
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCe-induced NiS bifunctional catalyst transformation: enhancing urea oxidation coupled with hydrogen electrolysis†
Yingzhen
Zhang‡
 abc,
Wei
Zhang‡
a,
Jianying
Huang
*a,
Weilong
Cai
*ab and
Yuekun
Lai
*ab
abc,
Wei
Zhang‡
a,
Jianying
Huang
*a,
Weilong
Cai
*ab and
Yuekun
Lai
*ab
aNational Engineering Research Center of Chemical Fertilizer Catalyst (NERC-CFC), College of Chemical Engineering, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: yklai@fzu.edu.cn; wlcai@fzu.edu.cn; jyhuang@fzu.edu.cn
bQingyuan Innovation Laboratory, Quanzhou 362801, P. R. China
cSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, Singapore 637459, Singapore
First published on 13th August 2024
Abstract
The treatment of urea-containing wastewater is crucial for sustainable environmental development, given its low theoretical thermodynamic barrier (0.37 V), which can effectively replace the OER process in water electrolysis and enhance hydrogen production efficiency. Nevertheless, designing dual-functional catalysts capable of effectively performing catalytic tasks remains a challenge. Herein, in this work a cerium-doped nickel sulfide (Ce–NiS) catalyst is synthesized by an electrodeposition method, which is used as a bifunctional catalyst for electrolytic hydrogen production from urea-containing wastewater. Ce–NiS exhibits a higher Faradaic efficiency (FE, 91.39%) compared to NiS (67.52%) for hydrogen production from simulated urea-containing wastewater. In situ Raman spectroscopy reveals that Ce doping induces the reconstruction of NiS into high-valence nickel species (NiOOH), which is considered the actual active center for the electrochemical UOR process. Notably, the apparent electrochemical activation energy for the UOR decreased from 8.72 kJ mol−1 (NiS) to 5.68 kJ mol−1 (Ce–NiS), indicating that doping with Ce significantly reduces the energy barrier for the UOR and enhances the catalytic urea oxidation capability. This study employs a strategy of rare-earth metal (Ce) doping to enhance the efficiency of urea-coupled electrolytic hydrogen production, providing promising insights for energy recovery from urea-containing wastewater and the development of high-performance dual-functional catalysts.
Broader contextUtilizing electrochemical technology to oxidize urea in wastewater is an effective means to achieve both waste reuse and enhanced efficiency of hydrogen production by water electrolysis. However, urea is a complex substance containing both electron-withdrawing carbonyl groups and electron-donating amino groups, posing significant challenges to an in-depth study of urea oxidation reactions. In this work, we employed a strategy of doping a rare-earth metal, specifically incorporating Ce, to modulate the morphology and electronic structure of a NiS electrocatalyst. This modulation aimed to optimize the catalytic capability of the active centers post-electrochemical reconstruction for the urea oxidation reaction and to improve the efficiency of hydrogen production from the electrolysis of urea-containing wastewater. This study opens up an attractive avenue for the reuse of urea-containing wastewater and low-energy water electrolysis for hydrogen production, contributing to the global sustainable development of the environment. |
1. Introduction
Electrochemical production of green hydrogen through water electrolysis is considered an effective pathway for developing clean energy.1,2 However, its hydrogen production efficiency is constrained by the sluggish kinetics of the anodic oxygen evolution reaction (OER, E = 1.23 V).3 In response to the global demand for clean energy and the imperative of environmental protection, developing a low-energy, economically viable hydrogen production technology from urea-containing wastewater via electrolysis has become crucial. Urea, a nitrogen-rich pollutant widely found in human urine, as well as industrial and agricultural wastewater,4 is notable for its low electrochemical oxidation potential (E = 0.37 V).5 Coupling the urea oxidation reaction (UOR) with the cathodic hydrogen evolution reaction (HER) in the electrolysis process (UOR//HER) not only reduces hydrogen production energy consumption (theoretically saving 70% energy compared to conventional water electrolysis),6 but also enables the effective treatment of urea-containing wastewater, offering a dual-benefit solution. Nevertheless, the UOR process under alkaline conditions is a six-electron transfer process (CO(NH2)2 + 6OH− → N2 + 5H2O + CO2 + 6e−),7 which produces complex reaction intermediates and poses significant challenges for the targeted design of highly active and stable UOR catalysts.Nickel is an abundant transition metal on Earth, and is renowned for its affordability and exceptional conductivity, making it a focal point in electrochemical water splitting,8 the nitrogen reduction reaction,9 the ammonia oxidation reaction,10 and so on. Nickel-based catalysts undergo a self-oxidation process during electrochemical oxidation reactions, where low-valent nickel species are converted to high-valent nickel species (NiOOH), which is considered the active species in electrocatalysis.11,12 However, pure NiOOH exhibits inadequate activity and stability in the UOR. It is worth noting that defects in electrocatalysts can alter the electronic properties of materials, create more active sites, and promote enhanced charge transfer, thereby improving catalytic performance.13 Cerium (Ce) is a rare earth metal with a unique tetravalent electron structure, and has been shown to induce oxygen defects in Ni(OH)2 upon doping, shifting the Ni d-band center towards the Fermi level and thereby lowering the electrochemical reaction barrier.14 Additionally, Fu et al. reported that Ce can activate Co sites through gradient orbital coupling, enhancing the electrochemical activity of Co-based catalysts.15 Despite these advancements, synthesis of dual-functional catalysts capable of catalyzing both the UOR and HER remains challenging.
Herein, Ce-doped nickel sulfide (Ce–NiS) materials were grown on the carbon cloth (CC) using a one-pot electrodeposition method, creating a bifunctional electrocatalyst for hydrogen production via the electrolysis of urea-containing wastewater. Ce doping modulated the surface electron cloud density of NiS, promoted electrochemical reconstruction, and exposed more Ni3+ active sites, thereby enhancing the catalytic activity for the UOR process. Ce–NiS shows a working voltage of 1.39 V at a current density of 50 mA cm−2, which is 30 mV lower than that of NiS. As a bifunctional catalyst, Ce–NiS displays exceptional hydrogen production capability in the UOR//HER system, with a hydrogen production rate of 75.73 μmol h−1, remarkably surpassing that in the OER//HER system (47.52 μmol h−1). Furthermore, in situ Raman spectroscopy confirmed that the true active site of Ce–NiS is NiOOH in the UOR process. The addition of Ce induces the conversion of NiS to the active phase NiOOH, which inherently contributes to the superior catalytic activity of Ce–NiS in the urea oxidation reaction. This work provides a promising strategy for the directional design of highly active electrocatalysts and hydrogen production from urea-containing wastewater.
2. Results and discussion
2.1 Materials characterization
Fig. 1a depicts that Ce-doped NiS materials were grown in situ on carbon cloth substrates by the electrodeposition method. The crystal structures of the catalysts and substrate carbon cloth were analyzed via XRD (Fig. 1b). Both pure NiS and Ce–NiS exhibited identical diffraction peaks at 2θ = 18.4°, 30.3°, 32.2°, 35.7°, 40.5°, and 48.8°, corresponding to the tripartite phase NiS (acicular nickel ores, PDF#12-0041) in the (110), (101), (300), (021), (211), and (131) planes,16–18 respectively. This phenomenon indicates that the crystalline structure of NiS remains unaltered upon the introduction of Ce. SEM was utilized to probe the morphology of the as-synthesized catalysts, and Ce–NiS (Fig. 1c) showed a looser surface morphology than pure NiS (Fig. 1d), indicating that Ce doping facilitates an increase in the porosity of NiS. Elemental maps (Fig. 1e) and the energy dispersive spectrum (EDS) (Fig. S1, ESI†) demonstrated the homogeneous distribution of Ni, S, and Ce elements on Ce–NiS. Furthermore, TEM was employed to further unveil the internal structure of the as-synthesized catalysts. Compared to pure NiS (Fig. 1f), Ce–NiS exhibited a distinct cavity structure (Fig. 1g), strongly proving that Ce doping promotes the formation of internal pore structures within NiS (corroborating the loose structure observed in SEM). This enhanced porosity facilitates electrolyte penetration during electrochemical processes. Additionally, the surface wettability of the as-prepared catalysts was assessed using contact angle (CA) measurements. Ce–NiS demonstrated superhydrophilicity (with a CA of 0°, the inset in Fig. 1c), while the pure NiS exhibited weak-hydrophilicity (with a CA of 131.1°, the inset in Fig. 1d). Superhydrophilic properties enhance the contact between the catalyst and the electrolyte in subsequent electrochemical processes,19,20 thereby increasing the solid–liquid contact area. Based on the TEM and CA analysis, the superior surface wettability of Ce–NiS was related to the structural changes caused by Ce doping and the formation of voids. | ||
| Fig. 1 (a) Schematic illustration of samples. (b) XRD patterns of bare CC, NiS, and Ce–NiS samples. SEM images of (c) Ce–NiS and (d) NiS. (e) SEM images and the corresponding elemental maps of Ni, S, and Ce of Ce–NiS. TEM images of (f) NiS and (g) Ce–NiS. | ||
X-ray photoelectron spectroscopy (XPS) was used to examine the surface composition and valence states of Ce–NiS composites. The XPS survey spectrum reveals the presence of three elements in the Ce–NiS materials, including Ce, Ni, and S (Fig. 2a). In the spectrum of S 2p (Fig. 2b), three main peaks located around 163.4 eV (S 2p3/2), 164.11 eV (S 2p1/2), and 165.2 eV were observed, corresponding to the metal–sulfur bond (Ni–S), low coordination S2− and C–S bond,21,22 respectively. Beyond that, the characteristic peak at 169.1 eV belongs to the S–O bond, which comes from the oxidation of the electrocatalyst when exposed to air.23–25 The Ni 2p spectrum (Fig. 2c) was divided into Ni 2p1/2 and Ni 2p3/2, accompanied by two shakeup satellite peaks (861.6 eV and 879.9 eV).26,27 The two peaks located at 855.9 eV and 873.4 eV are attributed to Ni2+ and the other two peaks at 857.2 eV and 875.3 eV correspond to Ni3+. Notably, compared to pure NiS, the binding energy of Ni2+ of Ce–NiS up-shifts by about 0.51 eV (Fig. 2c).28,29 Ce doping increases the binding energy of Ni 2p orbitals in NiS, indicating that Ni atoms lose some electrons. This process of electron loss is conducive to the conversion of bivalent Ni into trivalent Ni, which lays a good foundation for the subsequent electrooxidation behavior. The Ce 3d spectrum (Fig. 2c) was divided into two peaks, Ce3+ (886.9 eV, 882.6 eV) and Ce4+ (884.9 eV), respectively. Ce3+ has a larger peak area than Ce4+ and there is a free conversion between them,28 resulting in the easy formation of sulfur-rich defects and vacancies in NiS. To verify the density of S-vacancies in NiS by EPR, Fig. 2d shows a sharp characteristic signal at g = 2.003, attributed to the S-vacancies.30,31 Compared to pure NiS, Ce–NiS exhibits higher peak intensity, indicating that the introduction of Ce facilitates the generation of sulfur defects in NiS. Both XPS and EPR analyses confirm that Ce doping can indeed modify the electronic structure of NiS.
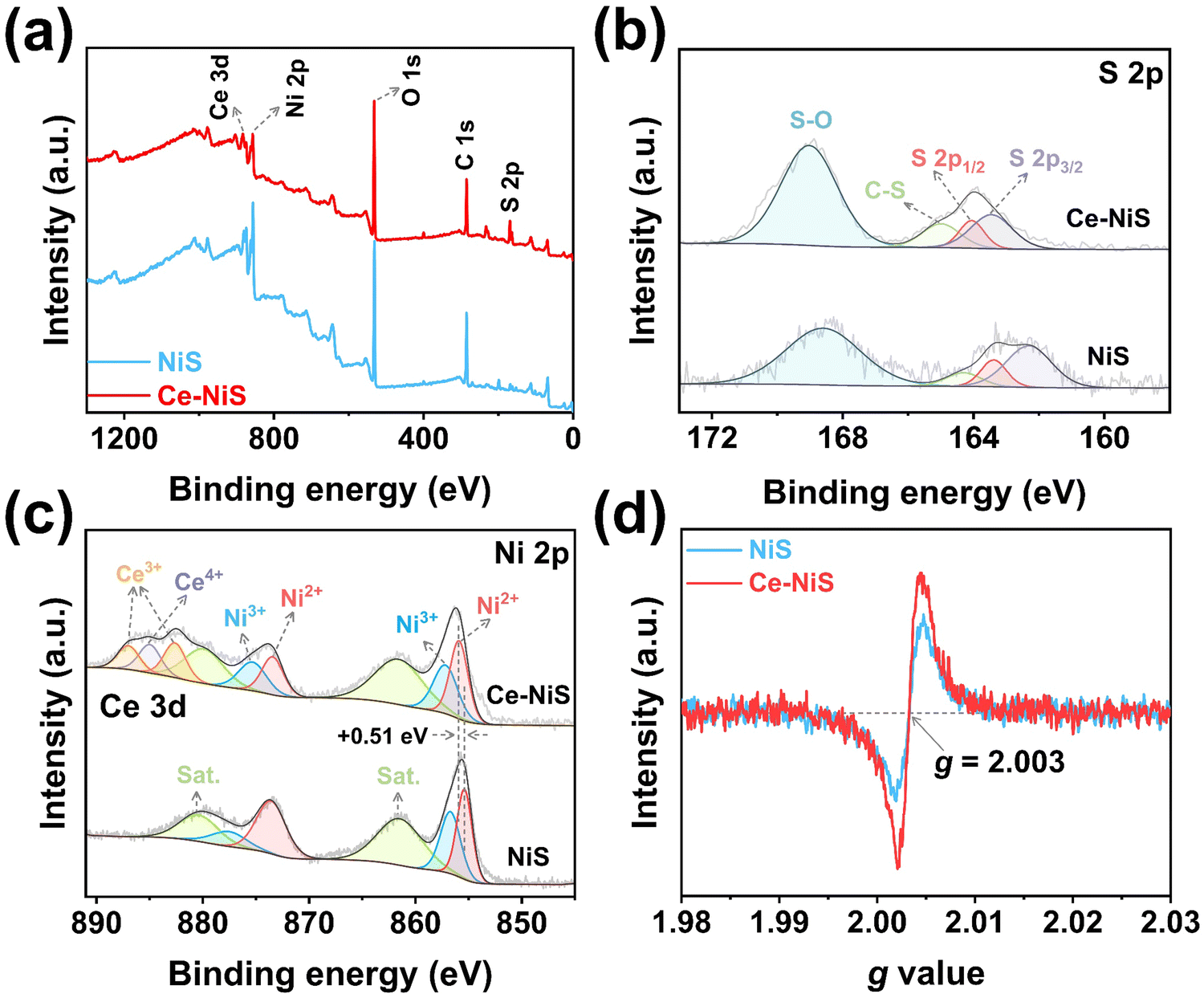 | ||
| Fig. 2 High-resolution XPS spectrum of Ce–NiS: (a) survey spectra, (b) S 2p, and (c) Ni 2p for Ce–NiS and NiS. (d) EPR spectrum of Ce–NiS and NiS electrocatalysts. | ||
2.2 UOR electrochemical performances of Ce–NiS
To exclude the influence of electrochemical activity of the carbon cloth, the OER and UOR activities of the carbon cloth were characterized. It was found that the pure carbon cloth had almost no catalytic activity on urea (Fig. S2, ESI†). The UOR electrochemical activity of different Ce-doped NiS samples was evaluated using linear sweep voltammetry (LSV). Fig. 3a demonstrates that pure Ce has almost no UOR activity, but doping Ce into NiS could significantly enhance the UOR activity of NiS. As shown in Fig. S3 (ESI†), Ce doping significantly enhances the UOR electrocatalytic activity of NiS, with 3% Ce–NiS exhibiting the highest activity, followed by 5% Ce–NiS and 1% Ce–NiS. Thus, the appropriate introduction of Ce into NiS improves UOR performance. Consequently, the 3% Ce–NiS catalyst (denoted as Ce–NiS) was selected as the catalyst with the best performance and was used for further electrochemical performance studies. Moreover, the Tafel slope (Fig. 3b) of Ce–NiS (51.16 mV dec−1) was lower than that of pure NiS (72.19 mV dec−1) for the UOR process, further proving that Ce doping was conducive to increasing the UOR kinetic rate of NiS.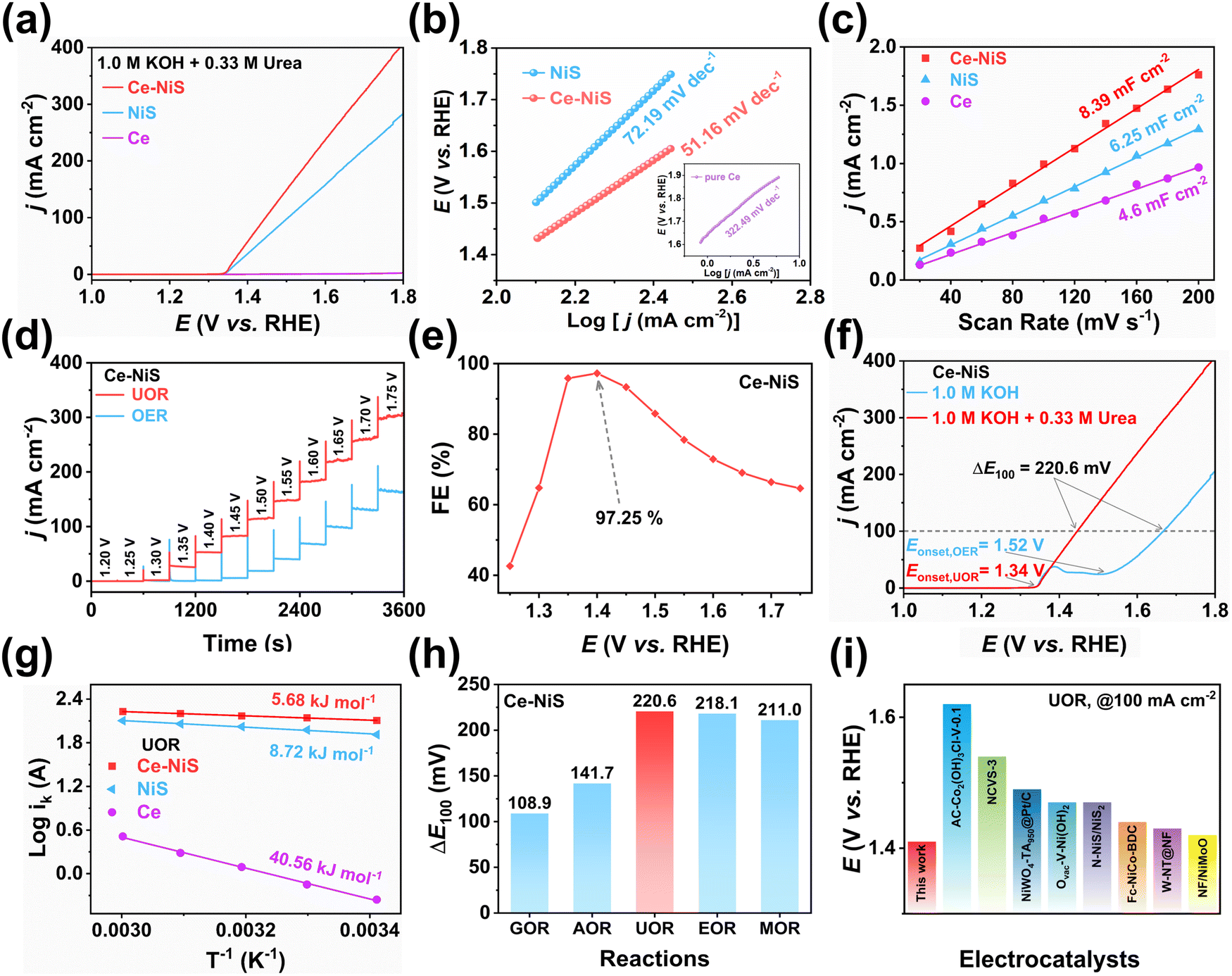 | ||
Fig. 3 (a) LSV curves and (b) Tafel slopes of Ce, NiS, and Ce–NiS in 1.0 M KOH with 0.33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M urea. (c) Cdl values of Ce, NiS, and Ce–NiS. (d) Differential pulse voltammetry curves of Ce–NiS towards the UOR and OER. (e) Faraday efficiency of Ce–NiS at different potentials in the UOR. (f) LSV curves of Ce–NiS in 1.0 M KOH with and without 0.33 M urea. (g) Ea of Ce–NiS, NiS, and Ce towards the UOR. (h) Selectivity of Ce–NiS for electrochemical oxidation of different small molecules (urea, glucose, ammonia, methanol, and ethanol). (i) Comparison of potentials of different catalysts at 100 mA cm−2. M urea. (c) Cdl values of Ce, NiS, and Ce–NiS. (d) Differential pulse voltammetry curves of Ce–NiS towards the UOR and OER. (e) Faraday efficiency of Ce–NiS at different potentials in the UOR. (f) LSV curves of Ce–NiS in 1.0 M KOH with and without 0.33 M urea. (g) Ea of Ce–NiS, NiS, and Ce towards the UOR. (h) Selectivity of Ce–NiS for electrochemical oxidation of different small molecules (urea, glucose, ammonia, methanol, and ethanol). (i) Comparison of potentials of different catalysts at 100 mA cm−2. | ||
Electrochemically active surface area (ECSA) and the charge passed on the catalyst surface are the important factors for unveiling the exposure of active sites on a catalyst. Fig. 3c displays that the Cdl value of Ce–NiS (8.39 mF cm−2) was greater than those of pure NiS (6.25 mF cm−2) and pure Ce (4.46 mF cm−2). And, Ce–NiS has the highest ECSA value of 209.8 cm2 (Fig. S4, ESI†), greatly exceeding those of the pure NiS (156.2 cm2) and pure Ce (116.8 cm2). This result indicates that the introduction of Ce was beneficial for increasing the ECSA of NiS, potentially facilitating the exposure of more catalytically active sites on NiS. Given the positive correlation between the number of catalytically active sites and the surface charge passed,32 the pulsed chronoamperometry method was further employed to accurately analyze the effect of Ce doping on the number of active sites. Ce–NiS exhibited a higher surface charge passing quantity compared to NiS (0.336 vs. 0.287 C, Fig. S5, ESI†). This finding aligns with the ECSA results, further validating that Ce doping enhances the exposure of active sites on NiS, thereby improving UOR electrochemical activity.
As the OER represents a potential competing reaction to the UOR, the differential pulse voltammetry method was employed to investigate the kinetic rates of these two electrochemical processes.33,34 As shown in Fig. 3d, a significant current jump (0 to 25 mA cm−2) was observed in the urea-containing electrolyte when the voltage exceeded 1.35 V, while in the pure KOH electrolyte, only a weak current jump (0 to 10 mA cm−2) occurred after the voltage exceeded 1.50 V. Moreover, Ce–NiS exhibited the highest Faraday efficiency (97.25%) at 1.4 V vs. the RHE (Fig. 3e). At the same current density (100 mA cm−2), the potential difference (ΔE100 = EUOR − EOER) between the UOR and the OER was as high as 220.6 mV (Fig. 3f), and the onset potential (Eonset) of the UOR (Eonset = 1.34 V) was much lower than that of the OER (Eonset = 1.52 V). These results substantiated that the kinetic rate of the UOR was faster than that of the OER (consistent with the Tafel plot). Furthermore, the current jump amplitude of pure NiS (0 to 5 mA cm−2) was notably weaker than that of Ce–NiS (Fig. S6, ESI†), further highlighting the positive impact of Ce doping on the UOR performance of NiS. Operating for 12 h at an applied voltage of 1.45 V, a significant decrease in current density was observed (Fig. S7, ESI†). Upon replacing the electrolyte involved in the reaction with a fresh urea-containing electrolyte, a remarkable recovery in current density was seen. The decline in current density during prolonged operation was attributed to the depletion of urea in the electrolyte, resulting in a reduced mass transfer rate. This issue could be mitigated by periodically refreshing the electrolyte.
Additionally, the LSV curves obtained in different temperature ranges (Fig. S8, ESI†) could be used to calculate the apparent electrochemical activation energy (Ea) of the catalyst in the OER and UOR processes using the Arrhenius equation,35 which is a classic method for revealing electrochemical thermodynamic barriers. As shown in Fig. S9 (ESI†), the Ea of Ce–NiS in the UOR process (5.68 kJ mol−1) was remarkably lower than that in the OER process (14.53 kJ mol−1). Moreover, the Ea of Ce–NiS in the UOR process was also lower than those of the pure NiS (8.72 kJ mol−1) and pure Ce (40.56 kJ mol−1), as shown in Fig. 3g. Both of these phenomena demonstrated that Ce doping was conducive to enhancing the kinetic rate of NiS and reducing the reaction barrier of NiS in the UOR process.
To further expand the application scenarios of Ce–NiS in the field of electrochemistry, the electrochemical oxidation performance of various substances (e.g., methanol, ethanol, ammonia, and glucose) was evaluated. As shown in Fig. S10 (ESI†), Ce–NiS delivers excellent electrochemical selectivity for the methanol oxidation reaction (MOR), ethanol oxidation reaction (EOR), ammonia oxidation reaction (AOR), and glucose oxidation reaction (GOR) with ΔE100 all exceeding 100 mV (Fig. 3h). This shows that Ce–NiS has great potential for application in electrochemical oxidation reactions. Furthermore, the UOR performance of Ce–NiS was superior to that of reported nickel-based electrocatalysts (Fig. 3i and Table S1, ESI†).
2.3 Investigation of the hydrogen evolution performance in the UOR//HER system
To further explore the feasibility of hydrogen production from electrolyzing urea-containing wastewater, the HER activity was tested in 1.0 M KOH containing 0.33 M urea. As shown in Fig. S11 (ESI†), Ce exhibits almost no HER performance, while both NiS and Ce–NiS show good HER performance, with Ce–NiS exhibiting the best HER performance. In addition, the influence of urea presence in the electrolyte on the HER activity of Ce–NiS could be neglected (Fig. S12, ESI†), which implies that Ce–NiS can be effectively used in the electrolysis system for hydrogen production from urea-containing wastewater. As Ce–NiS serves as both the cathode and anode, a urea electrolysis cell was constructed to explore the electrochemical activity via LSV for UOR//HER and OER//HER. As shown in Fig. 4a, Ce–NiS exhibits higher electrochemical activity for the UOR//HER system compared to the OER//HER system. Subsequently, the hydrogen generation rate (Fig. S13, ESI†) and Faraday efficiency (FE, Fig. 4b) were tested in water splitting (OER//HER) and simulated urea wastewater systems (UOR//HER). At 1.6 V, the simulated urea wastewater system demonstrated a higher hydrogen production rate (75.73 μmol h−1) and FE (91.39%) compared to the water-splitting system (47.52 μmol h−1 and 78.51%), underscoring the benefits of substituting the UOR for the OER. Moreover, abundant H2 bubble generation was observed on the surface of the Ce–NiS electrode (Fig. 4c). The hydrogen production rate and FE of Ce–NiS in the UOR//HER system were also markedly higher than those of the pure NiS (69.30 μmol h−1 and 67.52%), respectively. The aforementioned results insinuate that Ce doping expedites the UOR-coupled HER hydrogen production efficiency. Additionally, we have summarized the different small molecule oxidation reactions coupled with water electrolysis reported for hydrogen production in the field of electro/photocatalysis (as shown in Table S2, ESI†), confirming the significance of using small molecule (e.g., cyclohexanone, methanol, ethanol, glucose, ammonia, etc) oxidation reactions to replace the OER process for low-energy and high-efficiency hydrogen production.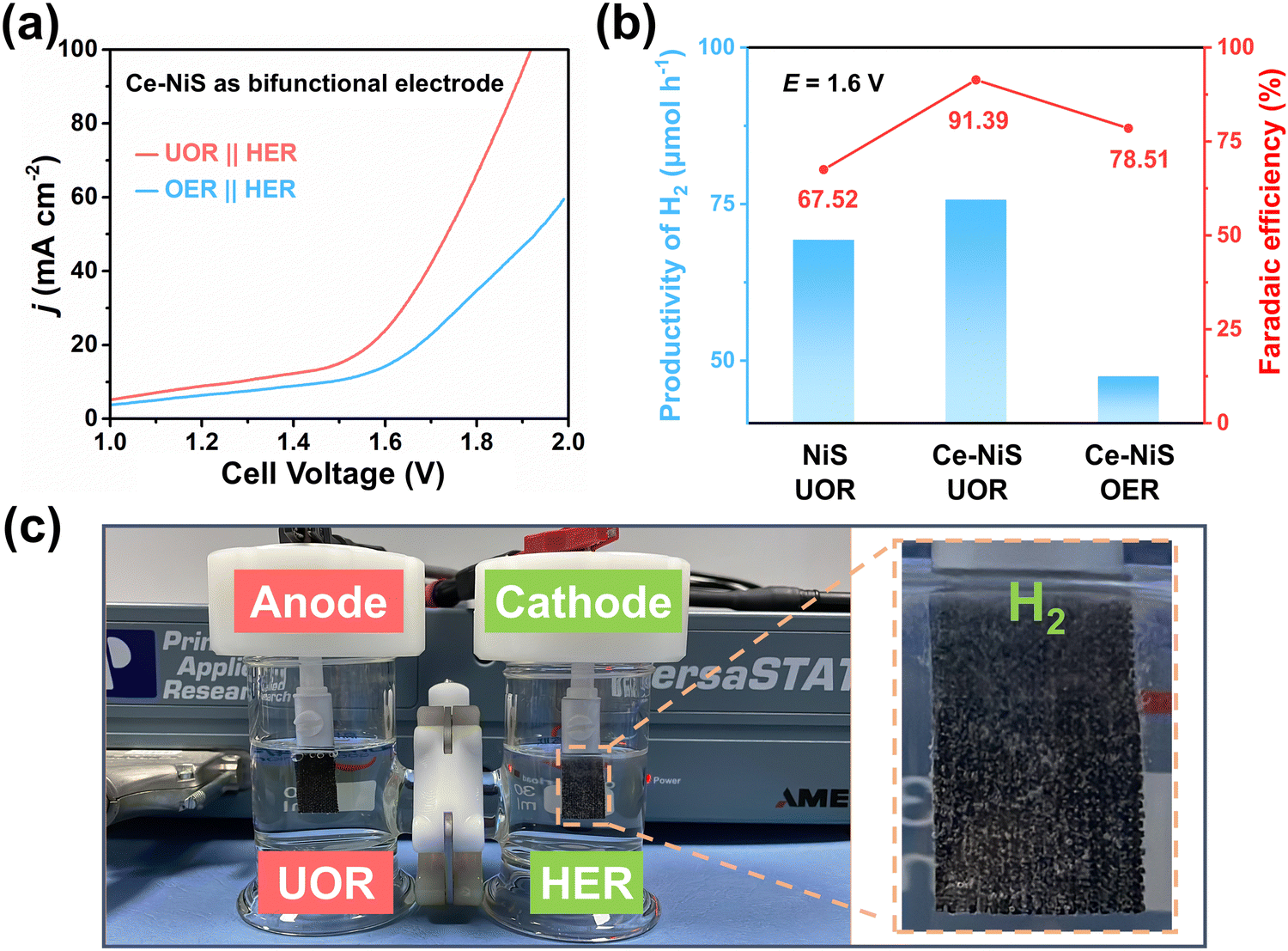 | ||
| Fig. 4 (a) The LSV profiles were evaluated using Ce–NiS as the anodic electrode and cathodic electrode, respectively. (b) Hydrogen generation rates and FE of NiS and Ce–NiS at UOR//HER and OER//HER, respectively. (c) Hydrogen production device, and the inset shows the optical image of the cathode. | ||
The durability of the catalyst is a key reference index in practical applications.36,37 Using Ce–NiS as a cathode and an anode, a urea electrolytic cell was assembled, and chronopotentiometry was used to characterize the electrochemical stability of the catalyst in the electrolytic simulation of the urea wastewater system (UOR//HER). As shown in Fig. S14 (ESI†), operating at 100 mA cm−2 for 3600 s (1 h), the electrochemical signal change was negligible. Additionally, SEM (Fig. S15, ESI†) revealed that the Ce–NiS morphology did not collapse after the UOR long-term stability test, which also proved that the Ce–NiS catalyst had good electrochemical stability.
2.4 Analysis of the UOR mechanism
To elucidate the genuine active sites of Ce–NiS, the evolving reconstruction behavior of samples during the UOR was tracked using in situ Raman spectroscopy (Fig. 5a). In Fig. S16a and b (ESI†), the in situ Raman spectra of NiS and Ce–NiS recorded at open circuit potential (OCP) to 1.6 V vs. RHE are shown. No Raman spectral band was observed at a lower potential (<1.40 V) in NiS, which was similar to the spectral feature at OCP. However, at 1.45 V and above, Ce–NiS exhibits two strong Raman bands at around 475.2 (δ-Ni–O) and 555.2 cm−1 (ν-Ni–O), corresponding to the Ni3+–O bending and stretching vibrations of NiOOH,38,39 respectively. Their presence indicates the transformation from NiS to NiOOH. In contrast to Ce–NiS, the Raman spectra of NiS during the reconstitution process do not exhibit the A1g stretching vibration peaks of NiOOH. Additionally, the Eg bending vibration peaks (473.3 cm−1) of NiS were red-shifted, and their intensities were lower compared to those of Ce–NiS. This phenomenon arises from the structural disorder and reduced crystallinity induced by Ce doping. It is particularly noteworthy that the introduction of Ce accelerates the reconstruction behavior of NiS: as the potential increases, the Raman signals related to NiOOH significantly strengthen. Both NiS (Fig. 5b) and Ce–NiS (Fig. 5c) exhibit a prominent absorption peak around 1000 cm−1 at OCP, which indicates that the adsorption of urea on the catalyst surface is a spontaneous process. The difference is that, as the voltage increases, the urea absorption peak disappeared for NiS, whereas the urea absorption peak persisted for Ce–NiS. This result indicates that the adsorption of urea on Ce–NiS was more favorable. Based on the aforementioned analysis, a mechanism was proposed for the catalysis of the UOR by Ce–NiS (Fig. 5d): (1) at open circuit potential, Ce–NiS maintains a stable initial structure. (2) When the potential was increased to 1.45 V, MII undergoes electrochemical oxidation to form active species (MIIIOOH).40 (3) Urea adsorbed on the active sites was oxidized. (4) The urea adsorbed on the active sites undergoes chemical oxidation reactions (6MIIIOOH + CO(NH2)2 + H2O ⇆ 6MIIOH + N2 + CO2).41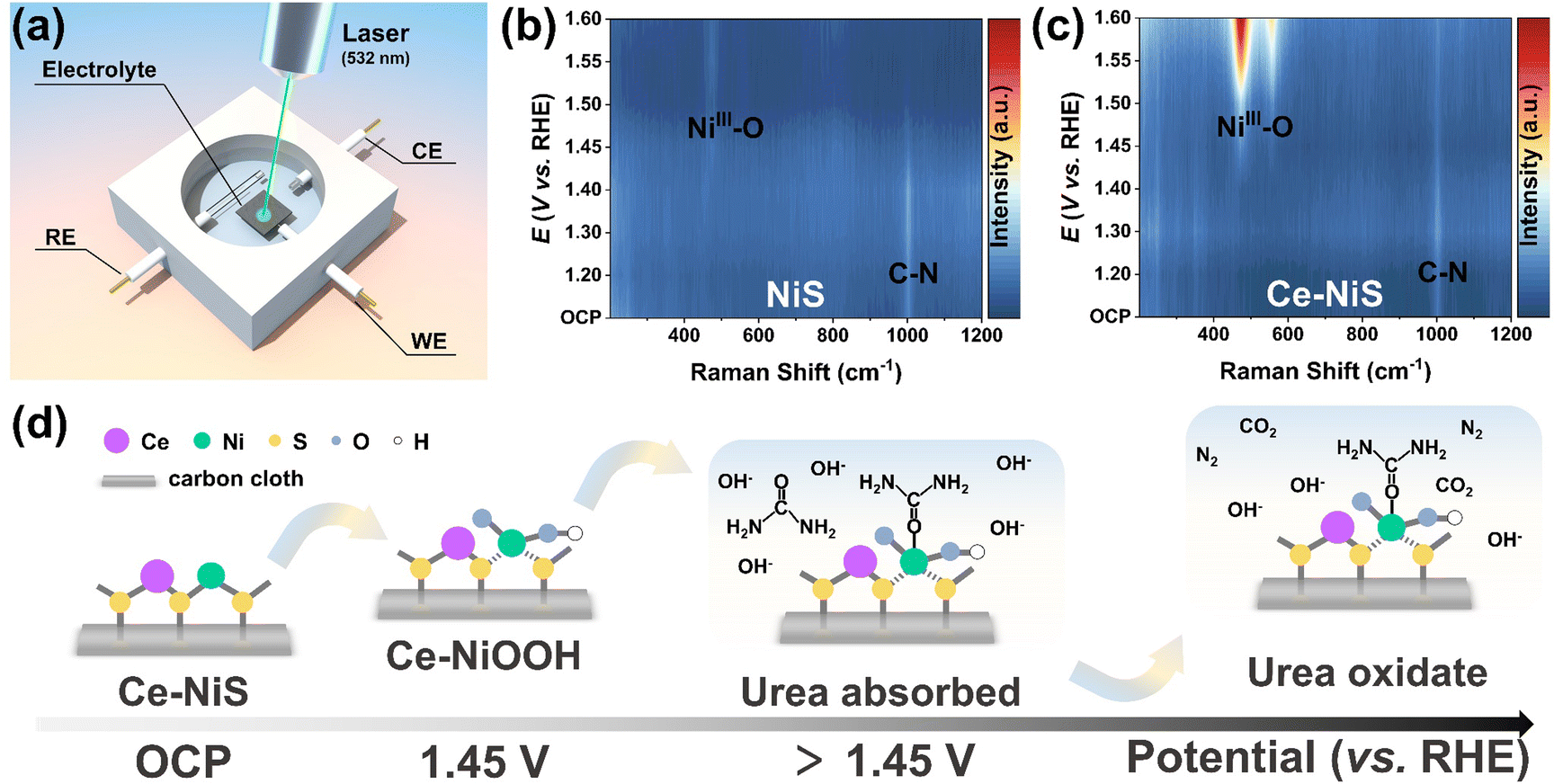 | ||
| Fig. 5 (a) Diagram of the in situ Raman device. In situ Raman spectra of (b) NiS and (c) Ce–NiS were recorded from OCP to 1.6 V vs. RHE in 1.0 M KOH with 0.33 M urea. (d) Schematic illustration of the UOR process on Ce–NiS. | ||
3. Conclusion
In summary, in this study a Ce-doped NiS electrocatalyst (Ce–NiS) was synthesized through a one-step electrodeposition method, growing in situ on a carbon cloth substrate. Ce–NiS features a loose surface, resulting in a high electrochemical surface area (209.8 cm2). Ce doping modifies the surface morphology and adjusts the electronic structure of NiS, increasing the density of S-vacancies. This enhancement promotes efficient catalytic reactions by accelerating mass transfer and improving charge transport efficiency. When utilized in an alkaline water electrolysis system, Ce–NiS exhibited exceptional catalytic activities for both the UOR and the HER, highlighting its potential for urea degradation and efficient hydrogen production (75.73 μmol h−1). Additionally, in situ Raman techniques revealed that the collaboration between Ce and NiS enhanced the electron distribution of the catalyst and accelerated the self-oxidative restoration process of the surface-active substance NiOOH, thereby significantly advancing the urea oxidation reaction. This study demonstrates that Ce doping with NiS produces a synergistic effect, remarkably enhancing the catalytic ability of the UOR and the HER. These findings offer a promising avenue for developing efficient catalysts suitable for hydrogen production systems driven by energy-efficient organic small molecules.Author contributions
Yingzhen Zhang carried out the catalyst syntheses, characterization, and data analysis. Wei Zhang did in situ Raman measurements and assisted in drawing a schematic diagram. Jianying Huang, Weilong Cai and Yuekun Lai directed the research, oversaw its development, and helped revising the manuscript.Data availability
All data generated or analyzed during this study are provided in the ESI† and are available from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from the International Cooperation and Exchanges NSFC (22361162607), National Key Research and Development Program of China (No. 2022YFB3804905), National Natural Science Foundation of China (22075046, 22378071, 22375047, 22378068), and Orientation Lab Project and Major Project of Qingyuan Innovation Laboratory (No. 00621005 and 00122006).References
- L. Guo, Q. Liu, Y. Liu, Z. Chen, Y. Jiang, H. Jin, T. Zhou, J. Yang and Y. Liu, Nano Energy, 2022, 92, 106707 CrossRef CAS.
- J. T. Ren, L. Chen, H. Y. Wang, W. W. Tian and Z. Y. Yuan, Energy Environ. Sci., 2024, 17, 49–113 RSC.
- Krishankant, Aashi, A. Jain, J. Sharma, R. Rani, C. Bera and V. Bagchi, ACS Appl. Energy Mater., 2024, 7, 1027–1036 CrossRef CAS.
- Y. Zhang, J. Huang and Y. Lai, Chin. J. Catal., 2023, 54, 161–177 CrossRef CAS.
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 CrossRef CAS PubMed.
- J. Chen, H. Chen, H. Yin, H. He, Z. Wang, D. Yu, J. Liang, Y. Huang, L. Qin and D. Chen, Chem. Eng. J., 2023, 477, 146885 CrossRef CAS.
- Y. Zhang, Y. Lei, Y. Yan, W. Cai, J. Huang, Y. Lai and Z. Lin, Appl. Catal., B, 2024, 353, 124064 CrossRef CAS.
- Z. Li, Y. Yao, S. Sun, J. Liang, S. Hong, H. Zhang, C. Yang, X. Zhang, Z. Cai, J. Li, Y. Ren, Y. Luo, D. Zheng, X. He, Q. Liu, Y. Wang, F. Gong, X. Sun and B. Tang, Angew. Chem., Int. Ed., 2024, 63, e202316522 CrossRef CAS PubMed.
- W. Liao, L. Qi, Y. Wang, J. Qin, G. Liu, S. Liang, H. He and L. Jiang, Adv. Funct. Mater., 2021, 31, 2009195 Search PubMed.
- Y. Zhang, D. Ma, Y. Lei, T. Zhu, J. Hu, Y. Tang, Z. Chen, J. Huang, Y. Lai and Z. Lin, Nano Energy, 2023, 117, 108896 CrossRef CAS.
- C. J. Huang, H. M. Xu, T. Y. Shuai, Q. N. Zhan, Z. J. Zhang and G. R. Li, Small, 2023, 19, 2301130 CrossRef CAS PubMed.
- Y. Zhang, W. Zhang, Y. Lei, J. Huang, Z. Lin and Y. Lai, Chem. Commun., 2023, 59, 14395–14398 RSC.
- J. Zhao, Y. Zhang, H. Guo, J. Ren, H. Zhang, Y. Wu and R. Song, Chem. Eng. J., 2022, 433, 134497 CrossRef CAS.
- S. F. Zai, X. Y. Gao, C. C. Yang and Q. Jiang, Adv. Energy Mater., 2021, 11, 2101266 CrossRef CAS.
- M. Li, X. Wang, K. Liu, Z. Zhu, H. Guo, M. Li, H. Du, D. Sun, H. Li, K. Huang, Y. Tang and G. Fu, Adv. Energy Mater., 2023, 13, 2301162 CrossRef CAS.
- M. Zhong, J. Yang, M. Xu, S. Ren, X. Chen, C. Wang, M. Gao and X. Lu, Small, 2024, 20, 2304782 CrossRef CAS PubMed.
- Q. Zhao, X. Li, C. Zhang, B. Ma, D. Jia, T. Chigan and P. Yang, J. Colloid Interface Sci., 2023, 652, 1250–1260 CrossRef CAS PubMed.
- Z. Tu, X. Liu, D. Xiong, J. Wang, S. Gong, C. Xu, D. Wu and Z. Chen, Chem. Eng. J., 2023, 475, 146253 CrossRef CAS.
- S. Jeong, U. Kim, S. Lee, Y. Zhang, E. Son, K. J. Choi, Y. K. Han, J. M. Baik and H. Park, ACS Nano, 2024, 18, 7558–7569 CrossRef CAS PubMed.
- Q. Li, F. Huang, S. Li, H. Zhang and X. Y. Yu, Small, 2022, 18, 2104323 CrossRef CAS PubMed.
- Q. Zeng, S. Tian, G. Liu, H. Yang, X. Sun, D. Wang, J. Huang, D. Yan and S. Peng, ACS Appl. Mater. Interfaces, 2022, 14, 6958–6966 CrossRef CAS PubMed.
- P. Chen, N. Zhang, S. Wang, T. Zhou, Y. Tong, C. Ao, W. Yan, L. Zhang, W. Chu, C. Wu and Y. Xie, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6635–6640 CrossRef CAS PubMed.
- G. Zhou, Y. Chen, H. Dong, L. Xu, X. Liu, C. Ge, D. Sun and Y. Tang, Int. J. Hydrogen Energy, 2019, 44, 26338–26346 CrossRef CAS.
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li, X. Zhong and J. Wang, Adv. Funct. Mater., 2023, 33, 2214488 CrossRef CAS.
- D. Yang, L. Cao, L. Feng, J. Huang, K. Kajiyoshi, Y. Feng, Q. Liu, W. Li, L. Feng and G. Hai, Appl. Catal., B, 2019, 257, 117911 CrossRef CAS.
- X. Han, L. Zhao, Y. Liang, J. Wang, Y. Long, Z. Zhou, Y. Zhang, Y. Li and J. Ma, Adv. Energy Mater., 2022, 12, 2202747 CrossRef CAS.
- X. Zhao, X. Wang, L. Chen, X. Kong, Z. Li, Y. Zhao, Z. Wu, T. Wang, Z. Liu and P. Yang, J. Energy Chem., 2022, 920, 116630 CAS.
- M. Li, X. Wu, K. Liu, Y. Zhang, X. Jiang, D. Sun, Y. Tang, K. Huang and G. Fu, J. Energy Chem., 2022, 69, 506–515 CrossRef CAS.
- Y. Cheng, L. Zhu and Y. Gong, Int. J. Hydrogen Energy, 2024, 58, 416–425 CrossRef CAS.
- M. Zhang, S. Xu, M. Boubeche, D. Decarolis, Y. Huang, B. Liu, E. K. Gibson, X. Li, Y. Wang, H. Luo, C. R. A. Catlow and K. Yan, Green Chem., 2022, 24, 9570–9578 RSC.
- Z. Lian, F. Wu, J. Zi, G. Li, W. Wang and H. Li, J. Am. Chem. Soc., 2023, 145, 15482–15487 CrossRef CAS PubMed.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- Z. Li, X. Li, H. Zhou, Y. Xu, S. M. Xu, Y. Ren, Y. Yan, J. Yang, K. Ji, L. Li, M. Xu, M. Shao, X. Kong, X. Sun and H. Duan, Nat. Commun., 2022, 13, 5009 CrossRef CAS PubMed.
- Z. Ma, H. Wang, H. Ma, S. Zhan and Q. Zhou, Fuel, 2022, 315, 123279 CrossRef CAS.
- W. Wang, Y. B. Zhu, Q. Wen, Y. Wang, J. Xia, C. Li, M. W. Chen, Y. Liu, H. Li, H.-A. Wu and T. Zhai, Adv. Mater., 2019, 31, 1900528 CrossRef PubMed.
- X. Ao, Y. Gu, C. Li, Y. Wu, C. Wu, S. Xun, A. Nikiforov, C. Xu, J. Jia, W. Cai, R. Ma, K. Huo and C. Wang, Appl. Catal., B, 2022, 315, 121586 CrossRef CAS.
- Z. Ji, Y. Song, S. Zhao, Y. Li, J. Liu and W. Hu, ACS Catal., 2022, 12, 569–579 CrossRef CAS.
- H. Qin, Y. Ye, J. Li, W. Jia, S. Zheng, X. Cao, G. Lin and L. Jiao, Adv. Funct. Mater., 2023, 33, 2209698 CrossRef CAS.
- J. Zhang, J. Zhu, L. Kang, Q. Zhang, L. Liu, F. Guo, K. Li, J. Feng, L. Xia, L. Lv, W. Zong, P. R. Shearing, D. J. L. Brett, I. P. Parkin, X. Song, L. Mai and G. He, Energy Environ. Sci., 2023, 16, 6015–6025 RSC.
- Y. Zhu, C. Liu, S. Cui, Z. Lu, J. Ye, Y. Wen, W. Shi, X. Huang, L. Xue, J. Bian, Y. Li, Y. Xu and B. Zhang, Adv. Mater., 2023, 35, 2301549 CrossRef CAS PubMed.
- Y. Zhou, Y. Wang, D. Kong, Q. Zhao, L. Zhao, J. Zhang, X. Chen, Y. Li, Y. Xu and C. Meng, Adv. Funct. Mater., 2023, 33, 2210656 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00119b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |