
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Harnessing enhanced lithium-ion storage in self-assembled organic nanowires for batteries and metal-ion supercapacitors†
Ievgen
Obraztsov
 *a,
Rostislav
Langer
b,
Jean G. A.
Ruthes
de,
Volker
Presser
def,
Michal
Otyepka
ab,
Radek
Zbořil
*ac and
Aristides
Bakandritsos
*ac
*a,
Rostislav
Langer
b,
Jean G. A.
Ruthes
de,
Volker
Presser
def,
Michal
Otyepka
ab,
Radek
Zbořil
*ac and
Aristides
Bakandritsos
*ac
aRegional Centre of Advanced Technologies and Materials (RCPTM), Czech Advanced Technology and Research Institute (CATRIN), Palacký University Olomouc, Šlechtitelů 27, Olomouc 77 900, Czech Republic. E-mail: ievgen.obraztsov@upol.cz; radek.zboril@upol.cz; a.bakandritsos@upol.cz
bIT4Innovations, VŠB-Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic
cNanotechnology Centre, Centre of Energy and Environmental Technologies, VŠB-Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic
dINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany
eDepartment of Materials Science and Engineering, Saarland University, Campus D2 2, 66123 Saarbrücken, Germany
fsaarene – Saarland Center for Energy Materials and Sustainability, Campus C4 2, 66123 Saarbrücken, Germany
First published on 15th October 2024
Abstract
Organic materials have emerged as highly efficient electrodes for electrochemical energy storage, offering sustainable solutions independent from non-renewable resources. In this study, we showcase that mesoscale engineering can dramatically transform the electrochemical features of a molecular organic carboxylic anode. Through a sustainable, energy-efficient and environmentally benign self-assembly strategy, we developed a network of organic nanowires formed during water evaporation directly on the copper current collector, circumventing the need for harmful solvents, typically employed in such processes. The organic nanowire anode delivers high capacity and rate, reaching 1888 mA h g−1 at 0.1 A g−1 and maintaining 508 mA h g−1 at a specific current of 10 A g−1. Moreover, it exhibits superior thermal management during lithiation in comparison to graphite and other organic anodes. Comprehensive electrochemical evaluations and theoretical calculations reveal rapid charge transport mechanisms, with lithium diffusivity rates reaching 5 × 10−9 cm2 s−1, facilitating efficient and rapid interactions with 24 lithium atoms per molecule. Integrated as the negative electrode in a lithium-ion capacitor, paired with a commercially available porous carbon, the cell delivers a specific energy of 156 W h kg−1 at a specific power of 0.34 kW kg−1 and 60.2 W h kg−1 at 19.4 kW kg−1, establishing a benchmark among state-of-the-art systems in the field. These results underscore the critical role of supramolecular organization for optimizing the performance of organic electrode materials for practical and sustainable energy storage technologies.
Broader contextAs we venture on a technological transformation towards improved sustainability and energy security, concerted efforts are required to develop critical raw material-free electrochemical energy storage technologies, while being more safe and effective. On this basis, organic molecules are promising due to their abundance, eco-friendly nature, processability, design versatility, and low cost. Research efforts to boost their performance have been focused on tailoring the design at molecular level, and on structural engineering involving their organization into crystalline frameworks. Here, going beyond the molecular or crystal structure, mesoscale morphological control of a small organic molecule offers another toolkit for reaching improved performances. The self-assembly of organic molecules into nanowires via a simple, fast, and environmentally benign method, lead to short lithium ion-diffusion lengths and rapid charge transport in the 3D organic nanowire network, leading to remarkable reversible capacities at slow and fast charging rates. Importantly, the self-assembled anode is prepared without organic solvents (a health and environmental bottleneck in the current battery industry), while displaying reduced exothermal footprint during charging. Its practical applicability is demonstrated by the high specific energy of the hybrid lithium metal-ion supercapacitor assembled using this anode and a commercial capacitive carbon. |
Introduction
The demand for mobile energy is quickly rising because of the growing number of electric vehicles and portable electronics. Combined with large-scale energy storage from renewable sources like solar and wind, this need has vastly mandated a global growth in lithium-ion battery production capacity. However, LIBs are approaching their maximum theoretical capacity,1 and raise concerns for their safety,2 their environmental impact,3,4 as well as for the rising costs and scarcity of the critical raw materials used in the cathode and the anode.5–7 As we venture further into the era of the fourth industrial revolution and technological transformation concerted efforts are required to develop electrochemical energy storage (EES) materials that overcome current limitations and comply with sustainability and energy security strategies.8,9In the quest of renewable electrodes, organic molecules are emerging as promising anodes or cathodes due to their abundance, eco-friendly nature, processability, design versatility, and low cost.10–12 A diverse range of organic materials, including carbonyl compounds,13 (bio)polymers,14,15 fused π-conjugated N-heteroaromatics,16,17 sulfonamides,18 peptides,19 and carboxylic compounds20 have been explored as cathodes and anodes. In the realm of anodes, terephthalic acid was reported to achieve an active material (AM) capacity of 300 mA h gAM−1 (similar to graphite, which is the current standard anode), combined with improved safety due to a three-fold lower heat release than graphite during lithiation.20 This discovery spurred further research into diverse organic carboxylic molecules as alternative anodes in LIBs, by leveraging the versatility of organic chemistry for controlling key features at the level of molecular design.10,21,22 For example, the transformation of carboxylic groups to carbothioates in naphthalene-2,6-dicarboxylate significantly improved Li+ storage, shifting the Li+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :COOH ratio from 1:1 to 1.5:1, and increasing the capacity from 198 mA h gEM−1 to 280 mA h gEM−1 (where EM represents the total electrode material mass).23 Oxocarbon salts (with ca. 300 mA h gEM−1 after 10 cycles),24 2,5-dihydroxyterephthalic acid (223 mA h gEM−1),25 or 4,4-(phenazine-5,10-diyl)dibenzoates (112 mA h gEM−1)26 were also investigated in the quest of efficient organic molecule/lithium interactions.
:COOH ratio from 1:1 to 1.5:1, and increasing the capacity from 198 mA h gEM−1 to 280 mA h gEM−1 (where EM represents the total electrode material mass).23 Oxocarbon salts (with ca. 300 mA h gEM−1 after 10 cycles),24 2,5-dihydroxyterephthalic acid (223 mA h gEM−1),25 or 4,4-(phenazine-5,10-diyl)dibenzoates (112 mA h gEM−1)26 were also investigated in the quest of efficient organic molecule/lithium interactions.
Structural design beyond the molecular level, involving the organization of organic molecules into crystalline frameworks, facilitated advances in performance.17,27,28 Two-dimensional triazine-based covalent organic nanosheets with carbon nanotubes as conductive additive achieved 581 mA h gEM−1 and 214 mA h gEM−1 specific capacity at 0.1 A g−1 and 5 A g−1, respectively, owing to the storage of up to 16 Li+ per organic molecule.29 The two-dimensional organic framework nanosheets were kinetically particularly effective, reaching Li-ion chemical diffusion coefficients as high as DLi+ ≈ 10−18 cm2 s−1. Zai et al. reported an organic framework anode incorporating hydroxyl and carbonyl functionalities, which was shown to deliver a reversible capacity of 764 mA h gEM−1 at 0.1 A g−1, 458 mA h gEM−1 at 5 A g−1 and 147 mA h gEM−1 at 10 A g−1.28
Mesoscale structural and morphological control is another toolkit to substantially modulate the macroscale properties and performance of organic electrode materials beyond their molecular or crystal structure.15,30 The previously discussed triazine/carbon nanotube-based frameworks29 almost doubled their capacity after their organization into hollow organic nanotubes,31 reaching 1021 mA h gEM−1 (1536 mA h gAM−1) at 0.1 A g−1. The supramolecular organization around the nanotubes enhanced Li+ diffusion by 6 orders of magnitude, reaching 2.5 × 10−12 cm2 s−1.31 However, this performance and capacity emerged only gradually after slow activation for 500 cycles and decayed to 215 mA h g−1 at 5 A g−1, indicating persisting ion diffusion limitations when the anode was subjected to faster discharging rates. A significant advancement in molecular organic carboxylic anodes occurred when maleic acid, combined with polyvinylidene fluoride (PVDF), self-assembled during drying from 1-methyl-2-pyrrolidone (NMP) into 5 nm nanocrystals encapsulated within a PVDF framework, delivering 1220 mA h gAM−1 at 0.15 A g−1.32,33 Itaconic acid, with two carboxyl groups, crystalized into a coral-patterned foam with ca. 100-nm walls, delivering 1074 mA h gAM−1 at 0.3 A g−1.34 Despite such advances, the continuous demand for safe and sustainable portable energy requires further developments in battery chemistry, electrode materials, and technologies.8
Herein, we report an high-performance organic anode based on mellitic acid (MelA), prepared by simple evaporation of an aqueous MelA solution on a commercial copper current collector. This leads to the self-assembly of MelA into nanowires via a simple, fast, and environmentally benign method, promoted by the presence of single copper metal ions from the current collector (Fig. 1a). The reversible capacity of MelA active material reaches 1888 mA h gAM−1 (or 1055 mA h gEM−1) at 0.1 A g−1, keeping 508 mA h gAM−1 at 10 A g−1, surpassing previous state-of-the-art materials. Short diffusion length and fast charge transport in the 3D MelA nanowire network of the anode enabled rapid Li+ diffusion rates (4 × 10−10 to 5 × 10−9 cm2 s−1), exceeding that of top performing organic anodes,31 and even of ultrafast Li-ion conductors.35,36 Importantly, the self-assembled MelA anode is prepared without any organic solvents (a bottleneck in the current battery manufacturing industry),37 and displays a reduced exothermal footprint during charging compared to graphite and other organic anodes.20 In addition to its performance as a LIB anode, a high-specific energy hybrid metal-ion supercapacitor assembled using the MelA anode and a capacitive carbon as cathode outperformed state-of-the-art organic, inorganic, and hybrid devices, delivering a specific energy of 156 kW h kgEM−1 at 0.1 A gEM−1 (throughout the text “gEM” represents the total materials mass on the electrode for the LIB anode, and the total materials mass on both electrodes for the case of LIC).
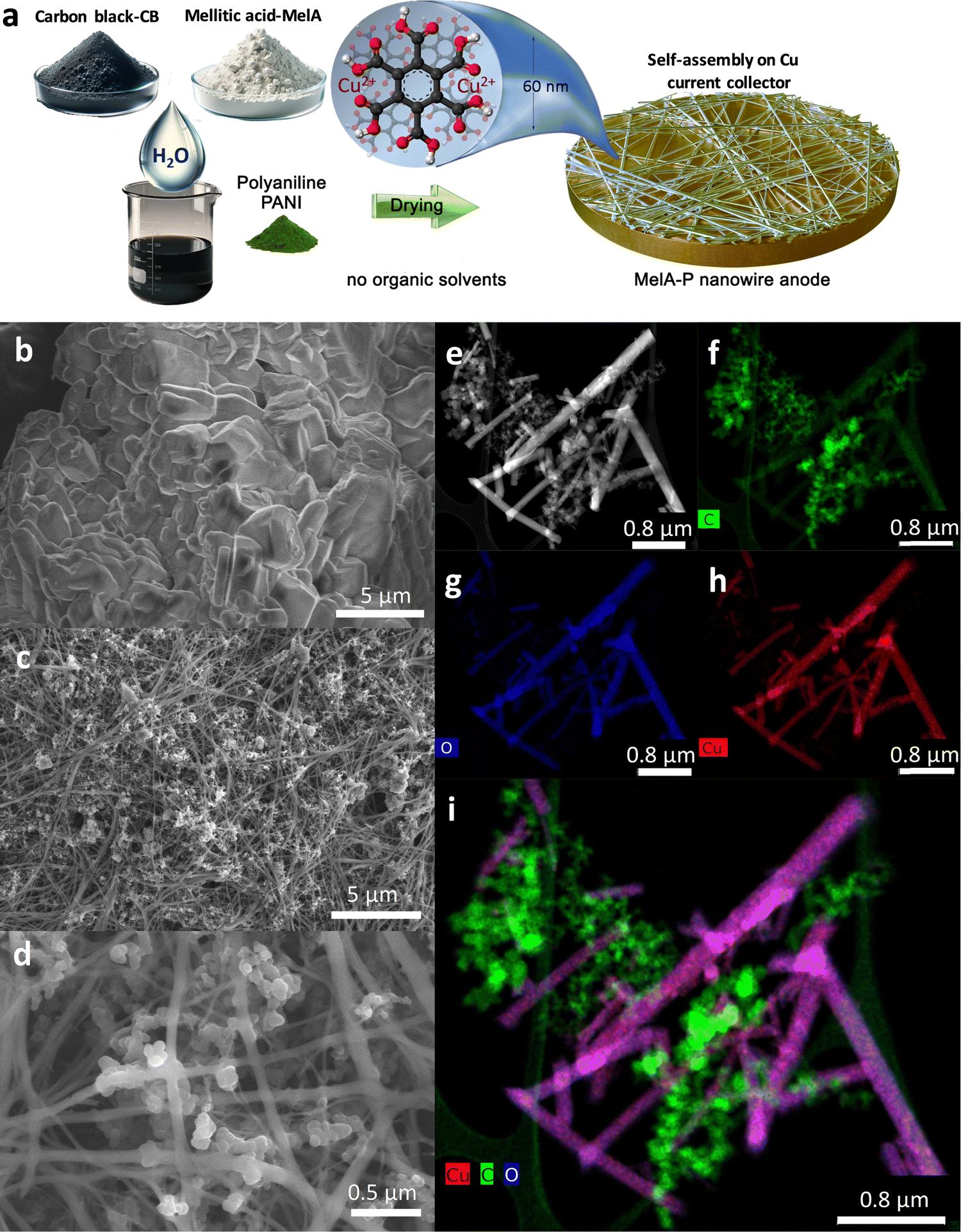 | ||
| Fig. 1 (a) Preparation via self-assembly of the MelA nanowire anode on the copper current collector. SEM images of (b) pristine MelA, and (c) and (d) MelA-nanowire electrode formed on the Cu current collector. (e) HAADF-STEM of MelA-nanowire anode, and (f)–(h) C, O, and Cu elemental mapping, as well as (i) combined C, O and Cu mapping. | ||
Results and discussion
The self-assembly of MelA electrode into a network of organic nanowires (called MelA-P) was achieved by casting aqueous slurries containing MelA (50 mass%), carbon black (CB, 40 mass%), and polyaniline (emeraldine base, PANI, 10 mass%) on a copper current collector, followed by slow drying at room temperature for 30 min and overnight in a vacuum oven at 100 °C (Fig. 1a). While pristine MelA is in the form of micrometer-sized particles or flakes, according to scanning electron microscopy (SEM, Fig. 1b), MelA-P is composed of a network of nanowires with a diameter of ca. 65 nm and length of ca. 25 μm, grown among the nanoparticles of CB (Fig. 1c, d and Fig. S1, ESI†). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, Fig. 1e) verified the growth of nanowires. Elemental mapping with STEM (Fig. 1f–i) revealed the presence of carbon in both MelA nanowires and CB particles, with a higher concentration in the latter, as anticipated. Oxygen was only seen in the MelA nanowires, verifying their chemical identity since only MelA contains oxygen in its carboxylic groups. Copper ions were present in the MelA nanowires due to the etching of the Cu current collector during the drying of the aqueous and acidic MelA-P slurry (Fig. 1h and i). To better understand the nanowire formation the MelA-P slurry was dried at 60 °C, instead of room temperature, keeping all other parameters the same. However, this slurry (MelA-P-hot, Fig. S2a and b, ESI†) leads to nanoparticle formation. This could be linked to fast slurry drying, dramatically affecting the self-assembly process. The latter typically requires time to avoid kinetic trapping to local energy minima before reaching the thermodynamically most favorable 3D organization. The absence of organic nanowires was also observed by performing the drying process using the solvent 1-methyl-2-pyrrolidone (NMP) instead of water (MelA-P-NMP, Fig. S2c and d, ESI†), which is the standard solvent used in battery manufacturing. This highlights the use of water as a key ingredient for the anode slurry, rendering the process much greener than the current NMP-based methodology, which must be performed under strict control due to the toxicity of NMP.37 To also probe the role of copper, the MelA-P slurry was deposited on a titanium current collector. Despite the slow drying from an aqueous slurry, exactly as in the case of the MelA-P deposition on Cu, no nanowires were grown (Fig. S2g and h, ESI†), highlighting the key role of Cu ions for nanowire formation. The absence of PANI did not affect the nanowire growth (Fig. S1c, MelA-C electrode, ESI†), while CB contents below 30 mass% resulted in the absence of nanowires (Fig. S3a–h, ESI†). A slurry made from 60% MelA and 40% CB without PANI displayed cracks, thus, the MelA-P formulation was selected for further studies.The MelA nanowire electrode on copper was mechanically very stable during bending and strongly adhered to the current collector. MelA deposited on copper was also extremely resistant to dissolution in the electrolyte, showing complete stability even after 40 electrochemical cycling tests and six-week storage in the cell (Fig. S3b and c, ESI†). While the X-ray diffractograms from pristine MelA revealed its typical crystal pattern, the MelA nanowire electrode was amorphous (Fig. S4, ESI†) and without copper oxides or copper nanoparticles. In contrast, the MelA electrode material collected from the Ti foil was crystalline with no nanowires, as the pristine MelA, highlighting the role of Cu ions. The content of Cu in the nanowires was 14.4 mass% Cu, as determined by inductively coupled plasma mass spectrometry. X-Ray photoelectron spectroscopy (XPS) showed the dominant presence of Cu2+ (Fig. S5c, ESI†), with 4.8 at% content. However, after electrochemical cycling of the MelA-P anode (discussed in the following), copper ions reduced to metallic copper clusters (Fig. S4c–e, ESI†).
MelA anodes were studied in a half-cell vs. Li metal in 1 M LiPF6 EC:DMC:EMC (1:1:1 by mass) as the electrolyte, with an addition of 10 vol% of FEC (for abbreviations, see ESI†). Pristine MelA electrode did not show electrochemical activity (ca. 20 mA h g−1 at 0.1 A g−1, Fig. S6a, ESI†). The optimal electrode material (EM) composition was determined by studying different component ratios (Fig. S6b–d, ESI†), which was 50:40:10 (MelA:CB:PANI) by mass. Lower contents of CB or absence of PANI did not yeld stable films (Fig. S3a–l, ESI†). Cyclic voltammograms recorded for MelA-P (Fig. 2a) showed three irreversible cathodic peaks in the initial scan: a double peak C1 at 1.6 V, C2 at 1.2 V, and a broad peak C3 at 0.5 V vs. Li+/Li. The C1 peak can be ascribed to the reduction of protons in carboxylic groups.38 The C2 and C3 peaks correspond to the solid–electrolyte interphase (SEI) formation involving FEC and EC.39 An irreversible anodic peak (A2) at the same potential as the hydrogen-evolution peak (C1) suggests its connection to the desorption of dihydrogen from the reduced mellitic acid. Reversible cathodic peaks C4, C5, and C6 emerging at 2.1 V, 1.0 V, and 0.7 V, respectively, and anodic peaks A1 and A3 at 2.6 V and 1.2 V vs. Li+/Li, respectively, correspond to redox processes of mellitic acid. A control electrode without PANI (MelA-C) showed an identical set of peaks (Fig. S7a, ESI†), indicating negligible or no effect of the 10 mass% of PANI on the observed electrochemical processes (in a few cases, the peaks are better resolved in presence of 10 mass% PANI suggesting improvement of charge-transfer processes).
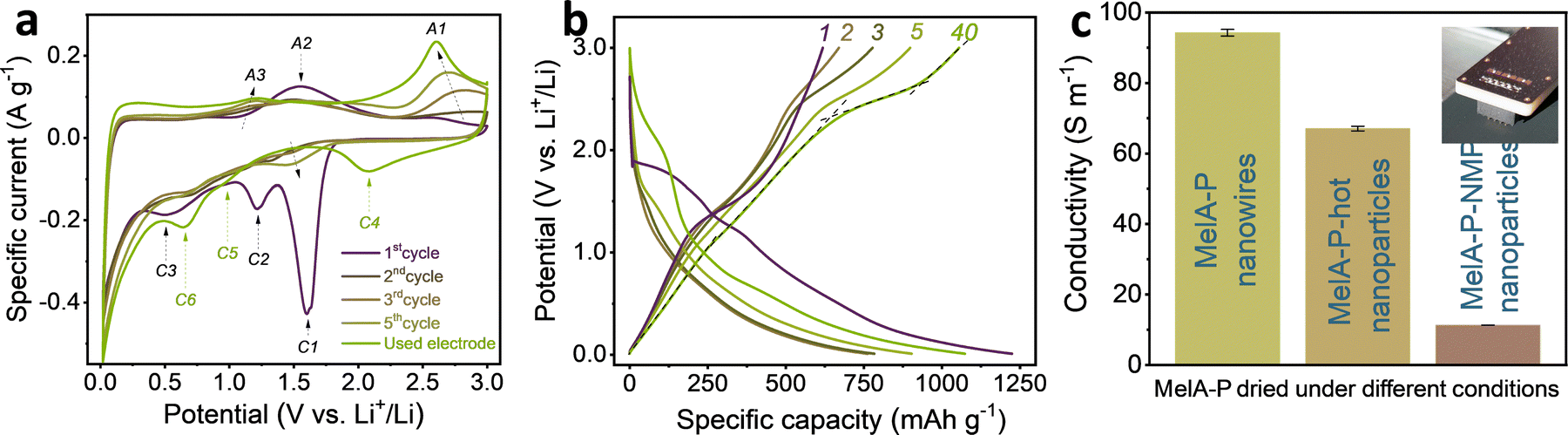 | ||
| Fig. 2 (a) Initial cyclic voltammograms of MelA-P anode at 0.1 mV s−1 potential sweep rate along with CV more cycling (used electrode), (b) five initial charge–discharge cycles in a MelA-P|Li half-cell at 0.1 A gEM−1 specific current, (c) electronic conductivity of MelA electrode materials. | ||
Galvanostatic charge–discharge curves for initial electrodes (Fig. 2b) align with the observations made during cyclic voltammetry. The initial lithiation has a characteristic dihydrogen evolution plateau38 in the 1.9–1.4 V vs. Li+/Li potential range (curve 1 in Fig. 2b) with a small activation overpotential, followed by the stepwise SEI formation with FEC and EC between 1.4–1.2 V and 0.8–0.4 V vs. Li+/Li. The peaks C1, C2, and C3 in the CVs at 1.6 V, 1.2 V, and 0.5 V vs. Li+/Li, respectively (Fig. 2a), align with the central parts of the plateaus. The delithiation (discharging) capacity of the MelA-P anode grew during the five initial charge–discharge cycles at 0.1 A g−1 from 618 mA h gEM−1 to 898 mA h gEM−1 (Fig. 2b) and to even higher values after rate tests (discussed below). The rising redox peaks in consecutively recorded cyclic voltammograms accompanied the slope changes of galvanostatic discharge curves (Fig. 2b). Four ranges could be identified: 0.01–1.0 V, 1.0–2.1 V, 2.4–2.6 V, and 2.7–3.0 V vs. Li+/Li (curve 40 in Fig. 2b). The asymmetric shape of the C4 and A1 peaks in the cyclic voltammograms suggests the presence of several overlapped electrochemical processes (due to the different interaction modes of Li with MelA, Fig. S19a, ESI†), desirable for high capacity redox-active energy storage materials. The control MelA-C anode showed identical behavior (Fig. S7, ESI†).
The current-rate performance of the MelA-P anode was tested in the 0.1–10 A g−1 range (Fig. 3a and b). The initial delithiation capacity of a fresh MelA-P anode at 0.1 A g−1 was 618 mA h gEM−1. It increased to 898 mA h gEM−1 during the next five cycles and grew to 934 mA h gEM−1 at 0.2 A g−1 over the subsequent five cycles (Fig. 3a). Even at 10 A g−1 the electrode retained 306 mA h gEM−1. The MelA-P anode regained its capacity after the current-rate test and delivered 1055 mA h gEM−1 at 0.1 A g−1 (Fig. 3a, cycles 35–40). Further stability testing at a high specific current of 2 A g−1 was performed in the consecutive 60 cycles, showing a small capacity decay from 796 mA h gEM−1 to 636 mA h gEM−1. However, it returned to 960 mA h gEM−1 upon three recovery cycles at 0.1 A g−1. The initial 50.5% Coulombic efficiency increased to 99.3% in the second cycle and exceeded 99.6% in consecutive cycles (Fig. 3a). To evaluate the effects of self-organization of the MelA-P electrode material, the MelA-P-hot, and the MelA-P-NMP (which were identical to MelA-P but without nanowire formation) showed dramatically inferior performances (Fig. S8, ESI†). The MelA-P-Ti anode (not deposited on Cu but on Ti current collector) also did not show nanowire formation and neither significant performance (Fig. S6a, ESI†). The formation of nanowires and creation of organized charge transport channels also contributed to improved electronic conductivity (Fig. 2c).
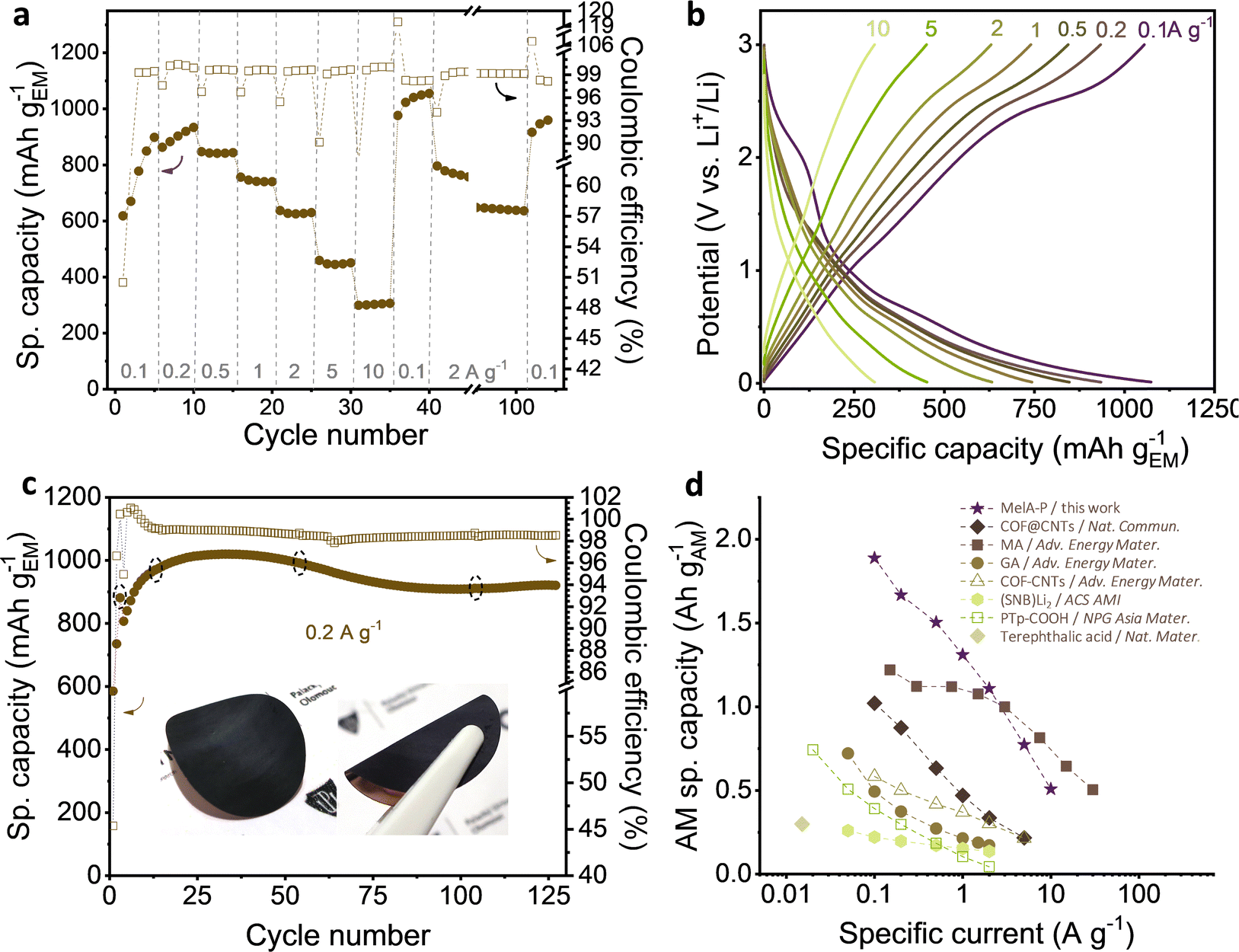 | ||
| Fig. 3 Electrochemical testing of MelA-P anode in half-cell vs. Li+/Li: (a) rate capability test; (b) 5th charge–discharge curves at increasing specific currents; (c) cycling stability at 0.2 A g−1 after three pretreatment cycles at 0.05 A g−1 (inset: MelA nanowire electrode material firmly attached on the copper current collector), and (d) performance comparison of organic anode active materials: COF@CNTs – imine-based COF layer over SWCNTs;31 MA – maleic acid;33 GA – graphene acid;40 COF-CNTs – exfoliated triazine-based COF nanosheets;29 (SNB)Li2 – sodium naphthalene-2,6-bis(carbothioate);23 PTp-COOH – carboxylated polythiophene.41 In panels (b) and (d), only the stabilized delithiation capacities of active materials are presented. Filled symbols in panels (a), (c) and (d) represent delithiation capacity. | ||
The stability of the MelA-P anode was evaluated by charge–discharge cycling at 0.2 A g−1 (Fig. 3c). After three initial SEI-forming cycles at 0.05 A g−1, the anode reached 881 mA h gEM−1 and 99.4% Coulombic efficiency. The capacity increased to 1055 mA h gEM−1 over the initial 30 cycles and stabilized at ∼920 mA h g−1 after 75 cycles. After considering the small capacity of pure CB and PANI electrodes (Fig. S9, ESI†), the capacity of MelA in the MelA-P electrode was 1055 mA h gEM−1 (or 1888 mA h gAM−1, Note S1, ESI†) at 0.1 A g−1, corresponding to 24 Li+ per MelA molecule (Note S2 and Fig. S10, ESI†). Although the active material capacity value is certainly not purely the property of MelA nanowires (as in all active-material-only capacity reports) since there is synergy between MelA, CB, and PANI, this value is commonly used for reporting and comparative reasons with other similar systems, as described in detail in comparative Table S1 (ESI†) and Fig. 3d.
Micrometer-sized terephthalic acid crystals delivered 300 mA gAM−1,20 (Table S1, entry 8, ESI†) corresponding to storing of two Li+ per terephthalic acid molecule and a two-electron process. In our comparative studies of terephthalic and trimesic acid, this value was not surpassed (Fig. S11a–c, ESI†). A three-electron electrochemical process was reported for sodium naphthalene-2,6-bis(carbothioate)23 (Table S1, entry 10, ESI†) delivering 260 mA h gAM−1 capacity at 0.05 A g−1. An improved capacity was reported with a carboxylated polythiophene (PTp-COOH) anode in the form of 200-nm-sized particles (508 mA h gAM−1 at 0.05 A g−1).41 Microflakes of poly(imine-anthraquinone) crystalized into sub-mm flakes with ionic channels showed high performance due to superlithiation effect and delivered 1291 mA h gAM−1 at 0.1 A g−1.42 Densely-functionalized graphene with carboxylic groups worked with a minimum amount of carbon additive but delivered a lower capacity (than MelA-P) 488 mA h gEM−1 at 0.1 A g−1.40 A five-nm thick COF synthesized around CNTs (Table S1, entry 12, ESI†) delivered 1021 mA h gAM−1 capacity at 0.1 A g−1 after very long electrochemical activation (320 cycles). At 1 A g−1, the capacity dropped to 519 mA h gAM−1 because of restricted diffusion in the material.31 On the contrary, the MelA-P retained 1310 mA h gAM−1 at 1 A g−1. Maleic acid anode (MA, Table S1, entry 2, ESI†) delivered a specific capacity of 1220 mA h gAM−1 at 0.15 A g−1.32 Fifty-nm flakes of itaconic acid (Table S1, entry 4, ESI†) was reported as another high-performance organic anode, with 1127 mA h gAM−1 at 0.3 A g−1,34 which is around 30% lower than MelA-P with 1504 mA h gAM−1 at even higher rate of 0.5 A g−1. Placing the performance of the MelA-P anode in context to the state-of-the-art highlights the substantially improved Li-storage properties in MelA-P, compared with previous advanced organic anodes.
Kinetic studies were performed to understand further the origin of the ultrahigh performance of MelA-P anode. Multi-rate CVs were recorded after the current rate test, showing both battery-type features with well-defined redox peaks and surface-controlled processes with a rectangular-shaped component (Fig. S12a, ESI†).42 The qualitative evaluation of kinetic processes in the MelA-P anode with the power law analysis (Note S3, ESI†) resulted in a b-value of 0.76 at 2.6 V (Fig. S12b, ESI†), which corresponds to the peak in the cyclic voltammogram and indicates co-presence of diffusion- and surface-controlled processes. A near-unity b-value was determined in the 1.2–1.8 V range, which reflects surface-controlled electrochemical processes (Fig. S12a and b, ESI†). Deconvolution of the recorded cyclic voltammograms by k1–k2 analysis to estimate the relative contributions of the diffusion- and surface-controlled processes (Note S3, ESI†) showed a mixed energy storage mechanism over the entire voltage range with 46% diffusion-controlled current at 0.1 mV s−1 potential sweep rate (Fig. S12c–f, ESI†). This fraction gradually decreased at higher potential sweep rates to 13% at 2.0 mV s−1. This charge component most likely corresponds to the redox reaction of carboxyl groups and Li plating/stripping at potentials below 0.5 V vs. Li+/Li, since only carboxylic groups-related diffusion-controlled current is expected in the ∼1.0–2.5 V potential range. Protons of MelA are reduced in the first charge–discharge cycle (peak C1, Fig. 2c), therefore, emeraldine salt formed during electrode preparation returns back to emeraldine base. In aprotic media, the emeraldine base is in situ p-doped with Li+ of the electrolyte.43 Finally, Li emeraldate can be formed in the 2.5–3.0 V range, contributing to the effective conductivity of the electrode material.44 The capacity of Li-emeraldate alone in this potential range is ∼25 mA h gPANI−1, corresponding to the contribution of 2.5 mA h g−1 to the MelA-P electrode material capacity. The difference between MelA-P and MelA-C anodes is 219 mA h gEM−1 at 0.1 A g−1; therefore, PANI promotes the redox activity of MelA. The electrochemical behavior of this MelA-C was qualitatively identical to MelA-P (Fig. S13a–e, ESI†), with lower diffusion-controlled current, ranging between 37–10% (Fig. S13e, ESI†). This originates from the rapid diffusion rate and short diffusion pathways of ions. The latter is defined by the ca. 32-nm average radius of MelA nanowires (Fig. S1b, ESI†).
As deduced from the analysis of the results with respect to state-of-the-art (Table S1, ESI†), MelA-P displays high capacity and rate performance. Electrochemical impedance spectroscopy (EIS) and galvanostatic intermittent titration technique (GITT) were employed to determine the charge transfer kinetics and Li+ diffusion coefficient (DLi+), respectively. The electrochemical impedance spectra (Fig. 4a and b) were recorded for the assembled cell at the open-circuit potential (2.64 V) before cycling, after three charge–discharge conditioning cycles at 0.05 A g−1, and during cycling stability test at 0.2 A g−1 (Fig. 3c). A uniform SEI was formed around the nanowires and carbon black particles (Fig. S14, ESI†). The charge transfer resistance (Rct) determined by fitting Nyquist plots using the equivalent circuit (Fig. S15, ESI†) was 70.2 Ω cm−2 for a fresh MelA-P anode, and drastically decreased to 12.9 Ω cm−2 after three formation cycles (Fig. 4c). This large change accompanied the growth of the specific capacity of the anode (585–881 mA h gEM−1; Fig. 3c) and indicated the occurrence of an electrochemical activation step, similar, yet much shorter to that observed in some covalent organic frameworks.28,29,31 The Rct further decreased to 9.2 Ω cm−2 (after 10 cycles), 2.8 Ω cm−2 (after 50 cycles), and 1.6 Ω cm−2 (after 100 cycles), showing fast electrochemical kinetics of the MelA-P anode. On the contrary, the Rct for the MelA-P-hot anode (Fig. S16a–d, ESI†) was 90.7 Ω cm−2 before cycling, 8.1 Ω cm−2 after three cycles at 0.05 A g−1 and 5.7 Ω cm−2 after ten cycles at 0.2 A g−1. Despite slightly faster activation, the specific capacity of the MelA-P-hot was lower, highlighting the importance of nanowire formation (Fig. 2c and Fig. S17a, b, ESI†).
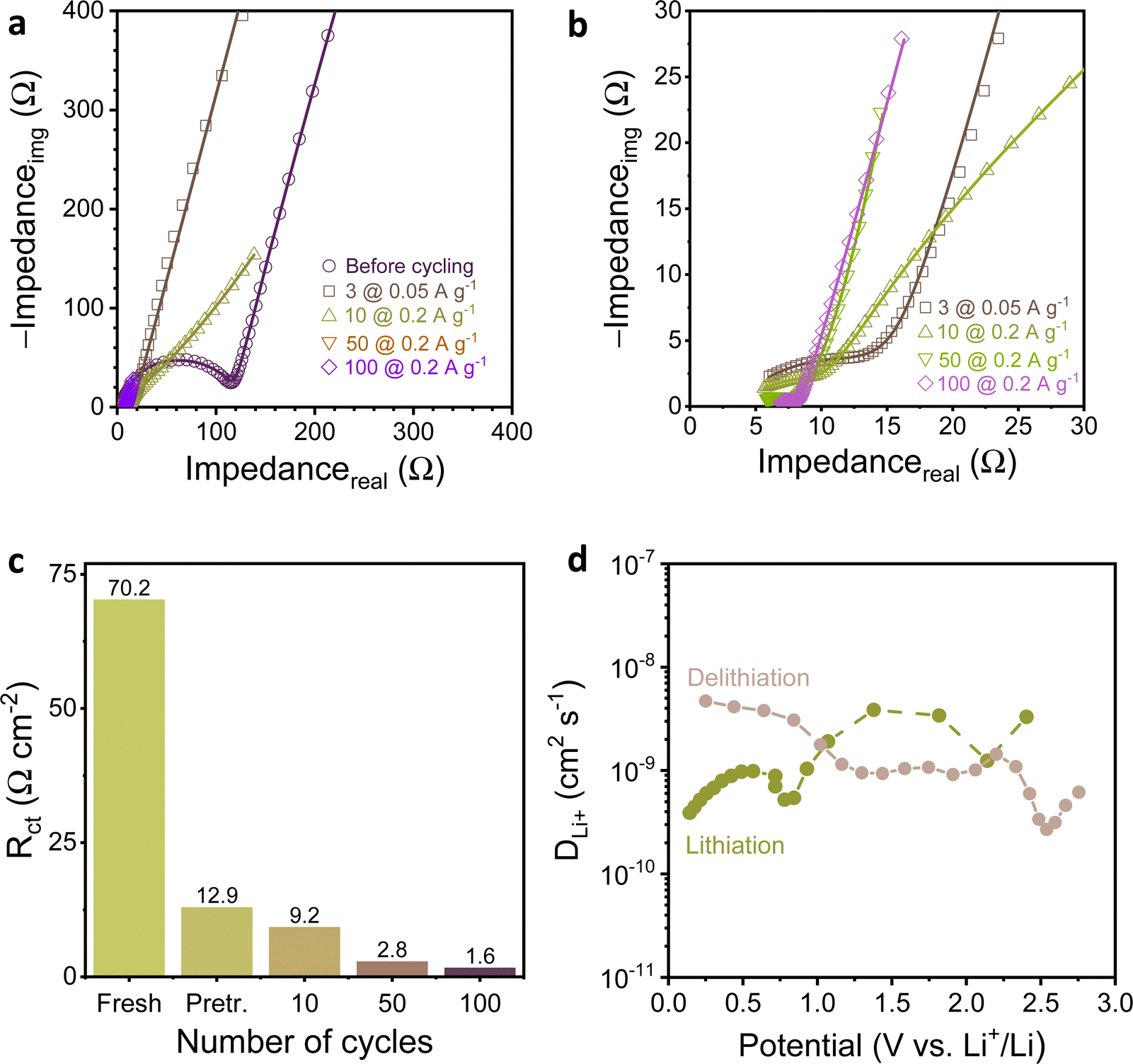 | ||
| Fig. 4 (a) Nyquist plots for EIS spectra recorded for a fresh electrode and after cycles of the stability test in the Fig. 3c, (b) magnified Rct region of the curves from panel “a”, (c) change of charge transfer resistance during cycling, and (d) diffusion coefficient determined by GITT. | ||
The apparent Li+ diffusion coefficient (Fig. 4d, Fig. S18 and Note S4, ESI†) was determined in the 4 × 10−10 to 5 × 10−9 cm2 s−1 range, depending on the potential and the current direction. The values are three orders of magnitude higher than the in-plane Li diffusivity in lithiated graphite (ca. 10−12 below 0.2 V).45,46 Despite that inorganic materials are better Li conductors than organic ones,47 the Li-ion diffusivity in MelA-P anode exceeded that of advanced Li-ion conductors such as lanthanum and aluminum LiCoO2 (4.7 × 10−11 cm2 s−1 to 1.2 × 10−10 cm2 s−1)36 and single-crystal lithium iron phosphate microrods (1.65 × 10−1 cm2 s−1).35 Thus, the ultrahigh electrochemical performance of MelA-P anode is due to a combination of all three factors: short diffusion path length in nanowires, fast electrochemical/charge transport kinetics, and efficient diffusion of Li+. The high diffusion coefficient suggests a lower diffusion barrier and activation energy of lithium-ion diffusion.48,49 However, the primary factor contributing to the performance is the nanowire structure. This is evident when comparing two samples: MelA-P (nanowires) with 4.8 at% Cu and MelA-P-hot (particles) with 3.3 at% Cu. Despite MelA-P having only 36% more copper, it exhibits a 108% improvement in capacity at 10 A g−1, highlighting the significant influence of the nanowire morphology (results obtained from Fig. S8, ESI†).
Theoretical calculations were carried out to better understand a mechanism of lithium atom binding to mellitic acid. First, calculations using a finite model containing one mellitic acid molecule and one Li atom were performed (at an unrestricted ωb97xd/def2SVP level50,51) to illustrate and validate the various modes of interactions of Li with mellitic acid observed in the periodic model calculations: (i) replacement of the H atom by Li showing the highest interaction (Fig. S19a, ESI†); (ii) less favorable interaction of Li between the carboxyl groups; (iii) Li interacting with a hollow site of the C6 aromatic ring. Furthermore, the radial distribution function, g(r), and coordination number of Li atoms around C and O atoms of mellitic acid were calculated at the lower (1.71 Li per molecule) and at higher (22.86 Li per molecule) Li loading to demonstrate the superlithiation effect. While Li atoms interacted dominantly with O atoms at lower Li loading, at high Li concentration interaction with C atoms significantly contributed, indicating on the superlithiation (Fig. S19b–e, ESI†). A periodic model of mellitic acid composed of seven molecules in a supercell interacting through a network of hydrogen bonds was used in computations. The lithium content in computations was from 0.14 to 22.8 Li atoms per mellitic acid molecule (Fig. S20, ESI†). At low lithiation states, the Li atoms preferred to replace hydrogen atoms of carboxylic groups. They were located between carboxyl groups interacting with three or four oxygen atoms with binding energies ranging from −146.4 to −136.3 kcal mol−1 per Li atom (Fig. S20, ESI†). Further lithiation resulted in additional modes of interaction between the Li atom and mellitic acid, i.e., (i) Li bridging between carboxyl groups of two molecules interacting with two oxygen atoms, (ii) Li binding to a carboxyl group and (iii) Li stacking over a hollow site of the C6 aromatic ring. All these possible interactions resulted in binding energy values up to −59.4 kcal mol−1 for the system containing 22.8 Li atoms per mellitic acid molecule (Fig. S20, ESI†). Thus, Li atoms prefer to interact with oxygen atoms of carboxyl groups. At the same time, superlithiation also contributes to their binding to mellitic acid nanowires at high Li loading.
Motivated by the high contribution of surface-controlled processes in MelA-P and rapid charge transport, we assembled a Li-ion capacitor (LIC) using the electrochemically prelithiated MelA-P anode and porous carbon cathode (PC) to demonstrate high-power energy storage. The cathode was characterized in a half-cell in the 2.0–4.5 V vs. Li+/Li voltage range before assembling the LIC (Fig. S21, ESI†). For the LIC assembling, the MelA-P anode was activated by three charge–discharge cycles at 0.05 A g−1 and ten cycles at 0.2 A g−1 followed by electrochemical lithiation to 0.01 V. The charge–discharge cycling of the MelA-P|PC LIC with the 1:3.8 (A:C) mass ratio was performed in the cell voltage range of 2.0–4.5 V and 1.0–4.5 V at 0.1 A g−1 to 10 A g−1 currents range (Fig. 5a–c). The initial specific energy of the LIC was 155.9 W h kgEM−1 after five cycles at 0.34 kW kgEM−1 and 60.2 W h kgEM−1 at 19.4 kW kgEM−1.
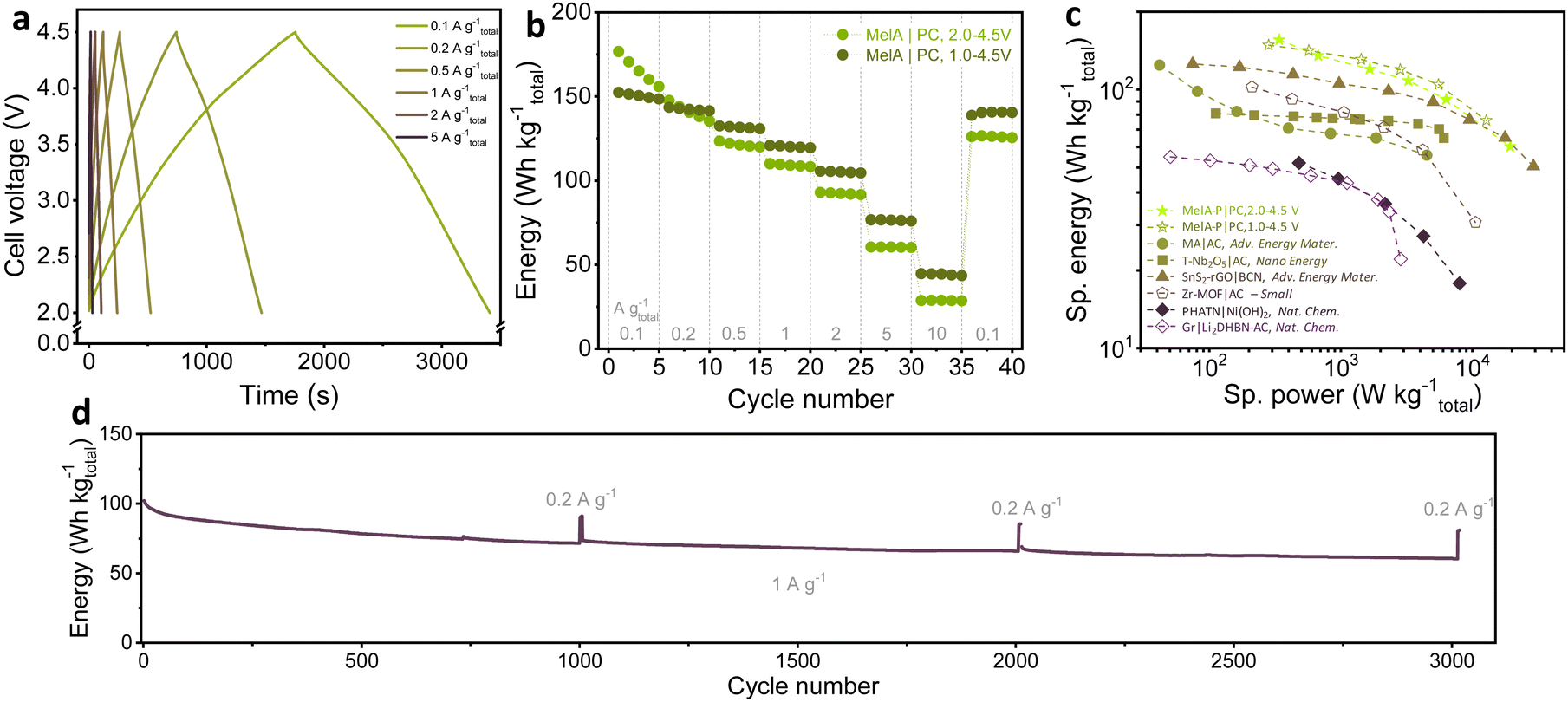 | ||
| Fig. 5 Electrochemical characterization of high-energy lithium-ion capacitor with MelA-P anode and porous carbon (PC): (a) charge–discharge profiles for the MelA-P|PC LIC, (b) specific energy with respect to the total mass of both electrode materials at different current rates in 2.0–4.5 V and 1.0–4.5 V voltage ranges, (c) performance comparison of high-energy LICs; values were normalized to the total mass of both anode and cathode electrode materials where necessary, and (d) stability test at 1 A g−1 in the 2.0–4.5 V voltage range, performed after the rate tests. (MA – maleic acid, AC – activated carbon;33 T-Nb2O5 – orthorhombic niobium oxide nanoparticles;52 SnS2-rGO – layer structured SnS2 growth on reduced graphene oxide, BCN – boron/nitrogen-doped carbon nanosheets;53 Zr-MOF – Zr4+ metal ions and tetrathiafulvalene-base ligands MOF;54 PHATN – perylene diimide-hexaazatrinaphthylene;17 Gr – graphite, Li2DHBN – 3,4-dihydroxybenzonitrile dilithium salt55). | ||
As shown in Table S3 and Fig. S5c (ESI†), MelA-P LIC showed a consistently higher performance than other advanced LICs based on organic or inorganic systems (Table S3, ESI† and Fig. 5c). The ultrahigh-performing LIC with maleic acid microbeads33 prelithiated to 0.0 V and Kuraray carbon cathode cycled in the 0.0–4.5 V range showed 124.5 W h kgEM−1 specific energy at a low specific power of 0.042 kW kgEM−1 (Note S5 for calculations, ESI†). Maleic acid LIC retained a very high specific energy of 55.7 W h kg−1 even at 4.51 kW kg−1 specific power.33 Our MelA-P|PC LIC shows a substantially higher performance at both low and high-power conditions (155.9 W h kg−1 at 0.34 kW kg−1 and 60.2 W h kg−1 at 19.4 kW kg−1). Few-nm particles of SnS2 uniformly spread in reduced graphene oxide facilitated the construction of a LIC with B/N-co-doped carbon nanosheet as a cathode.49 This material showed a performance of ∼125.2 W h kg−1 at 0.074 kW kg−1,49 and competed with MelA-P|PC LIC only at very high rates (Fig. 5c). Zero-dimensional T-Nb2O5 11-nm nanoparticles uniformly distributed on carbon microtubes served as a ground-breaking anode for LIC with P/N-co-doped carbon, delivering 80.9 W h kg−1 at 0.11 kW, and 64.9 W h kg−1 at 6.07 kW kg−1.50 The MelA-P LIC also substantially outperformed sophisticated organic electrodes of perylene diimide-hexaazatrinaphthylene (PHATN),17 3,4-dihydroxybenzonitrile dilithium salt (Li2DHBN|Ni(OH)2),51 or zirconium metal–organic frameworks,52 as described in detail in Table S3 (ESI†). Stability test of the LIC at 1 A gtotal−1 performed after the rate tests showed cyclability of LIC exceeding 3000 cycles (Fig. 5d).
Besides the performance advantages of the MelA-P anode, its production only with water as solvent (in contrast to the standard method requiring toxic organic solvents such as NMP) makes this approach especially beneficial from environmental, health safety, and sustainability perspectives. Moreover, unlike the standard graphite anode (responsible for uncontrolled energy release and thermal runaway) and other organic acids, the MelA-P electrode material released considerably lower heat (as probed with differential scanning calorimetry DSC, Fig. S22, ESI†), which is a key step for mitigating fire hazards in secondary batteries.
Conclusions
In this study, we demonstrate the potential of mesoscale engineering for transforming a simple organic molecule into a sustainable and high-performance electrode for energy storage. Utilizing an energy-efficient, environmentally friendly self-assembly process in water, we synthesized a network of organic nanowires directly on the copper current collector. Significantly, producing this anode avoids using organic solvents, addresses a critical challenge in battery manufacturing, and exhibits reduced exothermic reactions during charging, underscoring its superior safety profile. The obtained mellitic acid nanowire anode operated under mixed diffusion- and surface-controlled mechanisms, demonstrating unprecedented capacities and rate capability of 1888 mA h gAM−1 at 0.1 A g−1, keeping 508 mA h gAM−1 at 10 A g−1. The formation of nanowires leads to rapid charge transport kinetics, including lithium diffusivity rates reaching 5 × 10−9 cm2 s−1, facilitating the favorable accommodation of 24 lithium atoms per mellitic acid molecule, as in-depth electrochemical and theoretical studies revealed. The lithiated organic nanowire anode, coupled with a commercial porous carbon in a lithium-ion capacitor, outperforms state-of-the-art organic, inorganic, and hybrid systems, achieving a specific energy of 155.9 W h kg−1 at 0.34 kW kg−1 and 60.2 W h kg−1 at 19.4 kW kg−1. Together, the findings demonstrate the potential of organic materials for next-generation sustainable energy storage solutions and mark the avenue for discovering unprecedented properties in materials for electrochemical energy storage via mesoscale design.Author contributions
I. O. led this project, wrote the original draft, performed experiments, and analyzed data. R. L. performed simulations and M. O. supervised the simulations and reviewed the manuscript. J. G. A. R. performed EIS analyses, revised the manuscript, and contributed to the discussion. V. P. supervised J. G. A. R. and revised the manuscript, and contributed to the discussion. R. Z. supervised the project and reviewed the manuscript. A. B. revised the original draft and supervised the project. All authors read and approved the manuscript.Data availability
Data for this article are available at https://zenodo.org/ open repository and can be found under the same title as in this publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding of the joint Czech-German project GRAPHMAX by the German Research Foundation (DFG, Deutsche Forschungsgemeinschaft; J. G. A. R. and V. P.; grant no. PR-1173/29;) and the Czech Science Foundation (GACR, Grantová agentura České republiky; I. O. and A. B.; grant no. 22-27973K). We also acknowledge financial support of the European Union under the REFRESH – Research Excellence For Region Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition from the Ministry of the Environment of the Czech Republic. Support from the ERDF/ESF project TECHSCALE (no. CZ.02.01.01/00/22_008/0004587). R. L. acknowledges the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (ID:90254). We thank Jiri Hosek and Dr. Eirini Ioannou for SEM, Ondrej Tomanec for HR-TEM, Dr. Josef Kaslik for XRD, and Dr. Aby Cheruvathoor Poulose for XPS measurements.References
- W.-J. Kwak, et al., Lithium–Oxygen Batteries and Related Systems: Potential, Status, and Future, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Materials for lithium-ion battery safety, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- M. Chen, et al., Recycling End-of-Life Electric Vehicle Lithium-Ion Batteries, Joule, 2019, 3, 2622–2646 CrossRef CAS.
- G. Harper, et al., Recycling lithium-ion batteries from electric vehicles, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Performance and cost of materials for lithium-based rechargeable automotive batteries, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- Y. Yang, et al., On the sustainability of lithium ion battery industry – A review and perspective, Energy Storage Mater., 2021, 36, 186–212 CrossRef.
- C. Bauer, et al., Charging sustainable batteries, Nat. Sustain., 2022, 5, 176–178 CrossRef.
- J. W. Choi and D. Aurbach, Promise and reality of post-lithium-ion batteries with high energy densities, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- F. Duffner, et al., Post-lithium-ion battery cell production and its compatibility with lithium-ion cell production infrastructure, Nat. Energy, 2021, 6, 123–134 CrossRef CAS.
- P. Poizot, et al., Opportunities and Challenges for Organic Electrodes in Electrochemical Energy Storage, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed.
- Y. Lu and J. Chen, Prospects of organic electrode materials for practical lithium batteries, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS PubMed.
- J. Kim, et al., Organic batteries for a greener rechargeable world, Nat. Rev. Mater., 2023, 8, 54–70 CrossRef.
- B. Häupler, A. Wild and U. S. Schubert, Carbonyls: Powerful organic materials for secondary batteries, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- G. Milczarek and O. Inganäs, Renewable Cathode Materials from Biopolymer/Conjugated Polymer Interpenetrating Networks, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed.
- T. Zhu, et al., Formation of hierarchically ordered structures in conductive polymers to enhance the performances of lithium-ion batteries, Nat. Energy, 2023, 8, 129–137 CrossRef CAS.
- C. Peng, et al., Reversible multi-electron redox chemistry of π-conjugated N-containing heteroaromatic molecule-based organic cathodes, Nat. Energy, 2017, 2, 1–9 Search PubMed.
- J. C. Russell, et al., High-performance organic pseudocapacitors via molecular contortion, Nat. Mater., 2021, 20, 1136–1141 CrossRef CAS PubMed.
- J. Wang, et al., Conjugated sulfonamides as a class of organic lithium-ion positive electrodes, Nat. Mater., 2021, 20, 665–673 CrossRef CAS PubMed.
- T. P. Nguyen, et al., Polypeptide organic radical batteries, Nature, 2021, 593, 61–66 CrossRef CAS PubMed.
- M. Armand, et al., Conjugated dicarboxylate anodes for Li-ion batteries, Nat. Mater., 2009, 8, 120–125 CrossRef CAS PubMed.
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Design Strategies toward Enhancing the Performance of Organic Electrode Materials in Metal-Ion Batteries, Chem, 2018, 4(12), 2786–2813 CAS.
- L. Fédèle, F. Sauvage and M. Bécuwe, Hyper-conjugated lithium carboxylate based on a perylene unit for high-rate organic lithium-ion batteries, J. Mater. Chem. A, 2014, 2, 18225–18228 RSC.
- J. Wang, H. Zhao, L. Xu, Y. Yang, G. He and Y. Du, Three-Electron Redox Enabled Dithiocarboxylate Electrode for Superior Lithium Storage Performance, ACS Appl. Mater. Interfaces, 2018, 10(41), 35469–35476 CrossRef CAS PubMed.
- Q. Zhao, et al., Oxocarbon Salts for Fast Rechargeable Batteries, Angew. Chem., Int. Ed., 2016, 55, 12528–12532 CrossRef CAS PubMed.
- S. Wang, et al., Organic Li4C8H2O6 Nanosheets for Lithium-Ion Batteries, Nano Lett., 2013, 13, 4404–4409 CrossRef CAS PubMed.
- G. Dai, et al., A Dual-Ion Organic Symmetric Battery Constructed from Phenazine-Based Artificial Bipolar Molecules, Angew. Chem., Int. Ed., 2019, 58, 9902–9906 CrossRef CAS PubMed.
- T. Sun, J. Xie, W. Guo, D.-S. Li and Q. Zhang, Covalent–Organic Frameworks: Advanced Organic Electrode Materials for Rechargeable Batteries, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- L. Zhai, et al., 30 Li+-Accommodating Covalent Organic Frameworks as Ultralong Cyclable High-Capacity Li-Ion Battery Electrodes, Adv. Funct. Mater., 2022, 32, 2108798 CrossRef CAS.
- Z. Lei, X. Chen, W. Sun, Y. Zhang and Y. Wang, Exfoliated Triazine-Based Covalent Organic Nanosheets with Multielectron Redox for High-Performance Lithium Organic Batteries, Adv. Energy Mater., 2019, 9, 1801010 CrossRef.
- Y. Ham, et al., 3D periodic polyimide nano-networks for ultrahigh-rate and sustainable energy storage, Energy Environ. Sci., 2021, 14, 5894–5902 RSC.
- Z. Lei, et al., Boosting lithium storage in covalent organic framework via activation of 14-electron redox chemistry, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- Y. Wang, et al., Ultrahigh-Capacity Organic Anode with High-Rate Capability and Long Cycle Life for Lithium-Ion Batteries, ACS Energy Lett., 2017, 2, 2140–2148 CrossRef CAS.
- Z. Hu, et al., Self-Assembled Binary Organic Granules with Multiple Lithium Uptake Mechanisms toward High-Energy Flexible Lithium-Ion Hybrid Supercapacitors, Adv. Energy Mater., 2018, 8, 1802273 CrossRef.
- Y. Wang, et al., A high-capacity organic anode with self-assembled morphological transformation for green lithium-ion batteries, J. Mater. Chem. A, 2019, 7, 22621–22630 RSC.
- L. Hong, et al., Two-dimensional lithium diffusion behavior and probable hybrid phase transformation kinetics in olivine lithium iron phosphate, Nat. Commun., 2017, 8, 1194 CrossRef PubMed.
- Q. Liu, et al., Approaching the capacity limit of lithium cobalt oxide in lithium ion batteries via lanthanum and aluminium doping, Nat. Energy, 2018, 3, 936–943 CrossRef CAS.
- D. Larcher and J.-M. Tarascon, Towards greener and more sustainable batteries for electrical energy storage, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- L. Fédèle, et al., Mesoscale Texturation of Organic-Based Negative Electrode Material through in Situ Proton Reduction of Conjugated Carboxylic Acid, Chem. Mater., 2019, 31, 6224–6230 CrossRef.
- T. Hou, et al., The influence of FEC on the solvation structure and reduction reaction of LiPF6/EC electrolytes and its implication for solid electrolyte interphase formation, Nano Energy, 2019, 64, 103881 CrossRef CAS.
- I. Obraztsov, et al., Graphene Acid for Lithium-Ion Batteries—Carboxylation Boosts Storage Capacity in Graphene, Adv. Energy Mater., 2022, 12, 2103010 CrossRef CAS.
- H. Numazawa, K. Sato, H. Imai and Y. Oaki, Multistage redox reactions of conductive-polymer nanostructures with lithium ions: potential for high-performance organic anodes, NPG Asia Mater., 2018, 10, 397–405 CrossRef CAS.
- T. S. Mathis, et al., Energy Storage Data Reporting in Perspective—Guidelines for Interpreting the Performance of Electrochemical Energy Storage Systems, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS.
- K. S. Ryu, et al., Electrochemical and physical characterization of lithium ionic salt doped polyaniline as a polymer electrode of lithium secondary battery, Synth. Met., 2000, 110, 213–217 CrossRef CAS.
- P. Jiménez, et al., Lithium n-Doped Polyaniline as a High-Performance Electroactive Material for Rechargeable Batteries, Angew. Chem., Int. Ed., 2017, 56, 1553–1556 CrossRef PubMed.
- H. Yang, H. J. Bang and J. Prakash, Evaluation of Electrochemical Interface Area and Lithium Diffusion Coefficient for a Composite Graphite Anode, J. Electrochem. Soc., 2004, 151, A1247 CrossRef CAS.
- M. Schroeder, et al., Considerations about the influence of the structural and electrochemical properties of carbonaceous materials on the behavior of lithium-ion capacitors, J. Power Sources, 2014, 266, 250–258 CrossRef CAS.
- J. Wan, et al., Ultrathin, flexible, solid polymer composite electrolyte enabled with aligned nanoporous host for lithium batteries, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef CAS PubMed.
- W. Li, et al., Li+ ion conductivity and diffusion mechanism in α-Li3N and β-Li3N, Energy Environ. Sci., 2010, 3, 1524–1530 RSC.
- J. Pokharel, et al., Manipulating the diffusion energy barrier at the lithium metal electrolyte interface for dendrite-free long-life batteries, Nat. Commun., 2024, 15, 3085 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Hemmati, et al., 3D N-doped hybrid architectures assembled from 0D T-Nb2O5 embedded in carbon microtubes toward high-rate Li-ion capacitors, Nano Energy, 2019, 56, 118–126 CrossRef CAS.
- Y. Hao, S. Wang, Y. Shao, Y. Wu and S. Miao, High-Energy Density Li-Ion Capacitor with Layered SnS2/Reduced Graphene Oxide Anode and BCN Nanosheet Cathode, Adv. Energy Mater., 2020, 10, 1902836 CrossRef CAS.
- W. Yan, et al., High-Performance Lithium-Ion Capacitors Based on Porosity-Regulated Zirconium Metal–Organic Frameworks, Small, 2021, 17, 2005209 CrossRef CAS PubMed.
- P. Jeżowski, et al., Safe and recyclable lithium-ion capacitors using sacrificial organic lithium salt, Nat. Mater., 2018, 17, 167–173 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S20, Tables S1–S3, Notes S1–S5, and ref. 1–30. See DOI: https://doi.org/10.1039/d4ee02777a |
| This journal is © The Royal Society of Chemistry 2024 |