin and M. Liu, Nat. Commun., 2022, 13, 2075 CrossRef CAS PubMed
- A. Han, X. Wang, K. Tang, Z. Zhang, C. Ye, K. Kong, H. Hu, L. Zheng, P. Jiang, C. Zhao, Q. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19262–19271 CrossRef CAS PubMed
- L. Hu, C. Dai, L. Chen, Y. Zhu, Y. Hao, Q. Zhang, L. Gu, X. Feng, S. Yuan, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 27324–27329 CrossRef CAS PubMed
- L. Peng, J. Yang, Y. Yang, F. Qian, Q. Wang, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. N. Waterhouse, Adv. Mater., 2022, 34, 2202544 CrossRef CAS PubMed
- J. Masa, A. Zhao, W. Xia, M. Muhler and W. Schuhmann, Electrochim. Acta, 2014, 128, 271–278 CrossRef CAS
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS
- P. Peng, L. Shi, F. Huo, C. Mi, X. Wu, S. Zhang and Z. Xiang, Sci. Adv., 2019, 5, eaaw2322 CrossRef CAS PubMed
- Z. Miao, X. Wang, M.-C. Tsai, Q. Jin, J. Liang, F. Ma, T. Wang, S. Zheng, B.-J. Hwang, Y. Huang, S. Guo and Q. Li, Adv. Energy Mater., 2018, 8, 1801226 CrossRef
- M. Lefèvre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106, 8705–8713 CrossRef
- P. Song, Y. Wang, J. Pan, W. Xu and L. Zhuang, J. Power Sources, 2015, 300, 279–284 CrossRef CAS
- H. Shen, E. Gracia-Espino, J. Ma, H. Tang, X. Mamat, T. Wagberg, G. Hu and S. Guo, Nano Energy, 2017, 35, 9–16 CrossRef CAS
- N. Delaporte, E. Rivard, S. K. Natarajan, P. Benard, M. L. Trudeau and K. Zaghib, Nanomaterials, 2020, 10, 1947 CrossRef CAS PubMed
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefèvre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J.-P. Dodelet, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed
- X. Ao, W. Zhang, Z. Li, J.-G. Li, L. Soule, X. Huang, W.-H. Chiang, H. M. Chen, C. Wang, M. Liu and X. C. Zeng, ACS Nano, 2019, 13, 11853–11862 CrossRef CAS PubMed
- J. Li, L. Jiao, E. Wegener, L. L. Richard, E. Liu, A. Zitolo, M. T. Sougrati, S. Mukerjee, Z. Zhao, Y. Huang, F. Yang, S. Zhong, H. Xu, A. J. Kropf, F. Jaouen, D. J. Myers and Q. Jia, J. Am. Chem. Soc., 2020, 142, 1417–1423 CrossRef CAS PubMed
- J. Li, S. Ghoshal, W. Liang, M.-T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. Ma, X. Yuan, Z.-F. Ma, S. Mukerjee and Q. Jia, Energy Environ. Sci., 2016, 9, 2418–2432 RSC
- T. Mineva, I. Matanovic, P. Atanassov, M.-T. Sougrati, L. Stievano, M. Clémancey, A. Kochem, J.-M. Latour and F. Jaouen, ACS Catal., 2019, 9, 9359–9371 CrossRef CAS
- N. Zhang, T. Zhou, M. Chen, H. Feng, R. Yuan, C. A. Zhong, W. Yan, Y. Tian, X. Wu, W. Chu, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 111–118 RSC
- J. Wang, L. Li and Z.-D. Wei, Acta Phys.-Chim. Sin., 2016, 32, 321–328 Search PubMed
- W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L.-J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 11755–11759 CrossRef CAS PubMed
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS
- V. Armel, S. Hindocha, F. Salles, S. Bennett, D. Jones and F. Jaouen, J. Am. Chem. Soc., 2017, 139, 453–464 CrossRef CAS PubMed
- K. Song, Y. Feng, W. Zhang and W. Zheng, J. Energy Chem., 2022, 67, 391–422 CrossRef CAS
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC
- J. Li, H. Zhang, W. Samarakoon, W. Shan, D. A. Cullen, S. Karakalos, M. Chen, D. Gu, K. L. More, G. Wang, Z. Feng, Z. Wang and G. Wu, Angew. Chem., Int. Ed., 2019, 58, 18971–18980 CrossRef CAS PubMed
- S. Hu, W. Ni, D. Yang, C. Ma, J. Zhang, J. Duan, Y. Gao and S. Zhang, Carbon, 2020, 162, 245–255 CrossRef CAS
- M. Chen, F. Kong, H. Yao, Y. Chen, G. Meng, Z. Chang, C. Chen, H. Tian, L. Wang, X. Cui and J. Shi, Chem. Eng. J., 2023, 453, 139820 CrossRef CAS
- S.-H. Yin, J. Yang, Y. Han, G. Li, L.-Y. Wan, Y.-H. Chen, C. Chen, X.-M. Qu, Y.-X. Jiang and S.-G. Sun, Angew. Chem., Int. Ed., 2020, 59, 21976–21979 CrossRef CAS PubMed
- X. Ao, W. Zhang, Z. Li, L. Lv, Y. Ruan, H.-H. Wu, W.-H. Chiang, C. Wang, M. Liu and X. C. Zeng, J. Mater. Chem. A, 2019, 7, 11792–11801 RSC
- E. F. Holby and C. D. Taylor, Sci. Rep., 2015, 5, 9286 CrossRef PubMed
- S. Bhalla, D. T. Melnekoff, A. Aleman, V. Leshchenko, P. Restrepo, J. Keats, K. Onel, J. R. Sawyer, D. Madduri, J. Richter, S. Richard, A. Chari, H. J. Cho, J. T. Dudley, S. Jagannath, A. Laganà and S. Parekh, Sci. Adv., 2021, 7, eabg9551 CrossRef CAS PubMed
- R. Wang, L. Zhang, J. Shan, Y. Yang, J. F. Lee, T. Y. Chen, J. Mao, Y. Zhao, L. Yang, Z. Hu and T. Ling, Adv. Sci., 2022, 9, e2203917 CrossRef PubMed
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed
- X. Yang, D. Xia, Y. Kang, H. Du, F. Kang, L. Gan and J. Li, Adv. Sci., 2020, 7, 2000176 CrossRef CAS PubMed
- F. Wang, Y. Zhou, S. Lin, L. Yang, Z. Hu and D. Xie, Nano Energy, 2020, 78, 105128 CrossRef CAS
- Y. Wang, Y.-J. Tang and K. Zhou, J. Am. Chem. Soc., 2019, 141, 14115–14119 CrossRef CAS PubMed
- E. F. Holby and P. Zelenay, Nano Energy, 2016, 29, 54–64 CrossRef CAS
- Y. Lin, K. Liu, K. Chen, Y. Xu, H. Li, J. Hu, Y.-R. Lu, T.-S. Chan, X. Qiu, J. Fu and M. Liu, ACS Catal., 2021, 11, 6304–6315 CrossRef CAS
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC
- H.-T. Lien, S.-T. Chang, P.-T. Chen, D. P. Wong, Y.-C. Chang, Y.-R. Lu, C.-L. Dong, C.-H. Wang, K.-H. Chen and L.-C. Chen, Nat. Commun., 2020, 11, 4233 CrossRef CAS PubMed
- J. Li, M. Chen, D. A. Cullen, S. Hwang, M. Wang, B. Li, K. Liu, S. Karakalos, M. Lucero, H. Zhang, C. Lei, H. Xu, G. E. Sterbinsky, Z. Feng, D. Su, K. L. More, G. Wang, Z. Wang and G. Wu, Nat. Catal., 2018, 1, 935–945 CrossRef CAS
- X. X. Wang, D. A. Cullen, Y.-T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed
- L. Cao, Q. Luo, W. Liu, Y. Lin, X. Liu, Y. Cao, W. Zhang, Y. Wu, J. Yang, T. Yao and S. Wei, Nat. Catal., 2019, 2, 134–141 CrossRef CAS
- X. Wan, Q. Liu, J. Liu, S. Liu, X. Liu, L. Zheng, J. Shang, R. Yu and J. Shui, Nat. Commun., 2022, 13, 2963 CrossRef CAS PubMed
- Y. Lei, F. Yang, H. Xie, Y. Lei, X. Liu, Y. Si and H. Wang, J. Mater. Chem. A, 2020, 8, 20629–20636 RSC
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng, Z. Qi, C. Wang, R. Long, M. Chen, J. Zhu, P. Gao, L. Song, J. Jiang and Y. Xiong, Chem, 2019, 5, 2865–2878 CAS
- N. Zhang, T. Zhou, J. Ge, Y. Lin, Z. Du, C. A. Zhong, W. Wang, Q. Jiao, R. Yuan, Y. Tian, W. Chu, C. Wu and Y. Xie, Matter, 2020, 3, 509–521 CrossRef
- H. Li, X. Shu, H. Xi, Z. Lv, H. Tian, J. Zhang and H. Xia, J. Mater. Chem. A, 2022, 10, 14828–14837 RSC
- Y. X. Zhao, J. H. Wen, P. Li, P. F. Zhang, S. N. Wang, D. C. Li, J. M. Dou, Y. W. Li, H. Y. Ma and L. Xu, Angew. Chem., Int. Ed., 2023, 62, e202216950 CrossRef CAS PubMed
- Y. Liu, F. Tu, Z. Zhang, Z. Zhao, P. Guo, L. Shen, Y. Zhang, L. Zhao, G. Shao and Z. Wang, Appl. Catal., B, 2023, 324, 122209 CrossRef CAS
- D. Yu, Y. Ma, F. Hu, C.-C. Lin, L. Li, H.-Y. Chen, X. Han and S. Peng, Adv. Energy Mater., 2021, 11, 2101242 CrossRef CAS
- X. Zhong, S. Ye, J. Tang, Y. Zhu, D. Wu, M. Gu, H. Pan and B. Xu, Appl. Catal., B, 2021, 286, 119891 CrossRef CAS
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan, Y. Qin, L. Fang, R. Feng, J. Zhou, P. Zhang, J. Dong, G. Yu, Y. Liu, X. Zhang and X. Fan, Sci. Adv., 2020, 6, eaba6586 CrossRef CAS PubMed
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed
- K. Song, Y. Feng, X. Zhou, T. Qin, X. Zou, Y. Qi, Z. Chen, J. Rao, Z. Wang, N. Yue, X. Ge, W. Zhang and W. Zheng, Appl. Catal., B, 2022, 316, 121591 CrossRef CAS
- Z. Sun, H. Zhang, L. Cao, X. Liu, D. Wu, X. Shen, X. Zhang, Z. Chen, S. Ru, X. Zhu, Z. Xia, Q. Luo, F. Xu and T. Yao, Angew. Chem., Int. Ed., 2023, 62, e202217719 CrossRef CAS PubMed
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed
- M. Bauer, C. Lei, K. Read, R. Tobey, J. Gland, M. M. Murnane and H. C. Kapteyn, Phys. Rev. Lett., 2001, 87, 025501 CrossRef
- C. Puglia, A. Nilsson, B. Hernnäs, O. Karis, P. Bennich and N. Mårtensson, Surf. Sci., 1995, 342, 119–133 CrossRef CAS
- Q. Dang, H. Huang, L. Li, X. Lyu, S. Zhong, Y. Yu and D. Xu, Chem. Mater., 2021, 33, 5690–5699 CrossRef CAS
- P. Cui, L. Zhao, Y. Long, L. Dai and C. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218269 CrossRef CAS PubMed
- M. Fan, J. Cui, J. Wu, R. Vajtai, D. Sun and P. M. Ajayan, Small, 2020, 16, 1906782 CrossRef CAS PubMed
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed
- M. Fan, Q. Yuan, Y. Zhao, Z. Wang, A. Wang, Y. Liu, K. Sun, J. Wu, L. Wang and J. Jiang, Adv. Mater., 2022, 34, 2107040 CrossRef CAS PubMed
- P. Wei, X. Li, Z. He, X. Sun, Q. Liang, Z. Wang, C. Fang, Q. Li, H. Yang, J. Han and Y. Huang, Chem. Eng. J., 2021, 422, 130134 CrossRef CAS
- Z. Yao, M. Hu, Z. Iqbal and X. Wang, ACS Catal., 2020, 10, 160–167 CrossRef CAS
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS PubMed
- F. Xiang, X. Zhao, J. Yang, N. Li, W. Gong, Y. Liu, A. Burguete-Lopez, Y. Li, X. Niu and A. Fratalocchi, Adv. Mater., 2023, 35, 2208533 CrossRef CAS PubMed
- S. Li, L. Xia, J. Li, Z. Chen, W. Zhang, J. Zhu, R. Yu, F. Liu, S. Lee, Y. Zhao, L. Zhou and L. Mai, Energy Environ. Mater., 2023, 6, e12560 CrossRef
- H. Hu, J. Wang, B. Cui, X. Zheng, J. Lin, Y. Deng and X. Han, Angew. Chem., Int. Ed., 2022, 61, e202114441 CrossRef CAS PubMed
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed
- X. Zhu, X. Tan, K.-H. Wu, C.-L. Chiang, Y.-C. Lin, Y.-G. Lin, D.-W. Wang, S. Smith, X. Lu and R. Amal, J. Mater. Chem. A, 2019, 7, 14732–14742 RSC
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W.-C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed
- Z. Chen, X. Su, J. Ding, N. Yang, W. Zuo, Q. He, Z. Wei, Q. Zhang, J. Huang and Y. Zhai, Appl. Catal., B, 2022, 308, 121206 CrossRef CAS
- Z. Chen, H. Niu, J. Ding, H. Liu, P.-H. Chen, Y.-H. Lu, Y.-R. Lu, W. Zuo, L. Han, Y. Guo, S.-F. Hung and Y. Zhai, Angew. Chem., Int. Ed., 2021, 60, 25404–25410 CrossRef CAS PubMed
- X. Li, X. Yang, L. Liu, H. Zhao, Y. Li, H. Zhu, Y. Chen, S. Guo, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2021, 11, 7450–7459 CrossRef CAS
- Y.-C. Wang, W. Huang, L.-Y. Wan, J. Yang, R.-J. Xie, Y.-P. Zheng, Y.-Z. Tan, Y.-S. Wang, K. Zaghib, L.-R. Zheng, S.-H. Sun, Z.-Y. Zhou and S.-G. Sun, Sci. Adv., 2022, 8, eadd8873 CrossRef CAS PubMed
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 17832–17852 CrossRef CAS PubMed
- E. Luo, Y. Chu, J. Liu, Z. Shi, S. Zhu, L. Gong, J. Ge, C. H. Choi, C. Liu and W. Xing, Energy Environ. Sci., 2021, 14, 2158–2185 RSC
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, npj Comput. Mater., 2019, 5, 78 CrossRef
- M. T. M. Koper, Phys. Chem. Chem. Phys., 2013, 155, 1399–1407 RSC
- Y. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS
- H. Schulenburg, S. Stankov, V. Schünemann, J. Radnik, I. Dorbandt, S. Fiechter, P. Bogdanoff and H. Tributsch, J. Phys. Chem. B, 2003, 107, 9034–9041 CrossRef CAS
- L. Gubler, S. M. Dockheer and W. H. Koppenol, J. Electrochem. Soc., 2011, 158, B755–B769 CrossRef CAS
- L.-N. Chen, W.-S. Yu, T. Wang, X.-D. Yang, H.-J. Yang, Z.-X. Chen, T. Wang, N. Tian, Z.-Y. Zhou and S.-G. Sun, Sci. China: Chem., 2020, 63, 198–202 CrossRef CAS
- M. Lefèvre and J.-P. Dodelet, Electrochim. Acta, 2003, 48, 2749–2760 CrossRef
- C. H. Choi, C. Baldizzone, J.-P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC
- J. Wu, Y. Nabae, A. Muthukrishnan and T. Ohsaka, Electrochim. Acta, 2016, 214, 307–312 CrossRef CAS
- R. Chenitz, U. I. Kramm, M. Lefèvre, V. Glibin, G. Zhang, S. Sun and J.-P. Dodelet, Energy Environ. Sci., 2018, 11, 365–382 RSC
- D. Banham, S. Ye, K. Pei, J.-I. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS
- S. Baranton, C. Coutanceau, C. Roux, F. Hahn and J. M. Léger, J. Electroanal. Chem., 2005, 577, 223–234 CrossRef CAS
- C. H. Choi, C. Baldizzone, G. Polymeros, E. Pizzutilo, O. Kasian, A. K. Schuppert, N. Ranjbar Sahraie, M.-T. Sougrati, K. J. J. Mayrhofer and F. Jaouen, ACS Catal., 2016, 6, 3136–3146 CrossRef CAS
- J.-Y. Choi, L. Yang, T. Kishimoto, X. Fu, S. Ye, Z. Chen and D. Banham, Energy Environ. Sci., 2017, 10, 296–305 RSC
- J. Herranz, F. Jaouen, M. Lefèvre, U. I. Kramm, E. Proietti, J.-P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, P. Bertrand, T. M. Arruda and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 16087–16097 CrossRef CAS PubMed
- K. Strickland, R. Pavlicek, E. Miner, Q. Jia, I. Zoller, S. Ghoshal, W. Liang and S. Mukerjee, ACS Catal., 2018, 8, 3833–3843 CrossRef CAS
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Electrochim. Acta, 2010, 55, 2853–2858 CrossRef CAS
- H. Tian, A. Song, P. Zhang, K. Sun, J. Wang, B. Sun, Q. Fan, G. Shao, C. Chen, H. Liu, Y. Li and G. Wang, Adv. Mater., 2023, 35, 2210714 CrossRef CAS PubMed
- X.-C. Sun, K. Yuan, J.-H. Zhou, C.-Y. Yuan, H.-C. Liu and Y.-W. Zhang, ACS Catal., 2022, 12, 923–934 CrossRef CAS
- Y.-C. Wang, L. Huang, P. Zhang, Y.-T. Qiu, T. Sheng, Z.-Y. Zhou, G. Wang, J.-G. Liu, M. Rauf, Z.-Q. Gu, W.-T. Wu and S.-G. Sun, ACS Energy Lett., 2017, 2, 645–650 CrossRef CAS
- Y.-C. Wang, P.-F. Zhu, H. Yang, L. Huang, Q.-H. Wu, M. Rauf, J.-Y. Zhang, J. Dong, K. Wang, Z.-Y. Zhou and S.-G. Sun, ChemElectroChem, 2018, 5, 1914–1921 CrossRef CAS
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS PubMed
- X. Tang, Y. Wei, W. Zhai, Y. Wu, T. Hu, K. Yuan and Y. Chen, Adv. Mater., 2023, 35, 2208942 CrossRef CAS PubMed
- Y. Zhao, Z. Zhang, L. Liu, Y. Wang, T. Wu, W. Qin, S. Liu, B. Jia, H. Wu, D. Zhang, X. Qu, G. Qi, E. P. Giannelis, M. Qin and S. Guo, J. Am. Chem. Soc., 2022, 144, 20571–20581 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |