
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrodialysis and nitrate reduction (EDNR) to enable distributed ammonia manufacturing from wastewaters†
Jinyu
Guo
 a,
Matthew J.
Liu
a,
Chloe
Laguna
a,
Dean M.
Miller
a,
Kindle S.
Williams
a,
Brandon D.
Clark
a,
Carolina
Muñoz
a,
Sarah J.
Blair
a,
Adam C.
Nielander
b,
Thomas F.
Jaramillo
ab and
William A.
Tarpeh
*ab
a,
Matthew J.
Liu
a,
Chloe
Laguna
a,
Dean M.
Miller
a,
Kindle S.
Williams
a,
Brandon D.
Clark
a,
Carolina
Muñoz
a,
Sarah J.
Blair
a,
Adam C.
Nielander
b,
Thomas F.
Jaramillo
ab and
William A.
Tarpeh
*ab
aDepartment of Chemical Engineering, Stanford University, Stanford, CA 94305, USA
bSUNCAT Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, Menlo Park, CA, USA
First published on 18th October 2024
Abstract
Underutilized wastewaters containing dilute levels of reactive nitrogen (Nr) can help rebalance the nitrogen cycle. This study describes electrodialysis and nitrate reduction (EDNR), a reactive electrochemical separation architecture that combines catalysis and separations to remediate nitrate and ammonium-polluted wastewaters while recovering ammonia. By engineering operating parameters (e.g., background electrolyte, applied potential, electrolyte flow rate), we achieved high recovery and conversion of Nr in both simulated and real wastewaters. The EDNR process demonstrated long-term robustness and up-concentration that recovered >100 mM ammonium fertilizer solution from agricultural runoff that contained 8.2 mM Nr. EDNR is the first reported process to our knowledge that remediates dilute real wastewater and recovers ammonia from multiple Nr pollutants, with an energy consumption (245 MJ per kg NH3–N in simulated wastewater, 920 MJ per kg NH3–N in agricultural runoff) on par with the state-of-the-art. Demonstrated first at proof-of-concept and engineered to technology readiness level (TRL) 4–5, EDNR shows great promise for distributed wastewater treatment and sustainable ammonia manufacturing.
Broader contextThe nitrogen cycle is severely imbalanced by anthropogenic activities: reactive nitrogen removal occurs at half the rate of reactive nitrogen synthesis, constantly contaminating the environment. We developed a novel electrochemical reactive separation process to convert wastewater ammonium and nitrate into ammonia products. When powered by renewable electricity, this electrochemical architecture enables sustainable water treatment and fertilizer production, especially for communities not served by conventional centralized manufacturing. |
Introduction
The nitrogen cycle is in urgent need of re-engineering. Nitrogen (N) pollution is widespread—the US Environmental Protection Agency considers it “one of the costliest, most difficult environmental problems we face in the 21st century”.1 This pollution originates from imbalances between reactive nitrogen (Nr) production and its removal as N2 in the incumbent nitrogen management system. Most anthropogenic Nr comes from Haber–Bosch ammonia synthesis. The Haber–Bosch process successfully supplied sufficient fertilizer to solve the early 20th century global hunger challenge, but also presents several sustainability challenges for upcoming generations. Ammonia (NH3) is synthesized from inert N2 and steam reformed H2 at ∼700 K and ∼100 atm; these extreme conditions and reliance on fossil fuels contribute to 1–2% of global energy consumption and 1.2% of greenhouse gas emissions.2–5 In contrast, anthropogenic removal of reactive nitrogen (Nr) from the environment (often as N2) is only half the rate of its production, leading to costly Nr pollution that has exceeded critical thresholds for environment and human welfare and caused direct damage worth 0.3–3% of annual global gross domestic product.6,7 Even with universal adoption of known Nr mitigation actions (e.g., efficient fertilizer application and livestock management), environmental discharges of Nr are projected to surpass 95 million tons per year in 2050.3,7 This perilous gap between Nr production and mitigation calls for transformative technologies that can remove Nr from the environment and that can produce Nr products with low associated emissions.Compared to technologies solely targeting Nr removal (Nr to N2, e.g., denitrification) or sustainable Nr production (N2 to Nr, e.g., electrified ammonia synthesis), wastewater refining can shortcut the inert N2 intermediate and directly convert Nr pollutants to Nr products.8,9 Globally, wastewater contains a yearly stream of 35–78 million tons Nr, which could offset 15–34% of total Nr required by 2050.7,8 Over 90% of wastewater Nr exists in municipal wastewater and agricultural runoff, and nitrate (NO3−) and ammonium (NH4+) are the dominant aqueous Nr pollutants that threaten the health of both ecosystems (e.g., eutrophication) and humans (e.g., methemoglobinemia).10–13 Therefore, approaches are needed that valorize both NO3− and NH4+ from municipal wastewater and agricultural runoff to NH3 product, whereas most current technologies only target on either of the two major pollutants. Targeting both species can also address the Nr cascade problem, where Nr species interconvert to continue harming the environment.4,14 Electrochemical methods are uniquely positioned to facilitate replacement of fossil fuels with renewable energy inputs and enable distributed implementation that matches the distributed nature of our target wastewaters. Ultimately, electrochemically refining wastewater NO3− and NH4+ to NH3 can (1) remediate historically accumulated Nr pollution in the environment, (2) recover valuable Nr resources, and (3) reduce the need for virgin Nr production and related emissions from Haber–Bosch facilities.
Achieving the full potential of wastewater Nr refining requires overcoming challenges intrinsic to decentralized wastewater feedstocks, including dilute mixed Nr pollutants (typically below 10 mM), low total ionic conductivity, and complex and variable background matrices. In contrast, electrochemical NH3 recovery and nitrate reduction reaction (NO3RR) systems are often demonstrated with simplistic matrices with a single Nr species at higher concentrations (usually above 10 mM), well-controlled pH, and high ionic conductivity to operate efficiently.15 This mismatch in decentralized wastewater characteristics and electrochemical Nr recovery system requirements leads to low efficiency when real wastewater is directly used as the electrolyte. Therefore, we leverage electrochemical reactive separations, where separation and reaction are co-located within the same reactor and occur in tandem.8,14,16 Unlike processes with discrete reactant separation and catalysis steps, reactive separations utilize separations to create favorable and stable reaction environments from complex feedstocks, and reactions to produce product mixtures that inform separations. Electrochemical reactive separations have been demonstrated to recover carbon (reactive carbon capture),17–21 sulfur,22–26 and lithium,27–32 but have rarely been used to recover NH3 from NH4+-containing33–35 and from NO3−-containing36–39 wastewaters, and even more rarely for wastewaters containing both NH4+ and NO3−.
In this study, we developed a novel electrochemical reactive separation unit process, electrodialysis and nitrate reduction (EDNR), to recover and synthesize NH3 from dilute NH4+ and NO3−-polluted wastewaters. EDNR consists of three sub-unit processes: (1) electrodialysis to separate influent NH4+ and NO3− from wastewater, (2) deprotonation of NH4+ with electrochemically in situ generated OH− to recover NH3, and (3) electrocatalytic reduction of NO3− to synthesize NH3 using polycrystalline titanium (Ti) foil electrodes. This unit process is the first to our knowledge that targets multiple Nr pollutants and recovers NH3 from both dilute wastewater NH4+ and NO3− using electrochemical reactive separations. We achieved high Nr conversion (84 ± 10%) and recovery (111 ± 12%) in simulated wastewater by engineering operating parameters. Furthermore, we systematically studied effects of feedstock compositions and tested real wastewaters that span two orders of magnitude in total ionic concentration (well water, agricultural runoff, reverse osmosis retentate). The EDNR process showed excellent stability over 60-hour operation and recovered 12-fold concentrated ready-to-apply NH3 fertilizer solution from agricultural runoff, with an energy consumption (920 MJ per kg NH3–N) on par with the state-of-the-art (18–101 MJ per kg NH3–N from NH4+ and 168–31400 MJ per kg NH3–N from NO3−). Our efforts advanced beyond proof-of-concept to achieve TRL 4–540 (validation in a relevant environment, i.e., real wastewater), demonstrating that EDNR can be implemented as an individual module or part of a treatment train to enable integrated distributed water treatment and sustainable NH3 production.
Methods
Electrodialysis and nitrate reduction (EDNR) reactor and operation
The EDNR reactor is a three-chamber cell, with an anion exchange membrane (AEM) (Table 1) separating the NH3 synthesis (left) and influent (middle) chambers, and a cation exchange membrane (CEM, CMI-7000, Membranes International) separating the influent (middle) and NH3 recovery (right) chambers (Fig. 1a). All three chambers have the dimensions: 3.15 cm (H) × 1.8 cm (W) × 1.19 cm (D) (Fig. S1 in the ESI†). The geometric surface area of all electrodes and membranes used in the EDNR experiments was 5.7 cm2. Semi-batch mode was used, and electrolytes were recirculated between the electrochemical reactor chambers and their corresponding electrolyte reservoirs (total electrolyte volume of 50 mL for each chamber) using peristaltic pumps.| Proof-of-concept | Optimized NR | |
|---|---|---|
| Influent | 13.9 mM (NH4)2SO4 + 1.61 mM KNO3 | |
| NH3 synthesis chamber electrolyte | 0.1 M KClO4 | 1 M NaClO4 |
| NH3 recovery chamber electrolyte | 0.1 M KClO4 | 1 M NaClO4 |
| AEM | General (AMI-700) | Monovalent-selective (Selemion AMVN) |
| ED current density | 2.63 mA cm−2 | 3.95 mA cm−2 |
| ED duration | 60 min | |
| NR potential | −0.6 V vs. RHE, potentiostatic | −0.8 V vs. RHE, pulsed (10 s at reduction potential, followed by 10 s at open circuit) |
| NR flow rate | 30 mL min−1 | 100 mL min−1 |
| NR duration | 120 min | |
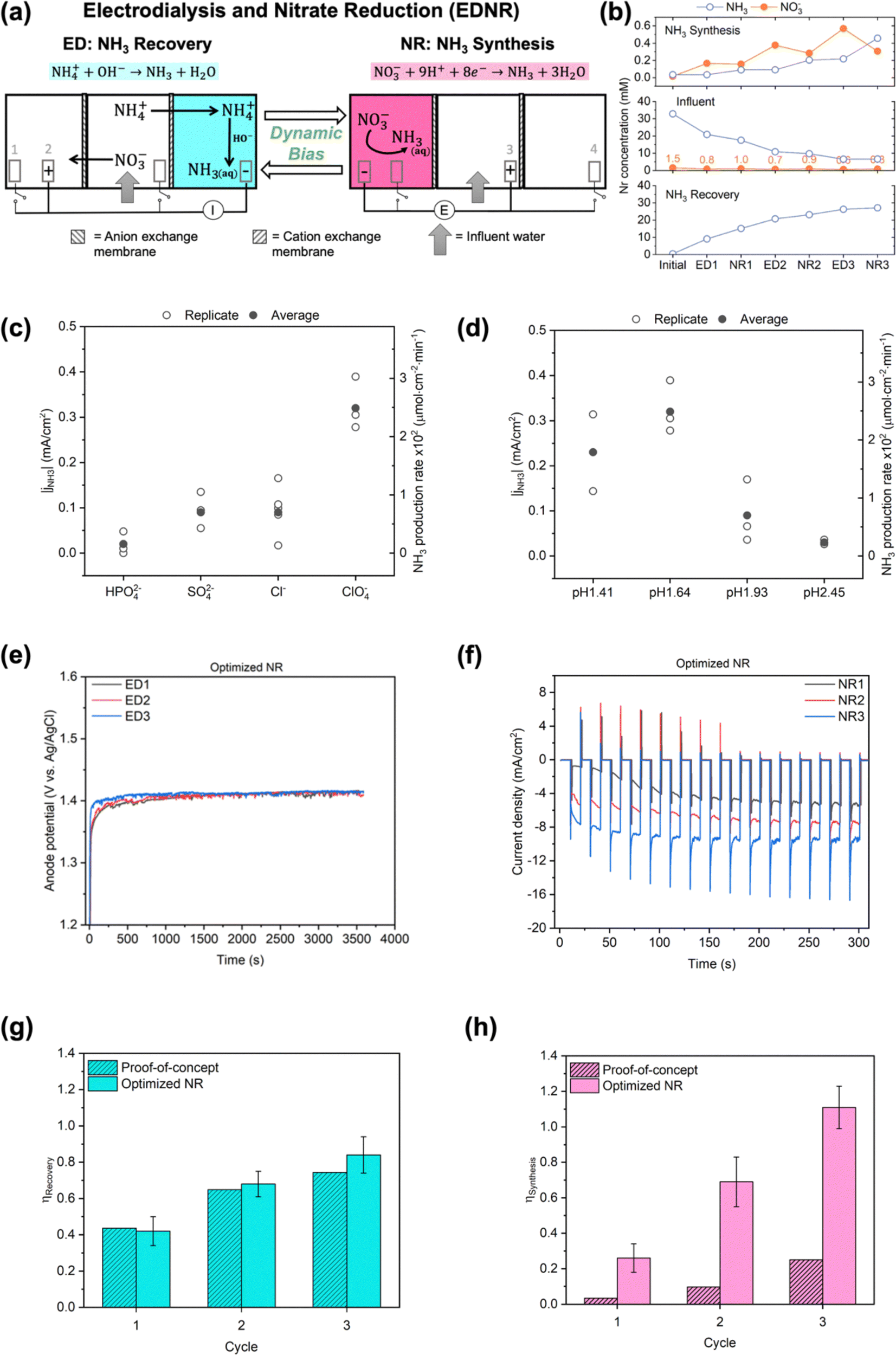 | ||
| Fig. 1 (a) Schematics of the EDNR process. Electrode (1) Ti foil, (2) and (3) IrO2–Ta2O5/Ti mesh, (4) Pt foil. I and E represent controlled applied current and potential in ED and NR stages, respectively. More experiment details (electrolyte composition, volume, flow rate, applied current/potential, stage duration) are shown in Table 1 and ESI† Fig. S1 and Table S1. (b) Trends of NH3 and NO3− concentrations in proof-of-concept experiment. NO3− concentrations in the influent chamber are enumerated to highlight their values on the large scale used for NH3 concentrations. (c) Magnitude of NH3 partial current density (left y-axis) and production rate (right y-axis) in background electrolytes with a variety of anions: 0.5 M Na2HPO4, 0.5 M Na2SO4, 1 M NaCl, and 1 M NaClO4, pH adjusted to 1.72 with 2 M HClO4. (d) Magnitude of NH3 partial current density (left y-axis) and production rate (right y-axis) in 1 M NaClO4 with a variety of initial pH: 1.41, 1.64, 1.93, and 2.45, adjusted by adding 2 M HClO4. All experiments in (c) and (d) were conducted in two-chamber reactors under static potential of −0.8 V vs. RHE for 30 min, with additional details shown in ESI† Section S1.3; partial current density is defined by eqn (S9) (ESI†). Open symbols represent results from each replicate experiment, and filled symbols represent the average values. (e) Anode (IrO2–Ta2O5/Ti mesh electrode in the NH3 synthesis chamber) potentials as functions of time in ED stages and (f) total current density in the first 5 min into each NR stage from one representative optimized NR experiment. The corresponding full chronoamperometry is shown in Fig. S13 (ESI†). Comparison of (g) NH3 recovery and (h) NH3 synthesis efficiencies in proof-of-concept and optimized NR experiments. Error bars represent ± one standard deviation from triplicate experiments for optimized NR. Only one replicate for proof-of-concept was performed to prioritize efforts to improve NR performance. | ||
The EDNR process operates in two stages, referred to as the electrodialysis (ED) stage and the nitrate reduction (NR) stage (Fig. S2 and S3, ESI†). In each ED stage, controlled current is applied to IrO2–Ta2O5/Ti mesh electrode (anode) in the NH3 synthesis chamber and platinum electrode (cathode) in the NH3 recovery chamber. Influent NO3− and NH4+ are separated via electromigration into the NH3 synthesis and NH3 recovery chambers, respectively; NH4+ combines with the electrochemically-generated OH−, and NH3 is recovered in the NH3 recovery chamber (NH4+ + OH− → NH3 + H2O). In each NR stage, controlled potential is applied to the Ti electrode (cathode) in the NH3 synthesis chamber and IrO2–Ta2O5/Ti mesh electrode (anode) in the influent chamber; NH3 is synthesized from the electrochemical NO3RR (NO3− + 8e− + 9H+ → NH3 + 3H2O) in the NH3 synthesis chamber. The two consecutive stages complete one EDNR cycle, and multiple cycles can be conducted to achieve treatment goals (e.g., complete removal and recovery of influent Nr).
Detailed experimental descriptions (e.g., reagents, instrumentation, procedures) are given in ESI† Section S1.1 and S1.2. Reactor schematics and operation procedures of two-chamber NR reactor, long-term EDNR, and membrane stripping experiments are described in ESI† Section S1.3 and S1.4. Electrolyte compositions and operating parameters (e.g., electrolyte flow rate, applied current/potential, stage duration) used in all EDNR experiments are summarized in Table S1 (ESI†).
Product analysis and key performance metrics
Electrolyte aliquots from all three electrolyte reservoirs were sampled for pH measurement and aqueous product analysis before and after each stage (ED or NR). Due to acid–base equilibria, we reported the sum concentrations of weak conjugate acid–base pairs using nitrite (NO2−) to represent the sum of anionic nitrite and nitrous acid (pKa 3.16 at 25 °C), and ammonia (NH3) to represent the sum of cationic ammonium and ammonia (pKa 9.25 at 25 °C) for brevity. NO3− and NO2− concentrations were quantified using anion chromatography, and NH3 concentrations were quantified using spectrophotometric flow injection analysis. See ESI† Section S1.5 for detailed sample analysis methods.To evaluate efficiency of the EDNR process in recovering NH3 from influent Nr, we defined the following two metrics:
NH3 recovery efficiency (ηRecovery):
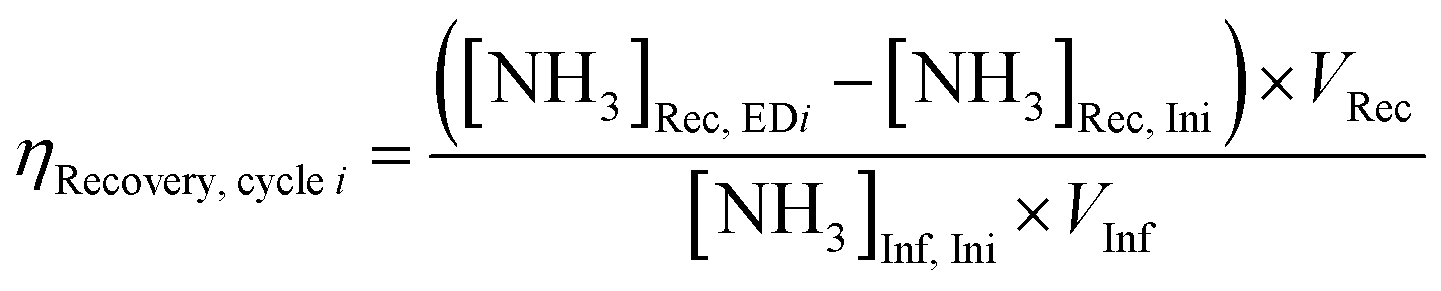 | (1) |
NH3 synthesis efficiency (ηSynthesis):
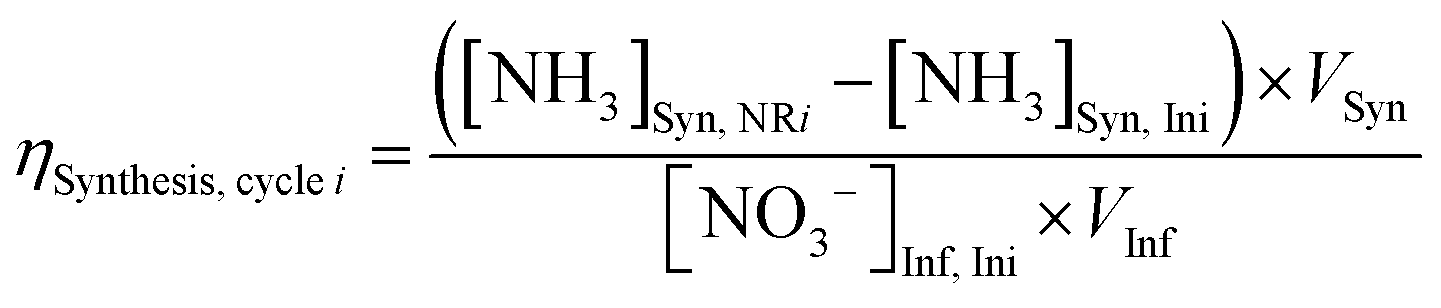 | (2) |
Definitions of the other performance metrics for the ED stage (NH4+ and NO3− ED current efficiency, and NO3− ED flux), NR stage (total current density, NH3 partial current density, time-averaged NR NO3− removal rate, faradaic efficiency), and energy consumption are defined in ESI† Section S1.7. All current densities shown were calculated using the electrode geometric area.
Results and discussion
Proof-of-concept EDNR
As proof-of-concept, we used simulated wastewater with relatively simple compositions and intermediate Nr concentrations between high values used in typical fundamental research (Fig. S7, ESI†) and our target wastewater feedstocks as the EDNR influent (13.9 mM (NH4)2SO4 + 1.6 mM KNO3). During ED stages, influent NH4+ and NO3− were separated into the NH3 recovery and NH3 synthesis chambers, respectively (Fig. 1b), and favorable pH environments were achieved by electrochemical water oxidation and reduction reactions: pH > 9 in the NH3 recovery chamber to recover NH4+ as NH3, and pH < 3 in the NH3 synthesis chamber to prepare for NR. Ti was chosen as a generic NO3RR electrocatalyst because it is selective to NH3, abundant, and corrosion resistant, all of which are suitable characteristics for treating real wastewater.41–43 Ti also exhibits higher nitrate reduction activity in acidic environments, making it well-suited for EDNR.41,43 In the following NR stages, NH3 was synthesized from Ti-catalyzed reduction of the electromigrated NO3−. By repeating ED and NR stages for multiple cycles, we removed increasing amounts of NH4+ and NO3− from the influent. At the end of three EDNR cycles, >70% of influent NH4+ was recovered (defined as NH3 recovery efficiency, ηRecovery, eqn (1)), and 25% of influent NO3− was converted to NH3 (defined as NH3 synthesis efficiency, ηSynthesis, eqn (2)). The total nitrogen balance in the system was also very well closed (−11.2% to +1.3% among all stages, Fig. S8d, ESI†). Although the process functioned as designed, ηSynthesis was consistently lower than ηRecovery, indicating more complete recovery from NH4+ than conversion from NO3− despite the much higher influent NH4+ concentration. The poor ηSynthesis resulted from low NO3RR activity and NH3 selectivity (Fig. S12, ESI†), which necessitates improving the NR process to extract NH3 more completely from wastewater Nr.Engineering of EDNR operating parameters
The EDNR process leverages several key operating parameters (e.g., background electrolyte, applied current/potential, stage duration, electrolyte flow rate) to flexibly adapt to treatment goals. To improve the low ηSynthesis in proof-of-concept experiments, we employed NR electrolyte engineering, which has been shown to substantially influence the activity and selectivity of electrocatalytic reactions44–47 including NO3RR.41,48,49 Rather than directly conducting NO3RR in complex, dynamic decentralized wastewaters, the EDNR reactor separates the NH3 synthesis chamber from the influent using an AEM. This design allows for flexible selection of background electrolyte, as well as conditioning of the NR electrolyte through preceding ED stages. Although high concentration of background electrolyte43 and acidic pH41,43 are known to enhance NO3RR activity and NH3 selectivity on Ti, effects of anion identity and specific optimal pH are not well understood. To address this knowledge gap, we varied the NR background electrolyte anion identity and initial pH in an isolated two-chamber reactor to identify the optimal NR environment (see ESI† Section S1.3). The background electrolyte concentration was fixed as 1 M (cation concentration) to ensure high NO3RR activity,36,43 and the cation identity was fixed as Na+. First, we found that weakly adsorbing ClO4−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,49,50 outperformed other anions commonly used in electrocatalysis studies and present in wastewater (HPO42−, SO42− and Cl−) and exhibited the highest NH3 partial current density (jNH3, eqn (S9), ESI† and Fig. 1c) (see ESI† Section S3.2.1 for detailed discussion). Second, the highest jNH3 occurred in an optimal initial pH around 1.6 (Fig. 1d), above which Ti electrode showed little activity (total current density jtotal < 0.2 mA cm−2, Fig. S10b, ESI†) and below which hydrogen evolution reaction (HER) and Ti hydride formation42 dominated electrode reactions (>50% faradaic efficiency, eqn (S11) and Fig. S10d, ESI†). In addition to electrolyte engineering, we previously found that changing the applied potential pattern from static to pulsed can periodically replenish the local electrolyte acidity and increase the ammonia-to-nitrite selectivity.43 When we applied a pulsed potential to this two-chamber system, jNH3 was successfully doubled (Fig. S11, ESI†).
10,49,50 outperformed other anions commonly used in electrocatalysis studies and present in wastewater (HPO42−, SO42− and Cl−) and exhibited the highest NH3 partial current density (jNH3, eqn (S9), ESI† and Fig. 1c) (see ESI† Section S3.2.1 for detailed discussion). Second, the highest jNH3 occurred in an optimal initial pH around 1.6 (Fig. 1d), above which Ti electrode showed little activity (total current density jtotal < 0.2 mA cm−2, Fig. S10b, ESI†) and below which hydrogen evolution reaction (HER) and Ti hydride formation42 dominated electrode reactions (>50% faradaic efficiency, eqn (S11) and Fig. S10d, ESI†). In addition to electrolyte engineering, we previously found that changing the applied potential pattern from static to pulsed can periodically replenish the local electrolyte acidity and increase the ammonia-to-nitrite selectivity.43 When we applied a pulsed potential to this two-chamber system, jNH3 was successfully doubled (Fig. S11, ESI†).
Therefore, we engineered the following EDNR operating parameters to enhance NR performance (Table 1): (1) chose 1 M NaClO4 as the NH3 synthesis chamber background electrolyte to maximize NH3 partial current density, (2) used a monovalent-selective AEM to limit the disturbance in NR activity from multivalent and strongly adsorbing anions in wastewater, (3) increased ED stage applied current from 2.63 mA cm−2 to 3.95 mA cm−2 to achieve optimal bulk pH before subsequent NR stages, (4) applied pulsed potential (reduction potential of −0.8 V vs. RHE) in NR stages to enhance NH3 selectivity, and (5) increased NR stage electrolyte flow rate to accelerate nitrate removal.43 We conducted triplicate 3-cycle EDNR experiments using the same simulated wastewater (13.9 mM (NH4)2SO4 + 1.61 mM KNO3) as the influent; this set of experiments is referred to as optimized NR in the following text (Fig. 1e and f). Compared to proof-of-concept, we successfully increased the FENH3 by 1.2–2.9 times (to around 20%) and jNH3 by 6–14 times (to −0.6 to −1.2 mA cm−2, Fig. S14, ESI†). Although FENH3 and jNH3 observed in optimized NR were lower than values reported in the NO3RR literature,10 we note that they were achieved in realistically dilute NO3− concentration and could be improved when using higher NO3− feedstocks.41,43 FENH3 and jNH3 remained steady across all cycles, suggesting that a favorable acidic pH in the NH3 synthesis chamber was repeatedly achieved through preceding ED stages. Importantly, optimized NR closed the gap between NH3 synthesis and NH3 recovery by achieving near-unity efficiency for both metrics at the end of 3 cycles (0.84 ± 0.10 for ηRecovery, and 1.11 ± 0.12 for ηSynthesis, Fig. 1g and h).
Next, we examined ED performance after implementing NR reaction environment engineering. High removal was achieved for both NH4+ (87%) and NO3− (84%) at the end of 3 cycles. But unlike the steady NR performance, ED performance decayed as more Nr was removed from the influent: current efficiency for NH4+ dropped from 57% (ED1) to 25% (ED3), and from 4.5% (ED1) to 1.0% (ED3) for NO3− (proportional to ionic flux, Fig. S15a and S16a, ESI†). Such decay in NH4+ and NO3− current efficiency coincided with the decreasing Nr concentrations in the influent chamber, and thus we identified that the ED ionic fluxes were likely controlled by transport from the influent to AEM/CEM, rather than transport across the membranes (see ESI† Section S3.2.2). Within the same cycle, the substantial difference in current efficiency between NH4+ and NO3− was caused by their abundance and conductivity relative to coexisting ions (transference number) in the influent. In the simulated wastewater, NH4+ has an initial transference number of 0.95 and was the major charge-carrying cation across the CEM, whereas NO3− has an initial transference number of 0.025 due to its low concentration and was a minor charge-carrying anion across the AEM (see ESI† Section S2). Compared to proof-of-concept, NO3− ED flux was improved by 0.4–3.7 times in optimized NR, confirming that the monovalent-selective AEM exhibits favorable selectivity towards NO3− (Fig. S15b and S16b, ESI†).
To enhance ED performance, we first tried shortening the ED duration to avoid operating under low transport driving force (low influent Nr concentrations, see ESI† Section S3.2.3). We found that halving the ED duration (i.e., halving the total charge passed) did not significantly impact the current efficiency nor flux for NH4+ and NO3− transport but lowered ηRecovery (Fig. S18, ESI†). The shortened ED duration also led to higher than optimal pH in the NH3 synthesis chamber and consequently impaired ηSynthesis (Fig. S19, ESI†). The adverse effects that shortened ED duration exhibited on NR performance underscore the intimate connection between separation and reaction in EDNR: separation influences subsequent reaction by conditioning the reaction environment. Aside from shortening ED duration, we tried enhancing NH4+ and NO3− transport by increasing the electrolyte flow rate during the shortened ED stages (to the same flow rate as in NR, 100 mL min−1). The higher electrolyte flow rate helped restore a high ηRecovery, but the NH3 synthesis chamber pH was not significantly altered, and ηSynthesis remained low (Fig. S18 and S19, ESI†). Therefore, we concluded that while the combination of shortened ED stage duration and high electrolyte flow rate could generate high ηRecovery, sufficient ED stage duration (charge passed) is critical to achieving the optimal NR reaction environment and associated high ηSynthesis. In the following experiments, operating parameters from optimized NR were applied unless otherwise specified.
Impacts of influent compositions on EDNR performance
As the target feedstocks for EDNR, decentralized wastewaters exhibit a wide range of compositions dependent on the source location and time;11,51–53 however, feedstock composition impacts have rarely been studied in electrochemical Nr conversion and recovery literature. We have demonstrated that NO3RR activity and selectivity is particularly prone to background electrolyte composition and initial pH. To further inform high-TRL EDNR implementation, we systematically studied influent composition effects on the unit process level using increasingly realistic feedstocks. First, we deconvoluted effects of common wastewater components, NO3−, SO42−, and Cl−, by independently increasing their concentration in the simulated wastewater matrix. Then, we moved on to using three real wastewater feedstocks: well water (Stanford, CA), agricultural runoff (Salinas, CA), and reverse osmosis (RO) retentate (from full advanced treatment of municipal wastewater, Silicon Valley Clean Water, Redwood City, CA) (Fig. 2a). In the following discussion, we (1) analyze impacts of each scenario on NH3 synthesis, (2) discuss generalizable implications of each scenario on NH3 recovery, and (3) identify strategies for EDNR to adapt to different feedstock compositions.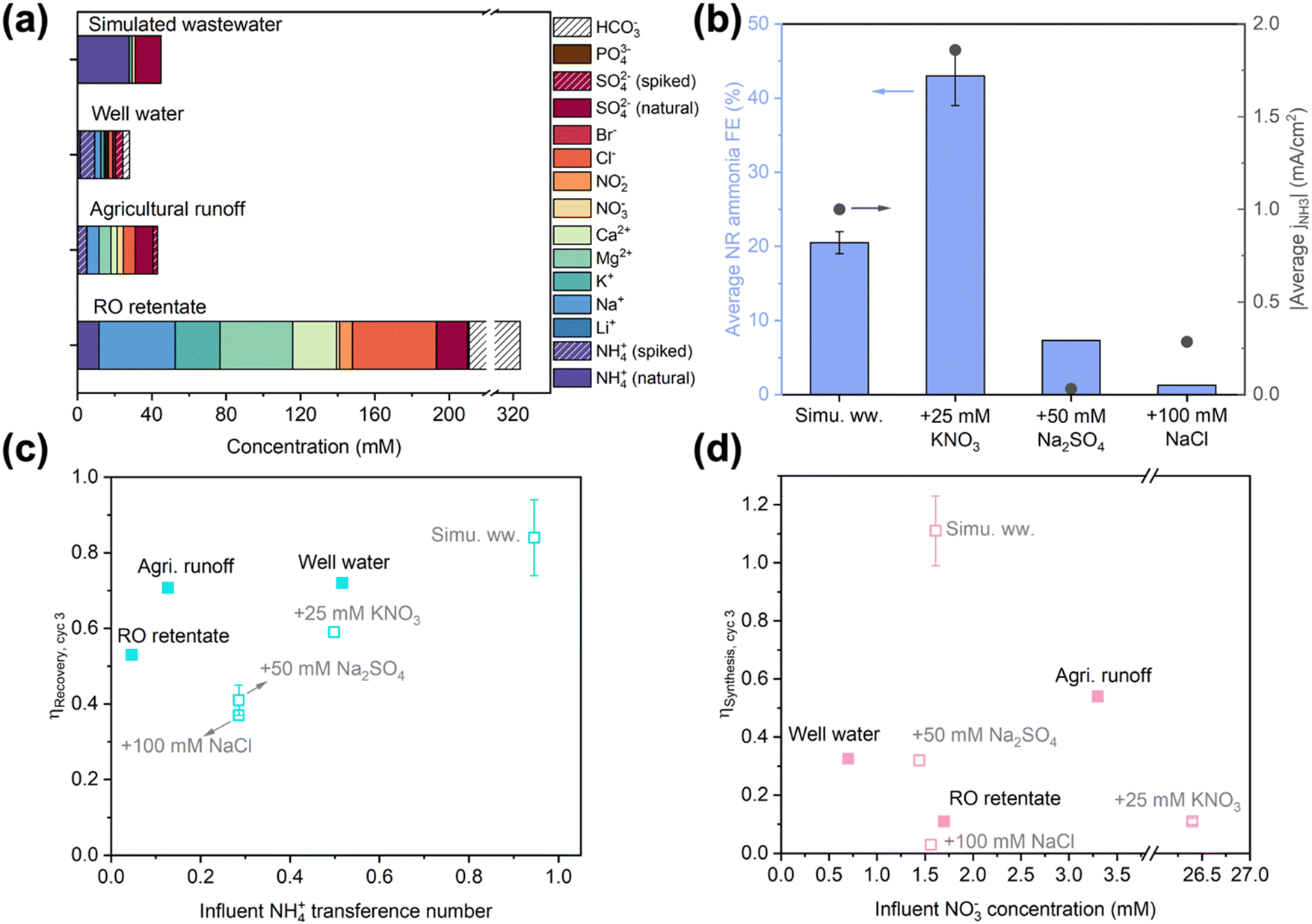 | ||
| Fig. 2 (a) Composition of different wastewaters used as influents in EDNR experiments. Total organic and inorganic carbon contents are shown in Fig. S26 (ESI†). (b) Effects of influent composition on average NH3 faradaic efficiency (left y-axis) and NH3 partial current density (right y-axis) in NR stages. Influents: simulated wastewater (13.9 mM (NH4)2SO4 + 1.6 mM KNO3), and simulated wastewater +25 mM KNO3 (NO3−-laden), or +50 mM Na2SO4 (SO42−-laden), or +100 mM NaCl (Cl−-laden). (c) End-of-run NH3 recovery efficiency as a function of influent NH4+ transference number, and (d) end-of-run NH3 synthesis efficiency as a function of influent NO3− concentration in EDNR experiments using different modified simulated (open symbols) and real (filled symbols) wastewaters. Error bars represent ± one standard deviation. | ||
Beyond NO3−, we introduced SO42− and Cl− because they are the most common divalent and monovalent anions in wastewaters. We added high concentrations to the baseline simulated wastewater (13.9 mM (NH4)2SO4 + 1.6 mM KNO3 + 50 mM Na2SO4 or 100 mM NaCl) as the influent to amplify their effects. Amidst these concentrated coexisting anions, NO3− transference number decreased by an order of magnitude (ESI† Section S2). The NO3− ED flux in both scenarios was lowered significantly in cycle 1, but gradually converged towards optimized NR, leading to similar NO3− concentrations in the NH3 synthesis chamber starting from cycle 2 (Fig. S23a and b, ESI†). The monovalent-selective AEM largely blocked SO42− from entering the NH3 synthesis chamber in the SO42−-laden scenario, and selectively transported Cl− to maintain charge neutrality in the Cl−-laden scenario (Fig. S23c–f, ESI†). As shown in NO3RR electrolyte engineering experiments, additional SO42− and Cl− suppressed FENH3 and lowered jNH3 (Fig. 2b). In the Cl−-laden scenario, the insufficient acidity at the beginning of each NR stage (caused by chlorine evolution reaction during ED, Fig. S24b, ESI†) further impaired NR performance. Consequently, end-of-run ηSynthesis decreased from 1.11 ± 0.12 in baseline simulated wastewater to 0.32 and 0.03 in SO42−-laden and Cl−-laden scenarios, respectively (Fig. 2b and Fig. S22a, ESI†). The sensitivity of ηSynthesis to influent coexisting anions highlights that to improve the adaptability of EDNR to treat a wide range of wastewaters, future efforts should develop NO3−-selective AEMs that enable targeted separation of NO3− from complex influent matrices.
In contrast to the composition-specific ηSynthesis, ηRecovery exhibited a generalizable trend with respect to the initial NH4+ transference number in the influent (Fig. 2c, open symbols). With a higher coexisting cation concentration, the NH4+ ED current efficiency decreased along with its transference number (Fig. S25, ESI†). Experimentally, we observed that the end-of-run ηRecovery nearly monotonically decreased with decreasing NH4+ transference number: 0.84 in baseline simulated wastewater, 0.59 in NO3−-laden, 0.41 in SO42−-laden, and 0.37 in Cl−-laden scenarios. Therefore, to restore nearly complete NH3 recovery in the presence of coexisting cations, we could extend ED stage duration (pass more charge) or increase ED state electrolyte flow rate (intensify the ED mass transport).
For NR performance, end-of-run ηSynthesis in all three wastewaters was far below that of the baseline simulated wastewater and did not correlate with influent NO3− concentration (Fig. 2d). jtotal in RO retentate was similar to in the baseline simulated wastewater, but significantly lower in well water and agricultural runoff (Fig. S29a, ESI†). Based on the influent composition effects observed in modified simulated wastewaters, we attributed the cause of impaired NR to unique compositions of each wastewater. RO retentate contains comparable concentrations of NO3− and SO42− to simulated wastewater, with additional NO2− (6.9 mM), Cl− (45.2 mM), and HCO3− (estimated 113.3 mM). While NO2− could also be reduced and produce NH3 during NR, it was counterbalanced by adverse effects from Cl− and possibly HCO3− (competitive adsorption,56,57 electrochemical deprotonation,58 or electrode surface scaling with divalent cations52), leading to significantly lower jNH3 (ca. 50% of optimized NR). Well water contains about half as much NO3− as simulated wastewater (0.7 vs. 1.6 mM), leading to lower jNH3 (15–33% of optimized NR). In contrast, agricultural runoff contains the highest NO3− concentration (3.3 mM) among the real wastewaters tested and exhibited higher FENH3 (45–63%) and similar jNH3 compared to simulated wastewater (Fig. S29b–d, ESI†). Therefore, to compensate for the coexisting cations and elevated NO3− concentration in agricultural runoff, we increased the number of EDNR operation to 4 cycles and acquired end-of-run ηRecovery (>0.77) and ηSynthesis (>0.70, Fig. S30, ESI†) that approached values in simulated wastewater. Achieving similar efficiencies in real wastewater compared to simulated wastewater shows the significance of EDNR for accelerating wastewater valorization: employing reactive separations based on systematic studies of electrolyte and operating parameters to understand and mitigate the effects of complex wastewater feedstock compositions.
Long-term EDNR and product purification to treat agricultural runoff
Despite being crucial to implementation, long-term studies conducted under realistic operating conditions are rarely reported for electrochemical Nr recovery processes.53 Similarly, energy consumption is not always reported in the literature but highly desired by practitioners.59 Thus, we examined the long-term stability and energy consumption of the EDNR unit process in treating real wastewater. We selected NH4+-enriched agricultural runoff (2.4 mM (NH4)2SO4 was manually added to agricultural runoff) as the target feedstock because among the wastewaters we tested, it exhibits moderate Nr concentration, moderate total ionic concentration, and diverse ionic species. Applying operating parameters slightly altered from optimized NR (detailed in ESI† Section S3.4), we conducted 5 batches of 4-cycle EDNR experiments that processed 50 mL of fresh influent per batch (i.e., every 4 EDNR cycles). To demonstrate generation of pure wastewater-derived NH3 products, we coupled the EDNR process with membrane stripping and formed a near-neutral ammonia phosphate fertilizer solution. The membrane stripping process also enabled reuse of electrolytes in NH3 recovery and synthesis chambers, with only minimal fresh electrolyte addition to offset loss from sampling (8 mL per batch). The integrated process was conducted for 5 consecutive days and processed a total of 250 mL NH4+-enriched agricultural runoff (experimental protocols in ESI† Section S1.4).The EDNR process demonstrated exceptional long-term robustness. Despite the complex composition of agricultural runoff, end-of-batch NH3 recovery and synthesis efficiencies approached values achieved in baseline simulated wastewater (0.77 ± 0.11 for ηRecovery, 0.66 ± 0.10 for ηSynthesis) and did not show appreciable decay with extended operation (Fig. 3a and b, except for batch 3). NH4+ and NO3− ED current efficiencies remained steady over time and unimpaired by observed membrane fouling (Fig. S31 and S34 and Table S6, ESI†), corroborating our conclusion that Nr ionic fluxes were controlled by transport from the influent to membranes. The steady ED performance repeatedly created favorable electrolyte environments for NR, as evidenced by nearly overlapping trends of pH and Nr ion movements across all batches (Fig. S35–S37, ESI†). High activity and selectivity were maintained during NR (Fig. S39, ESI†), with total current density at ca. 2 mA cm−2 and FENH3 > 40% across all batches. Within each batch, the FENH3 in cycle 4 (final cycle) was the lowest due to the low NO3− concentrations and loss of volatile NH3 from the alkaline electrolyte. Starting from the second batch, FENH3 in cycle 1–3 increased to >60%. In contrast to the more commonly observed loss in electrode activity and selectivity over time, the Ti electrode exhibited an ‘activated’ NH3 selectivity induced by the first batch of EDNR (8 h total in NR) and overnight air exposure (10 h exposed in an empty cell open to air). Based on our previous study, the near-surface of the Ti electrode in contact with the electrolyte likely converted to TiH2 after the first EDNR batch; however, TiH2 exhibits similar nitrate reduction activity and selectivity to unamended Ti.42 Therefore, we hypothesized that increased NH3 selectivity arose from altered surface morphology60–62 or partially oxidized TiH2/Ti.63–65 To summarize long-term performance, ED and NR stages showed excellent resilience to real wastewater over extended operation, achieving stable, high NH3 recovery and synthesis that enable future scale-up.
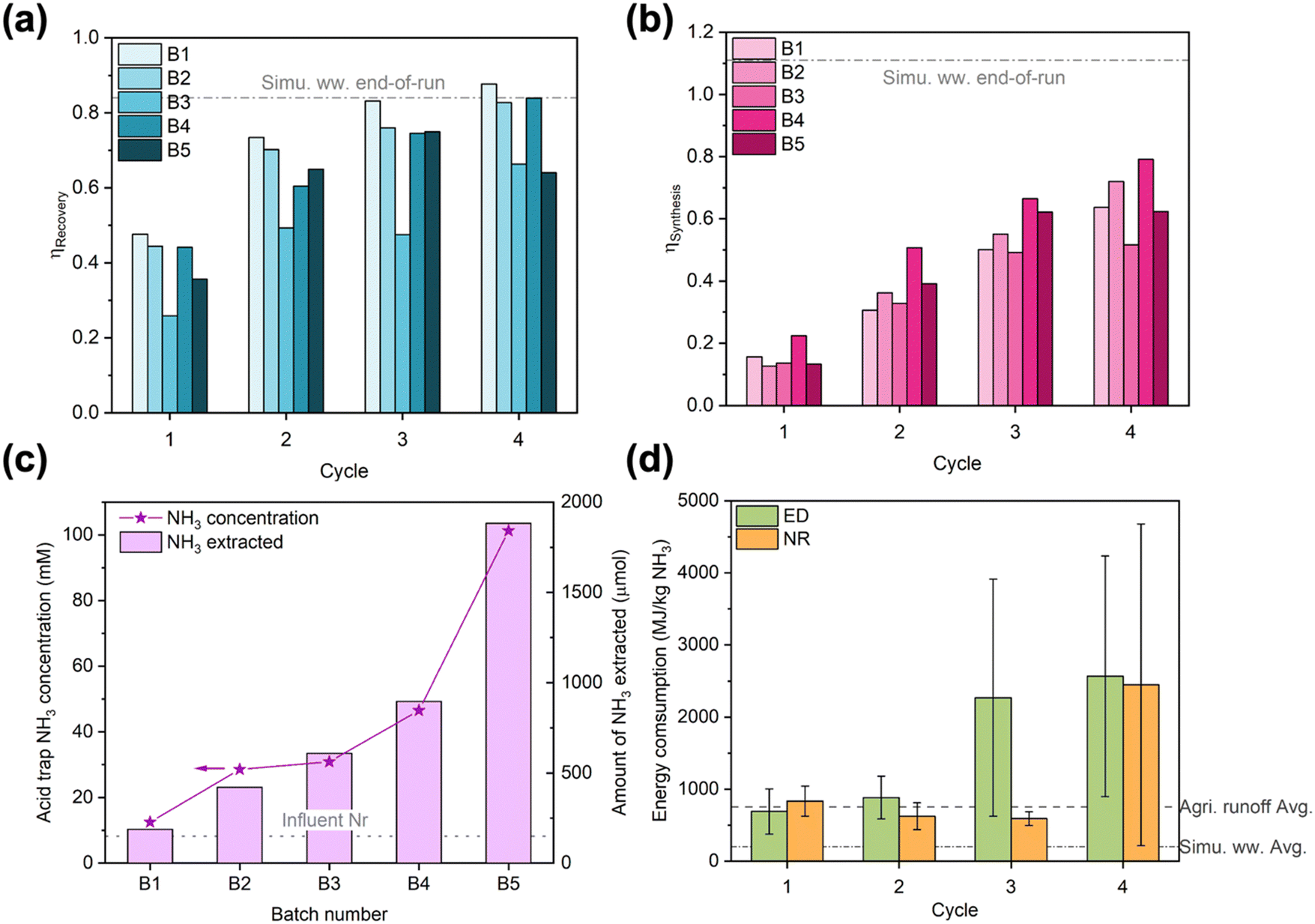 | ||
| Fig. 3 (a) NH3 recovery efficiency and (b) NH3 synthesis efficiency in long-term EDNR experiments using NH4+-enriched agricultural runoff. The few instances where efficiencies decreased with increasing cycle number in B3 and B5 were caused by decreasing NH3 concentration in corresponding chambers, possibly due to NH3 evaporation. Dash-dot lines represent the average end-of-run NH3 recovery and synthesis efficiencies in simulated wastewater feedstock. (c) Concentration (left y-axis) and total amount (right y-axis) of NH3 extracted into the acid trap through membrane stripping. Dotted line represents total Nr concentration in NH4+-enriched agricultural runoff influent. (d) Energy consumption in NH3 recovery (ESI† eqn (S7)) and synthesis (ESI† eqn (S12)). Large error bars in cycle 3 and cycle 4 resulted from negative NH3 recovery/synthesis in B3 and B5. Because pumping energy typically contributes minimally to the overall energy consumption for electrochemical wastewater treatment processes(<5%33,36), we based our calculations solely on electrical energy consumed in the EDNR process. Error bars represent ± one standard deviation. Dash and dash-dot lines represent the average energy consumption for NH3 production (ESI† eqn (S13)) in NH4+-enriched agricultural runoff and simulated wastewater feedstocks, respectively. | ||
To extract and concentrate EDNR-recovered and synthesized NH3 from background electrolytes, we combined EDNR with a low-energy passive separation process, membrane stripping, to recover a single NH3 product stream. After 5 batches, the acid trap chamber recovered 101 mM NH3 as a mixture of NH4H2PO4 and (NH4)2HPO4 (pH 6.42), which contained minimal metal contamination (Na, Mg, K, Ca, Cu below 10 ppb on inductively coupled plasma optical emission spectroscopy) and can be directly applied as a fertilizer (mono- and di-ammonium phosphate, MAP and DAP fertilizers, typical application concentration 43–454 mM NH4+).66,67 Importantly, this combined solution was 12.3 times more concentrated than the influent (8.2 mM total Nr, Fig. 3c). Note that this up-concentration factor can be further increased by using (1) a higher ratio of influent to NH3 synthesis/recovery chamber background electrolyte volume, (2) a higher ratio of NH3 synthesis/recovery chamber background electrolyte to acid trap volume, or (3) more batches of EDNR operation. From 250 mL agricultural runoff that contains dilute and unusable level of Nr, we recovered a concentrated fertilizer solution that can serve 50 cm2 of vegetative stage crops, highlighting the suitability of EDNR for decentralized nutrient recovery. We note that Na+ and ClO4− migrated from electrolytes in NH3 synthesis and recovery chambers into the wastewater influent (ESI† Table S7). Although there is no maximum contaminant level set by the US Environmental Protection Agency (EPA) for Na+, the ClO4− concentration exceeded its maximum contaminant level (5.6 × 10−4 mM). Considering that organic pollutants are also not treated by the EDNR process, downstream treatment steps for other pollutants might be needed to supplement the nitrogen removal and recovery of EDNR. For municipal wastewater, EDNR can be implemented as part of the treatment train (e.g., extract residual dilute Nr after electrochemical stripping using urine feedstock53); for agricultural runoff, which is not currently collected and treated, EDNR can become the first step in the treatment train that includes further polishing with established technologies ClO4− (e.g., ion exchange combined with biodegradation68) and organic contaminants (e.g., advanced oxidation processes using UV/H2O269).
To inform distributed NH3 manufacturing from wastewaters, we evaluated EDNR energy consumption and identified opportunities for future improvements (Fig. 3d). In the first two cycles, ED and NR stages consumed similar amounts of energy per kg NH3 produced. Starting from cycle 3, much more energy was consumed in ED to recover the marginal amount of residual influent NH4+ due to the significantly lower current efficiency (Fig. S34a, ESI†). NR energy consumption only increased in the last cycle due to the low FENH3 (Fig. S39b, ESI†). Accounting for both ED and NR stages, the average energy consumption using the NH4+-enriched agricultural runoff was 920 MJ per kg N. In comparison, the average energy consumption in simulated wastewater was 245 MJ per kg N (Fig. S40a, ESI†). We attributed the 3.75 times higher energy consumption in real wastewater to its significantly lower NH4+ concentration (5 times lower) and CEM scaling caused by divalent cations (leading to higher cell voltage, Fig. S40b, ESI†). These energy consumption values are among those for state-of-the-art electrochemical NH3 manufacturing technologies using similarly dilute Nr feedstocks (Table S8, ESI†). But distinct from most literature reports, the feedstock used in this work was a complex real wastewater with dilute Nr (vs. simplistic electrolytes with concentrated Nr), and a purified product stream was recovered with very low energy input (vs. products not separated from the influent or requiring downstream energy/chemical-intensive separation). Although the EDNR energy consumption is several times higher than traditional wastewater Nr removal (e.g., nitrification/denitrification, anammox; 10–100 MJ per kg N)13,70 and NH3 manufacturing technologies (e.g., Haber–Bosch, 31.6 MJ per kg N),53 this electrochemical reactive separation unit process enables highly tunable and robust wastewater refining at the point of wastewater generation. Future work can reduce the energy consumption by: (1) reducing the number of cycles to avoid operating in low mass transport driving force regions, when near-complete removal and recovery are not required; (2) employing more active ED (e.g., finer IrO2–Ta2O5/Ti mesh with higher surface area, nickel-stabilized ruthenium dioxide,71 barium doped cobalt(II,III) oxide72) and NR (e.g., polypyrrole-protected Cu nanoparticles,73 Fe2Co metal–organic framework,74 FeAu alloy75) electrode materials to lower overpotential; and (3) adding antifoulants or other mitigation strategies into the NH3 recovery chamber to prevent CEM fouling.
Conclusions
This study demonstrated that EDNR is a highly tunable and robust reaction separation process to recover and synthesize NH3 from dilute, Nr-polluted wastewaters. We found that engineering the NR reaction environment via electrochemical separations (electrolyte compositions and applied potential) plays a crucial role in improving electrocatalytic NH3 synthesis. In wastewater feedstocks, NH4+ transference number largely determines the NH3 recovery efficiency, while NO3− concentration as well as coexisting anion identity and concentration together influence the NH3 synthesis efficiency. Due to their complex compositions, real wastewaters tested in this study generally exhibited lower efficiency and higher energy consumption compared to simulated wastewater. Demonstrated using generic electrode and membrane materials here, the EDNR reactor can be used as a platform to benchmark high-performance materials tailored to feedstock conditions. Development of more active NR electrodes, monovalent-selective CEM, NO3−-selective AEM, and engineering strategies will advance the EDNR process to become more energy-efficient and compatible with an even wider range of feedstocks. Shown as a prototype here, EDNR can remediate impaired feedstocks and valorize Nr pollutants in a distributed manner. The process has great viability in scenarios not served by conventional manufacturing (farms, remote communities), and regions like sub-Saharan Africa,76 where limited access to centralized infrastructure and raw chemical inputs inhibits access to clean water and fertilizer. These issues of scale and access extend beyond the context of Nr recovery, which underscores the potential utility of EDNR as a modular architecture that enables wastewater refining by leveraging reactive separation and valorization of other ionic pollutants in wastewater (e.g., sulfide oxidation, sulfate reduction). By enabling distributed ammonia manufacturing, EDNR lies at the intersection of the food-energy-environment nexus, especially because it uses electricity to generate ammonia fertilizers and fuels while reducing waterborne discharges, circularizing the nitrogen cycle, and sustaining chemical manufacturing for future generations. Future efforts will advance towards this vision by assessing the technoeconomic viability of the EDNR process and advancing the scale and TRL of the process for realistic scenarios.Data availability
Data for this article, including raw current/potential, aqueous chemical concentrations, and calculated performance metrics, are available in the Stanford Data Repository (submitted, DOI forthcoming and will be available during review process).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to several funders of this work. J. G. acknowledges the National Science Foundation EFRI program (Award 2132007) and the Chemical Engineering Department at Stanford University. M. J. L. acknowledges support from the National Aeronautics and Space Administration (NASA) Space Technology Graduate Research Opportunities fellowship (Award 80NSSC20K1207) and Northern California Chapter of the ARCS Foundation (Rhoda Goldman Memorial Scholarship). D. M. M. acknowledges support from the National Aeronautics and Space Administration (NASA) Space Technology Graduate Research Opportunities fellowship (Award 80NSSC22K1191). K. S. W. acknowledges the Sustainability Accelerator within the Doerr School of Sustainability at Stanford University. SEM imaging and EDS mapping were performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-2026822. The authors thank Silicon Valley Clean Water for providing wastewater samples; Ouriel Ndalamba and Kristy Chan for support on conducting experiments; and the Tarpeh, Jaramillo, Maher, Mauter, and Lobell groups for critical feedback on the project. This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, Catalysis Science Program to the SUNCAT Center for Interface Science and Catalysis.References
- Committee on Science for EPA's Future, Board on Environmental Studies and Toxicology, Division on Earth and Life Studies, National Research Council, Science for Environmental Protection: The Road Ahead, National Academies Press, Washington, D.C., 2012 DOI:10.17226/13510.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, A Roadmap to the Ammonia Economy, Joule, 2020, 4(6), 1186–1205, DOI:10.1016/j.joule.2020.04.004.
- J. Lim, C. A. Fernández, S. W. Lee and M. C. Hatzell, Ammonia and Nitric Acid Demands for Fertilizer Use in 2050, ACS Energy Lett., 2021, 6(10), 3676–3685, DOI:10.1021/acsenergylett.1c01614.
- J. N. Galloway and E. B. Cowling, Reactive Nitrogen and The World: 200 Years of Change, ambi, 2002, 31(2), 64–71, DOI:10.1579/0044-7447-31.2.64.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Beyond Fossil Fuel–Driven Nitrogen Transformations, Science, 2018, 360(6391), eaar6611, DOI:10.1126/science.aar6611.
- W. Steffen, K. Richardson, J. Rockström, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpenter, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sörlin, Planetary Boundaries: Guiding Human Development on a Changing Planet, Science, 2015, 347(6223), 1259855, DOI:10.1126/science.1259855.
- B. L. Bodirsky, A. Popp, H. Lotze-Campen, J. P. Dietrich, S. Rolinski, I. Weindl, C. Schmitz, C. Müller, M. Bonsch, F. Humpenöder, A. Biewald and M. Stevanovic, Reactive Nitrogen Requirements to Feed the World in 2050 and Potential to Mitigate Nitrogen Pollution, Nat. Commun., 2014, 5(1), 3858, DOI:10.1038/ncomms4858.
- D. M. Miller, K. Abels, J. Guo, K. S. Williams, M. J. Liu and W. A. Tarpeh, Electrochemical Wastewater Refining: A Vision for Circular Chemical Manufacturing, J. Am. Chem. Soc., 2023, 145(36), 19422–19439, DOI:10.1021/jacs.3c01142.
- M. Yang, L. Chen, J. Wang, G. Msigwa, A. I. Osman, S. Fawzy, D. W. Rooney and P.-S. Yap, Circular Economy Strategies for Combating Climate Change and Other Environmental Issues, Environ. Chem. Lett., 2023, 21(1), 55–80, DOI:10.1007/s10311-022-01499-6.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Electrocatalytic Reduction of Nitrate: Fundamentals to Full-Scale Water Treatment Applications, Appl. Catal., B, 2018, 236, 546–568, DOI:10.1016/j.apcatb.2018.05.041.
- B. Hansen, L. Thorling, J. Schullehner, M. Termansen and T. Dalgaard, Groundwater Nitrate Response to Sustainable Nitrogen Management, Sci. Rep., 2017, 7(1), 8566, DOI:10.1038/s41598-017-07147-2.
- E. Abascal, L. Gómez-Coma, I. Ortiz and A. Ortiz, Global Diagnosis of Nitrate Pollution in Groundwater and Review of Removal Technologies, Sci. Total Environ., 2022, 810, 152233, DOI:10.1016/j.scitotenv.2021.152233.
- J. Theerthagiri, J. Park, H. T. Das, N. Rahamathulla, E. S. F. Cardoso, A. P. Murthy, G. Maia, D. N. Vo and M. Y. Choi, Electrocatalytic Conversion of Nitrate Waste into Ammonia: A Review, Environ. Chem. Lett., 2022, 20(5), 2929–2949, DOI:10.1007/s10311-022-01469-y.
- V. A. Niemann, P. Benedek, J. Guo, Y. Xu, S. J. Blair, E. R. Corson, A. C. Nielander, T. F. Jaramillo and W. A. Tarpeh, Co-Designing Electrocatalytic Systems with Separations To Improve the Sustainability of Reactive Nitrogen Management, ACS Catal., 2023, 13(9), 6268–6279, DOI:10.1021/acscatal.3c00933.
- S. Meng, Y. Ling, M. Yang, X. Zhao, A. I. Osman, A. H. Al-Muhtaseb, D. W. Rooney and P.-S. Yap, Recent Research Progress of Electrocatalytic Reduction Technology for Nitrate Wastewater: A Review, J. Environ. Chem. Eng., 2023, 11(2), 109418, DOI:10.1016/j.jece.2023.109418.
- W. A. Tarpeh and X. Chen, Making Wastewater Obsolete: Selective Separations to Enable Circular Water Treatment, Environ. Sci. Ecotechnology, 2021, 5, 100078, DOI:10.1016/j.ese.2021.100078.
- A. M. Zito, L. E. Clarke, J. M. Barlow, D. Bím, Z. Zhang, K. M. Ripley, C. J. Li, A. Kummeth, M. E. Leonard, A. N. Alexandrova, F. R. Brushett and J. Y. Yang, Electrochemical Carbon Dioxide Capture and Concentration, Chem. Rev., 2023, 123(13), 8069–8098, DOI:10.1021/acs.chemrev.2c00681.
- M. C. Freyman, Z. Huang, D. Ravikumar, E. B. Duoss, Y. Li, S. E. Baker, S. H. Pang and J. A. Schaidle, Reactive CO2 Capture: A Path Forward for Process Integration in Carbon Management, Joule, 2023, 7(4), 631–651, DOI:10.1016/j.joule.2023.03.013.
- R. E. Siegel, S. Pattanayak and L. A. Berben, Reactive Capture of CO2: Opportunities and Challenges, ACS Catal., 2023, 13(1), 766–784, DOI:10.1021/acscatal.2c05019.
- G. Gadikota, Multiphase Carbon Mineralization for the Reactive Separation of CO2 and Directed Synthesis of H2, Nat. Rev. Chem., 2020, 4(2), 78–89, DOI:10.1038/s41570-019-0158-3.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Direct Capture of CO2 from Ambient Air, Chem. Rev., 2016, 116(19), 11840–11876, DOI:10.1021/acs.chemrev.6b00173.
- P. K. Dutta, K. Rabaey, Z. Yuan, R. A. Rozendal and J. Keller, Electrochemical Sulfide Removal and Recovery from Paper Mill Anaerobic Treatment Effluent, Water Res., 2010, 44(8), 2563–2571, DOI:10.1016/j.watres.2010.01.008.
- K. Jangam, Y.-Y. Chen, L. Qin and L.-S. Fan, Perspectives on Reactive Separation and Removal of Hydrogen Sulfide, Chem. Eng. Sci.: X, 2021, 11, 100105, DOI:10.1016/j.cesx.2021.100105.
- E. Blázquez, D. Gabriel, J. A. Baeza, A. Guisasola, S. Freguia and P. Ledezma, Recovery of Elemental Sulfur with a Novel Integrated Bioelectrochemical System with an Electrochemical Cell, Sci. Total Environ., 2019, 677, 175–183, DOI:10.1016/j.scitotenv.2019.04.406.
- L.-F. Zhai, W. Song, Z.-H. Tong and M. Sun, A Fuel-Cell-Assisted Iron Redox Process for Simultaneous Sulfur Recovery and Electricity Production from Synthetic Sulfide Wastewater, J. Hazard. Mater., 2012, 243, 350–356, DOI:10.1016/j.jhazmat.2012.10.046.
- N. Sergienko and J. Radjenovic, Manganese Oxide-Based Porous Electrodes for Rapid and Selective (Electro)Catalytic Removal and Recovery of Sulfide from Wastewater, Appl. Catal., B, 2020, 267, 118608, DOI:10.1016/j.apcatb.2020.118608.
- L. Wu, C. Zhang, S. Kim, T. A. Hatton, H. Mo and T. D. Waite, Lithium Recovery Using Electrochemical Technologies: Advances and Challenges, Water Res., 2022, 221, 118822, DOI:10.1016/j.watres.2022.118822.
- G. Luo, X. Li, L. Chen, Y. Chao and W. Zhu, Electrochemical Lithium Ion Pumps for Lithium Recovery: A Systematic Review and Influencing Factors Analysis, Desalination, 2023, 548, 116228, DOI:10.1016/j.desal.2022.116228.
- G. Liu, Z. Zhao and L. He, Highly Selective Lithium Recovery from High Mg/Li Ratio Brines, Desalination, 2020, 474, 114185, DOI:10.1016/j.desal.2019.114185.
- S. Yang, F. Zhang, H. Ding, P. He and H. Zhou, Lithium Metal Extraction from Seawater, Joule, 2018, 2(9), 1648–1651, DOI:10.1016/j.joule.2018.07.006.
- J. Yu, D. Fang, H. Zhang, Z. Y. Leong, J. Zhang, X. Li and H. Y. Yang, Ocean Mining: A Fluidic Electrochemical Route for Lithium Extraction from Seawater, ACS Materials Lett., 2020, 2(12), 1662–1668, DOI:10.1021/acsmaterialslett.0c00385.
- Z. Zhao, G. Liu, H. Jia and L. He, Sandwiched Liquid-Membrane Electrodialysis: Lithium Selective Recovery from Salt Lake Brines with High Mg/Li Ratio, J. Membr. Sci., 2020, 596, 117685, DOI:10.1016/j.memsci.2019.117685.
- W. A. Tarpeh, J. M. Barazesh, T. Y. Cath and K. L. Nelson, Electrochemical Stripping to Recover Nitrogen from Source-Separated Urine, Environ. Sci. Technol., 2018, 52(3), 1453–1460, DOI:10.1021/acs.est.7b05488.
- F. Ferrari, M. Pijuan, S. Molenaar, N. Duinslaeger, T. Sleutels, P. Kuntke and J. Radjenovic, Ammonia Recovery from Anaerobic Digester Centrate Using Onsite Pilot Scale Bipolar Membrane Electrodialysis Coupled to Membrane Stripping, Water Res., 2022, 218, 118504, DOI:10.1016/j.watres.2022.118504.
- H. Dong, C. M. Laguna, M. J. Liu, J. Guo and W. A. Tarpeh, Electrified Ion Exchange Enabled by Water Dissociation in Bipolar Membranes for Nitrogen Recovery from Source-Separated Urine, Environ. Sci. Technol., 2022, 56(22), 16134–16143, DOI:10.1021/acs.est.2c03771.
- J. Sun, S. Garg and T. D. Waite, A Novel Integrated Flow-Electrode Capacitive Deionization and Flow Cathode System for Nitrate Removal and Ammonia Generation from Simulated Groundwater, Environ. Sci. Technol., 2023, 57(39), 14726–14736, DOI:10.1021/acs.est.3c03922.
- K. Kim, A. Zagalskaya, J. L. Ng, J. Hong, V. Alexandrov, T. A. Pham and X. Su, Coupling Nitrate Capture with Ammonia Production through Bifunctional Redox-Electrodes, Nat. Commun., 2023, 14(1), 823, DOI:10.1038/s41467-023-36318-1.
- J. Gao, N. Shi, Y. Li, B. Jiang, T. Marhaba and W. Zhang, Electrocatalytic Upcycling of Nitrate Wastewater into an Ammonia Fertilizer via an Electrified Membrane, Environ. Sci. Technol., 2022, 56(16), 11602–11613, DOI:10.1021/acs.est.1c08442.
- M. J. Liu, D. M. Miller and W. A. Tarpeh, Reactive Separation of Ammonia from Wastewater Nitrate via Molecular Electrocatalysis, Environ. Sci. Technol. Lett., 2023, 10(5), 458–463, DOI:10.1021/acs.estlett.3c00205.
- Technology Readiness Assessment (TRA) Guidance. https://apps.dtic.mil/sti/citations/ADA554900, (accessed 2024-06-08).
- J. M. McEnaney, S. J. Blair, A. C. Nielander, J. A. Schwalbe, D. M. Koshy, M. Cargnello and T. F. Jaramillo, Electrolyte Engineering for Efficient Electrochemical Nitrate Reduction to Ammonia on a Titanium Electrode, ACS Sustainable Chem. Eng., 2020, 8(7), 2672–2681, DOI:10.1021/acssuschemeng.9b05983.
- M. J. Liu, J. Guo, A. S. Hoffman, J. H. Stenlid, M. T. Tang, E. R. Corson, K. H. Stone, F. Abild-Pedersen, S. R. Bare and W. A. Tarpeh, Catalytic Performance and Near-Surface X-Ray Characterization of Titanium Hydride Electrodes for the Electrochemical Nitrate Reduction Reaction, J. Am. Chem. Soc., 2022, 144(13), 5739–5744, DOI:10.1021/jacs.2c01274.
- J. Guo, P. Brimley, M. J. Liu, E. R. Corson, C. Muñoz, W. A. Smith and W. A. Tarpeh, Mass Transport Modifies the Interfacial Electrolyte to Influence Electrochemical Nitrate Reduction, ACS Sustainable Chem. Eng., 2023, 11(20), 7882–7893, DOI:10.1021/acssuschemeng.3c01057.
- B. Deng, M. Huang, X. Zhao, S. Mou and F. Dong, Interfacial Electrolyte Effects on Electrocatalytic CO2 Reduction, ACS Catal., 2022, 12(1), 331–362, DOI:10.1021/acscatal.1c03501.
- J. C. Bui, C. Kim, A. J. King, O. Romiluyi, A. Kusoglu, A. Z. Weber and A. T. Bell, Engineering Catalyst–Electrolyte Microenvironments to Optimize the Activity and Selectivity for the Electrochemical Reduction of CO2 on Cu and Ag, Acc. Chem. Res., 2022, 55(4), 484–494, DOI:10.1021/acs.accounts.1c00650.
- B. N. Ruggiero, K. M. Sanroman Gutierrez, J. D. George, N. M. Mangan, J. M. Notestein and L. C. Seitz, Probing the Relationship between Bulk and Local Environments to Understand Impacts on Electrocatalytic Oxygen Reduction Reaction, J. Catal., 2022, 414, 33–43, DOI:10.1016/j.jcat.2022.08.025.
- G. Kastlunger, L. Wang, N. Govindarajan, H. H. Heenen, S. Ringe, T. Jaramillo, C. Hahn and K. Chan, Using pH Dependence for Understanding Mechanisms in Electrochemical CO Reduction, ACS Catal., 2022, 12(8), 4344–4357, DOI:10.1021/acscatal.1c05520.
- Q. Hu, K. Yang, O. Peng, M. Li, L. Ma, S. Huang, Y. Du, Z.-X. Xu, Q. Wang, Z. Chen, M. Yang and K. P. Loh, Ammonia Electrosynthesis from Nitrate Using a Ruthenium–Copper Cocatalyst System: A Full Concentration Range Study, J. Am. Chem. Soc., 2023, 146(1), 668–676, DOI:10.1021/jacs.3c10516.
- G. E. Dima, A. C. A. de Vooys and M. T. M. Koper, Electrocatalytic Reduction of Nitrate at Low Concentration on Coinage and Transition-Metal Electrodes in Acid Solutions, J. Electroanal. Chem., 2003, 554–555, 15–23, DOI:10.1016/S0022-0728(02)01443-2.
- G. Horányi and E. M. Rizmayer, Role of Adsorption Phenomena in the Electrocatalytic Reduction of Nitric Acid at a Platinized Platinum Electrode, J. Electroanal. Chem. Interfacial Electrochem., 1982, 140(2), 347–366, DOI:10.1016/0022-0728(82)85178-4.
- M. A. Alkhadra, X. Su, M. E. Suss, H. Tian, E. N. Guyes, A. N. Shocron, K. M. Conforti, J. P. de Souza, N. Kim, M. Tedesco, K. Khoiruddin, I. G. Wenten, J. G. Santiago, T. A. Hatton and M. Z. Bazant, Electrochemical Methods for Water Purification, Ion Separations, and Energy Conversion, Chem. Rev., 2022, 122(16), 13547–13635, DOI:10.1021/acs.chemrev.1c00396.
- A. Atrashkevich, A. S. Fajardo, P. Westerhoff, W. S. Walker, C. M. Sánchez-Sánchez and S. Garcia-Segura, Overcoming Barriers for Nitrate Electrochemical Reduction: By-Passing Water Hardness, Water Res., 2022, 225, 119118, DOI:10.1016/j.watres.2022.119118.
- A. Kogler, N. Sharma, D. Tiburcio, M. Gong, D. M. Miller, K. S. Williams, X. Chen and W. A. Tarpeh, Long-Term Robustness and Failure Mechanisms of Electrochemical Stripping for Wastewater Ammonia Recovery, ACS Environ. Au, 2024, 4(2), 89–105, DOI:10.1021/acsenvironau.3c00058.
- O. Q. Carvalho, R. Marks, H. K. K. Nguyen, M. E. Vitale-Sullivan, S. C. Martinez, L. Árnadóttir and K. A. Stoerzinger, Role of Electronic Structure on Nitrate Reduction to Ammonium: A Periodic Journey, J. Am. Chem. Soc., 2022, 144(32), 14809–14818, DOI:10.1021/jacs.2c05673.
- W. de Vries, J. Kros, J. C. Voogd and G. H. Ros, Integrated Assessment of Agricultural Practices on Large Scale Losses of Ammonia, Greenhouse Gases, Nutrients and Heavy Metals to Air and Water, Sci. Total Environ., 2023, 857, 159220, DOI:10.1016/j.scitotenv.2022.159220.
- J. Makover, D. Hasson, R. Semiat and H. Shemer, Electrochemical Removal of Nitrate from High Salinity Waste Stream in a Continuous Flow Reactor, J. Environ. Chem. Eng., 2020, 8(3), 103727, DOI:10.1016/j.jece.2020.103727.
- W. Huang, M. Li, B. Zhang, C. Feng, X. Lei and B. Xu, Influence of Operating Conditions on Electrochemical Reduction of Nitrate in Groundwater, Water Environ. Res., 2013, 85(3), 224–231, DOI:10.2175/106143012X13418552642047.
- G. Marcandalli, K. Boterman and M. T. M. Koper, Understanding Hydrogen Evolution Reaction in Bicarbonate Buffer, J. Catal., 2022, 405, 346–354, DOI:10.1016/j.jcat.2021.12.012.
- A. Kogler, M. Farmer, J. A. Simon, S. Tilmans, G. F. Wells and W. A. Tarpeh, Systematic Evaluation of Emerging Wastewater Nutrient Removal and Recovery Technologies to Inform Practice and Advance Resource Efficiency, ACS EST Eng., 2021, 1(4), 662–684, DOI:10.1021/acsestengg.0c00253.
- P. M. Krzywda, A. P. Rodríguez, L. Cino, N. E. Benes, B. T. Mei and G. Mul, Electroreduction of NO3− on Tubular Porous Ti Electrodes, Catal. Sci. Technol., 2022, 12(10), 3281–3288, 10.1039/D2CY00289B.
- J. Lim, C.-Y. Liu, J. Park, Y.-H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, Structure Sensitivity of Pd Facets for Enhanced Electrochemical Nitrate Reduction to Ammonia, ACS Catal., 2021, 11(12), 7568–7577, DOI:10.1021/acscatal.1c01413.
- J. F. Su, W.-F. Kuan, H. Liu and C. P. Huang, Mode of Electrochemical Deposition on the Structure and Morphology of Bimetallic Electrodes and Its Effect on Nitrate Reduction toward Nitrogen Selectivity, Appl. Catal., B, 2019, 257, 117909, DOI:10.1016/j.apcatb.2019.117909.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, Boosting Selective Nitrate Electroreduction to Ammonium by Constructing Oxygen Vacancies in TiO2, ACS Catal., 2020, 10(6), 3533–3540, DOI:10.1021/acscatal.9b05260.
- Insight into Hydrogenation Selectivity of the Electrocatalytic Nitrate-to-Ammonia Reduction Reaction via Enhancing the Proton Transport – Xu – 2022 – ChemSusChem - Wiley Online Library. https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cssc.202102450, (accessed 2024-01-30).
- Y. Chen, C. Yang, H. Li, Z. Ma, D. Wu, Y. Yao, X. Shen and D. Ma, Ti3+ Redox Dynamics Enabling Efficient Nitrate Reduction to Ammonia on Ti2O3, Chem. Eng. J., 2024, 481, 148857, DOI:10.1016/j.cej.2024.148857.
- soluMAP® - K + S Aktiengesellschaft. https://www.kpluss.com/en-us/our-business-products/agriculture/products/en-solumap/, (accessed 2024-01-31).
- Md. A. H. Chowdhury, T. Sultana, Md. A. Rahman, T. Chowdhury, C. E. Enyoh, B. K. Saha and W. Qingyue, Nitrogen Use Efficiency and Critical Leaf N Concentration of Aloe Vera in Urea and Diammonium Phosphate Amended Soil, Heliyon, 2020, 6(12), e05718, DOI:10.1016/j.heliyon.2020.e05718.
- L. Ye, H. You, J. Yao and H. Su, Water Treatment Technologies for Perchlorate: A Review, Desalination, 2012, 298, 1–12, DOI:10.1016/j.desal.2012.05.006.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Evaluation of Advanced Oxidation Processes for Water and Wastewater Treatment – A Critical Review, Water Res., 2018, 139, 118–131, DOI:10.1016/j.watres.2018.03.042.
- M. J. Liu, B. S. Neo and W. A. Tarpeh, Building an Operational Framework for Selective Nitrogen Recovery via Electrochemical Stripping, Water Res., 2020, 169, 115226, DOI:10.1016/j.watres.2019.115226.
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen, T.-W. Song, Z. Yin, H.-W. Liang, S. Zhang, G. Wang and H. Wang, Non-Iridium-Based Electrocatalyst for Durable Acidic Oxygen Evolution Reaction in Proton Exchange Membrane Water Electrolysis, Nat. Mater., 2023, 22(1), 100–108, DOI:10.1038/s41563-022-01380-5.
- N. Wang, P. Ou, R. K. Miao, Y. Chang, Z. Wang, S.-F. Hung, J. Abed, A. Ozden, H.-Y. Chen, H.-L. Wu, J. E. Huang, D. Zhou, W. Ni, L. Fan, Y. Yan, T. Peng, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, Doping Shortens the Metal/Metal Distance and Promotes OH Coverage in Non-Noble Acidic Oxygen Evolution Reaction Catalysts, J. Am. Chem. Soc., 2023, 145(14), 7829–7836, DOI:10.1021/jacs.2c12431.
- H. Chen, Z. Zhang, Y. Li, L. Yu, F. Chen and L. Li, Conductive Polymer Protection Strategy to Promote Electrochemical Nitrate Reduction to Ammonia in Highly Acidic Condition over Cu-Based Catalyst, Chem. Eng. J., 2024, 481, 148596, DOI:10.1016/j.cej.2024.148596.
- Y. Lv, S.-W. Ke, Y. Gu, B. Tian, L. Tang, P. Ran, Y. Zhao, J. Ma, J.-L. Zuo and M. Ding, Highly Efficient Electrochemical Nitrate Reduction to Ammonia in Strong Acid Conditions with Fe2M-Trinuclear-Cluster Metal–Organic Frameworks, Angew. Chem., 2023, 135(27), e202305246, DOI:10.1002/ange.202305246.
- J.-X. Liu, D. Richards, N. Singh and B. R. Goldsmith, Activity and Selectivity Trends in Electrocatalytic Nitrate Reduction on Transition Metals, ACS Catal., 2019, 9(8), 7052–7064, DOI:10.1021/acscatal.9b02179.
- C. A. Wong, D. B. Lobell and M. S. Mauter, Multicriteria Suitability Index for Prioritizing Early-Stage Deployments of Wastewater-Derived Fertilizers in Sub-Saharan Africa, Environ. Sci. Technol., 2023, 57(45), 17588–17597, DOI:10.1021/acs.est.3c05435.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; supporting tables; and additional experimental and simulation data. See DOI: https://doi.org/10.1039/d4ee03002h |
| This journal is © The Royal Society of Chemistry 2024 |