
Fast reductive defluorination of branched perfluorooctane sulfonic acids by cobalt phthalocyanine: electrochemical studies and mechanistic insights†
Maryam
Mirabediny
a,
Tsz Tin
Yu
 a,
Jun
Sun
a,
Matthew
Lee
b,
Denis M.
O'Carroll
b,
Michael J.
Manefield
b,
Björn
Åkermark
c,
Biswanath
Das
*c and
Naresh
Kumar
*a
a,
Jun
Sun
a,
Matthew
Lee
b,
Denis M.
O'Carroll
b,
Michael J.
Manefield
b,
Björn
Åkermark
c,
Biswanath
Das
*c and
Naresh
Kumar
*a
aSchool of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: n.kumar@unsw.edu.au
bSchool of Civil and Environmental Engineering, Water Research Centre, The University of New South Wales, Sydney, NSW 2052, Australia
cDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, Svante Arrhenius väg 16C, SE-10691 Stockholm, Sweden. E-mail: biswanath.das@su.se; das.biswanath85@gmail.com
First published on 6th November 2023
Abstract
Branched perfluorooctane sulfonic acid (PFOS) is recognized as a threatening environmental pollutant due to its high persistence and bioaccumulation in various environmental matrices as well as for its toxic effects on humans and wildlife even at very low concentrations. This study reports the first investigation of branched PFOS defluorination catalyzed by metal phthalocyanines. The reaction conditions were optimized using different reductants and temperatures. CobaltII phthalocyanine, when combined with TiIII citrate as a reducing agent, was able to defluorinate 10.9% of technical PFOS within 8 hours. In contrast, vitamin B12 only showed 2.4% defluorination during the same time period, under similar conditions. The defluorination mediated by the cobaltII phthalocyanine and TiIII citrate system corresponds to 54.5% of all branched PFOS isomers (br-PFOS isomers). Isomer-specific degradation was also investigated via high-resolution LC-orbitrap followed by their relative rates. The difference in catalytic efficacy of various phthalocyanine complexes is rationalized by their structures and electrochemical response. Lastly, a new defluorination mechanism is proposed based on the newly detected degradation products after the phthalocyanine treatment and previous studies.
Water impactThe presence of per and polyfluoroalkyl substances (PFASs) in groundwater has been a global issue, especially for perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA), due to their wide use and extremely high stability. There are several technologies for remediation of PFOS, but most of them suffer from the harsh reaction conditions that are needed to be effective or the high cost of the material. Vitamin B12 (VB12), one of the important components for organisms and animals, has been reported to slowly degrade (∼70% degradation after 5 days) branched PFOS (br-PFOS) in the presence of TiIII citrate at 70 °C. Here, we report for the very first time metal-phthalocyanines, a set of easy to prepare, nontoxic, high performing complexes for fast degradation of PFOS and the corresponding electrochemical investigations. Among these complexes, CoII-phthalocyanine exhibited superior activity in the presence of TiIII-citrate which resulted in 54% branched PFOS defluorination within the first 8 h at 65 °C. It is more than 5 times faster than that of vitamin B12 in the presence of TiIII-citrate. This CoII-phthalocyanine + TiIII-citrate system also showed superior reactivity within the temperature range of 25–45 °C in comparison to previously reported other porphyrins. We have also investigated isomer-specific PFOS degradation for VB12-TiIII citrate and CoPc-TiIII citrate systems. Being molecular and comparatively simpler systems, these phthalocyanines allow further structural alteration for molecular electrode fabrication that can result in much improved defluorination efficiency. |
Introduction
Per- and polyfluoroalkyl substances (PFASs) constitute a great number of manufactured halogenated organic chemicals that are of growing concern for public health and the environment.1 PFASs have been extensively produced since the 1940s and are used in a wide range of household and industrial applications, such as food processing, metal plating, textiles, firefighting/safety, and aviation.2,3 The strong bonds between carbon and fluorine atoms (around 485 kJ mol−1) in PFASs give these chemicals unique properties such as chemical and thermal stability, oleophobicity, and hydrophobicity. These properties have not been observed in fluorine-free alternatives so far.4Perfluorooctane sulfonate (PFOS) is one of the most widely used and studied perfluorosulfonic acids (PFSAs). It has been detected globally in different environmental matrices (air, soil, and water), and animal and human tissues.5 This chemical as a recalcitrant toxic pollutant could adversely affect human health and the environment. PFOS is now regulated as a persistent organic pollutant (POP).4 The United States and European Union have restricted the production of PFOS since 2000 but have not entirely banned it as it is still specifically used in hydraulic fluid additives, wetting agents, mist suppression, and in the manufacture of semiconductor chips in some regions.6 PFOS can easily be transported in terrestrial and aquatic systems, covering long distances from the source of its release.4 Moreover, the ultimate transformation of volatile PFAS precursors such as fluorotelomer and fluorosulfoamido alcohols to stable PFOS could be another reason for its intensified occurrence in the environment and in vivo.7 For example, PFOS concentrations in the groundwater of Tokyo were reported to be higher than that in the source of contamination, the urban runoff. The reason could be the conversion of PFAS precursors in the runoff into PFOS during groundwater infiltration through a soil column.8
Previous reports suggested that branched PFOS isomers (br-PFOS) account for a significant portion of the PFOS load in the aquatic environment due to the higher polarity of the branched structures compared to the linear isomers which preferentially absorb to soil and sediments.9 Ma et al. (2018) and Gao et al. (2019) reported that br-PFOS accounts for 40.8% and 60.1% of total PFOS in the river waters and groundwaters of China, respectively.10,11 Moreover, studies discovered that humans preferentially accumulate br-PFOS isomers over linear PFOS isomers (L-PFOS). Nian et al. (2019) showed that br-PFOS accounts for over 50% of total PFOS in the human blood serum.12–15 Accordingly, the development of remediation technologies to defluorinate br-PFOS from aquatic media and minimize human exposure is a very important task that needs to be addressed.
br-PFOS is susceptible to reductive cleavage of C–F bonds due to the high electronegativity of fluorine atoms.16–18 Ochoa-Herrera et al. (2008) and Park et al. (2017) evaluated the dehalogenation of technical PFOS (including 24.6% br-PFOS) catalyzed by a cobalt-containing biomolecule, vitamin B12 (VB12) using TiIII citrate and nanosized zerovalent zinc (nZn0) respectively as the reductants in an alkaline medium at 70 °C.18,19 CoI, a strong nucleophile, was produced by the reduction of CoIII/CoII in VB12, that attacked the polar C–F bond to initiate the reaction. According to Ochoa-Herrera et al. (2008), while 71% of total br-PFOS was defluorinated within 5 days, L-PFOS concentration remained unchanged.18 The stronger C–F bonds in L-PFOS compared to branched isomers make it even more unreactive.20 Efficient reductive cleavage of C–F bonds was previously reported using the VB12-TiIII citrate system.18 This study suggested that br-PFOS could be reduced under alkaline conditions at 70 °C. However, the defluorination of br-PFOS was not effective at ambient temperatures.18,19
There is only one study reporting the efficient catalytic defluorination of br-PFOS at room temperature using the reductive method. Sun et al. (2022) investigated the effective reductive defluorination of br-PFOS via porphyrin-based catalysts with experimental conditions identical to those reported by Ochoa-Herrera et al. (2008) (pH 9.0 and 65 °C).18,21 These molecular catalyst analogues of VB12 exhibited similar levels of PFOS defluorination after 8 h as VB12 achieved after 10 days. Moreover, they are known as low-temperature catalysts which displayed around 51% defluorination of br-PFOS within 1 day at 25 °C.21
Metal phthalocyanines (MPcs) are an abundant group of macrocyclic, organic compounds embracing a highly conjugated structure with a chelated metal ion at the centre.22 They are structurally related to porphyrin complexes. MPcs are extensively used in biological and natural environments for catalytic aerobic oxidations, reduction, and destruction of peroxides.22 Compared to porphyrin complexes, MPcs have received special attention in industrial use due to their thermal stability, cost-effectivity, and the possibility of straightforward large-scale production.22 Reductive reactions involving MPcs as catalysts have received less attention than oxidation reactions which may be due to their historically broader applications in industrial processes under aerobic conditions.22 So far, this class of catalysts was specifically investigated for the effective reduction of nitrite and nitrate while only limited studies are reported for the reductive dehalogenation of halogenated compounds.23,24 MPcs have been reported to induce the degradation of various other pollutants.25 Among oxidative reactions involving these catalysts, there are only two studies reporting the successful oxidative dehalogenation of chlorinated compounds using metal phthalocyanines.23,24 However, this catalyst was only active in non-aqueous media in the presence of toluene, a potentially harmful chemical.23 Iron tetrasulfophthalocyanine catalyzed the oxidative degradation of recalcitrant chlorinated phenols in aqueous solutions with partial mineralization.22 Comolban et al. (2014) reported efficient cleavage of C–F bonds in fluorinated aromatics under oxidative conditions via a diiron phthalocyanine complex.26 However, the ability of MPcs to remediate PFAS has not been investigated. Herein, the catalytic performance of various MPcs with different metal centres was studied to select the best-performing catalyst for PFOS defluorination in the aqueous system.
Herein, we report for the first time the catalytic reductive defluorination of branched PFOS via metal phthalocyanine derivatives in the presence of high electron affinity reductants. The electronic effect of these catalysts in PFOS defluorination was examined using different environmentally benign first-row transition metal centers and different substitutes on the conjugated organic system. The reason behind the difference in activity upon the structural changes of the catalysts is rationalized by the electrochemical investigations under similar reaction conditions. In order to optimize the conditions, the reactivity using different reductants and temperatures was tested. The reaction products were analyzed using LC/MS to investigate the rate of isomer-specific degradation. Based on the detected masses in the reductive defluorination of br-PFOS and previously reported results, a new mechanism for PFOS defluorination is proposed.
Materials and methods
Chemicals and solution preparation
Perfluorooctane sulfonate, PFOS (98%) (technical mixture with 20% branched isomers), was purchased from Matrix Scientific. The percentage of each isomer in the mixture is detailed in the ESI† (Table S3). Chemical structures of linear and branched isomers of PFOS are shown in Fig. S2.† TiCl3 (10–15% solution) and other chemicals such as sodium carbonate, sodium bicarbonate, ammonium acetate, and acetic acid (≥99.99% trace metals basis) from Sigma-Aldrich were used as received. LCMS-grade methanol and Milli Q water were used in this work. TiIII citrate was prepared according to the previous method reported by Liu et al. (2018).16 The detailed information and chemical structures of the catalysts used in this work such as 29H,31H-phthalocyanine (Pc), copperII phthalocyanine (CuPc), cobaltII phthalocyanine (CoPc), silicon phthalocyanine dichloride (SiPc), nickelII phthalocyanine (NiPc), ironII phthalocyanine (FePc), sodium cobaltII tetrasulfophthalocyanine (CoTSPc), cobaltII tetranitrophthalocyanine (CoTNPc), and vitamin B12 (VB12) are listed in the ESI† (Table S1). The synthesis and characterization of CoTSPc and CoTNPc are described in the ESI.† The preparation of the catalyst stock solutions is described in the ESI.†Batch experiment
The defluorination procedure of PFOS was derived from that reported by Ochoa-Herrera et al. (2008) and Sun et al. (2022).18,21 In an anaerobic chamber (Coy Lab Inc., >95% N2, <5% H2), a series of 20 mL borosilicate glass bottles were loaded with 10 mL reaction solution containing 0.1 mM technical PFOS. This concentration was chosen to ensure that any released fluoride by defluorination falls within the detection range of the ion chromatography (IC) detector, facilitating accurate analysis. 0.25 mM catalyst and 40 mM TiIII citrate were added as the reducing agent. The experiments were conducted in 80 mM carbonate buffer (at pH 9). In certain sets of experiments where the reaction was optimized using various reductants, equivalent concentrations of 40 mM of other strong reducing agents, such as sodium borohydride (NaBH4) and nanosized zerovalent zinc (nZn0), were employed. Each glass bottle was sealed using an aluminum crimp cap and transferred to an incubator shaker at the temperature of 65 °C except for the sets of experiments where the reaction was optimized using various temperatures (25, 45, and 65 °C). The majority of the experiments were analyzed for 24 h to quantify the PFOS defluorination. 300 μL samples were taken at different time intervals and centrifuged for 5 minutes (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) and then the upper liquid was kept in 2 mL autosampler vials to analyze the fluoride and PFOS isomer concentrations.
000 rpm) and then the upper liquid was kept in 2 mL autosampler vials to analyze the fluoride and PFOS isomer concentrations.
Sample analysis via LC-suppressed conductivity ion chromatography (IC) and LC-MS/MS
Fluoride concentration was measured with a Shimadzu Prominence Series IC equipped with a conductivity detector and ion suppressor. A 5 μL aliquot was loaded onto an IC-SA3 anion exchange column (4 mm i.d., 250 mm long) supported with a 228–41600−92 guard column. The mobile phase was 3.6 mM sodium carbonate. The measurements were done with a fixed flow rate of 0.8 mL min−1 at an oven temperature of 50 °C.
The fluoride concentration released in the aqueous solution was determined by subtracting the initial fluoride concentration (due to background F−) from fluoride detected at a later time point acquired by IC. The defluorination degree was calculated by using eqn (1) where CF− is the fluoride ion concentration (mM) and C0 is the initial equivalent fluoride concentration (mM) in the PFOS substrate.
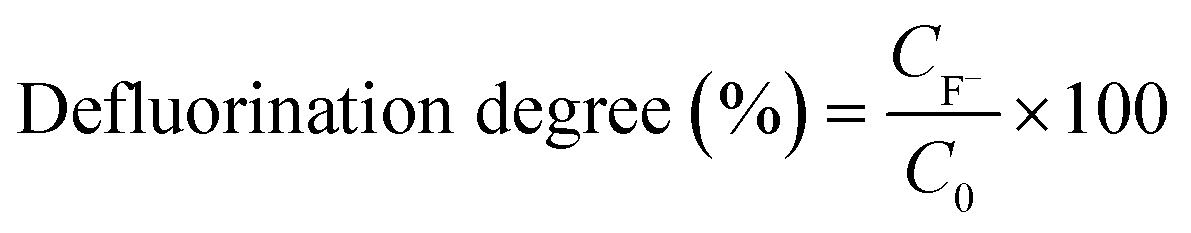 | (1) |
Results and discussion
Selection of the most reactive metal phthalocyanine complex (MPc) for PFOS defluorination
The reductive defluorination of technical PFOS (0.1 mM) by TiIII citrate (40 mM) was evaluated under the same experimental conditions as in previously reported studies (at pH 9 and 65 °C).16,18,21 The defluorination experiment was conducted using 29H,31H-phthalocyanine (Pc), and the corresponding complexes with various metal centres, such as Si, Cu, Ni, Co, and Fe. The chemical and structural information of the complexes is listed in Table S1.† The catalytic performance of these compounds was compared with VB12 as a well-known natural initiator for PFOS defluorination (Fig. 1). It showed that CoII phthalocyanine (CoPc) had superior reactivity, with 10.91 ± 0.41% defluorination within the first 8 h (4.6 times faster than VB12). Other Pc scaffolds showed less than 1% defluorination after 24 h (0.76, 0.33, 0.30, 0.81, and 0.39% defluorination corresponding to SiPc, CuPc, NiPc, FePc, and Pc, respectively). This demonstrates that the catalytic defluorination of PFOS largely depends on the central metal ion.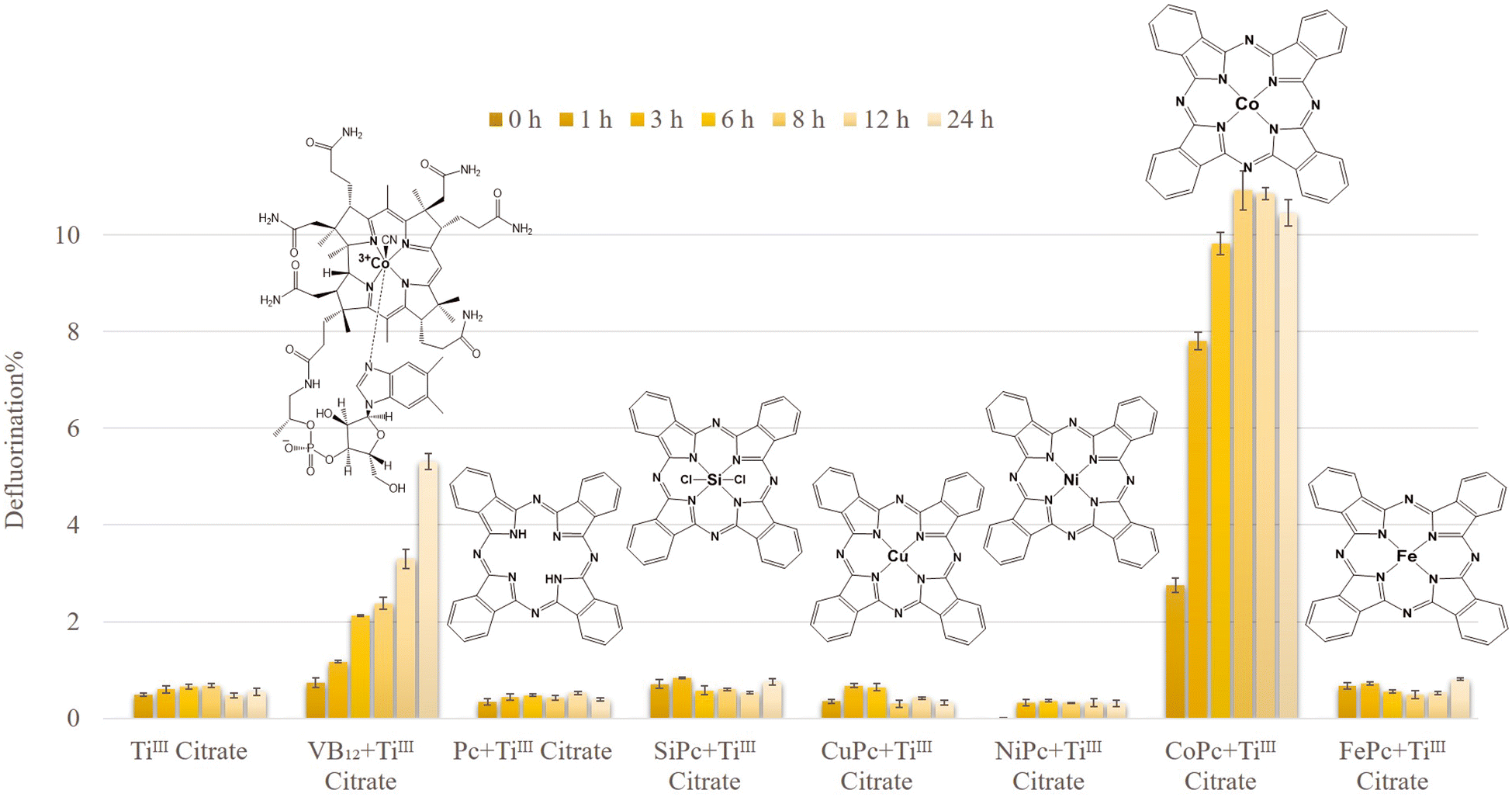 | ||
| Fig. 1 Time course of technical PFOS defluorination via various MPcs or VB12 in TiIII citrate at 65 °C and pH 9.0. Experimental conditions: technical PFOS (0.1 mM), catalyst (0.25 mM), TiIII citrate (40 mM), and carbonate buffer (85 mM) in Milli Q water. | ||
Interestingly, CoPc with limited solubility in the aqueous system (due to the presence of a hydrophobic organic Pc ligand) exhibited a significantly higher activity compared to that of water-soluble VB12. During the first 3 h at 65 °C, CoPc defluorinated technical PFOS 6.6 times faster than that of VB12. However, from Fig. 1 it is also clear that with CoPc, defluorination stops after 8 h. The reason is probably the decomposition of the catalyst under this harsh reductive environment. This can be further improved by covalently anchoring CoPc onto solid conductive surfaces. Similar strategies have been reported very recently to improve the stability of the electrocatalysts as well as the catalytic efficiency.27–29
No significant defluorination was observed when solely CoPc or TiIII citrate was applied to the synthetic PFOS solution (Fig. S3†), showing that both components are essential for effective defluorination.
It is worth mentioning that besides this promising and rapid defluorination performance of CoPc within the first few hours at 65 °C, in the temperature range of 25–45 °C, the CoPc-TiIII system also showed much higher defluorination activity than that of the VB12-TiIII system, even if it is compared after 24 h (Fig. 4).
We also studied other cobalt phthalocyanine derivatives, such as cobaltII tetrasulfophthalocyanine (CoTSPc) and cobaltII tetranitrophthalocyanine (CoTNPc) (Fig. S4†). CoTSPc with four negatively charged sulfonate functional groups on the phthalocyanine ring was expected to show higher defluorination compared to CoPc due to its improved solubility in the aqueous medium. However, its efficiency was observed to be less than 0.5% even after 24 h. The very low defluorination ratio can be attributed to the repulsive interactions between the negatively charged sulfonate groups in CoTSPc and the PFOS substrate. Another cobalt phthalocyanine derivative, CoTNPc, also showed a lower reactivity (4.24 ± 0.22% defluorination within 12 h) than CoPc, but better than CoTSPc. Although the neutral nitro groups lower the negative charge density in the periphery, this takes away electrons from the Pc center.
To investigate the possible reason for the notable defluorination performance of technical PFOS in the CoPc-TiIII system, the cyclic voltammograms (CVs) of those catalysts involved in this study were evaluated. The CVs of CoPc, VB12 (Fig. 2), CuPc, FePc (Fig. 3a), Pc, SiPc, NiPc (Fig. 3b), CoTNPc, and CoTSPc (Fig. 3c) were examined in a nonreactive DMSO solution. The CoII/CoI reduction process by CoPc occurred at −0.89 V vs. SCE while VB12, FePc, CuPc, and NiPc exhibited higher onset reduction potentials of −1.15, −1.17, −1.28, and −1.31 V vs. SCE, respectively.21,30 This explains the lower reactivity of the aforementioned catalysts compared to CoPc, which exhibits a more facile electron transfer. It should also be noted that no redox waves were observed for Pc under these experimental conditions (Fig. 3b). Moreover, silicon does not undergo metal electron transfer in the same manner as the transition metals due to its tendency to form covalent bonds with other elements, resulting in a stable complex structure in SiPc.31,32 Therefore, the two corresponding peaks observed in the reduction process of SiPc (−0.83 and −1.45 V vs. SCE) were potentially attributed to changes in the oxidation states of the ligands bound to Si (Fig. 3b). However, based on the results obtained from the PFOS defluorination test in the SiPc-TiIII system, this compound showed no evidence of fluoride release.
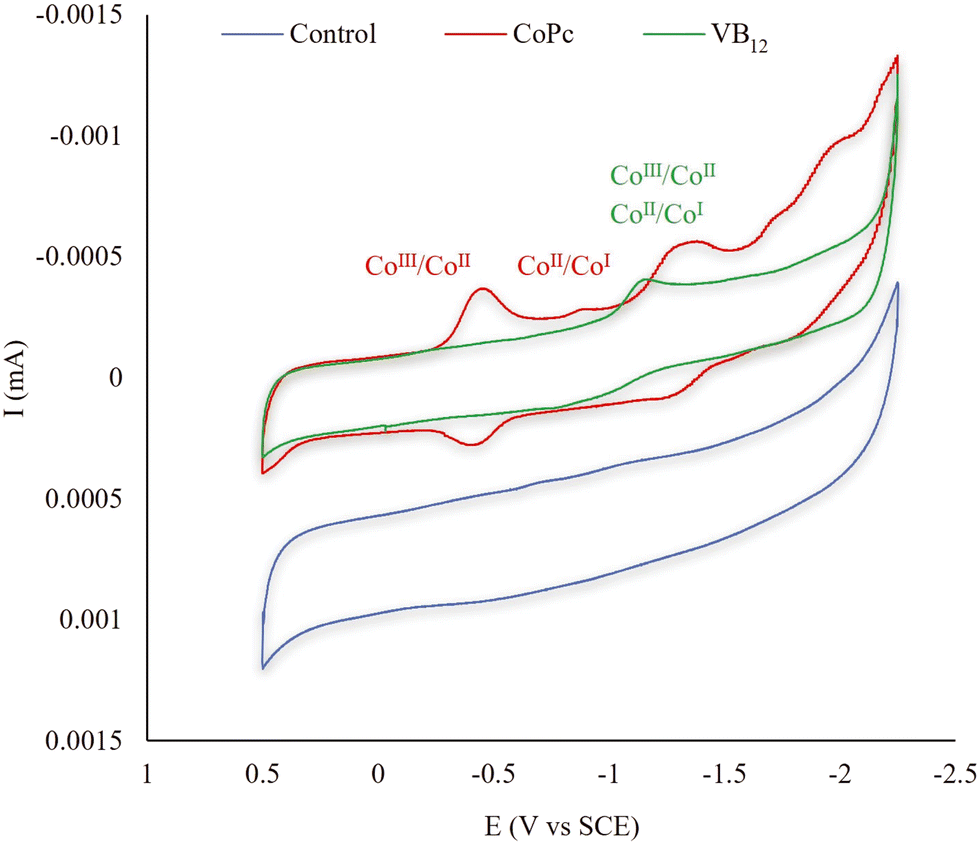 | ||
| Fig. 2 Cyclic voltammograms of 0.05 mM catalysts (CoPc and VB12) dissolved in a 0.1 M TBAP DMSO solution. The scan rate was set at 20 mV s−1. Glassy carbon and Pt wire were employed as the working and counter electrodes, respectively. A saturated calomel electrode (SCE) was used as the reference electrode. | ||
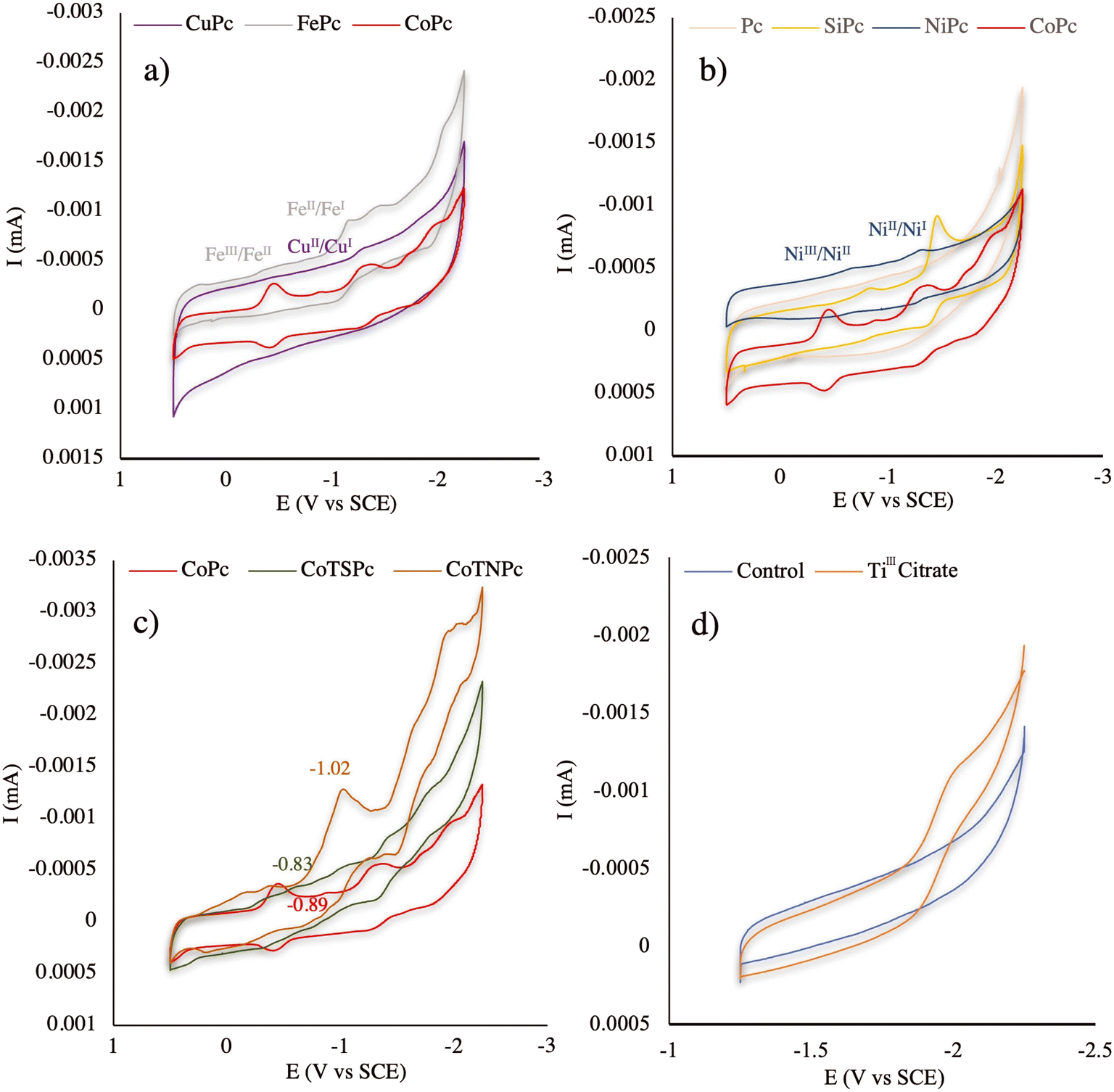 | ||
| Fig. 3 Cyclic voltammograms of 0.05 mM catalysts (a: CuPc, FePc, CoPc; b: Pc, SiPc, NiPc, CoPc; c: CoPc, CoTSPc, CoTNPc) and 0.05 mM TiIII citrate (d) dissolved in a 0.1 M TBAP containing DMSO solution. The scan rate was set at 20 mV s−1. Glassy carbon and Pt wire were employed as the working and counter electrodes, respectively. A saturated calomel electrode (SCE) was used as the reference electrode. | ||
The redox properties of CoPc were also compared with those of CoTNPc and CoTSPc (Fig. 3c). CoPc showed a CoII/CoI reduction process at −0.89 V vs. SCE while CoTNPc exhibited a higher onset reduction potential (−1.02 V vs. SCE) for the same process, indicating a more energy demanding conversion from CoII to CoI in CoTNPc. Additionally, the onset reduction potential of CoII/CoI in CoTSPc was lower (−0.83 V vs. SCE) than that of CoPc (−0.89 V vs. SCE). However, the lack of defluorination observed in the CoTSPc-TiIII system can be attributed to the repulsive interaction between negative charges on CoTSPc and PFOS.
Furthermore, the CV of TiIII citrate was examined under identical experimental conditions as the CVs of all the catalysts were tested (Fig. 3d). TiIII citrate exhibited an onset oxidation potential of −1.89 V vs. SCE, indicating a greater propensity for electron transfer in the CoPc system compared to the other catalysts with higher onset reduction potentials. The significant potential difference between TiIII citrate and CoPc created a more favorable driving force for reductive defluorination, thereby enhancing the efficiency of the CoPc-TiIII system.
CoPc is structurally composed of isoindole units linked by the nitrogen atoms in a ring with extended conjugation. The presence of this conjugate base structure is valuable for promoting the electron transfer between the reductant and cobalt in CoPc, facilitating the conversion of CoII to CoI to initiate PFOS defluorination. However, the electron-withdrawing groups, such as nitro or sulfonate groups, on the ring cause the attenuation in the conjugation and the electron transfer or repulsive interactions leading to lower efficiency.
The effect of varying temperatures
The activities of VB12 and CoPc were examined at 25, 45, and 65 °C to optimize the PFOS defluorination reaction (Fig. 4). Increasing the temperature from 25 to 65 °C increased the defluorination in the case of both VB12-TiIII citrate and CoPc-TiIII citrate systems. The PFOS defluorination by CoPc reached 8.09 ± 0.19% at room temperature within 8 h which was higher than the result reported by Sun et al. (2022) using CoII-TPP (around 6%).21 On the other hand, the VB12-TiIII citrate system reached only around 1% defluorination within 8 h at room temperature. This represents CoPc as the room-temperature catalyst which provides a potential applicability for the biodefluorination of PFOS. A previous report by Guerrero-Barajas and Field (2005) showed that cyanocobalamin and hydroxycobalamin enabled anaerobic degradation of chloroform in an unadapted methanogenic consortium.33 The use of microorganisms as natural and non-toxic sources of reductants potentially plays a significant role in the catalytic reductive degradation of environmental contaminants. These cultures require a temperature of around 35 °C to degrade the contaminants. In this study as well, VB12 did not show effective reactivity for PFOS defluorination at low temperatures (1.59% efficiency within 24 h at 45 °C). The high reactivity of CoPc at low temperatures (25 and 45 °C) within a short period of time opens the future investigation pathway for the catalytic bioremediation of fluorinated compounds in the environment.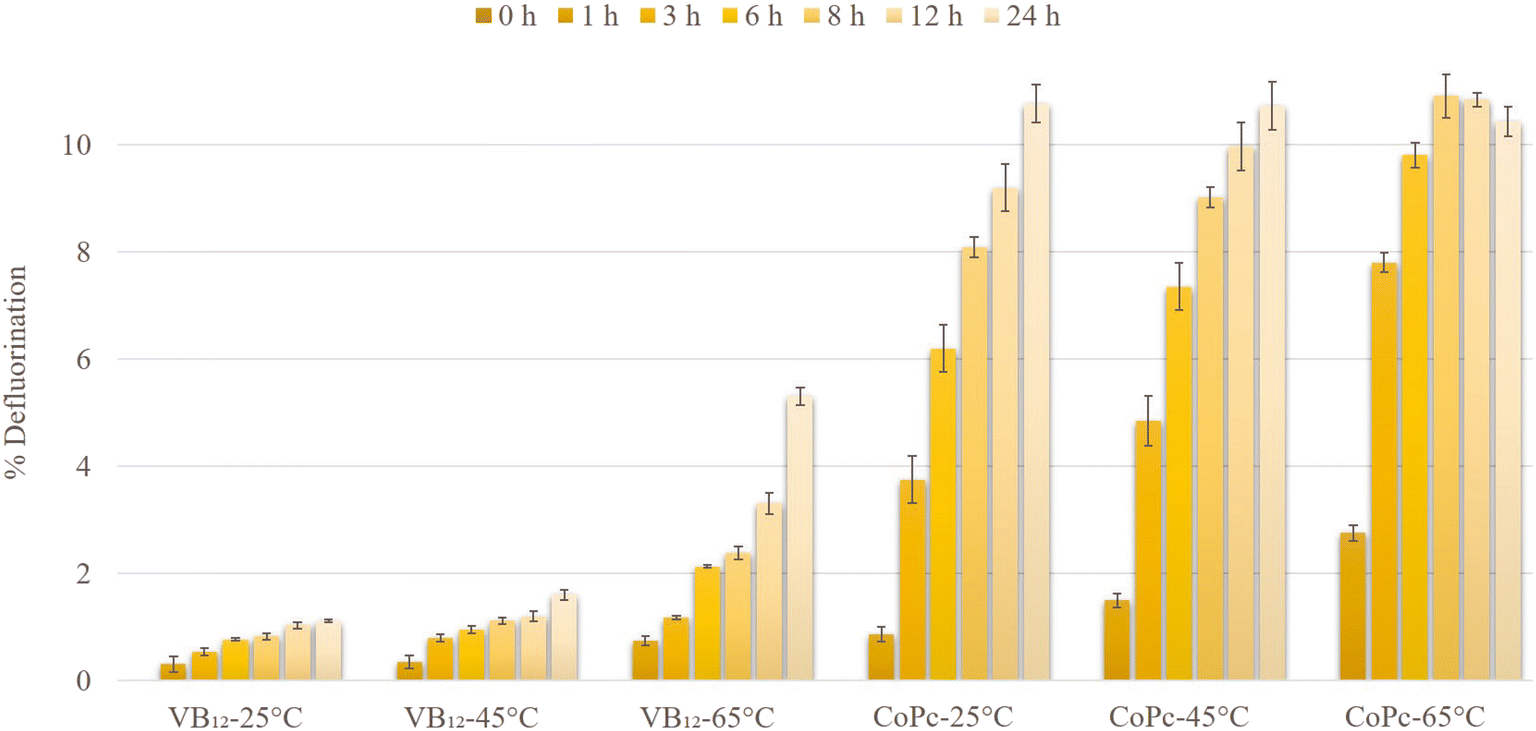 | ||
| Fig. 4 Time course of technical PFOS defluorination at different temperatures (at 25, 45, and 65 °C) via VB12 and CoPc at pH 9.0 over 24 hours. Experimental conditions: technical PFOS (0.1 mM), TiIII citrate (40 mM), and carbonate buffer (85 mM) in Milli Q water. | ||
The effect of different reductants
The defluorination of PFOS via VB12 reported in previous studies utilized a strong reducing agent, such as nZn0/TiIII citrate.18,19,21 To determine the best reductant for CoPc in the PFOS defluorination, apart from the reductants outlined above, the capability of sodium borohydride (NaBH4), a well-known strong reductant, was also investigated (Fig. 5). However, neither nZn0 nor NaBH4 showed significant defluorination with CoPc within 24 h (with only less than 1.4% defluorination). In the nZn0 system, the defluorination efficiency with CoPc was 1.08%, while this value increased to 3.64% within 24 h for VB12. The lower defluorination efficiency of CoPc could be due to the lower solubility of CoPc in the aqueous system compared to water-soluble VB12, as the reaction rarely occurs heterogeneously. Moreover, with the CoPc-NaBH4 system, only 1.34 ± 0.09% defluorination after 24 h was observed, which did not increase over 10 days. The lack of improvement of defluorination after 24 h could be due to the rapid deactivation of NaBH4 within the initial few minutes through reaction with water instead of activation of the catalyst.34 This also indicates that under these conditions, in situ formed hydrides are not effective defluorinating agents. TiIII citrate was identified as the best reductant for PFOS defluorination by CoPc due to the high solubility and stability in a water-based system. | ||
| Fig. 5 Time course of technical PFOS defluorination using different reductants such as NaBH4, nZn0, and TiIII citrate at 65 °C and pH 9.0. Experimental conditions: technical PFOS (0.1 mM), CoPc/VB12 (0.25 mM), the reductant (40 mM), and carbonate buffer (85 mM) in Milli Q water. | ||
This study was further extended to investigate the possible defluorination of PFOS using higher concentrations of TiIII citrate. Extra TiIII citrate was added to the reaction sample after 24 h (to reach a final concentration of 60 mM). The defluorination increased to 12.48% after 2 days and did not increase further with time. A similar result was observed in a previous study via CoII-5,10,15,20-tetraphenyl-21H,23H-porphyrin/CoIII-5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphyrin (CoII-TPP/CoIII-M-TPP).21
Kinetic rate constants of PFOS isomer-specific degradation
The concentrations of branched and linear PFOS isomers were measured by LC-MS/MS during the reductive defluorination via CoPc and VB12 at 65 °C (Fig. 6). 6-PFOS disappeared within 6 h while 89.82 ± 2.21% of 5-PFOS, 85.67 ± 0.11% of 4-PFOS, and 37.11 ± 1.45% of 3-PFOS were degraded within 8 h in the CoPc-TiIII citrate system. However, with the VB12-TiIII citrate system, only 6-PFOS showed significant degradation (55.89% degradation within 24 h). This clearly shows the promising performance of CoPc. Although the concentration of linear PFOS isomers remained unchanged during the reaction via CoPc or VB12, this study showed impressive defluorination reactivities of br-PFOS isomers by CoPc. The inactivity of L-PFOS can be attributed to (i) wider carbon-chain-carbon (CCC) angles in L-PFOS compared to the branched PFOS isomers14 and (ii) lack of electron density variation in the perfluoroalkyl chain which electronically limited its interaction with the catalyst.19 Promising results of the degradation of branched isomers using the CoPc-TiIII citrate system were also observed at lower technical PFOS concentrations (Fig. S5†), offering valuable insights into the practical applications of this study.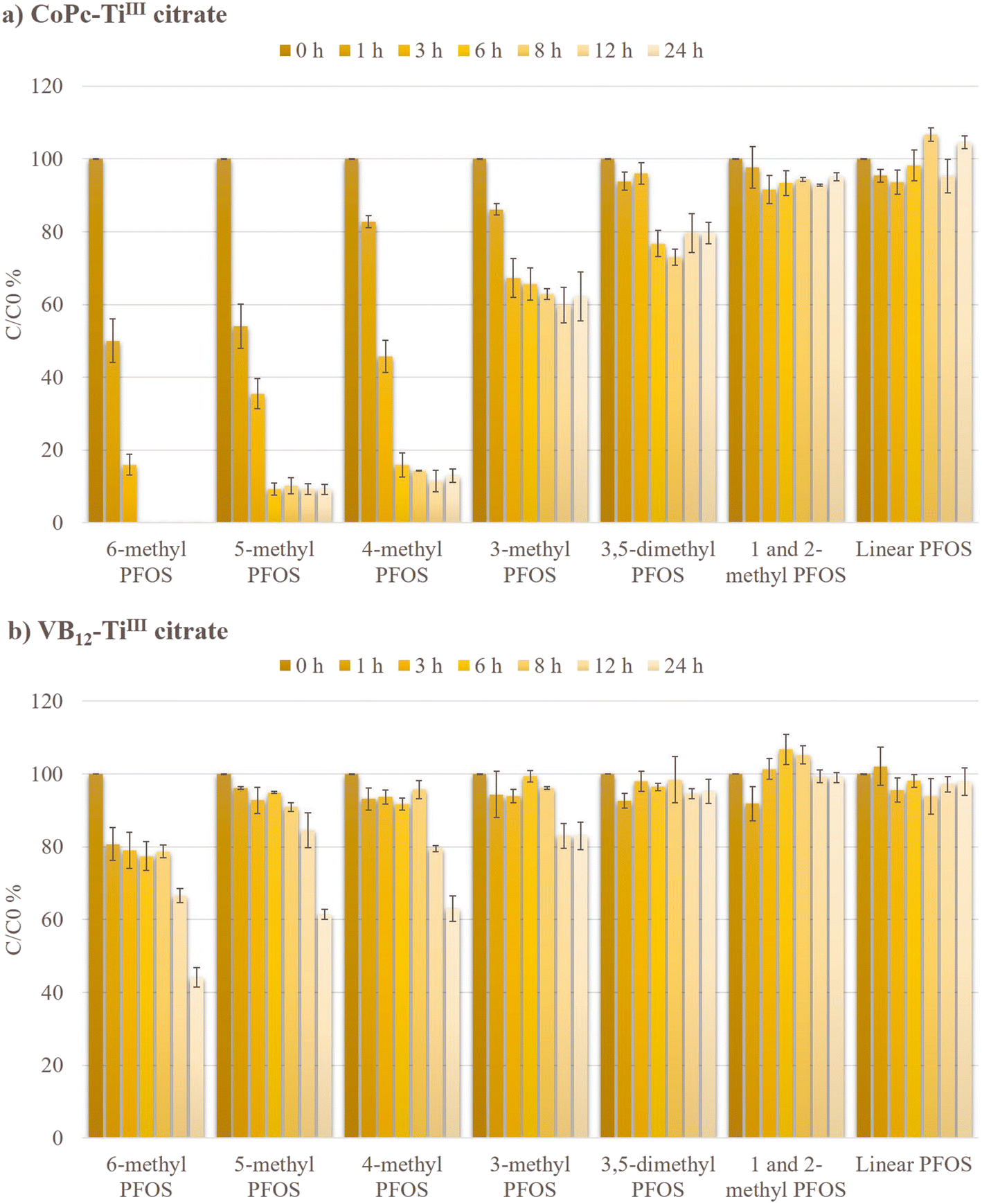 | ||
| Fig. 6 Isomer-specific degradation of PFOS for a) CoPc-TiIII citrate system and b) VB12-TiIII citrate system. Experimental conditions: technical PFOS (0.1 mM), CoPc/VB12 (0.25 mM), TiIII citrate (40 mM), and carbonate buffer (85 mM) at 65 °C. | ||
Under optimized conditions, the kinetic rate of isomer-specific degradation during defluorination by CoPc was studied using different time courses, owing to the difference in reactivities of the branched isomers (Fig. S6†). The degradation rate constants were measured for 3-PFOS for 8 h, 4-PFOS and 5-PFOS for 6 h, and 6-PFOS for 3 h. The mean values were compared with the related VB12-TiIII citrate system. The rates of 6-PFOS, 5-PFOS, 4-PFOS, and 3-PFOS degradation by the CoPc-TiIII system were 18.3, 17.9, 15.5, and 6.7 times higher respectively than those of the VB12 system (Table 1). The differences in the branched isomer reactivities could be due to the differences in the polarizability and electron density distribution in different branched isomers as they were previously reported to have localized LUMOs, making them more prone to be involved in redox reactions.14 The highest degradation rate of 6-PFOS and 5-PFOS suggested that branching in the vicinity of the terminal CF3 group increases the reactivity of the corresponding isomer. This result was consistent with the previous reports by Sun et al. (2022) and Park et al. (2017) who studied CoII-TPP-TiIII citrate and VB12-nZn0 systems respectively.19,21
| PFOS branched isomer | Degradation rate constant in VB12-TiIII system (1 h−1) | Degradation rate constant in CoPc-TiIII system (1 h−1) |
|---|---|---|
| 6-Methyl PFOS | 0.033 | 0.605 |
| 5-Methyl PFOS | 0.021 | 0.376 |
| 4-Methyl PFOS | 0.020 | 0.311 |
| 3-Methyl PFOS | 0.008 | 0.054 |
Overall reaction mechanism
The reaction mechanism for the reductive defluorination of br-PFOS was determined based on the detected masses in the system and the proposed mechanism is displayed in Fig. 7. 6-PFOS was used as the model molecule in the proposed defluorination pathway. The degradation products generated in the system were analyzed by the UPLC-Q Exactive Plus and are listed and numbered in Table 2. Ion chromatographs and spectra of the degradation products were interpreted via Thermo Scientific Xcalibur software and are presented in Fig. S7.†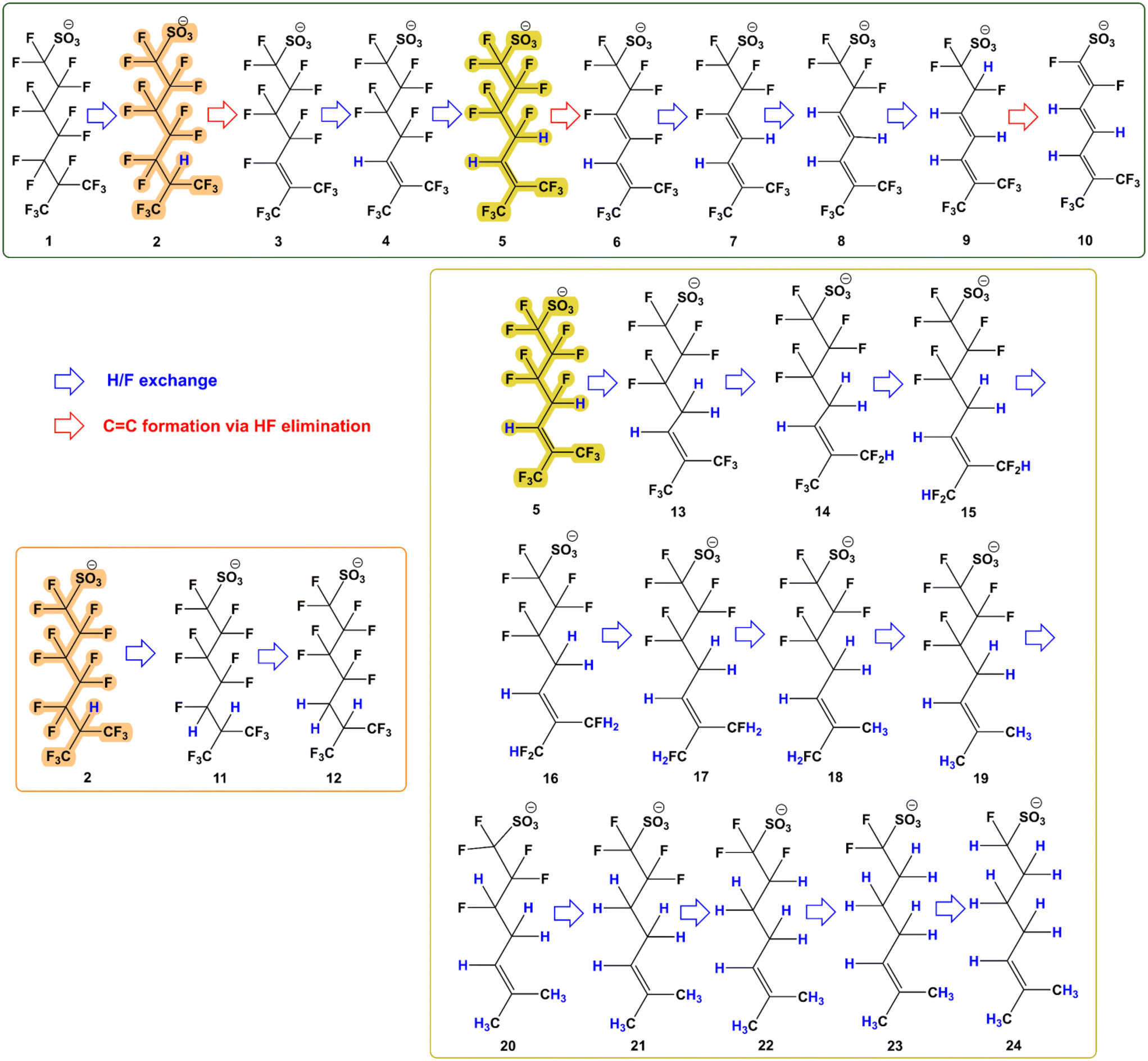 | ||
| Fig. 7 Proposed mechanism for the defluorination of 6-PFOS. | ||
| No. | Chemical formula | Measured mass in sample (Da) | Theoretical mass (Da) | DE on massesa (ppm) | Measured isotope mass in sample (Da) | Theoretical isotope mass (Da) |
|---|---|---|---|---|---|---|
| a Difference error (DE): mass difference between measured and theoretical masses. | ||||||
| 1 | C8F17SO3 | 498.9299 | 498.9291 | 1.6 | 499.9330 | 499.9325 |
| 2 | C8HF16SO3 | 480.9392 | 480.9385 | 1.4 | 481.9427 | 481.9419 |
| 3 | C8F15SO3 | 460.9333 | 460.9323 | 2.1 | 461.9367 | 461.9357 |
| 4 | C8HF14SO3 | 442.9424 | 442.9417 | 1.5 | 443.9447 | 443.9451 |
| 5 | C8H2F13SO3 | 424.9519 | 424.9512 | 1.6 | 425.9555 | 425.9545 |
| 6 | C8HF12SO3 | 404.9454 | 404.9449 | 1.2 | 405.9496 | 405.9483 |
| 7 | C8H2F11SO3 | 386.9551 | 386.9544 | 1.8 | 387.9586 | 387.9577 |
| 8 | C8H3F10SO3 | 368.9644 | 368.9638 | 1.6 | 369.9680 | 369.9671 |
| 9 | C8H4F9SO3 | 350.9740 | 350.9732 | 2.2 | 351.9773 | 351.9765 |
| 10 | C8H3F8SO3 | 330.9678 | 330.9670 | 2.4 | 331.9714 | 331.9703 |
| 11 | C8H2F15SO3 | 462.9489 | 462.9480 | 1.9 | 463.9521 | 463.9513 |
| 12 | C8H3F14SO3 | 444.9579 | 444.9574 | 1.1 | 445.9611 | 445.9607 |
| 13 | C8H3F12SO3 | 406.9612 | 406.9606 | 1.4 | 407.9648 | 407.9639 |
| 14 | C8H4F11SO3 | 388.9708 | 388.9700 | 2.0 | 389.9743 | 389.9734 |
| 15 | C8H5F10SO3 | 370.9802 | 370.9794 | 2.1 | 371.9836 | 371.9828 |
| 16 | C8H6F9SO3 | 352.9897 | 352.9888 | 2.5 | 353.9929 | 353.9922 |
| 17 | C8H7F8SO3 | 334.9991 | 334.9983 | 2.3 | 336.0026 | 336.0016 |
| 18 | C8H8F7SO3 | 317.0084 | 317.0077 | 2.2 | 318.0118 | 318.0110 |
| 19 | C8H9F6SO3 | 299.0179 | 299.0171 | 2.6 | 300.0213 | 300.0205 |
| 20 | C8H10F5SO3 | 281.0273 | 281.0265 | 2.8 | 282.0306 | 282.0299 |
| 21 | C8H11F4SO3 | 263.0367 | 263.0360 | 2.6 | 264.0401 | 264.0393 |
| 22 | C8H12F3SO3 | 245.0462 | 245.0454 | 3.2 | 246.0496 | 246.0487 |
| 23 | C8H13F2SO3 | 227.0557 | 227.0548 | 3.9 | 228.0589 | 228.0582 |
| 24 | C8H14FSO3 | 209.0651 | 209.0642 | 4.3 | 210.0685 | 210.0676 |
The exact pathway cannot be confirmed without isolation and NMR investigation of the products of the defluorination, even better with a smaller model PFOS type substrate. This is beyond the scope of the present study. However, some reasonable suggestions are possible. While proposing the current reaction mechanism based on the mass spectroscopic results, we have considered the following:
(i) Tertiary C–F bonds are easier to defluorinate than secondary and primary C–F bonds;16
(ii) SO3− groups in PFOS do not activate neighbouring C–F bonds;35
(iii) Although allylic C–F bonds are activated, vinylic C–F bonds undergo faster reductive defluorination;36
In the current study, a H/F exchange occurs at the tertiary position and a fluoride unit from 6-PFOS (1, C8F17SO3) is replaced by hydrogen, leading to the generation of intermediate 2 (C8HF16SO3). This is the most probable way to start the defluorination driven by the dissociation of the tertiary C–F bond in 6-PFOS as the bond dissociation energies (BDEs) of PFAS increase in the order: tertiary C–F bonds < secondary C–F bonds < primary C–F bonds.16,21,35 Then, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C formation occurred via HF elimination to produce intermediate 3 (C8F15SO3). This is followed by a series of H/F exchanges. Since the double bond will lead to a decrease in the bond energies at the allylic positions, it seems reasonable to assume that these positions are involved. However, the reductive replacement of vinylic C–F bonds by hydrogen appears to be facile. This is clearly shown by the cobalamin catalysed defluorination of tetrafluoropropene.36 According to this report, first, the vinylic fluorine is replaced by hydrogen, followed by the replacement of one allylic hydrogen, which is also a favoured reaction. This leads to the production of intermediates 4 (C8HF14SO3), 5 (C8H2F13SO3), 6 (C8HF12SO3), 7 (C8H2F11SO3), 8 (C8H3F10SO3), 9 (C8H4F9SO3), and 10 (C8H3F8SO3) via H/F exchange and HF elimination under high pH conditions. These detected intermediate products (2, 3, 4, 5, 7, 8, and 9) were also previously identified by Sun et al. (2022).21 However, Sun et al. (2022) could not identify intermediates 6 and 10 and these two intermediates are reported for the first time in this study, confirming HF elimination as the potentially new pathway in the catalytic defluorination of PFOS.21 Sun et al. (2022) previously proposed the FF elimination pathway in the defluorination for 6-PFOS to produce intermediate products such as C8H4F7SO3.21 However, a mass related to this intermediate (313) was not found in the current system. This suggested that CoPc could have impacted the reaction pathway.
C formation occurred via HF elimination to produce intermediate 3 (C8F15SO3). This is followed by a series of H/F exchanges. Since the double bond will lead to a decrease in the bond energies at the allylic positions, it seems reasonable to assume that these positions are involved. However, the reductive replacement of vinylic C–F bonds by hydrogen appears to be facile. This is clearly shown by the cobalamin catalysed defluorination of tetrafluoropropene.36 According to this report, first, the vinylic fluorine is replaced by hydrogen, followed by the replacement of one allylic hydrogen, which is also a favoured reaction. This leads to the production of intermediates 4 (C8HF14SO3), 5 (C8H2F13SO3), 6 (C8HF12SO3), 7 (C8H2F11SO3), 8 (C8H3F10SO3), 9 (C8H4F9SO3), and 10 (C8H3F8SO3) via H/F exchange and HF elimination under high pH conditions. These detected intermediate products (2, 3, 4, 5, 7, 8, and 9) were also previously identified by Sun et al. (2022).21 However, Sun et al. (2022) could not identify intermediates 6 and 10 and these two intermediates are reported for the first time in this study, confirming HF elimination as the potentially new pathway in the catalytic defluorination of PFOS.21 Sun et al. (2022) previously proposed the FF elimination pathway in the defluorination for 6-PFOS to produce intermediate products such as C8H4F7SO3.21 However, a mass related to this intermediate (313) was not found in the current system. This suggested that CoPc could have impacted the reaction pathway.
Intermediates where two and three H/F exchanges occur before the first CC formation were found in the reaction sample to generate intermediates 11 and 12. Intermediate 12 as the result of three H/F exchanges is reported for the first time in this study and was not identifiable by Sun et al. (2022).21
Lastly, intermediate 5 can undergo further H/F exchanges to give previously identified intermediates 13–20.21 However, the formation of intermediates 21–24, which are identified for the first time in this study, needs to be further investigated since H/F exchange is occurring at the inactivated positions. Further investigation on this species supported by the NMR studies can provide us with information on how to defluorinate the linear PFASs. This is beyond the scope of the current study. The ultimate degradation product detected has a molecular mass of 209 (C8H14FSO3) and contains only one fluoride atom with the other 16 F atoms removed from 6-PFOS while Sun et al. (2022) only managed to detect a degradation product with molecular mass as small as 277 (related to C8H6F5SO3).21 The current results demonstrate that br-PFOS is mineralized by CoPc to a much higher extent than by porphyrin catalysts. However, elucidating the accurate trend for the defluorination of br-PFOS using UPLC-Q Exactive Plus is impossible as it could not differentiate between different isomers which showed different reactivities in our system.
Conclusion
The investigation of reductive PFOS defluorination catalyzed by various metal phthalocyanine derivatives is the focus of this study. The reactions were optimized using different reductants and temperatures. Among various metal phthalocyanines and reductant combinations, CoII phthalocyanine (CoPc), in the presence of TiIII citrate, exhibited significantly higher activity, leading to the degradation of 54% of total br-PFOS isomers within the first 8 hours at 65 °C. This corresponds to overall 10.91 ± 0.41% PFOS defluorination which is around 5 times faster than VB12. The faster br-PFOS degradation activity remains valid within the temperature range of 25–65 °C over a duration of the first 24 h. The difference in reactivity among the metal phthalocyanines is rationalized by cyclic voltammetry experiments. The degradation of different br-PFOS isomers was investigated by LC-MS/MS and a new defluorination pathway is proposed based on the detected degradation products corroborated with previously published reports.Author contributions
Maryam Mirabediny: methodology, investigation, writing – original draft, visualization. Tsz Tin Yu: investigation, writing – review & editing. Jun Sun: methodology. Matthew Lee: writing – review & editing. Denis M O'Carroll: conceptualization, resources, supervision, project administration, funding acquisition, writing – review & editing. Michael J. Manefield: resources, project administration, funding acquisition, writing – review & editing. Björn Åkermark: writing – review & editing. Biswanath Das: conceptualization, visualization, supervision, funding acquisition, writing – original draft, writing – review & editing. Naresh Kumar: conceptualization, resources, supervision, project administration, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Australian Research Council's (ARC) special research initiatives (Project ID: SR180100030) provided at UNSW and Futura Foundation at Stockholm University.References
- U.S. Environmental Protection Agency, PFAS Explained|US EPA [Internet], 2021, Available from: https://www.epa.gov/pfas/pfas-explained Search PubMed.
- M. Marley, E. Moyer and R. Ball, Per- and poly-fluoroalkyl substances [Internet], 2017, vol. 28, pp. 52–55, Available from: https://ww2.health.wa.gov.au/Articles/F_I/Guidance-Statements-on-Perfluorinated-Chemicals Search PubMed.
- Multi-industry per- and polyfluoroalkyl substances (PFAS) study – Preliminary Report, United States Environmental Protection Agency (EPA), September 2021 Search PubMed.
- H. Li, A. L. Junker, J. Wen, L. Ahrens, M. Sillanpää, J. Tian, F. Cui, L. Vergeynst and Z. Wei, A recent overview of per- and polyfluoroalkyl substances (PFAS) removal by functional framework materials, Chem. Eng. J., 2023, 452, 139202 CrossRef CAS.
- G. W. Olsen, J. M. Burris, D. J. Ehresman, J. W. Froehlich, A. M. Seacat, J. L. Butenhoff and L. R. Zobel, Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers, Environ. Health Perspect., 2007, 115(9), 1298–1305 CrossRef CAS PubMed.
- Department of Water and Environmental Regulation (DWER), Government of Western Australia, PFAS Investigations in Western Australia, 2020 Search PubMed.
- E. F. Houtz and D. L. Sedlak, Oxidative conversion as a means of detecting precursors to perfluoroalkyl acids in urban runoff, Environ. Sci. Technol., 2012, 46(17), 9342–9349 CrossRef CAS PubMed.
- M. Murakami, N. Sato, A. Anegawa, N. Nakada, A. Harada, T. Komatsu, H. Takada, H. Tanaka, Y. Ono and H. Furumai, Multiple evaluations of the removal of pollutants in road runoff by soil infiltration, Water Res., 2008, 42(10–11), 2745–2755 CrossRef CAS.
- K. Schulz, M. R. Silva and R. Klaper, Science of the total environment distribution and effects of branched versus linear isomers of PFOA, PFOS, and PFHxS: A review of recent literature, Sci. Total Environ., 2020, 733, 139186 CrossRef CAS PubMed.
- X. Ma, G. Shan, M. Chen, J. Zhao and L. Zhu, Riverine inputs and source tracing of perfluoroalkyl substances (PFASs) in Taihu Lake, China, Sci. Total Environ., 2018, 612, 18–25 CrossRef CAS.
- Y. Gao, Y. Liang, K. Gao, Y. Wang, C. Wang, J. Fu, Y. Wang, G. Jiang and Y. Jiang, Levels, spatial distribution and isomer profiles of perfluoroalkyl acids in soil, groundwater and tap water around a manufactory in China, Chemosphere, 2019, 227, 305–314 CrossRef CAS PubMed.
- M. Nian, Q. Q. Li, M. Bloom, Z. M. Qian, K. M. Syberg, M. G. Vaughn, S. Q. Wang, Q. Wei, M. Zeeshan, N. Gurram, C. Chu, J. Wang, Y. P. Tian, L. W. Hu, K. K. Liu, B. Y. Yang, R. Q. Liu, D. Feng, X. W. Zeng and G. H. Dong, Liver function biomarkers disorder is associated with exposure to perfluoroalkyl acids in adults: Isomers of C8 health project in China, Environ. Res., 2019, 172, 81–88 CrossRef CAS.
- W. D. Hollander, P. De Voogt, W. De Coen and L. Bervoets, Rev. Environ. Contam. Toxicol., 2010, 208, 179–215 Search PubMed.
- F. J. Torres, V. Ochoa-Herrera, P. Blowers and R. Sierra-Alvarez, Ab initio study of the structural, electronic, and thermodynamic properties of linear perfluorooctane sulfonate (PFOS) and its branched isomers, Chemosphere, 2009, 76(8), 1143–1149 CrossRef CAS PubMed.
- K. Londhe, C. S. Lee, C. A. McDonough and A. K. Venkatesan, The need for testing isomer profiles of perfluoroalkyl substances to evaluate treatment processes, Environ. Sci. Technol., 2022, 56(22), 15207–15219 CrossRef CAS.
- J. Liu, D. J. V. Hoomissen, T. Liu, A. Maizel, X. Huo, S. R. Fernández, C. Ren, X. Xiao, Y. Fang, C. E. Schaefer, C. P. Higgins, S. Vyas and T. J. Strathmann, Reductive defluorination of branched per- and polyfluoroalkyl substances with cobalt complex catalysts, Environ. Sci. Technol. Lett., 2018, 5(5), 289–294 CrossRef CAS.
- M. Mirabediny, J. Sun, T. T. Yu, B. Åkermark, B. Das and N. Kumar, Effective PFAS degradation by electrochemical oxidation methods-recent progress and requirement, Chemosphere, 2023, 321, 138109 CrossRef CAS PubMed.
- V. Ochoa-Herrera, R. Sierra-Alvarez, A. Somogyi, N. E. Jacobsen, V. H. Wysocki and J. A. Field, Reductive defluorination of perfluorooctane sulfonate, Environ. Sci. Technol., 2008, 42, 3260–3264 CrossRef CAS PubMed.
- S. Park, C. De Perre and L. S. Lee, Alternate reductants with VB12 to transform C8 and C6 perfluoroalkyl sulfonates: limitations and insights into isomer-specific transformation rates, products and pathways, Environ. Sci. Technol., 2017, 51, 13869–13877 CrossRef CAS PubMed.
- Y. W. Alsmeyer, W. V. Childs, R. M. Flynn, G. G. I. Moore and J. C. Smeltzer, Organofluorin Chemistry, 1994, pp. 121–143 Search PubMed.
- J. Sun, S. Jennepalli, M. Lee, A. Jones, D. M. O'Carroll, M. J. Manefield, M. Bhadbhade, B. Åkermark, B. Das and N. Kumar, Efficient reductive defluorination of branched PFOS by metal-porphyrin complexes, Environ. Sci. Technol., 2022, 56(12), 7830–7839 CrossRef CAS PubMed.
- A. B. Sorokin, Phthalocyanine metal complexes in catalysis, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- T. E. Youssef, Catalytic reductive dehalogenation catalyzed by cobalt phthalocyanine, Lett. Org. Chem., 2017, 14, 419–426 CAS.
- J. Shao, A. Thomas, B. Han and C. A. Hansen, DDT-reductive dechlorination catalyzed by cobalt phthalocyanine, 2,3- and 3,4-tetrapyridoporphyrazine complexes in non-aqueous media, J. Porphyrins Phthalocyanines, 2010, 14(2), 133–141 CrossRef CAS.
- T. Nyokong and V. Ahsen, Photosensitizers in Medicine, Environment, and Security, 2012, pp. 1–662 Search PubMed.
- C. Comolban, E. V. Kudrik, P. Afanasiev and A. B. Sorokin, Catalytic defluorination of perfluorinated aromatics under oxidative conditions using N-bridged diiron phthalocyanine, J. Am. Chem. Soc., 2014, 136, 11321–11330 CrossRef.
- B. Das, E. A. Toledo-Carrillo, L. Li, F. Ye, J. Chen, A. Slabon, O. Verho, L. Eriksson, M. Göthelid, J. Dutta and B. Åkermark, Cobalt electrocatalyst on fluorine doped carbon cloth – a robust and partially regenerable anode for water oxidation, ChemCatChem, 2022, 14, e202200538 CrossRef CAS.
- Q. T. Nguyen, E. Rousset, V. T. H. Nguyen, V. Colliere, P. Lecante, W. Klysubun, K. Philippot, J. Esvan, M. Respaud, G. Lemercier, P. D. Tran and C. Amiens, Covalent grafting of ruthenium complexes on iron oxide nanoparticles: hybrid materials for photocatalytic water oxidation, ACS Appl. Mater. Interfaces, 2021, 13(45), 53829–53840 CrossRef CAS PubMed.
- B. Das, E. A. Toledo-Carrillo, G. Li, J. Ståhle, T. Thersleff, J. Chen, L. Li, F. Ye, A. Slabon, M. Göthelid, T. C. Weng, J. A. Yuwono, P. V. Kumar, O. Verho, M. D. Kärkäs, J. Dutta and B. Åkermark, Bifunctional and regenerable molecular electrode for water electrolysis at neutral pH, J. Mater. Chem. A, 2023, 11, 13331–13340 RSC.
- J. Zagal, M. Páez, A. A. Tanaka, J. R. dos Santos and C. A. Linkous, Electrocatalytic activity of metal phthalocyanines for oxygen reduction, J. Electroanal. Chem., 1992, 339(1–2), 13–30 CrossRef CAS.
- M. T. Rayez, A. Fritsch, J. C. Rayez, H. Fournier, C. Brochon and A. Soum, Structures and Si-N bond strengths of some cyclodi- and cyclotrisilazanes, J. Mol. Struct.: THEOCHEM, 1999, 487(3), 241–250 CrossRef CAS.
- K. Mitra and M. C. T. Hartman, Silicon phthalocyanines: synthesis and resurgent applications, Org. Biomol. Chem., 2021, 19(6), 1168–1190 RSC.
- C. Guerrero-Barajas and J. A. Field, Riboflavin- and cobalamin-mediated biodegradation of chloroform in a methanogenic consortium, Biotechnol. Bioeng., 2005, 89(5), 539–550 CrossRef CAS.
- Y. C. Lee, Y. P. Chen, M. J. Chen, J. Kuo and S. L. Lo, Reductive defluorination of perfluorooctanoic acid by titanium(III) citrate with vitamin B12 and copper nanoparticles, J. Hazard. Mater., 2017, 340, 336–343 CrossRef CAS PubMed.
- M. J. Bentel, Y. Yu, L. Xu, Z. Li, B. M. Wong, Y. Men and J. Liu, Defluorination of per- and polyfluoroalkyl substances (PFASs) with hydrated electrons: structural dependence and implications to PFAS remediation and management, Environ. Sci. Technol., 2019, 53(7), 3718–3728 CrossRef CAS PubMed.
- J. Im, G. E. Walshe-Langford, J. W. Moon and F. E. Löffler, Environmental fate of the next generation refrigerant 2,3,3,3-tetrafluoropropene (HFO-1234yf), Environ. Sci. Technol., 2014, 48(22), 13181–13187 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ew00612c |
| This journal is © The Royal Society of Chemistry 2024 |