
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uniting activity design principles of anode catalysts for direct liquid fuel cells†
Daniel J.
Zheng‡
 a,
Jiayu
Peng‡
*a,
Kaylee
McCormack
b,
Hongbin
Xu
a,
Jin Soo
Kang§
bc,
Zhenshu
Wang
b,
Zhichu
Ren
a,
Ju
Li
ad,
Yuriy
Román-Leshkov
*b and
Yang
Shao-Horn
*ace
a,
Jiayu
Peng‡
*a,
Kaylee
McCormack
b,
Hongbin
Xu
a,
Jin Soo
Kang§
bc,
Zhenshu
Wang
b,
Zhichu
Ren
a,
Ju
Li
ad,
Yuriy
Román-Leshkov
*b and
Yang
Shao-Horn
*ace
aDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: jypeng@mit.edu; shaohorn@mit.edu
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: yroman@mit.edu
cResearch Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
dDepartment of Nuclear Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
eDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 2nd August 2024
Abstract
Direct liquid fuel cells have advantages over hydrogen-based fuel cells and lithium-ion batteries for portable and mobile applications due to their high volumetric energy density and the convenient storage or refueling of liquid fuels. Unfortunately, the electrochemical oxidation of liquid fuels (such as methanol, ethanol, and formic acid) currently corresponds to ∼50% of the energy losses of these devices at operating conditions. Moreover, state-of-the-art catalysts for such critical reactions are generally composed of precious metals such as Pt and Pd, hindering the cost-effective implementation of these technologies. The development of novel catalyst design principles for electrochemical liquid fuel oxidation has been constrained by its complex, structure-sensitive reaction energetics that can involve multiple parallel, competitive reaction intermediates and pathways. In this review, we aim to dissect and bridge the understanding of fundamental energetics and the materials engineering of novel catalysts for the electrochemical oxidation of various liquid fuels. By deconvoluting these reactions into the energetics of different critical elementary steps, we define essential descriptors that govern the activity and selectivity of electrochemical liquid fuel oxidation. Several universal and fundamental design principles are proposed to optimize the catalytic performance of state-to-the-art and emerging electrocatalysts by tuning the chemistry and electronic structure of active sites. This review aims to provide a unique perspective connecting the electro-oxidation energetics of different liquid fuels with mechanistic and materials-centric studies to provide a holistic picture connecting the fundamental surface science with materials engineering for the rational design of electrocatalysts for liquid fuel oxidation.
Broader contextDirect liquid fuel cells and liquid fuels, with their high volumetric energy density and ease of refueling, are particularly suitable for portable applications where size can be the main limitation. However, the oxidation of liquid fuels is particularly challenging, constituting about ∼50% of the voltage losses found in these devices, even with high loading of catalysts. While there has been considerable work into both the understanding and engineering liquid fuel oxidation electrocatalysts, the benchmark materials have remained stagnant for nearly 50 years. This review aims to bridge the fundamental surface science and mechanistic understandings with electrocatalyst engineering for methanol, ethanol, and formic acid oxidation in acidic media. By understanding the energetics of the key elementary steps, descriptors for the activity and selectivity of liquid fuel oxidation are proposed. Design strategies aimed at materials with optimal descriptors are discussed with selected examples to highlight the effectiveness of these strategies. Finally, unexplored but potentially promising avenues for liquid fuel oxidation electrocatalysis are discussed. Taken altogether, this review provides a holistic picture that connects the fundamental surface studies for liquid fuel oxidation with the materials design and engineering studies to provide insight into the rational design of liquid fuel oxidation electrocatalysts. |
1. Introduction: promise and challenges in liquid fuel oxidation
Reducing the carbon footprint of our global energy consumption requires novel, cost-effective technologies that store and use low-cost electricity from renewable sources to meet our energy needs at scale and on demand.1 Low-cost solar and wind energy can be stored in the chemical bonds of energy carriers, such as hydrogen, hydrocarbons, and liquid fuels. Moreover, electrical energy can be regenerated through the oxidation of such energy carriers using fuel cells.2 Such technologies are advantageous over other clean energy storage and conversion technologies, such as Li-ion batteries, as these energy carriers store greater energy per unit weight and only involve earth-abundant elements in their chemical bonds.3 In contrast, Li-ion batteries require a transition metal ion to store one electron and are thus constrained by the availability of critical minerals (e.g., cobalt and nickel).4 Powering the planet with fuel cell technologies can enable efficient renewable energy utilization in diverse applications.Direct liquid fuel cells (DLFCs) possess many advantages over hydrogen-fed polymer electrolyte membrane fuel cells (PEMFCs). While hydrogen is flammable, explosive, and typically stored at high pressures (up to 700 bar), liquid fuels are much safer to handle and do not need high pressure for their transport and storage.5 More importantly, liquid fuels have comparable thermodynamic oxidation potentials but significantly higher volumetric energy density compared to compressed hydrogen, making DLFCs particularly promising for applications that require compact configurations and are primarily constrained by volume, e.g., portable electronic, space, and military applications.5 Based on our calculations (Note S1, ESI†), while these liquid fuels and hydrogen have similar oxidation potentials (Fig. 1a and Table 1), methanol (4820 W h L−1) and ethanol (6310 W h L−1) have markedly higher volumetric energy densities than hydrogen (e.g., 1320 W h L−1 at 700 bar). Moreover, alcohols with larger molecular weights (e.g., propanol and ethylene glycol) have even higher volumetric energy densities (Fig. 1a), but their complete oxidation is challenging due to the possibility of multiple partial oxidation by-products.5 Formic acid, dimethyl ether, and hydrazine have also been used as liquid fuels in DLFCs. However, dimethyl ether and hydrazine have safety concerns due to their flammability and toxicity.6 On the other hand, formic acid is generally regarded to be safer to handle than most liquid fuels.5 Although the volumetric energy density of formic acid is relatively low (2100 W h L−1) compared to alternatives, its electro-oxidation potential (−0.22 V vs. the reversible hydrogen electrode, VRHE) is also lower than most liquid fuels (Fig. 1a), which affords a greater theoretical cell potential (∼1.45 V) than the other DLFCs. From the perspective of thermodynamics, selectivity, and safety, methanol, ethanol, and formic acid have been considered the most desirable fuels for DLFCs.5,6 Therefore, we mainly focus on the electrochemical oxidation of these three liquid fuels in acidic conditions for this review.
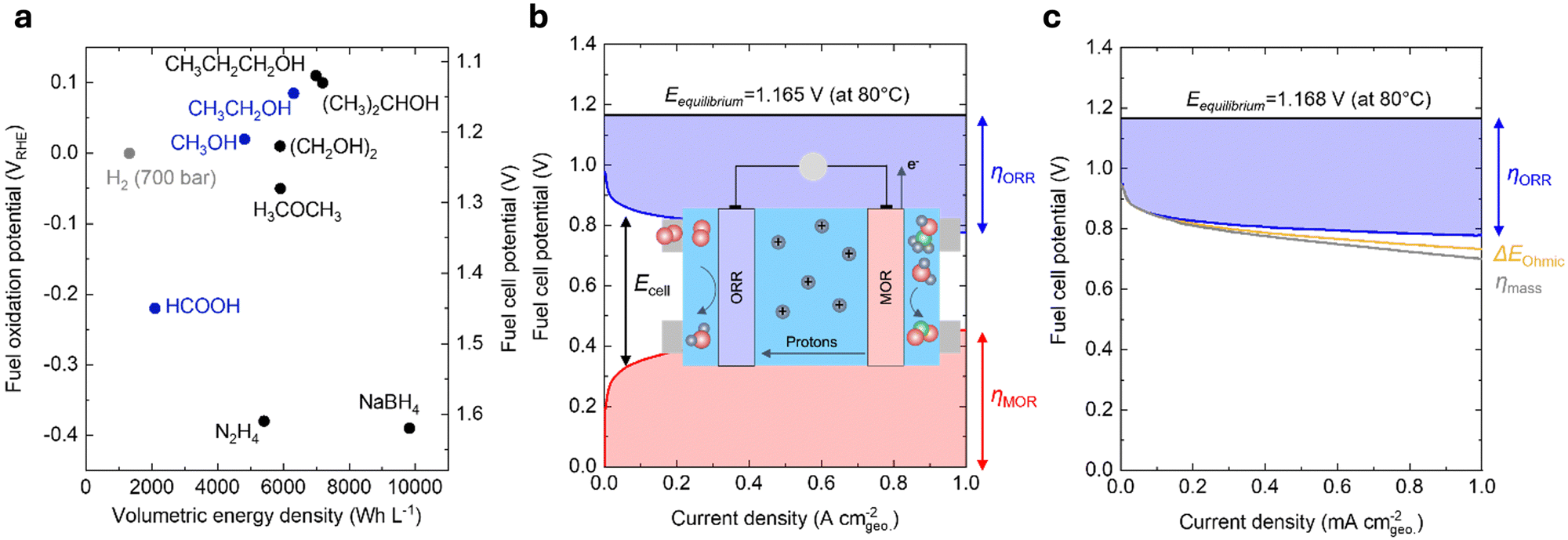 | ||
| Fig. 1 Thermodynamics and kinetics of liquid fuel electro-oxidation. (a) Comparison of the volumetric energy density and oxidation potential of hydrogen (700 bar) and various liquid fuels at 25 °C and 1 atm. The theoretical fuel cell potential was calculated using the difference between the fuel oxidation potential and the oxygen reduction potential at 1.23 VRHE. (b) Contribution of various voltage losses to the overall cell voltage of a practical DMFC. The losses are dominated by the sluggish kinetics of MOR (red) and ORR (blue). This figure has been adapted from ref. 7 with permission from John Wiley & Sons, Inc., copyright 2008. The inset schematic shows the typical operating procedure of a DMFC device. Methanol is fed into the anode and oxidized to CO2, producing electrons and protons. Oxygen (or air) is fed into the cathode, which reacts with protons that have been shuttled through a polymer electrolyte and electrons that have traveled through an external circuit to be reduced to H2O. O: red, H: blue, C: green. (c) Contribution of various voltage losses to the overall cell voltage of a practical H2/air-fed PEMFC.3 The losses are dominated by the sluggish ORR kinetics (blue) and are also led by minor contributions from the ohmic resistance (yellow) and the poor O2 mass transport (grey). DMFC conditions: 80 °C; 1.0 mg cmgeo.−2 PtRu/C anode, 1 M CH3OH; 0.4 mg cmgeo.−2 Pt/C cathode, air-fed (1 bar). PEMFC conditions: 80 °C; 0.4 mg cmgeo.−2 Pt/C anode, hydrogen-fed (1.5 bar); 0.4 mg cmgeo.−2 Pt/C cathode, air-fed (1.5 bar). Adapted from ref. 8. Copyright 2005 with permission from Elsevier. | ||
| Fuel | Anodic half-cell reaction | Oxidation potential (VRHE) | Specific energy (W h kg−1) | Energy density (W h L−1) |
|---|---|---|---|---|
a Potentials and energy densities were calculated using Gibbs free energy of formation and density data available from the NIST standard reference database and chemical handbooks.9–11
b Device-level gravimetric energy density was calculated by considering the mass of all reactants and products. For PEMFCs, the device-level gravimetric energy density was limited by the mass of the liquid product H2O (3660 W h kg−1), which is significantly lower than the gravimetric energy density of pure H2 fuel (33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 W h kg−1). 300 W h kg−1).
|
||||
| Hydrogen | H2 → 2H+ + 2e− | 0 | 33300 (fuel) |
1320 (700 bar) |
| 3660 (device)b | ||||
| Methanol | CH3OH + H2O → CO2 + 6H+ + 6e− | 0.02 | 6090 | 4820 |
| Ethanol | C2H5OH + 3H2O → 2CO2 + 12H+ + 12e− | 0.09 | 8000 | 6310 |
| Formic acid | HCOOH → CO2 + 2H+ + 2e− | −0.22 | 1725 | 2100 |
| 1-Propanol | C3H7OH + 5H2O → 3CO2 + 18H+ + 18e− | 0.1 | 8960 | 6990 |
| 2-Propanol | C3H7OH + 5H2O → 3CO2 + 18H+ + 18e− | 0.11 | 8880 | 7190 |
| Ethylene glycol | C2H6O2 + 2H2O → 2CO2 + 10H+ + 10e− | 0.01 | 5320 | 5850 |
| Dimethyl ether | C2H6O + 3H2O → 2CO2 + 12H+ + 12e− | ∼0.05 | 8960 | 5910 |
| Hydrazine | N2H4 → N2 + 4H+ + 4e− | ∼0.38 | 5390 | 5410 |
Compared to hydrogen-fueled PEMFCs, DLFCs have much higher kinetic losses at comparable current densities (Fig. 1b for direct methanol fuel cells (DMFCs) and Fig. 1c for hydrogen-fueled PEMFCs), which can be attributed mainly to the slow kinetics of electrochemical liquid fuel oxidation. For example, in a DMFC, at least 50% of the overall potential losses (i.e., a total loss of ∼0.9 V at 1 A cmgeo.−2, Fig. 1b) results from the sluggish kinetics of the anodic methanol oxidation reaction (MOR, CH3OH + H2O → CO2 + 6H+ + 6e−), while the cathodic oxygen reduction reaction (ORR, O2 + 4H+ + 4e− → 2H2O) primarily accounts for the remaining voltage losses, even with 2.5× anode catalyst loading compared to the cathode (i.e., 10× loading compared to the anode catalyst in a hydrogen-fueled PEMFC).7 In contrast, in state-of-the-art hydrogen-fueled PEMFCs, the ORR process gives rise to a ∼0.35 V potential loss at 1 A cmgeo.−2, whereas the kinetics of anodic hydrogen oxidation reaction are extremely facile with only millivolts of overpotential (Fig. 1c).3,8 Apart from high voltage losses, the crossover of liquid fuels from the anodes to the cathodes in DLFCs can lead to reduced fuel utilization, decreased cell potential, and cathode catalyst deterioration due to CO poisoning.5 In the past decades, many efforts have been focused on increasing the energy efficiencies and power densities of these devices. While many top-performing ORR cathode catalysts have been developed with enhanced activity,3,12 stability,13,14 and tolerance for liquid fuels,15,16 only limited progress has been achieved for the design of anode catalysts for liquid fuel oxidation,17–21 which can be attributed to complex reaction pathways, surface intermediate interactions, and parasitic by-products of liquid fuel oxidation reactions.22–24 More importantly, while many efforts have been devoted to either the elucidation of the mechanism or the materials engineering of novel catalysts for liquid fuel oxidation, there still exists a knowledge gap between previous mechanistic studies on single-crystal surfaces22–26 and materials-centric studies on nanostructured catalysts.17–21 Such a lack of a systematic and unified understanding across these two types of studies makes it challenging to establish meaningful activity descriptors and catalyst design principles for liquid fuel oxidation.
This review aims to construct a framework bridging fundamental energetics with catalyst designs for liquid fuel electrochemical oxidation in acidic media. There has been considerable work on the oxidation of liquid fuels in alkaline-based media due to the higher oxidation activity compared to acidic conditions and a wider range of stable elements in alkaline media.27–33 However, the low ionic conductivity of alkaline-based membranes34 and the formation of carbonate/bicarbonate (CO32−/HCO3−) precipitates that can block the porous structure of the electrodes and membranes limit the practicality of such devices.35 Furthermore, the local pH changes near the electrode associated with the CO32−/HCO3 species can make it difficult to draw fundamental conclusions on the energetics of the reaction due to an ill-defined interface. Therefore, we elected to focus on MOR, the ethanol oxidation reaction (EOR), and the formic acid oxidation reaction (FAOR) in acidic media and start by highlighting the critical reaction steps and surface intermediates in these processes (please refer to the following references for some reviews of liquid fuel oxidation in alkaline conditions36–39). We deconvolute the reaction pathways of MOR, EOR, and FAOR into the energetics of elementary steps, including initial dehydrogenation, C–C bond cleavage, and CO electro-oxidation, from which descriptors that govern the activity and selectivity of electrochemical liquid fuel oxidation are proposed. Based on these descriptors and energetics, we summarize key strategies for optimizing state-of-the-art catalysts and designing new materials for liquid fuel oxidation.
2. Electro-oxidation mechanisms of liquid fuels and state-of-the-art catalysts
The electrochemical oxidation reactions of liquid fuels, such as methanol, ethanol, and formic acid, are known to proceed via competitive, multi-pathway mechanisms.22–26 Overall, the electro-oxidation reactions start with the adsorption and dehydrogenation of liquid fuel molecules. After initial dehydrogenation, two major types of reaction pathways can be potentially involved in the further oxidation of liquid fuels, including the indirect and direct pathways. Specifically, in the indirect pathway, *CO is the key intermediate in the rate-limiting step and can lead to CO poisoning on the catalyst surface. On the contrary, the direct pathway proceeds without a *CO intermediate. Additionally, for liquid fuels with C–C bonds (e.g., ethanol), C–C bond cleavage is a critical step for full oxidation, which can have large barriers and lead to challenges in product selectivity.2.1 Methanol oxidation
The indirect pathway of the MOR gives rise to the formation of CO2 after the transfer of six electrons per methanol molecule (CH3OH + H2O → CO2 + 6H+ + 6e−), while formaldehyde (CH2O) and formic acid (HCOOH) are the main products of the direct pathway after the transfer of 2 and 4 electrons, respectively (Fig. 2a).24,40,41 The oxidation pathway on a catalyst surface is strongly dependent on the initial dehydrogenation step.24 If the cleavage of a C–H bond occurs first, MOR occurs via the indirect pathway.42 In this pathway, the dehydrogenation of methanol continues via the sequential dissociation of the C–H bonds, giving rise to *CHOH species, *HCO or *COH species, and eventually *CO intermediates.24,40,41 The electro-oxidation of *CO has been regarded as rate-limiting for the indirect pathway of MOR, particularly for the most active catalysts such as Pt-based alloys,43–45 where further oxidation can occur via a Langmuir–Hinshelwood pathway between *CO and *OH46 or an Eley–Rideal pathway between *CO and OH− species in the electrolytes.47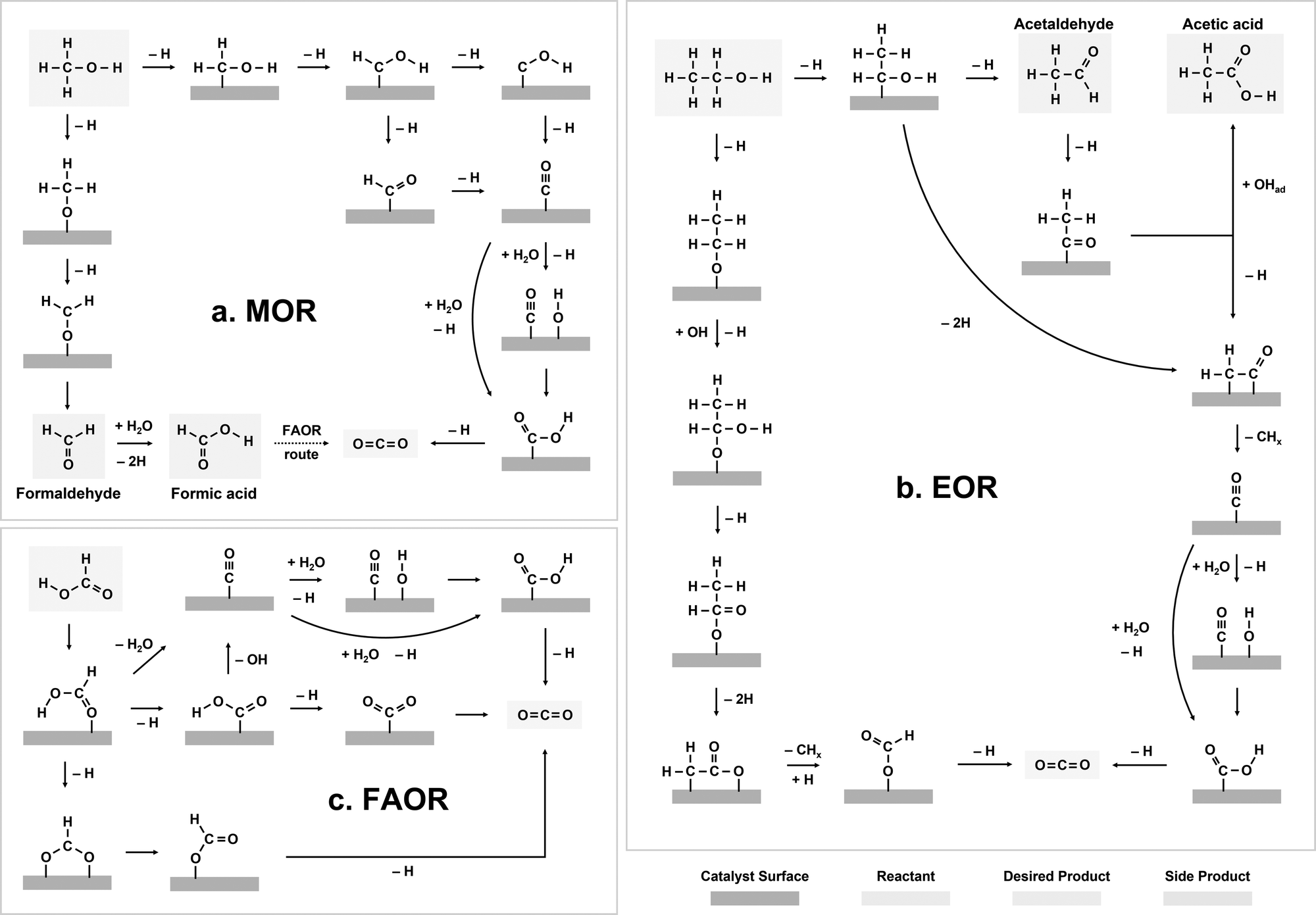 | ||
| Fig. 2 Electro-oxidation reaction mechanisms of liquid fuels. (a) Methanol oxidation. (b) Ethanol oxidation. (c) Formic acid oxidation. | ||
On the other hand, if the first dehydrogenation step occurs via the cleavage of the O–H bond on the catalyst surface, MOR proceeds through the direct pathway, where CH2O first forms via dehydrogenation of methanol and can be further oxidized to HCOOH through the chemical reaction with water.24 The re-adsorption of formate (HCOO−) species has been proposed to potentially lead to the production of CO2 on Pt,48 but the significance of this process is still controversial, as the observed HCOO− might only be a spectator.49 For DMFCs, the indirect MOR pathway is preferable as it enables the complete utilization of the high energy density of methanol molecules, since the CH2O and HCOOH species formed through the direct pathway often desorb from the catalyst surface.
State-of-the-art MOR catalysts for fuel cells have centered around Pt, with the most active materials typically consisting of PtRu or PtRu-based alloys. Typically, MOR on Pt and Pt-based materials primarily follows the indirect pathway and shows activity at moderate overpotentials (Fig. 3a, onset potential of ∼0.6 VRHE on Pt), but is severely limited by CO oxidation due to the poor affinity towards *OH species. Therefore, Ru has been alloyed with Pt to aid in the activation of H2O to form *OH species and increase the rate of CO oxidation. As such, PtRu is often regarded as the benchmark catalyst for MOR, achieving a specific activity of ∼0.1–0.4 mA cmPt−2 (normalized to the hydrogen underpotential deposition region-derived surface area, Fig. 3a)50,51 and mass activity of ∼0.05–0.2 A mgcatalyst−1 at 0.6 VRHE when measured using cyclic voltammetry51–54 in acidic electrolytes (H2SO4-based electrolytes generally leads to a lower MOR activity compared to HClO4 electrolytes on the same material due to the specifically adsorbing nature of SO42− ions26). When measured using chronoamperometry (CA), the MOR activity of PtRu is typically slightly lower than the measurements from CV.50,55 Furthermore, the measured current during CA usually decreases with time, which has been attributed to the progressive poisoning of the catalyst surface due to organic residues.56 The addition of Ru can increase the MOR reaction rate as much as 15–30×,50,57 with a 1:1 bulk ratio of Pt:Ru showing the highest activity (Fig. 3b). However, reports indicate that the surface may be closer to a 9:1 Pt:Ru ratio.50 As it appears that Pt is largely inevitable when it comes to highly active MOR catalysts, many recent reports have sought to ensure a high noble metal utilization. For example, Poerwoprajitno et al. dispersed Pt islands grown on Ru-branched nanoparticles to create highly dispersed Pt atoms (“single-atoms”) within a Ru matrix, achieving a high specific activity of 0.52 mA cmPt−2 and Pt-mass activity of 1.1 A mgPt−1, respectively, at 0.6 VRHE.58 However, due to Ru acting as the support matrix, a relatively low mass activity based on total metal loading of ∼0.09 A mgmetal−1 was achieved.58 To note, the polarization curve in Fig. 3 may appear to be different than the one found in Fig. 1b, which can be primarily attributed to the loading difference between the two measurements. In a fuel cell device, the loading of the catalyst is typically at least one to two orders of magnitude higher than in the three-electrode setup used in Fig. 3a, leading to a much higher effective surface area and higher current at a given potential. Unfortunately, to the best of our knowledge, there has not been a way to translate results obtained in the smaller-scale three-electrode setup to a real fuel cell device, even for reactions such as the ORR, which has had more systematic studies than liquid fuel oxidation.59
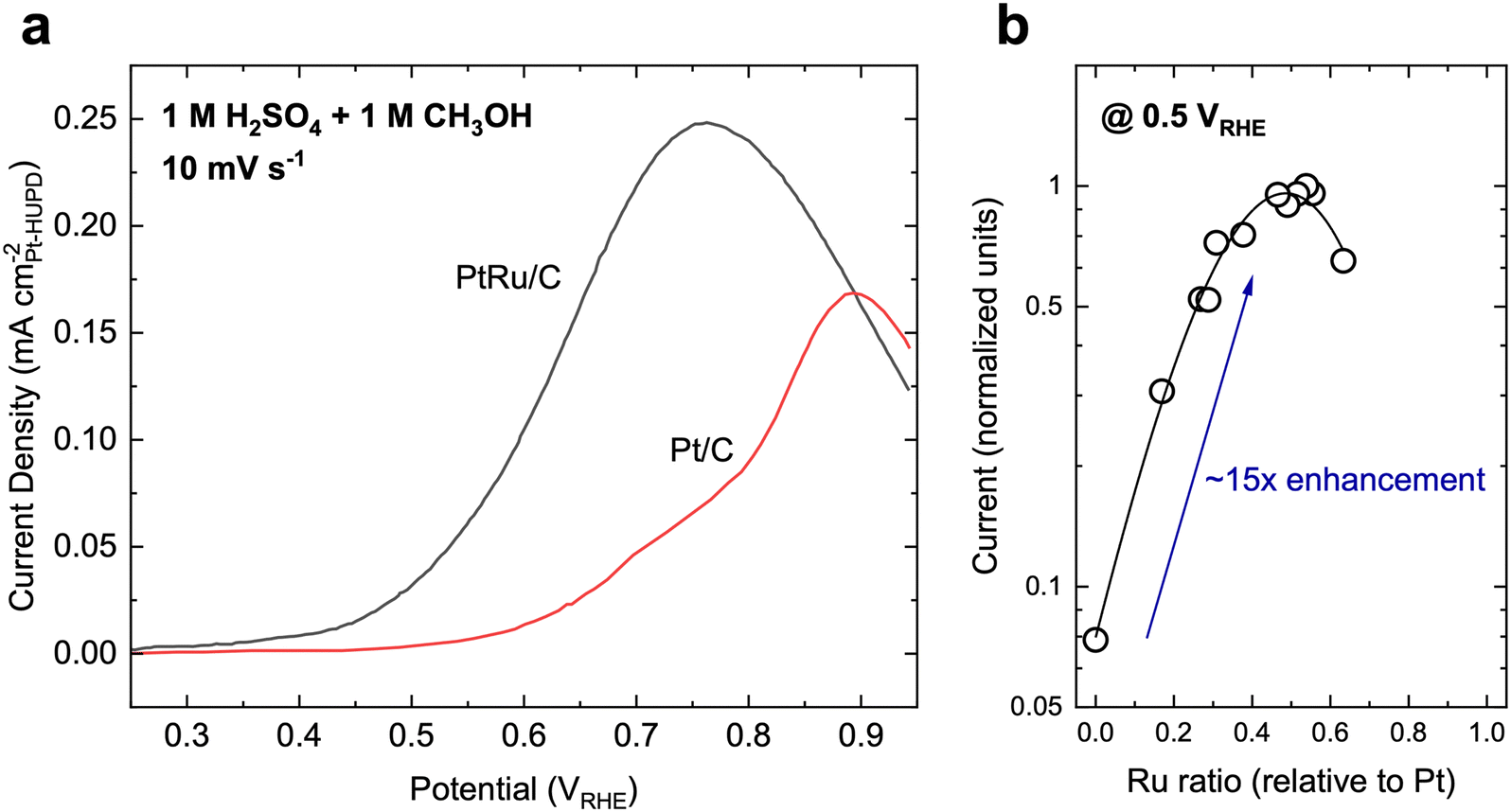 | ||
| Fig. 3 Methanol oxidation on typical reference catalysts. (a) Cyclic voltammetry polarization curves on Pt/C and PtRu/C catalysts, which are the typical benchmark catalysts for MOR. Adapted with permission from ref. 51. Copyright 2004, American Chemical Society. (b) The activity dependence of PtxRu1−x alloys. A maximum in MOR activity occurs when the bulk ratio of Pt:Ru is about 1:1, which can lead to a ∼15× enhancement in activity. Adapted from ref. 57. Copyright 1975 with permission from Elsevier. | ||
2.2 Ethanol oxidation
Compared with MOR, the mechanism of EOR is much less understood. EOR can involve a series of parallel and sequential reaction steps and a multitude of possible adsorbed species and oxidative products,23,60 making it challenging to rigorously determine the predominant adsorbed species and differentiate active intermediates from pure spectators. EOR can follow either the C1 or C2 pathway (Fig. 2b), which determines the product selectivity and fuel utilization efficiency. The C1 pathway is a 12-electron transfer process where ethanol fully oxidizes to CO2 (C2H5OH + 3H2O → 2CO2 + 12H+ + 12e−), while the C2 pathway leads to the partial oxidation of ethanol to acetaldehyde (CH3CHO) or acetic acid (CH3COOH) via the transfer of 2 or 4 electrons, respectively.23 In the C1 pathway, C–C bond cleavage can occur in ethanol or acetaldehyde after the initial dehydrogenation steps, leading to the formation of *CO and *CHx species, where these *CHx species can be further oxidized to *CO at high potentials.23,61 Similar to MOR, the electro-oxidation of *CO to form CO2 can occur via the Langmuir–Hinshelwood62 or Eley–Rideal pathways,47 which limits the EOR reaction rate. On the other hand, the C2 pathway is typically thought to initiate with a β-hydrogen dehydrogenation, producing acetaldehyde, which undergoes further oxidation to acetic acid.61 The acetaldehyde intermediate of the C2 pathway has also been proposed to be able to undergo C–C bond cleavage, at which it then follows the C1 pathway to complete oxidation.EOR is a difficult reaction to catalyze efficiently as both activity and selectivity are critical to maximize device performance. Pt is regarded as the most effective monometallic electrocatalyst for the EOR with a specific activity of ∼0.015 mA cmcatalyst−2 at 0.6 VRHE (Fig. 4a),63 but the faradaic efficiency towards CO2 has been measured to only be about 0.5–8%, even across a wide range of ethanol concentrations (Fig. 4b).64 Putting the selectivity in context, using the maximum selectivity of 8% towards CO2, and assuming the remaining products are entirely acetic acid (4 electrons transferred), then only ∼40% of the energy contained in ethanol can be extracted. Alloying Pt with Sn leads to a slight increase in activity at potentials near the onset of EOR (∼0.03 mA cmcatalyst−2 at 0.6 VRHE, Fig. 4a).63 On the other hand, alloying Pt with Ru significantly increases the overall EOR activity (Fig. 4a) (specific activity of ∼0.1 mA cmcatalyst−2 and mass activity ∼0.002 A mgmetal−1 at 0.6 VRHE). However, alloying Pt with either Ru or Sn has little effect on the selectivity towards the C1 pathway (Fig. 4c).65 Adzic et al. reported one of the highest activities towards EOR by growing Pt monolayers on heterometallic extended surfaces and found that Pt monolayers on Au(111) gave a specific activity of 1.57 mA cmPt−2 at 0.6 VRHE, but Pt monolayers supported on Au/C nanoparticles only gave a specific activity of 0.5 mA cmPt−2. However, in situ infrared reflection absorption spectroscopy revealed that only the C2 pathway was followed on these catalysts, suggesting that there is an activity/selectivity tradeoff for EOR catalysts.66
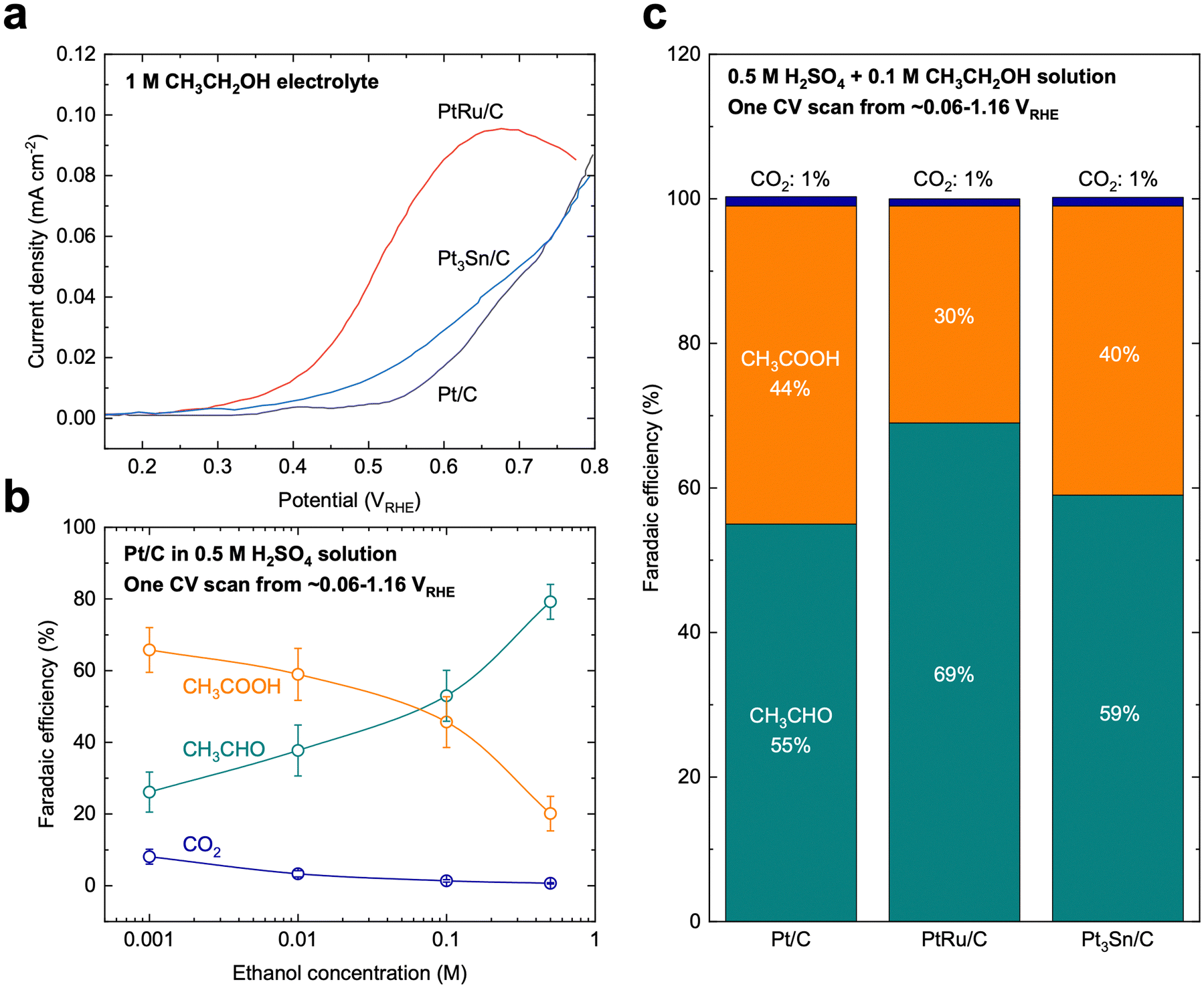 | ||
| Fig. 4 Ethanol oxidation on benchmark catalysts. (a) Cyclic voltammetry polarization curves on Pt/C, PtRu/C, and Pt3Sn/C catalysts, which are the typical benchmark catalysts for EOR. Adapted from ref. 63. Copyright 2006 with permission from Elsevier. (b) Selectivity of EOR on Pt/C catalysts as a function of ethanol concentration in the supporting electrolyte. Adapted with permission from ref. 64. Copyright 2004 American Chemical Society. (c) Selectivity of EOR on benchmark Pt/C, PtRu/C, and Pt3Sn/C catalysts. The current benchmark EOR catalysts still showing low selectivity (∼1%) towards full oxidation (CO2). Adapted from ref. 65. Copyright 2006 with permission from Elsevier. | ||
2.3 Formic acid oxidation
For FAOR, the oxidation of formic acid to CO2 (HCOOH → CO2 + 2H+ + 2e−) occurs via a dual-pathway mechanism, including an indirect (dehydration) pathway and a direct (dehydrogenation) pathway (Fig. 2c).22,24 While the indirect pathway involving the oxidation of HCOOH to *CO intermediates (which is followed by CO oxidation, similar to MOR and EOR) is relatively well-understood,67 a remaining question within the FAOR mechanism is the nature of active intermediates in the direct pathway,22 especially concerning the role of formate species. The first school of thought has proposed that formate is a key intermediate in the direct pathway of FAOR,68,69 while the second school of thought has argued that formate is only a spectator and the direct FAOR pathway proceeds without the formate species.70,71 Furthermore, some studies have suggested possible dual dehydrogenation pathways (e.g., due to comparable surface energetics for the formate-involving and formate-free pathways)72,73 or dual-species collaboration (e.g., due to non-negligible interactions between the adsorbed formate species and formate ions in solutions)74,75 for the oxidation of formic acid. Therefore, to the best of our knowledge, a final conclusion has not been drawn on the nature of active species in the direct pathway of FAOR.FAOR electrocatalyst engineering has primarily focused on Pt and Pd-based materials due to their high activity and relatively high stability. Pt is known to primarily follow the indirect pathway, whereby *CO can poison the surface, leading to deactivation.76 On the other hand, Pd is known to have an absence of *CO poisoning and typically follows the direct oxidation pathway,77 but has lower stability than Pt in acidic electrolytes. The measurements of FAOR catalysts are not as standardized as other reactions, and typically a wider range of values are found in literature. However, approximate ranges for the specific and mass activities for Pt are 1–5 mA cm−2 and 0.1–0.3 A mg−1 at 0.5 VRHE,76,78,79 respectively (Fig. 5a). The activity of Pd is typically higher than Pt, with specific and mass activities of approximately 2–10 mA cm−2 and 0.3–0.5 A mg−1 at 0.5 VRHE.79,80 As with MOR and EOR catalysts, improving FAOR electrocatalysts often relies on alloying Pt or Pd with other hetero-metals (Fig. 5b). As the rate-limiting step of FAOR is CO oxidation on Pt catalysts, state-of-the-art Pt-based FAOR electrocatalysts have focused on either preventing the indirect pathway from occurring on the Pt active sites or increasing the CO oxidation rate. For example, alloying Pt with Bi has been shown to increase the FAOR activity as Bi will cover the surface sites, diminishing the number of certain Pt ensembles that facilitate the indirect pathway.81–84 Furthermore, in addition to promoting MOR and EOR compared to pure Pt, PtRu has been known to increase the FAOR activity by about 2× through enhancing the CO oxidation step.85 Similarly, alloying Pd with elements such as Bi80,85 or Cu86 has also been shown to have the potential to boost FAOR activity and stability.
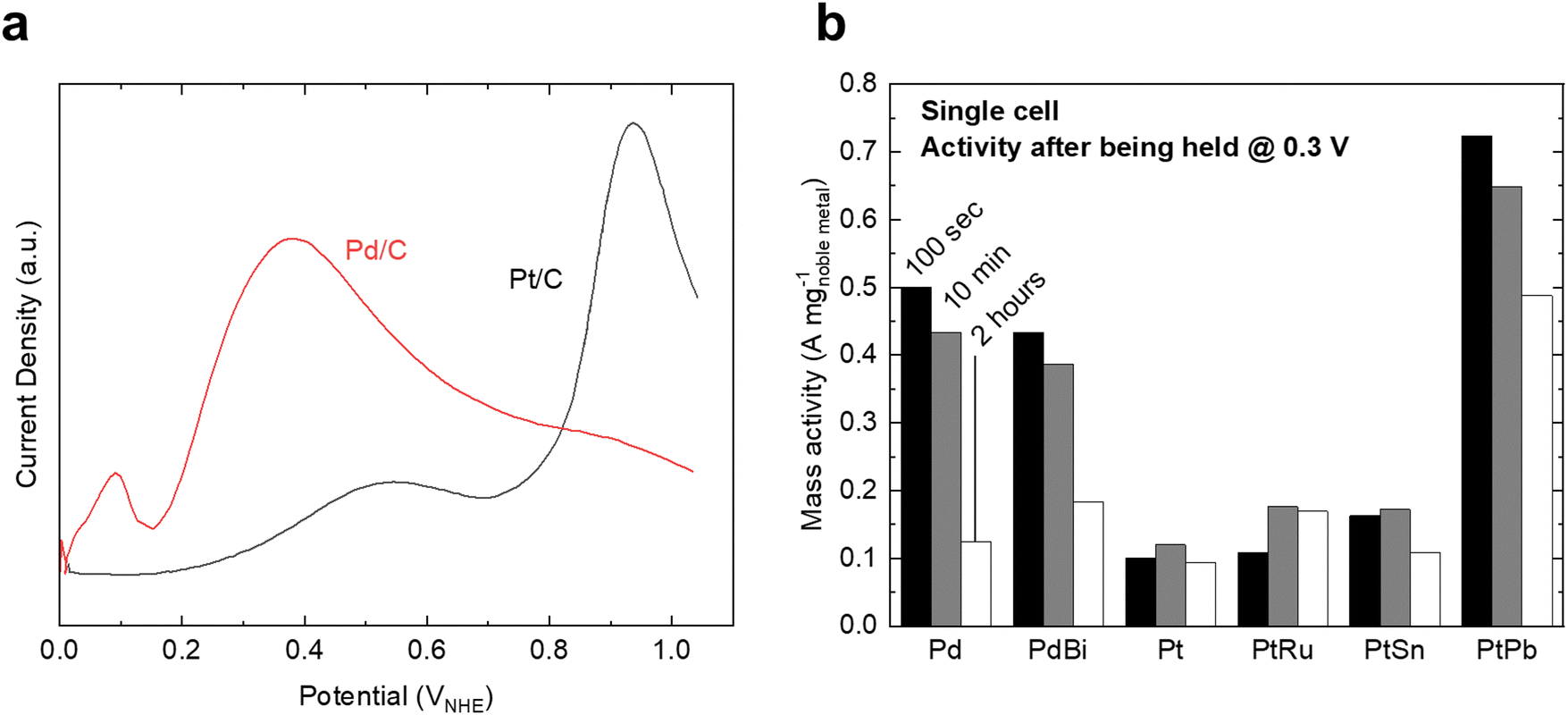 | ||
| Fig. 5 Formic acid oxidation on benchmark catalysts. (a) Polarization curves of Pt/C and Pd/C catalysts in 0.5 M HClO4 electrolyte. Due to different electrochemically active surface areas, the curves are normalized relative to the Pt/C curve. Adapted from ref. 78, Copyright (2007), with permission from Elsevier. (b) Mass activity of various Pd and Pt-based alloys for formic acid oxidation after being held at 0.3 V for a set amount of time in a single-cell test. Adapted from ref. 85. Reproduced with permission from Springer Nature. | ||
Based on these mechanisms, three critical elementary steps (i.e., dehydrogenation, C–C bond cleavage, and CO electro-oxidation) are involved in the oxidation energetics of liquid fuels. By further dissecting the overall energetics of liquid fuel oxidation into the fundamental energetics of these individual elementary steps in the next section, descriptors that determine such elementary energetics and, thus, govern the overall catalytic activity can be summarized.
3. Scaling relations and elementary energetics
The main role of a catalyst is to alter the energetic landscape of the reaction and lower the activation energy ΔEa for converting the reactants to the products without changing the overall reaction energy ΔEreaction (Fig. 6a). However, optimizing catalysts to achieve the maximum possible activity for a given liquid fuel oxidation reaction is extremely difficult, stemming from the fact that their reaction mechanism often involves many intermediates that have interactions with the catalyst surface that cannot be completely decoupled from one another.87 For example, during the MOR, many C-bound and O-bound intermediates interact with the surface of the catalyst, which leads to a multitude of energetics that would need to be optimized for the ideal catalyst. Fig. 6b shows the energetics of MOR on a Pt(111) surface,44 demonstrating the wide range of energetics associated with the reaction. While a wide variety of intermediates and pathways have been proposed for liquid fuel oxidation, the energetics of the various elementary steps are fundamentally connected,87 which prevents the tailoring of catalysts to tune the energetics of only a single step in the reaction. These fundamental connections among intermediates are broadly referred to as scaling relations, which states that there is a general correlation of adsorption energies among similar intermediates (e.g., bound by the same element) across different catalytic surfaces.87 For example, the adsorption energies of O-bound intermediates scale linearly with the OH binding energies (Fig. 7a), while the adsorption energies of C-bound species scale linearly with the CO binding energies on metal surfaces (Fig. 7b).44 Therefore, the overall thermodynamic reaction profiles of MOR can be described by the relative binding strength of *CO and *OH species.43–45 Likewise, the reaction intermediates in EOR88–90 and FAOR91,92 have similar linear scaling relations with the C and O binding energies, highlighting the importance of scaling relations in determining the energetics of liquid fuel oxidation. In addition to the scaling relations between adsorption energies, the transition state barriers in MOR,93 EOR,60 and FAOR94 scale with the free energy changes, which is known as the Brønsted–Evans–Polanyi (BEP) relation. For instance, such a BEP relation has been reported between the activation barriers and reaction energies of possible C–C, C–H, and O–H bond cleavage steps in EOR60 (Fig. 7c), showing that the thermodynamic profiles correlate strongly with the kinetics barriers and, therefore, the overall reaction rates. Using the scaling relations and BEP relation, we can further relate the energetics of dehydrogenation, C–C cleavage, and CO oxidation with the C and O binding energies (Fig. 8a) to develop fundamental intuitions on how the catalytic activity of liquid fuel electro-oxidation can be optimized via catalyst design. Generally, stronger C binding on catalyst surfaces promotes dehydrogenation and C–C cleavage but hinders CO oxidation (Fig. 8a top). In contrast, stronger O adsorption can result in less selectivity for C–C cleavage for CO2 formation but more favorable energetics for CO electro-oxidation (Fig. 8a bottom). Therefore, for MOR and FAOR, as state-of-the-art catalysts such as Pt are more limited by CO oxidation than dehydrogenation, we need to reduce the C binding and facilitate the O binding on these top-performing catalysts.43–45,90–92 Moreover, as EOR currently has challenges in both selectivity (C–C cleavage) and activity (CO oxidation), which have opposite trends in their energetics as a function of C or O binding (Fig. 8a), it is more challenging to develop ideal catalysts for EOR than for the other two anodic reactions in DLFCs. Common to MOR, EOR, and FAOR, dehydrogenation (C–H bond cleavage) is critical for full oxidation to CO2. Unfortunately, making a quantitative version of Fig. 8a is quite difficult as it would require benchmarking all the barriers with respect to the same parameters (such as ΔEC or ΔEO). Doing so would require further original experiments or computation as data from different sources can have different experimental or computational parameters, making such an endeavor outside the scope of this review. However, we hope that this review can motivate such work for future studies. Nonetheless, the discussion below will go into more detail about the scaling and BEP relations for each of these elementary steps individually.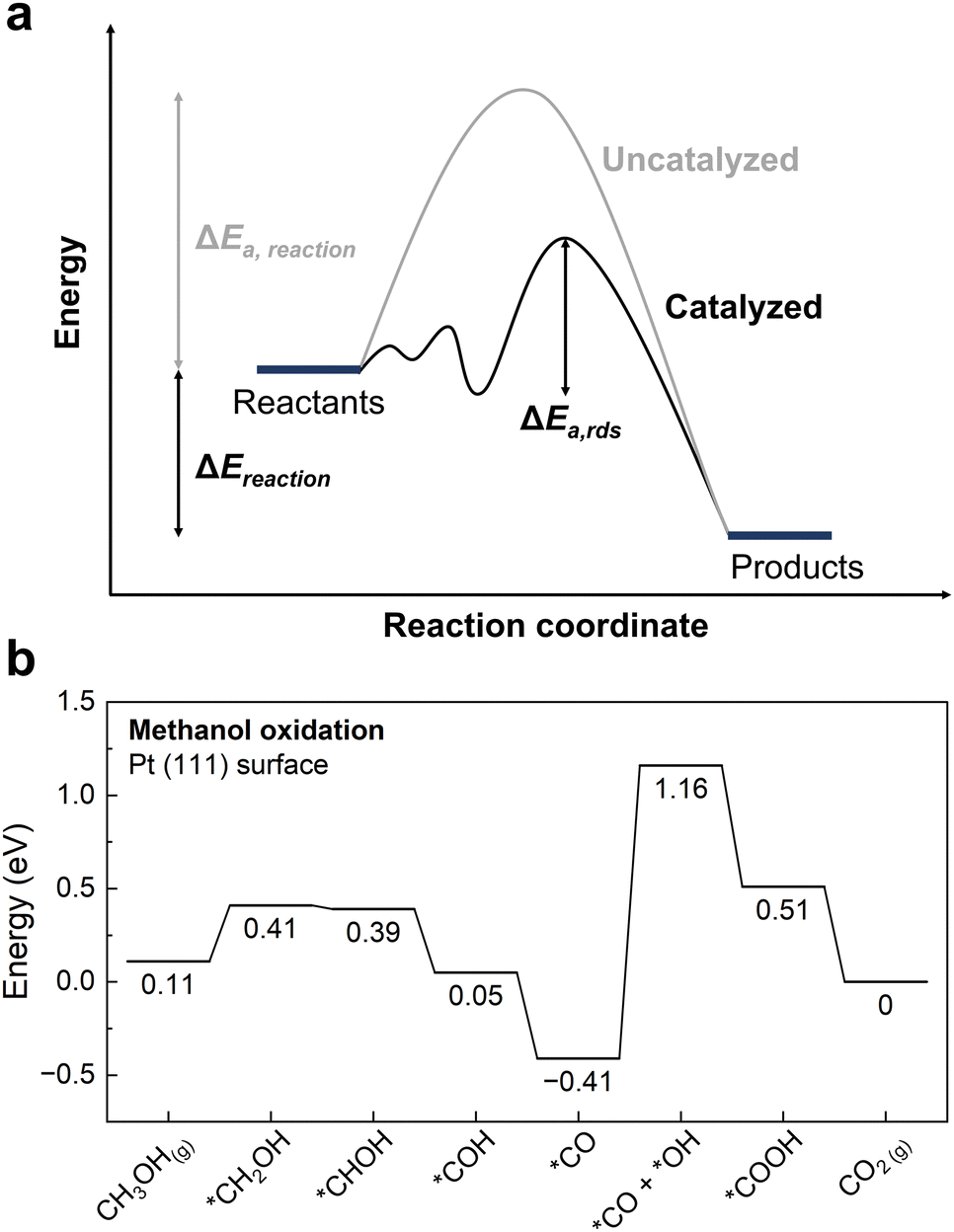 | ||
| Fig. 6 Energetics of catalysis. (a) Role of catalysts in altering the energetic landscape of the reaction. The uncatalyzed reaction (grey) has a higher activation energy ΔEa compared to the catalyzed reaction (black). The reaction energy ΔEreaction does not change. ΔEa,rds corresponds to the activation energy for the rate determining step in the catalyzed reaction. (b) Reaction energetics profile for MOR on Pt(111) following the indirect pathway. The CO oxidation steps typically are rate-limiting on Pt-based surfaces, as indicated by having the highest thermodynamic reaction barrier. Adapted with permission from ref. 44. Copyright 2012 American Chemical Society. | ||
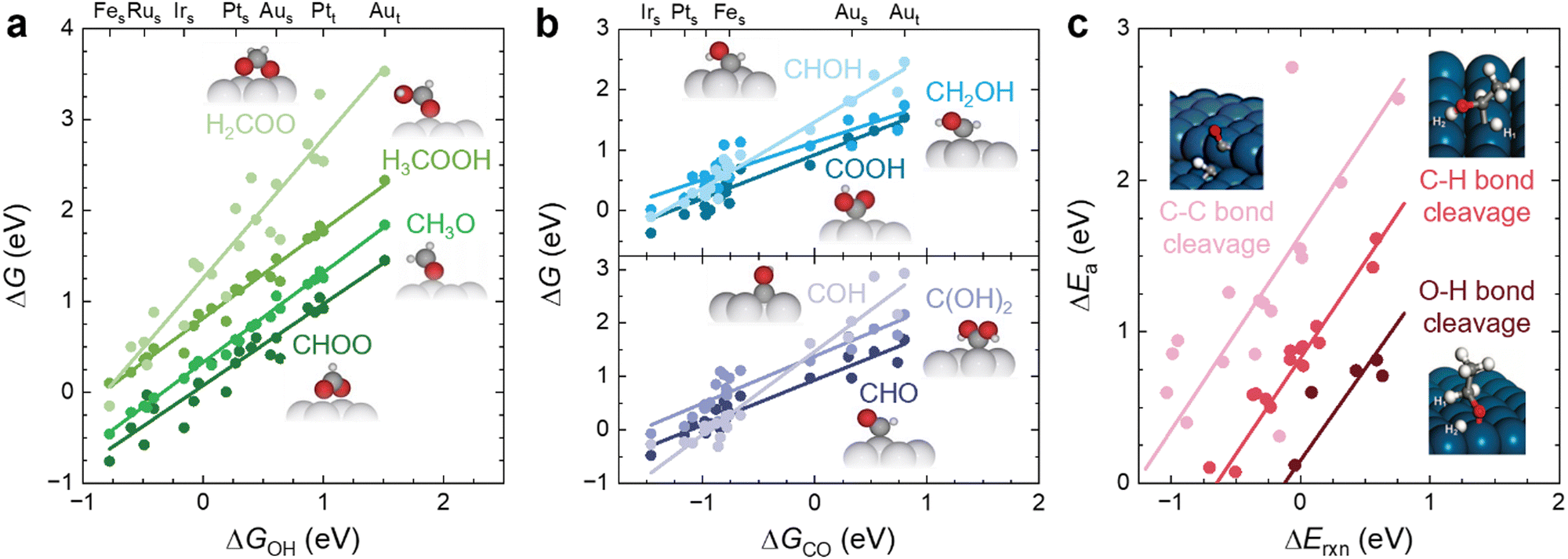 | ||
| Fig. 7 Scaling relations in liquid fuel electro-oxidation. (a) Linear scaling relations between the adsorption energies of O-bound MOR intermediates and the adsorption energies of OH on different metal surfaces. Adapted with permission from ref. 44. Copyright 2012 American Chemical Society. (b) Linear scaling relations between the adsorption energies of C-bound MOR intermediates and the adsorption energies of CO on different metal surfaces. Adapted with permission from ref. 44. Copyright 2012 American Chemical Society. Subscripts of s and t denote steps and terrace facets of each respective metal. (c) BEP relations between the activation barriers and reaction energies of possible bond-cleavage reaction steps in EOR on Pt(111) and Pt(211) surfaces. Adapted with permission from ref. 60. Copyright 2012 American Chemical Society. | ||
 | ||
| Fig. 8 C- and O-binding-dependent elementary energetics in liquid fuel electro-oxidation. (a) Energetics of the key elementary steps in liquid fuel oxidation as a function of C and O adsorption energies. (b) Dependence of the activation barriers of the initial dehydrogenation step of ethanol on the adsorption energies of ethanol on different metal surfaces. Adapted with permission from ref. 95. Copyright 2017 American Chemical Society. (c) Dependence of the energetic barriers of C–C bond cleavage and the side reaction (acetyl oxidation) in EOR on the adsorption energies of C and O on different metal surfaces. Adapted from ref. 89, Copyright (2018), with permission from Elsevier. (d) Dependence of the barriers for the key critical steps of CO oxidation in MOR on the adsorption energies of CO and O on different flat (triangles) and stepped (squares) metal surfaces. Adapted with permission from ref. 44. Copyright 2012 American hemical Society. | ||
First, the barriers for dehydrogenation can be lowered by increasing the binding of surface C, as it has been shown through experiments and computations to lead to higher rate constants of dehydrogenation of methanol to *CO species.96 Moreover, density functional theory (DFT) calculations have indicated that stronger adsorption of ethanol molecules on monometallic surfaces can lead to lower activation barriers for the initial dehydrogenation of ethanol via either O–H or α/β-C–H bond scission95 (Fig. 8b). Such a correlation has been supported by similar studies of DFT-computed dehydrogenated profiles of ethanol molecules on close-packed metal surfaces, where stronger C binding generally gives rise to more strongly adsorbed dehydrogenated derivatives and also smaller activation barriers,97,98 confirming the crucial role of C binding in determining the dehydrogenation energetics and selectivity of liquid fuels on catalyst surfaces. This trend originates from the scaling relations and BEP relation and can be extended to other processes, e.g., the dehydrogenation of hydrocarbons such as ethane99 and propane.100
Second, stronger C binding facilitates C–C bond cleavage, whereas stronger O binding inhibits C–C bond scission (Fig. 8a). This relation is supported by a recent study89 comparing and evaluating the competing energetics of C1 and C2 EOR pathways on monometallic surfaces. In the C2 pathway, the acetyl (*COCH3) intermediate is oxidized to acetate species (*O2CCH3) and finally becomes acetic acid, while in the C1 pathway, the fragmentation of acetyl species via C–C cleavage leads to the formation of *CO, *CH3, and finally CO2.88,97,101 Thus, the competition between acetyl oxidation and fragmentation can govern the product selectivity in EOR. In this study,89 it has been found that the barrier of acetyl fragmentation is reduced by increasing the C binding energy (blue in Fig. 8c), while stronger O binding and weaker C binding can lead to more favorable energetics for forming the side product (i.e., acetate, green in Fig. 8c). In other words, although C–C bond cleavage does not involve O-bound species, the side reaction with respect to C–C bond cleavage involves O-bound species. Thus, if O is more strongly bounded, the thermodynamic driving force of the side reaction is stronger, and C–C bond cleavage becomes less selective. Therefore, an ideal catalyst for C–C bond cleavage in EOR should have strong C binding but weak O binding, but among various metals, only Pt and Pd have such energetics.89 These trends for C–C bond cleavage have been corroborated by another recent work assessing the acetyl oxidation and fragmentation energetics on ternary Pt3RhM (M = Fe, Co, Ni, Cu, Ga, In, Sn, and Pb) nanoalloys.90 The authors have proposed slightly modified pathways, where the C–C bond cracking step can take place more easily in further dehydrogenated *COCH2 or *COCH intermediates, and the dissociation of water to *OH and *H can have a lower activation barrier than the combination of acetyl with *OH to form acetic acid. Nevertheless, this small change in reaction pathways does not alter the dependence of C–C bond cleavage energetics on the C and O binding energies. Enhancing the C adsorption can still facilitate the fragmentation of *COCHx, while reducing the O binding can hinder the energetics of both water dissociation and acetyl oxidation steps on catalyst surfaces.
Lastly, CO electro-oxidation (Fig. 8a) can be promoted by reducing the C binding and enhancing the O binding energies. Although state-of-the-art monometallic catalysts for liquid fuel oxidation (e.g., Pt and Pd) show high activity for dehydrogenation91,92 (Fig. 3b) and C–C cleavage89,95 (Fig. 8c), these catalysts can still have CO electro-oxidation as rate-limiting steps due to too strong C binding and too weak O binding.43–45,90–92 To promote CO oxidation, we can facilitate the formation of more *OH from water dissociation for CO electro-oxidation via the Langmuir–Hinshelwood pathway by increasing the O binding energy using more oxophilic elements such as Ru44 (orange in Fig. 8d). On the other hand, we can reduce the C binding energy to promote the combination of *CO and *OH to *COOH and the desorption of CO244 (red in Fig. 8d) by, for example, incorporating metals that interact more weakly with *CO, such as Au. Despite the low binding strength of *CO on Au (near reversible binding to the surface),26 Au exhibits a higher CO oxidation activity than Pt.102 The higher activity of Au compared to Pt for CO oxidation has been hypothesized to be from the direct formation of *COOH from *CO and H2O, due to the low binding strength of *CO on Au.103,104
The binding strength of various intermediates, and therefore the reaction rates of the aforementioned elementary steps, are largely dictated by the electronic structure of the surface,105 which acts as a strong lever for optimizing electrocatalytic activity. While quantum mechanical models of the surface–adsorbate interactions give accurate descriptions of such interactions, these models are computationally demanding and their results are often not generalizable to other similar systems.105 To address these limitations and aid in designing catalysts with features that increase activity or selectivity, most design strategies of liquid fuel oxidation electrocatalysts have revolved around characteristics that capture the scaling of the intrinsic activity with descriptors that are less computationally demanding or more intuitive to understand and control.12,106,107 One example of these descriptors commonly used for metallic surfaces is the relative energy difference between the Fermi level and the d-band center for metals (i.e., centroid energy of all d-states in a material), often referred to as the d-band model.105,108 This descriptor has been shown to scale linearly with the binding energy of adsorbates, in which a higher d-band center leads to stronger adsorbate–surface interactions. The physical basis of this scaling can intuitively be understood when the coupling between the adsorbate and surface electronic energetic states is considered. For the same adsorbate, a lower d-band center of the metal (surface) will lead to more anti-bonding states below the Fermi level after coupling with the adsorbate orbitals, leading to weaker interactions.108,109 Similar descriptors have also been established for other classes of materials, such as the eg orbital filling110,111 and O 2p band center112–114 for governing surface–adsorbate interactions on oxides. In the following section, we will review the various strategies that have been used to design electrocatalysts for liquid fuel oxidation. The rationale for the strategy will be explained and the current understanding of the physical origin of the activity changes will be discussed.
4. Design strategies for liquid fuel oxidation electrocatalysts
Design and engineering of liquid fuel oxidation electrocatalysts to optimize the energetics of the various elementary steps can be grouped into five fundamental approaches: (1) tuning of electronic structure through strain, (2) tuning of electronic structure through surface atomic structure control, (3) tuning of electronic structure through the ligand effect, (4) incorporation of different elements for the bifunctional effect, and (5) controlling the ensembles that constitute active sites. These approaches are not exclusive to liquid fuel oxidation and have found significant use in other electrochemical reactions (for example, hydrogen evolution/oxidation and oxygen reduction/evolution).3 In other words, these five approaches fundamentally affect the catalyst's surface atomic arrangement and/or electronic structure agnostic to the reaction. However, liquid fuel oxidation poses a unique problem that often involves both carbonaceous and oxygenated species in the reaction pathways. As the energetics to further oxidize the carbonaceous and/or oxygenated species typically scale in opposite ways for a catalyst, designing the optimal catalyst for these reactions is particularly difficult. In this section, a comprehensive overview of these design strategies along with the physical underlying mechanism for altering the intrinsic electrocatalytic activity via engineering of the barriers of dehydrogenation, C–C bond cleavage, and CO oxidation on the catalyst surface are discussed.4.1 Strain effect
Straining surfaces is a relatively straightforward way to tune the electronic structure of catalysts by altering the degree of overlap of atomic orbitals, and therefore band-width of the solid, between neighboring atoms, affording direct changes to the binding strength of adsorbates.115 In doing so, the d-band center of the catalyst can be tuned as a function of the lattice spacing. This technique for altering catalyst activity for liquid fuel oxidation has been demonstrated through DFT studies on strained Ru(0001), which have shown that varying the strain from −2 to +2% changes the d-band center linearly from −2.05 to −1.8 eV, respectively. Accompanying the d-band center change, the adsorption strength of CO on the surface also displayed a strong linear dependence from −1.99 to −2.04 eV, respectively (Fig. 9a, left).116 Similarly, Adzic et al.66 have strained Pt(111) monolayers on various close-packed transition metal surfaces, ranging from −8% (Cu) to +5% (Au). The potential required for MOR to proceed on these strained Pt(111) surfaces was calculated to range from +0.5 to −0.25 V, relative to the potential required for unstrained Pt(111) surfaces (Fig. 9a, right).66 This trend has been experimentally validated to apply beyond film catalysts by growing Pt monolayers on metal cores. In cyclic voltammetry measurements, Pt/Au/C (5% strain) particles showed nearly 10× higher peak current densities for MOR and about 3–4× higher peak current densities for EOR than the peak current densities measured with the Pt/C and Pt/Pd/C (−1.5% strain) particles. Recently, He et al.117 carefully controlled Pt strain by growing Pt(100) shells on expanded and contracted phosphorized Pd cores and found that the MOR activity displayed an M-shaped trend as a function of the strain. They explained this as with increasing tensile strain, adsorbed *OH species bind stronger and are more readily formed; however, adsorbed *CO species also bind stronger with tensile strain, so once about 5% strain is reached, the activity increased afforded by the increased OH binding is negated by the increased CO binding, leading to an overall decrease in MOR activity. Similarly, for increasing compressive strain, CO is bound less strongly, which leads to increased MOR activity, where at about −4% strain, the activity enhancement from decreased CO binding is negated by the decreased OH binding, leading to an overall rate decrease. Additionally, for ethanol oxidation, Strasser et al.118 have shown that strain can also enhance the selectivity of EOR towards the C1 pathway through straining Pt mono and sub-mono layer on Au nanoparticle cores. The selectivity changes have been attributed to the tensile strain of Pt due to the lattice mismatch with Au, which upshifts the d-band center, increasing the binding of adsorbates, and facilitating C–C bond cleavage. Overall, these results demonstrate the potential that strain engineering has for altering the fundamental electronic structure of electrocatalysts for tuning liquid fuel oxidation activity and selectivity. | ||
| Fig. 9 Design strategies for liquid fuel oxidation electrocatalysts. (a) (left) Dependence of CO adsorption energy and d-band center on strained Ru(0001) surfaces. Reprinted figure with permission from ref. 116. Copyright (1998) by the American Physical Society. (right) Dependence of the relative potential for CO oxidation on Pt monolayers grown on various transition metal surfaces as a function of the induced strain. Adapted with permission from ref. 66. Copyright 2012 American Chemical Society. (b) (left) Dependence of the CO oxidation and d-band center on various Pt facets and sites. In general, more under-coordinates sites lead to a more reactive surface. Adapted with permission from ref. 119. Copyright 1997 Springer Nature. (right) Measured experimental dependence of CO oxidation apparent rate constant as a function of applied potential on various Pt n(111) × (111) surfaces. In general, the rate of CO oxidation increases with increasing step density. kintrinsic represents the theoretical apparent rate constant of CO oxidation on a surface composed of entirely (110) steps. This figure has been adapted from ref. 26 with permission from John Wiley & Sons, Inc., copyright 2008. (c) The dependence of the Pt d-band center when a subsurface metal (“skin”) in introduced between the first and second surface layers. In general, more strongly interacting skins with Pt leads to lower Pt d-band centers. Adapted from ref. 120, with permission from AIP Publishing. (d) (top) Schematic diagram demonstrating the bifunctional effect, where two or more co-catalysts are combined such that each component can improve the rate of a different elementary step in the reaction mechanism. (bottom) DFT results showing the configuration of the bifunctional effect for MOR on Pt0.66Ru0.33 surfaces, where methanol preferentially dehydrogenates to form Pt bound CO* and water preferentially dissociates to form Ru bound OH*. Reprinted from ref. 121, Copyright (2003), with permission from Elsevier. (e) Schematic showing the ensemble effect for FAOR on Pt clusters in Au(111) the clusters on the left can catalyze FAOR via the direct pathway while the clusters on the right (or even larger) can catalyze the indirect pathway involving *CO species. Pt and Au are presented by grey and yellow, respectively. Adapted from ref. 122, Copyright (2015), with permission from Elsevier. | ||
4.2 Surface atomic structure
Another commonly used strategy for the design of liquid fuel oxidation catalysts is through controlling the surface structure of active sites.43,123,124 In general, more undercoordinated sites (e.g., high index facets) lead to sites that are more active and have higher binding of adsorbates. Norskov et al. have shown that by varying the Pt facet from (100) and (111) terraces to stepped (111) surfaces (i.e., (211) and (11 8 5)), a linear trend is observed among the various available binding sites of all the surfaces as a function of the Pt d-band center (Fig. 9b, left).119 Going from a (100) terrace site on the relaxed surface (hex-low site) to a highly undercoordinated (11 8 5) kink site, the d-band center shifts positive by 1 eV, along with a 1 eV increase in the CO binding strength. CO stripping experiments carried out on Pt(110), Pt(111), and various stepped Pt(111) surfaces [Pt n(111) × (111), n = 5, 10, and 30] further corroborated these computational results.26 Pt(110) and Pt(111) acted as theoretical upper and lower bounds for the activities of the stepped surfaces, respectively, as Pt(110) serves as a model system for the steps and Pt(111) serves as a model system for the terraces of the various Pt n(111) × (111) surfaces. The apparent rate constants of CO oxidation for the surfaces were calculated, which increased with increasing step density, where the rate constants of CO oxidation on all Pt n(111) × (111) surfaces all lie between those of Pt(110) and Pt(111) surfaces, confirming that undercoordinated steps are more active for CO oxidation than terraces (Fig. 9b, right).26 The enhanced CO oxidation activity of steps has primarily been attributed to the undercoordinated step sites being more active for water dissociation, allowing for surface *OH species to form at lower potentials, while the terraces, with their high rates of CO diffusion, can constantly supply *CO to the step sites.125 As CO oxidation is a key elementary step of MOR56 and EOR,126 the oxidation rates of these liquid fuels tend to increase with step density.56,127 For FAOR, Pt(100) exhibits higher activity than Pt(111),128,129 but it is still controversial as to if Pt(110) or Pt(100) displays a higher FAOR activity.76 When further extended to particles, the same trends hold where increasing the step density on Pt nanoparticle surfaces leads to increased MOR,55,130 EOR,131,132 and FAOR131,133 activity, due to the same mechanism as described for the well-defined Pt film electrodes.In addition to activity being facet dependent, selectivity is also highly dependent on the facet for these complex reactions.25,43,124 Koper et al.25 have used cyclic voltammetry coupled with mass spectrometry to study the MOR in 0.5 M H2SO4 and 0.1 M HClO4 electrolytes and reported that Pt(110) and Pt(111) surfaces primarily follow the direct pathway, while Pt(100) primarily follows the indirect pathway in H2SO4. On the other hand, in 0.1 M HClO4 electrolyte, where there is an absence of specifically adsorbing anions, Pt(110) is most active for the direct pathway, followed by Pt(100) and Pt(111), respectively. Using in situ IR spectroscopy to study low index Pt surfaces, it has been reported that C–C bond cleavage selectivity during EOR decreases in the order of (110) > (100) > (111), with the (111) surface showing a near absence of adsorbed surface CO during in cyclic voltammetry measurements.134–136 This behavior can be rationally explained similarly to that for the rate of CO oxidation: as (110) surfaces contain more undercoordinated sites relative to the other surfaces, these surfaces more readily cleavage the C–C bond due to their higher reactivity. For FAOR, where the indirect pathway is generally unfavored over the direct pathway due to CO poisoning the surface, the selectivity trend is mostly the opposite of that of the activity trend.26,137 Pt(111) shows the highest selectivity towards the direct pathway, where the inclusion of {100} and {110} family of steps does not alter the selectivity. On the other hand, steps of the {111} family tend to facilitate CO formation.128
4.3 Ligand effect
In addition to the mechano-electronic effects previously described, pure electronic effects for liquid fuel oxidation catalyst design typically involve the substitution of hetero-elements into a host lattice or subsurface layers of hetero-elements to alter the electronic structure of the host surface layers.138,139 This effect is often called the ligand effect due to the introduced elements altering the electronic environment around the host elements, leading to changes in the overall electronic structure of the material.95 However, the ligand effect often can be difficult to decouple from strain effects, as the substitution or addition of elements often results in local strain due to size or lattice mismatch.140 In efforts to isolate the ligand effect on surface electronic structure, Kitchin et al.120 have conducted computational studies of Pt(111) surfaces where a single layer of 3d transition metals was sandwiched between the first two Pt surface layers, which downshifted the d-band center of Pt by nearly 1 eV when the subsurface metal skin was varied towards the left of the periodic table. With the d-band center shift from the different 3d transition metal skins, the dissociative adsorption energies of O2 and H2 also decreased by approximately 1 eV (Fig. 9c), matching well with what is expected by d-band theory.120The underlying mechanism of this shift is related to the strength of bonding between the Pt and subsurface metal or substitute, leading to changes in the width of the d-bands.115 As the filling of the Pt d-states is essentially independent of the substituted metal, the main contribution of the change in the d-band center is related to the change in the width of the energy bands.115 For example, for elements that bond more strongly with Pt (i.e., higher Pt–X matrix coupling term; left side of the transition metals), the energy splitting will be larger leading to a wider d-band.120 To preserve the filling of energy states, the d states will undergo what is equivalent to a downshift in energy relative to the Fermi level, resulting in a lower d-band center.105,115,120
4.4 Bifunctional effect
Atomic substitution or alloying not only alter the electronic structure of the host lattice but can also increase the activity of liquid fuel oxidation electrocatalysts through the bifunctional effect, where the different elements within the alloy are specifically included to be active for different elementary steps in the reaction mechanism. For example, in PtRu alloys, one of the most active MOR catalysts,50,76 Pt is known to be active for the initial dehydrogenation steps of MOR but is relatively inactive for the water dissociation steps that are critical for CO oxidation. On the other hand, Ru, being quite oxophilic, is an excellent site for water dissociation. Alloying Pt with Ru combines the high activity of each component for different elementary steps (schematic in Fig. 9d),45,141 enhancing the overall rate of MOR. Using well-characterized PtRu surfaces, it has been suggested that the optimal surface composition of PtRu alloys for MOR is 90% Pt and 10% Ru, as this ratio maximizes the interface between three Pt atom ensembles in a triangle arrangement, which has been proposed to be necessary sites for dehydrogenation, and Ru single atom sites for water dissociation.50 While additional elements in an alloy also contribute to altering the Pt electronic structure via strain and/or ligand effects, through electrochemical nuclear magnetic resonance and temperature-programmed desorption measurements on Ru-decorated Pt surfaces, Wieckowski et al.121 have estimated that the bifunctional effect alters a catalyst's activity four times as much as the ligand effect does. The impact of the bifunctional effect has been further supported by computational studies on PtRu model surfaces, where it was found that *CO forms from methanol on Pt–Pt bridge sites, while OH* preferentially binds to Ru sites.142 Statistical models of surface atomic distribution between Pt and Ru on well-characterized PtRu surfaces have further corroborated these findings as the composition exhibiting the highest activity matches the predicted most active composition when assuming the bifunctional effect dominates the activity-composition relationship.50 Other similar systems that follow the same design principle of combining Pt with oxophilic co-catalysts to promote water dissociation and CO oxidation include PtSn for MOR and EOR,134,143,144 PtNi for MOR and EOR,145 PtBi for FAOR,18,146 and multicomponent catalysts,147–150 which can shift the onset potential for these reactions cathodically by over 100 mV (when compared at 0.1 mA cm−2) relative to that of only Pt.4.5 Ensemble effect
The local atomic coordination environment on a catalyst's surface can also strongly influence the reaction mechanism and ability to catalyze certain elementary steps in the reaction pathway, which is often referred to as the ensemble effect. For example, it is typically accepted that at least three contiguous sites in a triangle formation are required for the dehydrogenation of liquid fuels.151–155 One of the earliest reports of such an effect for the fuels focused on in this review is by Motoo and Furya,151 who measured formic acid oxidation on Pt(111), (110), and (100) surfaces and found comparable rates on (111) and (100), but slower rates on (100). Since (111) and (100) can easily accommodate three contiguous atoms, and (110) does not, they concluded that three contiguous atoms promote formic acid oxidation. Zhong et al.122 have computationally studied the geometries of Pt clusters on Au(111) and concluded that ensembles of less than three Pt atoms undergo the direct pathway to CO2 (Fig. 9e, left). On the other hand, Pt ensembles of three or more atoms will undergo the indirect pathway, unless the atoms are all co-linear or in an equilateral triangle (Fig. 9e, right), demonstrating that the geometry of the active sites also plays an important role in influencing the reaction pathways. Cuesta156 showed the first direct experimental evidence of the ensemble effect through the modification of a Pt(111) electrode with cyanide, which is known to specifically adopt a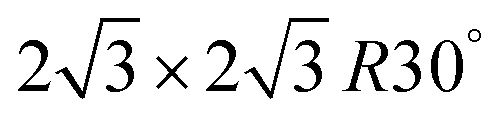 configuration on Pt(111), meaning the surface will not contain a single instance of three contiguous atoms that are free if saturated with cyanide. When tested for MOR activity, the polarization curve of the modified electrode in electrolyte with and without methanol showed no changes in the hydrogen underpotential deposition region, while in situ FTIR showed an absence of adsorbed CO up to 0.9 VRHE, indicating that the formation of CO requires three contiguous sites. Furthermore, work by Bagger et al.157 have demonstrated that the ensemble effect also strongly influences the reaction pathway that formic acid follows by influencing the adsorption of *H through comparing the electrochemical response during CV cycles on extended surfaces of Pt, PtBi, Pd, and single site catalysts of PtHg and PdHg. Using computations and changing the scan bounds to observe the changes in the hysteresis during CV scans, they suggest that FAOR will follow the indirect pathway (and be poisoned by *CO) due to a *H-mediated disproportion of *COOH to *CO and H2O. However, single Pt sites in PtHg or PtBi can destabilize the formation of *H, while Pd-based catalysts form hydrides, circumventing the formation of the *CO poison. The ensemble effect has also been investigated for EOR by controlling the probability of finding a Pt3Rh1 ensemble in a Pt–Rh–SnO2 system for maximizing the C2 pathway in ethanol oxidation.158
configuration on Pt(111), meaning the surface will not contain a single instance of three contiguous atoms that are free if saturated with cyanide. When tested for MOR activity, the polarization curve of the modified electrode in electrolyte with and without methanol showed no changes in the hydrogen underpotential deposition region, while in situ FTIR showed an absence of adsorbed CO up to 0.9 VRHE, indicating that the formation of CO requires three contiguous sites. Furthermore, work by Bagger et al.157 have demonstrated that the ensemble effect also strongly influences the reaction pathway that formic acid follows by influencing the adsorption of *H through comparing the electrochemical response during CV cycles on extended surfaces of Pt, PtBi, Pd, and single site catalysts of PtHg and PdHg. Using computations and changing the scan bounds to observe the changes in the hysteresis during CV scans, they suggest that FAOR will follow the indirect pathway (and be poisoned by *CO) due to a *H-mediated disproportion of *COOH to *CO and H2O. However, single Pt sites in PtHg or PtBi can destabilize the formation of *H, while Pd-based catalysts form hydrides, circumventing the formation of the *CO poison. The ensemble effect has also been investigated for EOR by controlling the probability of finding a Pt3Rh1 ensemble in a Pt–Rh–SnO2 system for maximizing the C2 pathway in ethanol oxidation.158
4.6 Design strategies to optimize state-of-the-art electrocatalysts for liquid fuel oxidation
There has been a significant amount of effort towards the rational design of nanostructured liquid fuel oxidation electrocatalysts to leverage the design strategies and further increase the activity of the current state-of-the-art catalysts for liquid fuel oxidation. For example, Solla-Gullon et al.125 have synthesized a range of Pt nanoparticles with controlled surface planes, including polyoriented Pt, (100) oriented nanocubes, (100)–(111) exposed hexagonal nanoparticles, and (111) exposed octahedra. Through CV and potentiostatic measurements, the activity was found to increase in the order of poly < (100) < (100)–(111) < (111) for both methanol and formic acid oxidation. In efforts towards exposing high index facets for increasing the liquid fuel oxidation activity, Lee et al.55 have reported decreasing the heat treatment temperature of Pt nanoparticles grown on multi-walled carbon nanotubes leads to an increase in the fraction of high index facets with high step density and that the methanol oxidation activity increased concomitantly with the fraction of density of high index fractions. Similarly, Tian et al.131 have developed a method for the controlled synthesis of tetrahexahedral Pt nanocrystals enclosed only by 24 high index facets (e.g. {730}, {210}, and {310} families) and found these nanocrystals to show about twice the current density at 0.25 V and 0.30 V for FAOR and EOR, respectively, compared to Pt nanospheres and commercial Pt/C (Fig. 10a). Since then, there have been many reports leveraging these strategies for creating nanoparticles with high index facets to increase the activity of MOR31,145,159–162 and FAOR,162,163 and both the activity and selectivity of EOR164,165 compared to Pt/C nanoparticles through advanced shape and morphology control of the catalyst. However, as high-index planes are typically more vulnerable to restructuring under the presence of CO,166,167 the stability of these materials will need to be carefully scrutinized and understood. In another vein to alter the surface properties of catalysts, Suntivich et al.168 have controlled the surface electronic structure of Pt by changing the heat treatment conditions of Pt0.5Au0.5 nanoparticles, which drastically changed the surface Pt to Au ratio. The optimal 68% Pt, 32% Au composition showed a 2× enhancement in MOR activity at 0.7 VRHE compared to commercial Pt nanoparticles (0.55 and 0.26 mA cmPt−2, respectively, as shown in Fig. 10b and c), which is attributed to Au's ability to electronically modify Pt and reduce the CO binding strength.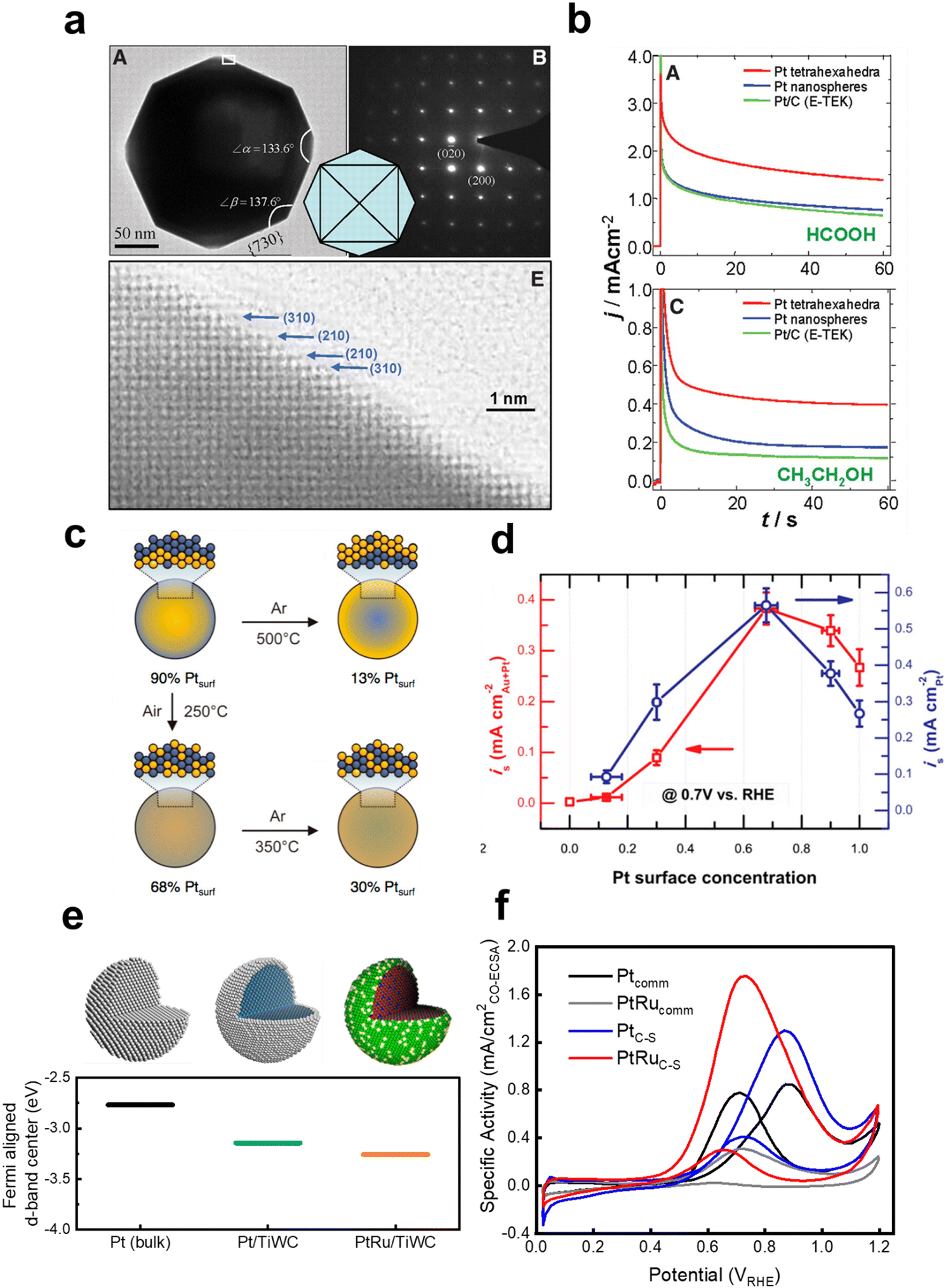 | ||
| Fig. 10 Examples of state-of-the-art catalysts for liquid fuel oxidation that have utilized modifying the surface electronic structure to increase activity. (a) Tetrahexahedral Pt nanoparticles that are composed bounded by high index facets {730} with vicinal {210} and {310} planes. From ref. 131. Reprinted with permission from AAAS. (b) Chronoamperometry measurements of the tetrahexahedral Pt nanoparticles for formic acid oxidation (top) and ethanol oxidation (bottom). From ref. 131. Reprinted with permission from AAAS. (c) Schematic showing how the surface Pt:Au ratio of a bulk Pt0.5Au0.5 particle can be controlled by heat treatment conditions. Reprinted with permission from ref. 168. Copyright (2013) American Chemical Society. (d) The methanol oxidation activity of the various surface Pt:Au ratios normalized to the total metal and Pt surface areas. Reprinted with permission from ref. 168. Copyright (2013) American Chemical Society. (e) Schematic illustrating the sequential design strategies starting from polycrystalline Pt, using the ligand effect with a Pt core and transition metal carbide core (Pt/TiWC), followed by incorporation of Ru in the Pt shell to impart a bifunctional effect with the changes to the Pt d-band electronic structure. Reprinted with permission from ref. 169. Copyright (2016) American Chemical Society. (f) Methanol oxidation cyclic voltammetry polarization curves of the core–shell particles compared to commercial Pt and PtRu benchmarks. From ref. 170. Reprinted with permission from AAAS. | ||
Ligand, strain, and bifunctional effects can be combined to engineer and optimize catalysts for liquid fuel electro-oxidation. For example, Abruña et al.171,172 have engineered PtFexCu1−x intermetallic nanoparticles by varying the Fe to Cu ratio and found that while Cu incorporation generally leads to a decreased MOR activity compared to bimetallic PtFe, Cu alters the ordering of the Pt, Cu, and Fe atoms at the surface and has a significant impact on the leaching rate of Fe atoms in acidic solutions. Kang et al.173 have synthesized Pt3Zn nanoparticles in the alloyed (disordered) and intermetallic phases (ordered) and found that the ordered phase is about 25% more active for methanol oxidation than the disordered phase or commercial Pt/C catalysts, indicating the local environment surrounding an active site also plays an important role in tuning the activity for liquid fuel oxidation. Furthermore, Román-Leshkov et al.14,170,174,175 have systematically studied precious metal overlayers on transition metal carbide core nanoparticles (Fig. 10e)169 and found that the unique core–shell structure of the nanoparticles not only significantly reduced the precious metal loadings, but also permitted more independent tuning of the bifunctional and ligand effect through the overlayer and the carbide core composition, respectively, to optimize the surface electronic structure. A 5× enhancement in MOR activity compared to that of commercial analogues (Fig. 10f) was observed for these core–shell particles,170 where the most active composition (Pt0.67Ru0.33/Ti0.11W0.89C) showed a specific activity of 0.88 mA cmPt−2 (surface area measured from CO stripping) and a mass activity of 0.43 A mgmetal−1 at 0.6 VRHE, which to the best of our knowledge, remains among the highest reported mass activity to date. Morphology control/nanostructuring is another widely employed method to maximize the utilization of noble metals by aiming to maximize the accessibility of all metal sites within a catalyst. For example, Zhao et al. have reported sub-4 nm, porous Pt72Ru28 nanoparticles that can achieve specific and mass activities of 2.1 mA cmPt−2 and 0.25 A mgmetal−1, respectively. Following similar strategies, other alloyed and intermetallic catalysts have been synthesized such as atomically dispersed Pt3Ti,176 Pt3V,176 PtSn,177 PtRuFe,178 PtBi/Pt core/shell,179 and PtFe/PtRuFe core/shell180 for the liquid fuel oxidation.
An excellent example of combining the ensemble effect with electronic modifications is the recent work of Duchesne et al.,181 who have demonstrated enhanced activity by engineering atomically dispersed Pt surface sites in a series of bimetallic PtAu catalysts (Fig. 11a). The CO binding energies on different sites were found to change based on the composition (Fig. 11b), which was attributed to changes in the electronic structure of the catalysts through the different ensembles (Fig. 11c). By combining the ligand effect of incorporating Au into the Pt lattice, which lowered the d-band center and minimized, if not prevented, CO poisoning, as well as surface engineering toward facilitating atomically dispersed Pt sites, the direct pathway of FAOR was promoted. The optimal Pt4Au96 composition afforded a nearly 20× enhancement in activity compared to Pt/C benchmark catalysts (Fig. 11d).
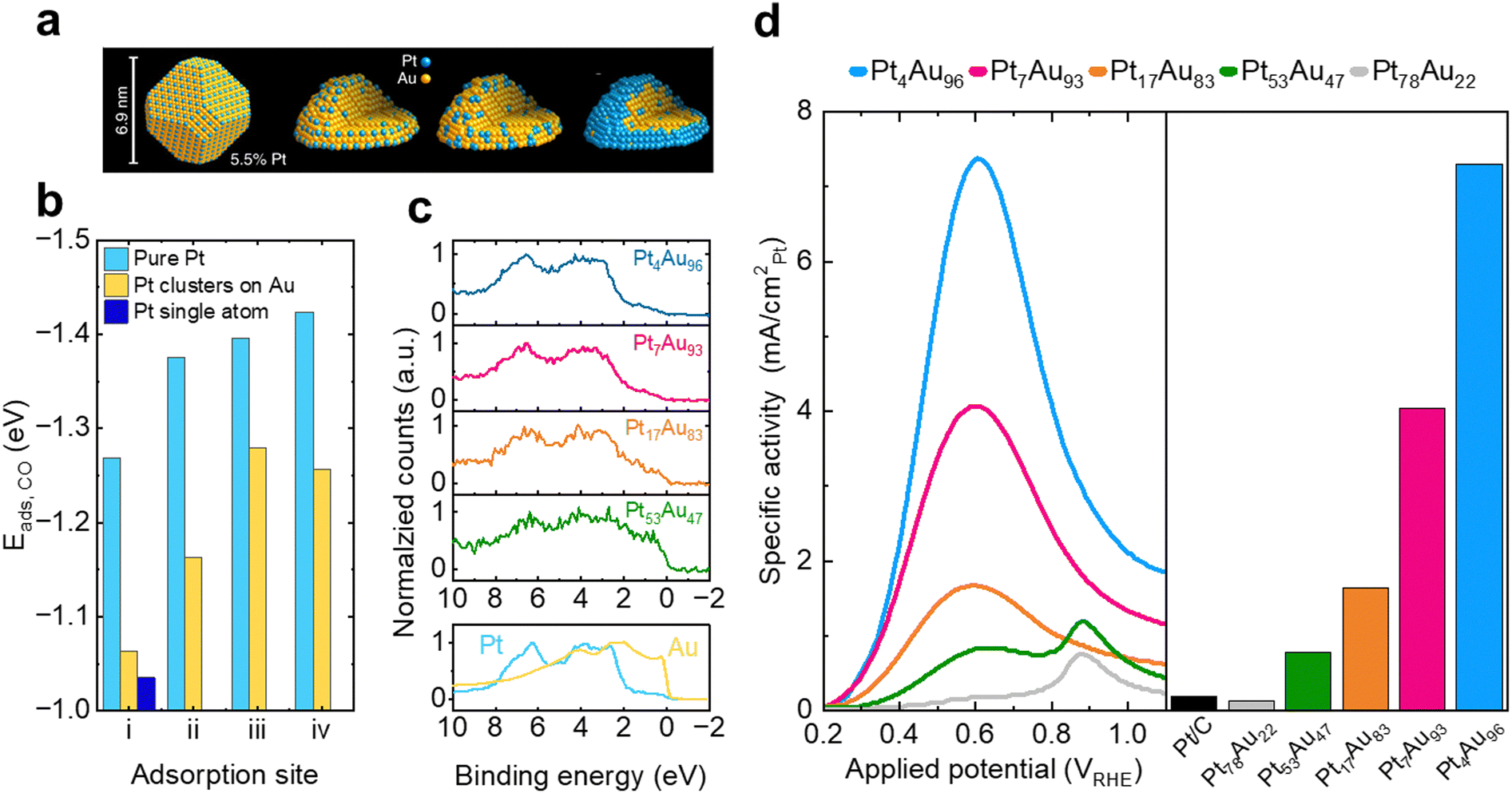 | ||
| Fig. 11 Optimization of FAOR catalysts (a) schematic of size, structure, and composition of PtAu single atom site catalysts. (b) DFT calculated CO adsorption energies on model (111) surfaces of pure, few-atom, and single-atom Pt ensembles for typical (i), bridging (ii), hexagonal close-packed hollow (iii) face-centered cubic hollow, and (iv) coordination sites. (c) XPS valence band spectra of Pt4Au96 (blue), Pt7Au93 (pink), Pt17Au83, (orange) and Pt53Au47 (green) samples compared with those of Pt (blue) and Au (yellow) foil references. (d) (left) FAOR anodic sweep voltammograms obtained from PtAu nanoparticle and commercial Pt/C catalysts, normalized by the ECSA of Pt, (right) Pt surface area normalized peak currents obtained from PtAu nanoparticle catalysts in 0.1 M HClO4 and HCOOH electrolytes. Adapted from ref. 181. Reproduced with permission from Springer Nature. | ||
4.7 Emerging strategies for optimizing liquid fuel oxidation electrocatalysts
There has been substantial progress in the design of electrocatalysts with high activity and/or selectivity for liquid fuel oxidation. However, the complex reaction pathways and interdependent surface energetics fundamental to liquid fuel oxidation reactions have cemented significant challenges in using traditional catalyst design strategies to break scaling relations. In recent years there have been significant breakthroughs in surface and electrocatalyst engineering through the advent of techniques that were largely previously unexplored, which have remained nearly untouched for liquid fuel oxidation electrocatalyst design. More specifically, we will discuss the potential advent of precious-metal-free catalysts, utilizing high entropy alloys to create active sites with a distribution of descriptors, and engineering the interfacial water structure to tune non-covalent interactions at the electrode surface. We highlight and give our perspective below as to how these newly reported strategies can potentially be applied to the future design of liquid fuel oxidation electrocatalysts for breaking scaling relations.There remains a lack of systematic searches or understanding for designing precious-metal-free liquid fuel oxidation electrocatalysts that are needed to break away from our reliance on Pt and other precious metals, which are typically required to form adsorbed CO species. While previous calculations have shown that first-row transition metals such as Co, Cu, and Ni can catalyze the initial dehydrogenation steps,43,44 their stability in acidic media limits their practical usage. Taking inspiration from other reactions that have relied heavily on precious metal catalysts in the past, precious-metal-free catalysts based on M–N4 macrocycles have proven to be promising candidates for the oxygen reduction reaction in acidic media.182,183 While to the best of our knowledge, there are currently no reports of any M–N4 macrocycles with high liquid fuel oxidation activity, these materials may be an interesting avenue worth exploring due to their wide potential tunability, low cost, and decent acid stability.184
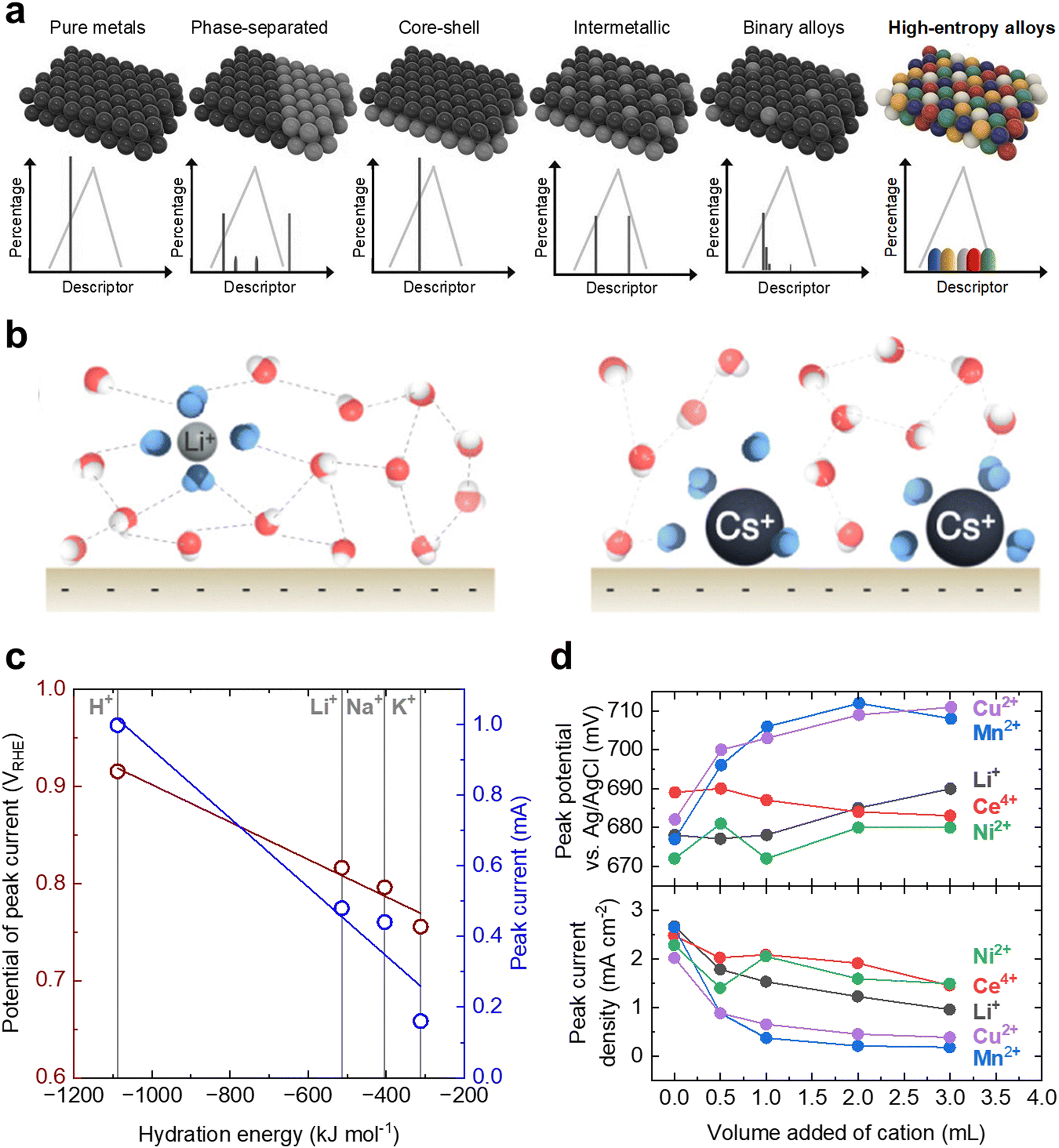 | ||
| Fig. 12 Promising design strategies for the future engineering of DLFC electrocatalysts. (a) Demonstration of the potential tunability of high entropy alloys compared to other design strategies. Due to the variation in local atomic environment, high entropy alloys can have a nearly continuous distribution of properties. This figure has been adapted from ref. 185 with permission from John Wiley & Sons, Inc., copyright 2021. (b) Difference in local water structure due to interactions with alkali metal cations: Li+ (left) and Cs+ (right). Reprinted with permission from ref. 197. Copyright (2021) American Chemical Society. (c) The dependence of the peak potential positions and peak current density of a Pt disk as a function hydration energy of the added cations. Adapted from ref. 198, Copyright (2019), with permission from Elsevier. (d) The dependence of the peak potential position (top) and peak current density (bottom) of a Pt disk as a function of the volume of cation solution added to 20 mL electrolyte reservoir. Adapted from ref. 199, Copyright (2006), with permission from Elsevier. | ||
While still few in number, these reports of high entropy alloys for liquid fuel oxidation demonstrate the potential of this material class and highlight the need for systematic understanding and design. To guide the rational design of high entropy alloys within the vast chemical space, the reaction mechanism of these high entropy alloys and their relation to the composition needs to be elucidated. Unfortunately, to date, the exact nature of the active sites within the complex surfaces is not well understood. However, it is not difficult to imagine that as a result of the high mixing of components within the high entropy alloy, active sites typically not accessible on the surface of traditional “low entropy” catalysts can be engineered, potentially allowing for a new avenue of tuning stable catalysts with an amalgamation of distinct active sites with unique electronic characteristics and geometries on the same surface.200 Due to the complex nature of liquid fuel oxidation reactions, high entropy alloys, with their wide chemical composition space and theoretically nearly infinite local atomic environments, appear to be a promising platform for future liquid fuel oxidation catalyst development.
Specifically for the oxidation of liquid fuels, Simlambarasan et al. have studied the effects of alkali cations on methanol oxidation in acidic media and found that cations with larger hydration energies correlate with lower peak potentials and peak current densities on a Pt disk electrode (Fig. 12c).204 Furthermore, Zhou et al.199 have studied the effects of various cations (Li+, Cu2+, Ni2+, Mn2+, and Ce4+) with different oxidation states as a function of concentration on methanol oxidation and found that they all generally decrease the methanol oxidation activity (Fig. 12d). A near monotonic decrease in peak current density as more cation is introduced was observed, with Li+ decreasing the peak current density from ∼2.6 mA cm−2 when no cation is added to ∼1 mA cm−2 when 3 mL of the cation solution is added. Out of the ones tested in the study, Mn2+ appears to have the largest effect on the peak current density, decreasing from ∼2.6 mA cm−2 when no cation is added to ∼0.2 mA cm−2 when 3 mL of the cation solution is added. Despite the general acceptance that non-covalent interfacial interactions can have large effects on interfacial reactions, there are few systematic studies on how interfacial non-covalent interactions can tune liquid fuel oxidation reaction rates in acidic media. All studies to date, to the best of our knowledge, only discuss empirical results with little fundamental understanding as to how these cations inherently affect the methanol oxidation thermodynamics and kinetics at catalyst surfaces. Given how most liquid fuel cells utilize a tank for storage of the fuel, the interfacial tuning additive would only need to be added to the fuel reservoir to increase the overall efficiency of the device; similarly, proton gates to control the interfacial pKa could be coated onto the membrane electrode assembly directly to increase device efficiency. As such, controlling the interfacial interactions at the electrified interface could provide an inexpensive yet potentially prodigious method for increasing the activity of liquid fuel oxidation catalysts.
5. Conclusions
Direct liquid fuel cells are primarily plagued by sluggish reaction kinetics and mixed selectivity found at the anode that currently prevents them from being competitive with the hydrogen fuel cell. However, due to their high volumetric energy density compared to hydrogen and batteries, they are prime candidates for transportation applications where the size of the device is the main limitation. In this review, we summarized the reaction mechanisms and scaling relations of the energetics within the three most widely studied liquid fuel oxidation reactions: methanol oxidation reaction, ethanol oxidation reaction, and formic acid oxidation. Given the complex reaction mechanisms of these reactions, we conclude that optimizing the activity and selectivity of liquid fuel oxidation electrocatalysts will require a careful yet precise balancing act between the C and O binding energies on the catalyst surface to facilitate key steps within the reaction pathway that will be unique to each reaction. While there has been significant attention towards the engineering of electrocatalysts using principles such as strain, surface structure, ligand effect, bifunctional effect, and ensemble engineering, there is still considerable innovation that is required to significantly decrease the overpotential or increase the selectivity of liquid fuel oxidation reaction. To that end, we believe potential avenues worth exploring for liquid fuel oxidation electrocatalysts are precious-metal-free catalysts to reduce our reliance on rare and expensive materials, exploration of high entropy alloys with their wide variety of active sites, and tuning interfacial hydrogen bonds and water structure through non-covalent interactions.Author contributions
Y. S.-H., Y. R.-L., D. J. Z., and J. P. conceived the work. D. J. Z., J. P., K. M., H. X., J. S. K., and Z. W. curated and compiled data and resources. D. J. Z., J. P., K. M., H. X., and J. S. K. visualized the data. Y. S.-H. and Y. R.-L. secured funding and administered the project. D. J. Z., J. P., K. M., H. X., J. S. K., Z. W., Y. R.-L., and Y. S.-H. wrote the original draft. All authors contributed to the review and editing of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Eni S.p.A. through the MIT Energy Initiative.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- K. Turcheniuk, D. Bondarev, V. Singhal and G. Yushin, Nature, 2018, 559, 467–470 CrossRef CAS PubMed.
- B. C. Ong, S. K. Kamarudin and S. Basri, Int. J. Hydrogen Energy, 2017, 42, 10142–10157 CrossRef CAS.
- U. B. Demirci, J. Power Sources, 2007, 169, 239–246 CrossRef CAS.
- H. A. Gasteiger and J. Garche, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2nd edn, 2008, p. 3111 Search PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, Boca Raton, FL, 92nd edn, 2011 Search PubMed.
- Perry's Chemical Engineers’ Handbook, ed. D. W. Green and R. H. Perry, McGraw-Hill Education, New York City, NY, 8th edn, 2008 Search PubMed.
- Chemical Properties Handbook: Physical, Thermodynamics, Environmental, Transport, Safety and Health Related Properties for Organic and Inorganic Chemicals, ed. C. L. Yaws, McGraw-Hill Education, New York City, NY, 1999 Search PubMed.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- P. P. Lopes, D. Li, H. Lv, C. Wang, D. Tripkovic, Y. Zhu, R. Schimmenti, H. Daimon, Y. Kang, J. Snyder, N. Becknell, K. L. More, D. Strmcnik, N. M. Markovic, M. Mavrikakis and V. R. Stamenkovic, Nat. Mater., 2020, 19, 1207–1214 CrossRef CAS PubMed.
- D. Göhl, A. Garg, P. Paciok, K. J. J. Mayrhofer, M. Heggen, Y. Shao-Horn, R. E. Dunin-Borkowski, Y. Román-Leshkov and M. Ledendecker, Nat. Mater., 2019, 19, 287–291 CrossRef PubMed.
- M. A. Ud Din, F. Saleem, B. Ni, Y. Yong and X. Wang, Adv. Mater., 2017, 29, 1–8 CrossRef PubMed.
- Y. Feng, Q. Shao, F. Lv, L. Bu, J. Guo, S. Guo and X. Huang, Adv. Sci., 2020, 7, 1800178 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Chem. Rev., 2014, 114, 12397–12429 CrossRef CAS PubMed.
- T. Shen, J. Zhang, K. Chen, S. Deng and D. Wang, Energy Fuels, 2020, 34, 9137–9153 CrossRef CAS.
- J. Bai, D. Liu, J. Yang and Y. Chen, ChemSusChem, 2019, 12, 2117–2132 CrossRef CAS PubMed.
- Y. Tong, X. Yan, J. Liang and S. X. Dou, Small, 2021, 17(9), 1904126 CrossRef CAS PubMed.
- K. Jiang, H.-X. Zhang, S. Zou and W.-B. Cai, Phys. Chem. Chem. Phys., 2014, 16, 20360–20376 RSC.
- Y. Wang, S. Zou and W.-B. Cai, Catalysts, 2015, 5, 1507–1534 CrossRef CAS.
- O. A. Petrii, Russ. J. Electrochem., 2019, 55, 1–33 CrossRef CAS.
- T. H. M. Housmans, A. H. Wonders and M. T. M. Koper, J. Phys. Chem. B, 2006, 110, 10021–10031 CrossRef CAS PubMed.
- M. T. M. Koper, S. C. S. Lai and E. Herrero, Fuel Cell Catal. A Surf. Sci. Approach, 2008, pp. 159–207 Search PubMed.
- J. Chang, G. Wang, C. Li, Y. He, Y. Zhu, W. Zhang, M. Sajid, A. Kara, M. Gu and Y. Yang, Joule, 2023, 7, 587–602 CrossRef CAS.
- J. Chang, G. Wang, X. Chang, Z. Yang, H. Wang, B. Li, W. Zhang, L. Kovarik, Y. Du, N. Orlovskaya, B. Xu, G. Wang and Y. Yang, Nat. Commun., 2023, 14, 1–15 Search PubMed.
- J. Chang, G. Wang, M. Wang, Q. Wang, B. Li, H. Zhou, Y. Zhu, W. Zhang, M. Omer, N. Orlovskaya, Q. Ma, M. Gu, Z. Feng, G. Wang and Y. Yang, Nat. Energy, 2021, 6, 1144–1153 CrossRef CAS.
- W. Huang, H. Wang, J. Zhou, J. Wang, P. N. Duchesne, D. Muir, P. Zhang, N. Han, F. Zhao, M. Zeng, J. Zhong, C. Jin, Y. Li, S. T. Lee and H. Dai, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li, H. Fei, Y. Zhu, R. Yu, J. Luo, K. Zang, Z. Lin, M. Ding, J. Huang, H. Sun, J. Guo, X. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef CAS.
- Y. Katayama, R. Kubota, R. R. Rao, J. Hwang, L. Giordano, A. Morinaga, T. Okanishi, H. Muroyama, T. Matsui, Y. Shao-Horn and K. Eguchi, J. Phys. Chem. C, 2021, 125, 26321–26331 CrossRef CAS.
- J. John, H. Wang, E. D. Rus and H. D. Abruña, J. Phys. Chem. C, 2012, 116, 5810–5820 CrossRef CAS.
- K. Scott, E. Yu, G. Vlachogiannopoulos, M. Shivare and N. Duteanu, J. Power Sources, 2008, 175, 452–457 CrossRef CAS.
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187–200 CrossRef CAS.
- L. L. Wang, Q. X. Li, T. Y. Zhan and Q. J. Xu, Adv. Mater. Res., 2014, 860–863, 826–830 CAS.
- P. A. Christensen, A. Hamnett and D. Linares-Moya, Phys. Chem. Chem. Phys., 2011, 13, 5206–5214 RSC.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654–2675 RSC.
- E. A. Monyoncho, T. K. Woo and E. A. Baranova, Ethanol electrooxidation reaction in alkaline media for direct ethanol fuel cells, 2019, vol. 15 Search PubMed.
- S. C. S. Lai, N. P. Lebedeva, T. H. M. Housmans and M. T. M. Koper, Top. Catal., 2007, 46, 320–333 CrossRef CAS.
- J. L. Cohen, D. J. Volpe and H. D. Abruña, Phys. Chem. Chem. Phys., 2007, 9, 49–77 RSC.
- D. Cao, G.-Q. Lu, A. Wieckowski, S. A. Wasileski and M. Neurock, J. Phys. Chem. B, 2005, 109, 11622–11633 CrossRef CAS PubMed.
- P. Ferrin and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 14381–14389 CrossRef CAS PubMed.
- G. A. Tritsaris and J. Rossmeisl, J. Phys. Chem. C, 2012, 116, 11980–11986 CrossRef CAS.
- J. Rossmeisl, P. Ferrin, G. A. Tritsaris, A. U. Nilekar, S. Koh, S. E. Bae, S. R. Brankovic, P. Strasser and M. Mavrikakis, Energy Environ. Sci., 2012, 5, 8335–8342 RSC.
- S. Gilman, J. Phys. Chem., 1963, 67, 1898–1905 CrossRef CAS.
- P. Urchaga, S. Baranton, C. Coutanceau and G. Jerkiewicz, Langmuir, 2012, 28, 13094–13104 CrossRef CAS PubMed.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS PubMed.
- K. Kunimatsu, H. Hanawa, H. Uchida and M. Watanabe, J. Electroanal. Chem., 2009, 632, 109–119 CrossRef CAS.
- H. A. Gasteiger, N. Marković, P. N. Ross and E. J. Cairns, J. Phys. Chem., 1993, 97, 12020–12029 CrossRef CAS.
- Z. Liu, J. Y. Lee, W. Chen, M. Han and L. M. Gan, Langmuir, 2004, 20, 181–187 CrossRef CAS PubMed.
- D. Y. Chung, K. J. Lee and Y. E. Sung, J. Phys. Chem. C, 2016, 120, 9028–9035 CrossRef CAS.
- H. Nitani, T. Nakagawa, H. Daimon, Y. Kurobe, T. Ono, Y. Honda, A. Koizumi, S. Seino and T. A. Yamamoto, Appl. Catal., A, 2007, 326, 194–201 CrossRef CAS.
- Z. Liu, X. Y. Ling, X. Su and J. Y. Lee, J. Phys. Chem. B, 2004, 108, 8234–8240 CrossRef CAS.
- W. L. Seung, S. Chen, W. Sheng, N. Yabuuchi, Y. T. Kim, T. Mitani, E. Vescovo and Y. Shao-Horn, J. Am. Chem. Soc., 2009, 131, 15669–15677 CrossRef PubMed.
- T. H. M. Housmans and M. T. M. Koper, J. Phys. Chem. B, 2003, 107, 8557–8567 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 267–273 CrossRef CAS.
- A. R. Poerwoprajitno, L. Gloag, J. Watt, S. Cheong, X. Tan, H. Lei, H. A. Tahini, A. Henson, B. Subhash, N. M. Bedford, B. K. Miller, P. B. O’Mara, T. M. Benedetti, D. L. Huber, W. Zhang, S. C. Smith, J. J. Gooding, W. Schuhmann and R. D. Tilley, Nat. Catal., 2022, 5, 231–237 CrossRef CAS.
- J. Fan, M. Chen, Z. Zhao, Z. Zhang, S. Ye, S. Xu, H. Wang and H. Li, Nat. Energy, 2021, 6, 475–486 CrossRef CAS.
- H.-F. Wang and Z.-P. Liu, J. Am. Chem. Soc., 2008, 130, 10996–11004 CrossRef CAS PubMed.
- S. C. S. Lai, S. E. F. Kleyn, V. Rosca and M. T. M. Koper, J. Phys. Chem. C, 2008, 112, 19080–19087 CrossRef CAS.
- S. Gilman, J. Phys. Chem., 1964, 68, 70–80 CrossRef CAS.
- F. Colmati, E. Antolini and E. R. Gonzalez, J. Power Sources, 2006, 157, 98–103 CrossRef CAS.
- H. Wang, Z. Jusys and R. J. Behm, J. Phys. Chem. B, 2004, 108, 19413–19424 CrossRef CAS.
- R. J. Wang, H. Jusys and Z. Behm, J. Power Sources, 2006, 154, 351–359 CrossRef.
- M. Li, P. Liu and R. R. Adzic, J. Phys. Chem. Lett., 2012, 3, 3480–3485 CrossRef CAS PubMed.
- M. Arenz, V. Stamenkovic, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, Phys. Chem. Chem. Phys., 2003, 5, 4242–4251 RSC.
- A. Cuesta, G. Cabello, C. Gutiérrez and M. Osawa, Phys. Chem. Chem. Phys., 2011, 13, 20091–20095 RSC.
- V. Grozovski, F. J. Vidal-Iglesias, E. Herrero and J. M. Feliu, ChemPhysChem, 2011, 12, 1641–1644 CrossRef CAS PubMed.
- Y.-X. Chen, M. Heinen, Z. Jusys and R. J. Behm, Langmuir, 2006, 22, 10399–10408 CrossRef CAS PubMed.
- J. Xu, D. Yuan, F. Yang, D. Mei, Z. Zhang and Y.-X. X. Chen, Phys. Chem. Chem. Phys., 2013, 15, 4367–4376 RSC.
- W. Gao, J. A. Keith, J. Anton and T. Jacob, J. Am. Chem. Soc., 2010, 132, 18377–18385 CrossRef PubMed.
- R. Zhang, H. Liu, B. Wang and L. Ling, J. Phys. Chem. C, 2012, 116, 22266–22280 CrossRef CAS.
- H.-F. Wang and Z.-P. Liu, J. Phys. Chem. C, 2009, 113, 17502–17508 CrossRef CAS.
- S. Brimaud, J. Solla-Gullón, I. Weber, J. M. Feliu and R. J. Behm, ChemElectroChem, 2014, 1, 1075–1083 CrossRef CAS.
- H. A. Gasteiger, N. Marković, P. N. Ross and E. J. Cairns, Electrochim. Acta, 1994, 39, 1825–1832 CrossRef CAS.
- X. Chen, L. P. Granda-Marulanda, I. T. McCrum and M. T. M. Koper, Nat. Commun., 2022, 13, 1–11 CAS.
- H. Li, G. Sun, Q. Jiang, M. Zhu, S. Sun and Q. Xin, Electrochem. Commun., 2007, 9, 1410–1415 CrossRef CAS.
- Z. Liu, L. Hong, M. P. Tham, T. H. Lim and H. Jiang, J. Power Sources, 2006, 161, 831–835 CrossRef CAS.
- T. Shen, S. Chen, R. Zeng, M. Gong, T. Zhao, Y. Lu, X. Liu, D. Xiao, Y. Yang, J. Hu, D. Wang, H. L. Xin and H. D. Abrunã, ACS Catal., 2020, 10, 9977–9985 CrossRef CAS.
- M. Shibata, N. Furuya, M. Watanabe and S. Motoo, J. Electroanal. Chem., 1989, 263, 97–108 CrossRef CAS.
- S.-C. Chang, Y. Ho and M. J. Weaver, Surf. Sci., 1992, 265, 81–94 CrossRef CAS.
- A. Sáez, E. Expósito, J. Solla-Gullón, V. Montiel and A. Aldaz, Electrochim. Acta, 2012, 63, 105–111 CrossRef.
- S. P. E. Smith, K. F. Ben-Dor and H. D. Abruna, Langmuir, 1999, 15, 7325–7332 CrossRef CAS.
- X. Yu and P. G. Pickup, J. Appl. Electrochem., 2011, 41, 589–597 CrossRef CAS.
- N. Yang, Z. Zhang, B. Chen, Y. Huang, J. Chen, Z. Lai, Y. Chen, M. Sindoro, A. L. Wang, H. Cheng, Z. Fan, X. Liu, B. Li, Y. Zong, L. Gu and H. Zhang, Adv. Mater., 2017, 29, 1–6 Search PubMed.
- J. Greeley, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 605–635 CrossRef PubMed.
- P. Ferrin, D. Simonetti, S. Kandoi, E. Kunkes, J. A. Dumesic, J. K. Nørskov and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 5809–5815 CrossRef CAS PubMed.
- E. A. Monyoncho, S. N. Steinmann, P. Sautet, E. A. Baranova and C. Michel, Electrochim. Acta, 2018, 274, 274–278 CrossRef CAS.
- L.-X. X. Dai, X.-Y. Y. Wang, S.-S. S. Yang, T. Zhang, P.-J. J. Ren, J.-Y. Y. Ye, B. Nan, X.-D. D. Wen, Z.-Y. Y. Zhou, R. Si, C.-H. H. Yan and Y.-W. W. Zhang, J. Mater. Chem. A, 2018, 6, 11270–11280 RSC.
- A. O. Elnabawy, J. A. Herron, J. Scaranto and M. Mavrikakis, J. Electrochem. Soc., 2018, 165, J3109–J3121 CrossRef CAS.
- A. O. Elnabawy, J. A. Herron, Z. Liang, R. R. Adzic and M. Mavrikakis, ACS Catal., 2021, 11, 5294–5309 CrossRef CAS.
- F. Mehmood, R. B. Rankin, J. Greeley and L. A. Curtiss, Phys. Chem. Chem. Phys., 2012, 14, 8644–8652 RSC.
- J. S. Yoo, F. Abild-Pedersen, J. K. Nørskov and F. Studt, ACS Catal., 2014, 4, 1226–1233 CrossRef CAS.
- H. Li and G. Henkelman, J. Phys. Chem. C, 2017, 121, 27504–27510 CrossRef CAS.
- J. Kang, S. Nam, Y. Oh, H. Choi, S. Wi, B. Lee, T. Hwang, S. Hong and B. Park, J. Phys. Chem. Lett., 2013, 4, 2931–2936 CrossRef CAS.
- J. E. Sutton and D. G. Vlachos, Ind. Eng. Chem. Res., 2015, 54, 4213–4225 CrossRef CAS.
- C.-Y. Syu, H.-W. Yang, F.-H. Hsu and J.-H. Wang, Phys. Chem. Chem. Phys., 2014, 16, 7481–7490 RSC.
- A. Hook, J. D. Massa and F. E. Celik, J. Phys. Chem. C, 2016, 120, 27307–27318 CrossRef CAS.
- M.-L. Yang, Y.-A. Zhu, X.-G. Zhou, Z.-J. Sui and D. Chen, ACS Catal., 2012, 2, 1247–1258 CrossRef CAS.
- E. A. Monyoncho, S. N. Steinmann, C. Michel, E. A. Baranova, T. K. Woo and P. Sautet, ACS Catal., 2016, 6, 4894–4906 CrossRef CAS.
- D. E. Hoare and A. D. Walsh, Trans. Faraday Soc., 1954, 50, 37–50 RSC.
- S. C. Chang, A. Hamelin and M. J. Weaver, J. Phys. Chem., 1991, 95, 5560–5567 CrossRef CAS.
- T. E. Shubina, C. Hartnig and M. T. M. Koper, Phys. Chem. Chem. Phys., 2004, 6, 4215–4221 RSC.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- W. T. Hong, K. A. Stoerzinger, Y. L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- B. Hammer, J. K. Norskov, P. J. Robinson and K. A. Holbrook, Nature, 1995, 376, 238–240 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough, Y. Shao-horn, F. Calle-vallejo, A. D. Oscar, M. J. Kolb, M. T. M. Koper, J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-horn, Science, 2011, 334, 2010–2012 CrossRef PubMed.
- Y. L. Lee, J. Kleis, J. Rossmeisl, S. H. Yang and D. Morgan, Energy Environ. Sci., 2011, 4, 3966–3970 RSC.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- A. Ruban, B. Hammer, P. Stoltze, H. L. Skriver, J. K. Nørskov, J. K. Ruban, A. Hammer, B. Stoltze, P. Skriver and H. L. Nørskov, J. Mol. Catal. A: Chem., 1997, 115, 421–429 CrossRef CAS.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- T. He, W. Wang, F. Shi, X. Yang, X. Li, J. Wu, Y. Yin and M. Jin, Nature, 2021, 598, 76–81 CrossRef CAS PubMed.
- R. Loukrakpam, Q. Yuan, V. Petkov, L. Gan, S. Rudi, R. Yang, Y. Huang, S. R. Brankovic and P. Strasser, Phys. Chem. Chem. Phys., 2014, 16, 18866–18876 RSC.
- B. Hammer, O. H. Nielsen and J. K. Nørskov, Catal. Lett., 1997, 46, 31–35 CrossRef CAS.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, J. Chem. Phys., 2004, 120, 10240–10246 CrossRef CAS PubMed.
- C. Lu, C. Rice, R. I. Masel, P. K. Babu, P. Waszczuk, H. S. Kim, E. Oldfield and A. Wieckowski, J. Phys. Chem. B, 2002, 106, 9581–9589 CrossRef CAS.
- W. Zhong, Y. Qi and M. Deng, J. Power Sources, 2015, 278, 203–212 CrossRef CAS.
- P. Ferrin, A. U. Nilekar, J. Greeley, M. Mavrikakis and J. Rossmeisl, Surf. Sci., 2008, 602, 3424–3431 CrossRef CAS.
- E. Herrero, K. Franaszczuk and A. Wieckowski, J. Phys. Chem., 1994, 98, 5074–5083 CrossRef CAS.
- J. Solla-Gullón, F. J. Vidal-Iglesias, A. López-Cudero, E. Garnier, J. M. Feliu and A. Aldaz, Phys. Chem. Chem. Phys., 2008, 10, 3689–3698 RSC.
- D. J. Tarnowski and C. Korzeniewski, J. Phys. Chem. B, 1997, 101, 253–258 CrossRef CAS.
- S. C. S. Lai and M. T. M. Koper, Faraday Discuss., 2008, 140, 399–416 RSC.
- S. Motoo and N. Furuya, Phys. Chem. Chem. Phys., 1987, 91, 457–461 CAS.
- C. Lamy, J. M. Leger, J. Clavilier and R. Parsons, J. Electroanal. Chem., 1983, 150, 71–77 CrossRef CAS.
- Y. Qi, T. Bian, S. Il Choi, Y. Jiang, C. Jin, M. Fu, H. Zhang and D. Yang, Chem. Commun., 2014, 50, 560–562 RSC.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- N. Tian, Z. Y. Zhou, N. F. Yu, L. Y. Wang and S. G. Sun, J. Am. Chem. Soc., 2010, 132, 7580–7581 CrossRef CAS PubMed.
- J. Geng, Z. Zhu, X. Bai, F. Li and J. Chen, ACS Appl. Energy Mater., 2020, 3, 1010–1016 CrossRef CAS.
- F. Colmati, G. Tremiliosi-Filho, E. R. Gonzalez, A. Berná, E. Herrero and J. M. Feliu, Faraday Discuss., 2008, 140, 379–397 RSC.
- F. Colmati, G. Tremiliosi-Filho, E. R. Gonzalez, A. Berná, E. Herrero and J. M. Feliu, Phys. Chem. Chem. Phys., 2009, 11, 9114–9123 RSC.
- J. Shin, W. J. Tornquist, C. Korzeniewski and C. S. Hoaglund, Surf. Sci., 1996, 364, 122–130 CrossRef CAS.
- X. H. Xia, H. D. Liess and T. Iwasita, J. Electroanal. Chem., 1997, 437, 233–240 CrossRef CAS.
- M. P. Hyman and J. W. Medlin, J. Phys. Chem. C, 2007, 111, 17052–17060 CrossRef CAS.
- T. Bligaard and J. K. Nørskov, Electrochim. Acta, 2007, 52, 5512–5516 CrossRef CAS.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 4–7 CrossRef PubMed.
- C. Roth, N. Benker, R. Theissmann, R. J. Nichols and D. J. Schiffrin, Langmuir, 2008, 24, 2191–2199 CrossRef CAS PubMed.
- S. Desai and M. Neurock, Electrochim. Acta, 2003, 48, 3759–3773 CrossRef CAS.
- H. Li, G. Sun, L. Cao, L. Jiang and Q. Xin, Electrochim. Acta, 2007, 52, 6622–6629 CrossRef CAS.
- F. Vigier, C. Coutanceau, F. Hahn, E. M. Belgsir and C. Lamy, J. Electroanal. Chem., 2004, 563, 81–89 CrossRef CAS.
- P. Yang, X. Yuan, H. Hu, Y. Liu, H. Zheng, D. Yang, L. Chen, M. Cao, Y. Xu, Y. Min, Y. Li and Q. Zhang, Adv. Funct. Mater., 2018, 28(1), 1704774 CrossRef.
- E. Casado-Rivera, Z. Gál, A. C. D. Angelo, C. Lind, F. J. DiSalvo and H. D. Abruña, ChemPhysChem, 2003, 4, 193–199 CrossRef CAS PubMed.
- P. Strasser, Q. Fan, M. Devenney, W. H. Weinberg, P. Liu and J. K. Nørskov, J. Phys. Chem. B, 2003, 107, 11013–11021 CrossRef CAS.
- F. G. Welsch, K. Stöwe and W. F. Maier, ACS Comb. Sci., 2011, 13, 518–529 CrossRef CAS PubMed.
- K. L. Ley, R. Liu, C. Pu, Q. Fan, N. Leyarovska, C. Segre and E. S. Smotkin, J. Electrochem. Soc., 1997, 144, 1543–1548 CrossRef CAS.
- H. Xu, D. J. Zheng, H. Iriawan, J.-H. Fang, J. Kim, X. Wang, Y. Román-Leshkov, J. Li and Y. Shao-Horn, Chem. Mater., 2024, 36(14), 6938–6949 CrossRef CAS.
- S. Motoo and N. Furuya, J. Electroanal. Chem., 1985, 184, 303–316 CrossRef CAS.
- B. Du and Y. Tong, J. Phys. Chem. B, 2005, 109, 17775–17780 CrossRef CAS PubMed.
- S. Motoo and N. Furuya, J. Electroanal. Chem. Interfacial Electrochem., 1986, 197, 209–218 CrossRef CAS.
- N. M. Marković, H. A. Gasteiger, P. N. Ross, X. Jiang, I. Villegas and M. J. Weaver, Electrochim. Acta, 1995, 40, 91–98 CrossRef.
- S. E. Evarts, I. Kendrick, B. L. Wallstrom, T. Mion, M. Abedi, N. Dimakis and E. S. Smotkin, ACS Catal., 2012, 2, 701–707 CrossRef CAS.
- A. Cuesta, J. Am. Chem. Soc., 2006, 128, 13332–13333 CrossRef CAS PubMed.
- A. Bagger, K. D. Jensen, M. Rashedi, R. Luo, J. Du, D. Zhang, I. J. Pereira, M. Escudero-Escribano, M. Arenz and J. Rossmeisl, Chem. Sci., 2022, 13, 13409–13417 RSC.
- M. Li, D. A. Cullen, K. Sasaki, N. S. Marinkovic, K. More and R. R. Adzic, J. Am. Chem. Soc., 2013, 135, 132–141 CrossRef CAS PubMed.
- N. Zhang, L. Bu, S. Guo, J. Guo and X. Huang, Nano Lett., 2016, 16, 5037–5043 CrossRef CAS PubMed.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- Z. Li, X. Jiang, X. Wang, J. Hu, Y. Liu, G. Fu and Y. Tang, Appl. Catal., B, 2020, 277, 119135 CrossRef CAS.
- X. Xu, X. Zhang, H. Sun, Y. Yang, X. Dai, J. Gao, X. Li, P. Zhang, H. H. Wang, N. F. Yu and S. G. Sun, Angew. Chem., Int. Ed., 2014, 53, 12522–12527 CrossRef CAS PubMed.
- Y. Lu and W. Chen, ACS Catal., 2012, 2, 84–90 CrossRef CAS.
- X. Y. Liu, Y. Zhang, M. X. Gong, Y. W. Tang, T. H. Lu, Y. Chen and J. M. Lee, J. Mater. Chem. A, 2014, 2, 13840–13844 RSC.
- J. M. Sieben and M. M. E. Duarte, Int. J. Hydrogen Energy, 2011, 36, 3313–3321 CrossRef CAS.
- M. Arenz, K. J. J. Mayrhofer, V. Stamenkovic, B. B. Blizanac, T. Tomoyuki, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2005, 127, 6819–6829 CrossRef CAS PubMed.
- D. Y. Chung, H. Shin, J. M. Yoo, K. S. Lee, N. S. Lee, K. Kang and Y. E. Sung, J. Power Sources, 2016, 334, 52–57 CrossRef CAS.
- J. Suntivich, Z. Xu, C. E. Carlton, J. Kim, B. Han, S. W. Lee, N. Bonnet, N. Marzari, L. F. Allard, H. A. Gasteiger, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef CAS PubMed.
- C. H. Hendon, S. T. Hunt, M. Milina, K. T. Butler, A. Walsh and Y. Román-Leshkov, J. Phys. Chem. Lett., 2016, 7, 4475–4482 CrossRef CAS PubMed.
- S. T. Hunt, M. Milina, A. C. Alba-Rubio, C. H. Hendon, J. A. Dumesic and Y. Román-Leshkov, Science, 2016, 352, 974–978 CrossRef CAS PubMed.
- J. Zhu, Y. Yang, L. Chen, W. Xiao, H. Liu, H. D. Abruña and D. Wang, Chem. Mater., 2018, 30, 5987–5995 CrossRef CAS.
- R. Zeng, Y. Yang, T. Shen, H. Wang, Y. Xiong, J. Zhu, D. Wang and H. D. Abruña, ACS Catal., 2020, 10, 770–776 CrossRef CAS.
- Y. Kang, J. B. Pyo, X. Ye, T. R. Gordon and C. B. Murray, ACS Nano, 2012, 6, 5642–5647 CrossRef CAS PubMed.
- A. Garg, D. S. Goncalves, Y. Liu, Z. Wang, L. Wang, J. S. Yoo, A. Kolpak, R. M. Rioux, D. Zanchet and Y. Román-Leshkov, ACS Catal., 2019, 9, 7090–7098 CrossRef CAS.
- Z. Wang, J. S. Kang, D. Göhl, P. Paciok, D. S. Gonçalves, H. K. Lim, D. Zanchet, M. Heggen, Y. Shao-Horn, M. Ledendecker and Y. Román-Leshkov, Adv. Energy Mater., 2024, 2304092, 1–9 Search PubMed.
- Z. Cui, H. Chen, M. Zhao, D. Marshall, Y. Yu, H. Abruña and F. J. Disalvo, J. Am. Chem. Soc., 2014, 136, 10206–10209 CrossRef CAS PubMed.
- W. Chen, Z. Lei, T. Zeng, L. Wang, N. Cheng, Y. Tan and S. Mu, Nanoscale, 2019, 11, 19895–19902 RSC.
- M. E. Scofield, C. Koenigsmann, L. Wang, H. Liu and S. S. Wong, Energy Environ. Sci., 2015, 8, 350–363 RSC.
- Y. Qin, M. Luo, Y. Sun, C. Li, B. Huang, Y. Yang, Y. Li, L. Wang and S. Guo, ACS Catal., 2018, 8, 5581–5590 CrossRef CAS.
- Q. Wang, S. Chen, P. Li, S. Ibraheem, J. Li, J. Deng and Z. Wei, Appl. Catal., B, 2019, 252, 120–127 CrossRef CAS.
- P. N. Duchesne, Z. Y. Li, C. P. Deming, V. Fung, X. Zhao, J. Yuan, T. Regier, A. Aldalbahi, Z. Almarhoon, S. Chen, D. en Jiang, N. Zheng and P. Zhang, Nat. Mater., 2018, 17, 1033–1039 CrossRef CAS PubMed.
- J. Zagal, M. Páez, A. A. Tanaka, J. R. dos Santos and C. A. Linkous, J. Electroanal. Chem., 1992, 339, 13–30 CrossRef CAS.
- D. K. Singh, V. Ganesan, D. K. Yadav and M. Yadav, Langmuir, 2020, 36, 12202–12212 CrossRef CAS PubMed.
- S. Yuan, J. Peng, Y. Zhang, D. J. Zheng, S. Bagi, T. Wang, Y. Román-Leshkov and Y. Shao-Horn, ACS Catal., 2022, 12, 7278–7287 CrossRef CAS.
- T. Löffler, A. Ludwig, J. Rossmeisl and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 26894–26903 CrossRef PubMed.
- Y. Xin, S. Li, Y. Qian, W. Zhu, H. Yuan, P. Jiang, R. Guo and L. Wang, ACS Catal., 2020, 10, 11280–11306 CrossRef CAS.
- C. Ng, S. Guo, J. Luan, S. Shi and C. T. Liu, Intermetallics, 2012, 31, 165–172 CrossRef.
- F. Otto, Y. Yang, H. Bei and E. P. George, Acta Mater., 2013, 61, 2628–2638 CrossRef CAS.
- M. P. Salinas-Quezada, J. K. Pedersen, P. Sebastián-Pascual, I. Chorkendorff, K. Biswas, J. Rossmeisl and M. Escudero-Escribano, EES Catal., 2024, 2, 941–952 RSC.
- C. F. Tsai, P. W. Wu, P. Lin, C. G. Chao and K. Y. Yeh, Jpn. J. Appl. Phys., 2008, 47, 5755–5761 CrossRef CAS.
- Y. Wang, Z. Zhang, T. Hu, J. Yang and Y. Li, Chem. Commun., 2024, 60, 4072–4075 RSC.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed.
- K. V. Yusenko, S. Riva, P. A. Carvalho, M. V. Yusenko, S. Arnaboldi, A. S. Sukhikh, M. Hanfland and S. A. Gromilov, Scr. Mater., 2017, 138, 22–27 CrossRef CAS.
- D. Wang, Z. Chen, Y. C. Huang, W. Li, J. Wang, Z. Lu, K. Gu, T. Wang, Y. Wu, C. Chen, Y. Zhang, X. Huang, L. Tao, C. L. Dong, J. Chen, C. V. Singh and S. Wang, Sci. China: Mater., 2021, 64, 2454–2466 CAS.
- D. Wang, Z. Chen, Y. Wu, Y. C. Huang, L. Tao, J. Chen, C. L. Dong, C. V. Singh and S. Wang, SmartMat, 2023, 4, 1–10 Search PubMed.
- X. Zuo, R. Yan, L. Zhao, Y. Long, L. Shi, Q. Cheng, D. Liu and C. Hu, J. Mater. Chem. A, 2022, 10, 14857–14865 RSC.
- B. Huang, R. R. Rao, S. You, K. Hpone Myint, Y. Song, Y. Wang, W. Ding, L. Giordano, Y. Zhang, T. Wang, S. Muy, Y. Katayama, J. C. Grossman, A. P. Willard, K. Xu, Y. Jiang and Y. Shao-Horn, JACS Au, 2021, 1, 1674–1687 CrossRef CAS PubMed.
- T. Wang, Y. Zhang, B. Huang, B. Cai, R. R. Rao, L. Giordano, S. G. Sun and Y. Shao-Horn, Nat. Catal., 2021, 4, 753–762 CrossRef CAS.
- X. Zhou, T. Lu, W. Xing and C. Liu, Electrochim. Acta, 2006, 52, 1688–1691 CrossRef CAS.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3, 834–845 CrossRef CAS.
- A. Goyal and M. T. M. Koper, Angew. Chem., Int. Ed., 2021, 60, 13452–13462 CrossRef CAS PubMed.
- A. C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M. T. M. Koper, Angew. Chem., Int. Ed., 2019, 58, 12999–13003 CrossRef CAS PubMed.
- S. Yu, H. Yamauchi, S. Wang, A. Aggarwal, J. Kim, K. Gordiz, B. Huang, H. Xu, D. J. Zheng, X. Wang, H. Iriawan, D. Menga and Y. Shao-Horn, Nat. Catal., 2024 DOI:10.1038/s41929-024-01197-2.
- K. Silambarasan, J. Joseph and S. Mayavan, Appl. Surf. Sci., 2019, 489, 149–153 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00100a |
| ‡ Denotes equal contribution. |
| § Present address: Department of Energy Systems Engineering, Seoul National University, Seoul 08826, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2024 |