
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBridging the structural gap of supported vanadium oxides for oxidative dehydrogenation of propane with carbon dioxide†
Li
Wang
ab,
Heng-Bo
Zhang
a,
Rongrong
Hu
 a,
Han-Qing
Ge
a,
Yong-Hong
Song
a,
Guo-Qing
Yang
*a,
Yuefeng
Li
b,
Zhao-Tie
Liu
a and
Zhong-Wen
Liu
*a
a,
Han-Qing
Ge
a,
Yong-Hong
Song
a,
Guo-Qing
Yang
*a,
Yuefeng
Li
b,
Zhao-Tie
Liu
a and
Zhong-Wen
Liu
*a
aKey Laboratory of Syngas Conversion of Shaanxi Province, School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi’an 710119, China. E-mail: gqyang@snnu.edu.cn; zwliu@snnu.edu.cn
bKaili Catalyst & New Materials Co., Ltd, Xi’an 710201, China
First published on 14th June 2024
Abstract
As an extensively used industrial catalyst for oxidation reactions, supported vanadium oxide (VOx) is a promising candidate for oxidative dehydrogenation of propane with carbon dioxide (CO2-ODP). Although the structure of VOx is found to be a key factor in determining the catalytic activity and stability of supported VOx for CO2-ODP, the essential reason still remains elusive at the molecular level. To shed some light on this fundamental issue, VOx/(−)SiO2 catalysts with narrow distributions of V loading while well-defined structures of VOx species, i.e., monomeric VOx, less polymeric VOx, highly polymeric VOx and V2O5 crystallites, were purposely synthesized by appropriate methods, including one-pot hydrothermal synthesis, incipient wetness impregnation and physical grinding. We found that the catalytic activity and stability of VOx species decrease in the order of monomeric VOx > less polymeric VOx > highly polymeric VOx > crystalline V2O5, which coincides with the ability for the re-oxidation of the correspondingly reduced VOx species by CO2. As a result of the most facile re-oxidation of the reduced monomeric VOx species by CO2, a well matched redox cycle of V5+/V4+ oxides during CO2-ODP can be maintained with increasing the time on stream, leading to an improved stability of the catalyst with more monomeric VOx. These mechanistic findings on the redox properties of VOx with different structures can be guidelines for developing a high-performance VOx-based catalyst for CO2-ODP.
Broader contextAlthough the oxidative dehydrogenation of propane with carbon dioxide (CO2-ODP) is a sustainable route for the efficient production of propene in tandem with CO2 reduction to CO, the rational design of high-performance catalysts for this green process is challenged by limited understanding of the nature of active sites and the reaction mechanism. Supported vanadium oxides (VOx) are promising for CO2-ODP considering their low price, environmental sustainability, and competitive catalytic performance, however the essential reason of VOx structural effects on the catalytic performance is still ambiguous from a mechanistic aspect. In present work, SiO2-supported VOx catalysts with narrow distributions of V loading while well-defined structures of VOx species, i.e., monomeric VOx, less polymeric VOx, highly polymeric VOx and V2O5 crystallites, were on-purpose synthesized. In addition to sophisticated catalytic tests for CO2-ODP, complementary characterization techniques were applied for deriving structural and mechanistic insights into catalyst functioning. We established that the catalytic activity and stability of different VOx species coincide well with the ability for the re-oxidation of correspondingly reduced VOx by CO2. These findings are important guidelines for purposeful development of efficient supported VOx catalysts for CO2-ODP. |
1. Introduction
Among the currently available methods, the production of propene from propane through either the direct dehydrogenation or the oxidative dehydrogenation reaction is the most competitively favorable route.1,2 However, only the direct propane dehydrogenation (PDH) technologies are commercialized on large scales, which characterize low propene productivity and high energy consumption.2,3 To overcome these shortcomings, the oxidative dehydrogenation of propane using carbon dioxide as a soft oxidant (CO2-ODP) has attracted much attention.3,4 In comparison with PDH, CO2-ODP can promote the equilibrium conversion of propane by removing the produced hydrogen via CO2 reduction. Moreover, CO2-ODP favors a higher propene selectivity than that by using O2 as an oxidant due to the soft oxidizing ability of CO2. Importantly, the efficient production of two value-added products of propene and CO from propane and the greenhouse gas of CO2, respectively, turns CO2-ODP into a charming tandem process from an environmental viewpoint.4,5 Moreover, although the separation of gaseous species resulting from the introduction of CO2 remains a significant challenge, the CO2-ODP product mixtures can be directly used as feeds to produce useful chemicals, such as the oxidative coupling of CO2 with alkenes to produce acrylic acids and acrylate in a one-step process, which is a more energy-saving and efficient technology than the conventional two-step methodology.6 As a result of these advantages, a number of catalysts including supported or bulk metal oxides e.g., Cr,7 V,8 In,9 Ga,10 and Zn,11 and supported metals, e.g., Pd,12 and Pt13,14 have been extensively studied for CO2-ODP. Among these potential catalysts, supported vanadium oxides (VOx) are promising for CO2-ODP considering their low price, environmental sustainability and competitive catalytic performance.2,15 However, the rational design of a high-performance supported VOx catalyst is still challenging due to limited understanding of the active and stable nature of VOx species as well as the reaction mechanism.Indeed, the apparent turnover frequency (TOF) of the supported VOx for activating propane during CO2-ODP has been revealed to be decreased with increasing the polymerization degree of VOx species.16,17 A similar conclusion is also valid for the oxidative dehydrogenation of ethylbenzene with CO2 reaction, in which monomeric VOx are more active and more stable than polymeric VOx and crystalline V2O5.18 Although these observations are tentatively correlated with the structural and chemical properties of VOx species with different structures, the essential reason in the mechanistic aspect, especially the activation of CO2 and the balance between the activations of C–O bonds in CO2 and C–H bonds in C3H8 molecules, is still ambiguous. In the cases of the reducible metal oxide catalysts, e.g., CrOx and VOx, the CO2-ODP reaction may follow the Mars–van-Krevelen redox mechanism.8,19 Thus, the redox cycles of the VOx species participated with CO2 and propane must proceed quickly in a well-matched manner, which is essential for a high-performance VOx. Specifically, the active oxygen species in VOx with a higher oxidation number of +5 is consumed by activating propane molecule to produce propene and water, accompanying the formation of VOx with a lower oxidation state (+4 and/or +3), which is re-oxidized to be V5+ by CO2 for the simultaneous formation of CO. If the weak oxidizing ability of CO2 is taken into account, the re-oxidation of V4+ and/or V3+ to V5+ by CO2 may be a key step in the redox cycle. With these considerations, the catalytic performance of different VOx species for CO2-ODP may be closely related to their different abilities for activating and converting CO2 molecules. However, the detailed mechanistic understandings are still of lacking, and only the partial re-oxidation of the reduced VOx species by CO2 is reported.20,21
As a common practice, supported VOx with the structures of monomeric VOx, less or highly polymeric VOx and even crystalline V2O5 can be achieved by adjusting the V loadings of the catalyst. However, in this case, the V loadings commonly vary in large ranges from about 0.3 wt% to 20 wt%,22–24 which makes it difficult to unambiguously correlate the catalytic performance of VOx catalysts with their structures. This can be reflected from different observations for oxidative dehydrogenation of propane with O2 catalyzed by VOx/Al2O3, i.e., the decreased25 and constant26 TOF of propane with increasing the V loadings. On the one hand, the catalyst with a very low V loading makes it difficult to perform the accurately qualitative and quantitative analysis of the characterization and reaction results, thus a lager error may be present. On the other hand, if the loading of V is too high, the intrinsic concentration of the adsorption and activation of feed components and reaction products on per active site of V on the catalyst surface during the reaction, may be affected by the steric constraint effect, leading to an inauthentic conclusion when the reaction results are discussed. Thus, the catalysts with different VOx structures while narrow V loading distributions are desired for better understanding the essential reason of the VOx structural effects on the catalytic performance of VOx for CO2-ODP.
As a matter of fact, in addition to adjusting V loadings, the catalysts with different VOx structures can be also obtained by changing the preparation methods.27,28 To date, the widely reported methods for preparing supported VOx catalysts are one-step direct synthesis and impregnation method.29,30 Generally, at a given loading of V, the VOx species prepared by one-step synthesis shows a higher dispersion than that synthesized by the impregnation method due to the incorporation of VOx species into the framework of the support.31 Additionally, supported crystalline V2O5 can be obtained by the directly physical grinding method due to the weakened interaction between the VOx precursor and the support.32,33
Based on these understandings, in this work, VOx/(−)SiO2 catalysts with 4–6 wt% V loadings are purposefully designed and prepared by different methods. As a result, well-defined structures of VOx species over the catalysts, i.e., monomeric VOx, less polymeric VOx, highly polymeric VOx and crystalline V2O5, are achieved as confirmed from the characterization results. More importantly, the structural impacts of different VOx species on the activations of C–O bonds in CO2 are rigorously revealed from the results of the pulse experiments, and the balance between the activations of C–O bonds in CO2 and C–H bonds in C3H8 molecules induced from different VOx species is established together with the reaction and characterization data. These understandings can provide important contributions to the essential reason of the structural impacts of VOx species on CO2-ODP, especially the activations of C–O bonds in CO2 and the balance between the activations of C–O bonds in CO2 and C–H bonds in C3H8 molecules, which are key factors in determining the catalytic activity and stability of the catalysts.
2. Experimental
2.1. Catalyst preparation
The one-pot hydrothermal synthesis29 was used to prepare the SiO2-supported VOx catalyst with a V loading of 4 or 6 wt%. In the detailed procedure, 1.74 g of cetyltrimethyl ammonium bromide (CTAB, ≥99.0%, Guanghua Sci-Tech Co., Ltd, Guangdong, China) and 0.25 g of sodium hydroxide (NaOH, ≥96.0%, Guanghua Sci-Tech Co. Ltd, Guangdong, China) were dissolved into 70 mL of deionized water. The ammonium metavanadate with desired amount (NH4VO3, ≥99.0%, Fuchen Chemical Reagent Co. Ltd, Tianjin, China) was dissolved into 25 mL of deionized water at 60 °C. Then, above solutions were completely mixed at 60 °C under magnetic stirring. After 30 min, 9.14 mL of tetraethyl orthosilicate (TEOS, 98.0%, Macklin Biochemical Co. Ltd, Shanghai, China) was slowly dropped into the mixture under magnetic stirring. After 2 h, the obtained mixture was transferred to a Teflon autoclave and crystallized at 120 °C for 2 days. The solids were centrifuged, washed several times by water and ethanol until the pH value of filtrate is neutral, and then dried at 120 °C for 6 h and calcined in air at 600 °C for 3 h. The obtained catalysts were named as 4VOx–SiO2-H and 6VOx–SiO2-H, respectively, where 4 and 6 are the weight percent of V (wt%), and H represents the hydrothermal synthesis method.The incipient wetness impregnation method was applied to prepare the SiO2-supported VOx catalyst with a V loading of 6 wt%. Before loading VOx, SiO2 with a BET surface area of 580 m2 g−1 and an average pore diameter of 3.0 nm (Fuji Silysia Chemical Ltd, Kasugai Aichi, Japan) was pre-treated in air at 600 °C for 3 h. After impregnating the desired amounts of aqueous solution of NH4VO3 assisted by oxalic acid (99.6%, Sinopharm Chemical Reagent Co. Ltd) at a molar ratio of 1/2, the sample was dried at 120 °C for 6 h and then calcined in air at 600 °C for 3 h. The obtained catalyst was named as 6VOx/SiO2-I, where 6 is the V loading (wt%), and I represents the impregnation method.
The SiO2-supported VOx catalyst with a V loading of 6 wt% was also prepared by the physical grinding method. The same SiO2 support (2 g) to that used in the impregnation experiment was directly blended with 0.275 g of NH4VO3 powders. After grinding for 30 min in an agate mortar, the sample was dried at 120 °C for 6 h and calcined in air at 600 °C for 3 h, leading to the catalyst of 6VOx/SiO2-P, where 6 is the V loading (wt%), and P represents the physical grinding method.
2.2. Catalyst characterizations
The loading of V over the catalyst was determined by inductively coupled plasma mass spectrometry (ICP-MS, Bruker Dalton-Aurora M90). Before the test, about 10.0 mg of sample was dissolved in a mixed solution of HNO3 (Sinopharm, 70%) and HF (Sinopharm, ≥30%) at 60 °C for 30 min.N2 physical adsorption/desorption isotherms were performed on a Bel-sorp-Max instrument at −196 °C. Before the measurement, each sample was pre-treated at 300 °C under vacuum for 10 h. The Brunauer–Emmett–Teller (BET) equation and Barrett–Joyner–Halenda (BJH) method were employed to calculate specific surface area and pore size distribution (PSD) of the catalysts, respectively.
Powder X-ray diffraction (XRD) patterns were obtained on an X-ray diffractometer equipped with Cu-Kα radiation (Bruker D8 Advance, 40 kV, 40 mA). The sample was scanned from 2θ value of 10° to 80° with a speed of 0.2 s per step.
Raman spectra of the fresh catalysts in the range of 200–1200 cm−1 were obtained by a confocal microprobe laser Raman spectrometer (HORIBA Jobin Yvon) equipped with the laser of 325 and 532 nm, respectively. The peak at 520.7 cm−1 of a silica standard was used to calibrate the obtained spectra. The sample was loaded into an in situ cell (Harrick, HVCDR2) and dehydrated at 400 °C under dried Ar (40 mL min−1) for 40 min. After cooling to ambient temperature in an Ar flow, Raman spectra were collected. The Raman spectra of the spent catalysts for CO2-ODP were directly recorded in the range of 1100–1700 cm−1 with the laser of 532 nm at ambient environment.
In situ UV-Vis diffuse reflectance spectra (UV-Vis DRS) were acquired on a PerkinElmer Lambda 950 spectrophotometer equipped with an in situ cell (Harrick, HVCDR2). The sample was previously diluted with BaSO4 (≥99.0%, Macklin Biochemical Co. Ltd, Shanghai, China), and then pre-treated at 400 °C under Ar (30 mL min−1) for 40 min. After cooling to room temperature, the spectra were recorded in the range of 200–800 nm by using BaSO4 as the background. The absorption edge energy (Eg) was determined by the intercept of the straight line, which is fitted through the low energy rise in [F(R∞)·hν]2versus the photon energy plot (hν), where F(R∞) is the Kubelka–Munk function.
Temperature-programmed reduction with H2 (H2-TPR) tests were carried out using an Autochem 2920 instrument (Micromeritics, USA) equipped with a thermal conductivity detector (TCD). 100.0 mg of sample was pre-treated at 400 °C for 30 min in an Ar flow (30 mL min−1). After cooling to 50 °C, the sample was heated from 50 to 800 °C with a rate of 10 °C min−1 in a 10 vol% H2/Ar flow (30 mL min−1). H2 consumption was monitored and determined by a pre-calibrated TCD.
X-ray photoelectron spectroscopy (XPS) measurements were performed on an Axis Ultra spectrometer (KRATOS Analytical Ltd) equipped with an Al-Kα radiation source (1486.6 eV). The C 1s spectrum at 284.6 eV was applied to calibrate the obtained spectra of the samples.
CO2 pulse experiments were performed on an AutoChem 2950 HP Chemisorption instrument (Micromeritics, USA). 0.25 g of catalyst was pre-reduced by 10 vol% H2/Ar (30 mL min−1) at 600 °C for 1 h. After purging with Ar (30 mL min−1) for 30 min, pulse of CO2 (0.5 mL loop) mixed with a flow of Ar (20 mL min−1) passed through the reduced catalyst. The temporal change of CO2 (m/z = 44) and the product of CO (m/z = 28) were monitored by an HPR QIC20 mass spectrometer (MS, Hiden Analytical, United Kingdom). Because the energy of ion source used in MS is relatively low (electron energy: 18.0 V; emission: 50 μA), the signal of 28 (m/z) from CO2 is negligible.
Thermo gravimetric and differential scanning calorimetry tests (TG-DSC) for the spent catalysts were carried out on a Q600SDT Thermoanalyzer System (TA Instruments). About 5.0 mg of sample was heated from 30 to 800 °C with a heating ramp of 10 °C min−1 in a flow of air (50 mL min−1).
2.3. Catalytic tests
Reaction tests were performed at 0.1 MPa in a quartz fixed-bed reactor (6 mm, i.d.). For each test, 0.25 g of catalyst (40–60 mesh) was loaded into the reactor. Firstly, the sample was pre-treated at 600 °C for 30 min in an Ar flow of 20 mL min−1, and then the gas mixture of Ar/C3H8/CO2 (32 mL min−1) in a molar ratio of 2/3/27 passed through the catalyst, where Ar is an internal standard gas. The products and unconvertible C3H8 and CO2 were analyzed by an online gas chromatograph (GC7920, Peking CEAULIGHT) equipped with TCD (TDX-01 column) and flame ionization detector (FID, Porapak Q column). Conversion of propane and CO2, and selectivity of different hydrocarbon products (CH4, C2H4, C2H6 and C3H6) were calculated by eqn (1)–(3), respectively.Carbon balances for all catalysts during the reaction calculated by eqn (4) were centered at 99.3 ± 0.7% (Fig. S1, ESI†), indicating a very small amount of coke deposits formed on the catalyst surface. Thus, by assuming that the carbon balance is 100%, the selectivity of CO produced from C3H8via the reforming of propane with CO2 reaction (CO2-RP) was estimated by subtracting the sum of the hydrocarbon products (CH4 + C2H4 + C2H6 + C3H6) from 100% (eqn (5)) according to ref. 13 and 34.
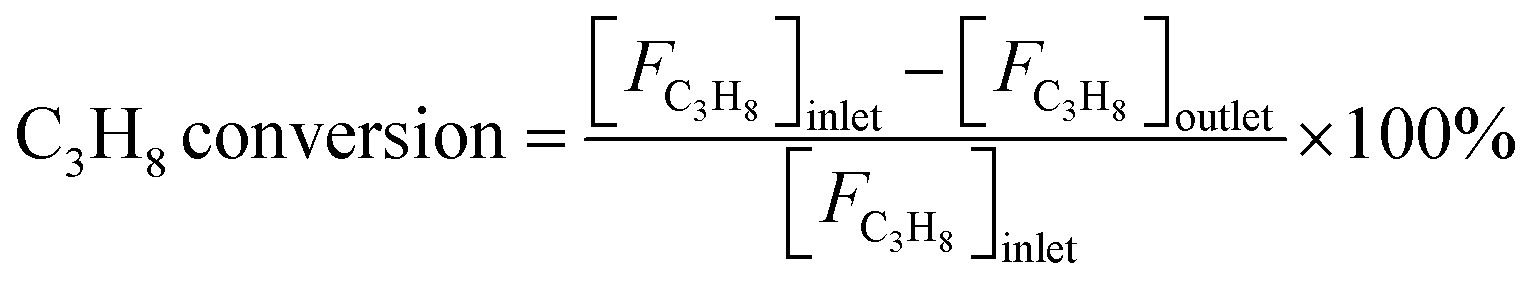 | (1) |
 | (2) |
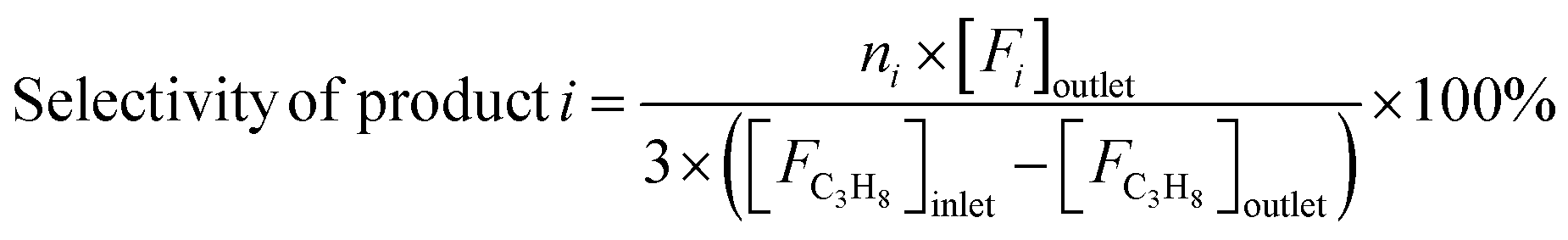 | (3) |
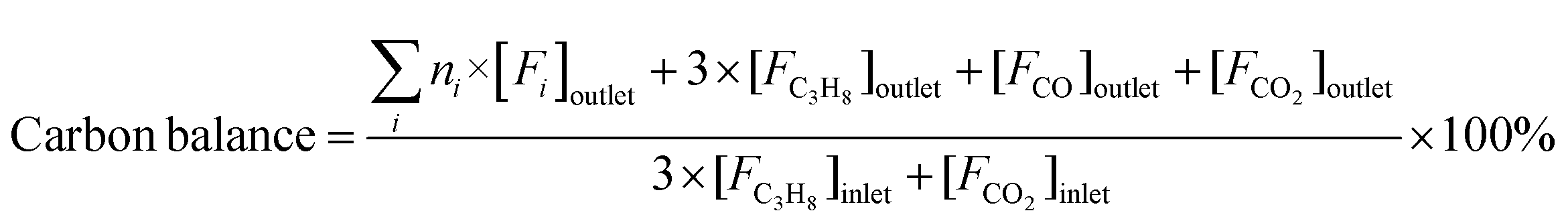 | (4) |
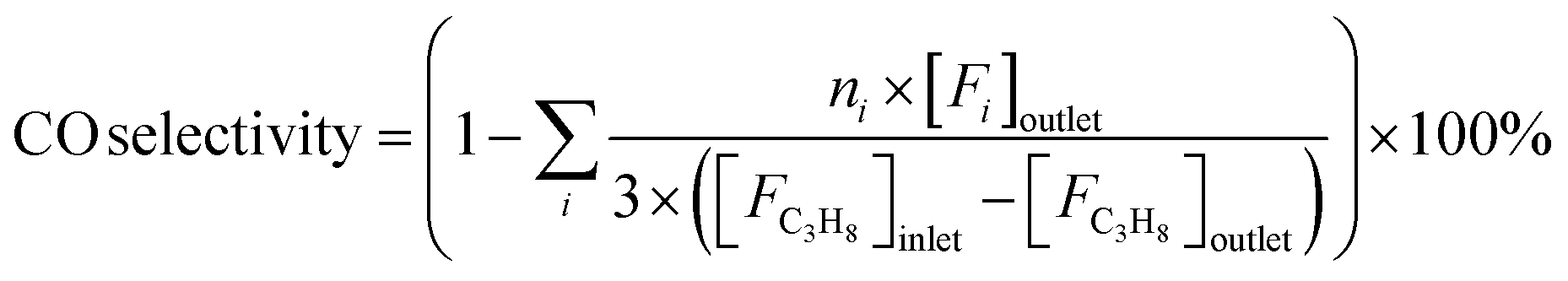 | (5) |
Relative deactivation rate (R) calculated by eqn (6) was used to evaluate the stability of catalyst.
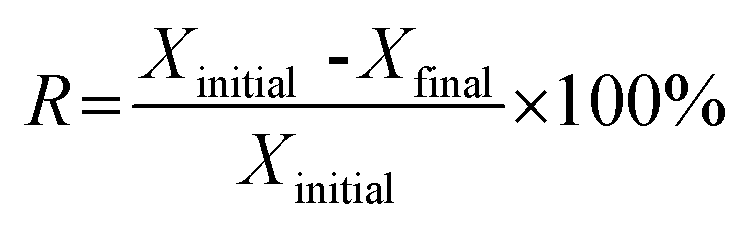 | (6) |
To estimate the effects of the internal or external diffusion limitations on the catalytic performance, the Weisz–Prater (CWP) and Mears’ criteria (CMM) were used for 6VOx–SiO2-H with the highest propane conversion according to ref. 35–37. The detailed calculations are given in Section 12 in the ESI.† The calculated CWP of 1.54 × 10−3 and CMM of 2.82 × 10−3 are significantly lower than those of the critical values of 1 and 0.15, respectively. This suggests that both internal and external diffusion limitations are negligible for the applied reaction conditions.
The propane consumption rate (r(C3H8), mmol g−1 min−1) was calculated using eqn (7). The apparent TOF(C3H8) defined as the number of C3H8 molecules converted per V atom per hour over the catalysts was calculated by eqn (8), and the TOF(CO2) was determined through the same method. The data were determined after a TOS of 5 min at a low propane/CO2 conversion below 10% by adjusting the amount of the loaded catalyst under the conditions of 600 °C, 0.1 MPa, Ar/C3H8/CO2/He molar ratio = 2/3/3/24, total flow rate = 32 mL min−1.
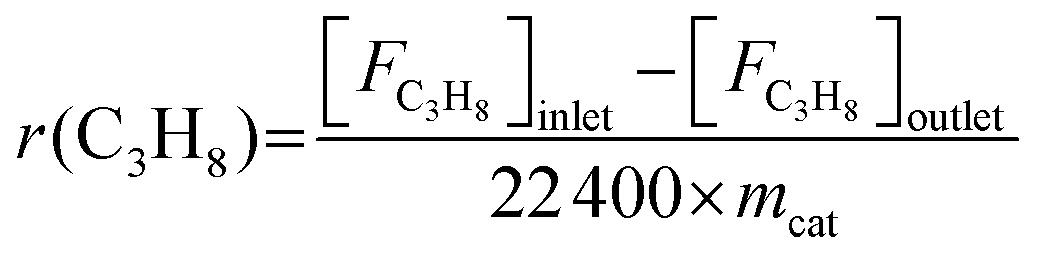 | (7) |
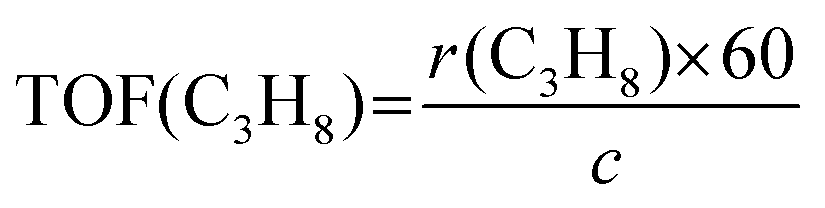 | (8) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, mcat and c stand for the molar volume at standard conditions (mL mol−1), catalyst mass (g), and the content of V over the catalyst (mmol g−1), respectively.
400, mcat and c stand for the molar volume at standard conditions (mL mol−1), catalyst mass (g), and the content of V over the catalyst (mmol g−1), respectively.
3. Results and discussion
3.1. Structural and chemical properties of the catalysts
The amounts of V over the 4VOx–SiO2-H, 6VOx–SiO2-H, 6VOx/SiO2-I and 6VOx/SiO2-P catalysts were determined by ICP-MS. According to the results given in Table 1, the nominal V loading of 4VOx–SiO2-H is exactly the same, to the measured value of 4.0 wt% within the experimental errors. In the cases of the catalysts with 6 wt% V, the measured values (5.7–5.8 wt%) are also very close to the nominal V loadings.| Catalyst | V loading (wt%) | Surface area (m2 g−1) |
|---|---|---|
| 4VOx–SiO2-H | 4.0 | 810 |
| 6VOx–SiO2-H | 5.7 | 809 |
| 6VOx/SiO2-I | 5.8 | 518 |
| 6VOx/SiO2-P | 5.8 | 496 |
XRD results of the catalysts are shown in Fig. 1a. For 4VOx–SiO2-H and 6VOx–SiO2-H, only a broad peak at about 22.6° corresponding to the SiO2 support with amorphous nature was detected, suggesting the presence of highly dispersed VOx species. Contrary to this, additionally weak reflections at 2θ of 20.3°, 26.2° and 31.1° were observed for 6VOx/SiO2-I, which are assigned to (001), (110), and (400) crystal planes of crystalline V2O5, respectively.18,22 In the case of 6VOx/SiO2-P, the more and stronger diffraction peaks assigned to crystalline V2O5 were observed. These results indicate that the structures of VOx over these catalysts are different, even though the V loadings are in a narrow distribution. The 4VOx–SiO2-H and 6VOx–SiO2-H catalysts prepared by one-pot hydrothermal synthesis show a higher dispersion of VOx.
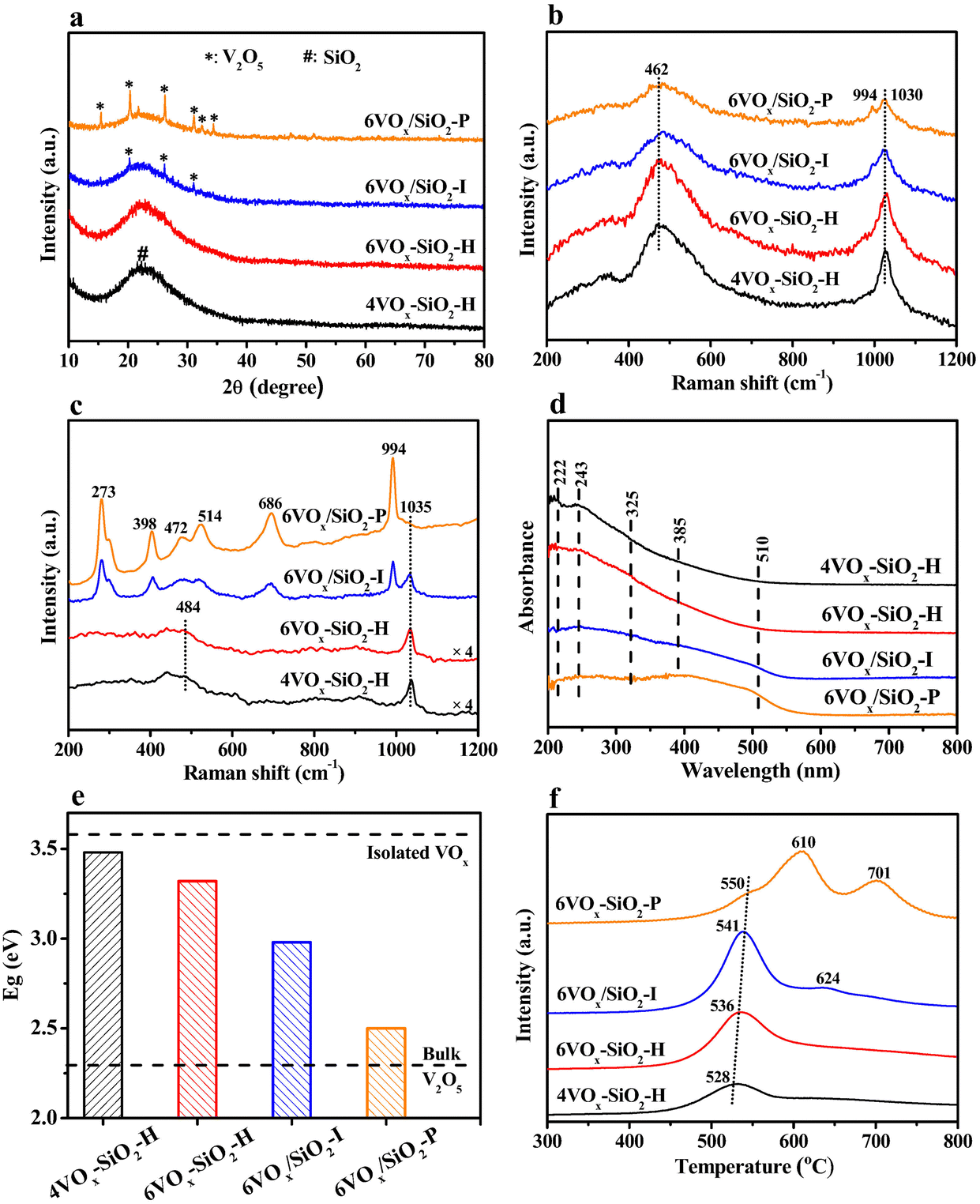 | ||
| Fig. 1 XRD patterns (a), in situ UV (b) and visible (c) Raman spectra, in situ UV-vis DRS spectra (d), calculated edge energy (e) and H2-TPR profiles (f) of the catalysts. | ||
Textural properties of the catalysts were analyzed by N2 adsorption–desorption isotherms. As given in Fig. S2a (ESI†), all catalysts exhibited the similar type-IV isotherms with an H1-type hysteresis loop, which are typical characteristics of the capillary condensation of N2 in mesoporous materials with uniform channels. This can be further reflected from the very narrow PSD peaks at about 3 nm determined by the BJH method (Fig. S2b, ESI†). From the calculated textural parameters summarized in Table 1, the specific surface areas of 4VOx–SiO2-H and 6VOx–SiO2-H were about ∼800 m2 g−1, which are clearly higher than that of 6VOx/SiO2-I and 6VOx/SiO2-P (∼500 m2 g−1). This commonly leads to a higher dispersion of VOx, coinciding with the XRD results (Fig. 1a).
To further identify the structure of VOx species over these catalysts, in situ UV and visible Raman were applied under dehydrated conditions. Generally, the former is more sensitive to detect isolated and polymerized VOx species, while the latter shows a higher sensitivity to crystalline V2O5.22,23,38 As shown in the Fig. 1b, two UV Raman bands at ∼460 and 1030 cm−1 were clearly observed over 4VOx–SiO2-H and 6VOx–SiO2-H, which can be assigned to V–O–Si and V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, respectively.38–40 However, an additionally weak Raman band at 994 cm−1 was detected for 6VOx/SiO2-I and 6VOx/SiO2-P, indicating the presence of crystalline V2O5.22,41 This can be more clearly reflected by the visible Raman spectra as given in Fig. 1c. Only the Raman band at ∼1035 cm−1 and the weak and broad band at about 484 cm−1 assigned to VO vibration and the SiO2 support,38,40 respectively, were observed in the spectra of 4VOx–SiO2-H and 6VOx–SiO2-H. However, the Raman bands at 273, 398, 472, 514, 686, and 994 cm−1 assigned to crystalline V2O5 were clearly observed in the case of 6VOx/SiO2-I.40 Furthermore, the intensity of these Raman bonds becomes more pronounced in the spectrum of 6VOx/SiO2-P, accompanying an almost disappearance of VO vibration at 1035 cm−1. This suggests the formation of dominated crystalline V2O5 over 6VOx/SiO2-P.
O stretching vibrations, respectively.38–40 However, an additionally weak Raman band at 994 cm−1 was detected for 6VOx/SiO2-I and 6VOx/SiO2-P, indicating the presence of crystalline V2O5.22,41 This can be more clearly reflected by the visible Raman spectra as given in Fig. 1c. Only the Raman band at ∼1035 cm−1 and the weak and broad band at about 484 cm−1 assigned to VO vibration and the SiO2 support,38,40 respectively, were observed in the spectra of 4VOx–SiO2-H and 6VOx–SiO2-H. However, the Raman bands at 273, 398, 472, 514, 686, and 994 cm−1 assigned to crystalline V2O5 were clearly observed in the case of 6VOx/SiO2-I.40 Furthermore, the intensity of these Raman bonds becomes more pronounced in the spectrum of 6VOx/SiO2-P, accompanying an almost disappearance of VO vibration at 1035 cm−1. This suggests the formation of dominated crystalline V2O5 over 6VOx/SiO2-P.
To estimate the polymerization degree of VOx over these catalysts, in situ UV-Vis DRS experiments were performed under dehydrated condition. As shown in Fig. 1d, a broad absorption band from about 200 to 550 nm was observed in all the spectra of the catalysts because of the ligand-to-metal charge transfer (LMCT) transitions and the d–d transitions of V ions. According to ref. 42 and 43 the coordination and polymerization degree of VOx species are closely connected with the band position of LMCT transitions. Specifically, the absorption features at 222 and 243 nm can be assigned to strongly and less distorted isolated VOx in tetrahedral coordination environment, respectively.31,44 The bands at 325 and 385 nm are generally assigned to oligomeric VOx species in either tetrahedral or octahedral coordination environment.37,45 Additionally, the octahedral V2O5 crystallites can be observed at ∼510 nm.37,45 For 4VOx–SiO2-H, the strongest bonds at 222 and 243 nm were observed. However, the intensity of these two bonds was decreased over 6VOx–SiO2-H, accompanying the enhanced signals at 325 and 385 nm. This indicates that the 4VOx–SiO2-H catalyst shows the most isolated VOx species, while more polymeric VOx was formed over 6VOx–SiO2-H. In the case of 6VOx/SiO2-I, an extremely weak band at 510 nm was observed, indicating the presence of both polymeric VOx and a small amount of crystalline V2O5. As for 6VOx/SiO2-P, the intensity of the band at 510 nm became more pronounced, suggesting the presence of higher amount of crystalline V2O5.
Furthermore, the edge energy (Eg) of these catalysts was determined based on the UV-Vis DRS results. As shown in Fig. S3 (ESI†) and 1e, the calculated Eg decreased in the order of 4VOx–SiO2-H (3.48 eV) > 6VOx–SiO2-H (3.32 eV) > 6VOx/SiO2-I (2.90 eV) > 6VOx/SiO2-P (2.50 eV), indicating an increased degree of polymerization or domain size of VOx.18,24 Moreover, the Eg for 4VOx–SiO2-H was very close to that for the bulk Na3VO4 (3.55 eV) only consisting of isolated VOx species.46 Thus, the isolated VOx species was dominated over 4VOx–SiO2-H. However, in the case of 6VOx/SiO2-P, the Eg was comparable with that of the bulk V2O5 (2.33 eV),46 suggesting the presence of dominated crystalline V2O5.
To study the redox property of the VOx species over the catalysts, H2-TPR was performed. As shown in Fig. 1f, only a reduction peak at around 530 °C was observed for 4VOx–SiO2-H and 6VOx–SiO2-H, which can be easily assigned to the reduction of highly dispersed VOx species.47,48 For 6VOx/SiO2-I, two peaks at 541 and 624 °C were observed. The former originates from the reduction of highly dispersed VOx species, while the weak peak at 624 °C can be attributed to the reduction of a small amount of microcrystalline V2O5.49 In the case of 6VOx/SiO2-P, three peaks at 550, 610 and 701 °C were present. The shoulder peak at 550 °C can be assigned to the reduction of a small amount of highly dispersed VOx species, while two sharp peaks at higher temperatures originate from the different stages for the reduction of bulk V2O5 from V5+ to V4+, and then to V3+.50 Moreover, the continuously increased peak temperatures in the order of 4VOx–SiO2-H (528 °C) < 6VOx–SiO2-H (536 °C) < 6VOx/SiO2-I (541 and 624 °C) < 6VOx/SiO2-P (550, 610 and 701 °C) indicate an increased difficulty in the reduction of the catalysts with a higher polymerization extent or a larger domain size of VOx species.29,49
As for the oxidation state of V over the fresh catalysts, a symmetric V 2p3/2 peak with a binding energy of 517.6 eV was observed in all XP spectra of the catalysts (Fig. S4, ESI†), suggesting that V5+ is dominated species on the catalysts surface.18,22 Taking this fact and the total amounts of H2 consumption during H2-TPR into account, the average oxidation state (AOS) of V over different catalysts after H2-TPR was calculated. As given in Table 2, the AOS of V decreased in the order of 4VOx–SiO2-H (+4.06) < 6VOx–SiO2-H (+3.88) < 6VOx/SiO2-I (+3.62) < 6VOx/SiO2-P (+3.35). This indicates that the reduction degree of VOx species increases with increasing its polymerization degree or domain size, leading to a more favorable reduction of V5+ to V3+ instead of V4+.18,22
| Catalyst | H2 consumptiona (μmol gcatalyst−1) | H/V ratioa | AOS of Va | Released COb (μmol gcatalyst−1) | Released COb (mmol gV−1) |
|---|---|---|---|---|---|
| a Calculated based on the H2-TPR results as given in Fig. 1f. b Cumulative amount of CO formed from the pulse of CO2 over the reduced catalyst at 600 °C as given in Fig. 4a–d. | |||||
| 4VOx–SiO2-H | 358 | 0.94 | +4.06 | 270 | 6.74 |
| 6VOx–SiO2-H | 621 | 1.12 | +3.88 | 353 | 5.89 |
| 6VOx/SiO2-I | 766 | 1.38 | +3.62 | 225 | 3.76 |
| 6VOx/SiO2-P | 915 | 1.65 | +3.35 | 36 | 0.61 |
Based on these complementary characterizations, the dominated structures of VOx over the four catalysts are proposed and illustrated in Scheme 1. It can be concluded that (1) monomeric VOx species is dominantly present over 4VOx–SiO2-H; (2) monomeric and less polymeric VOx coexist over the 6VOx–SiO2-H catalyst; (3) highly polymeric VOx and a small amount of crystalline V2O5 are present over 6VOx/SiO2-I; and (4) crystalline V2O5 is overwhelmingly dominated in the case of 6VOx/SiO2-P.
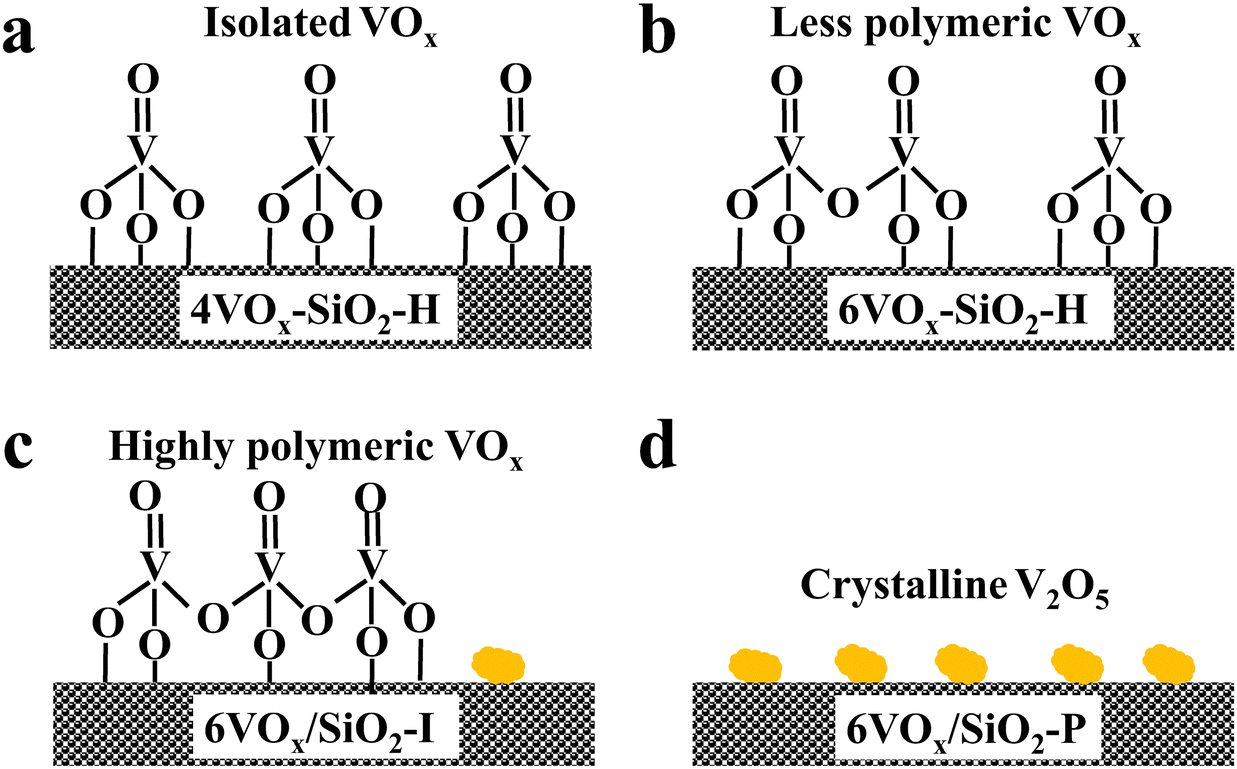 | ||
| Scheme 1 Illustration of the dominated VOx species over 4VOx–SiO2-H (a), 6VOx–SiO2-H (b), 6VOx/SiO2-I (c) and 6VOx/SiO2-P (d). | ||
3.2. Catalytic performance
The initial conversion of propane and CO2 at a TOS of 8 min over the catalysts for CO2-ODP is given in Fig. 2a. The propane conversion decreased from 41.0% to 11.9% in the order of 6VOx–SiO2-H > 4VOx–SiO2-H > 6VOx/SiO2-I > 6VOx/SiO2-P. If the CO2 conversion is compared, the same changing order can be observed. Considering the fact that the V loadings of these catalysts are very close, the results suggest that the structure of VOx plays an important role in determining the catalytic activity.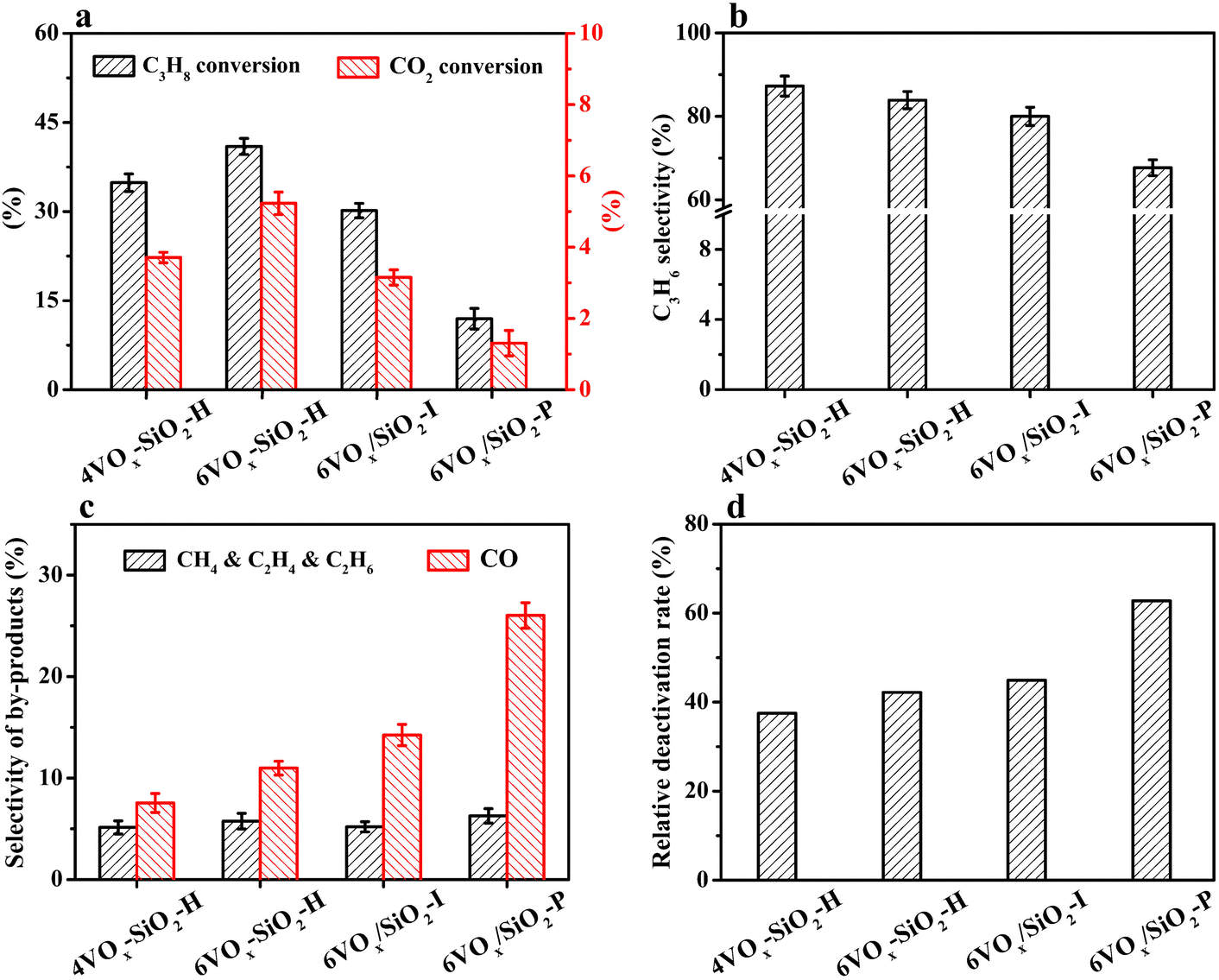 | ||
| Fig. 2 Initial conversion of C3H8 and CO2 (a), C3H6 selectivity (b) and by-products selectivity (c), and the relative deactivation rate (d) over the catalysts for CO2-ODP under the conditions of 600 °C, 0.1 MPa, Ar/C3H8/CO2 molar ratio = 3/2/27, total flow rate = 32 mL min−1, and GHSV = 7680 mL g−1 h−1. | ||
As show in Fig. 2b, the initial propene selectivity decreased from 87.3% to 67.7% in the order of 4VOx–SiO2-H > 6VOx–SiO2-H > 6VOx/SiO2-I ≫ 6VOx/SiO2-P. To understand the main side reactions, the initial selectivity of gaseous by-products was analyzed. As shown in Fig. 2c, CH4, C2H4 and C2H6 originating from the cracking and CO produced from the CO2-RP reaction were detected. For all catalysts, the total selectivity for the by-products of CH4, C2H4 and C2H6 was centered at around 5%. However, the selectivity of CO increased from 7.6% to 26.0% in the order of 4VOx–SiO2-H < 6VOx–SiO2-H < 6VOx/SiO2-I ≪ 6VOx/SiO2-P. The change order is inverse to that of the propene selectivity (Fig. 2b), suggesting that the formation of desired propene is strongly affected by the side reaction of CO2-RP during the reaction. The 4VOx–SiO2-H catalyst with the highest dispersion of VOx species, i.e., the dominantly monomeric VOx, showed the highest propene selectivity. With increasing the degree of polymerization or domain size of VOx, the amounts of active site over the catalyst which are favorable for CO2-RP involving the breaking of C–C bond increased, leading to the lowest propene selectivity over the 6VOx/SiO2-P catalyst with dominantly crystalline V2O5. A similar result is observed in the chemical looping oxidative dehydrogenation of propane catalyzed by VOx/TiO2, suggesting that the highly dispersed VOx is the main active species for the oxidative dehydrogenation of propane.51
In the case of the catalytic stability, an apparently decreased conversion of either propane or CO2 with increasing TOS was observed for all catalysts (Fig. S5a and b, ESI†). To quantitatively estimate the stability of the catalysts, the relative deactivation rate associated with propane conversion was calculated (Rpropane, see eqn (6)). As given in Fig. 2d, the stability of the catalysts varied in a lager extent, and Rpropane increased in the order of 4VOx–SiO2-H < 6VOx–SiO2-H < 6VOx/SiO2-I < 6VOx/SiO2-P. The same change pattern was observed when the relative deactivation rate was calculated based on the CO2 conversion (RCO2, Fig. S6, ESI†), resulting in the best stability of the 4VOx–SiO2-H catalyst.
3.3. Structural correlation of VOx with the catalytic activity
As revealed from the results in Sections 3.1 and 3.2, the catalytic activity of supported VOx for CO2-ODP is closely connected with the polymerization extent or domain size of VOx species. To further confirm this, the apparent TOF(C3H8) of the catalysts calculated by eqn (8) were correlated with the Eg values derived from the UV-vis DRS. As shown in Fig. 3, with decreasing the Eg, the TOF(C3H8) continuously decreased in the order of 4VOx–SiO2-H > 6VOx–SiO2-H > 6VOx/SiO2-I > 6VOx/SiO2-P, leading to the highest TOF(C3H8) of 4VOx–SiO2-H with dominantly monomeric VOx. In the case of the 4VOx–SiO2-H and 6VOx–SiO2-H catalysts, no crystalline V2O5 can be detected, and only two-dimensional VOx species is present. Moreover, the textural properties of 4VOx–SiO2-H and 6VOx–SiO2-H, i.e. the surface area and pore size distributions are almost the same (Table 1 and Fig. S2, ESI†), which commonly leads to the very similar V distributions on the SiO2 support. Thus, the decreased TOF(C3H8) can be ascribed to the increased in the polymerization degree of VOx.16,17 However, the crystalline V2O5 is formed over the 6VOx/SiO2-I and 6VOx/SiO2-P catalysts, in which not all V atoms can be touched with the reactants during the reaction. Thus, the decreased TOF(C3H8) from 6VOx/SiO2-I to 6VOx/SiO2-P is determined by both the increased polymerization degree of VOx and the formation of more crystalline V2O5. These analyses together with the structures of VOx illustrated in Scheme 1 indicate that the activity of VOx species for CO2-ODP decreases in the order of monomeric VOx > less polymeric VOx > highly polymeric VOx > crystalline V2O5. If the apparent TOF(CO2) of the catalyst was calculated and correlated with the Eg (Fig. S7, ESI†), exactly the same change pattern to that of the TOF(C3H8) was observed, and the highest value was obtained over 4VOx–SiO2-H, indicating the simultaneous activation of propane and CO2. Moreover, the TOF(CO2) of the catalyst is generally lower than its TOF(C3H8), which can be attributed to the weak ability for the activation of CO2 over the supported VOx species.52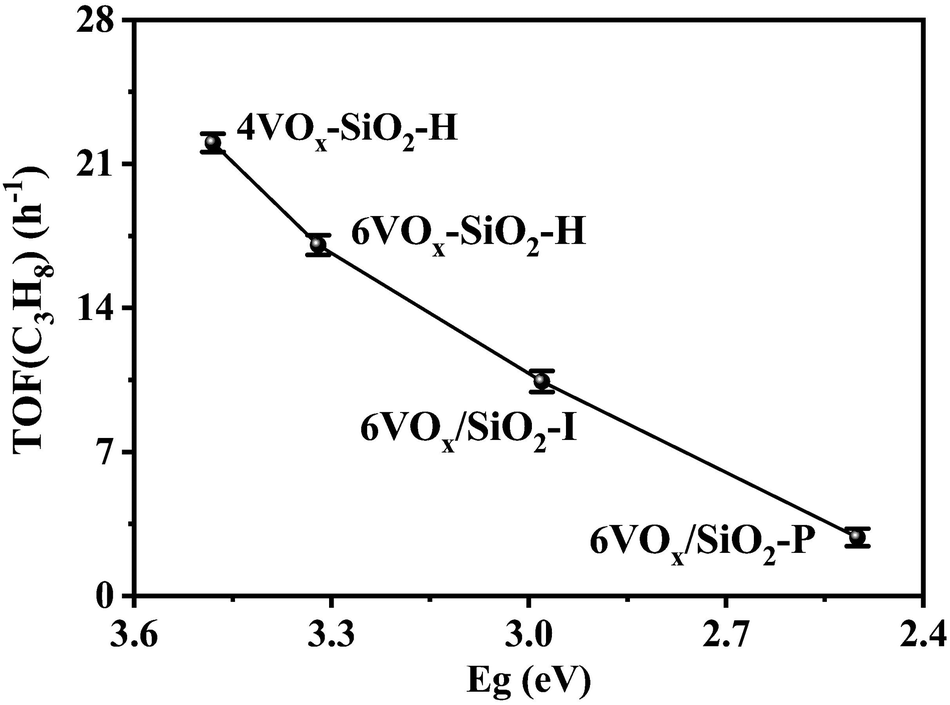 | ||
| Fig. 3 Relationship between TOF(C3H8) and Eg derived from UV-vis DRS of the catalysts. | ||
According to the Mars–van Krevelen redox mechanism for CO2-ODP catalyzed by supported VOx catalysts,8,53 the activated propane molecules abstract the active oxygen from the VOx species with a high V oxidation state (+5) to produce propene and water. Simultaneously, VOx at a reduced state is re-oxidized by the activated CO2 to complete the redox cycle of VOx with releasing CO. Thus, the re-oxidized ability of V3+/V4+ by CO2 may be crucial in determining the activity of the catalyst, if the facile redox property of supported VOx at high temperatures and the soft oxidizing ability of CO2 are taken into account. Following this understanding, the re-oxidized ability of the reduced catalysts by CO2 was investigated by CO2 pulse experiments at the reaction temperature of 600 °C. As shown in Fig. 4a–d, when CO2 (m/z = 44) was pulsed, CO (m/z = 28) was detected over all the catalysts pre-reduced by H2. Moreover, for pure SiO2 support prepared by hydrothermal synthesis method, no CO signal (m/z = 28) was detected (Fig. S8, ESI†). These results indicate that the reduced VOx species can be re-oxidized by CO2 to replenish the oxygen species consumed by H2 reduction. Moreover, as shown in Table 2, the amounts of released CO per gram of the catalyst varied in a large range, and decreased from 353 to 36 μmol gcatalyst−1 in the order of 6VOx–SiO2-H > 4VOx–SiO2-H > 6VOx/SiO2-I > 6VOx/SiO2-P. This indicates that the re-oxidized ability of reduced VOx species by CO2 is affected by the structure of VOx over the parent catalysts. To further confirm this, the amounts of released CO per gram of V over the catalyst were calculated (Table 2), and continuously decreased from 6.74 to 0.61 mmol gV−1 in the order of 4VOx–SiO2-H > 6VOx–SiO2-H > 6VOx/SiO2-I > 6VOx/SiO2-P. Thus, the re-oxidized ability of the reduced catalysts by CO2 decreases with increasing the polymerization extent or domain size of VOx over the parent catalysts, and 4VOx–SiO2-H with dominantly monomeric VOx shows the best ability for the activation of CO2 to release CO. As for the reason, on the one hand, the H2-TPR results have demonstrated that the V5+ oxide species with a higher polymerization extent or domain size over the parent catalysts was more easily reduced to a lower V oxidation state of V3+ rather than V4+ (Table 2), which is difficult to be re-oxidized by CO2 to V5+ if the soft oxidizing property of CO2 was taken into account.54,55 On the other hand, the CO2 pulse experiments were performed at 600 °C, however the 6VOx–SiO2-P catalyst, which exhibits a reduction peak at a higher temperature of about 700 °C during H2-TPR, may not be fully reduced at 600 °C. This also led to a lower amount of released CO when CO2 was pulsed. Moreover, the amount of released CO (270 μmol gcatalyst−1) during the CO2 pulse experiments over 4VOx–SiO2-H with the best ability for the activation of CO2 is still lower than the amount of H2 consumption (358 μmol gcatalyst−1) during the H2-TPR (Table 2). This indicates that the reduced VOx species cannot be fully re-oxidized by CO2 owning to the weak oxidizing ability of CO2.
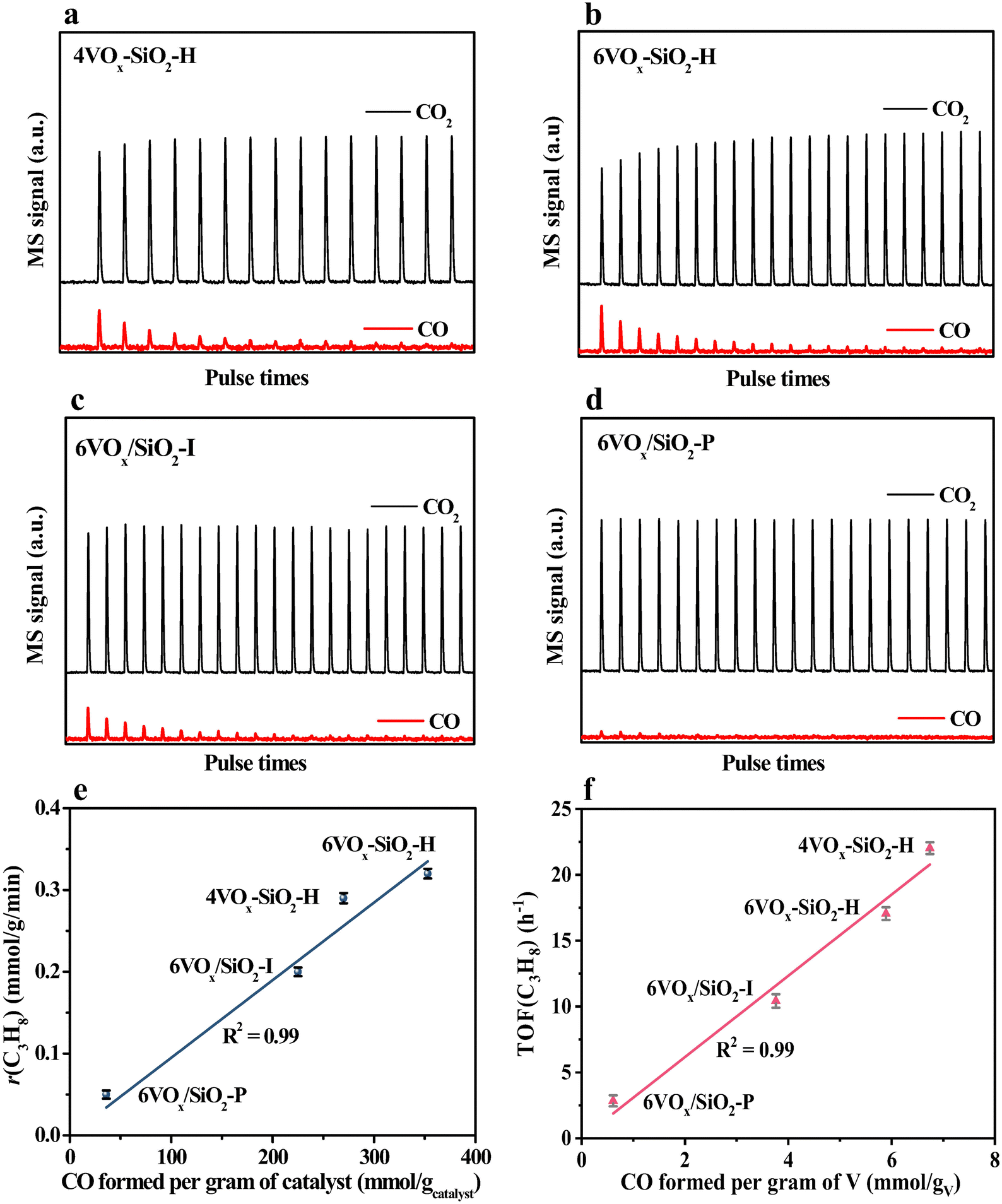 | ||
| Fig. 4 Transient MS response of the consecutive CO2 pulses over the pre-reduced catalysts at 600 °C (a)–(d), and correlating the propane consumption rate with the amount of cumulatively released CO per gram of catalyst (e) and the TOF(C3H8) with the amount of cumulatively released CO per gram of V (f) during the CO2 pulse experiments. | ||
Based on these understandings, the consumption rate of propane (r(C3H8), mmol g−1 min−1) was rationally correlated with the amount of formed CO per gram of catalyst (mmol gcatalyst−1) determined by CO2 pulse experiments. As shown in Fig. 4e, a directly proportional relationship was obtained, and r(C3H8) monotonously increased in the order of 6VOx/SiO2-P < 6VOx/SiO2-I < 4VOx–SiO2-H < 6VOx–SiO2-H with increasing the amount of formed CO. The higher amount of formed CO during the CO2 pulse experiments means the supplement of more active oxygen species over the reduced catalysts by CO2 reduction. Thus, this result clearly indicates that the activity of the parent catalyst is determined by the ability in activating CO2 to replenish the consumed oxygen species of the reduced catalyst, coinciding with the Mars–van-Krevelen redox mechanism. To further understand the relationship between the structure of VOx and the catalytic activity, the TOF(C3H8) was correlated with the amount of formed CO per gram of V. As shown in Fig. 4f, with increasing the amount of released CO per gram of V, the TOF(C3H8) increased in the order of 6VOx/SiO2-P < 6VOx/SiO2-I < 6VOx–SiO2-H < 4VOx–SiO2-H. The reduced 4VOx–SiO2-H catalyst with dominantly monomeric VOx species is the most easily re-oxidized by CO2, thereby the parent catalyst can well complete the redox cycle of VOx during CO2-ODP, resulting in the highest TOF(C3H8).
3.4. Insight into the catalytic stability
Generally, the coke deposition on the catalyst surface during the CO2-ODP reaction is associated with the catalyst deactivation. Thus, the spent catalysts after a TOS of 128 min were analyzed by TG, and the results are given in Fig. S9a–d (ESI†). According to ref. 11 and 16, the weight loss in the temperature range of 25–200 °C is attributed to the removing of adsorbed water, while the weight loss in the temperature range of 200–450 °C can be reasonably assigned to the burning of the deposited coke on catalyst surface. In our case, the coke content of the catalysts determined by the weight loss at 200–450 °C during TG was very low and varied in the range of 0.41 to 0.91% (Table 3). When the coke species was considered, visible Raman was performed for the spent catalysts at ambient environment. As shown in Fig. S10 (ESI†), typical Raman shifts at 1340 and 1600 cm−1, assigning to the disordered (D band) and graphitic carbon (G band),16,56 respectively, were clearly detected for all the spent catalysts. Generally, the graphitization degree of deposited coke can be estimated by the calculated intensity ratio of the D band to G band (ID/IG).16,56 As given in Table 3, the ID/IG was determined as 0.74 ± 0.08 for 4VOx–SiO2-H, 6VOx–SiO2-H and 6VOx/SiO2-I, indicating the similar coke species on the surface of catalysts. Contrary to this, a clearly lower ID/IG of 0.38 was obtained for 6VOx/SiO2-P, indicating the highest graphitization extent of coke species.7,16,56 As revealed from the analyses of the by-product selectivity, the 6VOx/SiO2-P catalyst with dominated V2O5 is the most favorable to break C–C bond via the CO2-RP and cracking reactions, while the remaining catalysts are more promising for CO2-ODP. Thus, the highest graphitization extent of coke species over 6VOx/SiO2-P may originate from the side reactions, which is favorable to form the coke species with a higher graphitization extent. This is consistent with previous work.57 Additionally, the coke content decreased in the order of 6VOx–SiO2-H > 4VOx–SiO2-H > 6VOx/SiO2-I > 6VOx/SiO2-P, which coincides well with their propane conversion (Fig. 2a), meaning that the catalyst which converts more propane shows a higher content of coke. Following this understanding, the apparent coking rate, which is defined as the amount of the coke by converting 1 mol propane (g mol−1) over the catalyst for CO2-ODP was calculated. As given in Table 3, the coking rate was determined as 0.45 ± 0.03 g mol−1 for 4VOx–SiO2-H, 6VOx–SiO2-H and 6VOx/SiO2-I, indicating a similar coke deposition behavior on the surface of catalysts. On the contrary, the significantly higher coking rate of 1.05 g mol−1 was obtained for 6VOx/SiO2-P, which coincides well with the highest graphitization extent of coke species (Table 3). It is worth noting that if the relative deactivation rate of the catalysts (Rpropane, Fig. 2d) was compared with either the coke content or the coking rate (Table 3), no obvious correlation of the catalytic stability with the deposited coke can be found, suggesting that the coke deposition may be not the critical factor in determining the stability of the catalysts.| Catalyst | Coke contenta (wt%) | I D/IGb | Coking ratec (g mol−1) | V oxidation state distributiond (%) | ||
|---|---|---|---|---|---|---|
| V5+ | V4+ | V3+ | ||||
| a Determined by the TG results as given in Fig. S9 (ESI). b Calculated based on the visible Raman results as given in Fig. S10 (ESI). c Calculated based on the amount of the coke by converting 1 mol propane. d Determined by the XP spectra results as given in Fig. 5b. | ||||||
| 4VOx–SiO2-H | 0.87 | 0.82 | 0.49 | 69.4 | 30.6 | 0 |
| 6VOx–SiO2-H | 0.91 | 0.74 | 0.50 | 61.8 | 31.1 | 7.1 |
| 6VOx/SiO2-I | 0.58 | 0.66 | 0.47 | 51.5 | 27.0 | 21.5 |
| 6VOx/SiO2-P | 0.41 | 0.38 | 1.06 | 25.1 | 29.1 | 45.8 |
According to the redox mechanism of CO2-ODP, the ability to replenish the active oxygen species by CO2 to complete the redox cycles of VOx plays an important role in determining the stability of the catalysts. To confirm this viewpoint, the relative deactivation rate (Rpropane) was correlated with the amount of released CO per gram of V over the catalyst. As shown in Fig. 5a, an almost linear relationship was obtained, i.e., a higher amount of released CO, a lower Rpropane. This indicates that the catalyst shows a better stability provided that the reduced VOx species over the catalyst is more easily re-oxidized by CO2. As a result, the best stability was obtained over the 4VOx–SiO2-H catalyst with dominantly monomeric VOx species. Taking the structural and chemical properties of different VOx species into account, the V5+ species with a higher polymerization extent or domain size over the parent catalysts was more easily reduced to V3+ (Table 2), which cannot be effectively re-oxidized by CO2 to V5+ to complete the redox cycles.18,54 Thus, the deep reduction of V5+ to V3+ during the reaction may be associated with the catalyst deactivation. To confirm this, the spent catalysts after CO2-ODP for a TOS of 128 min were characterized by XPS. As shown in Fig. 5b, the binding energy of V 2p3/2 over the spent catalysts shifted to a lower value in comparison with that of the fresh catalysts (517.6 eV, Fig. S4, ESI†), indicating an increased content of VOx with a lower oxidation state18,22 after the CO2-ODP reaction. To make a quantitative comparison, the V 2p3/2 spectra were deconvoluted. As given in Fig. 5b, three peaks at 515.7, 516.6 and 517.6 eV assigned to V3+, V4+ and V5+ species,22,58 respectively, were observed. The relative fractions of V3+, V4+ and V5+ over the spent catalysts were calculated by respective peak area. As shown in Table 3, the relative fraction of V5+ deceased from 69.4% to 25.1% in the order of 4VOx–SiO2-H > 6VOx–SiO2-H > 6VOx/SiO2-I > 6VOx/SiO2-P, the change pattern is consistent with that of their catalytic stability, i.e., the higher content of V5+ remained on the spent catalyst surface, the better stability of the catalyst was (Fig. S11a, ESI†). Contrary to this, when the relative fraction of V3+ was correlated with the stability of the catalysts, an opposite order was found (Fig. S11b, ESI†). These results clearly indicate that the deep reduction of V5+ to V3+ is the key reason for the deactivation of the catalysts in CO2-ODP. Thus, the redox rates of the VOx species participated with CO2 and propane are not very well matched, owning to the weak oxidizing ability of CO2. This is consistent with the CO2 pulse experiments and H2-TPR results (Table 2).
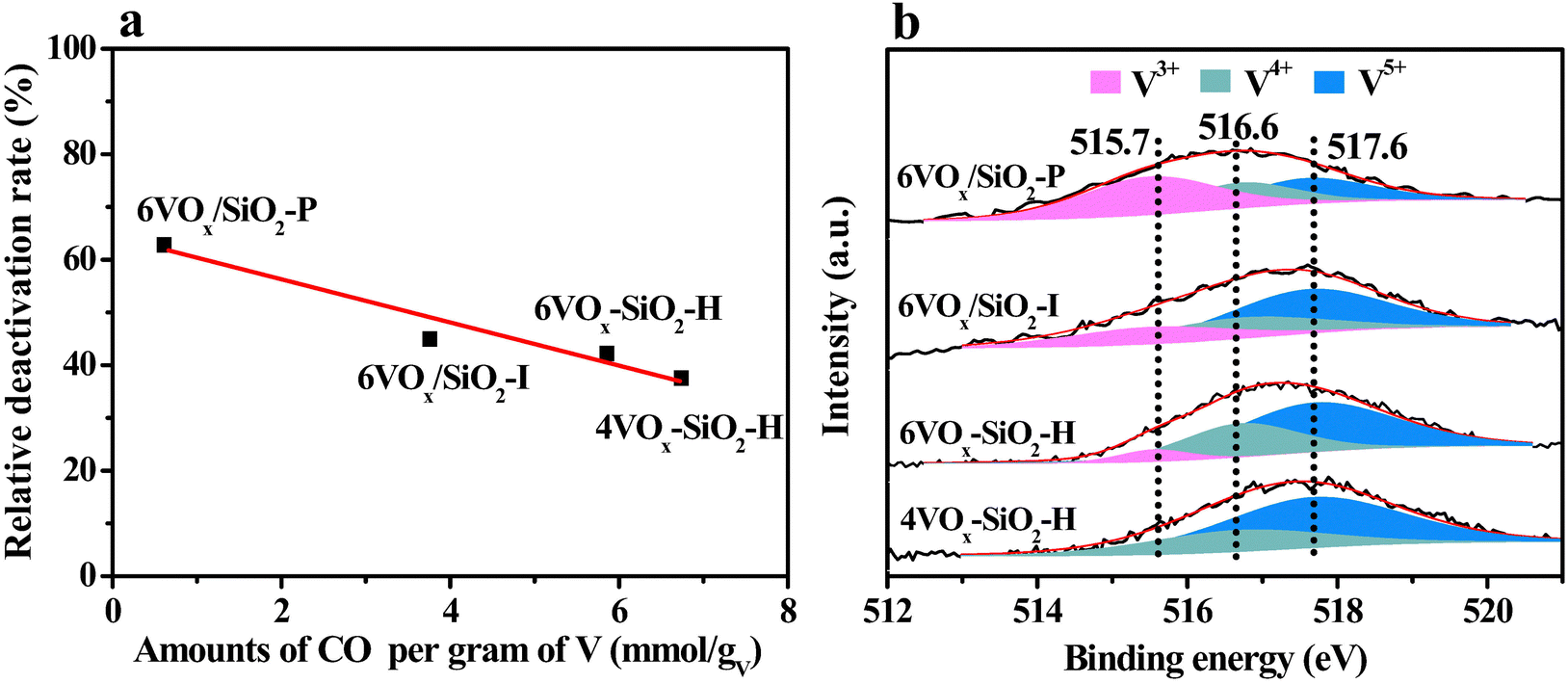 | ||
| Fig. 5 Correlation of relative deactivation rate (Rpropane) with the amount of formed CO per gram of V over the catalyst during CO2 pulse experiments (a), and V 2p3/2 XP spectra of the spent catalysts after CO2-ODP for a TOS of 128 min (b). | ||
Following the above discussion, the redox mechanism of CO2-ODP catalyzed by the SiO2-supported VOx catalysts with isolated VOx species is illustrated in Scheme 2. Propane reacts with the active oxygen of V5+ to produce propene and water, resulting in the formation of reduced VOx with an oxidation state of +4. To complete the redox cycle, CO2 reacts with the V4+ species to release CO and simultaneously replenish the active oxygen species. With increasing the polymerization extent or domain size of supported VOx species, the matching of the redox cycles of VOx during the CO2-ODP reaction was hindered, due to the formation of deeply reduced VOx species with a lower oxidation state of +3, which cannot be efficiently re-oxidized by CO2 to V5+. As a result of the highest dispersion of VOx species over 4VOx–SiO2-H, the best performance for CO2-ODP was obtained among the evaluated catalysts.
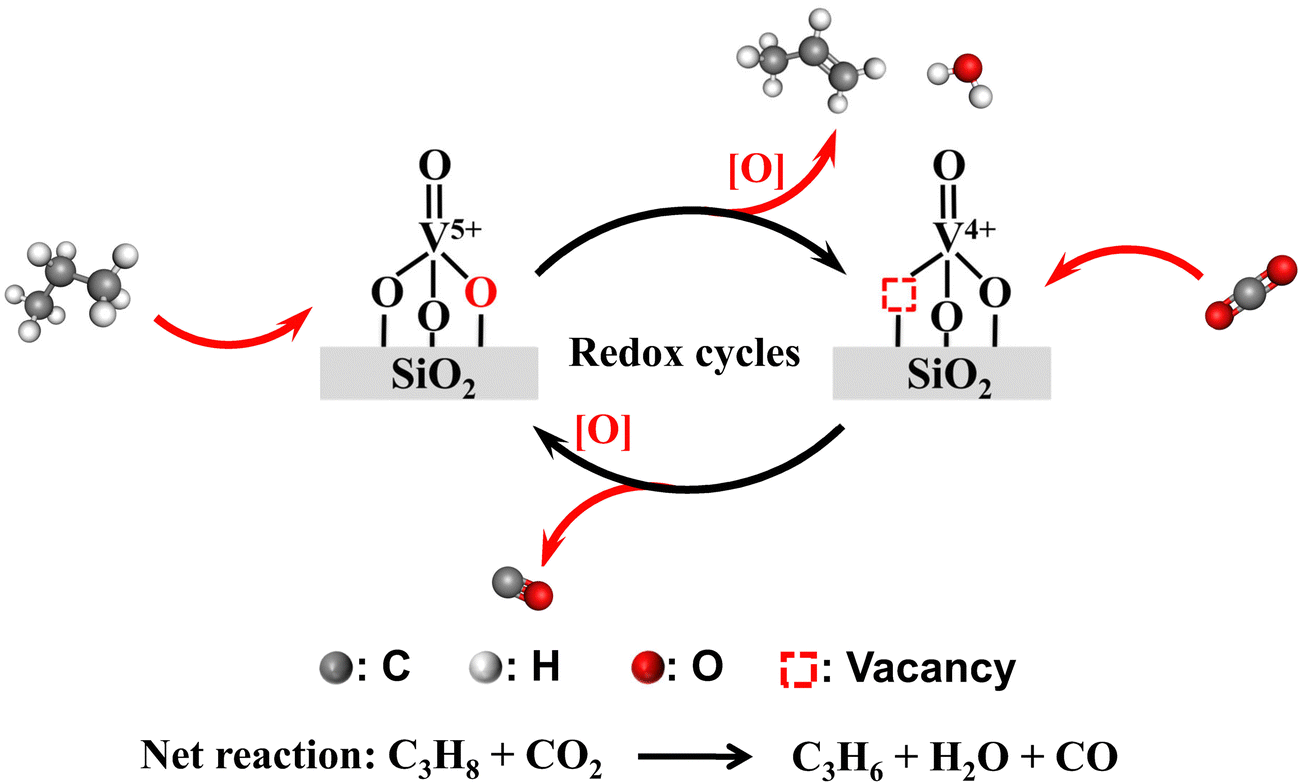 | ||
| Scheme 2 Redox mechanism over supported VOx catalyst for CO2-ODP. | ||
4. Conclusions
In summary, four SiO2-supported VOx catalysts with narrow distributions of V loading while well-defined structures of VOx species were synthesized by applying appropriate preparation methods. The dominated monomeric and less polymeric VOx were formed over the 4VOx–SiO2-H and 6VOx–SiO2-H catalysts, respectively, prepared by one-pot hydrothermal synthesis. In case of the 6VOx/SiO2-I catalyst prepared by simply impregnation method, the highly polymerized VOx with a small amount of V2O5 was present, while V2O5 crystallites were overwhelmingly predominant over the 6VOx/SiO2-P catalyst prepared by physic grinding method. The catalytic activity and stability for CO2-ODP decreased in the order of 4VOx–SiO2-H > 6VOx–SiO2-H > 6VOx/SiO2-I > 6VOx/SiO2-P, which can be explained by the ability for re-oxidation of the reduced VOx species with different structures by CO2. The reduced monomeric VOx is the most easily re-oxidized by CO2, and thus the reduction/oxidation cycles of V4+/V5+ species during the CO2-ODP reaction can be well completed, leading to the best performance. With increasing the polymerization extent or domain size of supported VOx species, the matching of the redox cycles of VOx during the reaction was hindered, due to the formation of deeply reduced VOx species with a lower oxidation state of +3, which cannot be efficiently re-oxidized by CO2. These understandings can provide important contributions to the essential reason of the structural impacts of VOx species on CO2-ODP, which are beneficial to promote the development of highly active and stable VOx-based catalysts for CO2-ODP.Author contributions
Li Wang: investigation, formal analysis, data curation, visualization, writing – original draft; Heng-Bo Zhang: conceptualization, investigation, formal analysis, data curation; Rong-Rong Hu and Han-Qing Ge: project administration; Guo-Qing Yang: conceptualization, investigation, methodology, writing – review & editing; Yuefeng Li, and Zhao-Tie Liu: investigation; Zhong-Wen Liu: conceptualization, supervision, writing – review & editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 22338010).References
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- T. Otroshchenko, G. Jiang, V. A. Kondratenko, U. Rodemerck and E. V. Kondratenko, Chem. Soc. Rev., 2021, 50, 473–527 RSC.
- X. Jiang, L. Sharma, V. Fung, S. J. Park, C. W. Jones, B. G. Sumpter, J. Baltrusaitis and Z. Wu, ACS Catal., 2021, 11, 2182–2234 CrossRef CAS.
- Y. Yuan, W. N. Porter and J. G. Chen, Trends Chem., 2023, 5, 840–852 CrossRef CAS.
- H. Guo, Z. Xie, X. Wang, J. G. Chen and P. Liu, EES Catal., 2023, 1, 17–25 RSC.
- E. Gomez, B. Yan, S. Kattel and J. G. Chen, Nat. Rev. Chem., 2019, 3, 638–649 CrossRef CAS.
- J. Wang, Y.-H. Song, Z.-T. Liu and Z.-W. Liu, Appl. Catal., B, 2021, 297, 120400 CrossRef CAS.
- S. Zhang, C.-a Zhou, S. Wang, Z. Qin, G. Shu, C. Wang, L. Song, L. Zheng, X. Wei, K. Ma and H. Yue, Chem. Eng. J., 2024, 481, 148231 CrossRef CAS.
- M. Chen, J. Xu, Y. Cao, H.-Y. He, K.-N. Fan and J.-H. Zhuang, J. Catal., 2010, 272, 101–108 CrossRef CAS.
- H. Xiao, J. Zhang, P. Wang, X. Wang, F. Pang, Z. Zhang and Y. Tan, Catal. Sci. Technol., 2016, 6, 5183 RSC.
- G.-Q. Yang, Y.-J. He, Y.-H. Song, J. Wang, Z.-T. Liu and Z.-W. Liu, Ind. Eng. Chem. Res., 2021, 60, 17850–17861 CrossRef CAS.
- E. Nowicka, C. Reece, S. M. Althahban, K. M. H. Mohammed, S. A. Kondrat, D. J. Morgan, Q. He, D. J. Willock, S. Golunski, C. J. Kiely and G. J. Hutchings, ACS Catal., 2018, 8, 3454–3468 CrossRef CAS.
- G.-Q. Yang, X. Ren, V. A. Kondratenko, H.-B. Zhang, E. V. Kondratenko and Z.-W. Liu, Nano Res., 2023, 16, 6237–6250 CrossRef CAS.
- F. Xing, Y. Nakaya, S. Yasumura, K.-I. Shimizu and S. Furukawa, Nat. Catal., 2022, 5, 55–65 CrossRef CAS.
- K.-X. Li, X. Cai, H.-B. Liu, X.-Y. Liu, Y. Shan, X. Feng and D. Chen, React. Chem. Eng., 2024, 9, 1292–1312 RSC.
- X. L. Xue, W. Z. Lang, X. Yan and Y. J. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 15408–15423 CrossRef CAS PubMed.
- Y.-L. Shan, H.-L. Sun, S.-L. Zhao, P.-L. Tang, W.-T. Zhao, J.-W. Ding, W.-L. Yu, L.-N. Li, X. Feng and D. Chen, ACS Catal., 2022, 12, 5736–5749 CrossRef CAS.
- G.-Q. Yang, H. Wang, T. Gong, Y.-H. Song, H. Feng, H.-Q. Ge, H.-B. Ge, Z.-T. Liu and Z.-W. Liu, J. Catal., 2019, 380, 195–203 CrossRef CAS.
- J. Wang, M.-L. Zhu, Y.-H. Song, Z.-T. Liu, L. Wang and Z.-W. Liu, J. Catal., 2022, 409, 87–97 CrossRef CAS.
- I. Takahara, M. Saito, M. Inaba and K. Murata, Catal. Lett., 2005, 102, 201–205 CrossRef CAS.
- H. Wang, W. Zhu, G.-Q. Yang, Y.-W. Zhang, Y.-H. Song, N. Jiang, Z.-T. Liu and Z.-W. Liu, Ind. Eng. Chem. Res., 2019, 58, 21372–21381 CrossRef CAS.
- G. Liu, Z.-J. Zhao, T. Wu, L. Zeng and J. Gong, ACS Catal., 2016, 6, 5207–5214 CrossRef CAS.
- Z. Wu, H.-S. Kim, P. C. Stair, S. Rugmini and S. D. Jackson, J. Phys. Chem. B, 2005, 109, 2793–2800 CrossRef CAS PubMed.
- U. Rodemerck, M. Stoyanova, E. V. Kondratenko and D. Linke, J. Catal., 2017, 352, 256–263 CrossRef CAS.
- K. Chen, A. T. Bell and E. Iglesia, J. Catal., 2002, 209, 35–42 CrossRef CAS.
- H. Tian, E. I. Ross and I. E. Wachs, J. Phys. Chem. B, 2006, 110, 9593–9600 CrossRef CAS PubMed.
- O. Schwarz, D. Habel, O. Ovsitser, E. V. Kondratenko, C. Hess, R. Schomäcker and H. Schubert, J. Mol. Catal. A: Chem., 2008, 293, 45–52 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- Z.-F. Han, X.-L. Xue, J.-M. Wu, W.-Z. Lang and Y.-J. Guo, Chin. J. Catal., 2018, 39, 1099–1109 CrossRef CAS.
- W. Wang, S. Chen, C. Pei, R. Luo, J. Sun, H. Song, G. Sun, X. Wang, Z.-J. Zhao and J. Gong, Science, 2023, 381, 886–890 CrossRef CAS PubMed.
- Q. Liu, J. Li, Z. Zhao, M. Gao, L. Kong, J. Liu and Y. Wei, Catal. Sci. Technol., 2016, 6, 5927–5941 RSC.
- H. Xiao, J. Zhang, P. Wang, X. Wang, F. Pang, Z. Zhang and Y. Tan, Catal. Sci. Technol., 2016, 6, 5183–5195 RSC.
- J. Wang, L. Liu, J. Feng, X. Zhang, X. Ju and P. Chen, Catal. Sci. Technol., 2022, 12, 5339–5348 RSC.
- M. Numan, E. Eom, A. Li, M. Mazur, H. W. Cha, H. C. Ham, C. Jo and S.-E. Park, ACS Catal., 2021, 11, 9221–9232 CrossRef CAS.
- R. J. Madon and M. Boudart, Ind. Eng. Chem. Fundam., 1982, 21, 438–447 CrossRef CAS.
- M. Zhang, M. Wang, B. Xu and D. Ma, Joule, 2019, 3, 2876–2883 CrossRef.
- Q.-X. Luo, X.-K. Zhang, B.-L. Hou, J.-G. Chen, C. Zhu, Z.-W. Liu, Z.-T. Liu and J. Lu, Catal. Sci. Technol., 2018, 8, 4864–4876 RSC.
- Z. Wu, S. Dai and S. H. Overbury, J. Phys. Chem. C, 2010, 114, 412–422 CrossRef CAS.
- J. T. Grant, C. A. Carrero, A. M. Love, R. Verel and I. Hermans, ACS Catal., 2015, 5, 5787–5793 CrossRef CAS.
- X. Gao, S. R. Bare, B. M. Weckhuysen and I. E. B. Wachs, J. Phys. Chem. C, 1998, 102, 10842–10852 CrossRef CAS.
- G. T.-M. Christian Hess and R. Herbert, J. Phys. Chem. C, 2007, 111, 9471–9479 CrossRef.
- X. Gao, J.-M. Jehng and I. E. Wachs, J. Catal., 2002, 209, 43–50 CrossRef CAS.
- M. D. Argyle, K. Chen, C. Resini, C. Krebs, A. T. Bell and E. Iglesia, J. Phys. Chem. B, 2004, 108, 2345–2353 CrossRef CAS.
- S. Dzwigaj, M. Matsuoka, M. Anpo and M. Che, J. Phys. Chem. B, 2000, 104, 6012–6020 CrossRef CAS.
- H. Zhang, S. Cao, Y. Zou, Y.-M. Wang, X. Zhou, Y. Shen and X. Zheng, Catal. Commun., 2014, 45, 158–161 CrossRef CAS.
- X. Gao and I. E. Wachs, J. Phys. Chem. B, 2000, 104, 1261–1268 CrossRef CAS.
- Y. Liu, W. Feng, T. Li, H. He, W. Dai, W. Huang, Y. Cao and K. Fan, J. Catal., 2006, 239, 125–136 CrossRef CAS.
- G.-Q. Yang, Y. Niu, V. A. Kondratenko, X. Yi, C. Liu, B. Zhang, E. V. Kondratenko and Z.-W. Liu, Angew. Chem., Int. Ed., 2023, 62, e202310062 CrossRef CAS PubMed.
- Y.-M. Liu, Y. Cao, N. Yi, W.-L. Feng, W.-L. Dai, S.-R. Yan, H.-Y. He and K.-N. Fan, J. Catal., 2004, 224, 417–428 CrossRef CAS.
- M. M. Koranne, J. G. Goodwin and G. Marcelin, J. Catal., 1994, 148, 369–377 CrossRef CAS.
- S. Chen, C. Pei, X. Chang, Z.-J. Zhao, R. Mu, Y. Xu and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 22072–22079 CrossRef CAS PubMed.
- F. Xing and S. Furukawa, Chem. – Eur. J., 2023, 29, e202202173 CrossRef CAS PubMed.
- I. Ascoop, V. V. Galvita, K. Alexopoulos, M.-F. Reyniers, P. Van Der Voort, V. Bliznuk and G. B. Marin, J. Catal., 2016, 335, 1–10 CrossRef CAS.
- M.-S. Park, V. P. Vislovskiy, J.-S. Chang, Y.-G. Shul, J. S. Yoo and S.-E. Park, Catal. Today, 2003, 87, 205–212 CrossRef CAS.
- Z. W. Liu, C. Wang, W. B. Fan, Z. T. Liu, Q. Q. Hao, X. Long, J. Lu, J. G. Wang, Z. F. Qin and D. S. Su, ChemSusChem, 2011, 4, 341–345 CrossRef CAS PubMed.
- H. Wang, G.-Q. Yang, Y.-H. Song, Z.-T. Liu and Z.-W. Liu, Catal. Today, 2019, 324, 39–48 CrossRef CAS.
- L. Wang, G.-Q. Yang, X. Ren and Z.-W. Liu, Nanomaterials, 2022, 12, 417 CrossRef CAS PubMed.
- S. Chen, L. Zeng, R. Mu, C. Xiong, Z.-J. Zhao, C. Zhao, C. Pei, L. Peng, J. Luo, L.-S. Fan and J. Gong, J. Am. Chem. Soc., 2019, 141, 18653–18657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00094c |
| This journal is © The Royal Society of Chemistry 2024 |