
Active site switching on high entropy phosphides as bifunctional oxygen electrocatalysts for rechargeable/robust Zn–air battery†
Ren
He‡
ab,
Shiqi
Wang‡
c,
Linlin
Yang‡
ab,
Sharona
Horta
 d,
Yang
Ding
ef,
Chong
Di
e,
Xuesong
Zhang
g,
Ying
Xu
e,
Maria
Ibáñez
d,
Yingtang
Zhou
h,
Stefan
Mebs
i,
Holger
Dau
i,
Jan Niklas
Hausmann
*j,
Wenyi
Huo
*kl,
Prashanth W.
Menezes
*jm and
Andreu
Cabot
*an
d,
Yang
Ding
ef,
Chong
Di
e,
Xuesong
Zhang
g,
Ying
Xu
e,
Maria
Ibáñez
d,
Yingtang
Zhou
h,
Stefan
Mebs
i,
Holger
Dau
i,
Jan Niklas
Hausmann
*j,
Wenyi
Huo
*kl,
Prashanth W.
Menezes
*jm and
Andreu
Cabot
*an
aCatalonia Energy Research Institute – IREC, Sant Adria de Besòs, 08930 Barcelona, Spain. E-mail: acabot@irec.cat
bEnginyeria Electrònica i Biomèdica Facultat de Física, Universitat de Barcelona, 08028 Barcelona, Spain
cJiangsu Key Laboratory of Advanced Metallic Materials, Southeast University, Nanjing 211189, China
dInstitute of Science and Technology Austria, Am Campus 1, 3400 Klosterneuburg, Austria
eHebei Key Lab of Optic-electronic Information and Materials, College of Physics Science and Technology, Hebei University, Baoding 071002, China
fSongshan lake materials laboratory, Dongguan 523820, China
gInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus de la UAB, 08193 Bellaterra, Spain
hNational Engineering Research Center for Marine Aquaculture, Marine Science and Technology College, Zhejiang Ocean University, Zhoushan, Zhejiang Province 316004, China
iDepartment of Physics, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany
jMaterials Chemistry Group for Thin Film Catalysis – CatLab, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany. E-mail: niklas.hausmann@helmholtz-berlin.de; prashanth.menezes@mailbox.tu-berlin.de
kCollege of Mechanical and Electrical Engineering, Nanjing Forestry University, Nanjing 210037, China. E-mail: wyhuo@njfu.edu.cn
lNOMATEN Centre of Excellence, National Centre for Nuclear Research, Otwock 05-400, Poland
mDepartment of Chemistry, Technische Universität Berlin, Straße des 17 Juni 135, Sekr. C2, 10623 Berlin, Germany
nICREA, Pg. Lluis Companys 23, 08010 Barcelona, Catalonia, Spain
First published on 22nd August 2024
Abstract
High-entropy materials (HEMs) offer a quasi-continuous spectrum of active sites and have generated great expectations in fields such as electrocatalysis and energy storage. Despite their potential, the complex composition and associated surface phenomena of HEMs pose challenges to their rational design and development. In this context, we have synthesized FeCoNiPdWP high entropy phosphide (HEP) nanoparticles using a low-temperature colloidal method, and explored their application as bifunctional electrocatalysts for the oxygen evolution and reduction reactions (OER/ORR). Our analysis provides a detailed understanding of the individual roles and transformations of each element during OER/ORR operation. Notably, the HEPs exhibit an exceptionally low OER overpotential of 227 mV at 10 mA cm−2, attributed to the reconstructed HEP surface into a FeCoNiPdW high entropy oxyhydroxide with high oxidation states of Fe, Co, and Ni serving as the active sites. Additionally, Pd and W play crucial roles in modulating the electronic structure to optimize the adsorption energy of oxygen intermediates. For the ORR, Pd emerges as the most active component. In the reconstructed catalyst, the strong d–d orbital coupling of especially Pd, Co, and W fine-tunes ORR electron transfer pathways, delivering an ORR half-wave potential of 0.81 V with a pure four-electron reduction mechanism. The practicality of these HEPs catalysts is showcased through the assembly of aqueous zinc–air batteries. These batteries demonstrate a superior specific capacity of 886 mA h gZn−1 and maintain excellent stability over more than 700 hours of continuous operation. Overall, this study not only elucidates the role of each element in HEMs but also establishes a foundational framework for the design and development of next-generation bifunctional oxygen catalysts, broadening the potential applications of these complex materials in advanced energy systems.
Broader contextRechargeable zinc–oxygen batteries represent a cutting-edge technology with promising applications ranging from large-scale stationary energy storage for renewable energy integration to powering electric vehicles and small-scale wearable devices. However, their performance is limited by the slow kinetics of oxygen redox reactions at the oxygen cathode, which significantly restricts their practical utility. To address these challenges, there is a pressing need for a new generation of oxygen redox catalysts. High entropy materials are emerging as powerful bifunctional oxygen electrocatalysts due to their quasi-continuous distribution of surface energy levels and the extensive flexibility they offer in tuning surface compositions. These properties are crucial for optimizing the adsorption energies of reactants, intermediates, and products involved in the redox processes. In this study, we present a solution-based synthesis route for producing FeCoNiPdWP nanoparticles, and demonstrate them to show outstanding combined performance in both oxygen reduction and evolution reactions. Besides, we have conducted a thorough investigation into the specific roles that each metal element plays in the redox process. Additionally, these high entropy phosphide nanoparticles are integrated as the air cathode in rechargeable zinc–air batteries, resulting in devices that exhibit unprecedented performance and stability. |
1. Introduction
Rechargeable aqueous zinc–air batteries (ZABs) are recognized for their exceptional energy densities, inherent safety, sustainability, and potential for cost-effectiveness across various applications, including electric vehicles and large-scale stationary energy storage systems.1 However, the performance of ZABs strongly relies on the activity, stability, and cost of the oxygen catalyst used at the cathode side to boost the oxygen evolution and reduction reactions (OER, ORR) during battery charging and discharging, respectively.2–4For decades, significant efforts have been directed toward developing high-performance bifunctional oxygen electrocatalysts. Traditionally, a physical mixture of noble metal catalysts, e.g. Pt/C for ORR and Ir/C for OER, has been used to accelerate the kinetics of the two reactions. Despite their relative effectiveness, the high cost, the scarcity of these noble metals, and the need to double the catalyst amount—since each material facilitates only one type of reaction—pose substantial barriers to the commercial viability of ZABs.5–8 To address this limitation, alternative bifunctional oxygen catalysts based on layered double hydroxides,9 perovskites,10 metallic alloys,11 sulfides,12 nitrides,13 phosphides,14 and their composites15–17 are intensively investigated.
Electrocatalysts based on high-entropy materials (HEMs) have emerged as particularly promising due to their vast compositional versatility and the ability to finely tune their surface properties to optimize performance. Their complex composition and a huge amount of different potential surface configurations also result in quasi-continuous surface energy levels able to suit the adsorption of reactants, intermediates, and products, facilitating enhanced catalytic activities across various reactions.18–23 The diverse range of surface sites provided by HEMs makes them ideal for catalyzing complex reactions and especially valuable as bifunctional catalysts. Despite these advantages, achieving a single HEM that excels in both OER and ORR remains a formidable challenge, primarily due to the divergent pathways and distinct intermediate adsorption energies required by these reactions.24,25
The vast range of potential HEM compositions makes trial-and-error processes hopeless to optimize them as dual-functional catalysts. Thus, to avoid a futile odyssey across the boundless expanse of HEMs, an educated selection of the HEMs characteristics leading to optimized catalytic performance is fundamental. This rational design must be supported by reliable structure–activity relationships and precise information on the distinct roles of each element in the OER and ORR mechanisms. However, gathering this information is especially difficult for HEMs having hundreds of thousands of possible atomic configurations. Besides, further complications arise with inevitable surface reconstructions caused by the strongly alkaline reaction conditions used.26,27 Therefore, it is essential to delve into the catalyst reconstruction and explore the unique contributions of each metal involved. This approach is crucial for a holistic comprehension of the intricate dynamics at play within HEMs during catalysis.
Herein, FeCoNiPdWP high entropy phosphide (HEP) nanoparticles are synthesized using a mild colloidal method. Subsequently, their OER and ORR performance is compared with a series of control phosphides and commercial reference catalysts. To gain deeper insights into the oxygen redox reaction mechanisms, the distinct roles played by each metal in HEPs are analyzed using electrochemical measurements, X-ray absorption spectroscopy (XAS), and density functional theory (DFT) calculations. Furthermore, the HEP surface reconstruction and the real active species are explored and identified. Last, rechargeable ZABs based on FeCoNiPdWP HEPs bifunctional oxygen catalysts as the air cathodes are assembled and tested.
2. Results and discussion
2.1 Design principles of HEP
A primary challenge in developing bifunctional catalysts for OER/ORR is ensuring the availability of proper catalytic sites. As illustrated in Scheme 1, an effective catalyst typically provides surface sites that strongly adsorb reactants and intermediates but only weakly bind to the products, thereby promoting high efficiency in driving the forward reaction. In contrast, this type of catalyst often shows much lower efficiency for the reverse reaction. The opposite is true for catalysts that favor strong adsorption of the products. HEMs offer a promising solution to this challenge. These materials combine a vast array of different active sites, providing a continuum of adsorption energies. This unique feature enables HEMs to efficiently catalyze both OER and ORR, making them highly effective as bifunctional oxygen catalysts.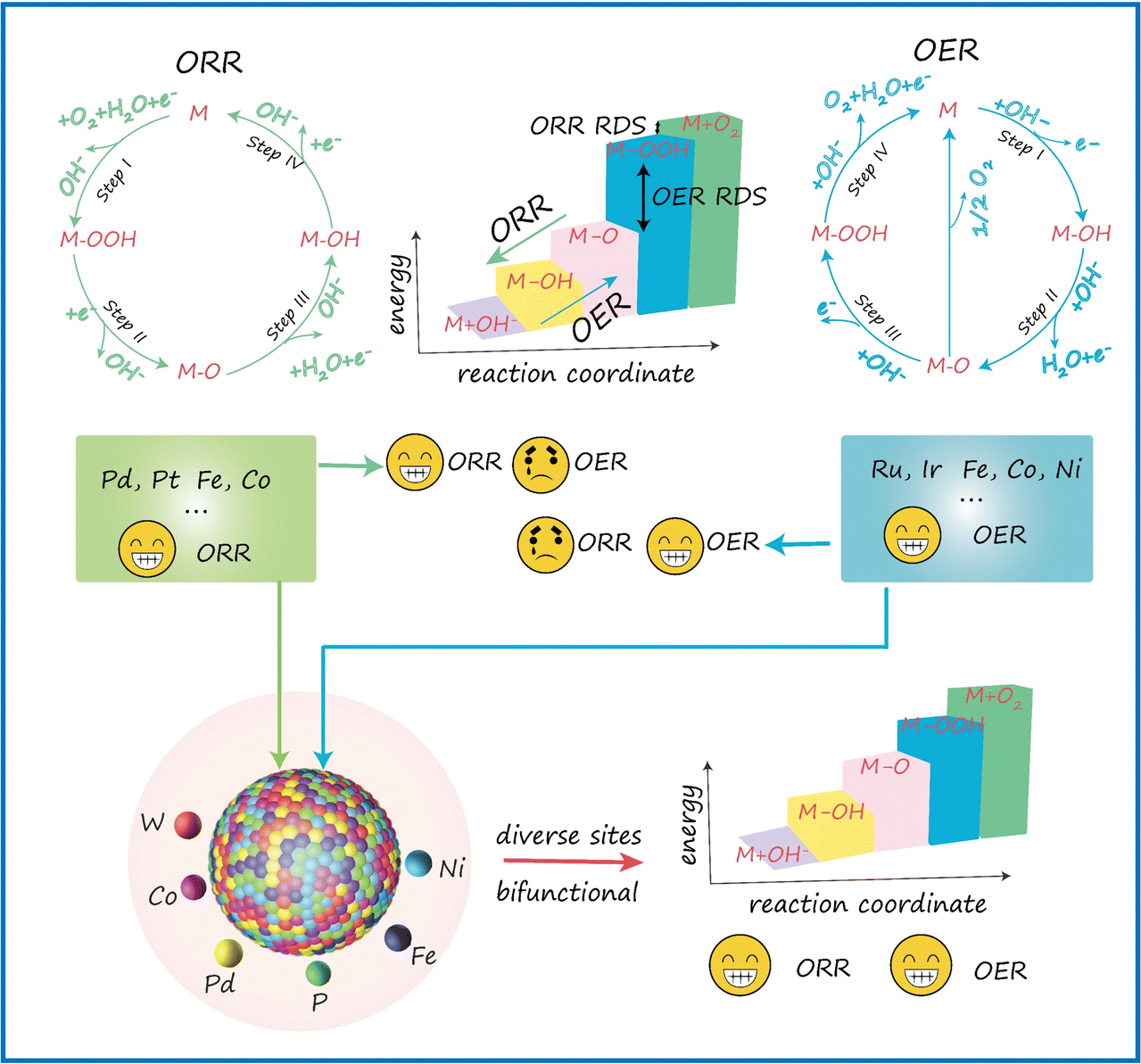 | ||
| Scheme 1 Schematic illustration of the design principles of FeCoNiPdWP HEPs as bifunctional oxygen catalysts in an alkaline electrolyte. | ||
Transition metal phosphides have attracted substantial attention due to their metallic character and associated high electrical conductivity, as well as their abundant reserves, cost-effectiveness, and stability.28–32 The higher electronegativity of phosphorus than transition metals triggers electron delocalization and leads to partially positively charged metal and negatively charged phosphorus, where transition metals could serve as the OH− acceptor to modulate the binding ability for OER/ORR. From the electronic band structure perspective, the presence of phosphorous modifies the electronic energy levels and d-band center, increasing the density-of-states near the Fermi level,33 which signifies abundant charge carriers similar to the noble metals for the OER/ORR process, thus frequently improving electrocatalytic performance.34–36 In addition, the covalent nature of the metal–phosphorus bond promotes the durability and long-term stability of the electrocatalysts. Indeed, the hybridization of d metal orbitals with the p orbital of P to form metal–phosphorus bonds has been demonstrated to be beneficial in boosting ORR performance.30 Therefore, the rational design and engineering of HEPs for ZAB applications is a worthy endeavor.
Based on our previous study, the combination of 3d/4d/5d metals significantly modulates the electronic state and surface electron density, altering the bond strength between the metal and oxygen species, which allows adjusting the adsorption/desorption ability of intermediates and products.18,37,38 The d orbitals of 5d metals are more extended and have higher energy and spatial coverage compared to those of 3d/4d metals. When 5d metals combine with 3d metals, hybrid orbitals are formed, adjusting the electron distribution and orbital overlap, thereby potentially providing more active sites and enhancing catalytic activity. The high electronegativity of the 5d metals allows for the regulation of the electron-filling states of the 3d/4d metals, optimizing the surface electronic structure of the catalysts. In addition, 5d metals provide strong electronic attraction and create new coordination environments, stabilizing reaction intermediates, and reducing poisoning. Thus, W, a 5d metal with a unique orbital filling state was incorporated into the HEP catalyst.
In terms of the 3d transition metals, Fe, Co, and Ni were selected as well-established effective OER active sites based on their cost-effectiveness, the theoretically calculated volcano graph curves between overpotential and intrinsic catalytic activity,39–43 and their rich valence state transitions, abundant electronic configuration, and adjustable spin state, which could effectively regulate the binding energy and modulate the spin flipping of the oxygen intermediates.44,45 On the other hand, while a large number of excellent ORR catalysts have been reported, the most effective catalysts are still based on Pt and Pd. Therefore, Pd was incorporated into the HEP to promote ORR performance.46,47 Besides, Pd easily forms alloys with other transition metals,48 facilitating the HEP formation. Notice also that within an HEM, the amount of Pd is minimized compared with catalysts based on elemental Pd or binary Pd-based alloys. Overall, FeCoNiPdWP HEPs are expected to simultaneously provide superior OER and ORR performance with expectable W-modulated Fe/Co/Ni as the OER active sites and Pd as the ORR active sites, making them a potentially ideal cathode material in ZABs.
2.2 HEP synthesis and characterization
FeCoNiPdWP HEPs and the related reference quaternary metal phosphides (FeCoNiPdP, FeCoPdWP, CoNiPdWP, FeNiPdWP, and FeCoNiWP) were synthesized using a heating-up colloidal synthesis approach (Fig. 1a) using a mixture of 1-octadecene and oleylamine as the solvent (see details in the experimental section in the ESI†). Triphenyl phosphite was selected as a low-cost, safe, and stable phosphorus source.49–51 Metal acetylacetonates and carbonyls were used as metal precursors. The reaction temperature was set to 290 °C. At this temperature, the phosphorus–oxygen bonds in triphenyl phosphite molecules are cleaved, releasing phosphorous atoms that react with the metal precursors to yield the HEP. Fig. S1–S6 (ESI†) show the results from the characterisation of the reference quaternary metal phosphides.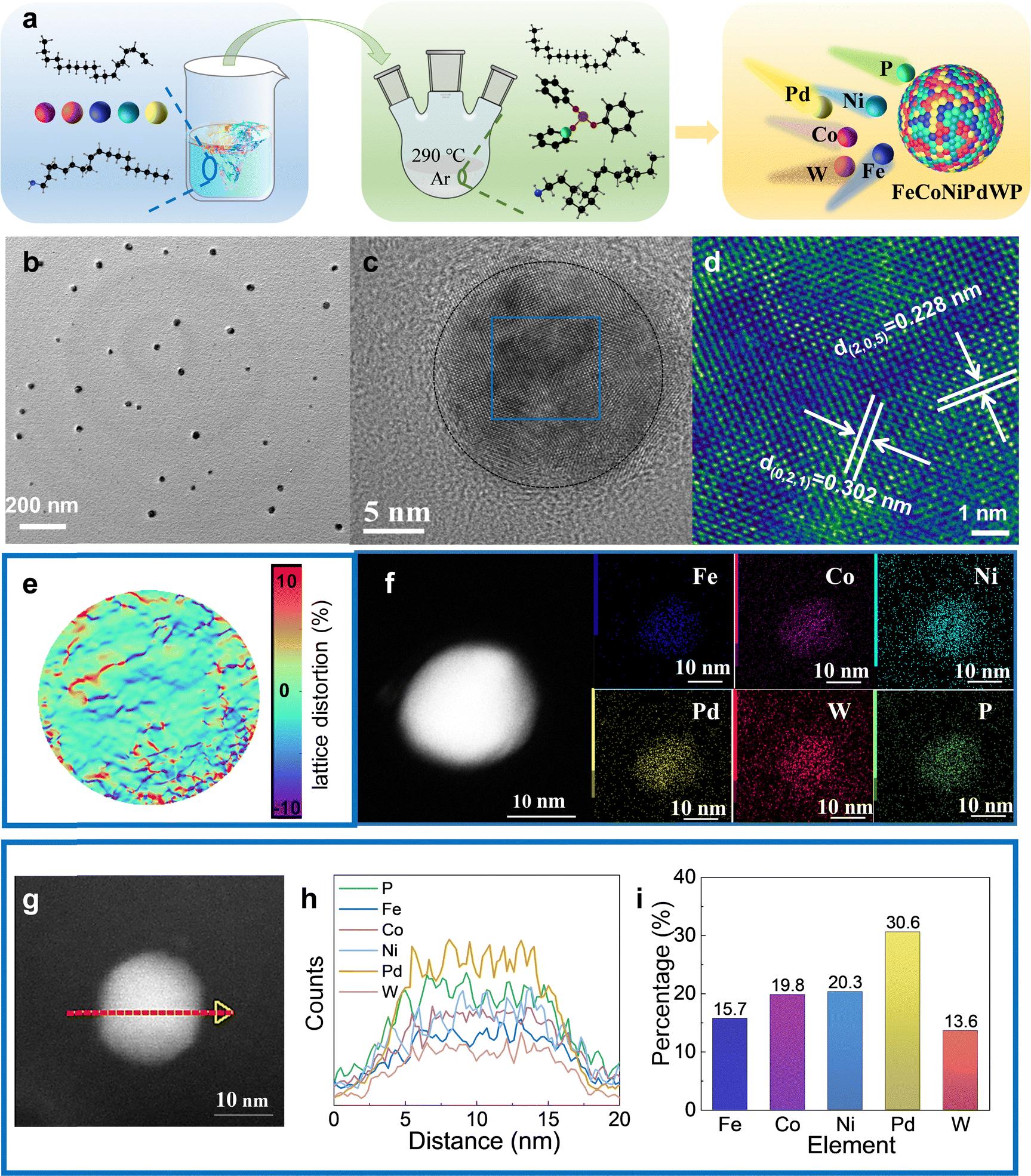 | ||
| Fig. 1 Chemical and structural characterization of FeCoNiPdWP nanoparticles. (a) Scheme of the synthesis approach used to produce FeCoNiPdWP HEPs. (b) TEM image. (c) AC-HRTEM image. (d) Magnified AC-HRTEM image and lattice distance analysis of the blue square in (c). Two crystal planes with lattice distances of 0.228 and 0.302 nm are indexed to the (205) and (021) crystal planes of the Pd15P2 rhombohedral phase, respectively. (e) GPA analysis. (f) HAADF-STEM micrograph and EDS compositional maps. (g) HAADF-STEM micrograph and (h) line scanning profiles. (i) Metal ratios obtained using ICP-OES. | ||
Transmission electron microscope (TEM) images show the FeCoNiPdWP HEP nanoparticles to have an average size of 20 nm (Fig. 1b). Spherical aberration-corrected high-resolution TEM (AC-HRTEM) analysis confirmed their high crystallinity (Fig. 1c and d). X-Ray diffraction (XRD) pattern determined the particles to have the Pd15P2 rhombohedral phase (JCPDS 01-071-0193) (Fig. S6a, ESI†). Geometric phase analysis (GPA) shows a random distribution of distortions induced by tensile and compressive stresses attributed to the different atomic radii and electronegativity of the constituent elements (Fig. 1e). High-angle annular dark field scanning TEM (HAADF-STEM) micrographs combined with energy-dispersive X-ray spectroscopy (EDS) compositional maps show a homogeneous distribution of the different elements within the HEP nanoparticles (Fig. 1f–h). The TEM-EDS metal ratios are consistent with those obtained from inductively coupled plasma optical emission spectroscopy (ICP-OES): Fe/Co/Ni/Pd/W = 15.7/19.8/20.3/30.6/13.6 (Fig. 1i).
2.3 OER
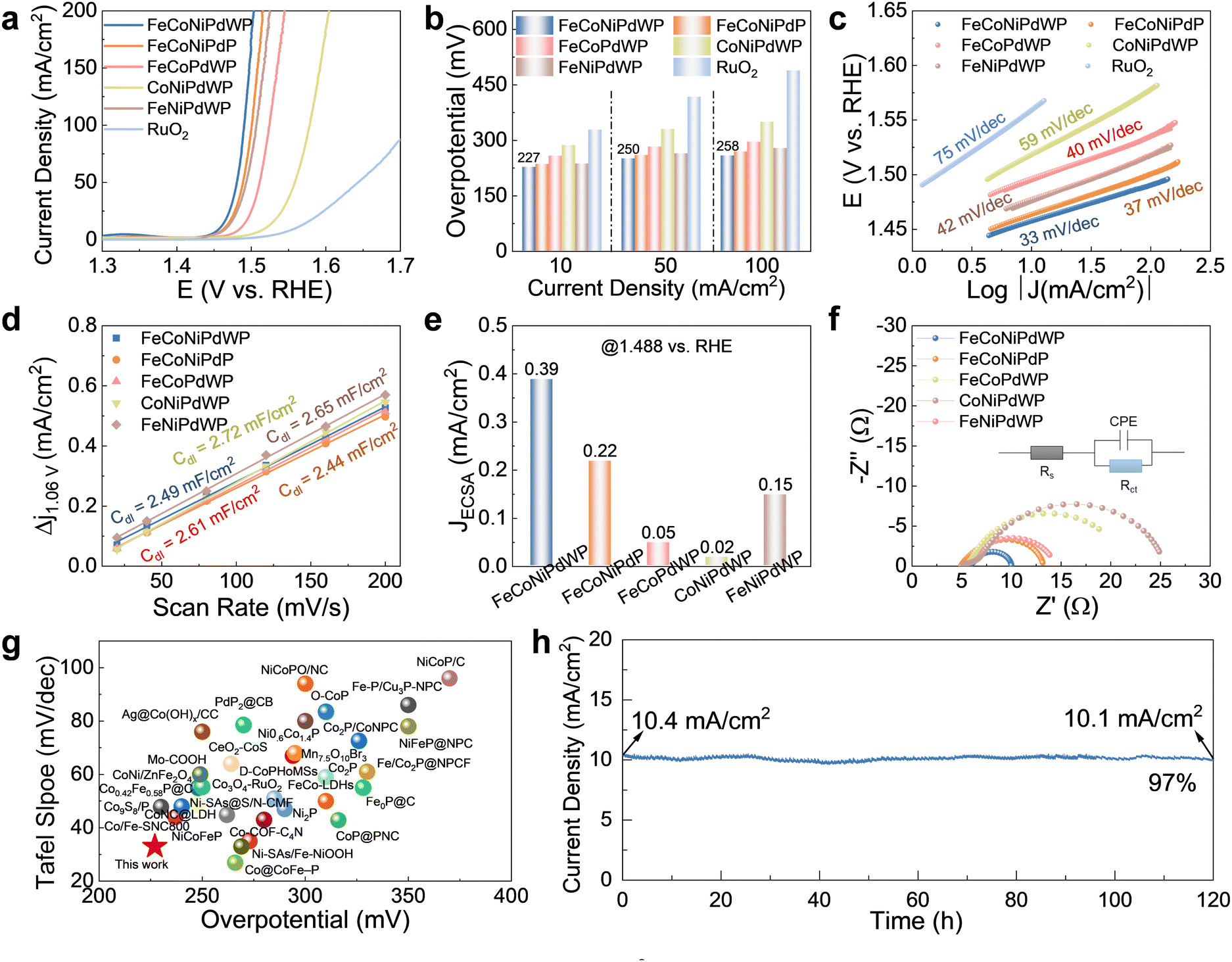 | ||
| Fig. 2 OER performance on GC electrodes with a loading of 0.5 mg cm−2. (a) LSV curves at 5 mV s−1. (b) Overpotential at different current densities. (c) Tafel fitting plots calculated from the LSV curves in (a), the open circles are measured data and the long solid lines are fitted data. (d) Cdl values calculated from the CV curves in Fig. S9 (ESI†). (e) Current density normalized by ECSA at 1.488 vs. RHE. (f) Nyquist plot of the EIS spectra of the different catalysts at 1.5 V vs. RHE. The inset shows the equivalent circuit model used to fit the experimental data (Table S1, ESI†), where Rs is the internal resistance of the cell, Rct is the charge-transfer resistance, and CPE represents the double-layer capacitance at the electrode–electrolyte interphase. (g) Comparisons of the OER performance of FeCoNiPdWP with other recently reported catalysts on GC. (h) CA curve of FeCoNiPdWP at 1.46 V vs. RHE. | ||
FeCoNiPdWP also shows the smallest Tafel slope (33 mV dec−1), well below that of the other metal phosphides (37–59 mV dec−1) and RuO2 (75 mV dec−1), demonstrating especially favorable OER kinetics (Fig. 2c). Cyclic voltammetry (CV) curves at different scan rates (Fig. S9, ESI†) and the derived double-layer capacitance (Cdl) values (Fig. 2d) show that all the quaternary and quinary phosphide catalysts have a similar electrochemical active surface area (ECSA), higher than that of PdPx (Fig. S10, ESI†). Normalization of the OER current densities by the ECSA values implies that FeCoNiPdWP has a higher intrinsic activity per active site (Fig. 2e). Electrochemical impedance spectroscopy (EIS) analysis confirms FeCoNiPdWP to display the smallest OER charge transfer impedance thus the most favorable reaction kinetics (Fig. 2f and Table S1, ESI†).
Overall, FeCoNiPdWP HEPs show superior OER performance when compared not only with the reference materials tested here but also with previously reported catalysts supported on GC, as shown in Fig. 2g and Table S2 (ESI†).4,52–84 Besides, FeCoNiPdWP shows excellent stability with a continuously applied potential of 1.46 V vs. RHE for 120 h resulting in just a 3% decrease in current density as determined by chronoamperometry (CA) measurements (Fig. 2h), well below the current density decay obtained from the quaternary metal phosphides (Fig. S11, ESI†)
XRD analysis of FeCoNiPdWP after the OER stability test revealed that the crystalline Pd15P2 rhombohedral phase disappeared during OER operation (Fig. S14, ESI†). Besides, EDS spectra (Fig. S15 and S16, ESI†) show that the P amount strongly decreased from 29% to 3% after OER. In situ Raman spectroscopy was used to explore the evolution of HEP with the applied potential (Fig. S17, ESI†). The spectra show Raman peaks at around 300 cm−1, and 400–700 cm−1, whose intensity increases with the applied voltage. This observation is related to the formation of layered Fe/Co/Ni (oxy)hydroxides.87,88 Thus, overall, XPS, XRD, and Raman results are consistent with the HEP being reconstructed into a high entropy oxyhydroxide (HEOOH) in OER conditions.
XAS on the Fe, Co, Ni K-edge was used to gain an understanding of the FeCoNiPdWP atomic structure and the reconstruction process taking place during OER for the as-prepared HEP nanoparticles, the fresh electrode, and the electrode with the completed reconstruction process. X-ray absorption near edge spectroscopy (XANES) analysis of the reconstructed samples revealed significant increases in the oxidation states of the metals during OER operation, with Fe increasing from 3.0 to 3.1, Co from 1.4 to 3.3, and Ni from 2.0 to 3.8 (Fig. 3a–f and Table S3, ESI†). Fe, Co, and Ni in oxidation states above three have been shown to facilitate water oxidation.89–94 Thus, the high Fe, Co, and Ni average oxidation states during the OER support the hypothesis that these three elements or their multimetallic neighbouring sites function as OER active sites.
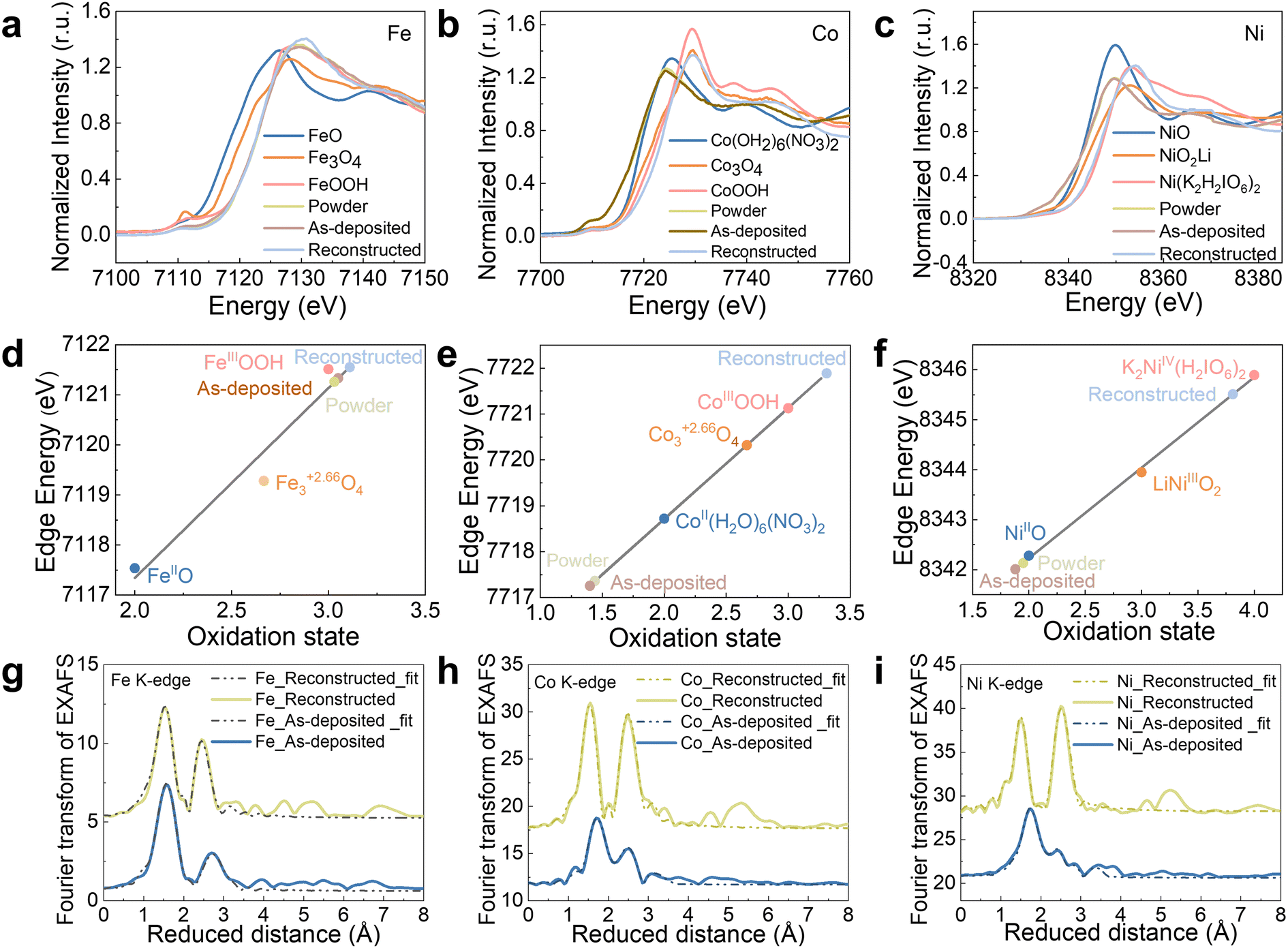 | ||
| Fig. 3 XAS characterization of HEPs. (a)–(c) Fe (a), Co (b), and Ni (c) XANES plots of the as-prepared powders, the as-deposited films, and reconstructed films, freeze-quenched at 1.53 V vs. RHE after 2 h OER operation. (d)–(f) Metal oxidation states of Fe (d), Co (e), and Ni (f) from the XANES. To quantify differences in the edge positions, the K-edge energy was calculated using the integral method,95 and the obtained values are plotted here as symbols. A linear regression including the three iron/cobalt/nickel oxide references was used to derive the oxidation states of the HEPs before and during the OER. (g)–(i) Fe (g), Co (h), and Ni (i) EXAFS spectra (Fig. S18, ESI†) and simulations (Table S4 for the simulation parameters, ESI†). The data at the bottom/up show the as-prepared/reconstructed electrodes. | ||
To gain insight into the local structure of Fe/Co/Ni, the extended X-ray absorption fine structure (EXAFS) data and k3-weighted EXAFS spectra were simulated, as shown in Fig. 3g–i and Fig. S18 (ESI†). For all the 3d metals, models of layers with edge-sharing [MO6] octahedra were applied to simulate the EXAFS spectra (see Table S4 for fitting parameters, ESI†). The model comprises three shells: (i) for the M–O bond of the [MO6] octahedra (1.87–2.02 Å), (ii) for the M–M distance of two adjacent [MO6] octahedra (2.81–2.98 Å), and (iii) a second, larger M–O distance (3.40–3.43 Å). This model is consistent with the FeCoNi oxyhydroxide structure or their deprotonated, oxidized, and potassium-intercalated relatives that have recently been proposed to form during OER.94 For Fe, two M–O bond distances (1.89 and 2.02 Å) were required, likely due to the presence of a Jahn–Teller distortion caused by the unevenly occupied t2g orbital of [FeIIIO6]. The presence of two different M–O bond lengths automatically leads to two different Fe–M bond distances as well, which were taken into account in the simulation. The short M–O bond distances (1.87–1.89 Å) are consistent with the high oxidation states extracted from the XANES data (Table S4, ESI†). Overall, results from XAS analyses reveal that, during OER, FeCoNiPdWP is fully reconstructed into layers of edge-sharing [MO6] octahedra with 3d metal oxidation states above three. These motifs are consistent with highly oxidized oxyhydroxides and related structures that are frequently observed during and after the OER.94,96–98
 | ||
| Fig. 4 DFT calculation results. (a) TDOS of FeCoNiPdWOOH, FeCoNiPdOOH, FeCoNiWOOH, FeCoPdWOOH, FeNiPdWOOH, and CoNiPdWOOH. (b) PDOS of FeCoNiPdWOOH. (c) Electronic density of states of the d-band for the metal atoms in various models. (d) PDOS for OH adsorption at various active sites. (e) COHP bonding analysis of M–O interactions (M in the surface site over FeCoNiPdWOOH and O in adsorbed OH). (f) Standard free energy diagram of the OER process at 0 V of FeCoNiPdWOOH for various active sites. (g) PDOS of key intermediates over Ni sites in FeCoNiPdWOOH. | ||
PDOS analysis was used to evaluate the adsorption behaviors over different sites. For the reactant OH− species, during the initial adsorption process, s/p bands were down-shifted (Fig. 4d), indicating electron transfer from the surface sites to OH− to stabilize adsorbed *OH. The improved adsorption capability favored the weakening of O–H bonds in *OH, thus triggering the subsequent dissociation processes. Compared with Fe, Co, and W sites, stronger p–d orbital couplings between the OH* and Ni/Pd sites were observed, which points to a more stable adsorption of OH* on the Ni/Pd sites within FeCoNiPdWOOH. This was experimentally demonstrated by the methanol probe molecule test of FeCoNiPdWP and FeCoNiWP in 1 M KOH and 1 M methanol, concluding that the presence of Pd significantly promoted the adsorption of OH− species (Fig. S26, ESI†). CDD results also showed that Ni sites presented noticeable electron depletion with OH− adsorption, demonstrating charge transfer during the adsorbing process (Fig. S27, ESI†). The integral projected crystal orbital Hamilton population value (ICOHP) provides the number of bonded electrons between the selected metal and O atoms in the active *OH and the corresponding bonding strength. Notably, the ICOHPs of Ni and Pd are larger than those of Fe/Co/W, demonstrating stronger OH adsorption in the formers (Fig. 4e).
Gibbs free energies were calculated considering various potential metal active sites to reveal the energy barriers of adsorption/desorption of key intermediates. As shown in Fig. 4f, the energy barriers (ΔG) for each step among various active sites were quite different. For the Fe/Co/Ni sites in the FeCoNiPdWOOH model, the third step (formation of *OOH from *O with OH− in the electrolyte) exhibited the largest barrier. On the other hand, for Pd and W sites, the ΔG values slightly increased at the second step and first step, respectively. Accordingly, Fe/Co/Ni active sites exhibited lower overpotentials than those of Pd and W sites, respectively, under the equilibrium potential (1.23 eV, Fig. S28, ESI†). Thus, in FeCoNiPdWOOH, activity was associated with Fe/Co/Ni active sites, and the third reaction step was the rate-determining step (RDS) during the four-electron transfer process. To visualize the electronic environment during the reaction coordinates of OER, the PDOS of the most stable binding of key intermediates (*OH, *O, *OOH, and *O2) is plotted in Fig. 4g. The σ components of O-2p orbitals facilitated the electron transfer of sequential adsorbed intermediates. From the initial OH species to the final product O2, the consecutive upshifting of the σ orbitals demonstrates efficient electron transfer with low barriers for intermediate transformation, which is consistent with the highly efficient OER reaction trend.
Notably, all of the Fe, Co, and Ni sites on the FeCoNiPdWOOH model showed smaller energy barriers than within the other models, demonstrating the significant effect of all the elements in the reconstructed sample (Fig. S29, ESI†). Consequently, Fe/Co/Ni are identified as the main active sites for OER, which is consistent with the experimental results. In particular, the strong and positive effect of Pd was further demonstrated by DFT calculations over FeCoNiPdWOOH and Pd-free FeCoNiWOOH models. Pd benefits the electron transfer (Fig. S30 and S31, ESI†), optimization of adsorption configuration (Fig. S32, ESI†), and electron interaction between the oxygen intermediates (Fig. S33, ESI†). Overall, the synergistic effect of the multiple metal atoms, materialized through an efficient p–d orbital coupling and moderate d-band center, optimized the adsorption/desorption of OER intermediates to promote OER kinetics.
In summary, the FeCoNiPdWOOH reconstructed from FeCoNiPdW HEP is highly active towards OER. In the reconstructed phase, Ni and Pd are favorable for the initial hydroxide adsorption, facilitating the subsequent electron transfer reaction steps. Subsequently, Fe/Co/Ni as the main active sites having favorable OER reaction intermediate adsorption energies, due to their electronic modification by Pd and W. Furthermore, XAS revealed unusually high oxidation states of Fe (+3.1), Co (+3.3), and Ni (+3.8) during the OER, which is crucial to promote the OER reaction.90–94
2.4 ORR
 | ||
| Fig. 5 ORR performance. (a) CV curves measured in Ar and O2 saturated electrolytes of FeCoNiPdWP. (b) LSV curves of FeCoNiPdWP at different rotation speeds in the range 400–2500 rpm. (c) LSV curves at 1600 rpm for different metal phosphides and Pd/C catalysts. (d) Half-wave potential comparison between metal phosphides and Pd/C catalysts prepared in this work and recently reported state-of-the-art ORR catalysts. (e) Stability of FeCoNiPdWP HEPs during 5000 cycles. | ||
An ORR electron transfer number (n) over 3.9 is obtained for FeCoNiPdWP, CoNiPdWP, FeCoPdWP, PdPx, and Pd/C, implying a 4 electron pathway (Fig. S46c, S47c and S48a, ESI†) at 0.5 V vs. RHE, where a stable limiting diffusion current density is measured. In contrast, FeCoNiPdP, FeNiPdWP, and FeCoNiWP are characterized by smaller n values, in the range 3.6–3.8, indicating the coexistence of the 4 electron and 2 electron pathways. The hydrogen peroxide (H2O2) yields further confirm the pathway assignment (Fig. S46d, S47d and S48b, ESI†). These results point at Pd, Co, and W as collectively assuming a more fundamental role in fine-tuning the reaction pathway, thereby amplifying the ORR performance of FeCoNiPdWP HEPs compared with the quaternary metal phosphides.
DFT calculations were further used to gain an understanding of the catalytic mechanism and element roles in the reconstructed HEPs during ORR. The reconstructed HEP model was built considering the experimental results, i.e. a HEP core with a ∼2 nm shell rich in Fe, Co, and Ni in a hydroxide environment and Pd and W in a phosphide environment. We denote this reconstructed HEP as FeCoNiPdWPOH (Fig. S49, ESI†). In the PDOS spectra, the good overlap between different metal elements-d orbitals and with the O-p orbitals implies possible p–p/p–d/d–d orbital coupling, which might further promote electron transfer efficiency (Fig. S50, ESI†).
The site-dependent PDOS of individual metal components located within or at the surface of the FeCoNiPdWPOH was also taken into consideration to appreciate their roles in the enhancement of ORR activity (Fig. 6a). From the reconstructed FeCoNiPdWPOH interior to the surface, the eg–t2g splitting of Fe-3d orbitals gradually decreased. The alleviated eg–t2g splitting could lead to higher valence states over the surface but also reduce the energy barrier of electron transfer from the electrocatalysts to the adsorbates to improve the ORR performance. Co sites displayed a site-independent electronic structure from the interior to the surface, which could endow the stable adsorption of intermediates and initial electroactive features during the ORR (Fig. 6b). On the other hand, the Ni-3d and Pd-4d bands gradually approach the EF from FeCoNiPdWPOH interior to the surface, suggesting improved electroactivity of the surface metal sites (Fig. 6c and d). In contrast, the W-5d orbitals demonstrated an upshifting trend away from EF, leading to decreasing valence states (Fig. 6e). This compensation of the PDOS trend, facilitated by the W-5d orbitals, contributes to the high electroactivity of the Pd/Ni/Co/Fe surface sites.
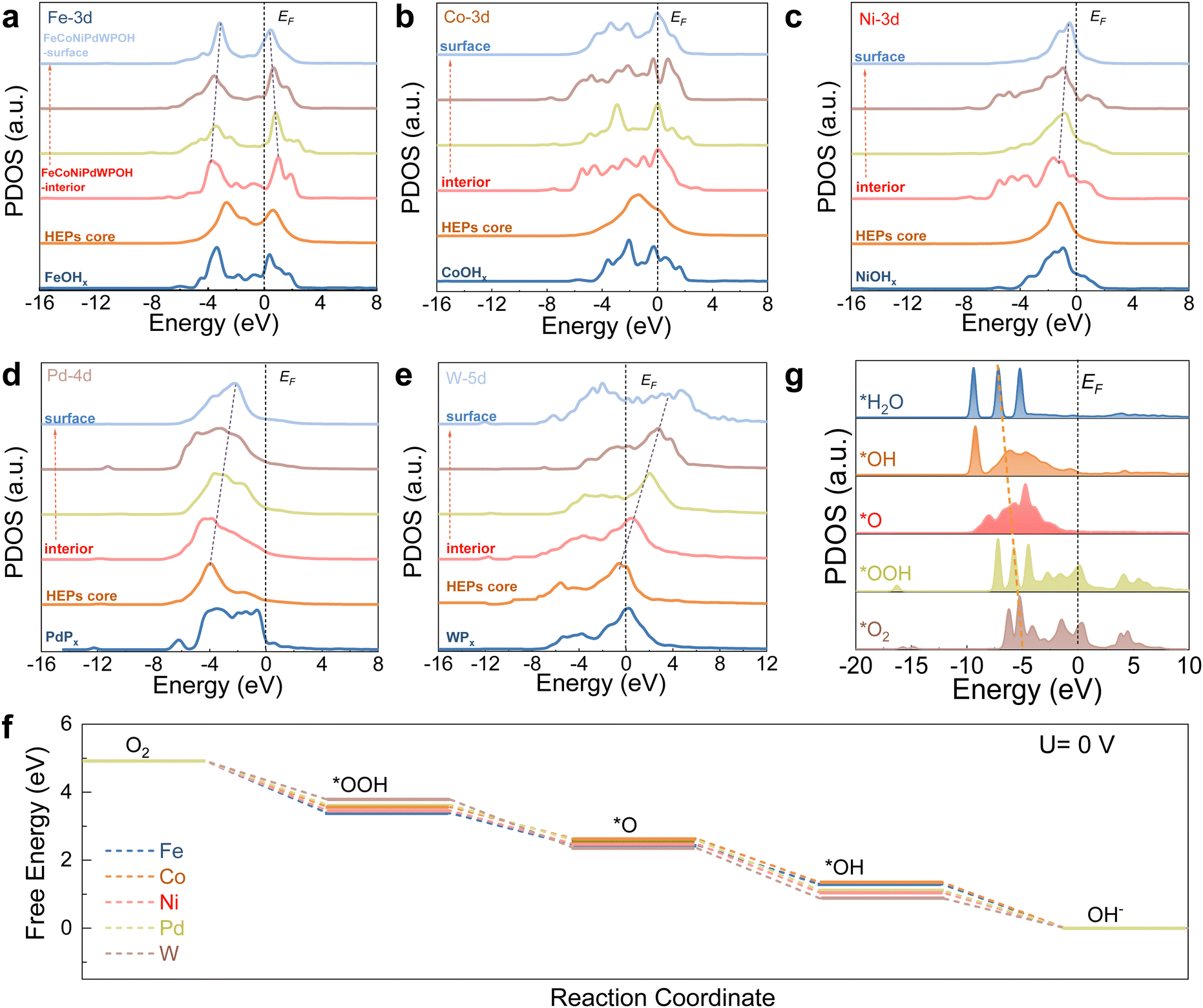 | ||
| Fig. 6 DFT calculations for ORR. (a)–(e) Site-dependent PDOSs of Fe, Co, Ni, Pd, and W sites in the FeCoNiPdWPOH structure. (f) Standard free energy diagram of the ORR process at 0 V of various active sites on the surface-reconstructed samples. (g) PDOS of key intermediates over Pd sites in FeCoNiPdWPOH. | ||
The ORR Gibbs energy barrier at various metal sites in FeCoNiPdWPOH was also calculated. As shown in Fig. 6f and Fig. S51 (ESI†), notably, among all of the various metal sites, Pd sites displayed the smallest energy barrier with just a 0.20 eV of the RDS for the conversion from *OOH to O*. Thus, Pd sites show the most thermodynamically favorable process, which is consistent with the experimental results. In addition, the PDOS of key ORR intermediates displayed a gradual downshifting of the O-p orbitals, indicating the reduction trends and highly efficient intermediate conversions for FeCoNiPdWPOH (Fig. 6g).
O2 adsorption results in strong local charge redistribution over the Pd active sites, causing electron migration from the surface to the adsorbed *O2 species (Fig. S52, ESI†). The PDOS for adsorbed O2 is down-shifted compared with that of the free O2 (Fig. S53, ESI†), further revealing the strong interaction between the Pd sites and O2. Besides, the final product H2O could easily escape from the surface of FeCoNiPdWPOH (Fig. S54, ESI†). The adsorption and desorption ability of the oxygen intermediates for Pd sites can be modulated by the introduction of other metals, as determined by the optimum dc values (Fig. S55, ESI†).
Overall, FeCoNiPdWP shows excellent ORR performance with large limiting current density, high half-wave potentials, four electron-transfer pathways, and long-term cycling performance. In FeCoNiPdWP, Pd was found to be the most active element among the five metals within the reconstructed FeCoNiPdWPOH leading to the excellent ORR catalytic performance. The strong d–d orbital coupling of different elements, especially Pd, Co, and W, fine-tunes ORR electron transfer pathways and the surface electron modulation of Pd with other metal elements decreases the energy barrier during the ORR.
2.5 HEP as bifunctional oxygen catalysts
A key performance parameter to evaluate the oxygen bifunctional catalytic performance is the potential gap (Egap) between the OER overpotential at 10 mA cm−2 (Ej = 10) and the ORR half-wave potential (E1/2). A smaller Egap value implies a better bifunctional performance, thus enabling ZABs with higher charge/discharge energy efficiency. Due to the excellent OER and ORR performance, the reconstructed FeCoNiPdWP shows an extremely low Egap value of 0.65 V in 0.1 M KOH, much lower than the combination of the benchmark OER and ORR catalysts, Pd/C and RuO2, resulting in an Egap of 0.75 V (Fig. 7a). Besides, the FeCoNiPdWP exhibits an outstanding low bifunctional Egap compared with state-of-the-art bifunctional oxygen catalysts, as shown in Fig. 7b and Table S6 (ESI†).4,52,54,56,78,108,109,111,112,117–121 The remarkable dual functionality of the reconstructed FeCoNiPdWP in both OER and ORR can be attributed to the distinct contributions and cooperative interactions of each metal involved. While OER and ORR typically necessitate varying strengths in binding with the oxygen intermediates, the distinctive roles played by each element within the reconstructed FeCoNiPdWP transcend these conventional constraints. In the OER process, Fe, Co, and Ni emerge as the primary active sites, whereas in ORR, Pd takes on the leading role, complemented by the involvement of Co and W in modulating the electron transfer pathway. The different metal elements play distinctive roles in OER and ORR, respectively, and their synergistic interaction maximizes the performance of the resulting HEM. | ||
| Fig. 7 Oxygen bifunctional properties and ZAB performance. (a) ORR/OER bifunctional LSV curves of different electrodes. (b) Comparison of the Egap values of FeCoNiPdWP and other state-of-the-art bifunctional oxygen catalysts. (c) ZAB schematic diagram. (d) OCP measurements, (e) power density plots, and (f) specific capacity comparisons between a FeCoNiPdWP- and a Pt/C&RuO2-based ZAB. Photograph of a red LED screen powered by a FeCoNiPdWP-based ZAB is also shown in (d). (g) Galvanostatic discharge–charge curves with 10 min discharge and 10 min charge cycles at a current density of 8 mA cm−2 of FeCoNiPdWP- and Pt/C&RuO2-based ZABs. (h) Enlarged galvanostatic discharge–charge cycles at the specific cycles (1st, 200th, 500th, 1000th, 1500th, and 2000th) for 20 min of the FeCoNiPdWP-based ZAB. | ||
2.6 ZAB performance
FeCoNiPdWP was then used as the oxygen cathode in aqueous rechargeable ZABs. Reference ZABs were also assembled with a combination of 20 wt% Pt/C and RuO2 (Pt/C&RuO2) as the cathode. Fig. 7c shows a schematic of the ZAB architecture. As shown in Fig. 7d, the ZAB with the FeCoNiPdWP-based air cathode exhibits an open-circuit potential (OCP) of 1.60 V, which is 96.4% of the theoretical limit (1.66 V) and higher than that of the reference Pt/C&RuO2-based ZAB at 1.56 V. The high OCP values could light a red-screen LED, proving its potential for practical application. The peak power density of the FeCoNiPdWP HEPs-based ZAB is 123 mW cm−2, significantly above that of the Pt/C&RuO2-based ZAB at 111 mW cm−2 (Fig. 7e). The specific capacity and energy density of the FeCoNiPdWP-based ZAB are 886 mA h gZn−1 and 1063.2 mW h gZn−1 at a current density of 8 mA cm−2, higher than those of the Pt/C&RuO2-based ZAB at 793 mA h gZn−1 and 911.9 mW h gZn−1 (Fig. 7f). The higher specific capacity and energy density are directly attributed to the improved ORR performance of the FeCoNiPdWP-based air cathode, which is the primary factor limiting the discharge process. The FeCoNiPdWP-based ZAB not only excels in performance compared with the reference ZAB tested here but also stands out when compared with state-of-the-art ZABs (Table S7, ESI†).14,52,54,56,103,105,108,109,119,122–132The rate performance of the FeCoNiPdWP-based ZAB was further evaluated. As shown in Fig. S56 and S57 (ESI†), the FeCoNiPdWP-based ZAB exhibits stable charge–discharge curves at the current density of 2–18 mA cm−2, with an energy efficiency in the range of 70–50%.
The long-term stability of the ZABs was also measured with continuous charge/discharge cycles at a current density of 8 mA cm−2. As shown in Fig. 7g, the FeCoNiPdW-based ZAB can be stable over 700 h, i.e. 1 month, of continuous cycling (over 2000 cycles). In contrast, the Pt/C&RuO2-based ZAB is stable for just about 100 h, i.e. 300 cycles. Fig. 7h shows the enlarged galvanostatic discharge–charge cycles of FeCoNiPdWP HEPs-based ZABs at the specific cycles at 1st, 200th, 500th, 1000th, and 2000th cycles. The initial charging/discharging potential gap is 1.0 V. With continuous cycling, the potential gap is decreased to 0.97 V at 200 cycles, and it remains in the range of 0.95–0.96 V after 1000–2000 cycles, further proving its good stability.
3. Conclusion
FeCoNiPdWP HEP nanoparticles were synthesized using a mild colloidal method and tested as bifunctional OER/ORR catalysts. These HEPs demonstrated exceptional OER performance, with a low overpotential of 227 mV at 10 mA cm−2 and a Tafel slope of 33 mV dec−1. The outstanding OER performance is attributed to a reconstructed FeCoNiPdWOOH surface rich in high-oxidation-state Fe, Co, and Ni (Fe = 3.1; Co = 3.3; Ni = 3.8) as main active sites, Pd aiding in OH− adsorption, and W optimizing oxygen intermediate adsorption via electronic structure modulation. For ORR, FeCoNiPdWP HEPs show a half-wave potential of 0.81 V and efficient four-electron transfer pathways. During ORR, the surface of the HEP was also slightly enriched in Fe, Co, and Ni and reconstructed into a FeCoNiPdWPOH, with Fe/Co/Ni found as oxyhydroxides but with Pd and W maintaining the phosphide chemical environment as observed by XPS analysis. This unique FeCoNiPdWPOH surface, together with the active role of Pd and the influence of Co and W in modulating the electron transfer pathways, significantly enhances the ORR performance. Overall, each element within FeCoNiPdWP HEPs plays a crucial role in enhancing both OER and ORR performances. The combination of all these elements playing distinctive roles and the switchable active sites in redox reactions transcends the usual constraints of bifunctional catalysts, which generally excel in only one of these reactions. As a result, FeCoNiPdWP shows an extremely low Egap value of 0.65 V and FeCoNiPdWP-based ZABs exhibit outstanding performance and stability for over 700 h. This work provides a platform to understand the different surface reconstruction behaviors of HEP catalysts and the distinct element roles in OER and ORR reactions, which paves the way for designing advanced bifunctional oxygen catalysis, potentially based on HEMs.Author contributions
Ren He (data curation: lead; formal analysis: lead; investigation: lead; methodology: lead; resources: lead; validation: lead; visualization: lead; writing – original draft: lead). Shiqi Wang (data curation: equal; software: lead; visualization: equal; writing – original draft: equal). Linlin Yang (data curation: equal; formal analysis: equal; validation: equal; writing – original draft: lead). Sharona Horta (formal analysis: equal; investigation: equal; resources: equal). Yang Ding (formal analysis: supporting; investigation: supporting). Chong Di (data curation: supporting; visualization: supporting). Xuesong Zhang (data curation: supporting; formal analysis: supporting). Ying Xu (data curation: supporting; resources: supporting). Maria Ibáñez (formal analysis: supporting; resources: supporting). Yingtang Zhou (formal analysis: supporting; validation: supporting). Stefan Mebs (formal analysis: supporting; investigation: supporting). Holger Dau (formal analysis: supporting; visualization: supporting). Jan Niklas Hausmann (data curation: equal; formal analysis: equal; writing – original draft: equal). Wenyi Huo (formal analysis: equal; software: lead; writing – original draft: equal; writing – review & editing: equal; supervision: equal). Prashanth W. Menezes (data curation: equal; formal analysis: equal; resources: equal; writing – original draft: equal). Andreu Cabot (conceptualization: lead; funding acquisition: lead; project administration: lead; resources: lead; writing – review & editing: lead).Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the SyDEC at project from the Spanish MCIN/AEI/FEDER (PID2022-136883OB-C22) and Generalitat de Catalunya 2021SGR01581. J. N. H. and P. W. M. acknowledge support from the German Federal Ministry of Education and Research in the framework of the project “Catlab” (03EW0015A/B). L. Yang thanks the China Scholarship Council (CSC) for the scholarship support (202008130132). This work was supported by the European Union Horizon 2020 research and innovation program (No. 857470) and the European Regional Development Fund via the Foundation for Polish Science International Research Agenda PLUS program (No. MAB PLUS/2018/8). The publication was created within the framework of the project of the Minister of Science and Higher Education, Poland “Support for the activities of Centres of Excellence established in Poland under Horizon 2020” under contract no. MEiN/2023/DIR/3795. H. D. and S. M. thank the German Federal Ministry of Education and Research (BMBF) for supporting the Live-XAS project (05K22KE1) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for support under Germany's Excellence Strategy – EXC 2008/1 – 390540038 – UniSysCat. The authors thank the Helmholtz-Zentrum Berlin (HZB) for beamtime allocation at the KMC-3 synchrotron beamline of the BESSY synchrotron in Berlin-Adlershof and Dr Ivo Zizak as well as Dr Michael Haumann for technical support.Notes and references
- X. Zhu, C. Hu, R. Amal, L. Dai and X. Lu, Energy Environ. Sci., 2020, 13, 4536–4563 RSC.
- J. N. Liu, C. X. Zhao, D. Ren, J. Wang, R. Zhang, S. H. Wang, C. Zhao, B. Q. Li and Q. Zhang, Adv. Mater., 2022, 34, e2109407 CrossRef PubMed.
- W. Niu, Z. Li, K. Marcus, L. Zhou, Y. Li, R. Ye, K. Liang and Y. Yang, Adv. Energy Mater., 2018, 8, 1701642 CrossRef.
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, J. Yu, X. Chen, B. Q. Li and Q. Zhang, Adv. Mater., 2021, 33, e2008606 CrossRef.
- T. Binninger and M. L. Doublet, Energy Environ. Sci., 2022, 15, 2519–2528 RSC.
- J. Chen, M. Aliasgar, F. B. Zamudio, T. Zhang, Y. Zhao, X. Lian, L. Wen, H. Yang, W. Sun, S. M. Kozlov, W. Chen and L. Wang, Nat. Commun., 2023, 14, 1711 CrossRef CAS PubMed.
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Adv. Mater., 2019, 31, e1802234 CrossRef PubMed.
- J. Zhang, J. Lian, Q. Jiang and G. Wang, Chem. Eng. J., 2022, 439, 135634 CrossRef CAS.
- C. Tang, H. S. Wang, H. F. Wang, Q. Zhang, G. L. Tian, J. Q. Nie and F. Wei, Adv. Mater., 2015, 27, 4516–4522 CrossRef CAS PubMed.
- L. Gui, Z. Wang, K. Zhang, B. He, Y. Liu, W. Zhou, J. Xu, Q. Wang and L. Zhao, Appl. Catal., B, 2020, 266, 118656 CrossRef CAS.
- C. Li, M. Wu and R. Liu, Appl. Catal., B, 2019, 244, 150–158 CrossRef CAS.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef CAS PubMed.
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi, X. Kang, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1502585 CrossRef.
- J. Li, Y. Kang, Z. Lei and P. Liu, Appl. Catal., B, 2023, 321, 122029 CrossRef CAS.
- L. Yang, X. Zeng, D. Wang and D. Cao, Energy Storage Mater., 2018, 12, 277–283 CrossRef.
- B. Q. Li, C. X. Zhao, S. Chen, J. N. Liu, X. Chen, L. Song and Q. Zhang, Adv. Mater., 2019, 31, e1900592 CrossRef PubMed.
- Y. Zhang, H. Jiang, Y. Lin, H. Liu, Q. He, C. Wu, T. Duan and L. Song, Adv. Mater. Interfaces, 2018, 5, 1800392 CrossRef.
- Y. Mei, Y. Feng, C. Zhang, Y. Zhang, Q. Qi and J. Hu, ACS Catal., 2022, 12, 10808–10817 CrossRef CAS.
- J. Kwon, S. Sun, S. Choi, K. Lee, S. Jo, K. Park, Y. K. Kim, H. B. Park, H. Y. Park, J. H. Jang, H. Han, U. Paik and T. Song, Adv. Mater., 2023, 35, e2300091 CrossRef PubMed.
- H. Zhu, S. Sun, J. Hao, Z. Zhuang, S. Zhang, T. Wang, Q. Kang, S. Lu, X. Wang, F. Lai, T. Liu, G. Gao, M. Du and D. Wang, Energy Environ. Sci., 2023, 16, 619–628 RSC.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3, 834–845 CrossRef CAS.
- Y. Chida, T. Tomimori, T. Ebata, N. Taguchi, T. Ioroi, K. Hayashi, N. Todoroki and T. Wadayama, Nat. Commun., 2023, 14, 4492 CrossRef CAS PubMed.
- A. Ostovari Moghaddam, A. Cabot and E. A. Trofimov, Int. J. Refract. Met. Hard Mater., 2021, 97, 105504 CrossRef CAS.
- R. He, L. Yang, Y. Zhang, D. Jiang, S. Lee, S. Horta, Z. Liang, X. Lu, A. O. Moghaddam, J. Li, M. Ibanez, Y. Xu, Y. Zhou and A. Cabot, Adv. Mater., 2023, 35, e2303719 CrossRef PubMed.
- R. He, L. Yang, Y. Zhang, X. Wang, S. Lee, T. Zhang, L. Li, Z. Liang, J. Chen, J. Li, A. Ostovari Moghaddam, J. Llorca, M. Ibáñez, J. Arbiol, Y. Xu and A. Cabot, Energy Storage Mater., 2023, 58, 287–298 CrossRef.
- L. Yang, R. He, X. Wang, T. Yang, T. Zhang, Y. Zuo, X. Lu, Z. Liang, J. Li, J. Arbiol, P. R. Martínez-Alanis, X. Qi and A. Cabot, Nano Energy, 2023, 115, 108714 CrossRef CAS.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- J. Liu, S. Wang, K. Kravchyk, M. Ibáñez, F. Krumeich, R. Widmer, D. Nasiou, M. Meyns, J. Llorca, J. Arbiol, M. V. Kovalenko and A. Cabot, J. Mater. Chem. A, 2018, 6, 10958–10966 RSC.
- M. I. Diez-Garcia, G. Montana-Mora, M. Botifoll, A. Cabot, J. Arbiol, M. Qamar and J. R. Morante, ACS Appl. Energy Mater., 2023, 6, 5690–5699 CrossRef CAS PubMed.
- J. Liu, X. Yu, R. Du, C. Zhang, T. Zhang, J. Llorca, J. Arbiol, Y. Wang, M. Meyns and A. Cabot, Appl. Catal., B, 2019, 256, 117846 CrossRef CAS.
- X. Wang, L. Yang, C. Xing, X. Han, R. Du, R. He, P. Guardia, J. Arbiol and A. Cabot, Nanomaterials, 2022, 12, 1098 CrossRef CAS.
- J. Liu, Z. Wang, J. David, J. Llorca, J. Li, X. Yu, A. Shavel, J. Arbiol, M. Meyns and A. Cabot, J. Mater. Chem. A, 2018, 6, 11453–11462 RSC.
- N. Khodayar, A. Noori, M. S. Rahmanifar, M. Moloudi, N. Hassani, M. Neek-Amal, M. F. El-Kady, N. B. Mohamed, X. Xia, Y. Zhang, R. B. Kaner and M. F. Mousavi, Energy Environ. Sci., 2024, 17, 5200–5215 RSC.
- D. Lai, Q. Kang, F. Gao and Q. Lu, J. Mater. Chem. A, 2021, 9, 17913–17922 RSC.
- Y. Li, Z. Dong and L. Jiao, Adv. Energy Mater., 2019, 10, 1902104 CrossRef.
- Y. Shi, M. Li, Y. Yu and B. Zhang, Energy Environ. Sci., 2020, 13, 4564–4582 RSC.
- H. Li, C. Zhang, W. Xiang, M. A. Amin, J. Na, S. Wang, J. Yu and Y. Yamauchi, Chem. Eng. J., 2023, 452, 139104 CrossRef CAS.
- G. Ke, F. Duan, B. Liu, X. Liu, J. Wen, B. Jia, X. Liu, H. He and Y. Zhou, J. Alloys Compd., 2023, 934, 167883 CrossRef CAS.
- K. Kim, K. Min, Y. Go, Y. Lee, S. E. Shim, D. Lim and S.-H. Baeck, Appl. Catal., B, 2022, 315, 121501 CrossRef CAS.
- Z. Wang, J. Ang, J. Liu, X. Y. D. Ma, J. Kong, Y. Zhang, T. Yan and X. Lu, Appl. Catal., B, 2020, 263, 118344 CrossRef CAS.
- W.-W. Tian, J.-T. Ren and Z.-Y. Yuan, Appl. Catal., B, 2022, 317, 121764 CrossRef CAS.
- Z. Li, M. Chen, L. Zhang, R. Xing, J. Hu, X. Huang, C. Zhou, Y. Zhou, T. Wågberg and G. Hu, J. Mater. Chem. A, 2023, 11, 2155–2167 RSC.
- C. He, L. Yang, X. Peng, S. Liu, J. Wang, C. Dong, D. Du, L. Li, L. Bu and X. Huang, ACS Nano, 2023, 17, 5861–5870 CrossRef CAS PubMed.
- L. Yang, R. He, M. Botifoll, Y. Zhang, Y. Ding, C. Di, C. He, Y. Xu, L. Balcells, J. Arbiol, Y. Zhou and A. Cabot, Adv. Mater., 2024, 36, e2400572 CrossRef.
- L. Wu, T. Guo and T. Li, iScience, 2021, 24, 102398 CrossRef CAS PubMed.
- H. Xie, S. Chen, J. Liang, T. Wang, Z. Hou, H. L. Wang, G. Chai and Q. Li, Adv. Funct. Mater., 2021, 31, 2100883 CrossRef CAS.
- F. Lin, F. Lv, Q. Zhang, H. Luo, K. Wang, J. Zhou, W. Zhang, W. Zhang, D. Wang, L. Gu and S. Guo, Adv. Mater., 2022, 34, e2202084 CrossRef PubMed.
- T. Wang, A. Chutia, D. J. L. Brett, P. R. Shearing, G. He, G. Chai and I. P. Parkin, Energy Environ. Sci., 2021, 14, 2639–2669 RSC.
- S. Wang, Y. Xie, K. Shi, W. Zhou, Z. Xing, K. Pan and A. Cabot, ACS Sustainable Chem. Eng., 2020, 8, 5920–5926 CrossRef CAS.
- J. Liu, Z. Luo, J. Li, X. Yu, J. Llorca, D. Nasiou, J. Arbiol, M. Meyns and A. Cabot, Appl. Catal., B, 2019, 242, 258–266 CrossRef CAS.
- J. Liu, M. Meyns, T. Zhang, J. Arbiol, A. Cabot and A. Shavel, Chem. Mater., 2018, 30, 1799–1807 CrossRef CAS.
- X. Yang, F. Wang, Z. Jing, M. Chen, B. Wang, L. Wang, G. Qu, Y. Kong and L. Xu, Small, 2023, 19, e2301985 CrossRef.
- Y. Guo, J. Tang, Z. Wang, Y. Sugahara and Y. Yamauchi, Small, 2018, 14, e1802442 CrossRef PubMed.
- H. Liu, J. Guan, S. Yang, Y. Yu, R. Shao, Z. Zhang, M. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, e2003649 CrossRef PubMed.
- P. He, X. Y. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2017, 56, 3897–3900 CrossRef CAS PubMed.
- Q. Shi, Q. Liu, Y. Ma, Z. Fang, Z. Liang, G. Shao, B. Tang, W. Yang, L. Qin and X. Fang, Adv. Energy Mater., 2020, 10, 1903854 CrossRef CAS.
- F. Luo, Q. Zhang, X. Yu, S. Xiao, Y. Ling, H. Hu, L. Guo, Z. Yang, L. Huang, W. Cai and H. Cheng, Angew. Chem., Int. Ed., 2018, 57, 14862–14867 CrossRef CAS PubMed.
- B. Qiu, L. Cai, Y. Wang, Z. Lin, Y. Zuo, M. Wang and Y. Chai, Adv. Funct. Mater., 2018, 28, 1706008 CrossRef.
- J. Wang and F. Ciucci, Appl. Catal., B, 2019, 254, 292–299 CrossRef CAS.
- C. Wang, W. Chen, D. Yuan, S. Qian, D. Cai, J. Jiang and S. Zhang, Nano Energy, 2020, 69, 104453 CrossRef CAS.
- Y. Deng, Y. Cao, Y. Xia, X. Xi, Y. Wang, W. Jiang, D. Yang, A. Dong and T. Li, Adv. Energy Mater., 2022, 12, 2202394 CrossRef CAS.
- G. Zhou, M. Li, Y. Li, H. Dong, D. Sun, X. Liu, L. Xu, Z. Tian and Y. Tang, Adv. Funct. Mater., 2019, 30, 1905252 CrossRef.
- R. Zhang, W. Liu, F.-M. Zhang, Z.-D. Yang, G. Zhang and X. C. Zeng, Appl. Catal., B, 2023, 325, 122366 CrossRef CAS.
- Y. Chen, J. Xu, M. Jiang, L. Wang, R. Ma, Y. Chen, Z. H. Xie, P. Munroe, F. Hu, L. Li and S. Peng, Adv. Energy Mater., 2023, 14, 2303450 CrossRef.
- Y. Gao, D. Zheng, Q. Li, W. Xiao, T. Ma, Y. Fu, Z. Wu and L. Wang, Adv. Funct. Mater., 2022, 32, 2203206 CrossRef CAS.
- T. Dai, X. Zhang, M. Sun, B. Huang, N. Zhang, P. Da, R. Yang, Z. He, W. Wang, P. Xi and C. H. Yan, Adv. Mater., 2021, 33, e2102593 CrossRef PubMed.
- S. Pan, H. Li, D. Liu, R. Huang, X. Pan, D. Ren, J. Li, M. Shakouri, Q. Zhang, M. Wang, C. Wei, L. Mai, B. Zhang, Y. Zhao, Z. Wang, M. Graetzel and X. Zhang, Nat. Commun., 2022, 13, 2294 CrossRef CAS PubMed.
- Z. Zhang, X. Li, C. Zhong, N. Zhao, Y. Deng, X. Han and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 7245–7250 CrossRef CAS PubMed.
- J. Wang, S.-J. Kim, J. Liu, Y. Gao, S. Choi, J. Han, H. Shin, S. Jo, J. Kim, F. Ciucci, H. Kim, Q. Li, W. Yang, X. Long, S. Yang, S.-P. Cho, K. H. Chae, M. G. Kim, H. Kim and J. Lim, Nat. Catal., 2021, 4, 212–222 CrossRef CAS.
- C. Chen, M. Sun, F. Zhang, H. Li, M. Sun, P. Fang, T. Song, W. Chen, J. Dong, B. Rosen, P. Chen, B. Huang and Y. Li, Energy Environ. Sci., 2023, 16, 1685–1696 RSC.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711 CrossRef PubMed.
- H. Liu, X. Xu, H. Xu, S. Wang, Z. Niu, Q. Jia, L. Yang, R. Cao, L. Zheng and D. Cao, Appl. Catal., B, 2021, 297, 120451 CrossRef CAS.
- L. Tang, L. Yu, C. Ma, Y. Song, Y. Tu, Y. Zhang, X. Bo and D. Deng, J. Mater. Chem. A, 2022, 10, 6242–6250 RSC.
- Y. Zhao, Y. Guo, X. F. Lu, D. Luan, X. Gu and X. W. D. Lou, Adv. Mater., 2022, 34, e2203442 CrossRef PubMed.
- S. Zhao, C. Tan, C.-T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Nat. Energy, 2020, 5, 881–890 CrossRef CAS.
- R. Yang, L. An, Y. Zhang, N. Zhang, T. Dai and P. Xi, ChemCatChem, 2019, 11, 6002–6007 CrossRef CAS.
- S. Li, B. Chen, Y. Wang, M. Y. Ye, P. A. van Aken, C. Cheng and A. Thomas, Nat. Mater., 2021, 20, 1240–1247 CrossRef CAS PubMed.
- Y. Li, Y. Liu, Q. Qian, G. Wang and G. Zhang, Energy Storage Mater., 2020, 28, 27–36 CrossRef.
- A. Dutta, A. K. Samantara, S. K. Dutta, B. K. Jena and N. Pradhan, ACS Energy Lett., 2016, 1, 169–174 CrossRef CAS.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- Y. Zhao, N. Dongfang, C. A. Triana, C. Huang, R. Erni, W. Wan, J. Li, D. Stoian, L. Pan, P. Zhang, J. Lan, M. Iannuzzi and G. R. Patzke, Energy Environ. Sci., 2022, 15, 727–739 RSC.
- P. Hou, D. Li, N. Yang, J. Wan, C. Zhang, X. Zhang, H. Jiang, Q. Zhang, L. Gu and D. Wang, Angew. Chem., Int. Ed., 2021, 60, 6926–6931 CrossRef CAS PubMed.
- X. Zheng, J. Yang, Z. Xu, Q. Wang, J. Wu, E. Zhang, S. Dou, W. Sun, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202205946 CrossRef CAS PubMed.
- W. Li, Y. Li, H. Fu, G. Yang, Q. Zhang, S. Chen and F. Peng, Chem. Eng. J., 2020, 381, 122683 CrossRef CAS.
- J. Duan, S. Chen, A. Vasileff and S. Z. Qiao, ACS Nano, 2016, 10, 8738–8745 CrossRef CAS PubMed.
- G. Zhang, G. Wang, Y. Liu, H. Liu, J. Qu and J. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- R. Beltrán-Suito, P. W. Menezes and M. Driess, J. Mater. Chem. A, 2019, 7, 15749–15756 RSC.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spori, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Perez-Ramirez, B. R. Cuenya, R. Schlogl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- L. Reith, J. N. Hausmann, S. Mebs, I. Mondal, H. Dau, M. Driess and P. W. Menezes, Adv. Energy Mater., 2023, 13, 2203886 CrossRef CAS.
- M. Gorlin, J. Ferreira de Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- J. N. Hausmann, S. Mebs, K. Laun, I. Zebger, H. Dau, P. W. Menezes and M. Driess, Energy Environ. Sci., 2020, 13, 3607–3619 RSC.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. Araujo, M. Gliech, D. Teschner, J. Zhu, W. X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- J. N. Hausmann, R. A. Khalaniya, C. Das, I. Remy-Speckmann, S. Berendts, A. V. Shevelkov, M. Driess and P. W. Menezes, Chem. Commun., 2021, 57, 2184–2187 RSC.
- R. D. L. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. Gonzalez-Flores and H. Dau, Nat. Commun., 2017, 8, 2022 CrossRef PubMed.
- R. D. L. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. González-Flores and H. Dau, Energy Environ. Sci., 2018, 11, 2476–2485 RSC.
- M. Risch, K. Klingan, J. Heidkamp, D. Ehrenberg, P. Chernev, I. Zaharieva and H. Dau, Chem. Commun., 2011, 47, 11912–11914 RSC.
- H. Li, M. Sun, Y. Pan, J. Xiong, H. Du, Y. Yu, S. Feng, Z. Li, J. Lai, B. Huang and L. Wang, Appl. Catal., B, 2022, 312, 121431 CrossRef CAS.
- J. Li, L. Zheng, B. Huang, Y. Hu, L. An, Y. Yao, M. Lu, J. Jin, N. Zhang and P. J. S. Xi, Small, 2022, 18, 2204723 CrossRef CAS PubMed.
- B. Bao, Y. Liu, M. Sun, B. Huang, Y. Hu, P. Da, D. Ji, P. Xi and C. H. J. S. Yan, Small, 2022, 18, 2201131 CrossRef CAS PubMed.
- R. He, T. Lu, N. Xu, G. Liu, Y. Zhang and J. Qiao, Chem. Eng. J., 2023, 461, 141843 CrossRef CAS.
- C. Yao, J. Li, Z. Zhang, C. Gou, Z. Zhang, G. Pan and J. Zhang, Small, 2022, 18, e2108094 CrossRef PubMed.
- H. Li, Q. Li, P. Wen, T. B. Williams, S. Adhikari, C. Dun, C. Lu, D. Itanze, L. Jiang, D. L. Carroll, G. L. Donati, P. M. Lundin, Y. Qiu and S. M. Geyer, Adv. Mater., 2018, 30, 1705796 CrossRef PubMed.
- J.-T. Ren, Y.-S. Wang, L. Chen, L.-J. Gao, W.-W. Tian and Z.-Y. Yuan, Chem. Eng. J., 2020, 389, 124408 CrossRef CAS.
- T. Wang, X. Cao, H. Qin, L. Shang, S. Zheng, F. Fang and L. Jiao, Angew. Chem., Int. Ed., 2021, 60, 21237–21241 CrossRef CAS PubMed.
- Y. Wang, J. Liu, T. Lu, R. He, N. Xu and J. Qiao, Appl. Catal., B, 2023, 321, 122674 Search PubMed.
- Q. P. Ngo, T. T. Nguyen, M. Singh, R. Balaji, N. H. Kim and J. H. Lee, Appl. Catal., B, 2023, 331, 122674 CrossRef CAS.
- D. Ren, J. Ying, M. Xiao, Y. P. Deng, J. Ou, J. Zhu, G. Liu, Y. Pei, S. Li, A. M. Jauhar, H. Jin, S. Wang, D. Su, A. Yu and Z. Chen, Adv. Funct. Mater., 2019, 30, 1908167 CrossRef.
- L. Yang, X. Zhang, L. Yu, J. Hou, Z. Zhou and R. Lv, Adv. Mater., 2022, 34, e2105410 CrossRef PubMed.
- Y. Lin, L. Yang, Y. Zhang, H. Jiang, Z. Xiao, C. Wu, G. Zhang, J. Jiang and L. Song, Adv. Energy Mater., 2018, 8, 1703623 CrossRef.
- C.-X. Zhao, J.-N. Liu, J. Wang, C. Wang, X. Guo, X.-Y. Li, X. Chen, L. Song, B.-Q. Li and Q. Zhang, Sci. Adv., 2022, 8, eabn5091 CrossRef CAS PubMed.
- Y. Zhou, Y. Yu, D. Ma, A. C. Foucher, L. Xiong, J. Zhang, E. A. Stach, Q. Yue and Y. Kang, ACS Catal., 2020, 11, 74–81 CrossRef.
- Z. You and I.-H. Jung, Metall. Mater. Trans. B, 2020, 51, 3108–3129 CrossRef.
- R. Ganesan, A. T. Dinsdale and H. Ipser, Intermetallics, 2011, 19, 927–933 CrossRef CAS.
- S. Boone and O. J. Kleppa, J. Chem. Thermodyn., 1991, 23, 1147–1151 CrossRef CAS.
- Q. Shi, Q. Liu, Y. Zheng, Y. Dong, L. Wang, H. Liu and W. Yang, Energy Environ. Mater., 2021, 5, 515–523 CrossRef.
- Y. Guo, P. Yuan, J. Zhang, Y. Hu, I. S. Amiinu, X. Wang, J. Zhou, H. Xia, Z. Song, Q. Xu and S. Mu, ACS Nano, 2018, 12, 1894–1901 CrossRef CAS PubMed.
- M. Ma, A. Kumar, D. Wang, Y. Wang, Y. Jia, Y. Zhang, G. Zhang, Z. Yan and X. Sun, Appl. Catal., B, 2020, 274, 119091 CrossRef CAS.
- H. Sun, M. Wang, S. Zhang, S. Liu, X. Shen, T. Qian, X. Niu, J. Xiong and C. Yan, Adv. Funct. Mater., 2020, 31, 2006533 CrossRef.
- S. Dresp, F. Luo, R. Schmack, S. Kühl, M. Gliech and P. Strasser, Energy Environ. Sci., 2016, 9, 2020–2024 RSC.
- E. Vijayakumar, S. Ramakrishnan, C. Sathiskumar, D. J. Yoo, J. Balamurugan, H. S. Noh, D. Kwon, Y. H. Kim and H. Lee, Chem. Eng. J., 2022, 428, 13115 CrossRef.
- J. Chen, C. Fan, X. Hu, C. Wang, Z. Huang, G. Fu, J. M. Lee and Y. Tang, Small, 2019, 15, e1901518 CrossRef PubMed.
- P. Yu, L. Wang, F. Sun, Y. Xie, X. Liu, J. Ma, X. Wang, C. Tian, J. Li and H. Fu, Adv. Mater., 2019, 31, e1901666 CrossRef PubMed.
- P. Peng, L. Shi, F. Huo, C. Mi, X. Wu, S. Zhang and Z. Xiang, Sci. Adv., 2019, 5, eaaw2322 CrossRef CAS PubMed.
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 1194 CrossRef.
- J. Han, X. Meng, L. Lu, J. Bian, Z. Li and C. Sun, Adv. Funct. Mater., 2019, 29, 1828872 Search PubMed.
- M. Moloudi, A. Noori, M. S. Rahmanifar, Y. Shabangoli, M. F. El-Kady, N. B. Mohamed, R. B. Kaner and M. F. Mousavi, Adv. Energy Mater., 2022, 13, 2203002 CrossRef.
- Z. Wang, J. Ang, B. Zhang, Y. Zhang, X. Y. D. Ma, T. Yan, J. Liu, B. Che, Y. Huang and X. Lu, Appl. Catal., B, 2019, 254, 26–36 CrossRef CAS.
- Z. Pan, H. Chen, J. Yang, Y. Ma, Q. Zhang, Z. Kou, X. Ding, Y. Pang, L. Zhang, Q. Gu, C. Yan and J. Wang, Adv. Sci., 2019, 6, 1900628 CrossRef PubMed.
- J. Qian, X. Guo, T. Wang, P. Liu, H. Zhang and D. Gao, Appl. Catal., B, 2019, 250, 71–77 CrossRef CAS.
- X. Zhao, X. Li, Z. Bi, Y. Wang, H. Zhang, X. Zhou, Q. Wang, Y. Zhou, H. Wang and G. Hu, J. Energy Chem., 2022, 66, 514–524 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee01912a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |