
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Active and highly durable supported catalysts for proton exchange membrane electrolysers†
Debora
Belami
a,
Matthew
Lindley
b,
Umesh S.
Jonnalagadda
 d,
Annie Mae
Goncalves Bullock
a,
Qianwenhao
Fan
d,
Wen
Liu
d,
Sarah J.
Haigh
b,
James
Kwan
c,
Yagya N.
Regmi
*a and
Laurie A.
King
*a
d,
Annie Mae
Goncalves Bullock
a,
Qianwenhao
Fan
d,
Wen
Liu
d,
Sarah J.
Haigh
b,
James
Kwan
c,
Yagya N.
Regmi
*a and
Laurie A.
King
*a
aFaculty of Science and Engineering, Manchester Metropolitan University, Chester Street, M1 5GD, UK. E-mail: y.regmi@mmu.ac.uk; l.king@mmu.ac.uk
bDepartment of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cDepartment of Engineering Science, University of Oxford, Parks Road, Oxford OX1 3PJ, UK
dSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 62 Nanyang Drive, 637459, Singapore
First published on 21st June 2024
Abstract
The design and development of supported catalysts for the oxygen evolution reaction (OER) is a promising pathway to reducing iridium loading in proton exchange membrane water electrolysers. However, supported catalysts often suffer from poor activity and durability, particularly when deployed in membrane electrode assemblies. In this work, we deploy iridium coated hollow titanium dioxide particles as OER catalysts to achieve higher Ir mass activities than the leading commercial catalysts. Critically, we demonstrate state-of-the-art durabilities for supported iridium catalysts when compared against the previously reported values for analogous device architectures, operating conditions and accelerated stress test profiles. Through extensive materials characterisations alongside rotating disk electrode measurements, we investigate the role of conductivity, morphology, oxidation state and crystallinity on the OER electrochemical performance. Our work highlights a new supported catalyst design that unlocks high-performance OER activity and durability in commercially relevant testing configurations.
Broader contextProton exchange membrane water electrolysers (PEM-ELs) are a promising technology that can produce low-cost green hydrogen. However, large scale commercialisation of PEM-ELs requires the use of high loadings (>2 mg cm−2) of expensive and scarce iridium to drive the oxygen evolution reaction. The ability to produce active and durable catalysts for long-term use without any Ir has long been a major challenge within this field. Many efforts have focused on reducing Ir (<75 wt%) by synthesising Ir-based supported catalysts. However, to date, such supported catalysts have shown insufficient stability under commercially relevant durability testing. To address this critical challenge, we report a unique water oxidation catalyst that deploys a Ir–AuPd–TiO2 OER catalysts which demonstrates excellent performance under acidic conditions. We assess the catalysts in both a half-cell rotating disk electrode setup as well as in a membrane electrode assembly. Impressively, after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 accelerated stress test cycles, our catalyst has a comparable degradation rate to commercial Ir catalysts. 000 accelerated stress test cycles, our catalyst has a comparable degradation rate to commercial Ir catalysts.
|
1. Introduction
The increasing environmental threats from rising global temperatures due to fossil fuel combustion has expedited the demand for clean renewable energy. However, the intermittent and fluctuating nature of renewable energy technologies such as wind and solar inherently requires coupling to energy storage technologies.1 Electrolytic hydrogen has been shown to be a sustainable energy carrier2 and can provide storage for renewable energy.3 Additionally, hydrogen is a feedstock in the chemical industry, as part of a mix of gases in steel production, fertiliser production, and can be used for heat and power generation. In 2021, the total annual production of hydrogen reached 94 million tonnes (Mt) globally.4 The majority (95%) of hydrogen is produced via hydrocarbon reforming which emits CO2.5,6 Indeed, the 90 Mt produced globally in 2020 emitted approximately 900 Mt CO2.7 The demand for hydrogen is expected to grow rapidly in the near future. Zero carbon methods of hydrogen production, such as electrolysis powered by renewable electricity, is a promising pathway to both store renewable energy and decarbonising such critical industrial chemical processes.8Several types of electrolysers exist today including proton exchange membrane water electrolysers (PEM-ELs) which are considered promising owing to their high efficiency and production of high purity H2.8 However, current commercial PEM-ELs require the use of expensive and scarce Ir-based catalysts at relatively high loadings (>2 mgIr cm−2)9,10 to achieve optimum activity and durability, largely due to the sluggish oxygen evolution reaction (OER).10,11 The high cost and low abundance of Ir represent a significant barrier to the scalability of the technology.12,13 Therefore, the development of high performance and cost effective OER electrocatalysts is critical to the successful widespread deployment of PEM-ELs.14
There have been many efforts focused on reducing Ir loading without compromising electrochemical activity and stability. For example, highly active electrocatalysts have been sought by employing mixed metal oxides (e.g. perovskites or pyrochlores), or by modifying the electrocatalyst's morphology to generate core–shell nanoparticles,15–17 nanorods,18,19 or nanowires.20 The development of supported Ir catalysts represents an alternative, and highly desirable pathway towards high-performance catalysts at drastically lower Ir loadings.21–24 To be viable in electrochemical applications, catalyst supports themselves must also have high chemical stability, sufficient electrical conductivity, high surface areas and be cost-effective.14,25 Metal oxides have recently received significant attention as supports for Ir catalysts for PEM-ELs, however, metal oxides inherently demonstrate low conductivity and so to mitigate this doped metal oxides have been explored.26,27 In particular, antimony doped tin oxide has gained attention due to its high conductivity and enhanced Ir utilisation, achieving high OER activity and stability under acidic environments.21,28,29 However, studies have shown that in many cases the dopant (Sb) leaches over time, thereby reducing the stability of the support and thus inducing the dissolution of the Ir catalysts.30,31 Consequently, exploring alternative strategies to develop high performance catalyst supports remains a critical challenge to fully exploit the potential of PEM-ELs at scale.
Amongst metal oxides, titanium dioxide (TiO2) is a very attractive OER catalyst-support because it has high resistance to corrosion and several TiO2 synthesis routes have been developed to prepare high surface area nanostructures.32 Despite these attributes, the low conductivity of TiO2 is a hinderance, with Ir loadings as high as 75 wt% required to achieve commercially relevant OER activities.33 Strategies to increase the conductivity of TiO2 have therefore been explored, including the synthesis of sub-stochiometric oxides.26,34,35 Nanostructuring strategies such as core–shells15,36–38 and high surface area TiO233 have also been employed to maximise Ir utilisation. Conductive layer coated supports (CCSs), where a conductive Pt interlayer is introduced between the TiO2 core and Ir catalyst, have recently shown 141% higher OER mass activity compared to commercial 75 wt% IrO2–TiO2 in a rotating disk electrode (RDE).9 The nanostructured conductive interlayer allows for a lower total platinum-group metal (PGM) loading and simultaneously enhanced conductivity and is thus a promising approach for lowering Ir catalyst loading in PEM-ELs.
In this study, we investigate a new catalyst-support morphology, deploying a hollow TiO2 support coated with alloyed AuPd nanoparticles (AuPd–TiO2). We evaluate the effects of AuPd nanoparticle loading (1 wt% and 5 wt%), Ir loading (25 wt% and 50 wt%) and annealing environment (reducing and oxidised) to decipher how these factors correlate with catalyst conductivity and OER activity. A previous study by Oakton et al. has shown that the conductivity precipitously decreases below 40 mol% Ir content.12 Thus, we selected a composition (25%) from low conductivity region and another from a region (50%) where conductivity starts to exponentially increase in this study. The electrochemical performance is assessed in both RDE half-cell setup and in a membrane electrode assembly (MEA) enabling comparisons between these two distinct electrochemical testing configurations. By RDE, we observe the highest mass activities (936 A gIr−1 at 1.65 VRHE) for 50 wt% Ir supported on 5 wt% AuPd–TiO2 annealed in a reducing atmosphere (50-WH5-H2) outperforming the 1 wt% AuPd–TiO2 and uncoated TiO2 supports. Interestingly, the highest performing catalyst tested in RDE was also the highest performing catalysts in MEAs when subjected to 10k accelerated stress test (AST) potential cycles. Specifically, 50-WH5-H2 reached 1 A cm−2 at 1.82 V whereas the less conductive 50 wt% Ir–1 wt% AuPd–TiO2 (50-WH1-H2) required 1.88 V to reach 1 A cm−2 after 10k AST cycles. Furthermore, the average voltage decay for the highest performing catalyst was 3 μV per cycle at 1 A cm−2 which is comparable to state-of-the-art IrO2 catalysts.39 Such high durabilities for supported catalysts demonstrate that our catalysts are state-of-the-art when compared against reported materials in the literature for PEM-ELs under analogous MEA architectures, operating conditions and accelerated stress test profiles. Analysis using X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and electron microscopy show that the Ir catalysts maintain the as-synthesised structural and electronic properties after AST in an MEA, which we hypothesise supports the material's high electrochemical stability.
2. Experimental section
2.1. Synthesis of hollow TiO2 and AuPd–TiO2
The synthesis of hollow TiO2 and AuPd–TiO2 nanostructures has been previously reported elsewhere.40 In brief, the TiO2 nanostructured supports were synthesised by a sol–gel templating method, whereby polystyrene beads were coated with TiOH gel and calcined at 500 °C. The TiO2 supports were subsequently decorated with AuPd alloy nanoparticles (Au/Pd w/w = 5:1). To integrate AuPd alloy nanoparticles, Au nanoparticles were initially synthesised by reducing HAuCl4·3H2O41 followed by alloying with Pd via a modified seed-mediated process.42 The AuPd nanoparticles were then added dropwise to a dispersion of the previously synthesised TiO2 while stirring at room temperature. After 2 hours, the powder was recovered by centrifugation and dried at 70 °C overnight. Two different nanoparticle loadings were prepared, which we label “WH1” and “WH5”. The AuPd loading was determined for each sample via ICP-OES (Table S1, ESI†) with WH1 the lower loading of 1.17 wt% Au and 0.26 wt% Pd and WH5 having the higher loading of 5.54 wt% Au and 1.08 wt% Pd.
2.2. Synthesis of Ir-WH1 and Ir-WH5
The deposition of iridium (Ir) onto the nanostructured TiO2 and AuPd–TiO2 supports (WH1 and WH5) was performed by a process adapted from a previously published incipient wetness impregnation synthesis.36 In brief, 134 mg of dihydrogen hexachloroiridate(IV) hydrate (H2IrCl6·6H2O, 40% Ir, Acros Organics) was mixed with 20 μL deionised H2O (18.2 MΩ, Milli-Q) and 40 μL acetic acid (99.7%, Fisher Scientific). Separately, each support (hollow TiO2, WH1 and WH5) was dispersed in 20 mL of ethanol to obtain a homogenous solution. The mass of the support was varied to enable control of Ir loading (25 wt% and 50 wt%). The prepared Ir and support dispersion were subsequently bath sonicated for 10 min, then stirred at 80–100 °C until dry.Each dried supported catalyst powder was collected and finely ground using a pestle and mortar. Each powder was placed into a tube furnace, ramped to 500 °C at 10 °C min−1 and held for 2 hours at temperature under a reducing atmosphere (5% H2 and 95% N2) at 50 sccm. The furnace was then allowed to cool to room temperature and a controlled passivation was conducted by flowing O2 (50 sccm) for 20 s. The resulting powders were then finely ground again. Each of the 50 wt% Ir supported catalysts were split into two batches and one half was subsequently annealed further in a box furnace (500 °C at 10 °C min−1 under atmospheric conditions for 30 minutes), cooled to room temperature and ground again using a pestle and mortar. The other half of these samples, which had only been annealed under a reducing atmosphere were tested as prepared.
2.3. Physical characterisation
The crystal structure of the catalyst-supports was examined by XRD using PANalytical X’pert powder X-ray diffractometer with a Cu Kα source (λ = 1.5406 Å). Diffraction peaks were recorded in the range 2θ = 20°–60° with a step size of 0.013°, a measurement time of 89 seconds per step and a sample rotation rate of 60 rpm. The reference patterns were identified using the Inorganic Crystal Structure Database (ICSD) and Scherrer analysis was performed on the 2θ peaks at 25.0° and 40.8° for Ir and Ti, respectively. XPS was performed using a Kratos Axis Supra system with a monochromated Al Kα1 radiation (hv = 1486.6 eV). The survey spectra are collected with a pass energy of 160 eV and region scans with a pass energy of 40 eV. All spectra were energy shifted to align to the C 1s peak at 284.8 eV. The morphologies of the supports were determined on a Zeiss Supra 40VP scanning electron microscopy (SEM) with EDAX 40VP energy dispersive X-ray (EDX) analyser. High resolution transmission electron microscopy (HRTEM) imaging of the catalysts was performed using an FEI Talos F200X and scanning TEM (STEM) was acquired using a probe aberration corrected FEI Titan G2 80-200 ChemiSTEM, with both microscopes operated at 200 kV. Particle sizes were determined using high-angle annular dark field (HAADF) STEM imaging, collected using 110 pA beam current, 21 mrad convergence angle, and a 48 mrad HAADF inner collection angle. STEM elemental mapping was performed by EDX spectroscopy using a Super-X quad silicon drift detector with a total collection angle of 0.7 sr. TEM samples were prepared by dry powder deposition onto holey carbon support films on 400 mesh Cu grids (Agar Scientific). The statistical analysis of Au and Ti particle sizes were determined using GraphPad Prism version 9.5.1 for Windows (GraphPad Software, San Diego, California USA). The statistical normally distributed data were compared with an unpaired t-test and non-normally distributed data were compared with Mann–Whitney U tests. The categorical frequency data were compared using the chi-squared test with post hoc Fisher's exact testing and results were deemed significant if p < 0.05. The elemental dissolution of the catalyst-supports was determined by analysing aliquots of electrolyte taken post electrochemical testing using inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7900). The metal loading of the supports was determined by inductively coupled plasma optical emission spectrometry (ICP-OES, Agilent 5800), where 10 mg of AuPd–TiO2 was digested in 10 mL of aqua regia, filtered and diluted to 50 mL.2.4. Conductivity measurements
The conductivities of the catalyst powders were measured in a 2-electrode conductivity cell developed in house, analogous to those previously reported in the literature.43–45 In brief, two copper electrodes were wrapped in a hole punched PTFE (Polyfon, thickness of 0.127 mm) to isolate the two electrodes. The PTFE hole acted as a sample holder for powder samples. The powder samples were then compressed between the electrodes under constant pressure at 500 psi. The conductivity cell was connected to a potentiostat (Metrohm, PGSTAT 204) to generate I–V curves using linear sweep voltammetry (LSV) from −0.4 to 0.4 V, to determine the electrical resistance and the conductivity calculations are discussed in the ESI.†2.5. Half-cell electrochemical characterisation
Half-cell electrochemical measurements were conducted on a RDE (Pine Research) and VSP-3e potentiostat from BioLogic Science Instruments in a three-electrode configuration. A 0.1 M HClO4 electrolyte (pH 1.1), gold disk working electrode (0.196 cm2), graphite rod counter electrode and Hg/HgSO4 reference electrode (OrigaSens, Alvatek) were used throughout. The synthesised supported catalysts were compared against commercial iridium(IV) oxide (IrO2, Premion 99.99%, Alfa Aesar). All potentials were adjusted to the reversible hydrogen electrode (RHE) as detailed in the ESI.†Catalysts were deposited onto the gold disk working electrode using a previously reported modified method.9,46 The catalyst inks were prepared by dispersing 2 mg of catalyst powder in 360 μL 1-propanol (NPA, 99%, Fisher Scientific), 120 μL deionised water (18.2 MΩ, Milli-Q) and 20 μL Nafion™ (5 wt%, Alfa Aesar). Inks were bath sonicated (FB15048, Fisherbrand) for 30 min prior to electrochemical testing followed by immediately drop casting onto the RDE Au disk at 200 rpm. The disk was subsequently rotated at 700 rpm for at least 30 minutes until dry. The theoretical Ir loading of each catalyst was 25.5 μgIr cm−2. Each synthesised catalyst ink was deposited and tested 3 times. Between each deposition, the electrode was polished with 0.05 μm alumina slurry, rinsed and sonicated for 30 seconds in deionised water.
Prior to electrochemical OER testing, potentiostatic electrochemical impedance spectroscopy (PEIS) measurements with 85% correction were conducted at 1.1 VRHE (100 mHz to 200 kHz) and used to iR correct the reported potentials. The catalyst deposited working electrode was conditioned using cyclic voltammetry (CV) between 0.025–1.0 VRHE for 50 cycles at 200 mV s−1. The OER activity was subsequently assessed at 2500 rpm using CV by sweeping the potential between 1.0–1.8 VRHE at 10 mV s−1 for 10 cycles. We used the performance of 10th CV cycle for analysis unless otherwise stated. To examine the stability in an RDE configuration, a CV was measured from 1.0–1.8 VRHE for 30 cycles at 10 mV s−1 and 2500 rpm.
2.6. Membrane electrode assembly
Catalyst inks were prepared following a method described elsewhere.47 Briefly, commercial 50 wt% Pt/C, deionised water, NPA and Nafion™ were mixed and bath sonicated for 30 minutes. The supported catalyst ink for the anode was mixed with deionised water, NPA and Nafion™ and horn sonicated (505, Fisherbrand) for 30 minutes. The vial was covered with parafilm and housed in an ice bath throughout sonication.The catalyst coated membranes (CCMs) were prepared by drop casting the catalyst inks onto PTFE films on a vacuum hot plate at 53 °C and left to dry. Decal transfer was achieved by sandwiching the Nafion membrane (N212, Chemours) between the catalyst deposited PTFE films using a hot press under 1 metric ton pressure at 130 °C for 3 minutes.48 The catalyst loading is determined via gravimetric measurements of the PTFE before and after the decal transfer. The anode catalyst loading ranged between 1.58–1.62 mgCat cm−2 and cathode catalyst loading was 0.7–0.84 mgCat cm−2.
The CCMs were integrated into a single cell electrolyser with Pt-plated single serpentine 5 cm2 titanium flow field on the anode, single serpentine 5 cm2 graphite flow field on the cathode, water feed at 120 mL min−1 on the anode and N2 (100% RH) at 50 sccm at the cathode. Platinised sintered titanium porous transport layers (PTLs) from Mott corporation USA were used on the anode and carbon cloth gas diffusion layers (GDLs) with mesoporous layer (MPL) from FuelCellStore on the cathode. The cell was equilibrated at 60 °C for an hour prior to collecting polarisation curves using a BioLogic potentiostat (VSP-3e) with a 20 A booster (VMP3B-20). PEM-EL polarisation curves were generated via chronoamperometry (CA) steps collected for 3 minutes at increments of 0.05 V from 1.2–2.0 V. For each voltage step, the average current density of the last 30 s was used for the polarisation curve. For the saw-tooth voltage cycling accelerated stress tests (ASTs), 10k CVs were collected at 50 mV s−1 between 1.20 and 2.00 V. Polarisation curves were collected after each 1k cycles to assess performance degradation due to AST CVs. The concentration of dissolved Ir from the supported catalysts post-AST were evaluated by conducting inductively coupled plasma mass spectrometry (ICP-MS) on a 5 mL aliquot of the anode water outlet at the end of the ASTs.
3. Results and discussions
3.1. Electrochemical half-cell performance
In this study, nanostructured hollow TiO2 catalyst supports were utilised as catalyst supports. The prepared nanostructures consisted of Ir deposited by wet impregnation onto AuPd–TiO2 supports (WH1 and WH5) and TiO2 supports (without AuPd). The catalyst powders were subjected to either: (1) a thermal reduction (5% H2/95% N2) (TiO2-H2, WH1-H2 and WH5-H2) or (2) a thermal reduction (5% H2/95% N2) followed by annealing in air (TiO2-Air, WH1-Air and WH5-Air). For each catalyst prepared, two different Ir loadings (25 wt% and 50 wt%) were prepared. The samples are labelled as follows, “Ir wt%-catalyst support-annealing environment” as summarised in Table 1 alongside key synthetic details.| Sample name | Ir wt% | Support | Annealing environment | Annealing programme |
|---|---|---|---|---|
| 50-TiO2-H2 | 50 | Open nanoshell TiO2 | 5% H2/95% N2 | 500 °C, 2 hours, 10 °C min−1 |
| 50-WH1-H2 | 50 | 1.1% Au/0.3% Pd–TiO2 | 5% H2/95% N2 | |
| 50-WH5-H2 | 50 | 5.5% Au/1.2% Pd–TiO2 | 5% H2/95% N2 | |
| 25-WH1-H2 | 25 | 1.1% Au/0.3% Pd–TiO2 | 5% H2/95% N2 | |
| 25-WH5-H2 | 25 | 5.5% Au/1.2% Pd–TiO2 | 5% H2/95% N2 | |
| 50-TiO2-Air | 50 | TiO2 | 1. 5% H2/95% N2 | 1. 500 °C, 2 hours, 10 °C min−1 |
| 2. Air | ||||
| 50-WH1-Air | 50 | 1.1% Au/0.3% Pd–TiO2 | 1. 5% H2/95% N2 | |
| 2. 500 °C, 0.5 hours, 10 °C min−1 | ||||
| 2. Air | ||||
| 50-WH5-Air | 50 | 5.5% Au/1.2% Pd–TiO2 | 1. 5% H2/95% N2 | |
| 2. Air |
The activity of the prepared catalyst-supports were initially assessed using a three-electrode RDE in acidic electrolyte (0.1 M HClO4). Typically, 0.5 M H2SO4 is used for electrochemical measurements however, within this work 0.1 M HClO4 was utilised to conduct the electrochemical half-cell measurements to mitigate the strong binding effect of SO4− to the catalyst which hinders the OER performance.49 It has been suggested that more electronegative anions have higher OER performance as it increases the likelihood of the ionic dissociation from the surface, thereby leaving more active sites exposed for OER.50 When catalysts are compared on a geometric current density basis, it is clear that the supported catalysts containing AuPd nanoparticles (WH1 and WH5) exhibited vastly superior OER performance over bare TiO2 (Fig. 1A). In addition to the presence of AuPd, thermal annealing conditions and iridium loading also had significant impacts on OER activities. Specifically, the average geometric current density at 1.65 VRHE for 50 wt% Ir loaded supported catalysts annealed in H2 is 21.2 mA cm−2. When the same material is subsequently annealed in air, the current density is reduced to 0.5 mA cm−2 (Fig. 1A). This is in agreement with previous literature whereby the annealing environment (air) led to decreased activities.51 Two distinct Ir loadings were synthesised and annealed under a reducing environment: 25 wt% and 50 wt% for both WH1 and WH5 supports. The average geometric area normalised current densities at 1.65 VRHE for 25 wt% Ir are 1.4 mA cm−2 and 21.2 mA cm−2 for 50 wt% Ir, respectively (Fig. 1A).
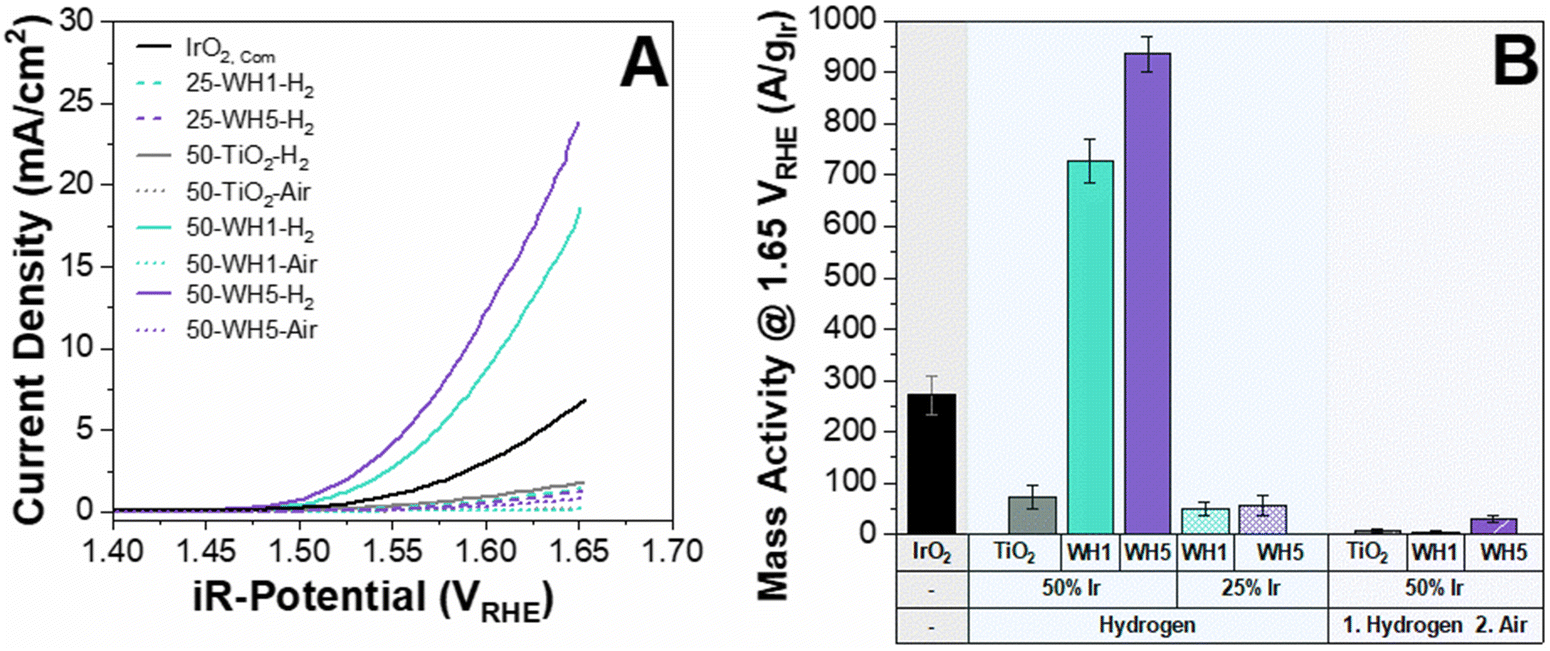 | ||
| Fig. 1 (A) Geometric area normalised OER activities of supported Ir catalysts and (B) OER activities normalised to the mass of Ir. All experiments were conducted at least in triplicates to generate error bars. The electrolyte was 0.1 M HClO4 electrolyte. Activities are reported for the 10th CV cycle with a theoretical Ir loading of 25.5 μgIr cm−2 and compared against unsupported commercial rutile IrO2 sample. | ||
The electrochemical impedance spectroscopy was measured between 100 mHz–200 kHz prior to the OER measurements to determine the ohmic resistance of each supported catalyst (Fig. S1, ESI†). The fitting is an estimation based on a modified Randles circuit.52,53 We observed that the ohmic resistance was <30 ohm for AuPd–TiO2 based samples annealed under H2 conditions compared to >30 Ohm for similar samples under air annealing. Interestingly, the Ohmic resistance of 25 wt% Ir supported catalysts was similar to its 50 wt% Ir counterpart despite lower OER performance (Fig. 1B).
To assess Ir utilisation, mass activities (A gIr−1) were calculated (Fig. 1B) using the theoretical Ir loading (details provided in the ESI†). Overall, catalysts with 50 wt% Ir have much higher mass activities than 25 wt%. For example, 50-WH5-H2 has a mass activity of 936 A gIr−1 compared to 25-WH5-H2 with 49 A gIr−1 at 1.65 VRHE. To evaluate the role of AuPd loading as a conductive additive to the support structure, two different AuPd loadings (WH1 and WH5) with 50 wt% Ir were compared at 1.65 VRHE. We observed that 50-WH5-H2 achieved a higher mass activity (936 A gIr−1) than 50-WH1-H2 (728 A gIr−1). By comparing these mass activities to 50-TiO2-H2 (72 A gIr−1 at 1.65 VRHE) we highlight that the AuPd significantly enhanced the electrochemical performance of our catalysts. When the prepared supported catalysts are benchmarked against unsupported commercial IrO2, we observed higher mass activities for 50-WH1-H2 and 50-WH5-H2 compared to commercial IrO2 (Fig. 1B). Furthermore, our highest performing OER catalysts demonstrated comparable mass activities to previously reported Ir-based catalysts prepared on TiO2-based supports when assessed in an RDE (Fig. 2 and Table S2, Fig. S2, ESI†).
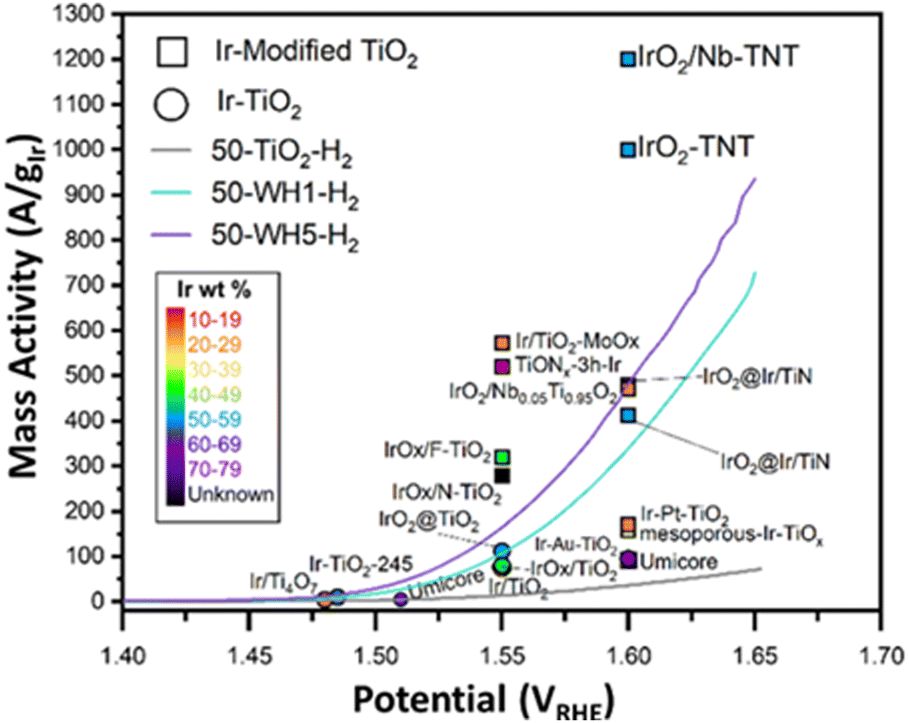 | ||
| Fig. 2 Mass activity of various Ir–TiO2 catalysts9,12,22,26,28,36,54–60 for the OER in acidic electrolyte compared to the results for the materials in this work (50-TiO2-H2, 50-WH1-H2, 50-WH5-H2). The circular symbols indicate Ir–TiO2 catalysts only, whereas the square symbols represent Ir-modified TiO2 (e.g., the addition of either metal or non-metal elements to the TiO2 support). The different colours detail the Ir wt%. | ||
To provide a preliminary screening of catalyst stability, RDE measurements were conducted. Firstly, the supported catalysts were conditioned by cycling between 0.025–1.0 VRHE at 200 mV s−1 for 50 cycles and subsequently by performing 10 CVs between 1.0–1.8 VRHE at 10 mV s−1 (Fig. S3, ESI†). For the thermally reduced supported catalysts with 25 and 50 wt% Ir, we observed an increase in activity during the first 10 CVs, irrespective of the AuPd loading (Fig. S3a, ESI†). It is interesting to highlight that the thermally oxidised supported catalyst 50-WH5-air showed minimal changes in activity during the first 10 CVs (Fig. S3b, ESI†). Conversely, thermally oxidised supported catalysts 50-WH1-air and 50-TiO2-air showed a deactivation of catalyst activity after 10 CV cycles. Similar dynamic behaviour of activation as a function of CV cycling has been observed in previously reported Ir-based OER catalysts, and has been attributed to the electrochemical growth of a hydrous Ir which can form during potential cycling.61,62 To assess the kinetics of the synthesised catalyst-support, Tafel analysis was performed (Fig. S4, ESI†). For each of the catalysts prepared, two distinct Tafel regions were observed corresponding to two distinct regions of the CV: low (<300 mV) and high overpotentials (>300 mV). This has previously been attributed to the presence of different active sites, or changes to the catalyst surface (reorganisation) at different applied potentials.9,63
3.2. Conductivity measurements
Triple phase boundaries (TPBs) are regions where the electrolyte, gas and catalyst are in contact leading to the OER to occur.64,65 These TPBs can enhance OER performance when there is high conductivity.65 To probe the role of conductivity on electrochemical OER activity, conductivity measurements of the synthesised supports and supported catalysts were conducted using an in-house developed cell (Fig. 3). It is important to note that owing to the nature of the conductivity setup, these values are not absolute conductivity values, but rather a method to enable in-house comparisons. All hollow TiO2 supports were found to have low conductivities irrespective of the AuPd loading or thermal annealing conditions (10−8–10−9 S cm−1). Interestingly, when Ir is deposited onto the catalyst-supports in combination with AuPd, a higher conductivity is observed for both Ir loadings (25 wt% and 50 wt%) and under both thermal treatments (reducing or oxidised) when compared to sibling samples prepared identically, but without AuPd. The presence of Au has previously been identified as a factor in increasing conductivity as the Au thickness increases.66,67 Similarly, in this work the catalysts with Ir deposited on WH5 have higher conductivities (10−2–10−7 S cm−1) compared to those supported on WH1 (10−3–10−8 S cm−1). Finally, when all other synthetic parameters remain constant, we observe significantly higher conductivity (10−2–10−8) for thermally reduced samples compared to the thermally oxidised supported catalysts (10−4–10−9 S cm−1).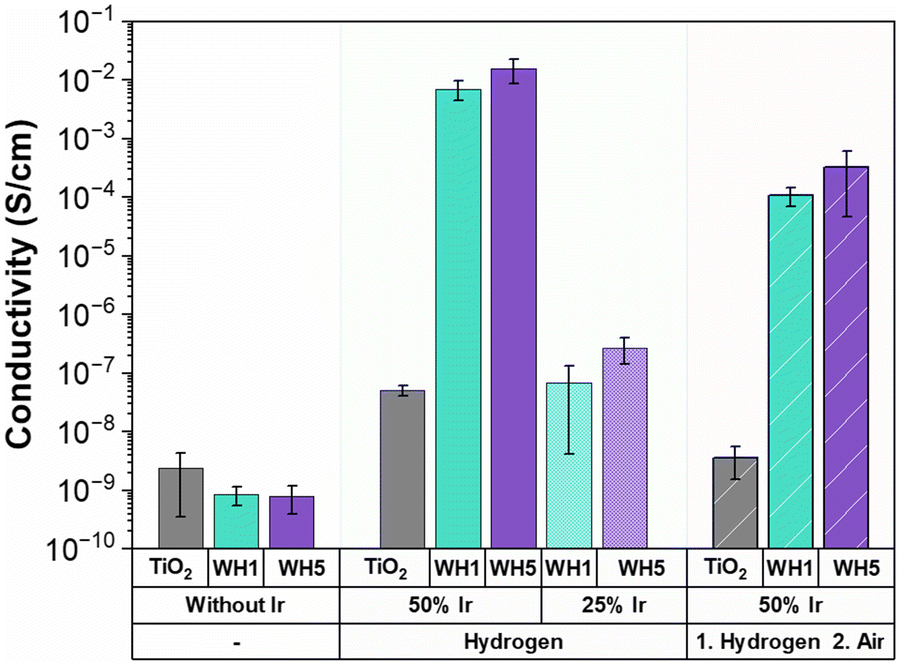 | ||
| Fig. 3 Conductivity of the various supports and catalyst-support motifs synthesised. Conductivity was measured with an in-house developed conductivity cell and are reported as an average of triplicate measurements. | ||
When analysing the OER performance (Fig. 1), it is noticeable that the two most conductive supported catalyst are indeed the two most active catalysts, suggesting that conductivity is a critical parameter for designing active OER catalysts. However, while the 50-WH1-air and 50-WH5-air samples have significantly higher conductivities compared to 25-WH1-H2 and 25-WH5-H2, their OER performance is rather similar. These results combined with the measured ohmic resistance (Fig. S14, ESI†) highlights the importance of obtaining optimal material composition with high conductivity to increase TPBs conditions leading to high OER performance. Thus, we can conclude that while reasonably high catalyst conductivity is essential for OER activity, such ex situ powder conductivity measurements are not the sole predictor for OER activity trends in these supported catalysts.
3.3. Catalyst-support materials characterisation
The hollow TiO2 supports are observed to have an open nanoshell morphology and are ∼270 nm in diameter (Fig. 4A and Table S3, ESI†). High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) imaging coupled with energy dispersive X-ray spectroscopy (EDS) was utilised to probe the morphology (Fig. 4B) and dispersion of Ir, Au, Pd and Ti (Fig. 4C–F and Fig. S6, S9–S11, ESI†). This confirmed the presence of spherical TiO2 motifs, and AuPd alloy nanoparticles non-uniformly distributed across TiO2 for the as synthesised AuPd–TiO2 supports (WH1 and WH5). It is notable that the Ir, Au, and Pd are distributed both within the hollow TiO2 (concave surface) as well as externally (convex surface). Analysis of lattice fringes visible in high resolution transmission electron microscopy (HRTEM) images of the AuPd nanoparticles on the WH1 support (Fig. S6e, ESI†), indicates a d-spacing of 0.24 nm, which can be attributed to the expected {111} lattice spacings of face centred cubic (FCC) AuPd alloy.68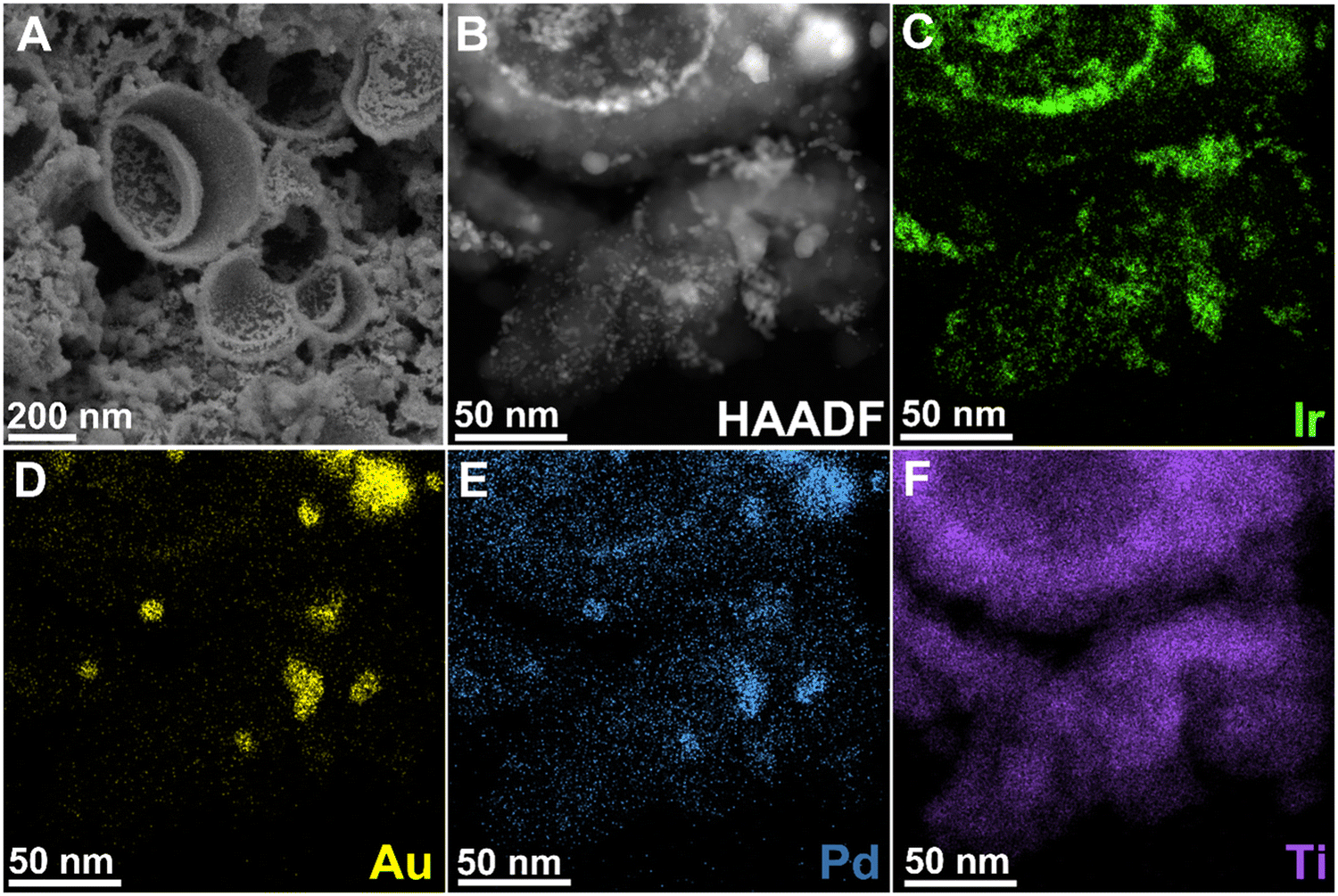 | ||
| Fig. 4 (A) SEM image of 50-WH5-H2, (B) HAADF-STEM image of 50-WH5-H2. (C)–(F) STEM-EDS elemental maps for Ir, Au, Pd and Ti, respectively. | ||
The average AuPd particle diameter in WH1 and WH5 is ∼8 nm (Table S3, ESI†) prior to Ir deposition (Fig. S6, ESI†). The Kolmogorov Smirnov tests of normality69 indicated that the size of AuPd particles in WH1 (as prepared) were not normally distributed whereas particles in WH5 showed normal distribution (Fig. S7, ESI†). Additional statistical analysis (chi-squared test) was used to identify the frequency of the particle sizes. By grouping the AuPd particles by size (<5, ≥5–<9, ≥9–<13, 13+), we observed a significant discrepancy (when p < 0.05) in the particle sizes distributions between WH1 and WH5 (p = 0.01). Therefore, post hoc Fisher's exact tests were conducted and identified that WH1 contained a significantly higher proportion of ≥5–<9 nm particles compared to WH5 which had a higher proportion of ≤5 nm particles (p = 0.025). The TiO2 hollow sphere particle sizes were measured from SEM images and show comparable average diameters of 270 nm in WH1 and 275 nm in WH5 (Table S3 and Fig. S7b, ESI†). Chi-squared analysis on TiO2 particle size proportion showed no significant difference (p = 0.53).
The morphology of the thermally reduced 25 and 50 wt% Ir loaded catalysts and the Au, Pd, Ir and Ti distributions were determined via SEM, HAADF-STEM imaging and STEM-EDS mapping (Fig. 4 and Fig. S9, respectively, ESI†). Using SEM, no discernible comparison could be obtained due to the limitations of electron microscopy on the small nanoparticles (Fig. S8, ESI†). A non-uniform distribution of Ir and AuPd particles across the TiO2 supports is observed post annealing via TEM. Generally, the AuPd nanoparticles appear to be co-located with Ir, while there are also some Ir agglomerates isolated from the AuPd. Additionally, we observe that the Ir appears to have been deposited both the inside (concave surface) and outside (convex surface) of the TiO2 open nanoshell.
The crystallinity of the supports and supported catalysts was assessed by XRD (Fig. 5A and Fig. S5, ESI†). For all samples, we observed 2θ peaks for crystalline anatase TiO2 at 25°, 48°, 54° and 55°. In all AuPd containing samples, peaks are observed at 38° and 44° corresponding to a FCC AuPd alloy. The thermally reduced samples contain a peak at 40° corresponding to FCC metallic Ir, whereas samples that were first reduced and subsequently thermally oxidised exhibited a mixture of FCC metallic Ir and low intensity rutile IrO2 (28° and 34.7°) (Fig. S5b, ESI†).
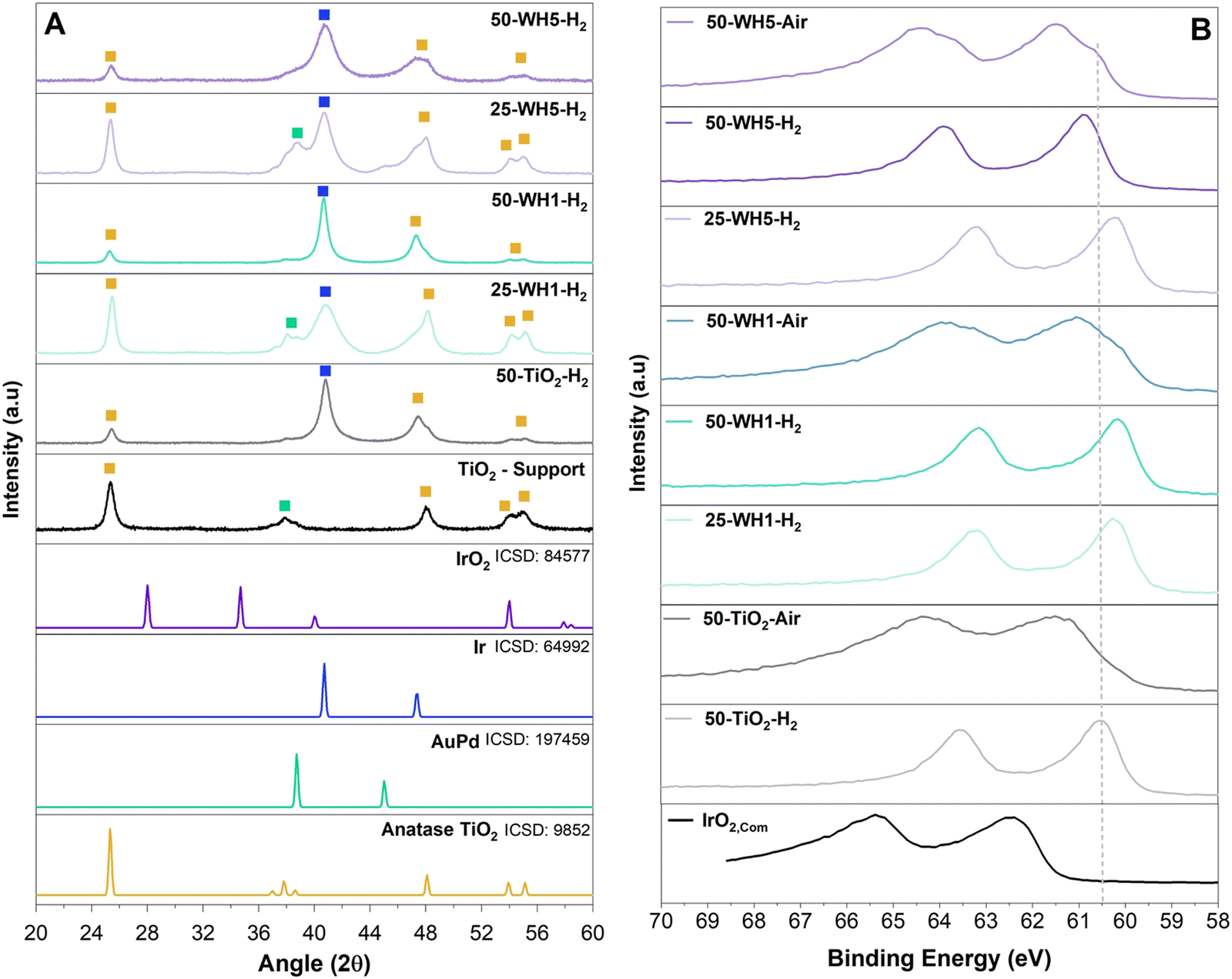 | ||
| Fig. 5 (A) XRD of the supported catalysts synthesised using 5% H2 in N2 atmosphere with reference patterns corresponding to anatase TiO2 (yellow), AuPd (green) and metallic Ir (blue) as shown by the symbols. The relevant ICSD collection codes are also provided. (B) XPS spectra of Ir 4f for the synthesised supported catalysts compared to a commercial IrO2. A dashed line is added as a guide for the reader. | ||
The crystallite size of the TiO2 support was assessed by the Scherrer equation and was shown to correspond approximately to the width of walls of the TiO2 nanospheres (∼14–16 nm). This remained constant irrespective of the composition or the synthesis environments (Table 2). Conversely, the crystallite size for Ir was found to vary across the samples. A temperature of 500 °C for H2 annealing was applied to the supported catalyst to ensure high crystallinity of the Ir. Crystalline Ir has been shown to improve the stability of the catalyst and adequately remove Cl− impurities.70–72 The additional annealing in air was conducted to form a thin layer of IrO2 to further enhance the stability of the catalyst as metallic Ir is more unstable than IrO2.73 In both annealing environments (H2 and air), broader metallic Ir peaks for 50-WH5 samples indicated the presence of smaller Ir crystallites (<5 nm). We estimate that annealing in air did not significantly impact the crystallinity of Ir as we do not see the presence of IrO2 potentially due to amorphous IrO2 formed as a result of low annealing time impacting the growth of crystalline IrO2. Narrower Ir XRD peaks were observed for 50-WH1 and 50-TiO2 indicating larger Ir metal crystallites of 10–14 nm. We speculate that the lower 25 wt% Ir in both WH1 and WH5 provides sufficient anchor sites to support these smaller Ir crystalline growth. Conversely, at the higher 50 wt% Ir loading, only WH5 has sufficient anchor sites to maintain the smaller crystallite size.
| Samples | Crystallite size (nm) | |
|---|---|---|
| Ir | TiO2 | |
| 25-WH1-H2 | 4 | 16 |
| 25-WH5-H2 | 5 | 15 |
| 50-TiO2-H2 | 10 | 14 |
| 50-TiO2-Air | 12 | 16 |
| 50-WH1-H2 | 13 | 16 |
| 50-WH1-Air | 14 | 16 |
| 50-WH5-H2 | 5 | 14 |
| 50-WH5-Air | 5 | 14 |
XPS analysis was performed on the as-synthesised supported catalysts and a commercial rutile IrO2 catalyst to probe the Ir oxidation state (Fig. 5B and Fig. S12, ESI†). For all of the synthesised samples, the observed Ir 4f doublets are shifted to lower binding energies compared to a typical rutile IrO2.74 Consistent with the XRD findings, the Ir in the thermally reduced samples is more metallic in nature (lower binding energies) compared to thermally oxidised samples. Furthermore, the thermally oxidised (air) samples show the clear presence of at least two doublets corresponding to a more oxidised (e.g., 4+) as well as reduced (metallic) Ir. Interestingly, no significant shift is observed in the Ir 4f region as a function of AuPd loading. The Au 4f region was also analysed. Typically, metallic Au 4f7/2 has a binding energy of 84.0 eV.75 A slight shift towards lower binding energies was observed for AuPd loaded supports (83.4 eV and 83.2 eV for WH1 and WH5, respectively) (Fig. S12a, ESI†). Upon addition of 25 wt% Ir (25-WH5-H2 and 25-WH1-air), the Au 4f region remains largely unchanged. However, with a higher Ir loading (50-WH5-H2 and 50-WH5-Air) a shift to lower Au binding energy <83.0 eV is observed, with 50-WH5 being slightly more oxidised compared to 50-WH1. Across all samples, the Ti 2p region remains unchanged, irrespective of AuPd, Ir loading and annealing environment (Fig. S12b, ESI†).76
3.4. Membrane electrode assembly performance
To understand the relevance of our catalyst design and RDE OER performance for PEM-ELs, we integrated our highest performing catalysts (50-WH1-H2 and 50-WH5-H2) into MEAs. The samples were benchmarked against an unsupported commercial IrO2 (Fig. 6). Nafion 212 membrane and a commercial 50 wt% platinum on carbon (Pt/C) cathode catalysts were used throughout. The anode catalyst loadings were 1.62 mgcat cm−2 for 50-WH1-H2 and 1.58 mgcat cm−2 for 50-WH5-H2. The unsupported IrO2 had a loading of 3 mgIrO2 cm−2. Fig. 6A and Fig. S11 (ESI†) shows the beginning of life (BoL) polarization curve for prior to AST potential cycling for the supported and unsupported catalysts. To reach 1 A cm−2, 1.79 V is required at the BoL for 50-WH5-H2 whereas 50-WH1-H2 requires 1.91 V. The commercial IrO2 only required 1.63 V to reach 1 A cm−2. The Ir utilisation was further evaluated by calculating Ir-mass activity (Fig. 6B and Fig. S11b, ESI†). We observe that both supported catalysts achieved superior mass activity compared to unsupported commercial IrO2 at 2 V (1.09 A mgIrO2−1). Specifically, 50-WH5-H2 achieved higher mass activities (2.42 A mgIr−1 at 2 V) compared to 50-WH1-H2 (1.60 A mgIr−1 at 2 V), indicating the critical role of the higher AuPd loading for OER activity. We also speculate that the smaller crystallite size present in 50-WH5-H2 leads to a higher performance MEA owing to a larger Ir surface area. More critically, we highlight that these mass activity trends in MEA agree with RDE mass activity measurements (Fig. 1 and Table S4, ESI†) where both supported catalysts exhibited higher mass activity than commercial IrO2.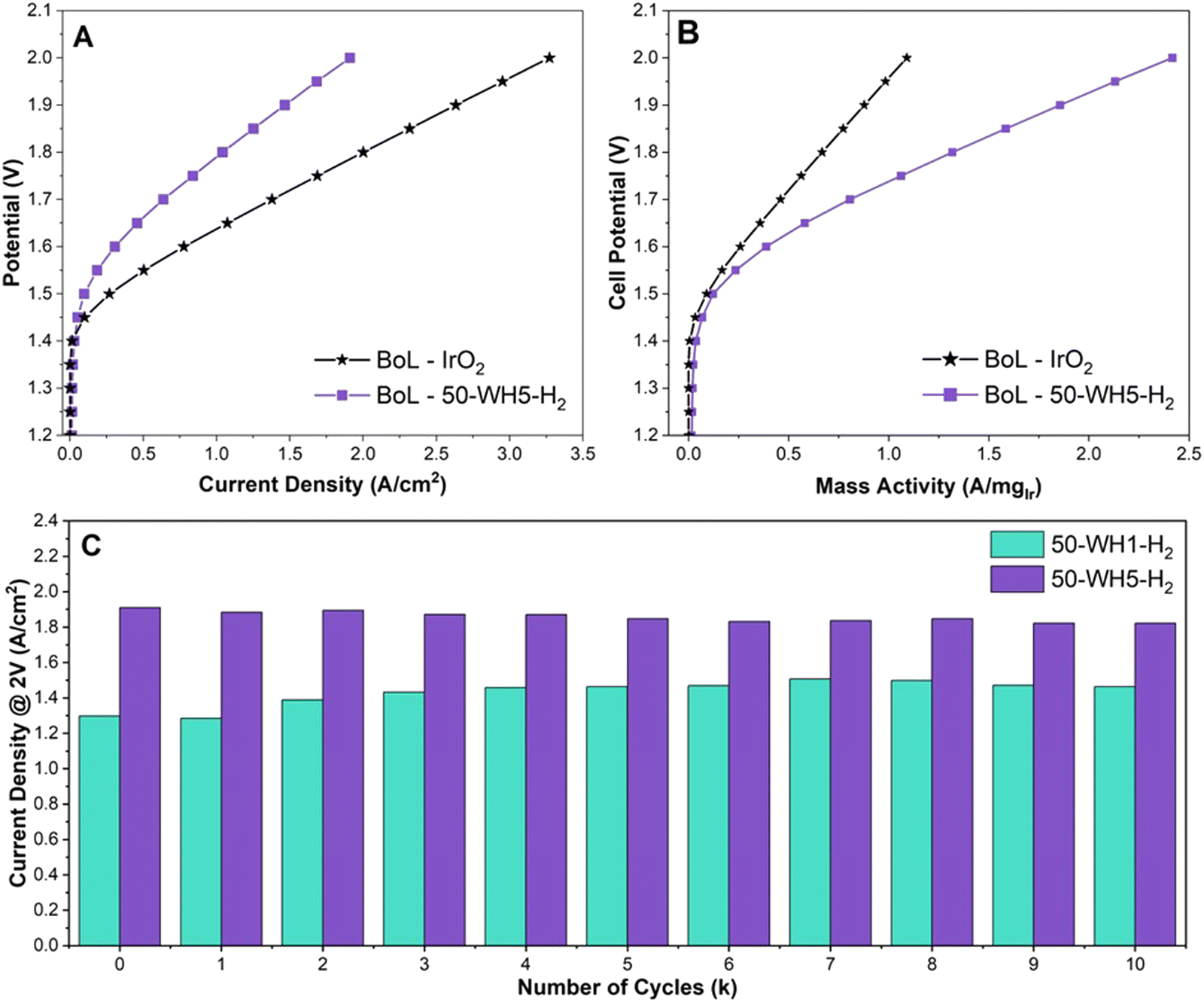 | ||
| Fig. 6 (A) Polarisation curve of proton exchange membrane water electrolyser deploying 50-WH5-H2 anode catalysts alongside a commercial rutile IrO2, (B) mass activity of the same catalysts and (C) performance of supported catalysts at 2 V after each thousandth cycle. Cell parameters: N212 membrane, 60 °C and N2 gas flow. OER catalyst loadings: 3 mgIrO2 cm−2 for IrO2 and 0.79 mgIr cm−2 for 50-WH5-H2. | ||
To assess catalyst stability in the single-cell MEA, we performed saw-tooth voltage cycling at 50 mV s−1 from 1.2–2 V for 10000 cycles (Fig. 6C and Fig. S13, ESI†). The current density to reach 2 V was analysed for both 50-WH1-H2 and 50-WH5-H2. 50-WH5-H2 showed a BoL current density of 1.91 A cm−2 and 1.82 A cm−2 after 10000 (10k) cycles (end of life, EoL). Conversely, 50-WH1-H2 at 2 V showed a BoL of 1.30 A cm−2 and EoL of 1.46 A cm−2 at 2 V. Interestingly, 50-WH1-H2 increased in performance at potentials >1.8 V after 10k cycles. Studies have shown that dissolution and redeposition of catalysts leads to improved OER performance due to the presence of a self-assembled catalyst layer.8–10 This could be due to either increase in Ir surface area or improved mass transport due to catalyst redeposition within the catalyst layer during AST potential cycling. Conversely, 50-WH5-H2 shows increased potential across current densities post AST. However, a voltage decay of 3 μV per cycle at 1 A cm−2 indicates minimal performance loss during the AST, suggesting that this catalyst is highly stable even in a MEA configuration. Direct comparison of MEA decay rates from literature are complicated by the differences in cell setup and AST profiles. The desired degradation rate for potential GW scale application of electrolysers is <6 μV h−1 and our results indicate that these TiO2 supported Ir catalysts exceed the desired degradation rates.39 However, chronopotentiometric holds exceeding thousands of hours at >1 A cm−2 instead of potential sweeps utilised here are necessary to empirically demonstrate such durabilities. Such long-term durabilities are deemed not viable in academic research laboratories and are better suited for commercial feasibility studies in the future.
The higher AuPd content in the support clearly has a positive influence on the performance and durability of the Ir catalysts when assessed in both MEA and RDE configurations. When benchmarked against previously reported supported Ir catalysts tested in an MEA (Table S4, ESI†), we observed that our catalysts required lower potentials to reach 1 A cm−2 compared to majority of the other catalysts, some with even higher anode catalyst loadings (>2 mgIr cm−2). However, we note that variations in cell temperature, membrane and testing conditions have a significant impact on the performance and thus a direct comparison (as we have done for RDE testing) is not possible.
To evaluate Ir dissolution in the MEAs, a sample of the test water was collected at the end of the AST and analysed using ICP-MS. We observed that Ir dissolution was highest for better performing 50-WH5-H2 (3.6 μg L−1) compared to 50-WH1-H2 (1.8 μg L−1) after 10k cycles. The increased Ir dissolution for 50-WH5-H2 may, at least to some extent, account for the decrease in activity during the AST 10k cycles (Fig. 6C). We hypothesise that the larger Ir crystallite size (∼13 nm) in 50-WH1-H2 could leach at slower rate than the smaller crystallites (∼5 nm) in 50-WH5-H2. It is also plausible that the larger Ir particles in 50-WH1-H2 could be undergoing nanostructuring leading to higher surface area under the prevailing electrochemical conditions, and thus may explain the observed improved performance post-AST. We note that Ir is potentially redeposited onto the support rather than irreversible leaching.77 It has been shown previously that most of the leached Ir is likely trapped within the membrane, transport layers and catalyst layers rather than entering the water feed, meaning that the majority of the trapped Ir will not be detected in our ICP analysis.78
To assess any changes in the oxidation state during the AST, we conducted EoL XPS analysis of the tested MEAs (post AST) which were compared to the as synthesised catalyst powders (Fig. S14, ESI†). In both supported catalysts, the Ti 2p region remains unchanged post AST when compared to the as synthesised catalyst powder (Fig. S14c, ESI†). As discussed previously, the Ir surface of the as synthesised 50-WH1-H2 and 50-WH5-H2 was predominantly metallic in nature (Fig. 5B). However, a slightly lower binding energy was observed for 50-WH1-H2 (∼60.2 eV) compared to 50-WH5-H2 (∼60.8 eV). Post AST, 50-WH1-H2 was shown to have shifted slightly to higher binding energy (∼0.2 eV) indicating a more oxidised surface. Conversely, 50-WH5-H2 Ir 4f peaks were shifted to lower binding energy (∼0.7 eV) suggesting a more reduced catalyst surface. Interestingly, the Au 4f7/2 (Fig. S14b, ESI†) region showed lower binding energies (more metallic) for 50-WH1-H2 compared to 50-WH5-H2 in the as prepared powders as well as post AST. For both samples, the peaks shift to a higher binding energy post AST indicating that the Au is oxidised.
4. Conclusion
We demonstrate a unique, hollow TiO2 structure decorated with AuPd nanoparticles as a high-performance catalyst-support for iridium-based OER electrocatalysts. We show that the AuPd, Ir loadings and thermal annealing environment are critical to OER performance. Furthermore, catalysts prepared with a higher AuPd and Ir loading outperform those prepared with lower loadings. Specifically, the highest performing catalyst-support exhibited 3-times higher mass activity at 1.65 VRHE (936 A gIr−1) compared to commercial IrO2 (271 A gIr−1) in half-cell RDE testing. The highest performing catalysts were integrated into a membrane electrode assembly and subjected to 10k accelerated stress test consisting of saw-tooth potential sweep cycles. The same trend in OER activity were found to translate from the half-cell measurements to full devices (50-WH5-H2 > 50-WH1-H2). Our highest performing catalyst (50-WH5-H2) by RDE testing showed the highest mass activity in an electrolyser achieving 2.42 A mgIr−1 at cell potential of 2 V compared to 1.09 A mgIrO2−1 for commercial IrO2. Even more impressively, our catalyst showed state-of-the-art device durabilities when compared to reported values under analogous electrolyser architecture, operating conditions and accelerated stress test protocols. Future studies will further investigate this class of OER catalysts through additional durability testing (chronopotentiometry and chronoamperometry in single cell and stack). Furthermore, intermediate compositions (between 25% and 50% Ir loading and higher Au/Pd content catalysts) will be prepared and investigated.Data availability
The data supporting this article will be included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
LK and YR are grateful for support from the UK Catalysis Hub funded by EPSRC grant reference EP/R026645/1. LK also acknowledges support from EPSRC (grant EP/X009734/1). SJH acknowledges support from EPSRC (grants EP/W03395X/1, EP/V056565/1 and EP/S021531/1) and from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant ERC-2016-STG-EvoluTEM-715502). We acknowledge the support of the Henry Royce Institute for DB through the Royce PhD Equipment Access Scheme. TEM access at the University of Manchester was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1 and EP/P025498/1 (equipment access). We also thank Mott Corporation USA for providing the titanium porous transport layers. We thank technical specialist Dr Gary Miller, for the XPS and XRD data collection.References
- P. Nikolaidis and A. Poullikkas, A comparative overview of hydrogen production processes, Renewable Sustainable Energy Rev., 2017, 67, 597–611 CrossRef CAS.
- M. A. Rosen and S. Koohi-Fayegh, The prospects for hydrogen as an energy carrier: an overview of hydrogen energy and hydrogen energy systems, Energy, Ecol. Environ., 2016, 1, 13–14 Search PubMed.
- M. T. Gur, Review of electrical energy storage technologies, materials and systems: challenges and prospects for large-scale grid storage, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- J. M. Bermudez, S. Evangelopoulou and F. Pavan, Hydrogen, https://www.iea.org/reports/hydrogen, (accessed 7 May 2023).
- L. Moriau, M. Smiljanić, A. Lončar and N. Hodnik, Supported Iridium-based Oxygen Evolution Reaction Electrocatalysts - Recent Developments, ChemCatChem, 2022, 14, 1–23 Search PubMed.
- I. E. Stephens and N. Athanassopoulou, Materials for the Energy Transition roadmap: Materials for Low-Carbon Production of Hydrogen and Related Energy Carriers and Chemical Feedstocks, England, 2020.
- International Energy Agency, Net Zero by 2050: A Roadmap for the Global Energy Sector, International Energy Agency, 2021, p. 224.
- J. Chi and H. Yu, Water electrolysis based on renewable energy for hydrogen production, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS.
- Y. N. Regmi, E. Tzanetopoulos, G. Zeng, X. Peng, D. I. Kushner, T. A. Kistler, L. A. King and N. Danilovic, Supported Oxygen Evolution Catalysts by Design: Toward Lower Precious Metal Loading and Improved Conductivity in Proton Exchange Membrane Water Electrolyzers, ACS Catal., 2020, 10, 13125–13135 CrossRef CAS.
- M. A. Hubert, L. A. King and T. F. Jaramillo, Evaluating the Case for Reduced Precious Metal Catalysts in Proton Exchange Membrane Electrolyzers, ACS Energy Lett., 2022, 7, 17–23 CrossRef CAS.
- S. Hu, S. Ge, H. Liu, X. Kang, Q. Yu and B. Liu, Adv. Funct. Mater., 2022, 32, 1–21 Search PubMed.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, IrO2-TiO2: A High-Surface-Area, Active, and Stable Electrocatalyst for the Oxygen Evolution Reaction, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- S. Geiger, O. Kasian, B. R. Shrestha, A. M. Mingers, K. J. J. Mayrhofer and S. Cherevko, Activity and Stability of Electrochemically and Thermally Treated Iridium for the Oxygen Evolution Reaction, J. Electrochem. Soc., 2016, 163, F3132–F3138 CrossRef CAS.
- A. S. Pushkarev, I. V. Pushkareva and D. G. Bessarabov, Supported Ir-Based Oxygen Evolution Catalysts for Polymer Electrolyte Membrane Water Electrolysis: A Minireview, Energy Fuels, 2022, 36, 6613–6625 CrossRef CAS.
- A. L. Strickler, M. Escudero-Escribano and T. F. Jaramillo, Core-Shell Au@Metal-Oxide Nanoparticle Electrocatalysts for Enhanced Oxygen Evolution, Nano Lett., 2017, 17, 6040–6046 CrossRef CAS PubMed.
- K. Yu, Z. Wu, Q. Zhao, B. Li and Y. Xie, High-temperature-stable Au@SnO2 core/shell supported catalyst for CO oxidation, J. Phys. Chem. C, 2008, 112, 2244–2247 CrossRef CAS.
- B. M. Tackett, W. Sheng, S. Kattel, S. Yao, B. Yan, K. A. Kuttiyiel, Q. Wu and J. G. Chen, Reducing Iridium Loading in Oxygen Evolution Reaction Electrocatalysts Using Core-Shell Particles with Nitride Cores, ACS Catal., 2018, 8, 2615–2621 CrossRef CAS.
- T. Ling, D. Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. Zheng, J. Mao, X. W. Du, Z. Hu, M. Jaroniec and S. Z. Qiao, Engineering surface atomic structure of single-crystal cobalt (II) oxide nanorods for superior electrocatalysis, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- J. Cheng, J. Yang, S. Kitano, G. Juhasz, M. Higashi, M. Sadakiyo, K. Kato, S. Yoshioka, T. Sugiyama, M. Yamauchi and N. Nakashima, Impact of Ir-Valence Control and Surface Nanostructure on Oxygen Evolution Reaction over a Highly Efficient Ir-TiO2 Nanorod Catalyst, ACS Catal., 2019, 9, 6974–6986 CrossRef CAS.
- G. Liu, J. Xu, Y. Wang and X. Wang, An oxygen evolution catalyst on an antimony doped tin oxide nanowire structured support for proton exchange membrane liquid water electrolysis, J. Mater. Chem. A, 2015, 3, 20791–20800 RSC.
- H. S. Oh, H. N. Nong and P. Strasser, Preparation of mesoporous Sb-, F-, and in-doped SnO2 bulk powder with high surface area for use as catalyst supports in electrolytic cells, Adv. Funct. Mater., 2015, 25, 1074–1081 CrossRef CAS.
- E. J. Kim, J. Shin, J. Bak, S. J. Lee, K. Hyun Kim, D. H. Song, J. H. Roh, Y. Lee, H. W. Kim, K. S. Lee and E. A. Cho, Stabilizing role of Mo in TiO2-MoOx supported Ir catalyst toward oxygen evolution reaction, Appl. Catal., B, 2021, 280, 1–10 Search PubMed.
- H. Ohno, S. Nohara, K. Kakinuma, M. Uchida, A. Miyake, S. Deki and H. Uchida, Remarkable Mass Activities for the Oxygen Evolution Reaction at Iridium Oxide Nanocatalysts Dispersed on Tin Oxides for Polymer Electrolyte Membrane Water Electrolysis, J. Electrochem. Soc., 2017, 164, F944–F947 CrossRef CAS.
- J. Liao, Y. Wang, M. Chen, M. Wang, J. Fan, H. Li, H. Wang, L. Zeng and T. Zhao, IrOXSupported onto Niobium-Doped Titanium Dioxide as an Anode Reversal Tolerant Electrocatalyst for Proton Exchange Membrane Fuel Cells, ACS Appl. Energy Mater., 2022, 5, 3259–3268 CrossRef CAS.
- A. Rabis, P. Rodriguez and T. J. Schmidt, Electrocatalysis for polymer electrolyte fuel cells: Recent achievements and future challenges, ACS Catal., 2012, 2, 864–890 CrossRef CAS.
- G. Li, X. Xu, H. Liu, X. Yang and M. C. Lin, Enhanced Electrocatalytic Performance of IrOx by Employing F-Doped TiO2 as Support towards Acidic Oxygen Evolution Reaction, ChemCatChem, 2022, 14, 1–7 Search PubMed.
- N. Roy, Y. Sohn, K. T. Leung and D. Pradhan, Engineered electronic states of transition metal doped TiO2 nanocrystals for low overpotential oxygen evolution reaction, J. Phys. Chem. C, 2014, 118, 29499–29506 CrossRef CAS.
- A. Hartig-Weiss, M. Miller, H. Beyer, A. Schmitt, A. Siebel, A. T. S. Freiberg, H. A. Gasteiger and H. A. El-Sayed, Iridium Oxide Catalyst Supported on Antimony-Doped Tin Oxide for High Oxygen Evolution Reaction Activity in Acidic Media, ACS Appl. Nano Mater., 2020, 3, 2185–2196 CrossRef CAS.
- Z. S. H. S. Rajan, T. Binninger, P. J. Kooyman, D. Susac and R. Mohamed, Organometallic chemical deposition of crystalline iridium oxide nanoparticles on antimony-doped tin oxide support with high-performance for the oxygen evolution reaction, Catal. Sci. Technol., 2020, 10, 3938–3948 RSC.
- S. Komini Babu, R. Mukundan, C. Wang, D. Langlois, D. A. Cullen, D. Papadias, K. L. More, R. Ahluwalia, J. Waldecker and R. Borup, Effect of Catalyst and Catalyst Layer Composition on Catalyst Support Durability, J. Electrochem. Soc., 2021, 168, 044502 CrossRef.
- R. Mohamed, T. Binninger, P. J. Kooyman, A. Hoell, E. Fabbri, A. Patru, A. Heinritz, T. J. Schmidt and P. Levecque, Facile deposition of Pt nanoparticles on Sb-doped SnO2 support with outstanding active surface area for the oxygen reduction reaction, Catal. Sci. Technol., 2018, 8, 2672–2685 RSC.
- A. Lavacchi, M. Bellini, E. Berretti, Y. Chen, A. Marchionni, H. A. Miller and F. Vizza, Titanium dioxide nanomaterials in electrocatalysis for energy, Curr. Opin. Electrochem., 2021, 28, 100720 CrossRef CAS.
- P. Mazúr, J. Polonský, M. Paidar and K. Bouzek, Non-conductive TiO 2 as the anode catalyst support for PEM water electrolysis, Int. J. Hydrogen Energy, 2012, 37, 12081–12088 CrossRef.
- R. E. Fuentes, J. Farell and J. W. Weidner, Multimetallic electrocatalysts of Pt, Ru, and Ir supported on anatase and rutile TiO2 for oxygen evolution in an acid environment, Electrochem. Solid-State Lett., 2011, 14, 5–7 CrossRef.
- F. C. Walsh and R. G. A. Wills, The continuing development of Magnéli phase titanium sub-oxides and Ebonex® electrodes, Electrochim. Acta, 2010, 55, 6342–6351 CrossRef CAS.
- C. Van Pham, M. Bühler, J. Knöppel, M. Bierling, D. Seeberger, D. Escalera-López, K. J. J. Mayrhofer, S. Cherevko and S. Thiele, IrO2 coated TiO2 core-shell microparticles advance performance of low loading proton exchange membrane water electrolyzers, Appl. Catal., B, 2020, 269, 118762 CrossRef.
- Y. T. Kim, P. P. Lopes, S. A. Park, A. Y. Lee, J. Lim, H. Lee, S. Back, Y. Jung, N. Danilovic, V. Stamenkovic, J. Erlebacher, J. Snyder and N. M. Markovic, Balancing activity, stability and conductivity of nanoporous core-shell iridium/iridium oxide oxygen evolution catalysts, Nat. Commun., 2017, 8, 1–8 CrossRef CAS.
- Z. Zhuang, W. Sheng and Y. Yan, Synthesis of monodispere Au@Co3O4 core-shell nanocrystals and their enhanced catalytic activity for oxygen evolution reaction, Adv. Mater., 2014, 26, 3950–3955 CrossRef CAS PubMed.
- K. Ayers, A. Soleymani, J. Jankovic, H. Yu and G. Mirshekari, Degradation Mechanisms in Advanced MEAs for PEM Water Electrolyzers Fabricated by Reactive Spray Deposition Technology, J. Electrochem. Soc., 2022, 169, 1–26 Search PubMed.
- U. S. Jonnalagadda, X. Su and J. J. Kwan, Nanostructured TiO2 cavitation agents for dual-modal sonophotocatalysis with pulsed ultrasound, Ultrason. Sonochem., 2021, 73, 1–9 CrossRef PubMed.
- S. Liu, G. Chen, P. N. Prasad and M. T. Swihart, Synthesis of monodisperse Au, Ag, and Au-Ag alloy nanoparticles with tunable size and surface plasmon resonance frequency, Chem. Mater., 2011, 23, 4098–4101 CrossRef CAS.
- C. J. Wrasman, A. Boubnov, A. R. Riscoe, A. S. Hoffman, S. R. Bare and M. Cargnello, Synthesis of Colloidal Pd/Au Dilute Alloy Nanocrystals and Their Potential for Selective Catalytic Oxidations, J. Am. Chem. Soc., 2018, 140, 12930–12939 CrossRef CAS PubMed.
- D. Xu, M. B. Stevens, M. R. Cosby, S. Z. Oener, A. M. Smith, L. J. Enman, K. E. Ayers, C. B. Capuano, J. N. Renner, N. Danilovic, Y. Li, H. Wang, Q. Zhang and S. W. Boettcher, Earth-Abundant Oxygen Electrocatalysts for Alkaline Anion-Exchange-Membrane Water Electrolysis: Effects of Catalyst Conductivity and Comparison with Performance in Three-Electrode Cells, ACS Catal., 2019, 9, 7–15 CrossRef CAS.
- B. Han, M. Risch, S. Belden, S. Lee, D. Bayer, E. Mutoro and Y. Shao-Horn, Screening oxide support materials for OER catalysts in acid, J. Electrochem. Soc., 2018, 165, F813–F820 CrossRef CAS.
- A. Celzard, J. F. Marêché, F. Payot and G. Furdin, Electrical conductivity of carbonaceous powders, Carbon, 2002, 40, 2801–2815 CrossRef CAS.
- S. M. Alia and N. Danilovic, Rotating Disk Electrode Standardization and Best Practices in Acidic Oxygen Evolution for Low-Temperature Electrolysis, Front. Energy Res., 2022, 10, 1–8 Search PubMed.
- Y. N. Regmi, X. Peng, J. C. Fornaciari, M. Wei, D. J. Myers, A. Z. Weber and N. Danilovic, A low temperature unitized regenerative fuel cell realizing 60% round trip efficiency and 10 000 cycles of durability for energy storage applications, Energy Environ. Sci., 2020, 13, 2096–2105 RSC.
- S. Shahgaldi, I. Alaefour, G. Unsworth and X. Li, Development of a low temperature decal transfer method for the fabrication of proton exchange membrane fuel cells, Int. J. Hydrogen Energy, 2017, 42, 11813–11822 CrossRef CAS.
- J. A. Arminio-Ravelo, A. W. Jensen, K. D. Jensen, J. Quinson and M. Escudero-Escribano, Electrolyte Effects on the Electrocatalytic Performance of Iridium-Based Nanoparticles for Oxygen Evolution in Rotating Disc Electrodes, ChemPhysChem, 2019, 20, 2956–2963 CrossRef CAS PubMed.
- L. Owe, M. Tsypkin and S. Sunde, The effect of phosphate on iridium oxide electrochemistry, Electrochim. Acta, 2011, 58, 231–237 CrossRef CAS.
- C. Daiane Ferreira Da Silva, F. Claudel, V. Martin, R. Chattot, S. Abbou, K. Kumar, I. Jiménez-Morales, S. Cavaliere, D. Jones, J. Rozière, L. Solà-Hernandez, C. Beauger, M. Faustini, J. Peron, B. Gilles, T. Encinas, L. Piccolo, F. H. Barros De Lima, L. Dubau and F. Maillard, Oxygen Evolution Reaction Activity and Stability Benchmarks for Supported and Unsupported IrOxElectrocatalysts, ACS Catal., 2021, 11, 4107–4116 CrossRef CAS.
- J. Van Der Merwe, K. Uren, G. Van Schoor and D. Bessarabov, Characterisation tools development for PEM electrolysers, Int. J. Hydrogen Energy, 2014, 39, 14212–14221 CrossRef CAS.
- C. Martinson, G. Van Schoor, K. Uren and D. Bessarabov, Equivalent electrical circuit modelling of a Proton Exchange Membrane electrolyser based on current interruption, Proc. IEEE Int. Conf. Ind. Technol., 2013, pp. 716–721 Search PubMed.
- G. Li, K. Li, L. Yang, J. Chang, R. Ma, Z. Wu, J. Ge, C. Liu and W. Xing, Boosted Performance of Ir Species by Employing TiN as the Support toward Oxygen Evolution Reaction, ACS Appl. Mater. Interfaces, 2018, 10, 38117–38124 CrossRef CAS PubMed.
- L. Wang, P. Lettenmeier, U. Golla-Schindler, P. Gazdzicki, N. A. Cañas, T. Morawietz, R. Hiesgen, S. S. Hosseiny, A. S. Gago and K. A. Friedrich, Nanostructured Ir-supported on Ti4O7 as a cost-effective anode for proton exchange membrane (PEM) electrolyzers, Phys. Chem. Chem. Phys., 2016, 18, 4487–4495 RSC.
- D. Bernsmeier, M. Bernicke, R. Schmack, R. Sachse, B. Paul, A. Bergmann, P. Strasser, E. Ortel and R. Kraehnert, Oxygen Evolution Catalysts Based on Ir–Ti Mixed Oxides with Templated Mesopore Structure: Impact of Ir on Activity and Conductivity, ChemSusChem, 2018, 11, 2367–2374 CrossRef CAS PubMed.
- W. Hu, S. Chen and Q. Xia, IrO2/Nb-TiO2 electrocatalyst for oxygen evolution reaction in acidic medium, Int. J. Hydrogen Energy, 2014, 6967–6976 CrossRef CAS.
- L. Moriau, M. Bele, Ž. Marinko, F. Ruiz-Zepeda, G. Koderman Podboršek, M. Šala, A. K. Šurca, J. Kovač, I. Arčon, P. Jovanovič, N. Hodnik and L. Suhadolnik, Effect of the Morphology of the High-Surface-Area Support on the Performance of the Oxygen-Evolution Reaction for Iridium Nanoparticles, ACS Catal., 2021, 11, 670–681 CrossRef CAS PubMed.
- G. Li, H. Jia, H. Liu, X. Yang and M. C. Lin, Nanostructured IrOx supported on N-doped TiO2 as an efficient electrocatalyst towards acidic oxygen evolution reaction, RSC Adv., 2022, 12, 28929–28936 RSC.
- R. V. Genova-Koleva, F. Alcaide, G. Álvarez, P. L. Cabot, H. J. Grande, M. V. Martínez-Huerta and O. Miguel, Supporting IrO2 and IrRuOx nanoparticles on TiO2 and Nb-doped TiO2 nanotubes as electrocatalysts for the oxygen evolution reaction, J. Energy Chem., 2019, 34, 227–239 CrossRef.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. J. Mayrhofer, Oxygen evolution activity and stability of iridium in acidic media. Part 2. - Electrochemically grown hydrous iridium oxide, J. Electroanal. Chem., 2016, 774, 102–110 CrossRef CAS.
- G. C. da Silva, S. I. Venturini, S. Zhang, M. Löffler, C. Scheu, K. J. J. Mayrhofer, E. A. Ticianelli and S. Cherevko, Oxygen Evolution Reaction on Tin Oxides Supported Iridium Catalysts: Do We Need Dopants?, ChemElectroChem, 2020, 7, 2330–2339 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion, Sci. Rep., 2015, 5, 1–21 Search PubMed.
- Q. Meyer, S. Liu, K. Ching, Y. Da Wang and C. Zhao, Operando monitoring of the evolution of triple-phase boundaries in proton exchange membrane fuel cells, J. Power Sources, 2023, 557, 232539 CrossRef CAS.
- J. Mo, Z. Kang, S. T. Retterer, D. A. Cullen, T. J. Toops, J. B. Green, M. M. Mench and F. Y. Zhang, Discovery of true electrochemical reactions for ultrahigh catalyst mass activity in water splitting, Sci. Adv., 2016, 2, e1600690 CrossRef PubMed.
- Z. Kang, J. Mo, G. Yang, Y. Li, D. A. Talley, S. T. Retterer, D. A. Cullen, T. J. Toops, M. P. Brady, G. Bender, B. S. Pivovar, J. B. Green and F. Y. Zhang, Thin film surface modifications of thin/tunable liquid/gas diffusion layers for high-efficiency proton exchange membrane electrolyzer cells, Appl. Energy, 2017, 206, 983–990 CrossRef CAS.
- G. Yang, S. Yu, Y. Li, K. Li, L. Ding, Z. Xie, W. Wang and F. Y. Zhang, Role of electron pathway in dimensionally increasing water splitting reaction sites in liquid electrolytes, Electrochim. Acta, 2020, 362, 137113 CrossRef CAS.
- L. Shi, A. Wang, T. Zhang, B. Zhang, D. Su, H. Li and Y. Song, One-step synthesis of Au-pd alloy nanodendrites and their catalytic activity, J. Phys. Chem. C, 2013, 117, 12526–12536 CrossRef CAS.
- P. Mishra, C. M. Pandey, U. Singh, A. Gupta, C. Sahu and A. Keshri, Descriptive statistics and normality tests for statistical data, Ann. Card. Anaesth., 2019, 22, 67–72 CrossRef PubMed.
- G. C. da Silva, N. Perini and E. A. Ticianelli, Effect of temperature on the activities and stabilities of hydrothermally prepared IrOx nanocatalyst layers for the oxygen evolution reaction, Appl. Catal., B, 2017, 218, 287–297 CrossRef CAS.
- T. Reier, I. Weidinger, P. Hildebrandt, R. Kraehnert and P. Strasser, Electrocatalytic Oxygen Evolution Reaction on Iridium Oxide Model Film Catalysts: Influence of Oxide Type and Catalyst Substrate Interactions, ECS Meet. Abstr., 2013, vol. MA2013-02, p. 711 Search PubMed.
- M. Bernt, C. Schramm, J. Schröter, C. Gebauer, J. Byrknes, C. Eickes and H. A. Gasteiger, Effect of the IrOx Conductivity on the Anode Electrode/Porous Transport Layer Interfacial Resistance in PEM Water Electrolyzers, J. Electrochem. Soc., 2021, 168, 084513 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J. P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, The electronic structure of iridium and its oxides, Surf. Interface Anal., 2016, 48, 261–273 CrossRef CAS.
- M. Khawaji and D. Chadwick, Au-Pd NPs immobilised on nanostructured ceria and titania: Impact of support morphology on the catalytic activity for selective oxidation, Catal. Sci. Technol., 2018, 8, 2529–2539 RSC.
- J. E. Gonçalves, S. C. Castro, A. Y. Ramos, M. C. M. Alves and Y. Gushikem, X-ray absorption and XPS study of titanium mixed oxides synthesized by the sol-gel method, J. Electron Spectrosc. Relat. Phenom., 2001, 114–116, 307–311 CrossRef.
- G. Bucci, K. Gadelrab and W. C. Carter, Mesoscale Model for Ostwald Ripening of Catalyst Nanoparticles, J. Electrochem. Soc., 2021, 168, 054515 CrossRef CAS.
- M. Milosevic, T. Böhm, A. Körner, M. Bierling, L. Winkelmann, K. Ehelebe, A. Hutzler, M. Suermann, S. Thiele and S. Cherevko, In Search of Lost Iridium: Quantification of anode Catalyst Layer Dissoluton in Proton Exchange Membrane Water Electrolyzers, ACS Energy Lett., 2023, 8, 2682–2688 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00026a |
| This journal is © The Royal Society of Chemistry 2024 |