
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic transformation of active sites in energy and environmental catalysis
Hao
Zhang†
 *ab,
Lei
Chen†
*c,
Feng
Dong†
de,
Zhiwen
Lu†
f,
Enmin
Lv
g,
Xinglong
Dong
g,
Huanxin
Li
*b,
Zhongyong
Yuan
*c,
Xinwen
Peng
*h,
Shihe
Yang
*d,
Jieshan
Qiu
i,
Zhengxiao
Guo
*e and
Zhenhai
Wen
*f
*ab,
Lei
Chen†
*c,
Feng
Dong†
de,
Zhiwen
Lu†
f,
Enmin
Lv
g,
Xinglong
Dong
g,
Huanxin
Li
*b,
Zhongyong
Yuan
*c,
Xinwen
Peng
*h,
Shihe
Yang
*d,
Jieshan
Qiu
i,
Zhengxiao
Guo
*e and
Zhenhai
Wen
*f
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: hzhchem@mit.edu
bDepartment of Chemistry, University of Oxford, Oxford OX1 3TA, UK. E-mail: hao.zhang@chem.ox.ac.uk; huanxin.li@chem.ox.ac.uk
cSchool of Materials Science and Engineering, Smart Sensing Interdisciplinary Science Center, Nankai University, Tianjin 300350, P. R. China. E-mail: chen-lei@mail.nankai.edu.cn; zyyuan@nankai.edu.cn
dGuangdong Key Lab of Nano-Micro Material Research, School of Advanced Materials, Shenzhen Graduate School, Peking University, Shenzhen 518055, P. R. China. E-mail: chsyang@pku.edu.cn
eCAS-HKU Joint Laboratory on New Materials & Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, P. R. China. E-mail: zxguo@hku.hk
fCAS Key Laboratory of Design and Assembly of Functional Nanostructures, and Fujian Provincial Key Laboratory of Materials and Techniques toward Hydrogen Energy, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China. E-mail: wen@fjirsm.ac.cn
gKey Laboratory of Materials Modification by Laser, Ion and Electron Beams, School of Materials Science and Engineering, Dalian University of Technology, Dalian 116024, P. R. China
hState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510641, P. R. China. E-mail: fexwpeng@scut.edu.cn
iCollege of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
First published on 26th July 2024
Abstract
Active sites play a pivotal role in photo/electrocatalysis, particularly in the transition from fossil fuels to clean, efficient and renewable energy sources. Precise identification of catalyst active sites and understanding of their dynamic transformation are crucial for engineering the activity, selectivity and stability of a catalyst for a specific reaction. Herein, we provide an in-depth and interdisciplinary overview of the recent advancements in dynamic transformation of active sites in photo/electrocatalysis. Firstly, we explore the underlying principles of the dynamic reconstruction, focusing on dynamic transformations in surface structure, composition and properties. Subsequently, advanced operando/in situ characterization for dynamic transformation is summarized, to provide mechanistic insights for the identification of such processes. In order to improve catalytic performance, we discussed comparatively the triggers and the corresponding reaction mechanisms of the dynamic process. Finally, we present an insightful analysis of the challenges and the future prospects for the applications of dynamic transformation of active sites in photo/electrocatalysis.
Broader contextIn recent years, there has been a surge in research focused on understanding the dynamic transformations of active sites in photo/electrocatalysis, driven by the urgent need for transition from fossil fuels to renewable energy sources. Our review provides an interdisciplinary overview of recent advancements in the area of energy and environmental catalysis, encompassing the origins, identification methods, and targeted construction of dynamic transformations. We commence our review by exploring the origins of dynamic reconstruction under reaction conditions, delving into dynamic transformations involving changes in surface properties, composition, and structure. Subsequently, we summarize advanced operando/in situ characterization for dynamic transformations, offering valuable insights for the identification and detailed investigation of these processes. To manipulate the dynamic conversion of active sites and improve catalytic performance, we discuss various triggers, corresponding reaction mechanisms, and the resulting photo/electrocatalytic performance. Additionally, we present an insightful analysis of the challenges and outline future prospects for the dynamic transformation of active sites in photo/electrocatalysis. |
1. Introduction
In the contemporary epoch, the burgeoning severity of global energy and environmental crises has considerably expedited the development of renewable energy technologies, encompassing notably hydroelectric, wind, and solar power.1–3 Among these, solar energy has led the way to the development of sustainable energy storage and conversion systems, turning its instability and intermittency to continuous and reliable utilization, e.g. via photovoltaics and photoelectrochemical catalysis.4,5 Meanwhile, electrochemistry has drawn great scientific interest for providing sustainable energy carriers, such as hydrogen (H2), as well as energy conversion and storage devices, including metal–air/ion batteries, electrolyzers and fuel cells.6,7 Such systems rely on various photo/electrocatalytic redox couples, such as the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), hydrogen evolution reaction (HER), carbon dioxide reduction reaction (CO2RR), nitrogen reduction reaction (NRR) and NOx reduction reaction (NOxRR).8–11 Catalysts play a critical role in activating inert reactants and determining reaction pathways, which are often influenced by the dynamic transformation of the active sites that adapt to the chemical potential environment under operational conditions.12,13 Therefore, comprehending the dynamic behavior of active sites is essential for the rational design of catalysts to ensure high-efficiency energy and environmental applications.Since Taylor introduced the concept of the ‘active site’ in 1925,14 research on fundamental catalysis has focused on the identification of catalytically active species and the comprehensive understanding of their operational mechanisms.15 Catalytically active sites, representing a small proportion of a catalyst's energetically favorable surface, comprise individual atoms or specific groups of atoms concentrated at surface facets and defects, such as edges, corners and amorphous regions.16 Generally, catalysts lower the activation energy for molecular conversion by chemically binding reactant molecules, thereby leading to proton and/or electron transfer between the reactants and the active sites.17 Therefore, catalytic processes, particularly those that necessitate energy-driven conditions, such as temperature, voltage or photo-irradiation, inevitably induce changes in the geometric and electronic structures of catalysts. These changes, referred to as dynamic reconstruction, encompass alterations in chemical valences, phases, structures, and coordination environments.18,19 For thermal catalysis, the charge transfer primarily occurs on the catalyst's surface and can be attributed to surface reconstruction resulting from interactions with reactants.20 However, electrocatalytic and photocatalytic processes generally involve charge transfer throughout the entire catalyst rather than being confined to surface active sites.21 Specifically, the absorption of photons triggers the generation of charge carriers in photocatalysts, and the application of an external voltage drives electron migration towards electrodes and surfaces during electrocatalysis.22 Active sites in photo(electro)catalysts are thermodynamically metastable and transiently arise from interactions with, under photo illumination and/or applied bias, electrolytes and reactants/intermediates.23 By investigating the dynamic transformation of catalysts, valuable information about active sites can be obtained, allowing us to uncover possible mechanisms for the activation and deactivation of (pre)catalysts.
The increasing global focus on sustainability has prompted the substitution of conventional thermal heterogeneous catalytic processes with renewable-powered photo/electrocatalytic processes.24Fig. 1 showcases the developments and discoveries of the reconstruction of various chemical compounds and materials. We observe a trend towards increasingly precise construction of such materials, transitioning from the elucidation of oxide chemical reconstructions to the precise modulation of reconstructions involving specific elements or individual atoms. This progression underscores notable advancements in our comprehension and characterization of the reconstruction mechanisms. In recent years, the development of in situ/operando characterization techniques has unveiled substantial dynamic transformation in catalysts during photo/electrocatalytic reactions.25 The dynamic transformations of catalysts span from atomic-scale surface property changes (a few Å) to phase transformations at a relatively larger scale (tens of nm), and even significant variations in morphologies (to mm). As illustrated in the summary plot of the relationship between the size of pre-catalysts and the thickness of reconstruction layers (Fig. 2), it is apparent that specific elements or nanostructures frequently correspond to varying scales of reconstruction. This phenomenon underscores our ability to strategically design catalysts with tailored reconstruction patterns. Generally speaking, these dynamic transformations are able to modulate photo/electrocatalytic behaviors, such as adsorption, activation and desorption, and can be engineered to enhance catalytic performance.26,27 However, it should also be acknowledged that the reconstructed photo/electrocatalysts may experience ongoing restructuring throughout a catalytic process, which may result in the loss of active components and the degradation in catalytic activity.28 This dynamic transformation also serves as a clear indication of the eventual deterioration of the catalyst, commonly referred to as catalyst corrosion.29,30 In this regard, optimizing the reconstruction process to produce a profusion of active sites with high intrinsic activity represents a powerful strategy for enhancing the catalytic performance of photo/electrocatalysts.31
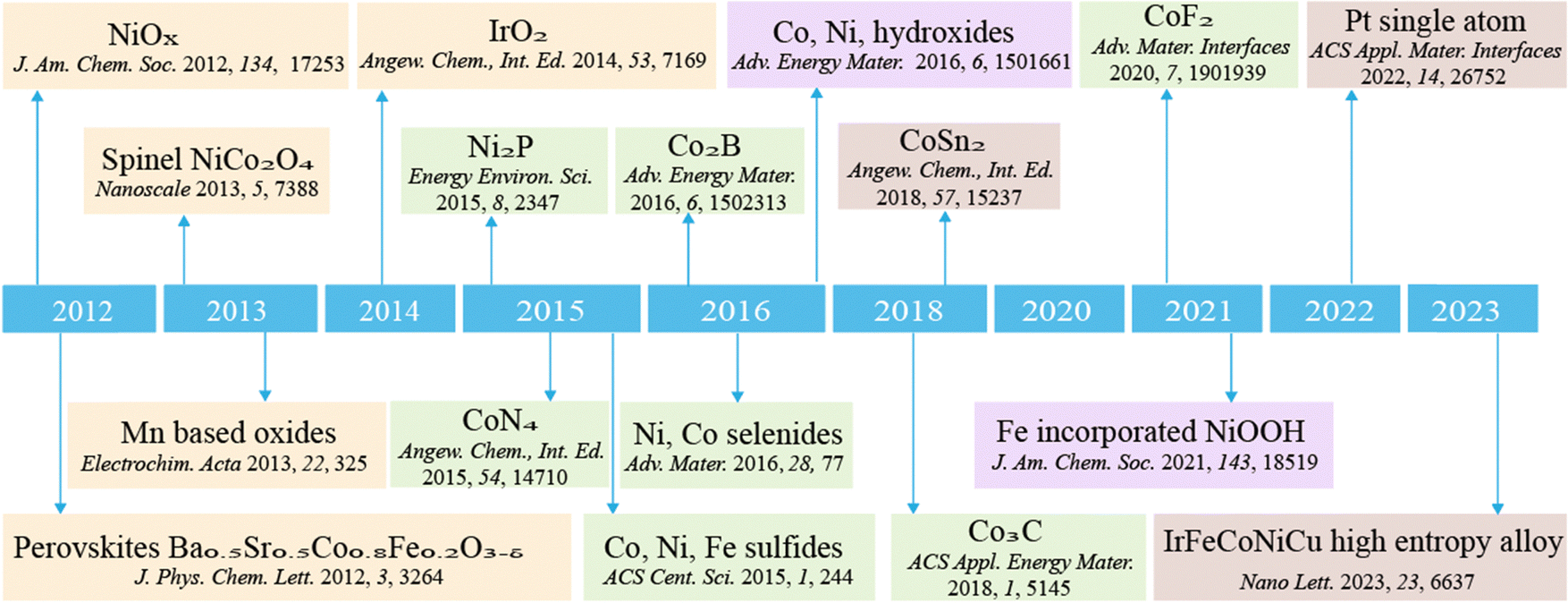 | ||
| Fig. 1 Chronogram of the developments in reconstructed catalysts. | ||
 | ||
| Fig. 2 Scatter plot of the relationship between the size of pre-catalysts and the thickness of reconstruction layers. | ||
To date, the existing reviews on the dynamic transformation of electrocatalysts mainly focus on either one or a limited number of energy and environmental applications, such as the OER, CO2RR and HER.32 In addition, they enumerate various types of restructuring catalysts without associating restructuring triggers with specific types of elements.33,34 It is highly desirable to rationalize the discovered correlations between restructuring triggers and different categories of reported elements for engineering the reconstruction from various perspectives (Fig. 3). This endeavor aims to facilitate more effective utilization of certain elements in constructing specific catalysts via restructuring processes. Recent studies on dynamic transformation have been found in diverse photo/electrocatalytic systems for energy and environmental applications, albeit with varied quantity of literature available across these fields.35 The emergence and maturity of nanotechnology have dissolved the boundaries of single-disciplinary fields and facilitated multidisciplinary investigations across diverse scientific domains.36 Undoubtedly, understanding the dynamic transformation mechanism of photo/electrocatalysts, optimizing the reconstruction processes, and uncovering the true structure–activity relationship call for interdisciplinary efforts in order to achieve a noteworthy breakthrough in formulating photo/electrocatalysts.37 In addition, this can form a robust foundation for comprehending the dynamic transformation of catalysts and serve as a pioneering reference in engineering multifunctional catalysts for photochemical and electrochemical reactions.
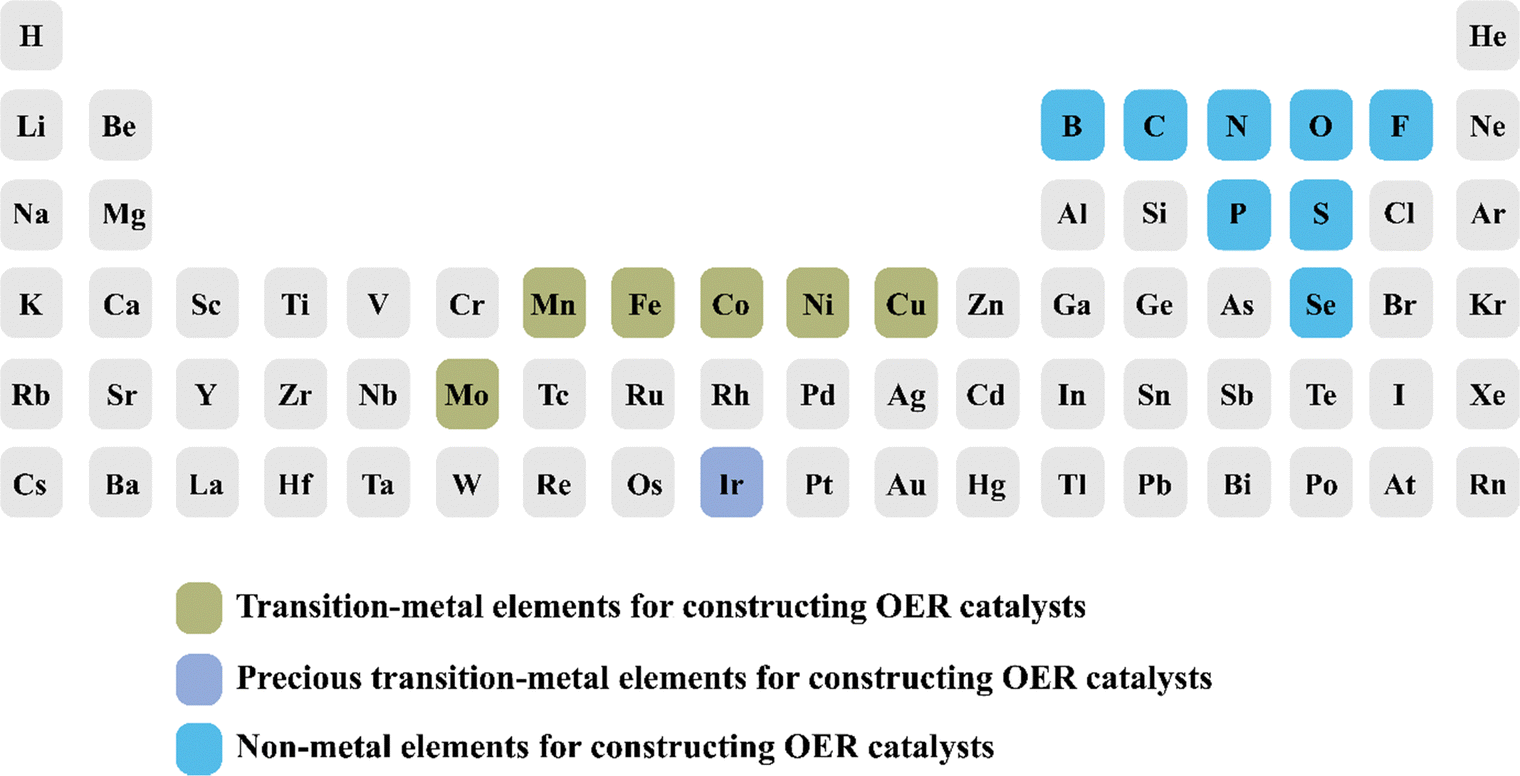 | ||
| Fig. 3 Reported elements for constructing transition metal-based OER catalysts. | ||
This review aims to present an in-depth analysis, critical assessment and new insight of the dynamic transformation into active sites in the realm of energy and environmental catalysis, including the origin and types of dynamic transformation, the underlying principles, the identification methods with related operando/in situ or multiangle/level analyses, and several triggers of dynamic transformation, which are categorized into single atom catalyst induction, defect engineering, heterostructure construction, leaching, and crystallinity modification, as well as the photo/electrocatalytic applications in energy and environmental domains, such as electrocatalytic HER/OER/CO2RR/NRR and photocatalytic pollutant degradation/HER/OER/CO2RR. Note that the focus is on the theory-structure–activity relationships and some optimization strategies of dynamic transformation, benefiting from the mitigation of the adverse effects resulting from these transformations and exploring potential positive effects that may emerge. Finally, we propose the existing challenges and novel opportunities that hold the promise of propelling the development of dynamic transformation in photo/electrocatalytic reactions and contribute to the rational design of high-performance earth-abundant photo/electrocatalysts.
2. Fundamentals of dynamic transformation
Catalytic systems involving dynamic transformation of active sites have emerged as a common occurrence in the scientific literature, garnering increasing attention and interest from scholars across various disciplines.38 An in-depth study of dynamic transformation enables precise customization of intrinsic properties and facilitates the discovery of new phenomena in nanomaterials. Theoretical insights into dynamic transformation are crucial for understanding reaction mechanisms and optimizing catalyst design for specific processes. The purpose of this section is to deliver a thorough introduction to the fundamentals of the dynamic transformation of photo/electrocatalysts, encompassing their origin, various types and identification methods (Fig. 4). | ||
| Fig. 4 Summary of the correlation of various factors that can affect the dynamic transformation during energy and environmental catalysis. | ||
2.1 Origins
To gain a deeper understanding of the dynamic evolution of active sites, it is crucial to trace their origin. It is widely acknowledged that the realization of high catalytic activity and selectivity in most catalytic reactions critically hinges upon the meticulous establishment of tailored reaction conditions. However, such stringent conditions may inevitably induce the dynamic structural evolution of catalysts.39 Generally, the energy input intensity (e.g., applied bias and light radiation) serves as the driving force to induce the dynamic transformation of photo/electrocatalysis.11 Variable testing conditions, including reaction medium, temperature, and reaction time, play a crucial role in modulating the energy input intensity, which can influence the stability of precatalysts and impact their degree of reconstruction.40In order to elucidate the electrocatalyst reconstruction behavior, electrocatalysts can be conceptually simplified as materials consisting of active sites and ligands. The active center, often composed of a metallic or nonmetallic element (e.g., carbon), exhibits electrocatalytic activity, while the ligands can encompass either metallic/nonmetallic elements or molecules capable of coordination. Then, the dynamic transformation of the electrocatalyst can be perceived as encompassing the oxidation/reduction and dissolution/redeposition of active sites and ligands, along with the incorporation of new ligands. Typically, electrocatalysts tend to undergo oxidation or partial oxidation when the applied potential surpasses the theoretical potential for electron loss from active sites or ligands, resulting in the dissolution and substitution of active sites or ligands. In contrast, at sub-theoretical electrode potentials for electron acquisition, electrocatalysts experience reduction or partial reduction. This can lead to ligand dissolution and substitution, but active site dissolution is generally not observed in this case. During potential cycling, active sites/ligands that have dissolved at high potential are subsequently redeposited onto the surface of the electrocatalyst when the potential is reduced.
The testing conditions can primarily influence specific redox potentials and govern the extent of electrocatalyst reconstruction. Under specific testing conditions (temperature, electrolyte and pressure), the reduction of initial potential affords the creation of highly reactive sites at low potential, thereby decreasing the overpotential of the electrocatalytic reaction. Among these testing conditions, the pH value and/or cation size of electrolytes have a large influence on the dynamic reconstruction of electrocatalysts. In the context of ligand substitution reconstruction, oxygen acts as a potential substituent within an acidic environment, while in an alkaline medium, a hydroxyl group could potentially undergo replacement. Furthermore, the cation sizes in the electrolyte exhibit a pronounced influence on the adsorption characteristics of the electrocatalyst. For example, through the synergistic effect of enlarging the alkali-metal cationic sizes within the electrolytic medium and incorporating subsurface oxygen species that promote the adsorption for CO2, a notable enhancement in the C–C coupling process on the reconstructed CuOx electrodes was observed (Fig. 5a).46
 | ||
| Fig. 5 (a) CO* formation free energy calculated in the absence and presence of Li*, Na*, K*, and Cs* on bare Cu(100) and Cu(100) with 2/9 ML OS. This figure has been reproduced from ref. 46 with permission from American Chemical Society, copyright 2018. (b) Schematic representation of the variation in the chemical state of Cu during photocatalysis. This figure has been reproduced from ref. 47 with permission from Nature, copyright 2019. (c) Structure and dynamics of the N–FeN4C10 moiety in the ORR with optimized geometries (the angles represent the deviation of iron from the N4-plane). This figure has been reproduced from ref. 48 with permission from Elsevier, copyright 2020. (d) Schematic representation of the catalyst reconstruction process: (I) the as-synthesized metallic Cu NPs, (II) the catalyst oxidation and dissolution of Cu ions into the electrolyte, (III) the redeposition of dissolved ions onto the catalyst when a reducing potential is applied, and (IV) the dissolution of smaller particles into transient species, whose nature remains unknown to date, that contribute to the growth of larger nanoparticles under a constant applied potential during cell operation. This figure has been reproduced from ref. 49 with permission from Wiley-VCH, copyright 2020. (e) SEM images and a high-resolution TEM image of pristine FeS/IF, FeS/IF after the HER and FeS/IF after the OER. This figure has been reproduced from ref. 50 with permission from Elsevier, copyright 2018. (f) SEM of images of the as-prepared (pristine) CoSx/NGF and the CoSx/NGF after the OER. This figure has been reproduced from ref. 51 with permission from American Chemical Society, copyright 2018. | ||
Beyond the photoinduced carriers, some reactants can also participate in the reconstruction of the photocatalysts. The interaction between the reactants and the photocatalyst surface can lead to the formation of new active sites, enhance the charge separation and transfer efficiency, and ultimately improve the photocatalytic performance of the system. For example, the surface of TiO2 can undergo atomic rearrangement when exposed to H2O molecules, and light irradiation further amplifies the extent of these structural modifications.54 Thin amorphous layers were formed, measuring approximately 1–2 atomic layers in thickness, on crystalline TiO2 nanoparticles subsequent to their exposure to ultraviolet irradiation and H2O vapor. In addition, molecular co-catalysts that exhibit strong affinity towards photocatalyst surfaces have shown remarkable potential in inducing electronic structural modifications, thus resulting in a mild surface reconstruction. The utilization of molecular co-catalysts, such as CF3CO2H and ethylenediamine, has exhibited commendable enhancement in photocatalytic performance.55,56
2.2 Types
The dynamic transformations of photo/electrocatalysts can be systematically classified based on the length scale of these transformations, spanning a diverse range that extends from the atomic-level alterations (a few Å) manifesting in the surface properties, to the macroscopic dimensions (tens of nm) associated with phase transformations, and even encompassing obvious structural reconstructions. The comprehensive elucidation is presented as follows.2.2.1.1 Chemical states. The surface properties of photo/electrocatalysts can transform as a consequence of intricate interactions among reactants, charge carriers and active sites. The chemical states of a catalyst can readily undergo alterations during the process of photo/electrocatalysis.57 Typically, the occurrence of more negative potential conditions and the generation of photogenerated electrons tend to facilitate the establishment of a metallic state. While exposed to relatively positive potentials or in contact with photogenerated holes, catalysts generally exhibit a preference for existing in an oxidized state.58 It is easily comprehensible that the initially present oxidized species of metals, such as titanium oxide or copper oxide, in catalysts can undergo reduction to a lower valence state by photogenerated electrons or by the application of cathodic potentials.59 It is crucial to emphasize that even in the case of a metal-based initial catalyst, rapid oxidation reactions can occur upon exposure to air or the electrolyte under open circuit potential conditions, resulting in the formation of a thin oxide layer on the surface of the metal catalyst.60 This oxide layer can also be reduced by photogenerated electrons or under cathodic potentials. The presence and characteristics of this reduced oxide layer have a pronounced influence on subsequent catalytic processes.61 One notable example is the reduction of Cu2O to metallic Cu observed under CO2RR conditions.62 This reduction process results in an increased metallicity of the copper electrode, which subsequently promotes C–C bond formation under high cathodic polarization. In addition, the PtO species of the PtO–TiO2 photocatalyst experienced partial reduction when exposed to light irradiation due to the presence of photogenerated electrons.63
The dynamic transformation of catalysts to an oxidized state generally occurs in oxidation reactions, which necessitate a significant driving force to overcome the slow kinetic barriers, such as the OER.64 Metal redox in oxide-hydroxide catalysts is feasible due to the mobility of lattice oxygenic ligands, enabling their exchange with water molecules.65 For example, a significant proportion of Co ions undergoes a valence state transition from Co3+ to Co4+ in response to voltage and time variations accompanied by spontaneous delithiation.66 The high OER activity is primarily attributed to the highly oxidized Co4+ site, as opposed to the Co3+ site or the oxygen vacancy site. NiOx species, commonly used as oxidation cocatalysts for photocatalytic water splitting, can be oxidized to form NiO(OH) when exposed to photogenerated holes during the early stages of the reaction.67 The formation of Ni3+ species played a crucial role in facilitating the OER process and contributed to the overall efficiency of the photocatalytic water-splitting reaction. Beyond metal oxides, high-valent metals have been found to be crucial in various OER catalyst systems, including metal phosphides, sulfides, phosphates, carbides, etc. Starting from cobalt phosphates, Co2+ in the pristine precatalyst can be oxidized to Co3+, which acts as the active species for the OER activity.68,69
2.2.1.2 Coordination environments. In empirical investigations and utilizing powdered samples, it has been substantiated that altering the spatial positioning of an individual metal atom on a support surface can result in a discernible change in the coordination environment surrounding the metal atom. The mobility of catalytic metal atoms across a surface or the removal of coordinated anions in the support, thereby creating a vacancy, can potentially lead to the emergence of an alternative coordination environment. The ensuing alterations in both geometric and electronic structures exert a significant influence on the catalytic activity. Taking the Cu–TiO2 catalyst composed of isolated CuII sites as a typical example, the presence of photogenerated electrons leads to the formation of CuI species encircled by a distorted support structure, arising from the partial reduction of TiIV to TiIII (Fig. 5b). Such modified CuI species serve as the active sites responsible for the photocatalytic HER.47
During the ORR process, there is often a dynamic transformation observed between planar and curved coordination geometries in the structural evolution of M–Nx sites. Near the ORR onset potential (0.9 V vs. RHE), a reduction in the Fe–N bond length occurs as the central Fe atom in N–FeN4C10 moves towards the N4-plane (Fig. 5c).48 This structural change was coupled with the transformation of Fe2+ from a high-spin (HS) to a low-spin (LS) electronic state with the formation of an O2–FeN5 intermediate. Conversely, during the formation of the O2–FeN4 intermediate at higher bias potentials (0.7 V and 0.5 V versus RHE), as the central Fe atom within the FeN4C12 species shifted away from the N4-plane, the Fe–N bond length underwent an expansion. This structural modification was concurrent with the conversion of the Fe2+ electronic state from LS to HS.
2.2.2.1 Element redistribution. Element redistribution is a prevalent occurrence in the reactions of bimetallic or multi-metallic catalysts, and one illustrative example of this phenomenon is found in alloy catalysts. Alloy nanoparticles have garnered widespread utilization in a multitude of electrocatalytic reactions, encompassing the ORR, alcohol oxidation and water oxidation. Notably, the distribution of metals within a catalyst after utilization can exhibit marked disparities when compared to the initial pristine or precatalyst state. For instance, PtM nanoparticles, where M typically represents first-row transition metals like Fe, Co, Ni, or Cu, undergo a transformation into core–shell structures during electrochemical processes, characterized by a PtM core enveloped by a Pt shell, owing to the electrochemical de-alloying. The de-alloyed bimetallic nanoparticles exhibit enhanced activity in comparison to their more homogeneous counterparts, which can be attributed to the distinctive electronic properties of the Pt shell, as well as the lattice strain that arises at the interface between the Pt shell and the alloyed core.70
The phenomenon of segregation in composite metal-oxide materials leads to the transformation of a uniform solid into one characterized by the presence of interfacial structures.71 This can be exemplified by considering perovskite materials, where certain metal ions have the propensity to undergo reduction during the process of high-temperature solid oxide fuel cells, resulting in the formation of metallic regions.72 In most reported works, the active catalyst is not a pristine starting (pre)catalyst but, instead, an in situ-formed structure. In some cases, chemical segregation can yield an active material, but it may also exert a detrimental influence on electrocatalytic performance. Taking MnCo spinel oxides as an example,73 Mn sites exhibited the ability to activate O2, while Co sites are proficient in activating H2O, in the alkaline ORR. However, when Mn or Co oxides were segregated into distinct surface patches, the cooperative interaction between these two different metals was compromised, resulting in an inefficient performance of the ORR.
2.2.2.2 Phase transformation. Under most circumstances, a pristine (pre)catalyst undergoes a crystal phase transformation when exposed to the catalytic reaction conditions. This transformation can involve transitioning into an amorphous phase or evolving into different chemical substances. Subsequently, the catalyst adopts a new crystal phase, enabling its continued participation in the catalytic reaction. Metal–organic frameworks (MOFs) represent a category of compounds characterized by the formation of coordination bonds between organic ligands and metal ions or clusters. Within MOFs, these metal components serve as active sites, exhibiting notable attributes such as high mono-dispersibility and atomic efficiency. Nonetheless, an inherent issue with MOF-based photocatalysts is their vulnerability to instability under reaction conditions. The presence of photogenerated electrons, holes, and radicals can lead to the degradation or collapse of the framework structures of MOFs. Accordingly, the application of MOF photocatalysts has frequently been associated with a decrease in crystallinity or a transition to amorphous structures.74 Several MOFs, such as NiCo–MOF, NiFe–MOF, and CoFe–MOF, have risen in prominence for their pivotal roles in electrochemical reactions, notably as effective electrocatalysts for the OER. Recent investigations have confirmed that the actual catalytic species for the OER in MOFs are derived from the reconstruction of oxyhydroxides rather than originating from the pristine metal nodes. A ligand substitution mechanism has been proposed to elucidate the reconstruction of ZIF-67 during the OER.75 Upon exposure to an electrolyte containing alkaline hydroxide ions, the tetrahedral Co-bonded mIM molecules on the surface layer of ZIF-67 would be substituted by hydroxide ions, resulting in the formation of Co(mIM)3OH, Co(mIM)2OH2, Co(mIM)OH3−, and eventually Co(OH)42−, often denoted as α-Co(OH)2. As the applied potential is increased, α-Co(OH)2 would suffer a phase transition, transforming into β-Co(OH)2. Then, the oxidation would occur in both α-Co(OH)2 and β-Co(OH)2, ultimately giving rise to CoOOH, which represented the authentic catalytic species involved in the OER.
Many oxide-based electrocatalysts for the OER undergo the formation of amorphous metal hydroxide layers during catalysis, which generally depends on the reaction conditions and material composition. The formation of amorphous surface layers commences at the surface and extends into the internal regions, suggesting that these amorphous layers exhibit porosity, enabling the diffusion of H2O and its subsequent reaction with the bulk oxide.76 For some metal (such as Fe, Co and Ni) oxides, the amorphous hydroxide layer is indeed produced through oxide hydration and exhibits a structural similarity to a layered double hydroxide (LDH).77 The high oxidation potential exhibited by CoIII or FeIII species within the amorphous layer is conducive to the dissociation of H2O.78 The dissolution and subsequent re-deposition of metal ions have been conspicuously observed in the realm of these composite oxide catalysts. In addition, perovskite materials can similarly undergo transformation into amorphous layers during catalysis, a phenomenon driven by element-specific dissolution mechanisms.79 As for other metal compounds, such as phosphides, carbides, nitrides or sulfides, the generation of hydroxide layers is attributed to the oxidation of the original catalyst by oxygen-containing species, followed by the hydrolysis of the formed oxide product. In fact, these metal-based compounds can also serve as photocatalysts or cocatalysts in photocatalytic water splitting. Similar to the electrocatalytic reaction process, these metal-based photocatalysts also inevitably experience phase transformation. For example, the metallic Ni, employed as a cocatalyst for photocatalytic HER, could undergo progressive oxidation initiated by photogenerated holes, ultimately dissolving in H2O and leading to the creation of a hollow NiO structure.80 In the context of NiOx species functioning as cocatalysts in the photocatalytic OER, these species could be oxidized to NiO(OH) when exposed to photogenerated holes, and these Ni(III) species in NiO(OH) were the essential active sites for the OER.67,80,81
2.2.3.1 Size. A prevalent structural change observed in materials during catalysis is the alteration of the particle size. The sintering of catalysts, which leads to a reduction in the specific surface area and potential change in the coordination environment, is a key factor in irreversible deactivation. In the context of metal nanoparticle sintering on a support, two principal mechanisms are proposed: the dissolution of metal into the electrolyte followed by its subsequent redeposition on the support, a phenomenon known as Ostwald ripening, or direct particle coalescence.82 When a Pt catalyst is employed in a proton-exchange membrane fuel cell, the impetus driving Ostwald ripening and particle coalescence stems from a redox transformation involving the conversion between metallic Pt0 and Pt2+/Pt4+, which is markedly expedited during voltage cycling and intricately linked to the catalyst's morphological characteristics.83 Specifically, Pt undergoes size-dependent oxidation to Pt2+ during electrocatalysis. Subsequently, the oxidized Pt species may dissolve in acidic electrolytes, and they can either migrate to the anode for reduction by H2, resulting in the formation of large nanoparticles, or they may undergo re-reduction at the cathode.
Likewise, the sintering of a metal co-catalyst can take place when subjected to light irradiation and H2O vapor. When compared to metal nanoparticles, subnanometer metal species, including isolated atoms or clusters, used as cocatalysts on semiconductors, exhibit enhanced photocatalytic performance.84,85 However, Pt atoms and clusters, commonly used as cocatalysts in the photocatalytic HER, may sinter, yielding more active Pt nanoparticles (∼1 nm) compared to isolated Pt atoms or larger counterparts.86,87 During the photocatalytic water splitting, the photogenerated electrons and hydrogen-containing intermediates can induce the reduction of Ptn+ cations to highly dispersed Pt nanoparticles, and the size of partially oxidized Pt co-catalysts can be maintained at less than 2 nm by precise control, contributing to the efficient photocatalytic overall water splitting by effectively inhibiting the reverse reaction between H2 and O2.75 Due to their high associated surface free energy, transition metal oxide photocatalysts are prone to sintering when particle sizes are less than 10 nm. As a typical example, considering CoO nanoparticles with sizes below 10 nm, the sintering phenomenon observed during photocatalytic overall water splitting can be attributed to the generation of amorphous Co(OH)2 on the particle surface and the subsequent dehydrative coalescence.88
In addition, the dissolution and redeposition process of the catalyst within the electrolyte exerts a significant impact on its size. The removal of atoms from the catalyst surface can directly dissolve into the electrolyte, resulting in a reduction in the catalyst size. Nevertheless, dissociated metal atoms located near the cathode have the potential to redeposit on the catalyst surface, leading to the enlargement of existing particles or the formation of new clusters. These clusters and catalyst fragments can aggregate, forming agglomerations of varying sizes and shapes. Furthermore, the Ostwald ripening triggers the transportation of atoms from small to larger nanoparticles, and even facilitates substance redistribution in the process. Taking the complete restructuring of Cu nanoparticles as an example, the process involved three sequential stages: the prompt oxidation to Cu2O upon exposure to the electrolyte, Cu ion dissolution during the open circuit potential state, the reintegration of aqueous Cu ions into the nanoparticle surface at the startup stage (0.1 VRHE). Ultimately, particle size redistribution occurs, resulting in the disappearance of smaller particles during the operational stage (Fig. 5d).49
2.2.3.2 Morphologies. The migration of just a small number of atoms within each crystal can result in the exposure of different facets on the particles, often accompanied by morphological changes. Notably, metal nanocrystals demonstrate varying selectivity and activity profiles on their distinct facets. As exemplified in the case of metal Cu catalysts employed in the electrocatalytic CO2RR, Cu (100) facets exhibited a preference for generating C2+ products, especially C2H4, whereas Cu (111) facets tended to favor CH4 production. Compared to Cu (111) facets. The lower coupling barrier and the higher adsorption energy of *CO endow Cu (100) facets with enhanced performance in facilitating C–C coupling through *CO dimerization and subsequent hydrogenation into C2+ products.89 When metal atoms partake in interactions with metal–CO2 and metal-intermediates, accompanied by electron transfer from metal to the bound CO2 or intermediate, it is plausible that the metal atoms may undergo positional changes on the catalyst surface. This dynamic process could potentially lead to an overall morphological transformation of the catalyst. For instance, under the CO2RR reaction conditions, the degradation of Cu nanocubes was noted to coincide with the vanishing of the (100) facets, which was associated with a decrease in C2+ selectivity since the (100) facets played a favorable role in promoting C–C coupling.90
In contrast to the evident morphological alterations observed in pure metallic catalysts, metal compounds undergo a transformation into amorphous oxide or oxyhydroxide layers, typically with a thickness ranging from 2 to 10 nm during the OER process. The discernibility of this change depends on the specific morphology of the precatalyst. As reported, the boronized metal sheets would undergo the dynamic evolution of metaborate-containing oxyhydroxide thin films (2–5 nm) during the OER process. Remarkably, these ultra-thin oxyhydroxide films exerted no significant impact on the overall morphology of the material. However, for mackinawite FeS nanosheets grown on iron foam with unique nanoarray structures, obvious morphology changes could be observed.91 As shown in Fig. 5e, core@shell iron@iron oxysulfide nanoparticles as the catalytically active phase are generated in situ on FeS nanosheets during the HER testing, and FeS nanosheets totally transform into a porous amorphous FeOx film during the OER testing.50 Strikingly, amorphous CoSx nanoboxes underwent a remarkable transformation into low-crystallinity CoOOH nanoplates due to the oxidizing conditions required for the OER51 (Fig. 5f).
In this section, we comprehensively outline the dynamic transformations involving alterations in surface properties, composition, and structure. These profound changes inherently encompass subtle, low-level transformations. In other words, structural evolution is frequently concomitant with variations in components. As these components change, the chemical state and coordination environment generally undergo corresponding alterations. It is worth noting that some studies have reported the reversibility of dynamic transformation on photo/electrocatalysts. As a typical example, the well-known MOF-74 demonstrated a “self-healing” phenomenon.26 It initially transformed into Ni0.5Co0.5(OH)2 and Ni0.5Co0.5OOH0.75 species under different applied potentials and then reverted to its original structure after 15 days in ambient air. In addition, Ni3N, an efficient OER catalyst, exhibited an intermediate conversion from hydroxide/oxide to oxyhydroxide.92 Importantly, the transformation from hydroxide/oxide to oxyhydroxide was reversible, contrasting with the irreversible nature of the transition between Ni3N and hydroxide/oxide. The complexity of photo/electrocatalysis introduces numerous variables affecting dynamic transformation reversibility. Variations in electrolytes and catalyst surfaces can introduce changes in dynamic evolution, even when considering the same material and reaction type. Therefore, reversibility in one case does not necessarily apply to an entire material class. Further research should focus on exploring the reversibility of photo/electrocatalysts.
2.3 Identification
The identification of the dynamic transformation of catalysts during the photo/electrocatalytic reaction can be achieved by a series of experiments and characterization studies. Over an extended past time frame, the dynamic transformation of photo/electrocatalysts before and after reactions has been primarily investigated through the utilization of ex situ characterization and electrochemical measurements. Nonetheless, these ex situ characterization studies frequently encounter challenges in delivering compelling results due to the scarcity of short-lived intermediates, the potential reversibility of reconstruction, and the inherent issues of catalyst aging, damage and contamination during the sample preparation and measurement procedures. Moreover, the indirect evidence presented by electrochemical measurements, including variations in oxidation peaks, changing trends in catalytic current density, and fluctuations in charge transfer resistance, may fall short of offering robust substantiation for catalyst reconstruction phenomena. With the rapid advancement of in situ/operando techniques, real-time information can be acquired to gain comprehensive insights into the intricate dynamic transformation mechanism of catalysts.93 In the following section, we will provide a concise overview of the key attributes of frequently utilized in situ/operando techniques and expound upon their contributions to the elucidation of as-formed active species, the assessment of reversibility phenomena, and the exploration of catalytic mechanisms at the atomic level.In theory, the XAS spectra comprise two regions: X-ray absorption near edge structure (XANES), offering element-specific electronic and bonding insights, and extended X-ray absorption fine structure (EXAFS), which unveils local structural characteristics, encompassing bond distances and coordination numbers surrounding the absorbing atoms. Recently, operando XAS spectra have been obtained to elucidate the sequential dynamic transformations from CuN4 to CuN4/Cu-nanoclusters (NC), and subsequently evolving into CuN3/CuNC during the electrocatalytic ORR process.95 Concretely, the XANES spectra (Fig. 6a) exhibited an outstanding negative shift of the white-line peak edge with the applied potential, suggesting the gradual reduction of Cu2+ to Cu+ during the cathodic ORR process. Furthermore, the primary Cu–N bond at approximately 1.5 Å could be evidently observed in the EXAFS spectra of the Cu K-edge. Furthermore, additional fitting curves illustrated a reduction in the coordination number of the Cu–N bond from 4.5 to around 2.8 during the ORR within the 0.9–0.3V potential range. Simultaneously, Cu–Cu bonds with a coordination number of approximately 3 emerged, suggesting the generation of Cu NC. In the realm of in situ/operando investigations, diffraction patterns serve as invaluable tools for analyzing the dynamic alterations in the phase structure and crystal size of bulk photo/electrocatalysts. As an illustrative example, in situ XRD patterns (Fig. 6b) of Ni(OH)2 provided compelling evidence of the presence of γ-NiOOH at distinct electrochemical potentials, as manifested by the conspicuous appearance of robust peaks.96 Despite the benefits of rapid detection and ease of operation offered by XRD techniques, they fall short in accurately discerning the dynamic transformation of surface-reconstructed species with an amorphous state. X-ray photoelectron spectroscopy (XPS) is a surface-sensitive, quantitative technique known for its ability to provide precise, element-specific insights into electronic states and chemical composition by precisely measuring the kinetic energy of photoelectrons. As illustrated by the operando XPS spectra in Fig. 6c, the XPS peak intensities corresponding to Co3+ and M–O of Co0.8Fe0.2P@C exhibited a noticeable increase, while a decline could be observed in the peak assigned to the P–O bond during electrochemical activation.97 This result substantiated the emergence of active CoOOH species, concomitant with the leaching of P. It is noteworthy that due to its sensitivity to catalyst surface properties, XPS is susceptible to the influence of surface impurities.
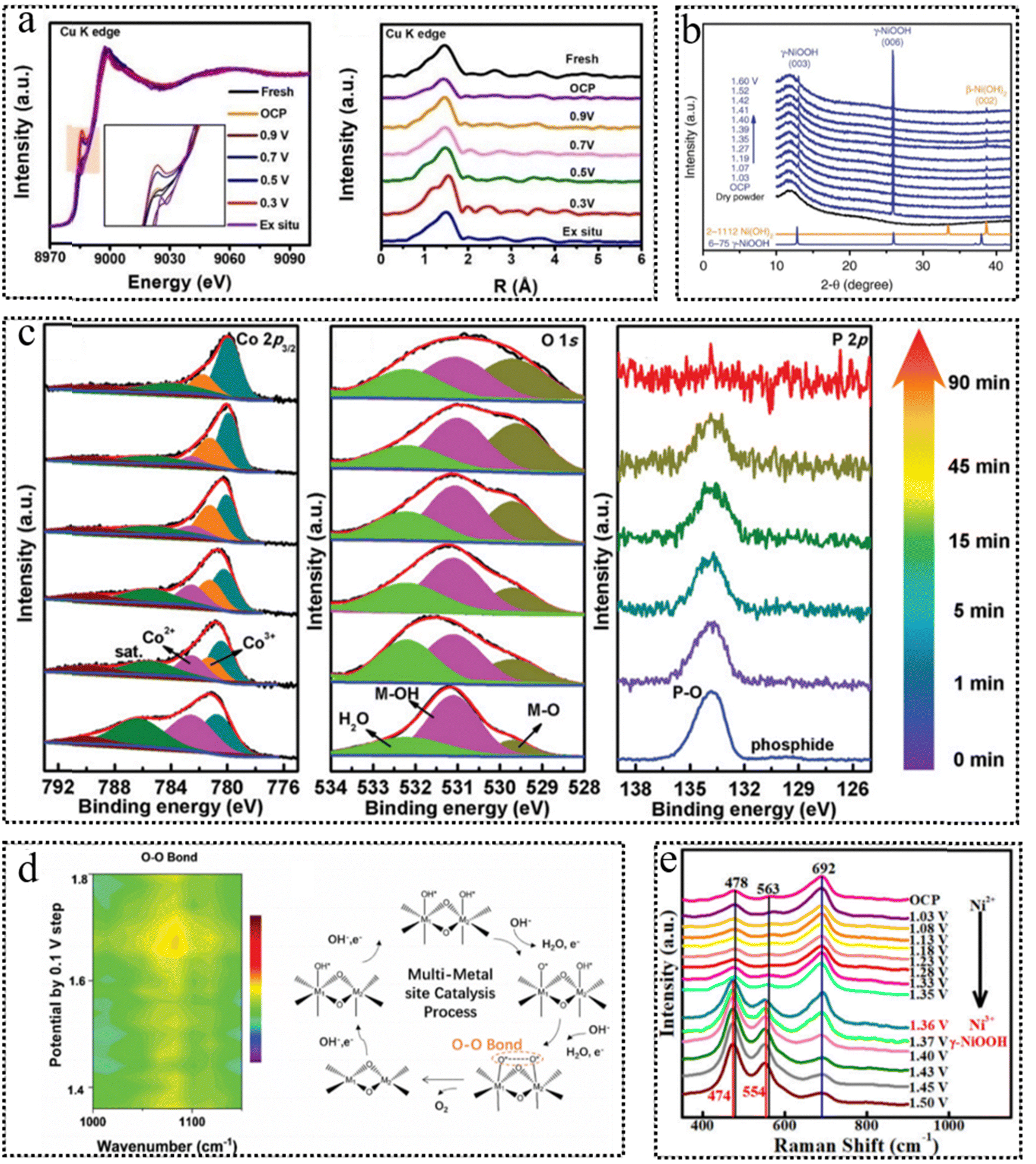 | ||
| Fig. 6 (a) Operando Cu K-edge XANES spectra at different states and Cu K-edge FT-EXAFS spectra of Cu–N–C/GC. This figure has been reproduced from ref. 95 with permission from Wiley-VCH, copyright 2022. (b) Operando XRD patterns of Ni(OH)2/NF at different potentials. This figure has been reproduced from ref. 96 with permission from Nature, copyright 2018. (c) Operando Co 2p3/2, O 1s and P 2p XPS spectra of Co0.8Fe0.2P@C. This figure has been reproduced from ref. 97 with permission from Wiley-VCH, copyright 2020. (d) 2D contour image of the operando ATR FT-IR spectrum for the OER (1.4–1.8 V vs. RHE using 1 M KOH in H216O) and schematic illustration of the multi-metal-site catalytic mechanism. This figure has been reproduced from ref. 98 with permission from Royal Society of Chemistry, copyright 2020. (e) Operando Raman spectra of NFO/NF with photothermal effect (i.e., irradiated with 808-nm NIR light) from OCP to 1.50 V vs. RHE in 1.0 M KOH. This figure has been reproduced from ref. 99 with permission from American Chemical Society, copyright 2021. | ||
Raman spectroscopy, when coupled with probing molecules, provides a means to access surface information about catalysts during catalysis. This technique exhibits a high sensitivity for monitoring the transformation of chemical bonds and functional groups within the bulk catalysts. Note that Raman spectroscopy can identify metal-related bonds and key intermediate species through low-wavenumber signals. The operando Raman measurements (Fig. 6e) were performed to detect the dynamic transformation of NiFe2O4 during the electrocatalytic OER process.99 The active vibrational modes spanning 400 to 700 cm−1 demonstrated potential-dependent behavior, suggesting dynamic phase transformations occurring at the electrode surface within the potential range from open circuit potential (OCP) to 1.50 V. Upon reaching a potential of 1.41 V, the appearance of peaks at 474 and 554 cm−1, which correspond to γ-NiOOH, indicates the generation of Ni3+ species on the electrode surface.
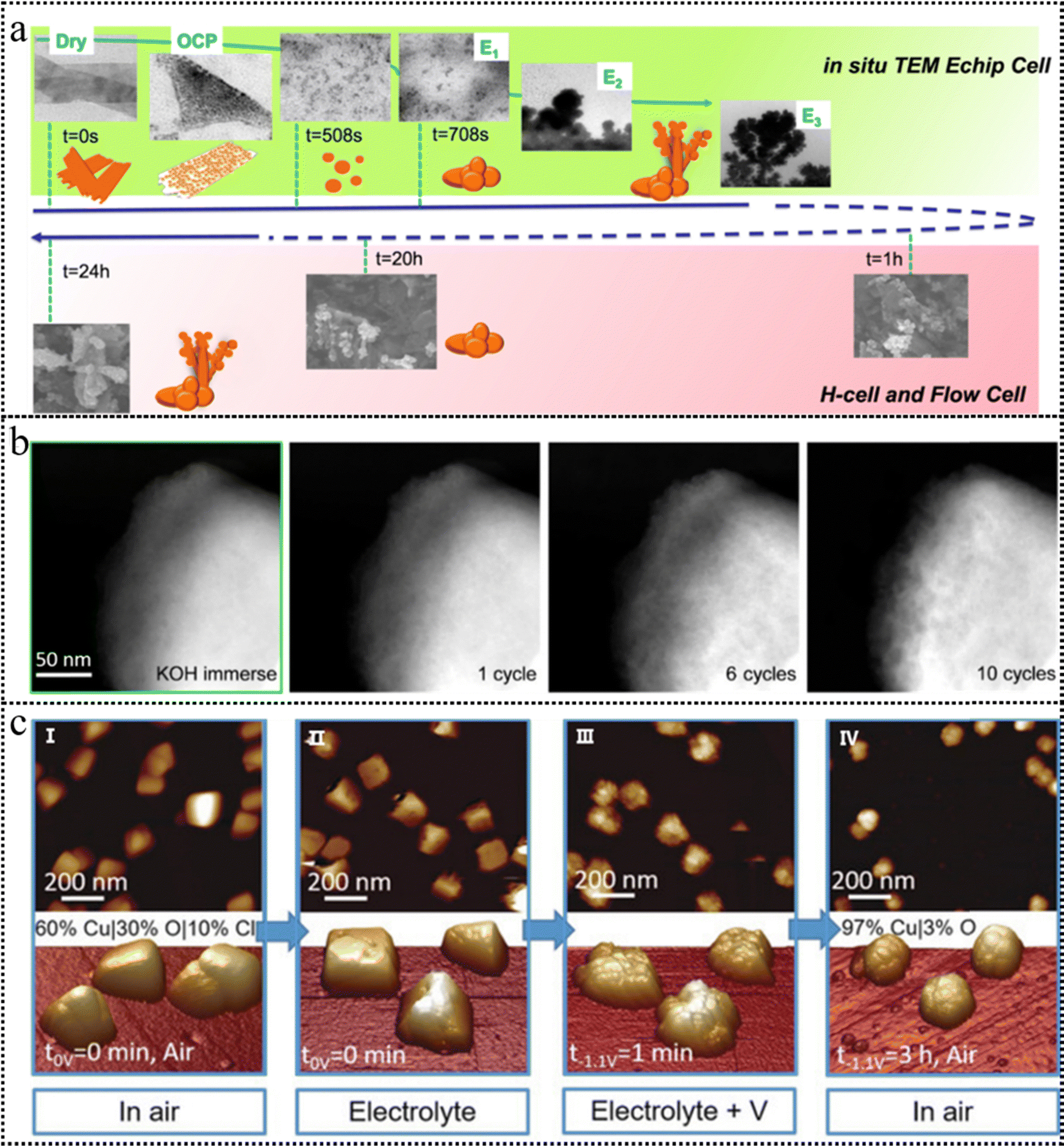 | ||
| Fig. 7 (a) A schematic timeline illustrating the experimentally observed evolution of the CuO NS morphology probed through an in situ TEM E-chip flow cell, H-cell, and flow cell electrolyzer. This figure has been reproduced from ref. 100 with permission from Nature, copyright 2021. (b) HAADF-STEM images of the BSCF catalyst after cyclic voltammetry, conducted within a potential range of 1.1–1.8 V vs. RHE at a scan rate of 10 mV s−1. This figure has been reproduced from ref. 101 with permission from American Chemical Society, copyright 2020. (c) AFM images of Cu cubes electrodeposited on highly oriented pyrolytic graphite (HOPG) acquired in air (I), and operando EC-AFM measurements in a CO2-saturated 0.1 M KHCO3 aqueous solution at open circuit potential (II), at −1.1 V versus RHE in the same electrolyte for 1 minute (III) and after 3h under the same conditions as in (III) and subsequent air exposure (IV). This figure has been reproduced from ref. 102 with permission from Wiley-VCH, copyright 2018. | ||
IL-TEM offers the advantage of monitoring the transformation of identical particles before and after the reaction, enabling precise comparative analysis. IL-TEM was conducted to monitor the morphological evolution of the Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) perovskite during the OER process. Although the IL-TEM images displayed an unaltered morphology of a BSCF particle, the high-resolution identical location high-angle annular dark-field STEM (IL-HAADF-STEM) images (Fig. 7b) exhibited the propensity of the BSCF surface to become a porous configuration.101 Notably, this porosity was observed to increase with an increase in the number of cycles. Due to the existence of a high-energy electron beam, TEM imaging has the potential to induce changes or even irreversible damage to the structure of low-crystalline catalysts. This raises the concern that the observed structural evolution during in situ experiments may be attributed to electron beam-induced irradiation effects.
AFM has emerged as a widely adopted non-contact technique for surface characterization in contemporary research. A tip with atomic sharpness scans across the surface, systematically probing variations in the interatomic forces between the tip and the surface atoms. Subsequently, the surface's atomic-scale topography is meticulously mapped by recording the positional adjustments of scanning points corresponding to the microcantilever. As depicted in Fig. 7c, the operando electrochemical AFM measurements provided clear evidence of the surface roughening of the Cu nanocubes, the vanishing of (100) facets, the emergence of pores, the depletion of Cu, and the reduction of CuOx species during the electrocatalytic CO2RR.102
The capabilities, advantages and disadvantages of the above-mentioned in situ/operando characterization techniques for identifying dynamic remodeling are summarized in Table 1 and Fig. 8. For the identification of dynamic reconstructions in surface chemical states and coordination environments, XAS, XPS, Raman and FT-IR spectroscopy are ideally suited due to their unique capabilities. XRD is instrumental in monitoring phase transformations and variations in crystal size during catalytic reactions. Larger-scale dynamic changes, such as those in size and morphology, can be observed using SEM, TEM and AFM. Additionally, SEM and TEM equipped with Energy Dispersive Spectroscopy (EDS) are useful for detecting elemental redistribution, while high-resolution TEM also excels in observing phase transformations. Theoretically, only the integration of multiple in situ/operando characterization techniques can furnish substantial evidence for the dynamic transformation occurring in active sites. Future research should place an emphasis on resolving the impact of instrumentation and operating conditions on catalysts during in situ/operando characterization.
| Types | Techniques | Capabilities | Advantages | Disadvantages |
|---|---|---|---|---|
| X-ray characterization | XAS | Chemical states and coordination environments | High precision and extensive applicability | Vulnerable to mutual elemental interference and overlapping peaks |
| XRD | Phase structure and crystal size | Rapid detection and straightforward operation | Not suitable for amorphous structure testing | |
| XPS | Elemental composition and chemical valence states | High sensitivity and excellent spectral repeatability | Fail to detect the bulk phase and sensitive to surface impurities | |
| Spectroscopic characterization | FT-IR | Group composition and molecular structure | High penetration | Limited sensitivity |
| Raman | Chemical bonds and groups | High sensitivity with rapid testing | Sensitive to optical parameters | |
| Microscopic characterization | SEM | Surface morphology | Stereoscopic and wide-field imaging | Low resolution and inability to show fine structure |
| AFM | Surface morphology and thickness | Stereoscopic and wide-field imaging | Low resolution and inability to show fine structure | |
| TEM | Surface morphology and hyperfine structure | High resolution and hyperfine structure clarity | Only captures two-dimensional information |
 | ||
| Fig. 8 In situ/operando characterization techniques for identifying internal electric fields and their specific functions. | ||
3. Triggers of reconstruction
Photo/electrocatalysis relies on catalysts, electrolytes, and applied bias as primary components. Catalysts possess intrinsic physicochemical attributes that govern their self-construction. This section delves into the myriad factors influencing this self-assembly to enhance overall.3.1 Single-atom catalysts
Single-atom catalysts, including cluster catalysts, have been intensively studied in catalysis due to their exceptional atomic efficiencies and heightened activities.103–105 However, prevailing research typically adheres to the synthesis–characterization–performance testing paradigm. Consequently, our understanding of catalyst active sites predominantly adopts a “static” perspective, disregarding the dynamic structural evolution of single atoms amidst catalyst activation and reaction conditions. This oversight poses critical challenges. On the one hand, single-atom catalysts may undergo reconstruction during catalytic reactions, creating disparities between the actual active sites and the initial catalytic configurations.106 Conversely, uncontrolled active site reconstruction might hasten catalyst degradation. Hence, leveraging the interaction between single atoms and their support becomes pivotal in regulating the extent of reconstruction.Small metal species like single atoms and nanoclusters possess high surface energy, predisposing them to aggregate, especially under reductive potential. This tendency often proves detrimental, leading to catalytic performance decline.107–110 Paradoxically, such aggregation might also facilitate catalytic reactions by transforming single atoms into nanoclusters, thereby assuming the role of actual active sites. Wei et al. reported a novel catalytic urea synthesis method, using Cu single atoms incorporated on a ceria support as the catalyst, to directly couple nitrate and carbon dioxide.111 The paper reveals the structural evolution of the catalyst during urea synthesis reaction by using XAS and IR spectroscopy techniques. From the operando XAS results depicted in Fig. 9a and b, it was found that Cu single atoms reconstitute into copper clusters at reducing potentials, and the copper clusters are the real active sites. Additionally, the schematic diagram in Fig. 9c illustrates the dynamic and reversible transformation of Cu4 into Cu1 configurations when switching the applied potential to the open-circuit potential. The paper also investigates the reaction mechanism of the catalyst by using density functional theory calculations and kinetic measurements and finds that copper clusters can effectively adsorb and activate nitrate and carbon dioxide, and significantly reduce the activation energy of O–O coupling. Another work done by Dilan Karapinar et al. shows the same phenomenon of Cu single atoms catalysts.112Operando XAS analysis reveals that the isolated sites are gradually transformed into metallic copper nanoparticles under working conditions. These metallic copper nanoparticles are believed to be the catalytically active species. Interestingly, from the XAS characterization of post-electrolysis samples without applying voltage, the disappearance of the metallic copper phase and the restoration of the original spectra illustrate that the restructuring behavior is reversible (Table 2).
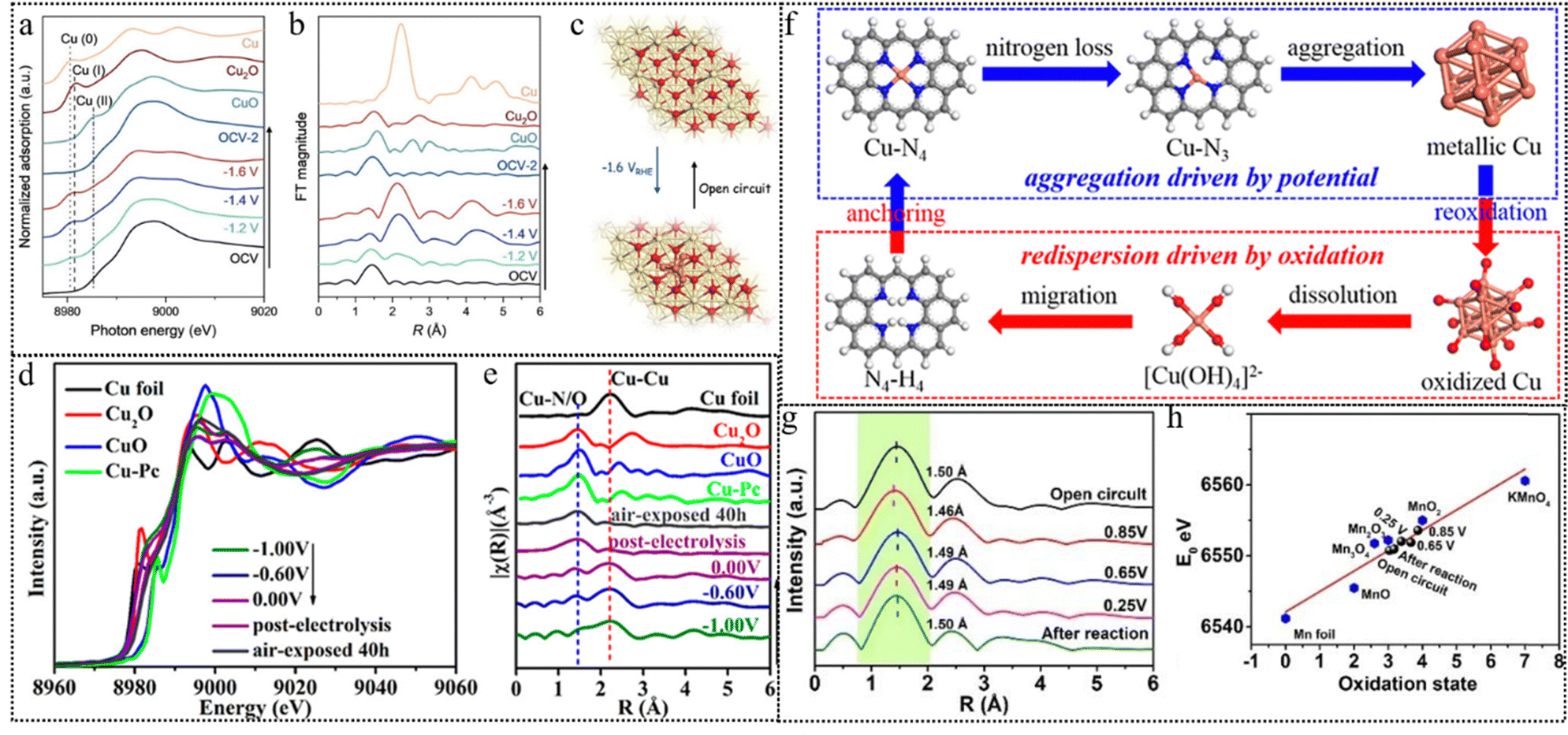 | ||
| Fig. 9 (a) Cu K-edge XANES spectra of Cu1–CeO2 recorded at different cathodic potentials during C–N coupling. (b) Cu R-space EXAFS spectra of Cu1–CeO2 recorded at different cathodic potentials during C–N coupling. (c) Schematic diagram of reconstitution of copper single-atoms to clusters suggested by the operando XAS measurements. This figure has been reproduced from ref. 111 with permission from Wiley-VCH, copyright 2023. (d) Cu K-edge XANES spectra at different potentials from −1.00 to 0.00 V vs. RHE. (e) Cu K edge FT-EXAFS spectra by switching potentials from −1.00 to 0.00 V vs. RHE. (f) Proposed mechanisms of aggregation driven by the applied potential and re-dispersion in the oxidation environment. This figure has been reproduced from ref. 113 with permission from American Chemical Society, copyright 2022. (g) Mn K-edge FT-EXAFS spectra at open circuit, different applied potentials, and after reaction. (h) Average oxidation state of Mn in the Mn single atom sites at open circuit, 0.85 V, 0.65 V, 0.25 V and after reaction compared with Mn foil, MnO, Mn2O3, Mn3O4, MnO2 and KMnO4. This figure has been reproduced from ref. 114 with permission from Wiley-VCH, copyright 2023. | ||
| Intrinsic triggers | Reconstruction types | Precatalysts | Reconstructed species | Reaction/electrolyte | Characterization | Ref. |
|---|---|---|---|---|---|---|
| Single-atom catalysts | Surface properties | Cu1–CeO2 | Cu4–CeO2 | Urea synthesis/0.1 M KHCO3 with 50 mM KNO3 | TEM, STEM, and Operando XAS | 111 |
| Surface properties | Cu–N4 | Metallic Cu nanoparticles | CO2RR/0.1 M CsHCO3 | TEM, STEM, and Operando XAS | 112 | |
| Surface properties | Cu–N4 | Metallic Cu nanoparticles | Reduction of nitrate to ammonia/0.1 M KOH including 0.1 M KNO3 | TEM, STEM, and Operando XAS | 113 | |
| Composition changes | Ru/Ni3V-LDH | Ru–O–Ni | OER&HER/1.0 M KOH | TEM, STEM, and Operando XAS/Raman | 115 | |
| Surface properties | Mn–N4 | Mn–N3/C, Mn–N2/C, and Mn–N/C | ORR/0.1 M KOH | TEM, STEM, and Operando XAS | 114 | |
| Defects engineering | Composition changes | n-type α-Ni–Fe LDH | p-type γ-Ni–Fe LDH | OER/1.0 M KOH | TEM, STEM, XAS, EPR, and FTIR | 116 |
| Surface properties | (110) facet of WO3 | 5-fold symmetry pentagonal column structure | — | Cs-STEM | 117 | |
| Composition changes | Co3O4 | Co(OH)2 intermediate with proton vacancies (Co(II)Ox(OH)y) | OER/1.0 M NaOH | TEM, STEM, XAS, and Operando Raman/ATR-IR/TEM | 118 | |
| Composition changes | NiFe LDH | γ-(NiFe)OOH | OER/1.0 M KOH | TEM, STEM, XAS, and Raman | 119 | |
| Composition changes | VNi-α-Ni(OH)2−x | NiOOH | OER/1.0 M KOH, UOR/1.0 M KOH + 0.33M urea | TEM, STEM, XAS, ESR, and PAS | 120 | |
| Composition changes | CoF2 porous nanowires | F–CoOOH nanosheets | OER/1.0 M KOH | TEM, STEM, and XAS | 121 | |
| Structural reconstruction | ||||||
| Composition changes | Ni1−xFexPS | Disordered MOOH@MxSOy@MxPOy(M = Ni, Fe) | OER/1.0 M KOH | TEM, XAS, and Raman | 122 | |
| Composition changes | Mo/N substituted CoO | CoOOH | OER/1.0 M KOH | Cs-STEM/TEM, real-time XPS | 123 | |
| Composition changes | Pr3Ir1−xMoxO7 | Ir-Obri-Mo species | OER/0.1 M HClO4 | TEM, EPR | 124 | |
| Heterostructure construction | Surface properties | NiSe2/FeSe2 heterostructure | N(III)–O–Fe(III) | OER/1.0 M KOH | TEM, XAS, and Raman | 125 |
| Composition changes | LaNiO3/NiO | LaNiO3/NiOOH | OER/1.0 M KOH | TEM, STEM, XAS, and Operando Raman | 126 | |
| Composition changes | Core/shell Pd/Fe3O4 nanoparticles | Core/shell Pd/FeOx(OH)2−2x nanoparticles | HER/0.1 M KOH | TEM, XAS | 127 | |
| Composition changes | Nii-P–O/MoOx | Ni–P–O | OER/1.0 M KOH | TEM, Raman, FTIR, and | 128 | |
| Composition changes | LaNiO3 | VLa-LaNiO3/NiOOH | OER/1.0 M KOH + 0.1 M H2SO4/0.1 M H2CO3/0.1 M HNO3 | TEM | 129 | |
| Structural reconstruction | ||||||
| Composition changes | Spinel Li2Co2O4 | Amorphous Li2−xCo2O4−δ(OH)δ (Co3+/Co4+) | OER/1.0 M KOH | TEM, STEM, XAS, and Raman | 130 | |
| Structural reconstruction | ||||||
| Surface properties | ||||||
| Composition changes | Co2(OH)3Cl | β-CoOOH | OER/1.0 M KOH/1.0 M PBS buffer | TEM, STEM, and XAS | 131 | |
| Composition changes | IrFeCoNiCu HEA | Ir rich shell | OER/0.1 M HClO4 | TEM and STEM | 132 | |
| Composition changes | Fe0.01–Ni/Ni0.2Mo0.8N | Fe0.01/Mo–NiO | HER/1.0 M KOH/1.0 M KOH + natural seawater | TEM and Operando Raman | 133 | |
| Composition changes | Co-MOF/Fe10 | CoFe LDH | OER/1.0 M KOH | TEM, STEM, and Raman | 134 | |
| Composition changes | NiFe PBA | NiOOH/FeOOH | OER/1.0 M KOH | TEM, STEM, Raman, and FTIR | 135 | |
| Composition changes | NiFeP | NiOOH/FeOOH | OER&HER/1.0 M KOH | TEM | 136 | |
| Composition changes | NiFe/NiSx | NiFe-LDH intercalated SO4, and CO3 anions | OER/1.0 M KOH | Raman and TOF-SIMS | 33 | |
| Surface properties | ||||||
| Composition changes | NiOOH | FexNiOOH | OER/1.0 M KOH + 10 uM Fe3+ | XAS and XFM | 137 | |
| Composition changes | NiO | NiOOH/NiO and NiFe LDH/NiO | 0.5 M borate buffer + 1 mM Fe(NO3)3 | EC-TEM, STEM, Operando XAS/Raman, and time-resolved ICP-MS | 138 | |
| Surface properties | ||||||
| Structural reconstruction | ||||||
| Composition changes | Bi-MOF | Bi2O2CO3 and metallic Bi | 0.1 M–1.0 M KHCO3 aqueous solution | TEM, STEM, FTIR, and OperandoRaman/ATR-SEIRAS | 139 | |
| Surface properties | ||||||
| Crystallinity | Structural reconstruction | Crystalline Ni3Fe | Amorphous Ni3Fe | 1 M ultra pure KOH | TEM, STEM, and Operando XAS | 140 |
| Composition changes | Amorphous NiFeMo | NiFeOOH | OER/0.1 M KOH | TEM, STEM, XAS, UPS, and Operando Raman | 141 | |
| Structural reconstruction | Amorphous SrCo0.85Fe0.1P0.05O3−δ | Reconstructed SCFP | OER&HER/1.0 M KOH | TEM and XAS | 142 | |
| Composition changes | Amorphous Pt nanospheres | Crystalline clusters with Pt–O ligand | HER/0.5 M H2SO4, HOR/0.1 M HClO4 | TEM, Raman, FTIR, and UV-vis | 143 | |
| Surface properties | ||||||
| Structural reconstruction | ||||||
| Composition changes | H–Co(OH)2@Fe(OOH)1−xClx | CoOOH@FeOOH | OER/1.0 M KOH | TEM, 57Fe isotope labeled ICP-MS | 144 | |
| Surface properties | ||||||
Yang et al. used operando XAS combined with transmission electron microscopy (TEM) to reveal that the synthesized Cu–N4 single-atom site underwent restructuring, forming nanoparticles of about 5 nm during the chemical process of nitrate reduction to ammonia.113 According to the operando XAS measurements in Fig. 9d and e, the reduction of Cu2+ to Cu+ and Cu0, followed by the aggregation of copper single atoms, aligned with the increase in the NH3 production rate induced by the applied potential, as the applied potential scanning from 0.00 V to −1.00 V. At −1.00 V, the catalyst achieved its peak NH3 production rate at 4.5 mg cm−2 h−1 (12.5 mol NH3 gCu−1 h−1), exhibiting a Faraday efficiency of 84.7%, surpassing the performance of most previously reported Cu catalysts. The proposed mechanism is depicted in Fig. 9f. When the applied potential was removed, the aggregated Cu nanoparticles (NPs) demonstrated a reversible transformation into single atoms, which subsequently reverted to the Cu–N4 structure upon exposure to the ambient atmosphere. This phenomenon typically obscures the potential-driven restructuring process that is challenging to observe during the reaction. These findings suggest that Cu NPs serve as the primary catalytic site for the reduction of nitrate to ammonia, corroborating the results obtained from post-deposited Cu NP catalysts and DFT calculations.
A single metal atom may also migrate instead of aggregation from one type of bonding site to another. Here is an example. The authors employ operando XAS to monitor the dynamic process of the Mn–N4 configuration and the valence state of Mn atoms throughout the oxygen reduction reaction (ORR).114 Through analysis of the Mn K-edge FT-EXAFS spectra in Fig. 9g, it is observed that the primary scattering peak, indicative of the Mn–N/C bond length, shifts successively from 1.50 to 1.46 Å, and then to 1.49 Å. This shift illustrates the transformation of the Mn–N4 configuration into Mn–N3C and subsequently into Mn–N2C under varying applied potentials. The linear fitting results according to the XANES spectra are presented in Fig. 9h. During the transformation of the coordination environment of Mn, the valence state of Mn changes from +3.0 to +3.8 and then to +3.2.
In addition to the involvement of single atom migration in the reconstruction, single-atom modification can also serve to anchor active sites, thereby preventing excessive reconstruction. For instance, Sun et al. demonstrated that a catalyst with Ru atoms atomically dispersed on a nickel–vanadium (Ni3V) layered double hydroxide (LDH) support, achieved through a robust atomic metal–support interaction, exhibited remarkable activity in both the HER and OER.115In situ XAS and operando Raman spectroscopy studies have shown that the presence of Ru atoms on the surface of nickel–vanadium LDH plays a crucial role in stabilizing the surface rich in dangling bonds, further leading to a non-reconstructed surface. Due to the robust metal–support interaction on nickel–vanadium LDH, the substantial interaction effectively increased the stability of the reactive atomic Ru sites, minimizing oxidation state fluctuations during the cathodic HER without necessitating reconstruction. In addition, Ru atomic sites can stabilize Ni sites, rendering them more resilient to bond contraction and structural distortion induced by oxidizing Ni sites during the anodic potential. This stabilization facilitates an increase in the oxidation state of Ni sites, thereby enhancing the catalyst's OER performance. Unlike other bifunctional catalysts that necessitate structural reconstruction or transformation to accommodate HER/OER cycles, the Ru/Ni3V LDH catalyst possesses stable dual reaction sites and exhibits a robust metal–support interaction (i.e., between Ru and Ni sites) during the water decomposition process.
3.2 Defect engineering
The deliberate introduction of defects significantly influences a catalyst's electronic configuration and the surrounding coordination environment of metal sites. This strategic maneuver accelerates photon/electron transfer, thereby augmenting catalytic performance.145,146 Defect introduction stands as an effective means to regulate the reconstruction of catalysts. For example, thermodynamically equilibrium defects such as point defects (vacancies and interstitials) exhibit mobility or reactivity under external stimuli, such as light or specific potentials during catalysis.22,147–149 This phenomenon promotes the formation of catalytic intermediates, rendering the system more stable through reconstruction. Conversely, line defects, though non-equilibrium defects themselves, often undergo spontaneous reconstruction. However, their exploration within the catalysis field remains limited and lies beyond the scope of this discussion. Surface defects, such as grain boundaries, represent another common avenue for modulating catalyst performance. By impeding atomic migration, these boundaries offer a means for controlled reconstruction, thereby allowing for the adjustment of catalyst performance. These defects play a pivotal role in modulating the adsorption energies of active species of the catalyst's surface, thereby selectively stabilizing specific active species and fine-tuning catalytic performance.150,151,152Inherent oxygen vacancies (VO) can facilitate the initiation of reconstruction during catalytic reactions by enhancing the photon/electron density around metal atoms and reducing the valence states of metal ions. Zhou et al. presented a novel strategy to synthesize ultrathin Ni–Fe LDH nanosheets with inherent VO by a coprecipitation method. They then examined the influence of these vacancies on the catalyst structure and the performance in catalytic OER performance.116 The study revealed that oxygen vacancies facilitate the surface reconstruction of Ni–Fe LDH, resulting in the generation of a p–n interface within the γ-Ni–Fe LDH/α-Ni–Fe LDH heterostructure as shown in the schematic diagram in Fig. 10a, which can enhance the electron density, reduce the oxidation state, optimize the OER reaction pathway, and lower the overpotential. VO on the surface can induce surface structure reconstruction as a result of an enhancement in surface catalytic activity. Using spherical aberration-corrected scanning transmission electron microscopy (Cs-STEM) with atomic-scale precision, Meng et al. directly observed the surface restructuring on the step areas or high-index facets of WO3.117 The area with a significant abundance of VO promotes the formation of a unique structure with 5-fold symmetry illustrated using the Cs-STEM image in Fig. 10b. As Fig. 10c shows the proposed reconstruction pathway, the presence of oxygen vacancies at the edge breaks the local symmetry and lowers the coordination number of W atoms outside. Consequently, these unsaturated coordinated W atoms tend to migrate into interstitial places to lower the surface energy finally forming such special structures. DFT calculations reveal that the step area requires much lower formation energy of oxygen vacancies which follows previous observation. This work presents that enhancement of surface reconstruction can be spontaneously achieved by high concentrations of oxygen vacancies. The introduction of oxygen vacancies often accompanies the structure reconstruction of the catalyst and the improvement of OER activity. As a skeleton mechanism of the OER, the LOM shows that the evolution of the O2 molecule involving lattice oxygen would generate oxygen vacancies that are occupied by the adsorbed OH− in the electrolyte in the later cycles.150 It means oxygen vacancies can affect and participate in structural reconstruction from the reaction mechanism.
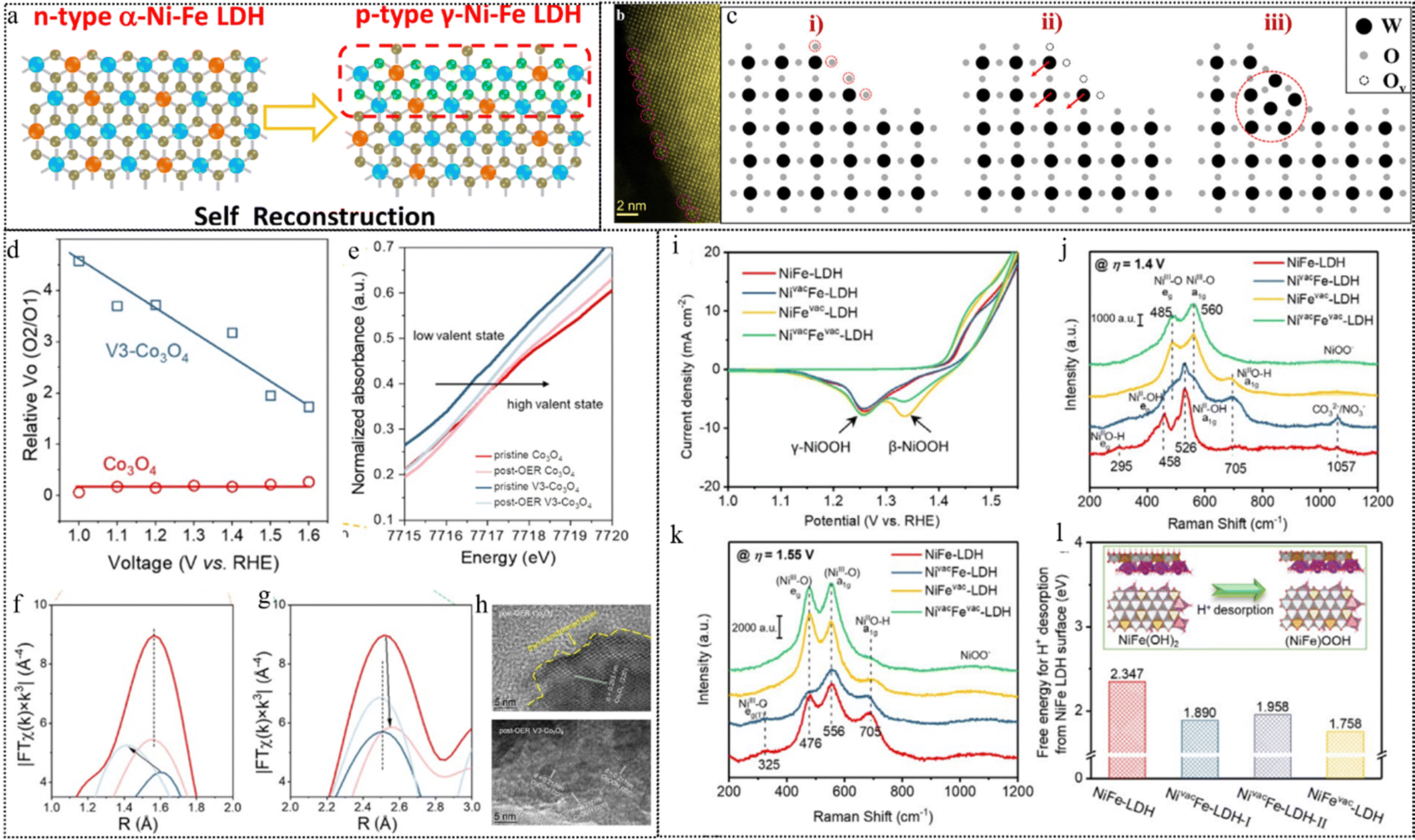 | ||
| Fig. 10 (a) Schematic diagram of a p–n interface via the reconstruction of p-type γ-Ni–Fe LDH on n-type α-Ni–Fe LDH. This figure has been reproduced from ref. 116 with permission from American Chemical Society, copyright 2021. (b) Atomic-resolution STEM-HAADF images of the surface atomic structure of WO3 with pentagonal columns. (c) The schematic images of surface reconstruction. This figure has been reproduced from ref. 117 with permission from American Chemical Society, copyright 2021. (d) Relative surface Vo (O2/O1) contents of Co3O4 and V3-Co3O4 in the ex situ XPS measurements under different applied potentials. (e) XANES spectra of Co3O4 and V3-Co3O4 before and after the OER. (f) and (g) EXAFS spectra of Co3O4 and V3-Co3O4 before and after the OER. (h) Post-OER HRTEM images of Co3O4 and V3-Co3O4. This figure has been reproduced from ref. 118 with permission from American Chemical Society, copyright 2023. (i) ECSA-normalized CV curves at a scan rate of 10 mV s−1. (j), (k) Raman spectra at applied potentials of (j) 1.4 V and (k) 1.55 V vs. RHE. (l) DFT calculated free energy of H+ desorption from the surface of different NiFe-LDH-based structural models. This figure has been reproduced from ref. 119 with permission from Wiley-VCH, copyright 2021. | ||
Zhou et al. investigated the impact of Vo on structural reconstructing under OER conditions using a Vo-rich spinel Co3O4 as a model catalyst.118 From their XPS results (Fig. 10d), they observed that the comparative surface Vo concentration (ratio of O2/O1 deconvoluted from XPS of O 1s) of Co3O4 remains nearly constant regardless of the working potential, suggesting the inactive nature of the inherent low concentration of Vo and their limited transformation. Conversely, the relative Vo content of V3-Co3O4 steadily decreases with increasing positive potential, indicating a significant transformation of the introduced Vo during activation and the OER processes. Additionally, changes in the valence state of Co are confirmed by XAS of the near-edge structure (Fig. 10e). Analysis of the EXAFS spectral results (Fig. 10f and g) reveals that the Co–O distances for Co3O4 exhibit a minimal variation after the OER, indicating the relatively stable spinel structure. In contrast, the Co–O distances decrease for V3-Co3O4 after the OER due to the formation of CoOOH. While disordered lattice fringes are observed only at the surface within a thin layer, indicating a low degree of surface reconstruction in Co3O4, V3-Co3O4 exhibits a highly disordered lattice arrangement and increased grain boundaries post-OER (Fig. 10h). The presence of a d spacing of 0.217 nm, indexed to the (200) facet of CoOOH, further confirms the facilitated structural transformation by the introduced Vo during the OER. Subsequent correlative operando characterization and electrokinetic analyses suggest that a moderate Vo density can alter the O–O bond formation pathway, favoring an intramolecular nucleophilic attack pathway over a water nucleophilic attack pathway, which is more kinetically favorable for water oxidation.
Likewise, the presence of cation-type vacancies in precatalysts can stimulate rapid surface reconstruction and facilitate achieving reactive and stable photo/electrocatalysts. Peng et al. presented a method to improve the activity and stability of NiFe-LDH materials for the oxygen evolution reaction (OER) in alkaline media by atomic cation-vacancy engineering, which inhibits the dissolution of metals and allows the pre-catalyst to rapidly restructure into a structure with high catalytic activity.119 The authors synthesized NiFe-LDH with Zn2+/Al3+ doped precursors by a hydrothermal method and then selectively etched Zn2+ and Al3+ with 1.0 M KOH solution to construct Zn2+ vacancies, Al3+ vacancies, or mixed with Zn2+ and Al3+ vacancies on the basal plane of LDH nanosheets. The authors investigated the structural evolution of the LDH catalysts during the OER by CV (Fig. 10i) and Raman spectroscopy (Fig. 10j and k). They show that the cation vacancies (especially cation(III) vacancies) expedite the formation of γ-(NiFe)OOH active sites from NiFe(OH)2 layers in the LDHs, and also change the structure evolution pathway from a direct conversion to a two-step conversion via the β-(NiFe)OOH intermediate (Fig. 10l). The researchers also show that the cation vacancies lower the free energy for deprotonation on the LDH surfaces, thus facilitating the OER process.
Another similar example is that He et al. reported Ni(OH)2 with controllable concentrations of nickel vacancies (VNi) that promote the formation of active species (NiOOH) by enhancing charge transfer and reducing formation energy.120 The photo/electrochemical measurements and DFT calculations support the correlation between VNi and the catalysts’ reconstruction ability during reactions. Specifically, the introduction of VNi leads to the emergence of new defect levels and an increase in hole density near the Fermi level, accelerating charge transfer and facilitating the generation of NiOOH.
Ion incorporation into pre-catalysts, as another type of conventional point defect, optimizes their electronic structure and promotes dynamic restructuring.153 This reconstruction is induced by doping with metal or non-metal heteroatoms, triggering a reorganization of atomic arrangements.154,155 Specifically, doping of F on the metal-based catalysts can form weak M–F bonds. These bonds are readily broken during the reconstruction phase, leading to the conversion into F-doped metal (oxy)hydroxides. Furthermore, doping of F adjusts the electronic configuration of metal sites and modulates the energy of adsorption, thereby enhancing catalytic activity.156 Chen et al. exploited the high electronegativity and weak metal–fluorine bond characteristics of fluorine anions, which enabled them to migrate dynamically from inside to outside and enrich on the surface of cobalt-based materials during electrochemical activation.121 The surface-enriched fluorine anions increased the hydrophilicity of the materials, facilitating the contact between reactants and active sites, as well as improving the photon/electron transfer ability and adsorption energy of reactants. The researchers conducted a series of structural characterization studies to investigate the effect of F− on the structure reconstruction of F–Co3O4 under OER conditions. As shown in Fig. 11a, F− are primarily concentrated on the surface of F–CoOOH nanosheets, and the surface hydrophilicity increased after fluorine doping, which facilitated the contact between water and active sites. The synthesis of the pure phase of Co3O4 was revealed from the XRD pattern of the F–Co3O4 sample post the OER test. The O K-edge XANES spectra of F–Co3O4 after OER tests (Fig. 11b) indicated the evolution of surface oxyhydroxides during the OER process, further supported by the Raman spectra of F–Co3O4 before and after OER tests (Fig. 11c). The missing F2g peak and the rightward shift of Eg and A1g peaks suggested a gradual conversion of the surface of F–Co3O4 into F–CoOOH during the OER process. In Fig. 11d, the ex situ HRTEM images and element mapping revealed the visual surface reconstruction phenomenon. These results suggested that F anions can dynamically migrate within the material to its surface during the photo/electrochemical activation process. This migration promotes the generation of catalytically active layers (F–CoOOH) and enhances the OER catalytic activity of cobalt-based electrode materials.
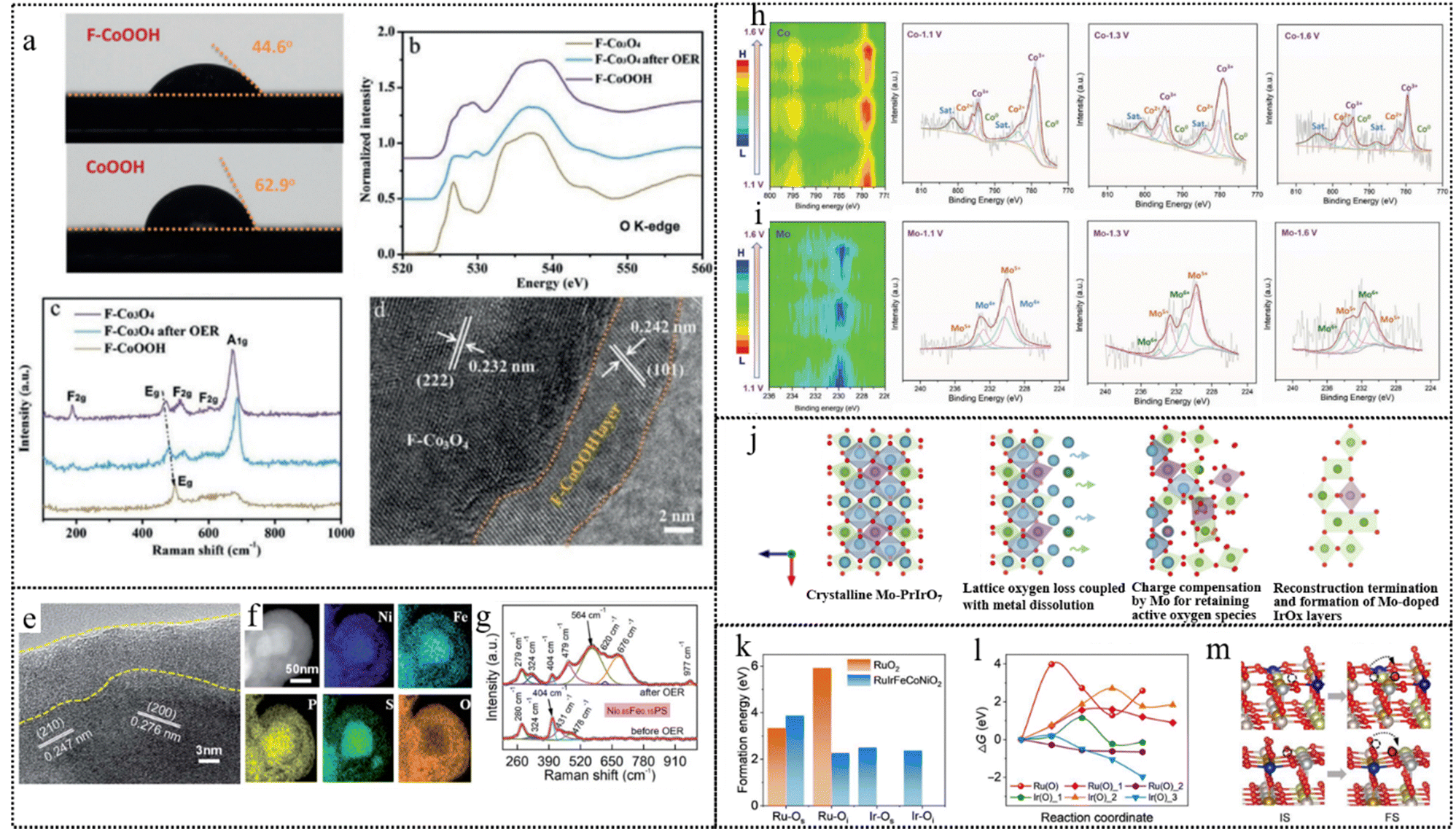 | ||
| Fig. 11 (a) Static water contact-angle measurements of F–CoOOH and CoOOH products. (b) The O K-edge XANES spectra and (c) Raman spectra of F–CoOOH, F–Co3O4, and F–Co3O4 after the OER test. (d) The ex situ HRTEM image of the F–Co3O4 product after OER tests. This figure has been reproduced from ref. 121 with permission from Wiley-VCH, copyright 2018. (e) HRTEM and (f) TEM-EDX maps of the Ni0.85Fe0.15PS after catalysis. (g) Raman spectra of Ni0.85Fe0.15PS before and after the OER. This figure has been reproduced from ref. 122 with permission from The Royal Society of Chemistry, copyright 2020. (h) and (i) Potential-dependent XPS structural evolution of (h) Co 2p and (i) Mo 3d in the CMN-500 catalyst at the reconstruction stage within the potential ranging from 1.1 to 1.6 V during the alkaline OER, and the corresponding fitted XPS spectra at the potentials of 1.1, 1.3, and 1.6 V. This figure has been reproduced from ref. 123 with permission from Wiley-VCH, copyright 2021. (j) Schematic illustration of chemical surface reconstruction over Mo–Pr3IrO7. This figure has been reproduced from ref. 157 with permission from Springer Nature Publishing Group, copyright 2023. (k) Formation energy of O vacancies in RuO2 and RuIrFeCoNiO2. Os and Oi refer to the surface and inner oxygen that coordinate with Ru/Ir, respectively. (l) Free energy diagram for oxygen diffusion of Ru-coordinated oxygen in RuO2 [Ru(O)] and RuIrFeCoNiO2 [Ru(O)_1, and Ru(O)_2], and Ir-coordinated oxygen in RuIrFeCoNiO2 [Ir(O)_1, Ir(O)_2, and Ir(O)_3]. (m) Representative models for IS and FS of Ru(O)_2 (top) and Ir(O)_3 (bottom) in l, the hazel, pale green, tan, cyaneous, gray, and red spheres in the atomic models represent Ru, Ir, Fe, Co, Ni, and O atoms, respectively, IS and FS represent the initial state and final state, respectively. This figure has been reproduced from ref. 124 with permission from American Association for the Advancement of Science, copyright 2023. | ||
Dopants can also enhance charge transfer and modulate the electronic structure in pre-catalysts, leading to changes in the oxidation state of metal atoms and influencing the kinetics of reconstruction. The team of Peng et al. found that a certain content of Fe doping in NiPS (Ni0.85Fe0.15PS) stimulates the activation of higher oxidation state NiFe (oxy)hydroxides, driving the irreversible surface reconstructing.122 The surface morphology and composition of Ni1−xFexPS compounds before and after the OER by HRTEM (Fig. 11e) and TEM-EDX maps (Fig. 11f) revealed that moderate Fe regulation in NiPS forms distortion on MOOH@MxSOy@MxPOy surfaces during the OER process. The authors used Raman spectroscopy to study the vibrational modes of the Ni1−xFexPS compounds (Fig. 11g) and found that Fe doping enhances the electron transfer between Ni and S atoms, which is beneficial for self-reconstruction in the OER. DFT calculations were conducted to investigate the electronic structure and mechanism of the Ni1−xFexPS compounds and found that new active sites formed by Fe doping after structure reconstruction exhibit higher nucleophilicity compared to Ni sites, rendering them more active for the oxygen evolution reaction (OER). This enhanced activity is attributed to the facile oxidation state change between Fe(II) and Fe(III).
Some dopant elements are relatively unstable under catalytic conditions, leading to reconstruction due to reactions, such as some main group elements (N, S, Se, P, etc.34,158,159) or transition metal elements containing multiple oxidation state forms. Bai et al. employed a cationic molybdenum-substitution strategy to prepare polymorphic cobalt with various phase compositions by a solid-state reaction on two-dimensional cobalt oxide (CoO) nanosheets. These compositions include CoO and a metallic cobalt phase with a face-centered cubic structure or hexagonal close-packing (hcp).123 The Mo substitution acts as an electron donor, stabilizing the metastable hcp metallic cobalt phase in polymorphic cobalt oxides. This process reduces the energy barrier of rapid surface changes, a critical factor in the generation and stabilization of active sites for the OER. The authors used the real-time XPS (Fig. 11h and i) technique to track the evolution of surface chemical states during the catalytic reaction, revealing the effects of different phase compositions on the initial surface reconstruction kinetics under OER conditions in an alkaline environment. The results showed that the hcp metallic cobalt phase in the molybdenum-promoted polymorphic cobalt catalyst could effectively facilitate surface reconstructing and generate durable active sites, thus reaching optimal OER catalysis performance. According to DFT calculations, the most rapid structural reconstruction kinetics and the lowest free energy barrier among the investigated catalysts are attributed to the co-existence of the mixed metallic phases and oxide phases of cobalt, as well as the presence of molybdenum and nitrogen.
Controlling reconstruction in a precise manner is pivotal for fabricating highly active catalysts, especially for noble metal catalysts with high activity and unsatisfactory stability, to prevent the excessive dissolution of active sites. Chen et al. presented a novel catalyst, orthorhombic Pr3IrO7 doped with high-valence Mo, which can catalyze water oxidation reactions under acidic conditions.157 The authors use density functional theory calculations and experimental methods to reveal the process of Mo-promoted surface reconstruction depicted in Fig. 11j, forming self-terminated Ir-Obri-Mo active species. Mo doping not only enhances the Ir–O covalency and lattice oxygen activity, leading to faster surface reconstruction but also buffers the charge imbalance caused by Pr leaching by increasing the Mo oxidation state, preventing excessive loss of lattice oxygen. TEM and XPS after photo/electrochemical cycling show that Mo doping can trigger deeper surface reconstruction with more exposure of catalytically active sites.
Uncontrollable reconstruction results in the instability of catalysts particularly severe in acidic water electrolysis. Grain boundary engineering (GBE) of a microstructure has been demonstrated to enhance the intergranular corrosion,160 oxidation,161 and other mechanical properties in various industrially relevant alloys, which shed light on increasing the stability of catalysts and rationally controlling the reconstruction. Hu et al. presented a method to design efficient OER catalysts by combining various components and grain boundaries (GB) which overcome the thermodynamic solubility limits of different metal elements, enhancing the catalyst's performance.124 The catalyst was prepared by a fast, nonequilibrium salt melt synthesis method, which introduced acid-resistant elements (Ir) and catalytically active elements (Fe, Co, and Ni) into the RuO2 lattice, forming a nanosheet structure with abundant grain boundaries (GBs). The authors show that the catalyst undergoes a structural evolution from RuO2 to hydrous RuOx and IrOx species under anodic potentials by using operando Raman spectroscopy. This structural transformation is responsible for the high activity and durability of the catalyst. The oxygen-forming energy (Fig. 11k) and diffusing energy barrier (Fig. 11l) of the O atom coordinated by Ru/Ir were calculated by DFT. The oxygen diffusing energy barrier of RuIrFeCoNiO2 is much lowered so that Ir/Ru tend to attract the O coordination from the surrounding Fe/Co/Ni atoms. The proposed scenario (Fig. 11m) depicted by DFT calculations shows that the defects O vacancies generated near the Ru/Ir atoms can be readily repaired by the diffusion of the neighboring Fe/Co/Ni coordination O atoms, prolonging catalyst durability. It can be summarized as that the incorporation of foreign elements and GB affects the charge redistribution, lowers the formation energy and diffusion energy barrier of O vacancies, and enhances the stability of surface oxygen.
3.3 Heterostructure constructions
Heterostructures that break both space-inversion and time-inversion symmetries provide opportunities to manipulate electronic and structural properties.162–164 Additionally, interfacial reconstruction, which depends closely on the symmetry breaking at the interface, is considered one of the primary driving forces behind the reconstruction of more functional structures.165–168Heterostructures, through their intricate interfaces, play a pivotal role in fostering the generation of active species. These interfaces serve as accelerators for photon/electron transfer, particularly in managing defects within these interface regions. Moreover, the robust interfacial electronic interactions exert a significant influence by reshaping the electronic configuration of reactive species. This modification effectively unveils additional active sites within pre-catalysts, thereby facilitating their reconstruction for enhanced reactivity. Furthermore, the localized bonding environments established within heterostructures play a crucial role in expediting the conversion of species, ensuring a swift and efficient catalytic process. For example, Tao et al. reported the synthesis and characterization of a novel heterostructure catalyst, NiSe2/FeSe2-NC, for the OER, achieving mutual advantages after reconstruction.125 By using in situ Raman spectroscopy (Fig. 12a) and operando XAFS, the authors show that the catalyst undergoes dynamic surface reconstruction during the OER, forming a Ni–O–Fe bonding configuration with both Ni and Fe in a trivalent state derived from the fitting results of XAFS (Fig. 12b), which act as the real active sites. They also show that the FeSe2 nanodomains are responsible for OH− adsorption and forming O–O− intermediates, while the NiSe2 compositions activate O–O coupling and facilitate the evolution of O2. DFT calculations illustrate that the heterojunction between NiSe2/FeSe2 improves the electron conductivity and modulates the Se vacancy formation energy, which activates transformation of NiSe2/FeSe2 into (oxy)hydroxides and maintains the stability of the main structure.
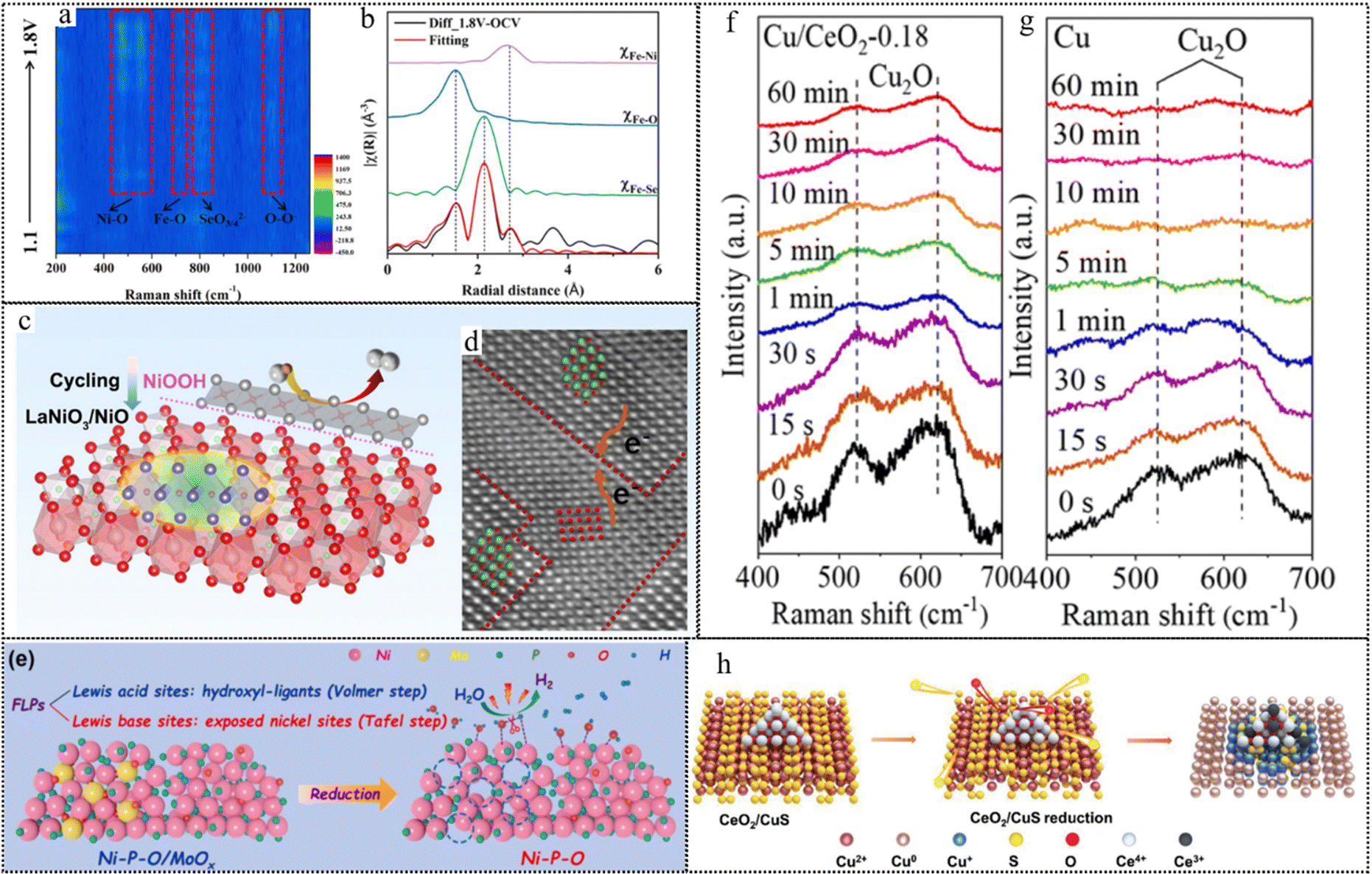 | ||
Fig. 12 (a) Raman intensity maps of NiSe2/FeSe2-NC. (b) Fitting results of diff_1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V-OCV and contributions from different paths. This figure has been reproduced from ref. 125 with permission from Elsevier, copyright 2023. (c) Schematic illustration of electrochemical heterostructure reconstruction. (d) Atomic HAADF-STEM image of LaNiO3/NiO. This figure has been reproduced from ref. 126 with permission from American Chemical Society, copyright 2022. (e) The mechanism of the electrochemical in situ reconstruction for Ni–P–O/MoOx during alkaline HER. This figure has been reproduced from ref. 128 with permission from Elsevier, copyright 2022. Operando Raman spectra of Cu/CeO2-0.18 (f) and Cu (g) evolution as a function of reaction time at open circuit potential (OCP). This figure has been reproduced from ref. 169 with permission from American Chemical Society, copyright 2022. (h) Schematic representation of the reconstruction process of CeO2/CuS. This figure has been reproduced from ref. 170 with permission from Wiley-VCH, copyright 2023. V-OCV and contributions from different paths. This figure has been reproduced from ref. 125 with permission from Elsevier, copyright 2023. (c) Schematic illustration of electrochemical heterostructure reconstruction. (d) Atomic HAADF-STEM image of LaNiO3/NiO. This figure has been reproduced from ref. 126 with permission from American Chemical Society, copyright 2022. (e) The mechanism of the electrochemical in situ reconstruction for Ni–P–O/MoOx during alkaline HER. This figure has been reproduced from ref. 128 with permission from Elsevier, copyright 2022. Operando Raman spectra of Cu/CeO2-0.18 (f) and Cu (g) evolution as a function of reaction time at open circuit potential (OCP). This figure has been reproduced from ref. 169 with permission from American Chemical Society, copyright 2022. (h) Schematic representation of the reconstruction process of CeO2/CuS. This figure has been reproduced from ref. 170 with permission from Wiley-VCH, copyright 2023. | ||
Another example is that Wei et al. reported a heterostructure constructed by deliberately making La deficiency in LaNO3 perovskite. This heterogeneous structure can facilitate the formation of the active site and composition reconstitution, forming a distinct boundary between the perovskite host and separation of the NiO phase for the OER.126 The authors found that the charge transfer on the interface causes the electron rearrangement of Ni ions, which leads to the in situ formation of an active NiOOH layer at a relatively low potential. The schematic process is illustrated in Fig. 12c and the heterostructure was confirmed by the atomic HAADF-STEM image (Fig. 12d). The LaNiO3/NiO heterostructure demonstrates a remarkable enhancement in OER activity and stability preventing La and Ni from dissolution under the OER conditions. The active centers and the surface reconstruction process of the LaNiO3/NiO heterostructure were investigated by operando Raman spectroscopy, quasi in situ XPS, and DFT calculations. They showed that the Ni sites at the interface are the main active sites for the OER, which can completely transform into NiOOH at a potential of ∼1.35 V vs. RHE. They also illustrated the photon/electron transfer path and delineated various oxidation states across different regions of the LaNiO3/NiO heterostructure through cyclic voltammetry (CV) analysis.
In certain heterostructure systems, heterostructural components that are initially inactive may transform functional species, thereby enhancing the catalytic activity by providing support to the active species. Liao et al. reported a novel method to create a multisite catalyst for enhancing the hydrogen evolution reaction (HER) on a nano-Pd surface in alkaline media.127 The catalyst is composed of core/shell Pd/FeOx(OH)2−2x nanoparticles (NPs), where the FeOx(OH)2−2x shell acts as a water dissociation site and the Pd core acts as a hydrogen formation site. The coverage of the FeOx(OH)2−2x shell on the Pd surface can be tuned by chemical cycling, which converts the inert Fe3O4 shell to FeOx(OH)2−2x and controls its removal. There remain challenges and limitations of existing non-noble alkaline HER catalysts impeded by poor intrinsic activity and inert catalytic sites. To deal with this issue, Xiao et al. synthesized a Ni–P–O/MoOx catalyst which consists of Ni(PO3)2–Ni2P nanoparticles coupled with MoOx on NiMoO4 nanorods for the alkaline hydrogen evolution reaction.128 The proposed mechanism of the chemical in situ reconstruction is illustrated in Fig. 12e. The catalyst undergoes structural evolution during the HER, as MoOx dissolves and exposes more nickel sites, and hydroxyl-ligands form on the surface of the nanoparticles according to the ex situ FTIR spectra and photo/electrochemical measurements. The DFT calculations reveal that the Ni–P–O catalyst integrates artificial frustrated Lewis pairs (FLPs) consisting of nickel sites as Lewis base sites and hydroxyl-ligands as Lewis acid sites, which can synergistically facilitate the Volmer and Tafel steps of the HER. The hydroxyl-ligands can strongly interact with OH* and lower the energy barrier for water dissociation, while the nickel sites can optimize the H* binding and desorption ability.
Notably, if reconstruction is not properly controlled, it may lead to an outrageous disintegration of active sites and degradation of the catalyst. However, the destructive effect of reconstruction can be mitigated by incorporating robust components. NiFe LDHs have garnered significant interests as noble-metal-free photo/electrocatalysts for the oxygen evolution reaction (OER) due to their abundant reserves and superior performance.171 However, their stability requires further enhancement. In a recent study, Yang et al. addressed this challenge by constructing a heterostructure that combines the following components: outstanding activity but unsatisfactory durability NiFe LDH and good structure stability but insufficient activity birnessite (Bir).172 The increased interlayer distance observed in XRD, along with the consequent relaxation of the proton bonding as indicated by the 1H solid-state NMR results of LDH Bir, would promote rapid electrolyte substance transport. This, in turn, would mitigate the localized pH decrease that typically triggers the loss of active sites. As a result, the catalyst remains exceptionally stable even when working under high current densities. In another case in CO2 reduction (CO2R), the specific structure of the catalyst is crucial for the selectivity of high-value-added products (C2+). Some literature indicates that the inherent characteristics of monometallic Cu restrict the selectivity for C2+.173–175 Therefore, reconstruction from Cu with a high selective structure during the catalytic process is often undesirable.
Wang et al. reported a novel synthesis of a copper/ceria heterostructure that can enhance the electrocatalytic conversion of carbon dioxide (CO2) to multicarbon (C2+) products.128 The authors induce strain in the Cu domain through the introduction of a ceria shell while simultaneously preventing structural changes in copper, which keeps favorable selectivity of the CO2R. The authors characterize the structure, morphology, and chemical state of the catalysts by XRD, SEM, TEM, EDS, XPS, AES, and XAS. The authors confirm that the dominant oxidation states for Cu and Ce are Cu+ and Ce4+, respectively. After analyzing the CO2R by HRTEM, XRD, and ATR-SEIRAS, the results reveal the intergrowth of Cu and CeO2 and that the cubic structure was preserved. From the in situ Raman spectra in Fig. 12f and g, after applying a cathodic potential (−0.7 V vs. RHE), the characteristic peaks of Cu2O gradually declined within 10 minutes for the pure Cu2O, indicating its complete reduction to Cu. However, in the Cu2O/CeO2−0.18 heterostructure, the characteristic Cu2O modes were retained for over 60 minutes, indicating enhanced durability of Cu+ under cathodic potential. Analysis of the Raman results suggests that Cu2O within the heterostructure exhibits greater resistance to cathodic reduction, attributed to the interaction between Cu2O and CeO2, favoring the formation of strained copper domains. A similar phenomenon exists in CuS/CeO2 reported by Yan et al.170 By using various in situ and ex situ techniques, such as XAS, Raman spectroscopy, FTIR, and DFT calculations. The authors propose that the presence of CeO2 inhibits the complete reduction of CuS and preserves the active Cu0/Cu+ species illustrated in Fig. 12h, which reduces the energy barrier of C–C coupling and increases the selectivity of C2+. From the analysis of XPS, the increase in the Ce3+ concentration in CeO2/CuS after CO2RR explains the stability of Cu+ in CeO2/CuS, which means CeO2 is used as a self-sacrificing agent. The heterostructure of CuS/CeO2 successfully undermines the undesired reconstruction from Cu+ to metallic Cu during the CO2RR.
3.4 Leaching
Reconstructions resulting from leaching processes encompass various distinct categories within material structures. Firstly, these include instances where unstable ions, such as specific sites within the perovskite structure contribute to leaching phenomena. Additionally, spontaneous leaching can be triggered by alkali metal ions or halide ions within the material matrix. Under catalytic conditions, pre-catalysts may undergo chemical reactions leading to leaching, commonly observed under oxidative conditions. Moreover, the interaction between the catalyst surface and the electrolyte engenders dynamic leaching and deposition processes, contributing to the evolving catalytic dynamics. These categories encapsulate the diverse mechanisms through which leaching induces reconstructions across different material systems, particularly in catalytic scenarios.In the majority of inorganic ABO3 or AB(OH)6 perovskites, alkaline metals and lanthanides typically fill the A-site, while a variety of transition metals such as Ni, Co, and Ir are found at the B-site.176 The process of reconstruction can lead to the leaching of A or B-site cations, which in turn can trigger the formation of active species.177 These include active OH− groups and reactive oxygen ligands, which can enhance their OER performance. Tang et al. presented a novel perovskite catalyst that can accelerate the surface reconstruction process by combining the leaching atoms and the oxyanions on the solid–liquid interface, thereby enhancing the oxygen evolution reaction efficiency.129 The spontaneous leaching of La into electrolytes, leaving plenty of vacancies, triggered the surface reconstruction as shown in Fig. 13a. The authors found that adding oxyanions such as sulfate or phosphate to the perovskite catalyst can reduce the adsorption energy of surface hydroxyls, facilitate the formation and migration of oxygen vacancies, and enhance the lattice oxygen oxidation mechanism. From Fig. 13b and c, the increase in the reduction peak area over time of immersion, coupled with a simultaneous enhancement in both OER performance and electrochemically active surface area (ECSA), suggest the generation of additional active sites during the immersing process of LaNiO3−δ. The HRTEM images revealed an amorphous layer generated on the surface while the bulk structure remained unchanged when soaked in a 1 M KOH solution for 24 hours. Based on the XAFS and XPS characterization (Fig. 13e), the reconstructed layer presented that the oxidation state of Ni reduced from 3+ to 2+. The results showed that the perovskite catalyst treated with sulfate had the best oxygen evolution reaction activity and stability.
 | ||
| Fig. 13 (a) The proposed mechanism for surface reconstruction immersed in electrolytes with oxyanions. (b) Backward scan polarization curves (1.2–1.7 V vs. RHE) of LaNiO3−δ. (c) The variation of current density (at 1.65 V vs. RHE) and double layer capacitance (Cdl) with immersed time. (d) STEM of pristine(upper) LaNiO3−δ being immersed in 1 M KOH for 24 hours (lower). (e) XAFS spectra of Ni L2,3-edges, this figure has been reproduced from ref. 129 with permission from Wiley-VCH, copyright 2023. (f) Co K-edge XANES of Li2Co2O4 electrodes with open circuit voltage (OCV) and at 1.60 V vs. RHE after 20 and 60 min in 1 M KOH. The inset shows the pre-edge peaks. (g) FT of the corresponding Co K-edge EXAFS. This figure has been reproduced from ref. 130 with permission from American Chemical Society, copyright 2019. (h) CV curves of 100 cycles in 1.0 M KOH with a scan rate of 100 mV s−1. (i) STEM images of AC-Co2(OH)3Cl. This figure has been reproduced from ref. 131 with permission from Wiley-VCH, copyright 2019. | ||
Alkali metal elements such as Li/Na/K and some halide elements such as F/Cl possess quite large reduction/oxidation potential, which are stable in their ion forms. When the layered materials contain the above elements, they tend to spontaneously dissolve to promote material reconstruction. Li et al. found that spinel Li2Co2O4 (LCO) undergoes a spontaneous delithiation process that triggers the formation of an amorphous layer of LixCo2O4−δ(OH)δ on the surface.130 In Fig. 13f, the operando XAS data show that the valence state of Co ions increases from 3+ to 3.3+ and the CoO6 octahedron became distorted under OER conditions. The EXAFS fitting data (Fig. 13g) also show that the average Co–O bond distance decreased and the Co–Co distance become disordered. These results suggest that the surface structure of LCO undergoes a dynamic reconstruction under OER conditions. The ex situ XRD and Raman spectra show that the overall crystal phase of LCO remains unaltered after CV scans, while the TEM and HAADF-STEM images reveal an amorphous structure on the surface area. The post-mortem XAS data demonstrate an increase in the concentration of Co4+ ions alongside the CV scans, as evidenced by the positive energy shift and the emergence of a peak at the Co L2,3-edge. The O K-edge XAS data show that the oxygen valence state also increases, as indicated by the increased intensity and the decreased energy position of the pre-edge peaks. The authors identify the simultaneous occurrence of Co4+ and oxidized O ions with the amorphous active layer accompanied by a spontaneous delithiation process that reduces the Li/Co ratio from 2 to 0.8, as confirmed by ICP-OES analysis. Another example is that Cui et al. used electrochemical activation to intentionally extract Li ions from LiCoO2.178 The delithiation process induces a phase transition from hexagonal to monoclinic Li0.5CoO2, which alters the oxidation state and covalency of Co and O atoms and makes the material more electrophilic and metallic.
Song et al. synthesized cobalt oxychloride (Co2(OH)3Cl) as the OER precatalyst.131 The removal of the Cl− lattice during the chemical process facilitates continuous reconstruction and enhanced OER activities. The Co2(OH)3Cl precatalyst shows an ever-increasing chemical surface area during the CV activation (Fig. 13h), indicating more active sites exposed for the reaction. The etching of the Cl− lattice is detected by the AgNO3 aqueous solution and the XPS spectra, suggesting the irreversible cleavage of the Co–Cl bond in Co2(OH)3Cl. The STEM images of activation-derived (AC)-Co2(OH)3Cl in Fig. 13i showed lots of defects. The operando XANES and EXAFS spectra reveal that the reconstruction-derived component has a similar structure framework to the β-CoOOH bulk, but with lower intensity of the white peak and higher intensity of the pre-edge peak, indicating structure distortion and the existence of defects consistent with the STEM results. The EXAFS spectra also show that the surface reconstruction-derived component has lower coordination numbers of the Co–O and Co–Co bonds than the β-CoOOH core part, demonstrating unsaturated sites for potential catalysis.
Many materials undergo reconstruction under catalytic conditions, especially at high oxidation potentials, due to the dissolution of some of their constituent elements. For instance, metal alloys can be partially dissolved by forming high-valent oxides, hydroxides, or oxyhydroxides. Similarly, transition metal compounds with main group elements (borides, carbides, nitrides, phosphides, and chalcogenides) can lose some of their anionic components by oxidation, leading to different degrees of dissolution. Moreover, materials with C, N, and O elements as the backbone, such as MOFs and PBAs, can also experience oxidation and dissolution of their framework structures, which facilitates the reconstruction process.
For alloy, Yang et al. reported the synthesis of IrFeCoNiCu high-entropy alloy (HEA) nanoparticles by a microwave-assisted shock synthesis method as catalysts for the acidic oxygen evolution reaction (OER).132 The HEA nanoparticles exhibit excellent activity and stability in 0.1 M HClO4 electrolyte, with a lower overpotential and a smaller Tafel slope than pure Ir and other Ir-based bimetallic alloys. The authors analyzed the structural evolution of the HEA nanoparticles under OER conditions by STEM-EDX, XRD, XPS, and other techniques, and found that an enriched Ir shell layer generates on the surface of the HEA nanoparticles after chemical activation, mainly due to the dissolution of 3d metals (Fe, Co, Ni, and Cu). The schematic reconstruction pathway is illustrated in Fig. 14a. The core of the nanoparticles preserves the homogeneous uniform phase of the HEA structure without notable phase separation or elemental segregation. The shell layer has a polycrystalline structure and a thin oxide layer on the surface confirmed by STEM images in Fig. 14b and c. The authors suggest that this reconstructed structure is relatively durable and does not undergo further dramatic structural degradation during stability tests. This article indicated that the near-surface structure of HEA nanoparticles exhibits a certain degree of structural dynamics under acidic working conditions.
 | ||
| Fig. 14 (a) Illustration of the structural evolution of IrFeCoNiCu-HEA nanoparticle under acidic OER conditions. (b) STEM-EDX maps showing the Ir-rich shell layer near the surface of the nanoparticle, as highlighted by the dashed lines (scale bar: 10 nm). (c) Near-surface structure of the nanoparticle showing the HEA core, polycrystalline alloy shell layer (∼2–6 nm), and surface oxide (indicated by white arrows) after electrochemical activation. This figure has been reproduced from ref. 132 with permission from American Chemical Society, copyright 2023. (d) CV scans and (e) Illustration of the electrochemical reconstruction for Fe0.01–Ni&Ni0.2Mo0.8N. (f) HRTEM image of Fe0.01&Mo–NiO after reconstruction. This figure has been reproduced from ref. 133 with permission from The Royal Society of Chemistry, copyright 2022. (g) Schematic illustration for the deep reconstruction of NiFeP nanoboxes into hierarchical NiOOH/FeOOH nanoboxes. (h) The relationship between the ratio of δ(Ni–O) to ν(Ni–O) and the applied potentials in the in situ Raman spectra. This figure has been reproduced from ref. 136 with permission from The Royal Society of Chemistry, copyright 2021. | ||
Ning et al. presented a method of preparing an efficient alkaline fresh water and seawater electrolysis catalyst based on photo/electrochemical reconstructing.133 The catalyst consists of Fe-doped Ni&Ni0.2Mo0.8N microrods, which are grown on the Ni foam by hydrothermal and ammonia reduction nitridation methods. The catalyst presents excellent HER performance at low Fe doping levels, but poor HER performance at high Fe doping levels. When the anodic potential is applied, the catalyst undergoes rapid chemical reconstruction confirmed by a dramatic change in the oxidation peak of CV (Fig. 14d), in which most of the Mo dissolves into the electrolyte, while Ni and Fe are oxidized to NiO and Fe2O3 illustrated in Fig. 14f. The reconstructed catalyst consists of NiO co-doped with Fe and Mo (Fex&Mo–NiO) as depicted in Fig. 14e and has comparable OER performance to the best OER catalysts. Therefore, the catalyst can serve as a bifunctional catalyst with both HER and OER activity.
By undergoing complete reconstruction from MOFs to (oxy)hydroxides in alkaline media, MOFs have emerged as potential alternative precatalysts for the OER. Du and co-workers presented the synthesized Fe-doped Co-MOF nanosheets (Co-MOF/Fe) undergoing an entire transformation from MOFs to CoFe-LDHs in the KOH electrolyte, which are the true catalytically active catalysts for the OER.134 Prussian blue analogs (PBAs) are also promising pre-catalysts for the OER, but most of them only undergo surface reconstruction and cannot completely transform into active metal (oxy)hydroxides. Yu et al. developed a chemical–physical synergistic etching strategy to enable the deep reconstruction of NiFe PBA nanocubes for efficient OER and Zn–air batteries.135 They demonstrated that the NiFe PBA nanoboxes have hollow structures and dual vacancies (Ni vacancies and CN vacancies), which facilitate the deep reconstruction and preserve a certain amount of iron species.
Another work conducted by this group demonstrated that the NiFeP nanoboxes (NBs) can deeply reconstruct into defective NiOOH/FeOOH nanosheet assembled NiOOH/FeOOH NBs during the OER process, which exhibit outstanding OER performance with a low overpotential of 228 mV at 10 mA cm−2 and excellent stability.136 A schematic illustration is shown in Fig. 14g for the formation of NiFeP NBs and deep reconstruction of NiFeP NBs into hierarchical NiOOH/FeOOH NBs. By analyzing the bending vibration δ(Ni–O) and tensile vibration ν(Ni–O) peak values of NiOOH obtained from the Raman spectrum (Fig. 14h), the high ratio of δ(Ni–O) to ν(Ni–O) indicated γ-NiOOH formation, which is believed to have higher OER activity. It revealed that the NiFeP NBs deeply reconstructed into defective NiOOH/FeOOH nanosheet assembled NBs, while the NiFeP NCs only partially reconstructed into NiOOH/FeOOH NCs with Ni(OH)2 remaining.
In addition, some elements leach out during the oxidation reaction and reassemble on the surface of the catalyst to form a polyanion layer. Constructing a polyanion layer as an electrostatic repulsion barrier within the active material surface not only effectively prevents Cl− adsorption but also does not compromise the exposure of active sites, which has been proven to effectively inhibit the CER reaction and achieve excellent stability of seawater electrolysis.33,179–181 Dai et al. presented a novel anode material for water splitting in alkaline seawater.33 The material consists of a NiFe hydroxide catalyst layer uniformly coated on a nickel sulfide (NiSx) layer, which is formed on porous nickel foam (NF) (abbreviated as Ni3). The material exhibits excellent catalytic activity and corrosion resistance in solar-driven alkaline seawater electrolysis, operating at industrially required current densities (0.4 to 1 A cm−2) for over 1000 hours. The authors found that during the anode activation process, the NiSx layer was oxidized to generate sulfate ions confirmed by the Raman spectra (Fig. 15a) and TOF secondary ion mass spectrometry (TOF-SIMS) mapping (Fig. 15b), which intercalated into the NiFe layer along with carbonate ions from the alkaline solution, forming a polyanion-rich passivation layer that repelled and blocked chloride ions from seawater, thus enhancing the anode corrosion resistance.
 | ||
| Fig. 15 (a) Raman spectra of Ni3 and NiSx/Ni after 12-h activation in 1 M KOH + 0.5 M NaCl. (b) TOF-SIMS map of SOx2−/1− and CO32−/1−fragments from a Ni3 and NiSx/Ni electrode surface after activation in 1 M KOH + 0.5 M NaCl at 400 mA cm−2. This figure has been reproduced from ref. 33 with permission from National Academy of Science, copyright 2023. (c) Pristine Ni(OH)2, Ni(OH)2 after 6 h CA measurement at 1.6 V vs. RHE in 1 M KOH containing 10 μM Fe3+. This figure has been reproduced from ref. 137 with permission from American Chemical Society, copyright 2021. (d) In situ STEM schematic setup and images and STEM/Fe EDX maps of NiO octahedra acquired in a 0.5 M borate buffer +1 mM Fe(NO3)3 solution. (e) Illustration of the evolution of Fe incorporated surface reconstruction. This figure has been reproduced from ref. 138 with permission from American Chemical Society, copyright 2023. In situ Raman spectra revealing the conversion from (f) Bi-MOF NR to Bi2O2CO3 NS on exposure to 0.1 M KHCO3 and (g) Bi2O2CO3 NS to Bi NS under CRR conditions. This figure has been reproduced from ref. 139 with permission from Wiley-VCH, copyright 2021. | ||
In addition to element leaching, elements in the electrolyte also restructure and redeposit on the photo/electrocatalyst surface to form a dynamic equilibrium, especially the influence of Fe ions in the electrolyte on Ni, Co-based catalysts cannot be ignored.182 Kuai et al. reported on water oxidation reaction catalysts, mainly investigating the effects of iron incorporation and distribution in nickel hydroxide on its catalytic activity.137 The authors also point out that Fe ions undergo dynamic exchange between the catalyst and the electrolyte under OER conditions, leading to chemical and structural changes in the catalyst surface. The authors used XRF microscopy as shown in Fig. 15c and XAS techniques to track the incorporation and distribution dynamics of iron in two-dimensional nickel hydroxide nanosheets, and found that iron incorporation mainly occurred at the edge sites of nanosheets, and was closely related to the water oxidation reaction process. The authors further used DFT calculations to indicate that iron incorporation can enhance the reducibility of nickel sites, thereby improving the catalytic activity of water oxidation reactions. This research provides a comprehensive approach to understanding and tuning the dynamic changes of metal hydroxide photo/electrocatalytic interfaces, which is of great significance for controlling catalytic processes.
Moreover, Cuenya and colleagues used in situ electrochemical (scanning) transmission electron microscopy (EC-(S)TEM) combined with energy-dispersive X-ray spectroscopy (EDX) mapping to observe how Fe impurities in the electrolyte affect the surface structure and composition of nickel oxide (NiO) catalysts during the OER in alkaline solution.138 The authors found that in a borate buffer solution containing 1 mM Fe, the NiO surface first formed a surface-rich NiFe double-layered hydroxide (NiFe-LDH) during the potential cycling process illustrated in the operando STEM image (Fig. 15d), which shifted the onset potential of the OER to the low potential direction. As shown in Fig. 15e, with the extension of the reaction time, when the LDH layer could no longer accommodate more Fe, Fe oxide (FeOx) aggregates began to appear on the surface, which reduced the active surface area of the catalyst. Under a constant anodic potential, the LDH layer also gradually degraded, leading to more FeOx formation and partial Fe returning to the electrolyte. This process not only further reduced the performance of the catalyst but also did not restore NiO to its original state.
Even at a low operating current (10 mA cm−2), the leaching of Fe from the electrode surface is inevitable, and this phenomenon becomes more severe at a high current density (200 mA cm−2). Investigations on the dynamic restructuring processes of Fe include in situ observations,137,138 as well as studies on the characteristics of Fe restructuring in Ni and Co systems. However, diverse perspectives exist regarding the description of the Fe restructuring process. The analysis of operando Mössbauer spectroscopy illustrated that a partial photo/electron transfer from Fe(III) occurs accompanied by the oxidation of Ni(II), generating higher oxidation states of Fe. These high-valent Fe species may significantly contribute to increasing OER activity. Subsequently, researchers employed in situ Mössbauer spectra to reveal the process of Fe(III) transforming into Fe(IV). The possibility of Fe(VI) has even been reported, as evidenced by cyclic voltammetry (CV) tests under non-aqueous conditions, indicating successive oxidation states of four, five, and six for NiFe LDH at elevated potentials. Researchers, such as Yanbo Li and colleagues proposed a self-healing mechanism involving Fe(VI) species.121 In subsequent work, they successfully enriched Fe(VI)O42-during continuous OER processes under aqueous conditions.121
Qiao et al. reported a comprehensive investigation into the controllable reconstruction of Bi-based MOFs(Bi-MOFs) for photo/electrochemical CO2 reduction reaction (CO2RR).139 By analyzing the in situ Raman spectroscopy results in Fig. 15f and g, the band at 820 cm−1 disappeared and a new band referred to the stretching vibration mode of C–O in Bi2O2CO3 appeared after immersing in 0.1 M KHCO3 for 20 min, which indicated this dynamic transformation from Bi-MOF to Bi2O2CO3. When applying potential from −0.5 to −1.3 V vs. RHE, the typical vibration modes of C–O and Bi![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Bi2O2CO3 faded gradually, in the meantime two new bands of Bi–Bi appeared at 71 and 97 cm−1. The authors revealed that the reconstruction process can be separated into two steps: (1) electrolyte-regulated conversion of Bi-MOF nanorods (NR) to Bi2O2CO3 nanosheets (NS) by bicarbonate-initiated ligand substitution, and (2) potential modulated reduction of Bi2O2CO3 NS to Bi NS. The first step governs the ultimate morphology and formation of defects, while the subsequent step determines the final composition and oxidation states.
O of Bi2O2CO3 faded gradually, in the meantime two new bands of Bi–Bi appeared at 71 and 97 cm−1. The authors revealed that the reconstruction process can be separated into two steps: (1) electrolyte-regulated conversion of Bi-MOF nanorods (NR) to Bi2O2CO3 nanosheets (NS) by bicarbonate-initiated ligand substitution, and (2) potential modulated reduction of Bi2O2CO3 NS to Bi NS. The first step governs the ultimate morphology and formation of defects, while the subsequent step determines the final composition and oxidation states.
3.5 Crystallinity
The amorphous photo/electrocatalysts benefit from abundant active sites, exotic electronic configuration, and flexible structure. These features expedite the adsorption of reaction mediators and facilitate photon/electron transfer between metal sites and intermediates, thereby promoting the transformation of precatalysts into active components.183,184The impact of crystallinity on reconstruction, especially concerning amorphous structures, is multifaceted. Amorphous structures exhibit numerous influential characteristics: they possess abundant active sites and structural flexibility due to their lack of defined arrangement, enabling enhanced adsorption crucial for catalysis. Moreover, the surplus of dangling bonds within these structures significantly reduces the energy barrier required for rapid reconstruction into active phases, while the absence of dislocations and grain boundaries further facilitates this transformation process. Additionally, the tunable energy levels inherent in amorphous structures play a pivotal role in expediting the transition into active phases, thereby promoting efficient catalytic reactions. For example, the active phases with high energy evolved from amorphous photo/electrocatalysts improve the inherent catalytic efficacy of their active sites. Cai et al. prepared amorphous NiFe alloy catalysts for the OER by a solution method at room temperature.140 An anodic potential was applied to activate the amorphous NiFe exposing more active sites due to the short-range order of the amorphous structure. The amorphization features of the synthesized NiFe alloy catalyst were confirmed by HAADF-STEM imaging (Fig. 16a) and the annealed NiFe alloy catalyst at 600 °C in nitrogen (Fig. 16b) was provided for comparison. Though both amorphous and crystalline catalysts show comparable electrochemically active surface area (Fig. 16c), the amorphous NiFe alloy achieved an overpotential of 265 mV (η10 mA cm−2), which is 100 mV lower than that of the NiFe crystalized alloy. According to the CV measurements in Fig. 16d, the redox peaks of the amorphous catalyst are 50 times stronger than those of the crystalline form. The reconstruction of amorphous catalysts greatly increases the number of active sites and thus significantly improves the water oxidation activity.
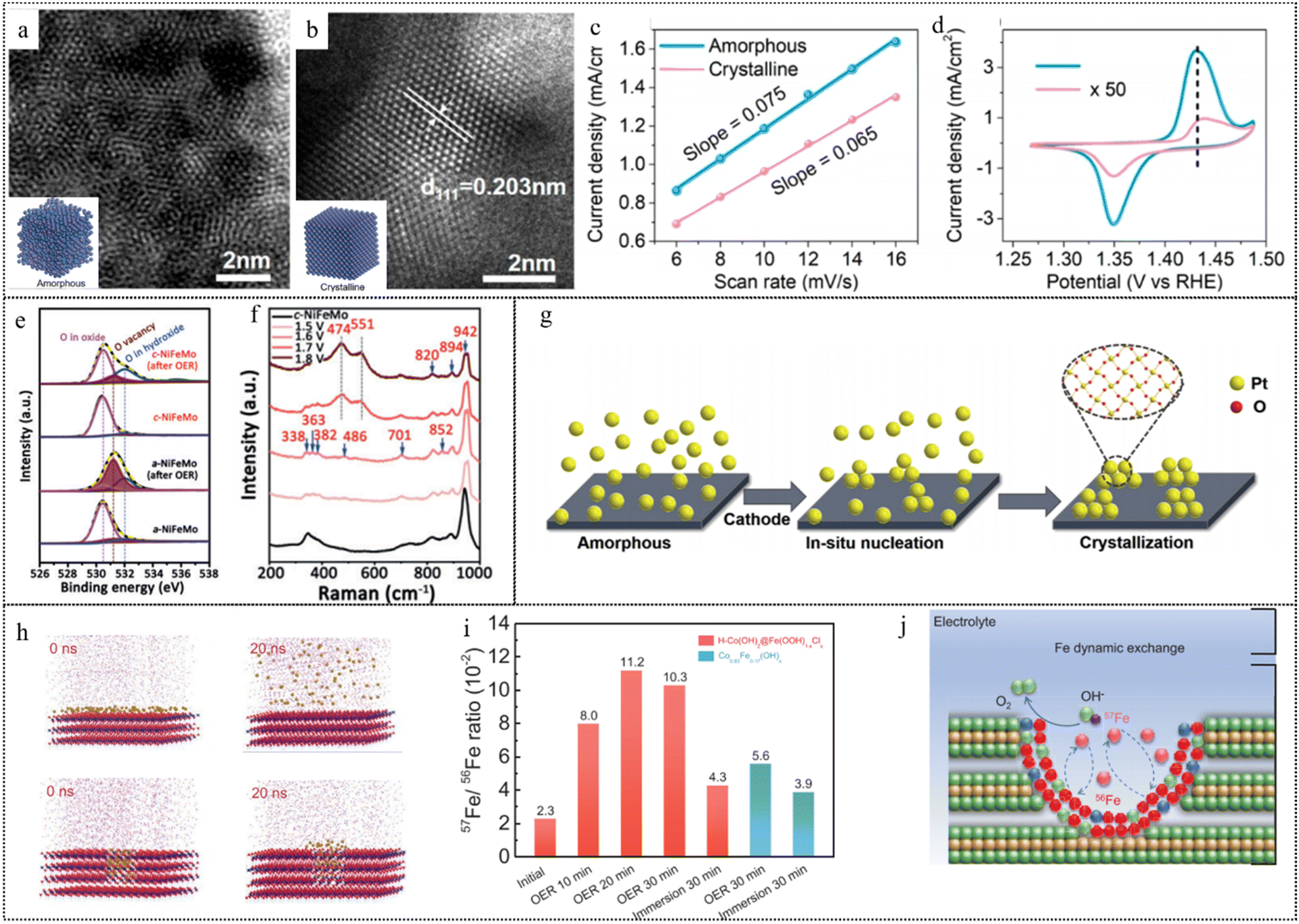 | ||
| Fig. 16 HAADF-STEM images of (a) the as-prepared amorphous nickel-iron alloy catalyst and (b) the annealed nickel–iron alloy catalyst at 600 °C in nitrogen. (c) Double-layer capacitance acquired at a potential from 1.01 to 1.05 V vs. RHE to determine the ECSA. (d) CV curves obtained at a scan rate of 10 mV s−1. This figure has been reproduced from ref. 140 with permission from American Chemical Society, copyright 2020. (e) O 1s XPS spectra of a-NiFeMo and c-NiFeMo before and after the OER. (f) Operando Raman spectroscopy measurements of a-NiFeMo. This figure has been reproduced from ref. 141 with permission from Wiley-VCH, copyright 2019. (g) Schematic illustration of the in situ crystallization and activation of A-Pt during the electrochemical cycling process. This figure has been reproduced from ref. 143 with permission from American Chemical Society, copyright 2021. (h) Snapshots of Fe3+ diffusion from the (0001) basal plane (upper) and a β-CoOOH nanohole (lower) in aqueous alkaline solutions at various times. (i) The 57Fe/56Fe ratios of the initial H-Co(OH)2@Fe(OOH)1−xClx and the catalysts after chronopotentiometric OER testing for various times as well as soaking for 30 min in an electrolyte containing 1 ppm 57Fe3+. (j) Schematic representation of the isotope-labeled Fe exchange mechanism in the OER. This figure has been reproduced from ref. 144 with permission from American Chemical Society, copyright 2023. | ||
Duan et al. reported a scalable production method of amorphous NiFeMo oxides, which can produce up to 515 grams of catalyst in one batch.141 From the O 1s XPS spectra depicted in Fig. 16e, it can be observed that the concentration of O vacancies in the a-NiFeMo catalyst after the OER is greater than that in the c-NiFeMo catalyst. The rapid reconstruction of the amorphous structure was revealed by Raman spectroscopy (Fig. 16f) that the a-NiFeMo oxide reconstructed rapidly on the surface under OER conditions, generating a metal oxy(hydroxide) active layer with an enrichment of O vacancies, while the c-NiFeMo oxide shows no significant structural change. The authors also demonstrate by DFT calculations that O vacancies can lower the dissociation barrier of *OOH intermediates, thus enhancing the OER activity.
In addition, transforming catalysts into an amorphous state is another effective strategy to enhance the precatalyst's contact area. The abundance of defects and disarrayed configurations resulting from amorphization allow for better electrolyte infiltration into the inner layer of precatalysts, thereby boosting the degree of reconstruction. This is a characteristic not typically observed in crystalline catalysts. For example, Chen et al. presented the utilization of fast reconstruction of amorphous nanofilm precatalysts for designing superior water-splitting catalysts.142 The authors amorphized SrCo0.85Fe0.1P0.05O3−δ (SCFP) nanofilm precatalysts with weakened chemical bonds by using high-energy argon plasma to destroy the crystal structure of composite oxides, and deposited them on conductive nickel foam (NF) substrates. Under water-splitting operation conditions, the amorphous SCFP-NF catalysts could rapidly reconstruct within 2 minutes, forming novel amorphous phases with high activity and stability. The SCFP-NF catalysts after reconstructing activation exhibited ultrahigh mass activity of up to 1000 mA mg−1 and excellent durability of up to 650 hours.
Amorphization structures typically exhibit unique and appealing catalytic performance, and the photo/electrochemical performance can be improved by in situ crystallization to obtain the optimized crystal structure. For example, Ma et al. developed a facile approach of the self-activation of amorphous platinum (A-Pt) nanospheres.143 As illustrated in Fig. 16g, A-Pt was in situ reconstructed to a crystalline phase and partially oxidized during the CV activation cycling. This process led to the self-activated enhancements of both HER and HOR activities. As the electrochemical cycling process progresses, A-Pt gradually transforms into crystalline clusters and is subsequently oxidized. The formed Pt–O connection emerges as a key factor contributing to the elevated activities of the HER and HOR. Fine facet engineering could also facilitate the dynamic reconstruction of Fe ions on the surface, creating more active sites for the OER and thus prolonging the stability of the catalyst. Xue and coworkers used 57Fe isotope labeled mass spectrometry (MS) to demonstrate that the catalyst achieved a local high-flux Fe dynamic equilibrium inside the nanoholes for the OER, which boosted the OER activity and reduced the Fe leaching amount.144 The authors used DFT and molecular dynamics simulations (Fig. 16h) to indicate that the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) facets on the nanohole walls exhibited stronger Fe adsorption than the (0001) basal plane. Additionally, the nanoholes were found to impede Fe diffusion from the catalyst to the electrolyte. The 57Fe isotope labeled ICP-MS measurement of the OER mechanism was tested to investigate the dynamic Fe interchanging mechanism of the catalyst under the OER conditions. Isotope 57Fe3+ as the tracer and was added to the electrolyte. As shown in Fig. 16i, the 57Fe content in the catalyst was about 5 times higher compared to that of the initial state after the OER process. Combining experimental and theoretical results, the proposed dynamic reconstruction is illustrated in Fig. 16j. The free Fe species exhibited a distinctive preference for selective and robust adsorption onto the (100) sites situated along the walls of the nanoholes, as opposed to the (0001) basal plane. Simultaneously, the nanohole structure exerted a constraining effect, impeding the dissolution of Fe from the catalyst to the electrolyte (Fig. 17).
0) facets on the nanohole walls exhibited stronger Fe adsorption than the (0001) basal plane. Additionally, the nanoholes were found to impede Fe diffusion from the catalyst to the electrolyte. The 57Fe isotope labeled ICP-MS measurement of the OER mechanism was tested to investigate the dynamic Fe interchanging mechanism of the catalyst under the OER conditions. Isotope 57Fe3+ as the tracer and was added to the electrolyte. As shown in Fig. 16i, the 57Fe content in the catalyst was about 5 times higher compared to that of the initial state after the OER process. Combining experimental and theoretical results, the proposed dynamic reconstruction is illustrated in Fig. 16j. The free Fe species exhibited a distinctive preference for selective and robust adsorption onto the (100) sites situated along the walls of the nanoholes, as opposed to the (0001) basal plane. Simultaneously, the nanohole structure exerted a constraining effect, impeding the dissolution of Fe from the catalyst to the electrolyte (Fig. 17).
 | ||
| Fig. 17 Triggers and transformation mechanisms of catalyst reconstruction behavior. | ||
4. Applications
4.1 Electrocatalysis
Reconstruction in electrocatalysis can be attributed to surface energy drive or potential drive, the former causes topological changes, and the latter causes oxidation or reduction reactions. (Fig. 18) The pre-catalysts can be tuned in their structural reconstruction, composition changes, and surface properties to demonstrate significantly enhanced conductivity, accelerated charge transfer kinetics, and decreased energy barriers required for the formation of active intermediates. Reconstruction is applied in novel material synthesis and other electrocatalysis (such as the OER/ORR, UOR, MOR, HER, NORR and CO2RR), and several representative examples are provided. | ||
| Fig. 18 Schematic diagram of electrocatalytic reactions and the related standard equilibrium potentials versus the reversible hydrogen electrode (RHE) under standard conditions. (Note: typical elements for specific electrocatalytic reaction and corresponding reconstruction are listed in elliptic boxes.) | ||
In the realm of electrocatalysis, reconstruction is a critical phenomenon akin to surface modification strategies. It refers to the spontaneous changes in the surface structure of a catalyst, which can impact both its lifespan and activity. The reconstruction of electrocatalysts is basically attributed to the applied cathodic/anodic potentials and the robust interactions with reactants or intermediates. In this section, we will introduce the representative applications of favorable catalyst dynamic transformation in the field of electrocatalysis.
4.1.1.1 Oxygen evolution reaction (OER). In the context of electrocatalytic water splitting, the OER is one of the half-reactions, and its kinetic process is typically sluggish. Therefore, the actual overpotential for the OER is often very high. In many previous studies, it has been found that high-valent transition metal species exhibit excellent OER catalytic activity. Taking the NiFe system as an example, some research suggests that high-valent Ni(IV) serves as the active site,185–187 while others propose high-valent Fe(IV)188,189 or even Fe(VI)190,191 as the active site. However, one point universally agreed upon is that the generation of high-valence active components during dynamic transformation is crucial for enhancing OER activity. Reconstruction during the OER process can be used not only to improve the catalytic activity of the catalyst but also to effectively design seawater electrolysis catalysts, improving the selectivity of the OER in the presence of Cl− as mentioned in the previous section.
4.1.1.2 Urea oxidation reaction (UOR). For the urea oxidation reaction (UOR), published works are focusing on developing the UOR process as a viable solution for wastewater treatment and converting it into energy. The UOR process has a thermodynamic potential of 0.37 V (vs. reversible hydrogen electrode, RHE), which is much lower than that of the energy-intensive oxygen evolution reaction (OER; 1.23 V vs. RHE). This makes the UOR process a promising candidate for replacing the OER process. Nickel hydroxides, acting as a typical and high-performance catalyst for the UOR, generate NiOOH and other active species by reconstruction.192 However, the production of these reactive species requires a significant amount of energy. Therefore, researchers are focusing on finding ways to reduce the energy barriers associated with the reconstruction process. One approach is to modulate the electronic state or ligand structure of the active site, which can reduce the generation energy of reactive species such as NiOOH. This modulation promotes the reconstruction reaction and improves the catalytic activity of the UOR process. Cao et al. reported the synthesis and characterization of amorphous nickel sulfoselenide and revealed that nickel sulfoselenide is transformed into nickel oxyhydroxide during the UOR. The in situ formed nickel oxyhydroxide has a high reaction rate and selectivity for urea oxidation, leading to a lower cell voltage than that required for overall water splitting.193
4.1.1.3 Methanol oxidation reaction (MOR). Methanol oxidation reaction has been applied in direct methanol fuel cells since the 1950s for the convenience and safety of ethanol transportation.194 Another application is that the MOR can be used to replace the OER for electrolyzers due to the lower thermodynamic equilibrium potential of 0.04 V vs. RHE. Though great progress has been made in the development of highly active Pt/Pd SACs for the MOR, further studies are needed to investigate the dynamic structural reconstruction of SACs under working conditions and break the scaling relation limit of DFT calculations,113,195 since SACs may aggregate into clusters as true catalytic active species and then return to the initial atomic dispersion under ambient exposure. As for non-noble metal-based catalysts, the onset potential is high enough to oxidize catalysts during reactions. Zhang et al. reported the construction of a bismuth oxyhydroxide (BiOx(OH)y)–Pt inverse interface via electrochemical reconstruction for the enhanced MOR.196 The electrochemical treatment illustrated in Fig. 19a can induce the structural reconstructing of PtBi catalysts, leading to the formation of the BiOx(OH)y–Pt inverse interface.
 | ||
| Fig. 19 (a) Schematic diagram of the preparation of BiOx(OH)y–Pt. This figure has been reproduced from ref. 113 and 195 with permission from American Chemical Society, copyright 2020. (b) In situ Raman spectra of Ni4Mo during the HER. This figure has been reproduced from ref. 197 with permission from The Royal Society of Chemistry, copyright 2019. (c) Schematic diagram of atomic structures of Cu(OH)F and the transformed Cu/CuxOF. This figure has been reproduced from ref. 198 with permission from The Royal Society of Chemistry, copyright 2021. (d) Schematic diagram of the Cu(I)/Cu(0) core/shell nano-disk for P-Cu1.65/Cu2OF. This figure has been reproduced from ref. 199 with permission from Elsevier, copyright 2023. | ||
4.1.2.1 Oxygen reduction reaction (ORR). As for the ORR, reconstruction in transition metal-based catalysts often implies a loss of stability, such as being prone to oxidation, aggregation or dissolution in the electrolyte.200–202 M–C–N (metal, metal-free) composites possess good qualities of low cost, high tolerance, high activity, and high stability.203,204 In other words, an electrocatalyst for the ORR depends more on the structural stability to offer stable oxygen adsorption sites, where the reconstruction process is usually unfavorable and requires control.
4.1.2.2 Hydrogen evolution reaction (HER). In the HER, both the reduction and oxidation processes of electrocatalysts are involved. For example, nickel sulfides stand out with high intrinsic activity and good electron transport capability.205 The Ni element whether in the Ni(II) or Ni(III) form can undergo reduction at cathodes during the HER. Zheng et al. showed that two-dimensional NiS2 was activated and transformed into nanosheets with metallic Ni.206 In addition to reduction during the HER, the operation conditions affect the oxidation of electrocatalysts, as the redox potentials of the elements involved are partly overlapped by the applied potentials. For Mo-based catalysts, the potential required for the conversion of Mo to MoO42− is lower than that required for water splitting.207 The dissolved MoO42− also plays a key role in electrocatalyst stability.197 As further confirmed by in situ Raman spectroscopy (Fig. 19b), MoO42− was re-adsorbed and polymerized to Mo2O72− on the electrode surface, mitigating the strong *H adsorption on Ni sites and enhancing the alkaline HER.
4.1.2.3 Nitrogen reduction reaction (NRR). Current electrocatalytic NRR processes are primarily centered on the production of NH3via the reduction reaction from N2, which is energy-intensive for the inert nature of N2. Meanwhile, a significant quantity of nitrate (NO3−), an alternative source for the production of NH3, is being released into surface water and underground aquifers, posing a risk to human health.208 Zhang et al. synthesized CuO nanowire arrays and used 15N isotope labeling confirming that the produced NH3 originates from NO3− reduction. In situ Raman and other ex situ analyses have demonstrated the electrochemical reduction from CuO to Cu/Cu2O during the electrocatalysis process, confirming the electron transfer at the Cu/Cu2O interface, which acts as the active phase.
4.1.2.4 CO2 reduction reaction (CO2RR). The microscopic states of active sites affect the CO2RR, so determining the active intermediates and monitoring their changes under working conditions is pivotal.209,210 Additionally, the CO2RR operates at more negative potentials, facilitating the reduction of Cu oxides or hydroxides in situ.211 Reconstruction has been employed to move the reduction reaction towards well-constructed chemical compositions of the catalyst surface.212 Halogen anions commonly exhibit robust interactions with copper atoms, substantially influencing the reconstruction processes of Cu. They are frequently employed to stabilize the highly active intermediate valence states of copper. Yang et al. used 2D Cu(OH)F nanoplates as a precursor and transformed them into Cu/CuxOF by electrochemical treatment under the same conditions as those used for the alkaline CO2RR (Fig. 19c).198 They revealed that the introduction of F not only modulated the exposed facets of Cu nanoparticles, but also stabilized the oxidized Cu species and induced the s,p-d coupling between the metal and hetero-anions, which enhanced the electroactivity and selectivity for the acetate formation via the CO2RR. Yang et al. conducted another study, where they synthesized a range of P-Cux/Cu2OF catalysts with varying Cu(I)/Cu(0) ratios.199 This was achieved through the pulsed potential method with Cu(OH)F as the precursor. Characterization of the morphology and structure confirmed a nanodisk morphology comprising a Cu(0) core and a satellite-like Cu2OF shell. This arrangement provided exposed sites of Cu(I), Cu(0), and the Cu(I)/Cu(0) interface (depicted in Fig. 19d). The interfacial region was found favorable to ethanol formation, yielding a high FE(C2+) of up to 80.2% and achieving a notable partial current density of up to 128 mA cm−2.
Currently, water electrolysis technology involves conventional alkaline water electrolyzer (AWE) as well as two emerging promising designs: a proton exchange membrane water electrolyzer (PEMWE) and an anion exchange membrane water electrolyzer (AEMWE) (Fig. 20a–c). The latter two can significantly reduce energy costs due to the compact configuration of their membrane electrode assemblies, low ionic resistance and reduced ohmic drop.216,217 The anodic reaction of PEMWE and AEMWE is affected by membrane properties and local pH, with the oxygen evolution reaction in acidic and alkaline environments, respectively. The PEMWE technology is well established and widely used, but faces the challenge of developing a non-precious metal OER catalyst. AEMWE, on the other hand, has advantages due to the applicability of a wider range of catalysts, nevertheless, membrane stability and electrolyzer design need to be improved to meet long-term operational requirements.218 These key factors in design such as membrane materials, binders, supports (Fig. 20d), etc. will be explored in further detail.
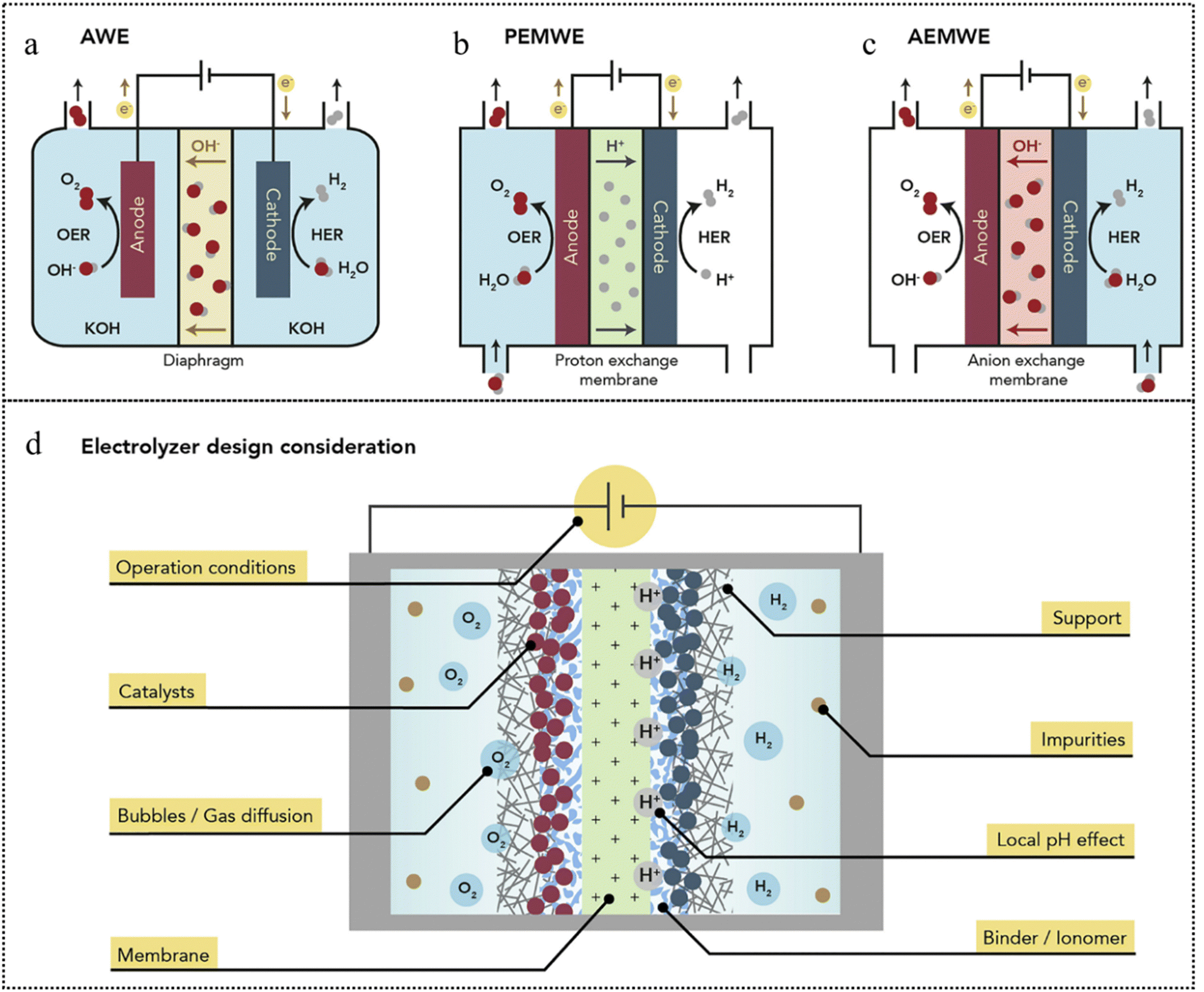 | ||
| Fig. 20 (a)–(c) Leading configurations of current water electrolyzers and design considerations for long-term stability, (d) Critical elements that need to be considered for fabricating electrolyzers with long-term stability. This figure has been reproduced from ref. 219 with permission from Elsevier, copyright 2021. | ||
4.1.3.1 Membranes and ionomers. Membrane stability is critical in electrolyzer design. Proton exchange membranes have been developed to commercial levels of durability, with Nafion membranes being widely used in PEMWE equipment, although membrane thinning and degradation may occur at high temperatures and current densities.220 This may be due to the formation of peroxides and free radicals caused by the crossing of oxygen on the cathode side, leading to membrane degradation under certain operating conditions. While anion exchange membranes have been improved to degrade only slightly over thousands of hours, improvements are still needed to create durable AEMWE devices.221 Improving the conductivity of hydroxide ions in anion exchange membranes is a challenge in long-term operation. The study proposes a new method for preparing MEA that enhances conductivity by creating an additional OH− transport pathway.222 Factors such as degradation of polymer functional groups, blockage of ion exchange pathways, and the effect of temperature and electrolyte on membrane stability need to be investigated in order to enhance membrane durability.
Ionomers are a key factor in electrolytic stability, acting as a binder and ionic conductor for the electrode catalyst. Their content needs to be optimized for the catalyst and operating conditions to avoid high ionic resistance or electrode clogging.223 The design of AEMWE's durable ionic polymers is more challenging than that of stable Nafion.
4.1.3.2 Electrodes and support materials. The selection of suitable support materials is critical for electrolytic stability. Studies have shown that the interaction between the catalyst and the support material affects the electronic structure and catalytic stability.224 For example, the acidic OER behaviors of IrOx nanoparticles on carbon black and antimony-doped tin oxide varies depending on the support. Different support materials (e.g., glassy carbon, gold, and boron-doped diamond) also lead to different Ir stability. The NiFe oxyhydroxide system exhibits a similar phenomenon under alkaline conditions.225,226 Differences in electronic structure and catalyst-support material interactions explain the importance of optimizing catalyst loading. Carbon-based support or gas diffusion layers may initiate carbon corrosion or oxidation, affecting catalytic stability, which is important for both practical electrolysis installations and laboratory-scale experiments.227 In addition, regeneration of catalytic electrodes and decomposition of impurities on electrodes on commercial electrolyzers can be achieved by high-temperature pulse annealing.228 These findings aid in the fabrication of durable OER electrolysis electrodes.
4.1.3.3 Operating conditions and local environments. When applying the results of laboratory-scale catalyst testing to commercial electrolyzers, it is important to consider the actual operating conditions. Studies have shown that the corrosion mechanisms of catalysts are related to voltage and current density, resulting in potentially different stability behavior in practical applications. Therefore, even if certain catalysts are stable in laboratory tests, they may not be able to withstand the high current densities and yield rates required by industry.229 In addition, there may be differences in local pH between laboratory three-electrode cells and MEA electrolyzers, the latter of which may develop extreme pH environments due to factors such as the ion transport properties of the membrane, reactant diffusion and product surface coverage.230
At high current densities, the bubbles generated by the OER may differ in physical properties from those under laboratory conditions, leading to problems such as interfacial supersaturation, high catalytic surface coverage, and increased ohmic internal resistance, all of which may contribute to operational instability.231In situ TEM observations have shown that bubble formation and collapse can lead to oscillations in the catalyst structure due to water uptake and precipitation of oxygen within the oxide.232 Therefore, care should be taken to monitor the operating conditions of the electrolyzer at high potentials and current densities to avoid catalytic instability.
4.1.3.4 Impurities. Electrolysis conditions in practical applications often differ from laboratory test environments, which typically use pure electrolytes for catalytic performance measurements, whereas in practice impure water sources and brines may be involved. This requires consideration of issues such as OER selectivity, catalytic site blockage and ion transport pathway blockage.233 NiFe-(oxy)hydroxide-based catalysts perform well in seawater decomposition and are suitable for use in alkaline AEMWE, but impurity deposition remains a challenge.234 Electrolytes, impurities, and factors such as membranes, ionomers, support materials, and electrodes need to be considered when designing actual electrolyzers for long-term operation.
4.1.3.5 Catalyst reconstruction in electrolyzers. Significant breakthroughs have been achieved in the study of electrolyzers under industrial conditions. Developing non-precious metal anode catalysts for PEM electrolyzers is both attractive and highly challenging. Non-precious metals such as Fe, Co, and Ni, which perform excellently in alkaline environments, tend to undergo irreversible dissolution. However, recent research by Francisco Pelayo and colleagues has shown great breakthrough using cobalt tungstate (CWO) treated in an alkaline environment to achieve efficient and stable PEMWE catalysts.231 The study demonstrated that the stable network of oxides and water hydroxides in CWO can achieve low overpotentials and high current densities. Under industrial conditions (80 °C), the CWO catalyst achieved a current density of 1.8 A cm−2 at 2 V and maintained stability at 1.77 V for 600 hours, showcasing significant performance improvements. These findings highlight the potential of non-precious metal catalysts for PEM electrolyzers, paving the way for more cost-effective and sustainable solutions in industrial applications.
As for commercial alkaline water electrolyzers, the systems typically operate at 80 °C to minimize energy consumption. However, one of the most promising anode candidates, nickel–iron alloy catalysts, faces a high solubility bottleneck at this temperature. Sun and co-authors found that the dissolution of NiFe-LDH during operation not only leads to the degradation of the anode itself but also deactivates the cathode, resulting in an overall decline in electrocatalytic performance.229 To mitigate this dissolution, oxygen anions were used as inhibitors in the electrolyte. Adding phosphates to the electrolyte was found to inhibit the loss of NiFe-LDH active sites at 400 mA cm−2, reducing the loss to one-third of its original level. This intervention decreased the performance degradation rate by 25 times. These findings underscore the importance of addressing the dissolution issue in high-temperature industrial applications, emphasizing the need for further research into effective inhibitors and stabilization strategies to enhance the longevity and performance of non-precious metal catalysts in commercial electrolyzers.
Other challenges remain in industrial applications. For example, in PEM systems, the quality of water required is very high. Impurities can lead to destructive catalyst reconstruction or degradation of the ion-exchange membrane, presenting critical challenges. Scholars from Graz University of Technology have revealed the fatal impact of chloride ion impurities in water on PEMWE under industrial conditions.224 They found that even a chloride ion concentration as low as 10 ppm can significantly affect the performance of the electrolyzer. After 50 hours of operation, the low-frequency resistance of the system increased from 0.23 Ω cm2 by 100% at 1.0 A cm−2. Additionally, they observed catalyst detachment and ion-exchange membrane degradation. This finding compels us to carefully consider the impact of using tap water, which contains residual chlorine, on the lifespan of PEM systems. While further purification of water could mitigate these effects, it would also increase operational costs. Therefore, balancing water quality and cost is a crucial issue that needs to be addressed for the sustainable operation of PEM systems.
4.2 Photocatalysis
Photocatalysis represents a sustainable technology aimed at mitigating the global energy crisis and environmental pollution through the utilization of abundant and renewable solar energy resources. Typically, photocatalytic reactions entail the generation of charges upon exposure to light, subsequent charge separation and transportation, and ultimate charge consumption through surface redox reactions. In certain cases, alongside the use of semiconductor photocatalysts, cocatalysts are required to suppress charge recombination and facilitate charge transport efficiently. It is inevitable that charge carriers and reactants interact with the semiconductor and/or cocatalysts upon contact, resulting in the dynamic transformation of catalysts during the photocatalytic reaction. This dynamic transformation not only gives rise to real active sites, resembling those in electrocatalysis, but also provides an avenue for precisely fine-tuning the energy band structure of photocatalysts. In this section, we will introduce the representative applications of favorable catalyst dynamic transformation in the field of photocatalysis.| 2H+ + 2e− → H2 (g), ΔE0 = −0.41 V | (1) |
| 2H2O (l) + 4h+ → O2 (g) + 4H+, ΔE0 = 0.82 V | (2) |
Overall reaction:
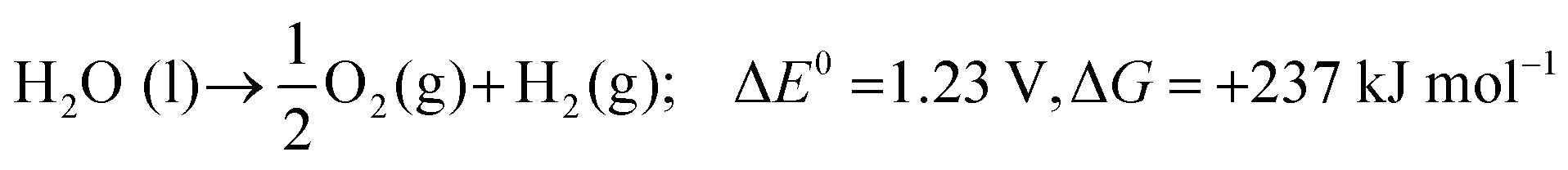 | (3) |
4.2.1.1 Co-catalysts. Photocatalytic water splitting is inherently an uphill reaction. To facilitate this process, the band structures of photocatalysts must adhere to specific criteria, including a conduction band (CB) minimum that is more negative than the H+/H2 reduction potential and a valence band (VB) maximum that is more positive than the O2/H2O oxidation potential. Certainly, meeting all these stringent requirements with a single photocatalyst poses a significant challenge, making it necessary to introduce cocatalysts to enhance the charge separation efficiency and reaction kinetics. In general, electrocatalysts exhibiting high efficiency in electrocatalytic water splitting, such as Pt,85 metal oxides, phosphides,235 sulfides, carbides, and others, have strong potential as cocatalysts for photocatalytic water splitting.236 These cocatalysts undergo dynamic transformations during the photocatalytic water-splitting process (Table 3).
| Catalysts | Dynamic transformation type | Application | Catalytic performance | Ref. |
|---|---|---|---|---|
| Pt/P90-C | Chemical state and coordination environment | HER | 1.4 μmol min−1 | 237 |
| Pt/PC500-C | Chemical state and coordination environment | HER | 1.6 μmol min−1 | 237 |
| Pt/PC500-R | Chemical state and coordination environment | HER | 2 μmol min−1, AQY 11% | 237 |
| PtO/TiO2 | Coordination environment | Overall water splitting | — | 63 |
| p-NiSe2/CN | Phase transformation | HER | 3.75 μmol h−1, | 238 |
| Ni@NiOx-SrTiO3 | Phase transformation and morphology | Overall water splitting | H2 yeild: 0.3 μmol min−1 g−1 | 67 |
| O2 yeild: 0.1 μmol min−1 g−1 | ||||
| ME-TiO2@Ru | Chemical state | HER | 323.2 μmol h−1 | 239 |
| Bi4TaO8Cl | Phase transformation | OER | 15 μmol h−1 | 240 |
| Bi4TaO8Br | Phase transformation | OER | 20 μmol h−1 | 240 |
| CdS/a-MoOx-32.6 wt% | Chemical state | HER | 19.3 mmol h−1 g−1 | 241 |
| TP-COFs-1 | Coordination environment | HER | 0.96 mmol g−1 h−1 | 242 |
| QDs-Ni(OH)2 | Phase transformation | HER | 56 μmol h−1 | 243 |
| Pt1/ZnNiTi-LDHs-E | Chemical state | CO2RR (Light irradiation: 200 mW cm−1) | CO yeild: 95.41 μmol g−1 h−1 | 244 |
| Pt1/ZnNiTi-LDHs-E | Chemical state | CO2RR (Light irradiation: 500 mW cm−1) | CO yeild: 109.89 μmol g−1 h−1 | 244 |
| C2H6 yeild: 29.84 μmol g−1 h−1 | ||||
| Pt1/ZnNiTi-LDHs-E | Chemical state | CO2RR (Light irradiation: 1000 mW cm−1) | CO yeild: 224.65 μmol g−1 h−1 | 244 |
| C2H6 yeild: 30.12 μmol g−1 h−1 | ||||
| CH4 yeild: 25.54 μmol g−1 h−1 | ||||
| CuZnTCPP/g-C3N4 | Phase transformation | CO2RR | CO yeild: 92.0 μmol g−1 h−1 | 219 |
| CH4 yeild: 11.3 μmol g−1 h−1 | ||||
| Cu-CuTCPP/g-C3N4 | Chemical state | CO2RR | CO yeild: 12.3 μmol g−1 h−1 | 245 |
| C2H6 yeild: 18.5 μmol g−1 h−1 | ||||
| CH4 yeild: 11.6 μmol g−1 h−1 | ||||
| U-BOC | Element redistribution | CO2RR | CO yeild: ∼ 21.36 μmol g−1 h−1 | 246 |
| S3-Co-600 | Phase transformation | CO2RR | CO yeild: 0.0924 mmol | 244 |
| ZnO/CuOx-C CNFs | Coordination environment | CO2RR | CH4 yeild: 241.6 μmol g−1 h−1 | 247 |
| 2%CuOx/TiO2-{001} | Chemical state | CO2RR | CH4 yeild: 1.19 μmol g−1 h−1 | 248 |
| 2%CuOx/TiO2-{100} | Chemical state | CO2RR | CH4 yeild: 0.41 μmol g−1 h−1 | 248 |
| 2%CuOx/TiO2-{101} | Chemical state | CO2RR | CH4 yeild: 0.87 μmol g−1 h−1 | 248 |
| TiO2@BiOCl | Chemical state and Coordination environment | CO2RR | CH4 yeild: 168.5 μmol g−1 h−1 | 249 |
| Pt/Ga–Zn–oxynitride | Phase transformation | Methanol reforming reaction | H2 yeild: 0.64 mmol g−1 h−1 | 250 |
| 2-ZnS/Zn–Al | Chemical state | Cr(VI) removal | 17.14 mg L−1 h−1 | 251 |
| ZnAl-3 | Morphology | Phenol degradation | 8 ppm h−1 | 252 |
| ZnGaAl-5% | Morphology | Phenol degradation | 5.3 ppm h−1 | 252 |
| ZnAlCe5% | Chemical state | Phenol degradation | 9 ppm h−1 | 253 |
Pt is frequently employed as a cocatalyst in the photocatalytic HER, and the beneficial impact of achieving a high dispersion of Pt in photocatalytic HER has been well-documented. However, these highly dispersed metal-based catalysts are susceptible to photochemical processes. During photocatalytic operando XAS experiments conducted on Pt single-atom catalysts (SACs), the typical trend observed in relation to the initial conditions is a reduction in Pt–O coordination, with coordination numbers decreasing from 4–5 to 2–3.237 This indicates a partial reduction of Pt species, and the SACs exhibited partial Pt–Pt clustering under the reaction conditions. The anatase-rich P90 titania support could lead to extensive clustering with a Pt–Pt coordination number of 7, and this was significantly suppressed by employing a precalcined Pt/PC500 anatase-rich support with high-specific surface areas and high surface defectiveness. Specifically, Pt/PC500 maintained an atomic Pt dispersion and a high Pt oxidation state consisting of single atoms, dimers, and trimers, with Pt–Pt coordination numbers of 1–2 during the photocatalytic HER. Notably, using the same support, a mild reducing pretreatment can yield near-neutral and highly dispersed Pt species with Pt–O and Pt–Ti bonds in the pre-reduced Pt/PC500, leading to improved photocatalytic HER performance and stability (0.14 mol h−1 g−1, a turnover frequency per Pt atom of 0.37 s−1, a photonic yield of 5.6%, and an apparent quantum yield of 11%). In addition to the photocatalytic HER, Pt-based co-catalysts can be employed with great efficiency in photocatalytic overall water splitting. Taking a PtO/TiO2 photocatalyst as an example, the Pt–O bond length extended from 2.07 to 2.13 Å, while the coordination number decreased from 4.0 to 2.5 during the photocatalytic overall water splitting.63 The dynamic transformation of the PtO cocatalyst is instrumental in unilaterally quelling undesirable hydrogen oxidation and achieving exceptional efficiency in the hydrogen evolution rate.
Transition metal compounds have been proven as efficient cocatalysts for photocatalytic water splitting when integrated with appropriate semiconductors. When NiSe2 with different phases were incorporated into carbon nitride (CN) to form a hybrid photocatalyst (m-NiSe2/CN and p-NiSe2/CN), these hybrids exhibited similar photocatalytic HER rates of 3.26 μmol h−1 and 3.75 μmol h−1, respectively.236 Strikingly, distinct surface conversion behaviors were observed in m-NiSe2/CN and p-NiSe2/CN during the photocatalytic process. After the photocatalytic HER, the chemical structure and morphology of m-NiSe2 were well retained, but partial p-NiSe2 would undergo conversion to NiOOH, resulting in different stability of m-NiSe2/CN and p-NiSe2/CN. Specifically, m-NiSe2/CN maintained stable H2 evolution, whereas p-NiSe2/CN experienced a significant 57.1% reduction in catalytic activity over 25 hours.
In addition, core–shell co-catalysts consisting of Ni@NiOx were employed to functionalize SrTiO3 (BSTO-1000-NiOx) for photocatalytic overall water splitting.67 Under the influence of humidity and aqueous conditions, the NiO phase underwent transformation into Ni(OH)2. In the early stages of illumination, excess hydrogen production with an H2:O2 yield ratio exceeding 2 implied the oxidation of Ni(OH)2 to NiOOH, serving as a cocatalyst for water oxidation. Upon discontinuation of illumination, a disproportionation reaction took place involving Ni and NiOOH, giving rise to the formation of Ni(OH)2 with residual embedded Ni. The subsequent reinitiation of illumination elucidated the recurring transients noted in the formation of excess hydrogen and oxygen (Fig. 21a).
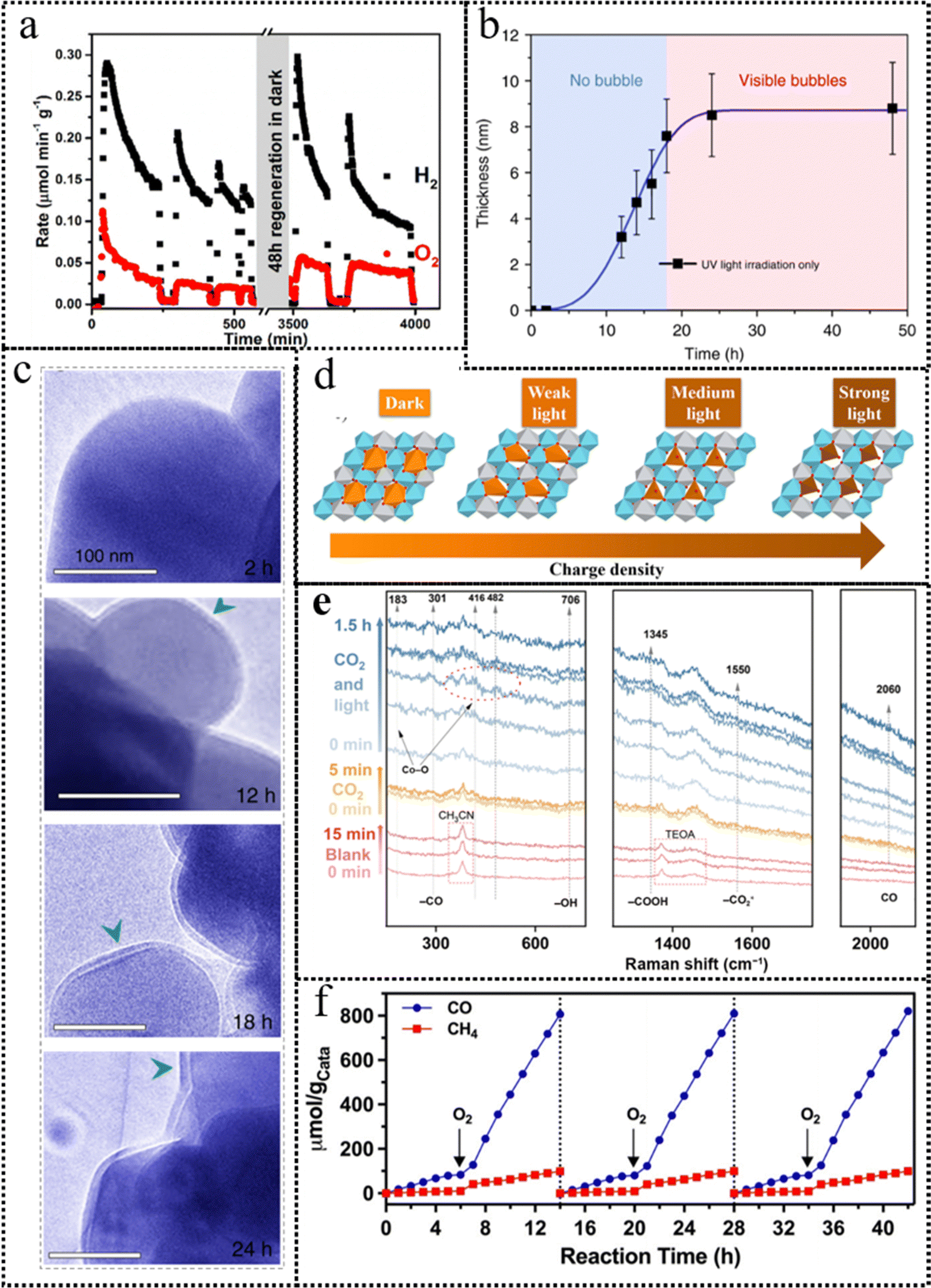 | ||
| Fig. 21 (a) Transient behavior in H2 and O2 evolution by using 25 mg of BSTO-1000-NiOx (black: H2; red: O2). This figure has been reproduced from ref. 67 with permission from American Chemical Society, copyright 2020. (b) Thickness of the surface shell on anatase TiO2vs. UV illumination time: experimental data (black squares) and fitted curve based on the KJMA equation 21 (blue line). Growth of the surface shell experienced a sudden decrease when bubbles became visible at around 18 hours (c) Photocatalysis experiments with different times of UV exposure. Each TEM image was recorded independently with fresh aqueous suspensions of TiO2 NPs controlled by the fluidic holder. This figure has been reproduced from ref. 254 with permission from Nature, copyright 2018. (d) Illustration of the dynamic transformation of Pt SAs under light irradiation. This figure has been reproduced from ref. 244 with permission from American Chemical Society, copyright 2022. (e) Pre-operando Raman spectra collected during the photocatalytic CO2 reduction. This figure has been reproduced from ref. 255 with permission from Springer, copyright 2023. (f) In the cyclic CO2 photoreduction experiment on pre-treated Cu-ZnTCPP/g-C3N4, the gas atmosphere was alternated between aerobic and anaerobic conditions in each cycle. This figure has been reproduced from ref. 256 with permission from Wiley-VCH, copyright 2023. | ||
4.2.1.2 Semiconductors. As one of the most significant semiconductor photocatalysts, TiO2 has garnered substantial interest and extensive research over several decades. Recently, the dynamic transformation of anatase TiO2 nanoparticles in photocatalytic H2 evolution was investigated by using liquid environmental TEM. As depicted in Fig. 21b and c, a self-hydrogenated shell was observed on the TiO2 surface prior to the emergence of hydrogen bubbles. This shell was established through subsurface diffusion of H atoms created via the reaction of water protons with photoinduced electrons, playing a critical role in promoting hydrogen generation by lowering the activation energy for H2 (H–H bond) formation.254 Similar dynamic transformation phenomena were also observed on the Bi4TaO8X photocatalyst.240 During the photocatalytic oxygen evolution process, photoinduced TaOx was in situ generated on the surface of the Bi4TaO8X photocatalyst at the start of irradiation and then underwent oxidation by the electron acceptor. The photoinduced TaOx could readily capture and react with hole scavengers or electron acceptors by virtue of the metastable chemical valence in an amorphous state, allowing photogenerated electrons and holes to participate in the photocatalytic HER and OER, respectively. As a result, the dynamic transformation process could not only yield active species for surface catalytic reactions but also enhance the separation of photogenerated charges, ultimately improving the overall efficiency of the photocatalytic reaction. However, dynamic transformation in metal oxide photocatalysts is not always beneficial to the reactions, such as the photocorrosion of copper oxide during the photocatalytic water splitting.257 The CB potential of Cu2O promoted the rapid transfer of photoinduced electrons for H2 evolution. However, photoinduced holes tended to accumulate and were responsible for severe Cu2O photocorrosion. This was caused by the relatively low VB energy level of Cu2O, which restricted the participation of photoinduced holes in water oxidation. The inability to efficiently remove photoinduced holes by adsorbed water molecules resulted in the self-oxidation of Cu2O. Introducing a hole scavenger with an appropriate oxidizing potential facilitated the transfer of photoinduced holes, thus enhancing Cu2O photostability.
Creating heterojunctions at the nanoscale is indeed an effective approach for improving the separation of photoinduced carriers, broadening the light absorption range, and facilitating mass transfer, all of which contribute to enhanced photocatalytic performance.258 For heterojunction photocatalysts, their dynamic transformation may be affected by interactions between components.259 In CdS/a-MoOx heterojunction photocatalysts, the initial photoexcited electrons in CdS demonstrated a robust reduction capacity and effectively transferred to a-MoOx, leading to the reduction of Mo6+ to Mo5+.241 This transformation initiated an in situ electronic structure reconstruction, and it was empirically demonstrated that this electronic reconstruction played a pivotal role in the progressive enhancement of photocatalytic H2 evolution performance. For the CdS/a-MoOx-32.6 wt% heterojunction catalyst, there was a substantial 16.8-fold increase in the rates of hydrogen production during the progression of the photocatalytic HER, elevating the rate from 1.1 mmol h−1 g−1 to 19.3 mmol h−1 g−1. It is noteworthy that this rate significantly outperformed the hydrogen evolution rate achieved by the CdS nanorod catalyst, which stood at 1.1 mmol g−1 h−1.
Covalent organic frameworks (COFs) composed of C, H, O, N, S and halogen elements represent a novel class of semiconducting polymeric materials for photocatalysis, which are gaining prominence by virtue of their exceptional attributes, including stable ordered crystal structures, high porosity and tunable light absorption, as well as versatile 2D or 3D network topologies. Very recently, the enol-to-keto dynamic transformation in the BT-COFs with imine linkages during the photocatalytic H2 evolution was studied.242 The enol-to-keto conversion in BT-COFs-1 was found to be thermodynamically favorable but kinetically sluggish, leading to the predominant presence of the enol form. Alkaline post-treatment effectively promoted dynamic transformation, leading to an increased population of keto-injectors within the structural building blocks, which could improve photocatalytic performance. The generation of keto-injectors not only rendered the CB level more negative but also introduced non-uniform charge distribution within the donor–acceptor molecular building blocks, thereby creating a significant intramolecular built-in electric field. Benefiting from the aforementioned advantages, TP-COFs-1, featuring a single keto group within its skeletal building blocks, exhibited excellent photocatalytic performance, yielding a remarkable H2 evolution rate of 0.96 mmol g−1 h−1.
To explore the dynamic transformation of single atom catalysts during the photocatalytic CO2RR, metal ion vacancy-rich ZnNiTi layered double hydroxide-etched (ZnNiTi-LDHs-E) supports were utilized to immobilize Pt single atoms (Pt SAs).244 As illustrated in Fig. 21d, the Pt SAs exhibited distinct photochemical responses, forming cationic Pt(IV), electron-rich Pt(II), and near-neutral Ptδ+ species under varying light intensities. The in situ generation of the various Pt SAs described above significantly influenced the product distribution in photocatalytic CO2 reduction. Under weak light conditions, Pt(IV) SAs failed to activate CO, thereby preventing further transformation into hydrocarbons. Moderate light exposure resulted in electron-rich Pt(II) SAs, which could collaborate with adjacent LDH surface sites (Ni2+/Ti4+) to enable the C–C coupling route, leading to C2H6 generation. The near-neutral Ptδ+ species play a crucial role in the direct hydrogenation of C1 to generate CH4. This research revealed the dynamic transformation of Pt SAs and deepened the comprehension of active site properties in artificial photosynthesis, offering valuable insights for the development of efficient SA photocatalysis.
The surface coordination structure of transition metal-based semiconductors is prone to alteration upon exposure to environmental O2 due to the coordinated nature of their 3d-orbital electrons. For example, the in situ formation of CoOx active sites on the Co (CoP) surface during the initial 4 cycles of testing resulted in an increase in transient activity and selectivity toward CO evolution.255 The pre-operando Raman spectrum in Fig. 21e simultaneously revealed the generation of a reconstructive Co–O bond and the existence of an intermediate specie CO, elucidating the dynamic interplay between the catalytic site structure and the photocatalytic properties. The electronic structure of the reconstructed surface sites played a crucial role in modulating the CO2 adsorption and CO desorption, ultimately reducing the energy barrier for the rate-determining step and enhancing CO2 reduction activity and selectivity. For some 2D semiconductors, the ultrathin structure can also promote the associated dynamic transformations during the photocatalytic process. Taking ultrathin BiOCl (U-BOC) as an example, the exposed {001} facets were terminated with chlorine atoms rather than the conventionally expected oxygen atoms.246 The ultrathin structure and low binding forces facilitated the outward migration of surface Cl atoms, leading to the formation of Bi, Cl, and O atoms with various valence states and corresponding chemical bond alterations in the lattice layers. During the photocatalytic CO2RR process, light irradiation initiated the retraction of surface Cl atoms, causing the elongation of Bi–Cl and Bi–O bonds in the lattice phase, along with the disappearance of multiple-valence Bi, Cl, and O atoms. With the assistance of the structural feature alteration described above, U-BOC exhibited outstanding photocatalytic CO2 reduction performance, approximately 21.36 μmol g−1 h−1, nearly 6.4 times higher than that of bulk BOC.
Constructing composite photocatalysts is an effective approach to enhance photocatalytic performance. Recently, a π–π stacking hybrid structure was formed between two highly conjugated materials: g-C3N4 and the 2D-MOF moiety of the hybrid photocatalyst employed tetrakis(4-carboxylpheny) porphyrin (TCPP) as the building block (Cu–CuTCPP), wherein g-C3N4 served as the photosensitizer to provide electrons and 2D-MOF Cu–CuTCPP acted as the interface to catalyze CO2 reduction.245 The node sites of the 2D-MOF were found to be crucial for C2H6 production, and a dynamic transformation process during photocatalysis was observed: the initial paddle-wheel CuII2(COO)4 node reconstructed into the partially reduced Cu1+δ2(COO)3. This reconstruction enhanced the capture of in situ generated CO and the synergistic effects of the dual Cu sites, ultimately leading to efficient C–C coupling for C2H6 production. Accordingly, this hybrid photocatalyst demonstrated remarkable efficiency in reducing CO2 to C2H6 with a selectivity of 44%, and the selectivity for total hydrocarbons (C2H6 and CH4) reached 71%, making it one of the most promising photocatalytic systems reported to date. Based on this work, a Cu–ZnTCPP/g-C3N4 hybrid photocatalyst was fabricated and it exhibited remarkable photocatalytic performance in converting CO2 into CO and CH4 under typical air conditions with 20% O2.219 Interestingly, the presence of O2 did not inhibit CO2 reduction; instead, it led to a five-fold increase in the reaction rate compared to when O2 was absent, as depicted in Fig. 21f. In the presence of O2, the Cu node in the 2D-MOF component experienced hydroxylation due to the hydroxyl radicals generated from O2 reduction. Under these conditions, the hydroxylated Cu node retained its structure throughout aerobic CO2 reduction but underwent structural changes and deactivation in the absence of O2. Further theoretical calculations verified a preference for CO2 reduction over O2 reduction on the hydroxylated Cu node (Fig. 22).
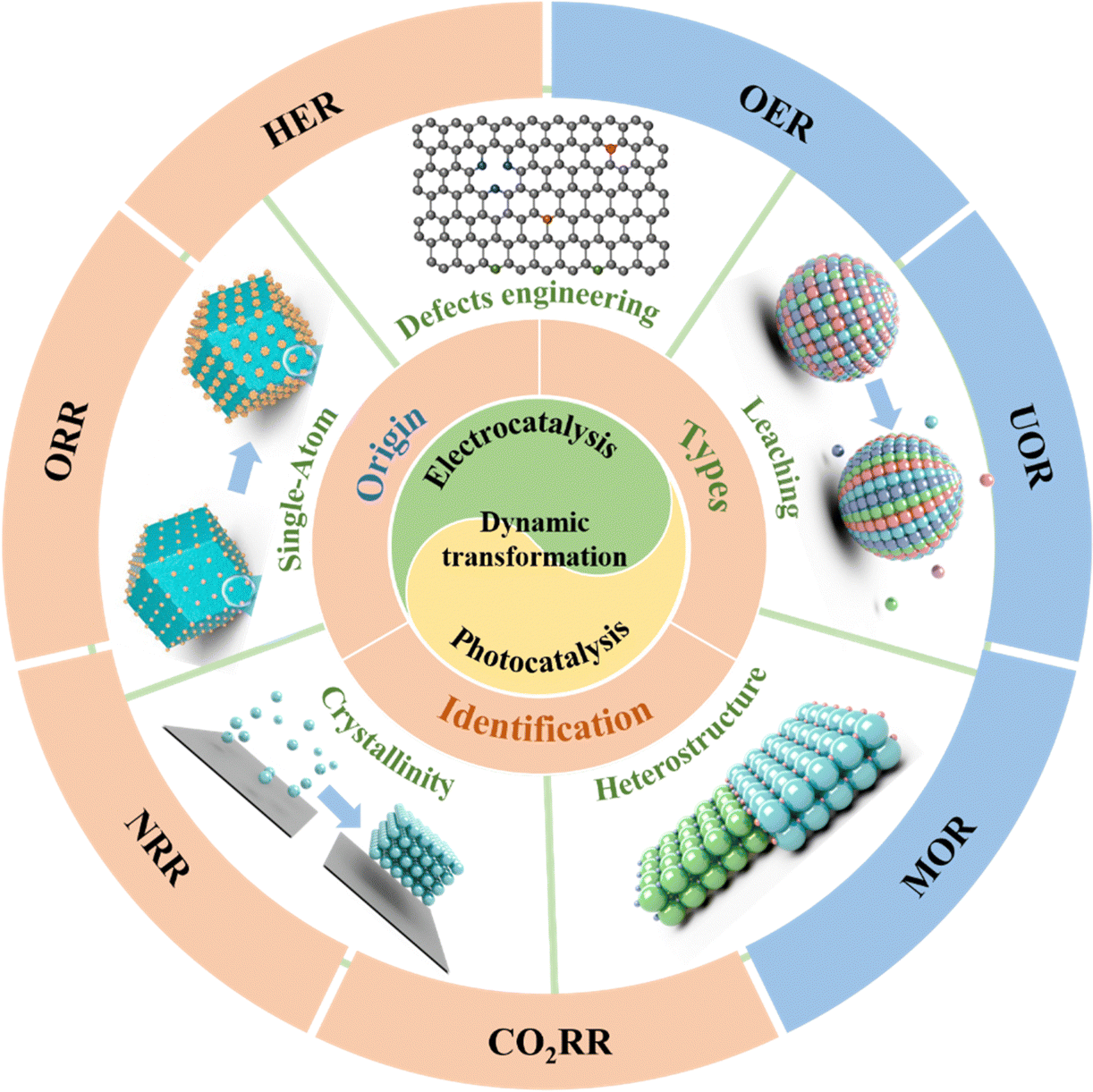 | ||
| Fig. 22 Overview of dynamic transformation of active sites in the field of photo/electrocatalysis, covering the origin and types of dynamic transformation, identification methods, various triggering factors and applications in photo/electrocatalysis. | ||
Comprehending and analyzing dynamic changes in heterojunction photocatalysts can be challenging, given the necessity to account for both the reaction environment and complex composition. For CuOx/TiO2 photocatalysts, the different crystals of TiO2 would influence the dynamic transformation of CuOx during the photocatalytic CO2RR.252 Under Xe light irradiation, CuO species on TiO2-{001} were photoreduced to stable Cu2O species, promoting CO2 reduction to CH4. However, CuOx species on TiO2-{100} and TiO2-{101} are photo-reduced to Cu0 species, hindering CH4 production. In addition, each constituent in a heterojunction photocatalyst may undergo dynamic transformations. A typical example is the TiO2@BiOCl heterojunction photocatalyst.249 Under light excitation, H-incorporated oxygen vacancies were formed on the defective TiO2 nanotube matrix to extend the light adsorption range, and concurrent reduction of BiOCl lamina resulted in the formation of an atomic heterojunction composed of BiOCl–Bi0–TiO2. Bi0 atoms directly transferred photogenerated electrons to Ti3+ within H-incorporated oxygen vacancies, facilitating efficient electron gathering and thus enhancing photocatalytic efficiency. The presence of H-incorporated oxygen vacancies and Bi–Ti dual metal sites optimized the CO2 reduction pathway, leading to high CH4 selectivity. With the assistance of the dynamic reconstruction, a remarkable photocatalytic CO2RR performance was achieved by TiO2@BiOCl with the optimal CH4 productivity rate of 168.5 μmol g−1 h−1 and CH4 selectivity of 99.4%.
In most cases, the comprehension of photocatalysis is attained by evaluating photocatalysts before and after photocatalytic reactions, thereby establishing an empirical foundation for deducing the photocatalytic processes. However, the photocatalytic mechanism remains elusive due to the involvement of high-energy electrons and holes, which act as “reactants” in photocatalytic reactions. These photogenerated charges not only participate in surface catalytic reactions at the outer surface but also potentially trigger reconstruction or self-activation of photocatalysts before reaching the outer surface. In comparison to electrocatalysts, dynamic transformation studies on photocatalysts are relatively limited and primarily concentrate on photocatalytic water splitting and the CO2RR. Future research endeavors should prioritize investigating dynamic changes in photocatalysts for a broad range of reactions. This approach will contribute to a comprehensive understanding of photocatalytic mechanisms.
5. Conclusion and perspectives
Over recent decades, there has been significant progress in the development of catalyst dynamic transformation, encompassing the exploration of the origins, underlying principles, identification methods, and targeted tailoring of the dynamic transformations. These advancements offer substantial potential for engineering the dynamic transformation to design efficient and durable photo/electrocatalysts. This comprehensive overview systematically explored such issues and identified several approaches and important triggering factors for the dynamic transformation and its applications in photo/electrocatalysis. It should be noted that reconstruction of catalysts is induced by single atom dopants, defects (vacancies, dopants, grain boundaries, etc.), heterostructures, leaching and crystallinity, which correspond to scales ranging from the atomistic defects to the mesoscopic scale crystalline structures of the materials. Despite the advancements, constructing photo/electrocatalysts with precisely controlled reconstructions for diverse photo/electrocatalytic reactions remains challenging. Enhancing the activity, stability and selectivity of these reconstructed photo/electrocatalysts for specific reactions poses ongoing challenges. Several critical questions remain:(1) Difficulty in in situ characterization: the dynamic environment of electrochemical reactions during catalyst restructuring poses significant challenges in accurately characterizing these changes. In particular, the study of active sites has focused on individual atoms, coordination structures, and specific crystal facets, so the corresponding characterization methods need to be further developed, which is conducive to the systematic description and understanding of the reconstruction mechanism. For example, researchers initially recognized the reconstruction process through changes in cyclic voltammetry curves. Subsequently, advancements in characterization techniques have evolved considerably, such as in situ TEM for observing surface morphological changes and in situ XAS for obtaining coordination environments and electronic structure information. The evolution of characterization techniques facilitates the correlation of dynamic changes in active sites with catalytic activity, product selectivity, and stability. This capability effectively guides the regulation of photo/electrocatalysts reconstruction.
(2) Catalyst restructuring involves complex chemical reaction pathways and intermediates. Identifying the true active sites and understanding the reaction mechanisms of dynamic processes are daunting and critical challenges in the field of catalysis. For instance, owing to the iron's inherent propensity for active reconstruction, the extensive research endeavors have been focused on unraveling the dynamic restructuring phenomena associated with iron, particularly within the realm of NiFe (hydroxide) oxides. Despite these efforts, a consensus on the dynamic transformation pathways of iron remains elusive, particularly concerning alterations in its oxidation states and coordination environments. Similarly, in the realm of photo/electrocatalysts for the CO2RR, predominantly Cu-based, the significance of discerning active sites cannot be overstated. These sites must efficiently adsorb CO2 molecules and facilitate the coupling of carbon atoms to yield multicarbon products, where material selectivity profoundly influences catalyst restructuring. Factors such as varying oxidation states of copper sites, distinct crystal facets, and specialized structures resulting from catalyst reconstruction, significantly impact the selectivity of reduction products. Hence, the imperative lies in recognizing the authentic catalytic sites and delving into the dynamics of the process. Such endeavors not only contribute to bolstering our comprehension of catalytic mechanisms but also pave the way for the development of more efficient and selective photo/electrocatalysts.
(3) Rationally controlling the degree of reconstruction in catalytic systems is of paramount importance for achieving a delicate balance between stability and activity. This equilibrium is crucial for optimizing catalytic performance and ensuring long-term efficacy. By strategically modulating the reconstruction process, one can tailor the surface structure and composition of photo/electrocatalysts to enhance their stability while preserving their catalytic activity. For example, noble metal oxide catalysts exhibit high activity in the OER due to their tendency to follow the LOM mechanism, which typically involves lower thermodynamic barriers. However, this heightened activity often leads to irreversible site dissolution, resulting in decreased stability of the catalyst. To address this issue, controlling the degree of self-reconstruction through modification of noble metal catalysts is crucial. Precise modulation of the reconstruction process allows for optimization of the surface structure and composition of the catalysts, thereby enhancing both activity and stability. Modification strategies may include introducing auxiliary species with higher stability, tuning the lattice structure or surface adsorption properties of the photo/electrocatalysts, and designing surface coatings resistant to dissolution.
(4) While there has been considerable discourse on the reconstruction mechanism in the electrocatalytic process, there is a notable scarcity of reports investigating the reconstruction mechanism under photocatalytic conditions. Moreover, the oxidation half-reaction is frequently overlooked in many instances, despite the equal significance of both the reduction and oxidation half-reactions. The rational and strategic utilization of dynamic transformation in the reduction reaction, particularly in photocatalysis, should be a focal point in future research efforts. Investigating the structure–performance relationship under operational conditions not only aids the identification of active sites/species but also enables the development of highly active and selective photo/electrocatalysts, leading to enhanced production rates and yields. For practical applications, the search for cost-effective, abundantly available, and atom-efficient catalytic materials is imperative, with a particular emphasis on single-atom/single-site catalysts, especially those based on non-precious metals such as Fe, Co, Ni, and Mn, to be guided by the further established structure–activity correlations.
(5) When considering the integration of catalysts into electrolyzers, exploring catalyst reconstruction involves the synergy of multiple factors. For instance, under alkaline industrial electrolysis conditions, where the electrolyte concentration typically ranges from 20 to 30 wt% KOH at temperatures of 50–80 °C, the catalyst's reconstruction characteristics become pivotal. Additionally, the evolution of catalyst-ionomer mixtures within MEA under electrolysis conditions represents another critical yet underreported issue. For photocatalysis, the development of a mature device for the large-scale application of photocatalytic reactions remains an ongoing challenge. In recent years, significant research has been dedicated to investigating various photocatalytic devices, each with its own unique reaction environment. For example, under controlled laboratory conditions, photocatalytic water splitting is frequently conducted using suspended powder photocatalysts directly in aqueous solutions.236–238 However, when transitioning to large-scale applications, it becomes imperative to deposit the catalyst onto a specific substrate. Such different reaction environments may cause differences in the dynamic transformation of photocatalysts during the reaction process.
(6) Integrating high-throughput experimentation and machine learning for studying catalyst reconstruction: high-throughput experimentation (HTE) enables the rapid screening and optimization of a large number of catalyst samples particularly in studying reconstruction behaviors of different elements and structures under various conditions. Machine learning algorithms can analyze vast datasets generated from HTE to uncover patterns and relationships that might be missed by traditional methods. By training AI models on experimental data, researchers can develop predictive models that provide insights into catalyst reconstruction behaviors and guide future experiments.
Author contributions
H. Zhang conceived the idea. H. Zhang, L. Chen and F. Dong co-wrote the paper. H. Zhang, Z. Lu, E. Lv, X. Dong, H. Li, Z. Yuan, X. Peng, S. Yang, J. Qiu, Z. Guo and Z. Wen discussed the results and revised the manuscript.Data availability
The authors declare that the data supporting the findings of this study are available in the article.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
H. Z. acknowledges the University of Oxford for the MPLS Enterprise and Innovation Fellowship and the support of Massachusetts Institute of Technology. Z. Y. Y. acknowledges the support of the National Natural Science Foundation of China (22179065, 22105108). X. W. P. acknowledges the support of the National Natural Science Foundation of China (31971614, 32301533). S. H. Y. acknowledges the support of the National Natural Science Foundation of China (21905006, 21972006). Z. X. G. acknowledges the support of the Hong Kong UGC-TRS (T23-713/22-R) award, the Environment and Conservation Fund (ECF 2021-152), the RGC-EU Collaborative Program initiative (Grand No. E-HKU701/23), and the “Hong Kong Quantum AI Lab Ltd” funded by the AIR@InnoHK, launched by the Innovation and Technology Commission (ITC). Z. H. W. acknowledges the support of the National Natural Science Foundation of China (22225902, U22A20436) and the National Key Research & Development Program of China (2022YFE0115900, 2021YFA1501500).References
- L. Liu, G. He, M. Wu, G. Liu, H. Zhang, Y. Chen, J. Shen and S. Li, Nat. Energy, 2023, 8, 870–880 CrossRef.
- M. Rastgar, K. Moradi, C. Burroughs, A. Hemmati, E. Hoek and M. Sadrzadeh, Chem. Rev., 2023, 123, 10156–10205 CrossRef CAS PubMed.
- M. A. Green, Nat. Energy, 2023, 8, 783–784 CrossRef.
- Q. Wang, C. Pornrungroj, S. Linley and E. Reisner, Nat. Energy, 2022, 7, 13–24 CrossRef.
- J. Ran, A. Talebian-Kiakalaieh, S. Zhang, E. M. Hashem, M. Guo and S.-Z. Qiao, Chem. Sci., 2024, 15, 1611–1637 RSC.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Nat. Energy, 2019, 4, 115–122 CrossRef CAS.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed.
- W. Fang, W. Guo, R. Lu, Y. Yan, X. Liu, D. Wu, F. M. Li, Y. Zhou, C. He and C. Xia, Nature, 2024, 626, 86–91 CrossRef CAS PubMed.
- M. Zheng, J. Zhang, P. Wang, H. Jin, Y. Zheng and S. Z. Qiao, Adv. Mater., 2024, 36, 2307913 CrossRef CAS PubMed.
- H. Jiang, Q. He, Y. Zhang and L. Song, Acc. Chem. Res., 2018, 51, 2968–2977 CrossRef CAS PubMed.
- Y. Li, X. Du, J. Huang, C. Wu, Y. Sun, G. Zou, C. Yang and J. Xiong, Small, 2019, 15, 1901980 CrossRef PubMed.
- H. S. Taylor, Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character, 1925, 108, 105–111.
- C. Pei, S. Chen, D. Fu, Z.-J. Zhao and J. Gong, Chem. Rev., 2024, 124, 2955–3012 CrossRef CAS PubMed.
- W. Chen, Y. Che, J. Xia, L. Zheng, H. Lv, J. Zhang, H.-W. Liang, X. Meng, D. Ma, W. Song, X. Wu and C. Cao, J. Am. Chem. Soc., 2024, 146, 11542–11552 CAS.
- M. N. Jackson, C. J. Kaminsky, S. Oh, J. F. Melville and Y. Surendranath, J. Am. Chem. Soc., 2019, 141, 14160–14167 CrossRef CAS PubMed.
- J.-W. Zhao, H.-Y. Wang, L. Feng, J.-Z. Zhu, J.-X. Liu and W.-X. Li, Chem. Rev., 2024, 124, 164–209 CrossRef CAS PubMed.
- D. Liu, Q. He, S. Ding and L. Song, Adv. Energy Mater., 2020, 10, 2001482 CrossRef CAS.
- R. Chen, Z. Ren, Y. Liang, G. Zhang, T. Dittrich, R. Liu, Y. Liu, Y. Zhao, S. Pang, H. An, C. Ni, P. Zhou, K. Han, F. Fan and C. Li, Nature, 2022, 610, 296–301 CrossRef CAS PubMed.
- T. Tang, X. Bai, Z. Wang and J. Guan, Chem. Sci., 2024, 15, 5082–5112 RSC.
- E. Pastor, M. Sachs, S. Selim, J. R. Durrant, A. A. Bakulin and A. Walsh, Nat. Rev. Mater., 2022, 7, 503–521 CrossRef CAS.
- J. Ren, H. Chi, L. Tan, Y.-K. Peng, G. Li, M. Meng-Jung Li, Y. Zhao and X. Duan, J. Mater. Chem. A, 2023, 11, 2528–2543 RSC.
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem., 2022, 6, 303–319 CrossRef CAS PubMed.
- H.-Q. Chen, L. Zou, D.-Y. Wei, L.-L. Zheng, Y.-F. Wu, H. Zhang and J.-F. Li, Chin. J. Catal., 2022, 43, 33–46 CrossRef CAS.
- S. Zhao, C. Tan, C.-T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang and H. Li, Nat. Energy, 2020, 5, 881–890 CrossRef CAS.
- B. Zhang, L. Wang, Z. Cao, S. M. Kozlov, F. P. García de Arquer, C. T. Dinh, J. Li, Z. Wang, X. Zheng and L. Zhang, Nat. Catal., 2020, 3, 985–992 CrossRef CAS.
- Q. Liu, Q. Jiang, L. Li and W. Yang, J. Am. Chem. Soc., 2024, 146, 4242–4251 CrossRef CAS PubMed.
- K. Seki, T. Higashi, Y. Kawase, K. Takanabe and K. Domen, J. Phys. Chem. Lett., 2022, 13, 10356–10363 CrossRef CAS PubMed.
- F.-M. Li, L. Huang, S. Zaman, W. Guo, H. Liu, X. Guo and B. Y. Xia, Adv. Mater., 2022, 34, 2200840 CrossRef CAS PubMed.
- L. Wang, Y. Hao, L. Deng, F. Hu, S. Zhao, L. Li and S. Peng, Nat. Commun., 2022, 13, 5785 CrossRef CAS PubMed.
- Y. Xie, Y. Sun, H. Tao, X. Wang, J. Wu, K. Ma, L. Wang, Z. Kang and Y. Zhang, Adv. Funct. Mater., 2022, 32, 2111777 CrossRef CAS.
- Y. Kuang, M. J. Kenney, Y. Meng, W.-H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li and L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- X. Liu, J. Meng, J. Zhu, M. Huang, B. Wen, R. Guo and L. Mai, Adv. Mater., 2021, 33, 2007344 CrossRef CAS PubMed.
- N. Ran, E. Song, Y. Wang, Y. Zhou and J. Liu, Energy Environ. Sci., 2022, 15, 2071–2083 RSC.
- K. Jeenmuang, P. Pornaroontham, M. F. Qureshi, P. Linga and P. Rangsunvigit, Chem. Eng. J., 2024, 479, 147691 CrossRef CAS.
- L. Liu and A. Corma, Nat. Rev. Chem., 2021, 5, 256–276 CrossRef CAS PubMed.
- J. Li, R. Chen, J. Wang, K. Wang, Y. Zhou, M. Xing and F. Dong, Angew. Chem., Int. Ed., 2024, 63, e202317575 CrossRef CAS PubMed.
- A. W. Heard, J. M. Suárez and S. M. Goldup, Nat. Rev. Chem., 2022, 6, 182–196 CrossRef PubMed.
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 CrossRef CAS.
- G. Chen, C. Wei, Y. Zhu and H. Huang, EcoMat, 2023, 5, e12294 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. A. Alsunni and C. B. Musgrave, Appl. Surf. Sci., 2023, 610, 155147 CrossRef CAS.
- X. Luo, X. Tan, P. Ji, L. Chen, J. Yu and S. Mu, EnergyChem, 2023, 5, 100091 CrossRef CAS.
- B. Yan, D. Krishnamurthy, C. H. Hendon, S. Deshpande, Y. Surendranath and V. Viswanathan, Joule, 2017, 1, 600–612 CrossRef CAS.
- D. Gao, I. T. McCrum, S. Deo, Y.-W. Choi, F. Scholten, W. Wan, J. G. Chen, M. J. Janik and B. Roldan Cuenya, ACS Catal., 2018, 8, 10012–10020 CrossRef CAS.
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim and S.-P. Cho, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed.
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang and Z. Hu, Chemistry, 2020, 6, 3440–3454 CrossRef CAS.
- J. Vavra, T. H. Shen, D. Stoian, V. Tileli and R. Buonsanti, Angew. Chem., Int. Ed., 2021, 133, 1367–1374 CrossRef.
- X. Zou, Y. Wu, Y. Liu, D. Liu, W. Li, L. Gu, H. Liu, P. Wang, L. Sun and Y. Zhang, Chemistry, 2018, 4, 1139–1152 CrossRef CAS.
- K. Fan, H. Zou, Y. Lu, H. Chen, F. Li, J. Liu, L. Sun, L. Tong, M. F. Toney and M. Sui, ACS Nano, 2018, 12, 12369–12379 CrossRef CAS PubMed.
- R. Chen, S. Pang, H. An, J. Zhu, S. Ye, Y. Gao, F. Fan and C. Li, Nat. Energy, 2018, 3, 655–663 CrossRef CAS.
- X. Song, Y. Wu, X. Zhang, X. Li, Z. Zhu, C. Ma, Y. Yan, P. Huo and G. Yang, Chem. Eng. J., 2021, 408, 127292 CrossRef CAS.
- L. Zhang, B. K. Miller and P. A. Crozier, Nano Lett., 2013, 13, 679–684 CrossRef CAS PubMed.
- W. Bi, X. Li, L. Zhang, T. Jin, L. Zhang, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Nat. Commun., 2015, 6, 8647 CrossRef CAS PubMed.
- J. Hu, J. Xie, W. Jia, S. Zhang, S. Wang, K. Wang and Y. Cao, Appl. Catal., B, 2020, 268, 118753 CrossRef CAS.
- A. Dutta, A. Kuzume, M. Rahaman, S. Vesztergom and P. Broekmann, ACS Catal., 2015, 5, 7498–7502 CrossRef CAS.
- S. Karasawa, K. P. Sharma, D. Yamamoto, T. Saida, S. Naritsuka and T. Maruyama, Chem. Phys. Lett., 2022, 804, 139889 CrossRef CAS.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu and H. Li, Nat. Energy, 2024, 1–11 Search PubMed.
- W. A. Waters, J. Am. Oil Chem. Soc., 1971, 48, 427–433 CrossRef CAS.
- Y. Pan, Y. Li, C.-L. Dong, Y.-C. Huang, J. Wu, J. Shi, Y. Lu, M. Yang, S. Wang and Y. Zou, Chemistry, 2023, 9, 963–977 CrossRef CAS.
- J.-J. Velasco-Velez, R. V. Mom, L.-E. Sandoval-Diaz, L. J. Falling, C.-H. Chuang, D. Gao, T. E. Jones, Q. Zhu, R. Arrigo and B. Roldan Cuenya, ACS Energy Lett., 2020, 5, 2106–2111 CrossRef CAS PubMed.
- Y. H. Li, C. Li and H. G. Yang, J. Mater. Chem. A, 2017, 5, 20631–20634 RSC.
- L. Quan, H. Jiang, G. Mei, Y. Sun and B. You, Chem. Rev., 2024, 124, 3694–3812 CrossRef CAS PubMed.
- S. Wang, W. Huo, F. Fang, Z. Xie, J. K. Shang and J. Jiang, Chem. Eng. J., 2022, 429, 132410 CrossRef CAS.
- J. Zhou, L. Zhang, Y.-C. Huang, C.-L. Dong, H.-J. Lin, C.-T. Chen, L. Tjeng and Z. Hu, Nat. Commun., 2020, 11, 1984 CrossRef CAS PubMed.
- K. Han, T. Kreuger, B. Mei and G. Mul, ACS Catal., 2017, 7, 1610–1614 CrossRef CAS PubMed.
- L. Chen, J.-T. Ren, Y.-S. Wang, W.-W. Tian, L.-J. Gao and Z.-Y. Yuan, ACS Sustainable Chem. Eng., 2019, 7, 13559–13568 CrossRef CAS.
- L. Chen, J.-T. Ren and Z.-Y. Yuan, ACS Sustainable Chem. Eng., 2021, 9, 1856–1866 CrossRef CAS.
- P. Strasser and S. Kühl, Nano Energy, 2016, 29, 166–177 CrossRef CAS.
- H. L. Xin, J. A. Mundy, Z. Liu, R. Cabezas, R. Hovden, L. F. Kourkoutis, J. Zhang, N. P. Subramanian, R. Makharia and F. T. Wagner, Nano Lett., 2012, 12, 490–497 CrossRef CAS PubMed.
- L. Zhu, B. Wei, Y. Zhang, Z. Lü, Z. Wang, X. Huang, Z. Cao, W. Jiang and Y. Li, Electrochim. Acta, 2015, 160, 89–93 CrossRef CAS.
- Y. Wang, Y. Yang, S. Jia, X. Wang, K. Lyu, Y. Peng, H. Zheng, X. Wei, H. Ren and L. Xiao, Nat. Commun., 2019, 10, 1506 CrossRef PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- W. Zheng, M. Liu and L. Y. S. Lee, ACS Catal., 2019, 10, 81–92 CrossRef.
- A. Corma, P. Concepción, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. López-Quintela and D. Buceta, Nat. Chem., 2013, 5, 775–781 CrossRef CAS PubMed.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik and Y. Zhu, Nat. Energy, 2020, 5, 222–230 CrossRef.
- M. Schwarz, F. Faisal, S. Mohr, C. Hohner, K. Werner, T. Xu, T. S. Skála, N. Tsud, K. C. Prince and V. Matolín, J. Phys. Chem. Lett., 2018, 9, 2763–2769 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin and L. Wiles, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- C. Sun, H. Zhang, H. Liu, X. Zheng, W. Zou, L. Dong and L. Qi, Appl. Catal., B, 2018, 235, 66–74 CrossRef CAS.
- T. K. Townsend, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2012, 5, 9543–9550 RSC.
- Y. Yu, H. L. Xin, R. Hovden, D. Wang, E. D. Rus, J. A. Mundy, D. A. Muller and H. D. Abruña, Nano Lett., 2012, 12, 4417–4423 CrossRef CAS PubMed.
- Z. Kou, Y. Yu, X. Liu, X. Gao, L. Zheng, H. Zou, Y. Pang, Z. Wang, Z. Pan and J. He, ACS Catal., 2020, 10, 4411–4419 CrossRef CAS.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H. L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- C. Dessal, L. Martínez, C. Maheu, T. Len, F. Morfin, J. L. Rousset, E. Puzenat, P. Afanasiev, M. Aouine and L. Soler, J. Catal., 2019, 375, 155–163 CrossRef CAS.
- L. Liu, D. M. Meira, R. Arenal, P. Concepcion, A. V. Puga and A. Corma, ACS Catal., 2019, 9, 10626–10639 CrossRef CAS PubMed.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng and Q. Yu, Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2019, 10, 1754–1768 CrossRef.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 130, 6300–6305 CrossRef.
- F. Guo, Y. Wu, H. Chen, Y. Liu, L. Yang, X. Ai and X. Zou, Energy Environ. Sci., 2019, 12, 684–692 RSC.
- J. Huang, Y. Li, Y. Zhang, G. Rao, C. Wu, Y. Hu, X. Wang, R. Lu, Y. Li and J. Xiong, Angew. Chem., Int. Ed., 2019, 131, 17619–17625 CrossRef.
- Y. Yang, Y. Xiong, R. Zeng, X. Lu, M. Krumov, X. Huang, W. Xu, H. Wang, F. J. DiSalvo and J. D. Brock, ACS Catal., 2021, 11, 1136–1178 CrossRef CAS.
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2020, 121, 882–961 CrossRef PubMed.
- G. Xing, M. Tong, P. Yu, L. Wang, G. Zhang, C. Tian and H. Fu, Angew. Chem., Int. Ed., 2022, 61, e202211098 CrossRef CAS PubMed.
- Z. Yan, H. Sun, X. Chen, H. Liu, Y. Zhao, H. Li, W. Xie, F. Cheng and J. Chen, Nat. Commun., 2018, 9, 2373 CrossRef PubMed.
- X. Han, C. Yu, Y. Niu, Z. Wang, Y. Kang, Y. Ren, H. Wang, H. S. Park and J. Qiu, Small Methods, 2020, 4, 2000546 CrossRef CAS.
- B. Wang, K. Zhao, Z. Yu, C. Sun, Z. Wang, N. Feng, L. Mai, Y. Wang and Y. Xia, Energy Environ. Sci., 2020, 13, 2200–2208 RSC.
- L. Gao, X. Cui, Z. Wang, C. Sewell, Z. Li, S. Liang and M. Zhang, Proceedings of the National Academy of 107 Sciences, 2021, 118, e2023421118.
- X. Wang, K. Klingan, M. Klingenhof, T. Möller, J. Ferreira de Araújo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl and H. Dau, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- T.-H. Shen, L. Spillane, J. Vavra, T. H. M. Pham, J. Peng, Y. Shao-Horn and V. Tileli, J. Am. Chem. Soc., 2020, 142, 15876–15883 CrossRef CAS PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. R. Cuenya, Angew. Chem., Int. Ed., 2018, 130, 6300–6305 CrossRef.
- Y. Gogotsi, Nat. Mater., 2015, 14, 1079–1080 CrossRef CAS PubMed.
- C. Ling, L. Shi, Y. Ouyang, Q. Chen and J. Wang, Adv. Sci., 2016, 3, 1600180 CrossRef PubMed.
- C. Xu, L. Wang, Z. Liu, L. Chen, J. Guo, N. Kang, X.-L. Ma, H.-M. Cheng and W. Ren, Nat. Mater., 2015, 14, 1135–1141 CrossRef CAS PubMed.
- L. Liu and A. Corma, Trends Chem., 2020, 2, 383–400 CrossRef CAS.
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan and J. Zhu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed.
- M. D. Marcinkowski, S. F. Yuk, N. Doudin, R. S. Smith, M.-T. Nguyen, B. D. Kay, V.-A. Glezakou, R. Rousseau and Z. Dohnálek, ACS Catal., 2019, 9, 10977–10982 CrossRef CAS.
- Z. Huang, T. Ban, Y. Zhang, L. Wang, S. Guo, C.-R. Chang and G. Jing, Appl. Catal., B, 2021, 283, 119625 CrossRef CAS.
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef CAS PubMed.
- X. Wei, Y. Liu, X. Zhu, S. Bo, L. Xiao, C. Chen, T. T. T. Nga, Y. He, M. Qiu and C. Xie, Adv. Mater., 2023, 35, 2300020 CrossRef CAS PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei and A. Zitolo, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su and L. Zhang, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS PubMed.
- M. Tong, F. Sun, G. Xing, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2023, 62, e202314933 CrossRef CAS PubMed.
- H. Sun, C.-W. Tung, Y. Qiu, W. Zhang, Q. Wang, Z. Li, J. Tang, H.-C. Chen, C. Wang and H. M. Chen, J. Am. Chem. Soc., 2021, 144, 1174–1186 CrossRef PubMed.
- Y. Zhou, W. Zhang, J. Hu, D. Li, X. Yin and Q. Gao, ACS Sustainable Chem. Eng., 2021, 9, 7390–7399 CrossRef CAS.
- J. Meng, Z. Lan, I. E. Castelli and K. Zheng, J. Phys. Chem. C, 2021, 125, 8456–8460 CrossRef CAS.
- D. Zhou, F. Li, Y. Zhao, L. Wang, H. Zou, Y. Shan, J. Fu, Y. Ding, L. Duan and M. Liu, ACS Catal., 2023, 13, 4398–4408 CrossRef CAS.
- L. Peng, N. Yang, Y. Yang, Q. Wang, X. Xie, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. Waterhouse, Angew. Chem., Int. Ed., 2021, 60, 24612–24619 CrossRef CAS PubMed.
- Q. He, Y. Wan, H. Jiang, Z. Pan, C. Wu, M. Wang, X. Wu, B. Ye, P. M. Ajayan and L. Song, ACS Energy Lett., 2018, 3, 1373–1380 CrossRef CAS.
- P. Chen, T. Zhou, S. Wang, N. Zhang, Y. Tong, H. Ju, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 15471–15475 CrossRef CAS PubMed.
- W. Peng, J. Li, K. Shen, L. Zheng, H. Tang, Y. Gong, J. Zhou, N. Chen, S. Zhao and M. Chen, J. Mater. Chem. A, 2020, 8, 23580–23589 RSC.
- J. Bai, J. Mei, T. Liao, Q. Sun, Z. G. Chen and Z. Sun, Adv. Energy Mater., 2022, 12, 2103247 CrossRef CAS.
- C. Hu, K. Yue, J. Han, X. Liu, L. Liu, Q. Liu, Q. Kong, C.-W. Pao, Z. Hu and K. Suenaga, Sci. Adv., 2023, 9, eadf9144 CrossRef CAS PubMed.
- S. Tao, G. Zhang, B. Qian, J. Yang, S. Chu, C. Sun, D. Wu, W. Chu and L. Song, Appl. Catal., B, 2023, 330, 122600 CrossRef CAS.
- Y. Wei, Y. Zheng, Y. Hu, B. Huang, M. Sun, P. Da, P. Xi and C.-H. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 25638–25647 CrossRef CAS PubMed.
- H. Liao, C. Wei, J. Wang, A. Fisher, T. Sritharan, Z. Feng and Z. J. Xu, Adv. Energy Mater., 2017, 7, 1701129 CrossRef.
- Z. Xiao, M. Yang, C. Liu, B. Wang, S. Zhang, J. Liu, Z. Xu, R. Gao, J.-J. Zou and A. Tang, Nano Energy, 2022, 98, 107233 CrossRef CAS.
- Y. Tang, C. Wu, Q. Zhang, H. Zhong, A. Zou, J. Li, Y. Ma, H. An, Z. Yu and S. Xi, Angew. Chem., Int. Ed., 2023, 135, e202309107 CrossRef.
- S. Zhang, S. Gu, Y. Wang, C. Liang, Y. Yu, L. Han, S. Zheng, N. Zhang, X. Liu and J. Zhou, ACS Catal., 2019, 9, 7389–7397 CrossRef CAS.
- H. Jiang, Q. He, X. Li, X. Su, Y. Zhang, S. Chen, S. Zhang, G. Zhang, J. Jiang and Y. Luo, Adv. Mater., 2019, 31, 1805127 CrossRef PubMed.
- A. L. Maulana, P.-C. Chen, Z. Shi, Y. Yang, C. Lizandara-Pueyo, F. Seeler, H. D. Abruña, D. Muller, K. Schierle-Arndt and P. Yang, Nano Lett., 2023, 23, 6637–6644 CrossRef CAS PubMed.
- M. Ning, F. Zhang, L. Wu, X. Xing, D. Wang, S. Song, Q. Zhou, L. Yu, J. Bao and S. Chen, Energy Environ. Sci., 2022, 15, 3945–3957 RSC.
- R. Yu, C. Wang, D. Liu, X. Wang, J. Yin and Y. Du, Inorg. Chem., 2022, 62, 609–617 CrossRef PubMed.
- L. Qiu, Q. Wang, P. Yan and X.-Y. Yu, J. Mater. Chem. A, 2022, 10, 21251–21259 RSC.
- P. Yan, Q. Liu, H. Zhang, L. Qiu, H. B. Wu and X.-Y. Yu, J. Mater. Chem. A, 2021, 9, 15586–15594 RSC.
- C. Kuai, C. Xi, A. Hu, Y. Zhang, Z. Xu, D. Nordlund, C.-J. Sun, C. A. Cadigan, R. M. Richards and L. Li, J. Am. Chem. Soc., 2021, 143, 18519–18526 CrossRef CAS PubMed.
- F. Yang, M. Lopez Luna, F. T. Haase, D. Escalera-López, A. Yoon, M. Rüscher, C. Rettenmaier, H. S. Jeon, E. Ortega and J. Timoshenko, J. Am. Chem. Soc., 2023, 145, 21465–21474 CrossRef CAS PubMed.
- D. Yao, C. Tang, A. Vasileff, X. Zhi, Y. Jiao and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 133, 18326–18332 CrossRef.
- W. Cai, R. Chen, H. Yang, H. B. Tao, H.-Y. Wang, J. Gao, W. Liu, S. Liu, S.-F. Hung and B. Liu, Nano Lett., 2020, 20, 4278–4285 CrossRef CAS PubMed.
- Y. Duan, Z. Y. Yu, S. J. Hu, X. S. Zheng, C. T. Zhang, H. H. Ding, B. C. Hu, Q. Q. Fu, Z. L. Yu and X. Zheng, Angew. Chem., Int. Ed., 2019, 58, 15772–15777 CrossRef CAS PubMed.
- G. Chen, Z. Hu, Y. Zhu, B. Gu, Y. Zhong, H. J. Lin, C. T. Chen, W. Zhou and Z. Shao, Adv. Mater., 2018, 30, 1804333 CrossRef PubMed.
- Z. Ma, C. Chen, X. Cui, L. Zeng, L. Wang, W. Jiang and J. Shi, ACS Appl. Mater. Interfaces, 2021, 13, 44224–44233 CrossRef CAS PubMed.
- Y. Wang, Y. Zhao, L. Liu, W. Qin, S. Liu, J. Tu, Y. Liu, Y. Qin, J. Liu and H. Wu, J. Am. Chem. Soc., 2023, 145, 20261–20272 CrossRef CAS PubMed.
- Z. P. Wu, X. F. Lu, S. Q. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS.
- F.-S. Zhang, J.-W. Wang, J. Luo, R.-R. Liu, Z.-M. Zhang, C.-T. He and T.-B. Lu, Chem. Sci., 2018, 9, 1375–1384 RSC.
- H. Li, S. Wang, H. Sawada, G. G. Han, T. Samuels, C. S. Allen, A. I. Kirkland, J. C. Grossman and J. H. Warner, ACS Nano, 2017, 11, 3392–3403 CrossRef CAS PubMed.
- E. Mosconi, D. Meggiolaro, H. J. Snaith, S. D. Stranks and F. De Angelis, Energy Environ. Sci., 2016, 9, 3180–3187 RSC.
- R. A. De Souza, V. Metlenko, D. Park and T. E. Weirich, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 174109 CrossRef.
- Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 17, 1926–1929 CrossRef PubMed.
- S. Zhang, J.-J. Shan, Y. Zhu, A. I. Frenkel, A. Patlolla, W. Huang, S. J. Yoon, L. Wang, H. Yoshida and S. Takeda, J. Am. Chem. Soc., 2013, 135, 8283–8293 CrossRef CAS PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- T. Tang, W.-J. Jiang, S. Niu, N. Liu, H. Luo, Y.-Y. Chen, S.-F. Jin, F. Gao, L.-J. Wan and J.-S. Hu, J. Am. Chem. Soc., 2017, 139, 8320–8328 CrossRef CAS PubMed.
- Y. Lin, L. Yang, H. Jiang, Y. Zhang, Y. Bo, P. Liu, S. Chen, B. Xiang, G. Li and J. Jiang, J. Phys. Chem. Lett., 2020, 11, 1746–1752 CrossRef CAS PubMed.
- C. Zhao, N. Li, R. Zhang, Z. Zhu, J. Lin, K. Zhang and C. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47858–47867 CrossRef CAS PubMed.
- M. Li, H. Liu and L. Feng, Electrochem. Commun., 2021, 122, 106901 CrossRef CAS.
- S. Chen, S. Zhang, L. Guo, L. Pan, C. Shi, X. Zhang, Z.-F. Huang, G. Yang and J.-J. Zou, Nat. Commun., 2023, 14, 4127 CrossRef CAS PubMed.
- Y.-P. Deng, Y. Jiang, R. Liang, S.-J. Zhang, D. Luo, Y. Hu, X. Wang, J.-T. Li, A. Yu and Z. Chen, Nat. Commun., 2020, 11, 1952 CrossRef CAS PubMed.
- Y. Zhao, N. Dongfang, C. A. Triana, C. Huang, R. Erni, W. Wan, J. Li, D. Stoian, L. Pan and P. Zhang, Energy Environ. Sci., 2022, 15, 727–739 RSC.
- C. Kaithwas, P. Bhuyan, S. Pradhan and S. Mandal, Mater. Charact., 2018, 145, 582–593 CrossRef CAS.
- C. Athreya, K. Deepak, D.-I. Kim, B. De Boer, S. Mandal and V. S. Sarma, J. Alloys Compd., 2019, 778, 224–233 CrossRef CAS.
- E. Bousquet, M. Dawber, N. Stucki, C. Lichtensteiger, P. Hermet, S. Gariglio, J.-M. Triscone and P. Ghosez, Nature, 2008, 452, 732–736 CrossRef CAS PubMed.
- S. Farokhipoor, C. Magén, S. Venkatesan, J. Íñiguez, C. J. Daumont, D. Rubi, E. Snoeck, M. Mostovoy, C. De Graaf and A. Müller, Nature, 2014, 515, 379–383 CrossRef CAS.
- A. Yadav, C. Nelson, S. Hsu, Z. Hong, J. Clarkson, C. Schlepütz, A. Damodaran, P. Shafer, E. Arenholz and L. Dedon, Nature, 2016, 530, 198–201 CrossRef CAS PubMed.
- J.-H. Kang, L. Xie, Y. Wang, H. Lee, N. Campbell, J. Jiang, P. J. Ryan, D. J. Keavney, J.-W. Lee and T. H. Kim, Nano Lett., 2018, 18, 6347–6352 CrossRef CAS PubMed.
- S. Cheng, C. Xu, S. Deng, M.-G. Han, S. Bao, J. Ma, C. Nan, W. Duan, L. Bellaiche and Y. Zhu, Sci. Adv., 2018, 4, eaar4298 CrossRef PubMed.
- H.-Y. Sun, G.-R. Xu, F.-M. Li, Q.-L. Hong, P.-J. Jin, P. Chen and Y. Chen, J. Energy Chem., 2020, 47, 234–240 CrossRef.
- Y. Liu, J. Wang, S. Kim, H. Sun, F. Yang, Z. Fang, N. Tamura, R. Zhang, X. Song and J. Wen, Nature, 2019, 570, 358–362 CrossRef CAS PubMed.
- H. Wang, H. Zhang, Y. Huang, H. Wang, A. Ozden, K. Yao, H. Li, Q. Guo, Y. Liu and A. Vomiero, ACS Nano, 2022, 17, 346–354 CrossRef PubMed.
- Z. Yang, D. Ji, Z. Li, Z. He, Y. Hu, J. Yin, Y. Hou, P. Xi and C. H. Yan, Small, 2023, 19, 2303099 CrossRef CAS PubMed.
- X. Long, S. Xiao, Z. Wang, X. Zheng and S. Yang, Chem. Commun., 2015, 51, 1120–1123 RSC.
- Z. Chen, M. Ju, M. Sun, L. Jin, R. Cai, Z. Wang, L. Dong, L. Peng, X. Long and B. Huang, Angew. Chem., Int. Ed., 2021, 60, 9699–9705 CrossRef CAS PubMed.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O’Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- X. Wang, C. Liu, C. Gao, K. Yao, S. S. M. Masouleh, R. Berté, H. Ren, L. D. S. Menezes, E. Cortés and I. C. Bicket, ACS Nano, 2021, 15, 10553–10564 CrossRef CAS PubMed.
- Z. Chen, X. Zhang and G. Lu, Chem. Sci., 2015, 6, 6829–6835 RSC.
- J.-W. Zhao, Z.-X. Shi, C.-F. Li, Q. Ren and G.-R. Li, ACS Mater. Lett., 2021, 3, 721–737 CrossRef CAS.
- H. J. Song, H. Yoon, B. Ju and D. W. Kim, Adv. Energy Mater., 2021, 11, 2002428 CrossRef CAS.
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CrossRef CAS PubMed.
- Y. Li, X. Wu, J. Wang, H. Wei, S. Zhang, S. Zhu, Z. Li, S. Wu, H. Jiang and Y. Liang, Electrochim. Acta, 2021, 390, 138833 CrossRef CAS.
- M. Yu, J. Li, F. Liu, J. Liu, W. Xu, H. Hu, X. Chen, W. Wang and F. Cheng, J. Energy Chem., 2022, 72, 361–369 CrossRef CAS.
- W. Liu, J. Yu, M. G. Sendeku, T. Li, W. Gao, G. Yang, Y. Kuang and X. Sun, Angew. Chem., Int. Ed., 2023, 135, e202309882 CrossRef.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- J. S. Kim, I. Park, E. S. Jeong, K. Jin, W. M. Seong, G. Yoon, H. Kim, B. Kim, K. T. Nam and K. Kang, Adv. Mater., 2017, 29, 1606893 CrossRef PubMed.
- H. Liang, A. N. Gandi, C. Xia, M. N. Hedhili, D. H. Anjum, U. Schwingenschlogl and H. N. Alshareef, ACS Energy Lett., 2017, 2, 1035–1042 CrossRef CAS.
- F. Dong, H. Duan, Z. Lin, H. Yuan, M. Ju, X. Du, J. Gao, J. Yu and S. Yang, Appl. Catal., B, 2024, 340, 123242 CrossRef CAS.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. R. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. Von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed.
- S. Lee, A. Moysiadou, Y.-C. Chu, H. M. Chen and X. Hu, Energy Environ. Sci., 2022, 15, 206–214 RSC.
- R. Mehmood, W. Fan, X. Hu, J. Li, P. Liu, Y. Zhang, Z. Zhou, J. Wang, M. Liu and F. Zhang, J. Am. Chem. Soc., 2023, 145, 12206–12213 CrossRef CAS PubMed.
- C. Feng, X. She, Y. Xiao and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202218738 CrossRef CAS PubMed.
- J. Kang, X. Qiu, Q. Hu, J. Zhong, X. Gao, R. Huang, C. Wan, L.-M. Liu, X. Duan and L. Guo, Nat. Catal., 2021, 4, 1050–1058 CrossRef CAS.
- D. Wang, W. Yan, S. H. Vijapur and G. G. Botte, J. Power Sources, 2012, 217, 498–502 CrossRef CAS.
- N. Chen, Y.-X. Du, G. Zhang, W.-T. Lu and F.-F. Cao, Nano Energy, 2021, 81, 105605 CrossRef CAS.
- A. S. Aricò, V. Baglio and V. Antonucci, Electrocatal. Direct Methanol Fuel Cells, 2009, 1–78 Search PubMed.
- X. Bai, X. Zhao, Y. Zhang, C. Ling, Y. Zhou, J. Wang and Y. Liu, J. Am. Chem. Soc., 2022, 144, 17140–17148 CrossRef CAS PubMed.
- X. Wang, M. Xie, F. Lyu, Y.-M. Yiu, Z. Wang, J. Chen, L.-Y. Chang, Y. Xia, Q. Zhong and M. Chu, Nano Lett., 2020, 20, 7751–7759 CrossRef CAS PubMed.
- J. Bau, S. Kozlov and L. Azofra, J. Mater. Chem. A, 2019, 7, 15031–15035 RSC.
- R. Cai, M. Sun, J. Ren, M. Ju, X. Long, B. Huang and S. Yang, Chem. Sci., 2021, 12, 15382–15388 RSC.
- R. Cai, M. Sun, F. Yang, M. Ju, Y. Chen, M. D. Gu, B. Huang and S. Yang, Chemistry, 2024, 10, 211–233 CrossRef CAS.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2015, 54, 10102–10120 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- Q. Ma, C. Hu, K. Liu, S.-F. Hung, D. Ou, H. M. Chen, G. Fu and N. Zheng, Nano Energy, 2017, 41, 148–153 CrossRef CAS.
- V. S. Saji and C. W. Lee, ChemSusChem, 2012, 5, 1146–1161 CrossRef CAS.
- L. Su, D. Han, G. Zhu, H. Xu, W. Luo, L. Wang, W. Jiang, A. Dong and J. Yang, Nano Lett., 2019, 19, 5423–5430 CrossRef CAS.
- C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Chem. Rev., 2019, 120, 1184–1249 CrossRef PubMed.
- C. Zhu, Z. Zhang, L. Zhong, C.-S. Hsu, X. Xu, Y. Li, S. Zhao, S. Chen, J. Yu and S. Chen, Chemistry, 2021, 7, 406–420 CrossRef CAS.
- Y. Wang, J. Liu and G. Zheng, Adv. Mater., 2021, 33, 2005798 CrossRef CAS PubMed.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- S. Martens, L. Asen, G. Ercolano, F. Dionigi, C. Zalitis, A. Hawkins, A. M. Bonastre, L. Seidl, A. C. Knoll and J. Sharman, J. Power Sources, 2018, 392, 274–284 CrossRef CAS.
- S. M. Alia, S. Shulda, C. Ngo, S. Pylypenko and B. S. Pivovar, ACS Catal., 2018, 8, 2111–2120 CrossRef CAS.
- H. A. El-Sayed, A. Weiß, L. F. Olbrich, G. P. Putro and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, F458–F464 CrossRef CAS.
- P. Lettenmeier, R. Wang, R. Abouatallah, S. Helmly, T. Morawietz, R. Hiesgen, S. Kolb, F. Burggraf, J. Kallo and A. S. Gago, Electrochim. Acta, 2016, 210, 502–511 CrossRef CAS.
- Y. Peng, K. Jiang, W. Hill, Z. Lu, H. Yao and H. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 3971–3977 CrossRef CAS PubMed.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi and E. D. Baca, Nat. Energy, 2020, 5, 378–385 CrossRef CAS.
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS.
- M. Chandesris, V. Médeau, N. Guillet, S. Chelghoum, D. Thoby and F. Fouda-Onana, Int. J. Hydrogen Energy, 2015, 40, 1353–1366 CrossRef CAS.
- E. J. Park, S. Maurya, M. R. Hibbs, C. H. Fujimoto, K.-D. Kreuer and Y. S. Kim, Macromolecules, 2019, 52, 5419–5428 CrossRef CAS.
- P. Thangavel, M. Ha, S. Kumaraguru, A. Meena, A. N. Singh, A. M. Harzandi and K. S. Kim, Energy Environ. Sci., 2020, 13, 3447–3458 RSC.
- M. Bernt and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, F3179 CrossRef CAS.
- H.-S. Oh, H. N. Nong, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schlögl, D. Teschner and P. Strasser, J. Am. Chem. Soc., 2016, 138, 12552–12563 CrossRef CAS PubMed.
- S. Geiger, O. Kasian, A. M. Mingers, S. S. Nicley, K. Haenen, K. J. Mayrhofer and S. Cherevko, ChemSusChem, 2017, 10, 4140–4143 CrossRef CAS PubMed.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. R. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert and H. Dau, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- J. P. Meyers and R. M. Darling, J. Electrochem. Soc., 2006, 153, A1432 CrossRef CAS.
- Q. Dong, T. Li, Y. Yao, X. Wang, S. He, J. Li, J. Luo, H. Zhang, Y. Pei and C. Zheng, Joule, 2020, 4, 2374–2386 CrossRef CAS.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 5054–5058 CrossRef CAS PubMed.
- S. Z. Oener, M. J. Foster and S. W. Boettcher, Science, 2020, 369, 1099–1103 CrossRef CAS PubMed.
- X. Zhao, H. Ren and L. Luo, Langmuir, 2019, 35, 5392–5408 CrossRef CAS PubMed.
- B. Han, K. A. Stoerzinger, V. Tileli, A. D. Gamalski, E. A. Stach and Y. Shao-Horn, Nat. Mater., 2017, 16, 121–126 CrossRef CAS PubMed.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- S. Dresp, T. N. Thanh, M. Klingenhof, S. Brückner, P. Hauke and P. Strasser, Energy Environ. Sci., 2020, 13, 1725–1729 RSC.
- L. Chen, J.-T. Ren and Z.-Y. Yuan, Sci. China Mater., 2022, 65, 2433–2444 CrossRef CAS.
- L. Chen, J.-T. Ren and Z.-Y. Yuan, Green Chem., 2022, 24, 713–747 RSC.
- L. Piccolo, P. Afanasiev, F. Morfin, T. Len, C. Dessal, J. L. Rousset, M. Aouine, F. Bourgain, A. Aguilar-Tapia and O. Proux, ACS Catal., 2020, 10, 12696–12705 CrossRef CAS.
- J. Yi, Z. Zhou, Y. Xia, G. Zhou, G. Zhang, L. Li, X. Wang, X. Zhu, X. Wang and H. Pang, Chin. Chem. Lett., 2023, 34, 108328 CrossRef CAS.
- H. Zhang, S. Zuo, M. Qiu, S. Wang, Y. Zhang, J. Zhang and X. W. Lou, Sci. Adv., 2020, 6, eabb9823 CrossRef CAS PubMed.
- X. Tao, W. Shi, B. Zeng, Y. Zhao, N. Ta, S. Wang, A. A. Adenle, R. Li and C. Li, ACS Catal., 2020, 10, 5941–5948 CrossRef CAS.
- B. Yan, J. Li, Z. Lin, C. Du and G. Yang, ACS Appl. Nano Mater., 2019, 2, 6783–6792 CrossRef CAS.
- S. Wu, C. Li, Y. Wang, Y. Zhuang, Y. Pan, N. Wen, S. Wang, Z. Zhang, Z. Ding and R. Yuan, Angew. Chem., Int. Ed., 2023, 135, e202309026 CrossRef.
- Y. Wang, Y. Ma, X.-B. Li, L. Gao, X.-Y. Gao, X.-Z. Wei, L.-P. Zhang, C.-H. Tung, L. Qiao and L.-Z. Wu, J. Am. Chem. Soc., 2020, 142, 4680–4689 CrossRef CAS PubMed.
- J. Fan, Y. Zhao, H. Du, L. Zheng, M. Gao, D. Li and J. Feng, ACS Appl. Mater. Interfaces, 2022, 14, 26752–26765 CrossRef CAS PubMed.
- S. Xie, Y. Li, B. Sheng, W. Zhang, W. Wang, C. Chen, J. Li, H. Sheng and J. Zhao, Appl. Catal., B, 2022, 310, 121320 CrossRef CAS.
- Y. Zhang, Z. Xu, Q. Wang, W. Hao, X. Zhai, X. Fei, X. Huang and Y. Bi, Appl. Catal., B, 2021, 299, 120679 CrossRef CAS.
- Y. Dou, A. Zhou, Y. Yao, S. Y. Lim, J.-R. Li and W. Zhang, Appl. Catal., B, 2021, 286, 119876 CrossRef CAS.
- F. Fang, Y. Liu, X. Sun, C. Fu, Y. P. Bhoi, W. Xiong and W. Huang, Appl. Surf. Sci., 2021, 564, 150407 CrossRef CAS.
- R. Li, Q. Luan, C. Dong, W. Dong, W. Tang, G. Wang and Y. Lu, Appl. Catal., B, 2021, 286, 119832 CrossRef CAS.
- Z. Pászti, S. Bálint, P. Németh, G. P. Szíjjártó, A. Tompos and E. Tálas, Mater. Res. Bull., 2016, 83, 65–76 CrossRef.
- X. Zheng, D. Liu, J. Wen and S. Lv, Sep. Purif. Technol., 2021, 275, 117934 CrossRef CAS.
- J. Prince, F. Tzompantzi, G. Mendoza-Damián, F. Hernández-Beltrán and J. S. Valente, Appl. Catal., B, 2015, 163, 352–360 CrossRef CAS.
- M. Suárez-Quezada, G. Romero-Ortiz, V. Suárez, G. Morales-Mendoza, L. Lartundo-Rojas, E. Navarro-Cerón, F. Tzompantzi, S. Robles, R. Gómez and A. Mantilla, Catal. Today, 2016, 271, 213–219 CrossRef.
- Y. Lu, W.-J. Yin, K.-L. Peng, K. Wang, Q. Hu, A. Selloni, F.-R. Chen, L.-M. Liu and M.-L. Sui, Nat. Commun., 2018, 9, 2752 CrossRef PubMed.
- K. Zhao, W. Pang, S. Jiang, C. Hu, P. Liu, D. Cui, X. An, B. Tian, C. Gao and P. Zhang, Nano Res., 2023, 16, 4812–4820 CrossRef CAS.
- S. Xie, C. Deng, Q. Huang, C. Zhang, C. Chen, J. Zhao and H. Sheng, Angew. Chem., Int. Ed., 2023, 62, e202216717 CrossRef CAS PubMed.
- C. Y. Toe, Z. Zheng, H. Wu, J. Scott, R. Amal and Y. H. Ng, Angew. Chem., Int. Ed., 2018, 57, 13613–13617 CrossRef CAS PubMed.
- L. Chen, J. T. Ren and Z. Y. Yuan, Adv. Energy Mater., 2023, 13, 2203720 CrossRef CAS.
- L. Chen, X.-L. Song, J.-T. Ren and Z.-Y. Yuan, Appl. Catal., B, 2022, 315, 121546 CrossRef CAS.
- H. Huang, H. Song, J. Kou, C. Lu and J. Ye, J. Energy Chem., 2022, 67, 309–341 CrossRef CAS.
- J. Li, W. Pan, Q. Liu, Z. Chen, Z. Chen, X. Feng and H. Chen, J. Am. Chem. Soc., 2021, 143, 6551–6559 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |