
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient CO2-to-CO conversion in dye-sensitized photocatalytic systems enabled by electrostatically-driven catalyst binding†
Vasilis
Nikolaou
 *a,
Palas Baran
Pati
a,
Hélène
Terrisse
*b,
Marc
Robert
*cd and
Fabrice
Odobel
*a
*a,
Palas Baran
Pati
a,
Hélène
Terrisse
*b,
Marc
Robert
*cd and
Fabrice
Odobel
*a
aNantes Université, CNRS, CEISAM, UMR 6230, F-44000 Nantes, France. E-mail: vasileios.nikolaou@univ-nantes.fr; fabrice.odobel@univ-nantes.fr
bInstitut des Matériaux de Nantes Jean Rouxel, IMN, Nantes Université, CNRS, F-44000 Nantes, France. E-mail: Helene.Terrisse@cnrs-imn.fr
cLaboratoire d’Electrochimie Moléculaire, Université Paris Cité, CNRS, F-75006 Paris, France. E-mail: marc.robert@sorbonne-universite.fr
dInstitut Universitaire de France (IUF), Paris, F-75005, France
First published on 23rd September 2024
Abstract
The development of noble metal-free dye-sensitized photocatalytic systems (DSPs) for CO2-to-CO conversion remains limited. Current literature primarily focuses on a single strategy: the simultaneous loading of both the photosensitizer (PS) and the catalyst (CAT) onto titanium dioxide nanoparticles (TiO2 NPs) using anchoring groups. Here, we introduce an innovative method through immobilizing a positively-charged molecular CAT onto negatively-charged PS–TiO2 NPs. Our approach yields promising results, including near-complete CO2-to-CO conversion (∼100% CO) and exceptional stability, achieving 1658 turnover numbers versus the CAT and an apparent quantum yield efficiency (AQY) of 16.9%.
Broader contextArtificial photosynthetic systems for producing chemicals, fuels, or materials offer a straightforward and cost-effective approach to CO2 recycling driven by solar energy. While these technologies are still in their early stages, they hold the potential for large-scale production of commodity chemicals and fuels in the future. Hybrid catalytic systems, which combine light-absorbing nano-objects like quantum dots and nanoparticles with catalysts such as molecular catalysts, have garnered significant interest. These systems efficiently convert CO2 into various products using catalytic processes powered by solar irradiation. However, designing systems that balance efficient photon absorption, catalytic efficiency, and long-term stability remains a major challenge. This work introduces an innovative strategy that leverages electrostatic interactions between negatively charged TiO2 nanoparticles, functionalized with a molecular photosensitizer, and a positively charged molecular catalyst, using only abundant metals like zinc and iron. Such systems demonstrate an apparent quantum yield (AQY) efficiency of CO formation at 525 nm close to 17% and exhibit good stability under operational conditions—a notable advancement toward achieving higher solar energy conversion efficiency. |
An auspicious strategy to convert carbon dioxide (CO2) emissions involves the utilization of solar-driven catalytic systems to produce valuable chemicals and fuels.1–4 Carbon monoxide (CO) is a valuable product of the CO2 reduction reaction (CO2RR), serving as a precursor in: (i) Fischer–Tropsch synthesis, (ii) methanol production, (iii) hydrocarbon synthesis, and (iv) manufacturing of various industrial materials.5–9 Within the landscape of solar-driven methodologies,10–14 dye-sensitized photocatalytic systems (DSPs) have garnered considerable scientific interest due to their high potential.15 More specifically, DSPs are extremely robust systems that are easy to fabricate and potentially low-cost. They offer exceptional tunability due to the versatile modifications possible in their individual components, including the photosensitizer, catalyst, and semiconductor nanoparticles.16
The common DSPs discussed in the literature consist of titanium dioxide (TiO2) nanoparticles (NPs) functionalized with a molecular catalyst (CAT) to facilitate CO2 reduction, along with a photosensitizer (PS). Upon absorption of a photon, the photosensitizer (PS) undergoes excitation, leading to the injection of electrons (e−) into the conduction band (CB) of TiO2. These electrons are subsequently transferred to the CO2 reduction catalyst (CO2R CAT), initiating the catalytic process. Upon accumulating two electrons, the molecular CAT drives the reduction of CO2 into CO, while the sacrificial electron donor (SED) regenerates the oxidized photosensitizer (PS+), restoring it into its initial ground state (Fig. 1). Several studies have explored various PSs, CATs, and metal-doped TiO2 NPs, and diverse strategies have been proposed to enhance both the stability and the efficiency of DSPs for H2 evolution.15,17,18 However, corresponding research into CO2R remains relatively scarce with pioneering contributions from Kang and co-workers.19–21 Recently, our research team explored the first noble metal-free DSPs for CO2-to-CO conversion.22 In all these examples, the catalyst was anchored to the TiO2 nanoparticles through a covalent linkage facilitated by either a carboxylic or phosphonic group. There is obviously significant room for further improving such systems, as well as for exploring new directions and concepts within this area of research.
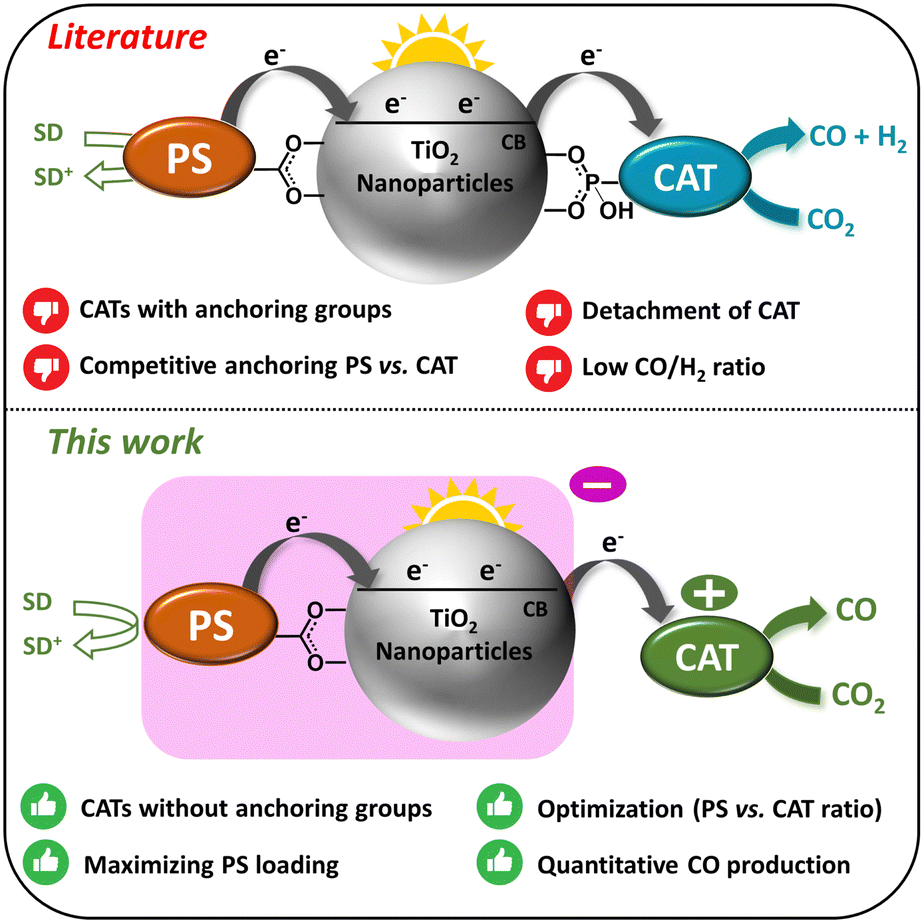 | ||
| Fig. 1 DSPs for CO2-to-CO reduction: (upper panel) previous examples from the literature; (lower panel) our strategy comprising a positively-charged molecular catalyst (CAT) and negatively-charged dye-sensitized TiO2 nanoparticles (PS–TiO2 NPs). | ||
Herein, we present an innovative strategy involving the utilization of electrostatic interactions between negatively-charged TiO2 NPs, functionalized with a PS, and a positively-charged molecular CAT. Although electrostatic interactions have been previously employed to position redox mediators near sensitizers in dye-sensitized solar cells (DSSCs) and photocatalytic systems,23–28 this technique has not yet been applied to the fabrication of DSPs, specifically for assembling catalysts onto sensitized TiO2 nanoparticles. As depicted in Fig. 1, this approach offers numerous advantages over the conventional CO2-to-CO reducing DSPs documented in the literature. Specifically, the absence of an anchoring group on the molecular catalyst eliminates the need for additional synthetic steps for its functionalization and facilitates maximal loading of the photosensitizer (PS). This is in contrast to previously reported DSPs, where the molecular CAT requires functionalization with an anchoring group, leading to competition with the chemical adsorption of the PS moiety. Furthermore, by employing electrostatic interactions, the ratio between the PS and the CAT can be easily tuned. In contrast, optimizing this ratio with the conventional approach depends on the relative affinity of each component's anchoring group (PS and CAT) with the TiO2 NPs. We report in this study novel DSPs, based on the electrostatic interaction approach, that exhibit outstanding CO selectivity (∼100% CO2-to-CO) and remarkable stability (1658 turnovers numbers, TONs vs. CAT), surpassing previous noble metal-free DSPs,22 where the PS and the CAT were functionalized with typical anchoring groups and covalently grafted to TiO2 particles.
Prior to photocatalytic studies, zeta potential experiments were conducted on the TiO2 NPs to verify their electrostatic properties (see ESI,† for detailed methodology). The experiments were performed in dimethylformamide (DMF) and acetonitrile (ACN) solutions, showing that the nanoparticles exhibit a negative charge, with zeta potential (ζ) = −21 ± 2 mV in DMF, and ζ = −29 ± 5 mV in ACN (entries 1 and 4, Table S1, ESI†). The anchoring of the PS further increases the negative zeta potential value of the TiO2 NPs, which reaches ζ = −30 ± 3 mV (DMF) and ζ = −39 ± 1 mV (ACN) (entries 2 and 5, Table S1, ESI†). Noteworthily, these studies were conducted in the presence of the sacrificial electron donor (SED) 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[D]imidazole (BIH), to closely mimic the conditions of photocatalytic experiments. Additionally, infrared spectroscopy experiments were performed to validate the successful synthesis of the DSPs, comparing the spectra of bare TiO2 and the functionalized photocatalytic NPs (see ESI,† and Fig. S2 and S3).
For our solar-driven CO2-to-CO conversion studies, we selected the positively charged Fe(o-TMA) iron porphyrin as a catalyst, one of the most efficient molecular CATs in the CO2RR.29–31 Moreover, this molecular catalyst has been extensively studied by various research groups, consistently demonstrating that the CO produced originates from CO2 rather than any side reactions or decomposition.29–33 Regarding the choice of the PS, three different dyes were employed: D35, N719 and ZnP (as illustrated in Fig. 2). While each of these dyes has demonstrated notable performance in TiO2-based solar cell34–42 and water splitting43–47 applications, it is worth highlighting that ZnP has proved to be an efficient PS in the CO2R, when anchored on the photocathodes48,49 and on TiO2-NPs.22 Our initial photocatalytic CO2 reduction experiments demonstrated that ZnP outperformed both the N719- and D35-sensitized systems reaching 124 mmol of CO per g of CAT in 96 hours of irradiation (Fig. S5 and ESI,† for experimental details). Remarkably, in all cases, no traces of H2 were detected, indicating quantitative CO2-to-CO conversion (∼100% selectivity).
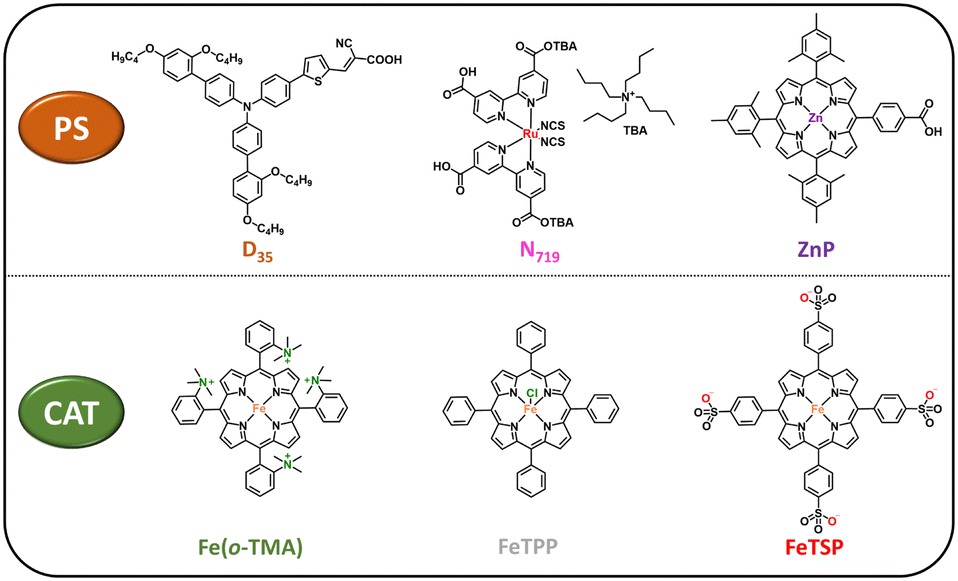 | ||
| Fig. 2 Photosensitizers (PS, upper panel) and molecular catalysts (CAT, lower panel) explored in this work. | ||
All three dyes exhibit absorption bands in the green region of the visible spectrum, but with significantly different absorption coefficients (Fig. S6a, ESI†), even though they can all be effectively excited by the green LED lamp (Fig. S6b, ESI†). Specifically, D35 exhibits the highest light collection efficiency, followed by N719, with ZnP showing the lowest absorption in this region. All three dyes have redox properties suitable for the two key processes: (i) electron injection into the conduction band (CB) of TiO2 and (ii) dye regeneration by the sacrificial electron donor (SED), BIH. As outlined in Table S2 (ESI†), the dyes exhibit similar driving forces for both electron injection (ΔGinj) and dye regeneration (ΔGreg). Consequently, the differences in redox and absorption properties alone are unlikely to account for the better photocatalytic performance of ZnP. Instead, this enhanced performance is likely due to other factors, such as the molecular organization on the TiO2 surface. Our current studies in DSPs for CO2 reduction demonstrate that ZnP consistently outperforms other sensitizers.50 This suggests that ZnP may possess a structural arrangement on the TiO2 surface that promotes more effective charge separation and transfer processes, although investigating these details is beyond the scope of this work. Overall, our observations strongly suggest that the superior performance of ZnP is not due to its redox properties and absorption characteristics, but rather to its specific organization, rendering the electronic interactions between TiO2 and the catalyst more favourable.
To assess the impact of the positively-charged molecular catalyst, we performed additional photocatalytic experiments upon using a neutral (FeTPP) and a negatively-charged (FeTSP) iron–porphyrin (Fig. 2). In both cases, the DSP studies indicated no conversion of CO2 to either CO or H2, further illustrating the key role of the electrostatic interactions between the negatively-charged ZnP–TiO2 NPs and the positively-charged Fe(o-TMA) CAT. Additional evidence supporting this conclusion was obtained from supplementary zeta potential experiments conducted in the presence of Fe(o-TMA). The negative charge of the ZnP–TiO2 nanoparticles (ζ = −30 ± 3 mV in DMF and ζ = −39 ± 1 mV in ACN) was reversed to positive upon the addition of Fe(o-TMA) to the solution (ζ = +21 ± 5 mV (DMF) and ζ = +24 ± 1 mV (ACN), entries 3 and 6, Table S1, ESI†), confirming the strong interaction (and adsorption) of the CAT onto the ZnP–TiO2 NPs (Fig. 3 and Fig. S4, ESI†).
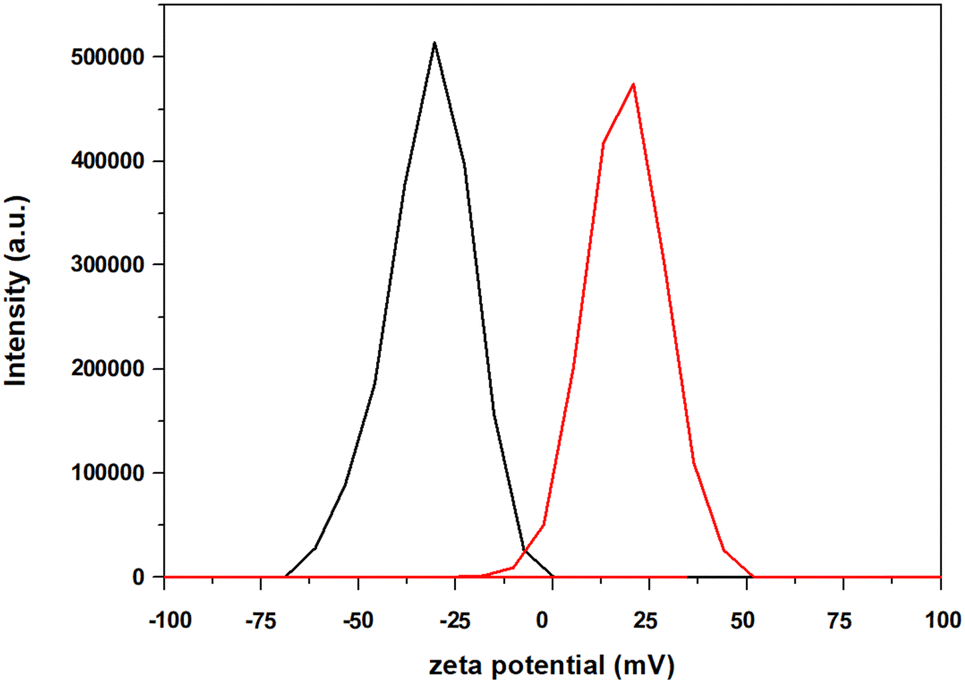 | ||
| Fig. 3 Zeta potential determination of ZnP@TiO2 (black) and ZnP@TiO2 + Fe(o-TMA) (red) in DMF solution containing 50 mM of BIH. See ESI,† for Experimental details. | ||
Encouraged by these results, we then optimized our DSPs. In our initial experiments, DMF was employed as the solvent and BIH as the SED. In a typical run, the ZnP–TiO2 NPs were dispersed in 4 mL of DMF solution, containing BIH (50 mM) and Fe(o-TMA) CAT. To determine the optimal ratio between the PS and the CAT, various combinations were investigated, assessing their corresponding TONs, selectivity, and CO production rates (Table S3, ESI†). First, control experiments revealed no catalytic activity (neither CO nor H2) in the absence of: (i) PS (entry 1, Table S3, ESI†), (ii) CAT (entry 2, Table S3, ESI†), (iii) SED (entry 3, Table S3, ESI†), and (iv) light irradiation (entry 4, Table S3, ESI†). Noteworthily, the combination of 0.24 μmol (0.2 mg) of ZnP and 0.032 μmol (0.05 mg) of Fe(o-TMA) resulted in quantitative CO selectivity (entry 5, Table S3, ESI†). Other tested PS and CAT ratios also exhibited high CO selectivity as well, ranging from 94% to 98% (entries 6–9, Table S3, ESI†). By decreasing the amount of Fe(o-TMA) to 0.016 μmol (0.025 mg), both stability (419 TONs) and CO production (268 mmol g−1) were significantly enhanced (entry 6, Table S3, ESI†). To investigate the effect of different PS amounts on electron transfer, we compared the results from entries 6 and 9 (Table S3, ESI†). In these experiments, all conditions were kept identical except for the PS concentration, which was 0.24 μmol in entry 6 and 0.12 μmol in entry 9. The photocatalytic results indicate that increasing the amount of PS does not hinder electron transfer. The highest CO production though in terms of μmols of CO, was attained using 0.12 μmol of ZnP (0.1 mg) and 0.032 μmol (0.05 mg) of Fe(o-TMA) (entry 8, Table S3, ESI†).
In subsequent experiments, we examined the influence of the proton (H+) source upon adding trifluoroethanol (TFE) and phenol. As listed in Table 1 (entries 1–3) the addition of either H+ source enhanced the photocatalytic parameters (CO yield, CO/H2 ratio, TONs, and CO production in mmol g−1). However, phenol was a more efficient H+ source compared to TFE, yielding 30.1 μmol of CO, with nearly 100% selectivity, 939 TONs, and 601 mmol g−1 of CO (entry 3, Table 1). Phenol serves as a more effective proton donor compared to weaker acids like TFE.51,52 Phenol can also engage more effectively in hydrogen bonding, whereas TFE's bulkier –CF3 group can introduce steric hindrance, diminishing its ability to stabilize intermediates and thereby reducing catalytic efficiency. The better performance of phenol as a proton source in our CO2-to-CO DSPs is attributed to a combination of its moderate acidity, and effective hydrogen bonding.
| Entry | Solvent | H+ sourcea | H2b | COb | TONs | mmol g−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
|---|---|---|---|---|---|---|
| a 0.1 M. b In μmol. c mmol of CO per gram of CAT. d 0.12 μmol (0.1 mg) of PS and 0.016 μmol of CAT (0.025 mg, C = 4 μM). | ||||||
| 1 | DMF | — | 0.19 | 8.0 | 250 | 160 |
| 2 | DMF | TFE | 0.16 | 10.3 | 321 | 205 |
| 3 | DMF | Phenol | 0.15 | 30.1 | 939 | 601 |
| 4 | ACN | — | 0.13 | 8.7 | 271 | 174 |
| 5 | ACN | Phenol | 0.80 | 45.7 | 1429 | 915 |
| 6d | ACN | Phenol | 1.48 | 27.1 | 1658 | 530 |
Overall, the optimum conditions for our DSPs were observed when ACN was used as a solvent (Fig. 4), a finding in excellent agreement with the zeta potential experiments (Fig. S4 and Table S1, ESI†). It is worth noting that the ζ potential of ZnP@TiO2 NPs in ACN was significantly more negative (ζ = −39 ± 1 mV) than the ζ potential of ZnP@TiO2 NPs in DMF (ζ = −29 ± 5 mV). In agreement with previous experiments, the addition of phenol significantly improved all the photocatalytic parameters (entries 4 and 5, Table 1), resulting in 1429 TONs and 915 mmol g−1 of CO. Furthermore, reducing the amount of Fe(o-TMA) to 0.016 μmol (0.025 mg, entry 6, Table 1) resulted in 1658 TONs.
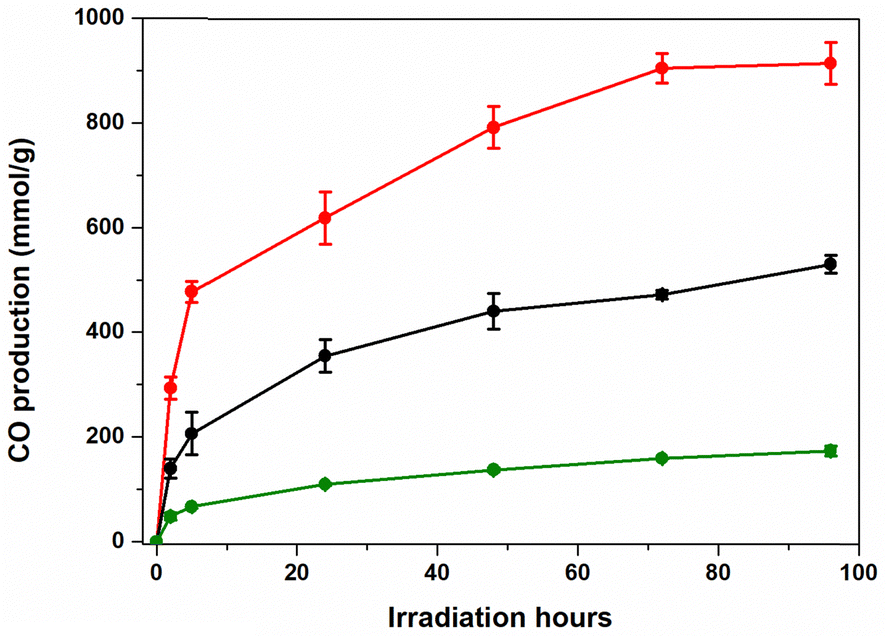 | ||
| Fig. 4 Photocatalytic CO2-to-CO conversion activities of ZnP@TiO2 NPs in 4 mL ACN solution containing 50 mM of BIH; (black): 0.12 μmol (0.1 mg) of PS, 0.032 μmol (0.05 mg) of Fe(o-TMA), (red): 0.12 μmol of PS (0.1 mg), 0.032 μmol (0.05 mg) of Fe(o-TMA) and 0.1 M of phenol, (green): 0.12 μmol of PS (0.1 mg), 0.016 μmol (0.25 mg) of Fe(o-TMA) and 0.1 M of phenol. The reported values are the average of three independently repeated experiments. | ||
To shed light on the factors limiting the reactivity of the DSPs during irradiation, we performed post-photocatalytic studies after 96 hours of irradiation, at which point CO production reaches a plateau. Interestingly, the NPs exhibited a negative charge with a ζ-potential of −36 ± 2 mV, in contrast to their initial value of +24 ± 1 mV before photocatalysis (Fig. S7 and Table S1, ESI†). This implies that the CAT was no longer attached to the surface of the NPs. Additionally, the absorption studies of the solution upon photocatalysis revealed porphyrin-based features (Fig. S8, orange line, ESI†). It remained however possible that the PS, the CAT, or both leached into the solution. Regeneration experiments made after 96 hours of irradiation revealed that the CO production could not be restored by adding either the PS or the CAT and purging the suspension with CO2. These observations suggest that the observed loss of activity may be attributed to both the PS and the CAT either leaching from the TiO2 NPs or undergoing degradation, as illustrated in Fig. S9 (ESI†).
To elucidate the role of TiO2 in our photocatalytic system, we performed additional measurements using the PS and the CAT in solution, without TiO2 nanoparticles. In the absence of TiO2, we detected a small amount of CO (2.4 μmol), which corresponds to low stability (77 TONs) and CO production (49 mmol g−1). This significantly highlights the importance of TiO2 as an electron mediator to shuttle electrons between the PS and the CAT, since the respective experiment (entry 4, Table 1) in the presence of TiO2 demonstrated superior catalytic efficiency, yielding significantly higher photocatalytic values (8.7 μmol of CO, 271 TONs, and 174 mmol g−1). Additionally, in a previous study we demonstrated fast and efficient photoinduced electron transfer from the zinc porphyrin excited state (ZnP*) to TiO2,22 therefore the direct through space electron transfer from the bound ZnP* to FeP is reasonably uncompetitive. As a further control experiment, we tested a mixture of TiO2, PS, and CAT. This experiment produced 2.9 μmol of CO, corresponding to 91 TONs and 58 mmol of CO per gram of CAT, which is significantly lower than the results obtained from the system based on electrostatic interactions (entry 4, Table 1). This notable difference further emphasizes the importance of anchoring both PS and CAT onto TiO2 for efficient electron transfer between them. Overall, TiO2 not only serves as a scaffold for anchoring PS, allowing the formation of a heterogeneous photocatalyst through electrostatic interactions with CAT, but it also plays a crucial role in facilitating the electron transfer processes necessary for efficient photocatalysis.
Conclusions
In our study, the solar-driven CO2-to-CO conversion operates through an oxidative quenching mechanism. Femtosecond transient absorption spectroscopy measurements revealed rapid regeneration of the radical cation of the PS (ZnP˙+) by the SED.8 Initially, photoexcitation of ZnP produces ZnP*, which undergoes ultrafast electron injection (11 ps) into TiO2. The ZnP˙+ formed is quickly regenerated by the SED, while the electron injected into TiO2 is captured by Fe(o-TMA) (see details in Scheme S1, ESI†). Consistent with literature reports, Fe(o-TMA) in our experiments also showed selective and nearly quantitative reduction of CO2 to CO, without forming additional reduction products.9 Importantly, BIH beyond serving as only a SED, could also function as second electron source, due to the formation of the highly reducing BI˙.13In summary, our study unveiled a strategy that combines negatively-charged PS–TiO2 NPs with a positively-charged iron-porphyrin catalyst for selective CO2-to-CO conversion. Our approach offers distinct advantages including easy assembling of the components of the photocatalytic system (Fig. 1). Table S4 and Fig. S10 (ESI†) provide a comparative overview of the TONs, CO production, and CO/H2 ratios between previous DSPs in the literature and this work. It should be noted that variations in light intensity and type of irradiation source across the different reports significantly influence parameters like stability and production rates, rendering direct comparisons challenging. Our electrostatic-interaction strategy demonstrates compelling results, evidenced by achieving ca. 1500 TONs and a CO production of 915 mmol g−1 (per g of CAT). Furthermore, the apparent quantum yield efficiency (AQY) of CO formation at 525 nm was determined to be 16.9% (see ESI,† for details). This value is higher compared to similar DSPs in the literature, which exhibit AQY values typically ranging from 0.1% to 3.9% (Table S4, ESI†).
In recent years, a plethora of positively-charged complexes have been employed as molecular catalysts in CO2 reduction. This includes iron-, cobalt-, and nickel-polypyridyl complexes,53 as well as noble metal-free dinuclear molecular catalysts.54 Currently, we are exploring additional combinations of charged nanoparticles and catalysts, aiming to develop more efficient systems and with the objective to eliminate the sacrificial reagent, so as to position DSPs as a viable solution in the field of CO2 photoconversion.
Data availability
The data supporting the findings of this study are available within the paper and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by HORIZON TMA MSCA Postdoctoral Fellowships – European Fellowships (HORIZON-TMA-MSCA-PF-EF) action program under the call MSCA Postdoctoral Fellowships 2021 ((HORIZON-MSCA-2021-PF-01)) to V. N., grant agreement no. 101064765, SOLAR-CAT. This study also received financial support under the EUR LUMOMAT project and the Investments for the Future program ANR-18-EURE-0012. Partial financial support to M. R. from the Institut Universitaire de France (IUF) is warmly acknowledged.Notes and references
- A. Gulzar, A. Gulzar, M. B. Ansari, F. He, S. Gai and P. Yang, Chem. Eng. J. Adv., 2020, 3, 100013 CrossRef CAS
.
- M. Robert, ACS Energy Lett., 2016, 1, 281–282 CrossRef CAS
- J. C. Wood, Z. Yuan and B. Virdis, Curr. Opin. Electrochem., 2023, 37, 101177 CrossRef CAS
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS PubMed
- T. A. Atsbha, T. Yoon, P. Seongho and C.-J. Lee, J. CO2 Util., 2021, 44, 101413 CrossRef CAS
- A. A. Khan and M. Tahir, J. CO2 Util., 2019, 29, 205–239 CrossRef CAS
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS
- T.-m Su, Z.-z Qin, H.-b Ji, Y.-x Jiang and G. Huang, Environ. Chem. Lett., 2016, 14, 99–112 CrossRef CAS
- L. Yang, A. U. Pawar, R. P. Sivasankaran, D. Lee, J. Ye, Y. Xiong, Z. Zou, Y. Zhou and Y. S. Kang, J. Mater. Chem. A, 2023, 11, 19172–19194 RSC
- Z. X. Bi, R. T. Guo, X. Hu, J. Wang, X. Chen and W. G. Pan, Nanoscale, 2022, 14, 3367–3386 RSC
- X. J. Liu, T. Q. Chen, Y. H. Xue, J. C. Fan, S. L. Shen, M. S. A. A. Hossain, M. A. Amin, L. K. Pan, X. T. Xu and Y. Yamauchi, Coord. Chem. Rev., 2022, 459, 214440 CrossRef CAS
- A. Mustafa, B. G. Lougou, Y. Shuai, Z. J. Wang and H. P. Tan, J. Energy Chem., 2020, 49, 96–123 CrossRef
- N. Nandal and S. L. Jain, Coord. Chem. Rev., 2022, 451, 214271 CrossRef CAS
- Q. S. Wang, Y. C. Yuan, C. F. Li, Z. R. Zhang, C. Xia, W. G. Pan and R. T. Guo, Small, 2023, 19, 2301892 CrossRef CAS PubMed
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC
- G. Reginato, L. Zani, M. Calamante, A. Mordini and A. Dessì, Eur. J. Inorg. Chem., 2020, 899–917 CrossRef CAS
- J.-F. Huang, Y. Lei, T. Luo and J.-M. Liu, ChemSusChem, 2020, 13, 5863–5895 CrossRef CAS PubMed
- L. Zani, M. Melchionna, T. Montini and P. Fornasiero, J. Phys. Energy, 2021, 3, 031001 CrossRef CAS
- M. S. Choe, S. Choi, H. S. Lee, B. Chon, J. Y. Shin, C. H. Kim, H. J. Son and S. O. Kang, ACS Appl. Mater. Interfaces, 2022, 14, 50718–50730 CrossRef CAS PubMed
- S. Choi, Y. J. Kim, S. Kim, H. S. Lee, J. Y. Shin, C. H. Kim, H. J. Son and S. O. Kang, ACS Appl. Energy Mater., 2022, 5, 10526–10541 CrossRef CAS
- H. J. Son, C. Pac and S. O. Kang, Acc. Chem. Res., 2021, 54, 4530–4544 CrossRef CAS PubMed
- V. Nikolaou, C. Govind, E. Balanikas, J. Bharti, S. Diring, E. Vauthey, M. Robert and F. Odobel, Angew. Chem., Int. Ed., 2024, 63, e202318299 CrossRef CAS PubMed
- H. Kumagai, R. Aoyagi, K. Kato, A. Yamakata, M. Kakihana and H. Kato, ACS Appl. Energy Mater., 2021, 4, 2056–2060 CrossRef CAS
- S. Nishioka, K. Hojo, L. Xiao, T. Gao, Y. Miseki, S. Yasuda, T. Yokoi, K. Sayama, T. E. Mallouk and K. Maeda, Sci. Adv., 2022, 8, eadc9115 CrossRef CAS PubMed
- L. Casarin, W. B. Swords, S. Caramori, C. A. Bignozzi and G. J. Meyer, Inorg. Chem., 2017, 56, 7324–7327 CrossRef CAS PubMed
- L. Mistry, L. Le-Quang, G. Masdeu, W. Björkman, H. Härelind and M. Abrahamsson, ChemCatChem, 2022, 14, e202200897 CrossRef CAS
- N. Yoshimura, A. Kobayashi, M. Yoshida and M. Kato, Chem. – Eur. J., 2020, 26, 16939–16946 CrossRef CAS PubMed
- W. B. Swords, G. Li and G. J. Meyer, Inorg. Chem., 2015, 54, 4512–4519 CrossRef CAS PubMed
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed
- V. C. C. Wang, ACS Catal., 2021, 11, 8292–8303 CrossRef CAS
- D. J. Martin and J. M. Mayer, J. Am. Chem. Soc., 2021, 143, 11423–11434 CrossRef CAS PubMed
- D. J. Martin, B. Q. Mercado and J. M. Mayer, Inorg. Chem., 2021, 60, 5240–5251 CrossRef CAS PubMed
- A. K. Surendran, G. L. Tripodi, E. Pluhařová, A. Y. Pereverzev, J. P. J. Bruekers, J. A. A. W. Elemans, E. J. Meijer and J. Roithová, Nat. Sci., 2023, 3, e20220019 CrossRef CAS
- D. P. Hagberg, X. Jiang, E. Gabrielsson, M. Linder, T. Marinado, T. Brinck, A. Hagfeldt and L. Sun, J. Mater. Chem., 2009, 19, 7232–7238 RSC
- X. Jiang, T. Marinado, E. Gabrielsson, D. P. Hagberg, L. Sun and A. Hagfeldt, J. Phys. Chem. C, 2010, 114, 2799–2805 CrossRef CAS
- J.-H. Yum, T. W. Holcombe, Y. Kim, J. Yoon, K. Rakstys, M. K. Nazeeruddin and M. Grätzel, Chem. Commun., 2012, 48, 10727–10729 RSC
- D. M. Niedzwiedzki, Phys. Chem. Chem. Phys., 2021, 23, 6182–6189 RSC
- K. Portillo-Cortez, A. Martínez, A. Dutt and G. Santana, J. Phys. Chem. A, 2019, 123, 10930–10939 CrossRef CAS PubMed
- B. Selvaraj, G. Shanmugam, S. Kamaraj, A. Gunasekeran and A. Sambandam, Inorg. Chem., 2021, 60, 1937–1947 CrossRef CAS PubMed
- H. Hayashi, T. Higashino, Y. Kinjo, Y. Fujimori, K. Kurotobi, P. Chabera, V. Sundström, S. Isoda and H. Imahori, ACS Appl. Mater. Interfaces, 2015, 7, 18689–18696 CrossRef CAS PubMed
- S. Hayashi, M. Tanaka, H. Hayashi, S. Eu, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 15576–15585 CrossRef CAS
- H. Imahori, Y. Matsubara, H. Iijima, T. Umeyama, Y. Matano, S. Ito, M. Niemi, N. V. Tkachenko and H. Lemmetyinen, J. Phys. Chem. C, 2010, 114, 10656–10665 CrossRef CAS
- B. D. Sherman, M. V. Sheridan, K.-R. Wee, S. L. Marquard, D. Wang, L. Alibabaei, D. L. Ashford and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 16745–16753 CrossRef CAS PubMed
- S. Gonuguntla, A. Tiwari, S. Madanaboina, G. Lingamallu and U. Pal, J. Hydrogen Energy, 2020, 45, 7508–7516 CrossRef CAS
- X. Yao, P.-Y. Ho, S.-C. Yiu, S. Suramitr, W.-B. Li, C.-L. Ho and S. Hannongbua, Dyes Pigm., 2022, 205, 110508 CrossRef CAS
- E. Agapaki, K. Ladomenou, V. Nikolaou and A. G. Coutsolelos, J. Porphyrins Phthalocyanines, 2023, 27, 479–489 CrossRef CAS
- V. Nikolaou, G. Charalambidis, G. Landrou, E. Nikoloudakis, A. Planchat, R. Tsalameni, K. Junghans, A. Kahnt, F. Odobel and A. G. Coutsolelos, ACS Appl. Energy Mater., 2021, 4, 10042–10049 CrossRef CAS
- Y. Kou, S. Nakatani, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, J. Catal., 2014, 310, 57–66 CrossRef CAS
- R. Nakazato, Y. Kou, D. Yamamoto, T. Shimada, T. Ishida, S. Takagi, H. Munakata, K. Kanamura, H. Tachibana and H. Inoue, Res. Chem. Intermed., 2021, 47, 269–285 CrossRef CAS
- Unpublished results.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC
- J.-W. Wang, D.-C. Zhong and T.-B. Lu, Coord. Chem. Rev., 2018, 377, 225–236 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00156g |
| This journal is © The Royal Society of Chemistry 2024 |