
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSalt precipitation and water flooding intrinsic to electrocatalytic CO2 reduction in acidic membrane electrode assemblies: fundamentals and remedies
Qianqian
Bai†
a,
Likun
Xiong†
b,
Yongjia
Zhang
a,
Mutian
Ma
a,
Zhenyang
Jiao
a,
Fenglei
Lyu
 *ac,
Zhao
Deng
ac and
Yang
Peng
*ac
*ac,
Zhao
Deng
ac and
Yang
Peng
*ac
aSoochow Institute for Energy and Material Innovations, College of Energy, Soochow University, Suzhou 215006, P. R. China. E-mail: fllv@suda.edu.cn; ypeng@suda.edu.cn
bSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai, 201418, China
cJiangsu Key Laboratory for Advanced Negative Carbon Technologies, Soochow University, Suzhou 215123, China
First published on 3rd September 2024
Abstract
Renewable electricity powered electrocatalytic CO2 reduction (eCO2R) is an emerging carbon-negative technology that upgrades CO2 into valuable chemicals and simultaneously stores intermittent renewable energy. eCO2R in anion exchange membrane (AEM)-based membrane electrode assemblies (MEAs) has witnessed high faradaic efficiency (FE). But severe CO2 crossover in AEMs results in low CO2 single-pass conversion (SPCCO2) and burdens the energy-intensive CO2 separation process. Utilizing cation exchange membranes (CEMs) and acidic anolytes, eCO2R in acidic MEAs is capable of addressing the CO2 crossover issue and overcoming the SPCCO2 limits in their AEM counterparts. Alkali metal cations such as K+/Cs+ are always adopted in acidic MEAs to suppress the competing hydrogen evolution reaction (HER) and boost eCO2R kinetics. However, K+/Cs+ accumulates and precipitates in the form of carbonate/bicarbonate salts in the cathode, which accelerates water flooding, deteriorates the gas-electrode–electrolyte interface, and limits the durability of acidic eCO2R MEAs to a few hours. In this mini-review, we discuss the fundamentals of salt precipitation and water flooding and propose potential remedies including inhibiting K+/Cs+ accumulation, decreasing local CO32−/HCO3− concentration, and water management in gas diffusion electrodes (GDEs). We hope that this mini-review will spur more insightful solutions to address the salt precipitation and water flooding issues and push acidic eCO2R MEAs toward industrial implementations.
Broader contextElectrocatalytic CO2 reduction (eCO2R) represents an emerging carbon-negative technology for the production of valuable chemicals from CO2, H2O, and renewable electricity. Benefiting from short CO2 diffusion length (∼50 nm), CO2 electrolysis in membrane electrode assemblies (MEAs) based on gas diffusion electrodes (GDEs) greatly boosts up the current density of eCO2R. MEAs utilizing anion exchange membranes (AEMs) have attained high eCO2R faradaic efficiency, but suffer from severe CO2 crossover and low carbon utilization efficiency (≤50%), which burdens the energy-intensive CO2 separation process. Utilizing cation exchange membranes (CEMs) and acidic electrolytes, acidic eCO2R MEAs address the CO2 crossover issue. However, alkali cations such as K+ and Cs+ are adopted to suppress the competing hydrogen evolution reaction (HER) and boost the eCO2R kinetics, which causes severe salt precipitation and water flooding and seriously limits the durability of acidic MEAs to a few hours. Herein, we discuss the fundamentals of salt precipitation and water flooding in acidic eCO2R MEAs and propose remedies that potentially overcome these issues, including mitigating/eliminating K+/Cs+ accumulation, reducing local CO32−/HCO3− concentration, and innovating the GDE structure. This mini-review may spur more inspiration to address salt precipitation and water flooding issues in acidic eCO2R MEAs. |
1. Introduction
Electrocatalytic CO2 reduction (eCO2R) driven by renewable electricity from sunlight and wind holds great promise to upcycle CO2 into valuable chemicals and simultaneously store intermittent renewable energy into chemical bonds.1–3 In virtue of the high energy efficiency, high production rate as well as feasible scalability at the industrial scale, CO2 electrolysis in zero-gap membrane electrode assemblies (MEAs) based on gas diffusion electrodes (GDEs) has drawn great attention.4–8 Benefiting from the much shorter CO2 diffusion length in GDEs (∼50 nm) than that in conventional H-type cells (∼50 μm),9 industrially relevant current densities (≥200 mA cm−2) with appreciable faradaic efficiencies (>90% for CO and >80% for C2H4) have been attained in CO2 MEAs utilizing anion exchange membranes (AEMs) and neutral anolytes (e.g. aqueous KHCO3/CsHCO3 solution).10,11 However, severe CO2 loss (≥50%) brought by carbonate/bicarbonate (CO32−/HCO3−) crossover in AEMs remains a significant challenge. As a result, the theoretical limits of the CO2 single-pass conversion (SPCCO2) in AEM-based MEAs are merely 50% for CO and 25% for C2H4, which seriously burdens the energy-intensive CO2 separation and purification process.12Conventional CO2 capture consumes energy from 170 to 390 kJ molCO2−1 depending on the CO2 source, which translates into a voltage loss of 0.88 V caused by CO2 recapture from the anode even with minimum energy consumption of 170 kJ mol−1 and 100% faradaic efficiency.13–15 The energy penalty caused by anode CO2 recapture for C2H4 is ∼1.55 times that of the C2H4 Gibbs free energy of reaction even with the maximum 25% SPCCO2, which makes the neutral MEAs untenable.16 Though techno-economic assessments demonstrate that the industrially relevant benchmarks of SPCCO2 are 30% for C1 and 15% for C2 products with CO2 crossover, the same model also shows an apparent cost reduction if no CO2 crossover exists.17 Therefore, it is imperative to solve the CO2 crossover issue to eliminate the energy cost for anode CO2 recapture. It is also noteworthy that SPCCO2 should not be the only target for eCO2R MEAs. 100% SPCCO2 may not be required since such high SPCCO2 brings additional issues of competing HER under the low CO2 available conditions.16,18 Maximizing the concentration of targeted eCO2R products in the cathode downstream, i.e., maximizing SPCCO2 without sacrificing the eCO2R selectivity, is a more reasonable and practical merit.19
eCO2R MEAs utilizing cation exchange membranes (CEMs) and acidic anolytes, namely acidic MEAs, are capable of addressing the CO2 crossover issue and potentially overcoming the theoretical SPCCO2 limit in neutral MEAs. In acidic MEAs, CO32−/HCO3− from the cathode catalyst layer (CL) reacts with H+ from the CEM and generates CO2 and H2O at the CL/CEM interface, eliminating the CO2 crossover to the anode.20
In zero-gap eCO2R MEAs, the cathode, ion exchange membrane, and anode are intimately assembled. The ion mobility of transported ionic species and ionic conductivity of the membrane dominate the ohmic loss between the cathode and anode. In acidic eCO2R MEAs, H+ exhibits much higher ionic mobility (3.62 × 10−7 m2 s−1 V−1) than CO32− (7.46 × 10−8 m2 s−1 V−1) in neutral eCO2R MEAs, rendering low ohmic loss in the ion exchange membrane and high energy efficiency.21 More importantly, the commercially available perfluorosulfonic acid-based CEMs with high ionic conductivity and long-term stability have been widely adopted in proton exchange membrane fuel cells and water electrolyzers, which paves the way for the industrial implementation of acidic eCO2R MEAs.
The proton source for eCO2R in acidic MEAs is H2O. While the proton source for the hydrogen evolution reaction (HER) is H3O+ under low overpotential and H2O under high overpotential. In acidic eCO2R MEAs with abundant H3O+, the Tafel step (*H + *H = H2 + 2*) or Heyrovsky (*H + H+ + e− = H2 + *) step is usually the rate-determining step for the HER with low overpotential, which shows fast kinetics with a low Tafel slope (30 mV dec−1 for Tafel step and 39 mV dec−1 for Heyrovsky step).22 In contrast, the activation of CO2 into *CO2− is both kinetically sluggish with a Tafel slope of 118 mV dec−1, and thermodynamically unfavorable. Moreover, the low CO2 solubility (∼34 mM) and high H3O+ accessibility in acidic electrolytes make the CO2 adsorption on the surface of conventional metallic eCO2R catalysts very difficult.23,24 The fast kinetics of the competing HER in the H+-rich microenvironment therefore leads to low eCO2R selectivity in acidic MEAs.25
In order to suppress the HER, alkali cations such as K+/Cs+ are added into the acidic electrolyte, in which they play a key role in modulating the eCO2R and HER kinetics (Fig. 1).26 Partially desolvated alkali cations stabilize the *CO2− intermediates via short range electrostatic interaction and medium range electric field-dipole interaction. The direct coordination in M+–O(CO2) also enhances the charge transfer to the CO2 unit.25 Alkali cations accumulated in the outer Helmholtz plane (OHP) modify the distribution of the electric field in the double layer and suppress the migration of H3O+.27 Moreover, OH− produced during eCO2R (eqn (1)) neutralizes the H3O+. Therefore, local high pH can be generated when the formation rate of OH− compensates the migration of H3O+.28 The alkali cations also play a key role in C–C bond formation. Partially desolvated alkali cations coordinate with *CO + *CO in the double layer, stabilize the key *OCCO intermediates and lower the energy barrier for C–C bond formation.29,30 Meanwhile, alkali cations induce a hydrophobic microenvironment, which tunes the structure of interfacial water and favors the C–C coupling.31,32
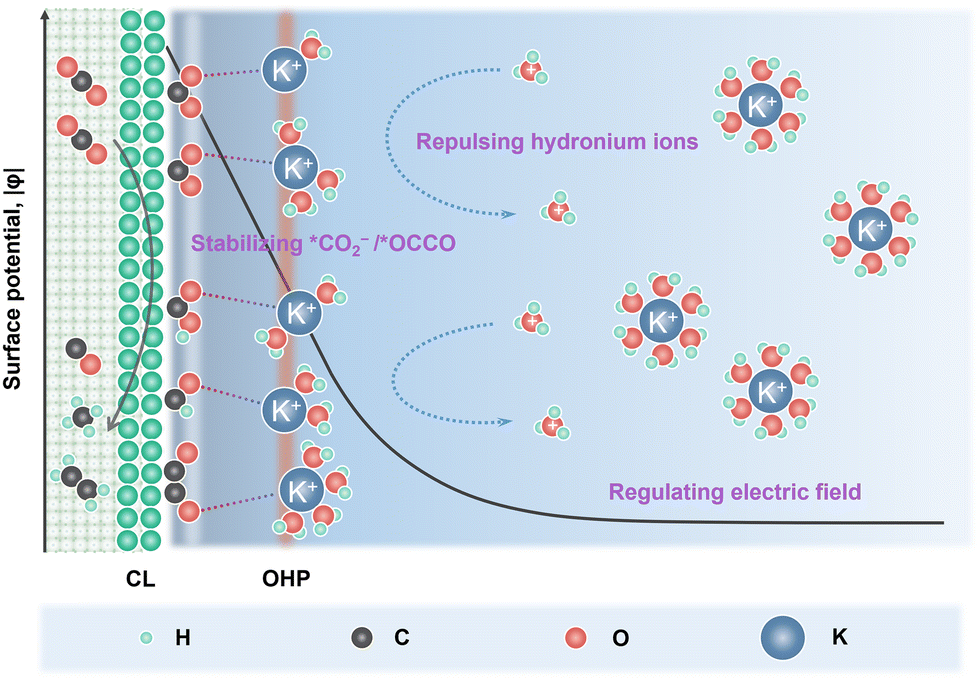 | ||
| Fig. 1 Schematic illustration of the alkali cation effects in acidic eCO2R. | ||
In acidic eCO2R MEAs, K+/Cs+ is usually added to the anolytes. K+/Cs+ is continuously dragged across the CEMs via the electric field and accumulates at the cathode CL, then combines with locally generated CO32−/HCO3−, and forms carbonate/bicarbonate salt precipitation. Salt precipitation destroys the hydrophobicity of GDEs and leads to severe water flooding, which not only blocks the transportation of gaseous CO2, but also deteriorates the gas–catalyst–electrolyte interface and eventually causes MEA failure.33
In this mini-review, we will discuss the origins of salt precipitation and water flooding in acidic eCO2R MEAs from a fundamental viewpoint. Then, we will summarize remedies that potentially overcome salt precipitation and water flooding issues, including mitigating/eliminating K+/Cs+ accumulation, reducing local CO32−/HCO3− concentration, and innovating the structure of GDEs. Perspectives on how to elevate the durability, SPCCO2 and energy efficiency of acidic eCO2R MEAs through the catalyst, ionomer, membrane and their interface engineering will be discussed in the end. We hope that this mini-review will enlighten more insightful thoughts in this field and push eCO2R in acidic MEAs toward industrial implementations.
2. Fundamentals of salt precipitation and water flooding in acidic eCO2R MEAs
2.1. Salt precipitation in acidic eCO2R MEAs
Alkali cations such as K+/Cs+ are capable of stabilizing the key *CO2− intermediates via electrostatic interaction and retard the H3O+ diffusion kinetics through the electric field effect, thus promoting eCO2R reactivity and suppressing the HER in acid.25 Since CEMs are conductive to K+/Cs+, plenty of K+/Cs+ from the anolyte is continuously transported across the CEMs via electroosmosis and accumulates at the cathode. Owing to the generation of OH− in eCO2R (eqn (1)) and the suppressed H3O+ diffusion, the surface of cathode CL can be highly alkaline although bulk anolyte is acidic. CO2 is chemically absorbed in the OH−-rich layer and transformed into HCO3− and CO32− (eqn (2) and (3)).| CO2 + H2O + 2e− → CO + 2OH− | (1) |
| CO2 + OH− → HCO3− | (2) |
| CO2 + 2OH− → CO32− + H2O | (3) |
As shown in Fig. 2, the combination of K+/Cs+ and CO32−/HCO3− generates K2CO3/Cs2CO3 or KHCO3/CsHCO3 salts, which precipitate in the microporous layer when their concentration exceeds the solubility limits (2.24 M for KHCO3, 3.49 M for CsHCO3, 7.93 M for K2CO3, and 8.01 M for Cs2CO3 at 20 °C in pure H2O).33 These porous and hydrophilic carbonate/bicarbonate salts accelerate electrolyte penetration into the GDEs, consequently speeding up the water flooding process. The salt precipitation strongly depends on the type and concentration of alkali cations, and the net flux of water (JH2O,net) in the cathode (eqn (4)). Carbonate/bicarbonate salts with higher solubility, low cation concentration in the anolyte, and low ion mobility have slower precipitation rates. Assuming that there is no crossover of anionic species such as HCO3−/CO32−, the JH2O,net is determined by the diffusion (Jdiff) from the anode to the cathode driven by concentration gradients, the electro-osmotic drag (Jeod) caused by solvated alkali cations and H3O+ from anode to cathode, the consumption of eCO2R (Jcon), and the back convection (Jbc) due to the repellence by the hydrophobic cathode GDE.34
| JH2O,net = Jdiff + Jeod − Jcon − Jbc | (4) |
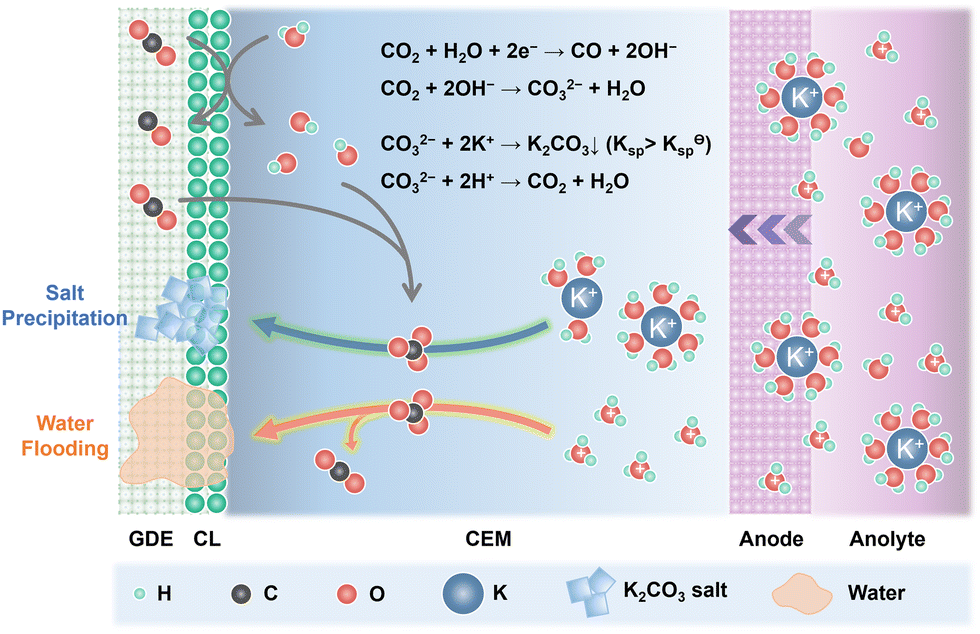 | ||
| Fig. 2 Schematic illustration of the salt precipitation and water flooding process in acidic eCO2R MEAs. | ||
J H2O,net is dominated by the interplay of the CEM, cathode GDE, and operational current density. CEMs with low water uptake have low Jdiff and Jeod. Operating at low current density and using solvated cations with low mobility decreases the Jeod and Jcon. Using cathode GDEs with high hydrophobicity and CEMs with low thickness favors the Jbc.
2.2. Water flooding in acidic eCO2R MEAs
Water flooding in GDEs remains a critical challenge in acidic eCO2R MEAs since it blocks the transportation of CO2 to the surface of catalysts and results in severe HER. Central to this issue is the gas/liquid flow scenario in porous structures. Assuming the GDE as a porous matrix with interconnected cylindrical pores (Fig. 3a–c), liquid transport in the GDE is driven by the capillary differential pressure (ΔP) between gas pressure (Pg) and liquid pressure (Pl), as shown in eqn (5) derived from the Young–Laplace equation.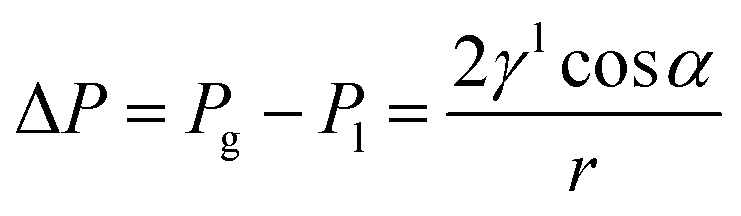 | (5) |
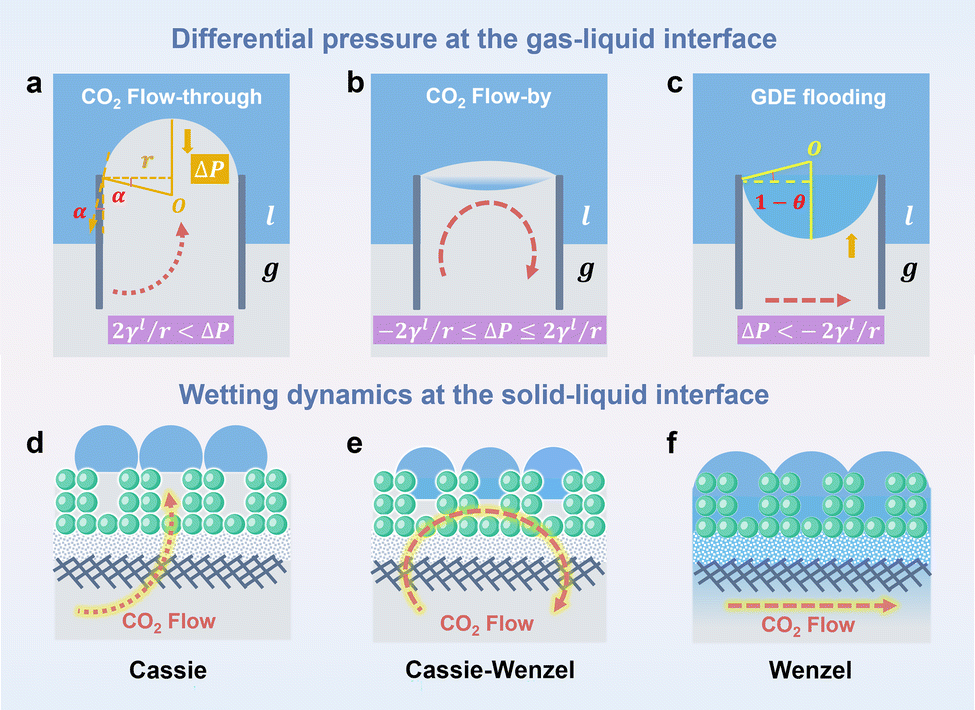 | ||
| Fig. 3 Schematic illustration of differential pressure at the gas–liquid interface and corresponding (a) CO2 flow-through, (b) CO2 flow-by and (c) GDE flooding modes. Wetting dynamics at the solid–liquid interface including (d) Cassie state, (e) Cassie–Wenzel coexistence state, and (f) Wenzel state. | ||
Wetting dynamics at the interface, defined by the contact angle between the solid surface and the liquid droplets, can be categorized into the Cassie, the Cassie–Wenzel coexistence, and the Wenzel state (Fig. 3d–f). Each state delineates a unique pathway for CO2 transportation through the GDE, corresponding to distinct flow patterns including flow-through (ΔP > 2γl/r), flow-by (−2γl/r ≤ ΔP ≤ 2γl/r), and GDE flooding (ΔP < −2γl/r).
ΔP is dynamically influenced by the high H2O flux from the anolyte to cathode GDE, which increases the Pl. In addition to H2O diffusion from the anolyte, H2O flux to the cathode GDE is amplified via the electroosmosis of H+ and K+/Cs+ in an acidic MEA because H+ and K+/Cs+ transfer is always in conjunction with the hydrated shells, i.e. H3O+ and K(H2O)m+/Cs(H2O)m+. Additionally, excessive H2O is generated at the CL/CEM interface during the regeneration of CO2 from HCO3− and CO32− (eqn (6) and (7)).
| HCO3− + H+ → CO2 + H2O | (6) |
| CO32− + 2H+ → CO2 + H2O | (7) |
Moreover, the imbalance between CO2 consumption and gas production rates at the interface, electrowetting, degradation of GDE hydrophobicity, and salt precipitation also contribute to the hydrostatic/hydrodynamic differential pressure by changing the Pg, γl and α.35 Such variations in the pressure balance can gradually reduce ΔP below the critical transition threshold, causing the CEM system to shift from the flow-by model to the GDE flooding model. We note that the triphasic gas–electrode–electrolyte interface under operando conditions is much more complicated. For example, gases are generated at the catalyst/liquid interface due to the Joule heating. The differential pressure might vary strongly among different parts due to the complex catalyst/hydrophobic moiety/carbon interface.
3. Remedies for salt precipitation and water flooding in acidic eCO2R MEAs
Salt precipitation and water flooding are always intertwined together, which severely deteriorates the gas–electrode–electrolyte interface and poses obstacles to the stability of acidic eCO2R MEAs. The accumulation of K+/Cs+, in situ formation of high-concentration CO32−/HCO3− on the cathode CL, and high H2O flux from the anolyte are the main culprits for severe salt precipitation and water flooding in acidic eCO2R MEAs. In this section, remedies including K+/Cs+, CO32−/HCO3−, and H2O management (Fig. 4) via engineering the electrodes, electrolyte, and membrane are proposed to mitigate these issues and enhance the durability of acidic MEAs.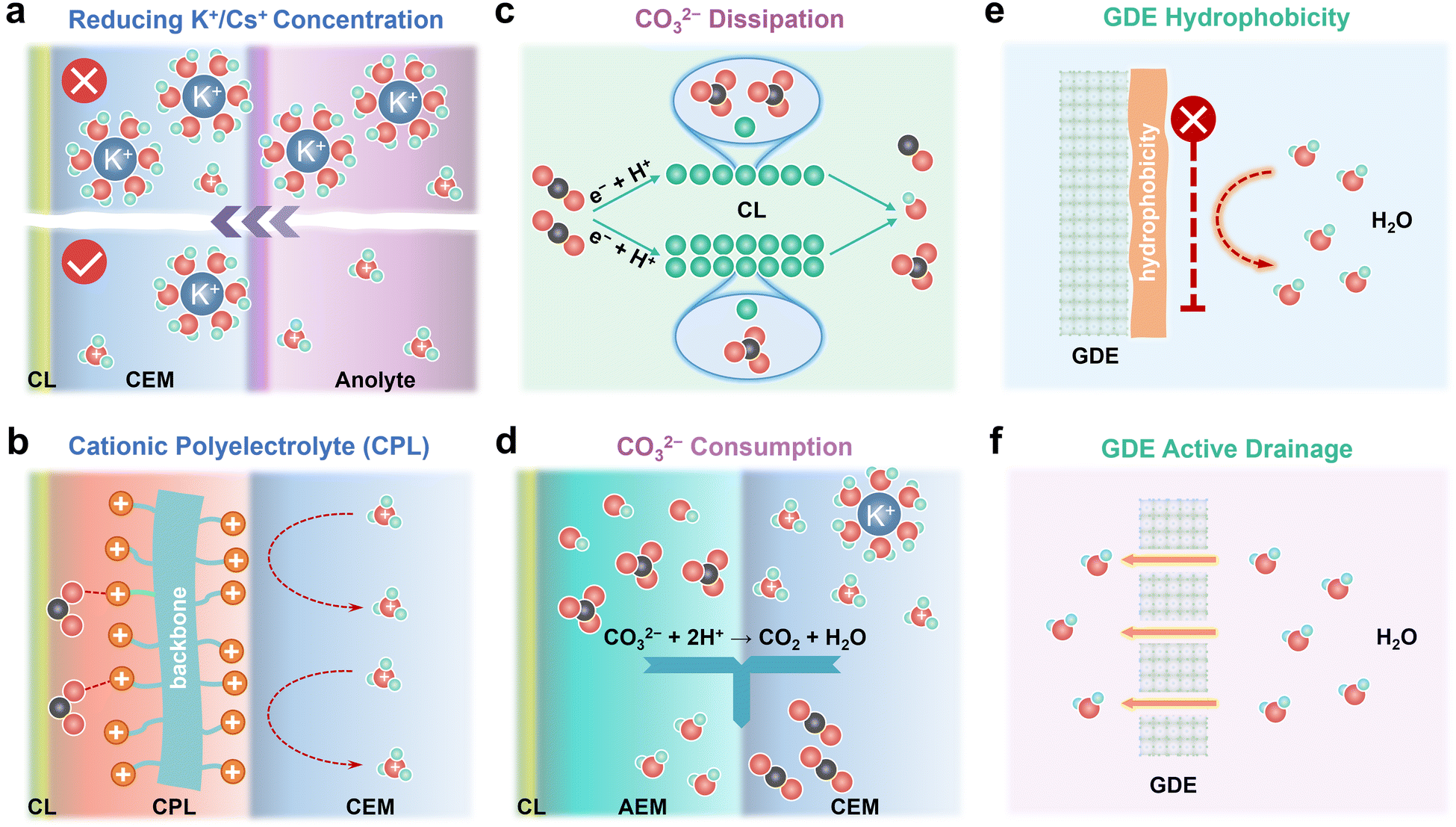 | ||
| Fig. 4 Remedies of mitigating salt precipitation and water flooding in acidic eCO2R MEAs including (a) reducing K+/Cs+ concentration in anolytes, (b) replacing K+/Cs+ with a cationic polyelectrolyte layer (CPL), (c) CO32− dissipation in the catalyst layer (CL), (d) accelerating CO32− consumption, (e) tailoring GDE hydrophobicity, and (f) GDE active drainage. | ||
3.1. Preventing K+/Cs+ accumulation or replacing K+/Cs+ with cationic polyelectrolytes
K+/Cs+ accumulated in the CL and microporous layer supplies cations for salt precipitation. Therefore, preventing K+/Cs+ accumulation represents a proactive solution to alleviate salt precipitation in acidic MEAs. Generally, the less K+/Cs+ migrates across the CEM, the longer the time for salts to precipitate. Reducing the concentration of K+/Cs+ in the anolyte is a straightforward way to limit the quantity of K+/Cs+ that electro-migrates from the anolyte to the cathode. In an ideal case, the concentration of K+/Cs+ in the anolyte should be as low as possible on the premise of high eCO2R selectivity (Fig. 4a). By doing so, the salt precipitation kinetics are effectively decelerated. Generalized modified Poisson–Nernst–Planck (GMPNP) modeling demonstrates that the concentration of cations (CM+) and the identity of M+ affect eCO2R by tuning the electric field strength in the stern layer. Increasing the CM+ and adding cations with smaller solvated sizes such as K+ and Cs+ results in higher partial current density (jCO). Meanwhile, the mass transport of H+ is inhibited when CM+ is not less than CH+. Decreasing the CK+ from 0.8 M to 0.02 M suppresses the KHCO3 precipitation in the acidic flow cell at 200 mA cm−2, but also leads to a low FECO.36 These results match with the report from Pan et al.18 In their work, the influence of Cs+ and H+ concentration in the anolyte on the jCO and faradaic efficiency of CO (FECO) using Ag/PTFE as the cathode GDE in acidic MEAs is systematically studied. Diluted anolyte (0.01 M H2SO4 + 0.01 M Cs2SO4) is optimal for high FECO (∼80%) at low current density (60 mA cm−2). While low pH and Cs+ concentration (0.2 M H2SO4 + 0.01 M Cs2SO4) are more favorable for FECO (75%) at high current density (140 mA cm−2). By using 0.01 M H2SO4 + 0.01 M Cs2SO4 as anolyte, 50-hour stability at 60 mA cm−2 with ∼90% SPCCO2 and 80% FECO is achieved. In a recent work, Kamiya et al. performed a quantitative analysis of how the transported alkali cations regulate the eCO2R selectivity in MEAs and suggested that continuously supplying a high amount of K+ is not necessary for C2+ formation.37 Intermittently supplying concentrated KHCO3 is able to extend the durability of MEAs by alleviating the salt precipitation, which provides an effective method to prevent K+/Cs+ accumulation.Salt precipitation and water flooding can be mitigated but not eliminated as long as alkali cations exist in anolytes. During continuous operation in acidic MEAs, anolyte pH gradually decreases because metal cation accumulation at cathode inhibits H+ transportation across CEMs and further induces H+ accumulation in the anolytes. Such imbalanced ion transportation also causes the unstable operation of acidic MEAs. Operating acidic eCO2R MEAs with pure acid or even pure water as anolytes potentially overcomes instability issues brought by imbalanced ion transportation and rapid salt precipitation. Recently, Dich et al. reported the using of pure water as the anolyte and periodically injecting Cs+ containing solution into the cathode chamber to provide cations for CO2 activation in a forward-bias bipolar membrane (f-BPM) with a porous anion exchange layer (AEL).38 A long stability of 200 hours at 100 mA cm−2 with FECO ∼80% is achieved because cation accumulation is prohibited by using pure water as the anolyte. The use of porous AEL for recovered CO2 recirculation in conjunction with CEMs also enables a maximum SPCCO2 of 52%.
Apart from using alkali cations, weakly coordinating organic cations, such as water-soluble tetraalkylammonium cations (PDDA), are intrinsically capable of supporting eCO2R on Au and Ag on par with alkali cations.39 Based on the modified Poisson–Boltzmann model, the electric field strength generated by the immobilized benzimidazole cationic group (CG) is in the same order as that generated by K+,40 which allows Cu to reach 80% C2+ in a pure acid electrolyte without K+ and to operate stably for 150 h. Therefore, a cationic polyelectrolyte layer (CPL) with sufficiently high charge density sandwiched between the cathode CL and the CEM is also capable of modulating the electrical field and stabilizing *CO2− as K+/Cs+ does (Fig. 4b).41
Polyelectrolytes with a high charge density usually have high solubility in water, which induces instability of the cathode CL/CPL/CEM interface because the water flux from the anode is increased during CO2 recovery (eqn (6) and (7)). To mitigate the loss of polyelectrolytes, Fan et al. demonstrated that immobilizing poly(diallyldimethylammonium) (PDDA) on graphene oxide (GO) via electrostatic interaction could displace metal cations for acidic eCO2R MEAs with pure acid or even pure water as the anolyte.42 A 50-hour stability at 100 mA cm−2 with a FECO of about 70% is demonstrated in the MEA fed with 0.01 M H2SO4. A FECO of 78% with an energy efficiency of 30% could be achieved using a PDDA-GO modified Ag catalyst at 100 mA cm−2 and 40 °C.
The key role of CPLs is activating CO2 without alkali cations, transporting CO32− and recovered CO2, and providing a local alkaline environment by suppressing the migration of H3O+. First, CPLs should have high ion exchange capacity (IEC) and maximize ionic conductivity, which favors CO2 activation and CO32− transportation. Second, CPLs should have moderate water uptake to manage the water content in the cathode CL since the low water uptake will cause water starvation in the cathode GDE and high water uptake will cause GDE flooding and fast H3O+ migration. Third, CPLs should have high mechanical and chemical stability, which not only can withstand both local acid and alkaline environments but also can mechanically stabilize the cathode CL/CPL/CEM interface. Fourth, CPLs should be permeable for gaseous CO2, which favors transporting recovered CO2 back to the cathode CL.
The knowledge gained from AEMs and anion exchange ionomers (AEIs) can be transferred to speed up the development of CPLs. Decent reviews on AEMs and AEIs43–46 have summarized the desired properties, such as OH− conductivity (60–100 mS cm−1), IEC (>1.5 meq g−1), tensile strain (20 MPa), and water uptake (50–80%), which can provide instructive knowledge for CPLs.45 In CPLs, cationic groups such as quaternary ammonium, pyrrolidonium, piperidinium and imidazolium groups are generally integrated into the mainchain or side chain of polymer backbones such as polystyrene, polybenzylimidazole, poly terphenyl, and polynorbornene. We summarize the molecular structure of AEMs/AEIs (Fig. 5) that have been proven to be feasible in pure water/pure acid-fed acidic eCO2R electrolyzers (c-PDDA, Sustainion, Aemion, and Piperion)16,47–49 or pure water fed neutral eCO2R MEAs (pTPN-Beim, Pention, QAPEEK, and QAPPT),10,50–52 which may guide the design of high-performance CPLs in the future.
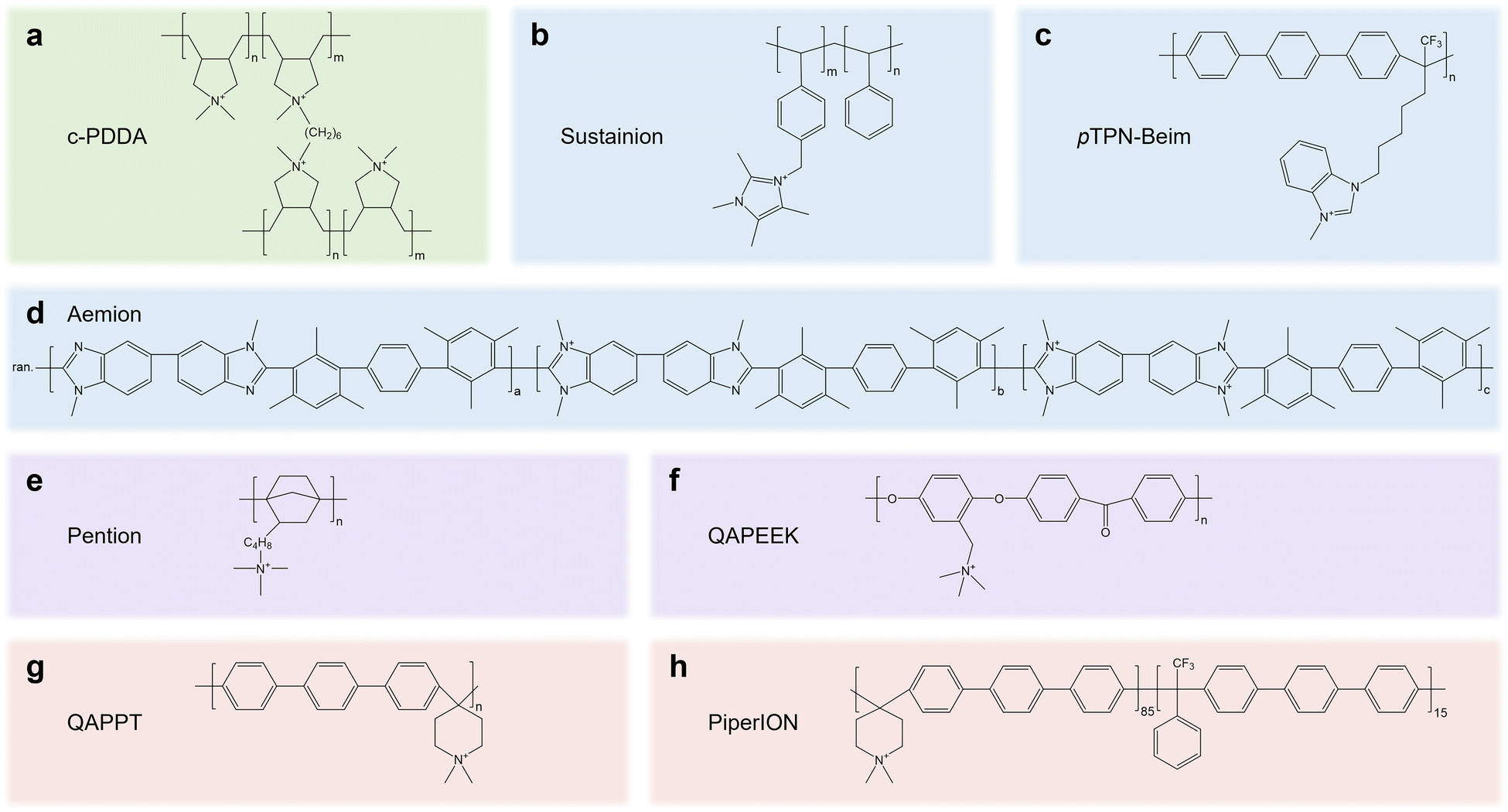 | ||
| Fig. 5 Molecular structures of AEMs/AEIs that are feasible in eCO2R electrolyzers fed with pure acid or pure water, including (a) c-PDDA, (b) Sustainion, (c) pTPN-Beim, (d) Aemion, (e) Pention, (f) QAPEEK, (g) QAPPT and (h) PiperION. | ||
3.2. Reducing local CO32−/HCO3− concentration by dissipation or rapid consumption
Reducing local CO32−/HCO3− concentration is also an important direction to decelerate salt precipitation kinetics since salt precipitation occurs only when the critical concentrations of metal cations and CO32−/HCO3− are achieved. Dissipation and rapid consumption present promising tactics to reduce the local CO32−/HCO3− concentration.As illustrated in eqn (1)–(3), local CO32−/HCO3− concentration strongly depends on the faradaic current at the cathode GDE. Increasing the amount of active site enabled by increasing the CL thickness or the density of the active site reduces the faradaic current generated per active site, thus reducing the local OH− generated per active site (Fig. 4c).53 In such a dissipation strategy, CO32−/HCO3− is kept at low concentrations. It should be noted that CO2 and ion transportation is slowed down when the CL is too thick. Therefore, the CL thickness should be subtly balanced. Mass-transport modelling shows that the local CO2 concentration depends on the CL thickness, porosity and operating current densities.9,54 Increasing the CL thickness from 1 μm to 5 μm decreases the local CO2 concentration from 20 mM to 17 mM at 100 mA cm−2. The local CO2 concentration reduced from 17 mM to 10 mM with CL thickness of 5 μm when the current density rises from 100 mA cm−2 to 300 mA cm−2.55 Increasing the CL porosity enhances the gas permeability and facilitates the gas transport. Increasing the CL porosity from 0.3 to 0.7 improves the current density by more than 100 mA cm−2 in a wetted CL.56
In the electric double layer (EDL), CO2, HCO3− and CO32− are in equilibrium. According to Henry's law, increasing input CO2 pressure (PCO2) results in increased CO2(eq), which tunes the CO2(eq)–CO32−–HCO3− equilibrium and potentially decreases the local CO32−/HCO3− concentration. Moreover, increasing PCO2 is also capable of boosting the eCO2R kinetics and suppressing the competing HER by enhancing the local CO2 concentration.57 When the same mole of CO2 is fed, higher CO2 pressure will increase the SPCCO2. However, if CO2 availability is not the limiting factor, increasing the CO2 pressure will not increase the SPCCO2 instead.
Rapid consumption of the CO32−/HCO3− generated in the CL represents another important solution to reduce the local CO32−/HCO3− concentration (Fig. 4d). In a f-BPM MEA, the regeneration of CO2 occurs at the AEM/CEM interface, where CO32−/HCO3− from the AEM is consumed by H+ from the CEM. Therefore, accelerating the transportation rate of CO32−/HCO3− from the CL to the AEM/CEM interface and enlarging the AEM/CEM interface potentially increases the consumption rate of CO32−/HCO3−, thereby decreasing the local CO32−/HCO3− in CL. In practice, this strategy can be achieved by constructing the intimate CL/AEM/CEM interface by the catalyst-coated membrane (CCM) and direct membrane deposition (DMD) method,58 which also has another benefit of reducing the transportation resistance of ions and increasing the overall energy efficiency.
3.3. Managing H2O content in cathode GDEs by surface hydrophobicity and active drainage
Salt precipitation and water flooding are generally trapped into a vicious cycle and finally block the CO2 transportation in GDEs. Though the salt precipitation problem can be potentially solved by Remedies 3.1 and 3.2, water flooding still exists because of the in situ formation of H2O during CO2 regeneration (eqn (6) and (7)). On the other hand, H2O plays a critical role as the proton source for eCO2R. Therefore, managing H2O content in cathode GDEs, which can be realized by tailoring surface hydrophobicity or actively draining, is significant to balance the selectivity and stability of the acidic eCO2R MEAs.The cathode GDE provides spaces for uniformly distributing CO2, H2O, and electrons. However, the transport pathways for CO2 and H2O are usually intertwined, which impedes CO2 diffusion. Hydrophobic treatment of the GDE is therefore essential to decouple CO2 and H2O transportation (Fig. 4e). In previous work, Wang et al. reported a hydrophobized integral GDE embedding undercoordinated Ni–N–C active sites (NiNF) for full-pH CO2 electroreduction in MEAs.59 By virtue of the integral architecture, hierarchical porosity, and high hydrophobicity, it not only enhances CO2 transportation but also mitigates salt precipitation and water flooding. NiNF exhibits a near-unity faradaic efficiency of CO and stable operation for more than 273 hours in neutral MEAs and a high single-pass CO2 conversion of 78% in acidic MEAs. Post-mortem characterizations reveal that the failure of MEAs is mainly attributed to the loss of hydrophobicity.
Conventional GDEs are composed of macroporous carbon fiber paper (CFP), microporous layer (MPL), and CL, among which micropores in the MPL are the most easily flooded due to electrochemical wetting by water. Active draining by innovating the GDE structure holds promise to enhance its tolerance to water flooding and extend its durability (Fig. 4f). For example, through tailoring the wettability gradient and introducing large-size pores by laser drilling in a Janus carbon-based GDE, water spontaneously transports from the hydrophobic side to hydrophilic side, enabling a remarkable antiflooding capability.60 In addition to conventional carbon-based GDEs, metal foam-based GDEs have recently drawn great attention because of the high conductivity of the metal foam and abundant large pores with microns in size. A hydrophobic catalytic layer can be in situ grown on metal foams through etching metal foams or electrodeposition.11,61 Such architecture not only enables a high CO2 diffusion rate by surface hydrophobicity but also enhances resilience to flooding because the large pores are favorable for drainage, offering great opportunities to mitigate GDE flooding.
In Remedy 3.1, reducing the concentration of K+/Cs+ helps to extend the stability of acidic MEAs and simultaneously utilizes the cation effect to keep the high eCO2R selectivity. However, the salt precipitation after long-term operation is unavoidable since the CEMs are permeable to K+/Cs+. Utilizing a CPL to replace K+/Cs+ can eliminate the salt precipitation issue. But how to rationally fabricate the CPL with high chemical and mechanical robustness, and stabilize the cathode CL/CPL/CEM interface still remains to be explored. In Remedy 3.2, increasing the CL thickness or the density of active sites can reduce the local concentration of CO32−, but will cause the problem of slow CO2 and ion transfer. Constructing an intimate cathode CL/AEM/CEM interface by the CCM or DMD method can boost the CO32− transfer and consumption, and increase the energy efficiency at the same time. The flux of H+, CO32−, and CO2 should be carefully balanced at the CL/AEM/CEM interface. In Remedy 3.3, increasing the hydrophobicity of GDEs can facilitate CO2 transportation and increase the resistance to water flooding. But the loss of hydrophobicity by electrowetting and loss of PTFE binder over time cannot be avoided. The active drainage of GDEs can effectively manage water content in GDEs. More efforts should be devoted to properly engineering the distribution of wettable areas since the flooding behavior and water distribution in cathode GDEs remain elusive, which may increase the fabrication of the GDEs. In addition, periodically adding cations or refreshing the anode electrolyte is necessary to compensate for the loss of cations brought by the water flow out of the electrolyte.
We note that some of these strategies including reducing the concentration of K+/Cs+, tailoring the porosity and thickness of cathode CL, constructing a cathode CL/membrane interface for favorable ion transfer, increasing the hydrophobicity or constructing wettable area for water management in the cathode GDE can be extended to the neutral/alkaline eCO2R MEA too. But the optimal conditions in these strategies vary since neutral/alkaline eCO2R MEAs manifest different local environments (such as local CO2 concentration and local pH) from acidic MEAs. The AEIs of neutral/alkaline eCO2R are critical for managing the CO2 gas and CO32− ion transfer in the cathode CL. But the CPLs in acidic eCO2R MEAs should be more challenging since they not only provide the gas and ion transfer pathway but also are capable of stabilizing the cathode CL/CPL/CEM interface during CO2 recovery.
4. Summary and perspective
Acidic MEAs have become an important direction in the development of eCO2R for industrial applications because of their high carbon utilization and energy efficiency. However, the durability issue of acidic eCO2R MEAs brought by salt precipitation and water flooding remains challenging. In this mini-review, we discuss the origins of salt precipitation and water flooding from the fundamental viewpoint and propose potential remedies including inhibiting K+/Cs+ accumulation, decreasing local CO32−/HCO3−, and water management in GDEs.Apart from the electrode, electrolyte, and membrane engineering, recovery steps/washing is necessary to recover the performance loss caused by salt precipitation and water flooding with relatively low operation cost. For example, washing the cathode GDE with water can dissolve the salt precipitates and maintain the moderate FECO (85%) during continuous electrolysis.62 Periodically activating and regenerating the cathode GDE with cation containing solution (1 M CsOH in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 isopropanol/water mixture) in the pure water-fed electrolyzer can maintain the stability for 200 h.38,63 In addition to the commonly used aqueous electrolyte, organic electrolyte can also be utilized to reduce the carbonation of aqueous electrolyte. By utilizing dimethyl sulfoxide as the solvent and acetic acid/acetate as the proton source in the aprotic solvent-based eCO2R system, Chu et al. demonstrated that attenuating the water content in an organic medium and using a nonnucleophilic buffer with a matched pKa simultaneously mitigates carbonation and the HER.64
:3 isopropanol/water mixture) in the pure water-fed electrolyzer can maintain the stability for 200 h.38,63 In addition to the commonly used aqueous electrolyte, organic electrolyte can also be utilized to reduce the carbonation of aqueous electrolyte. By utilizing dimethyl sulfoxide as the solvent and acetic acid/acetate as the proton source in the aprotic solvent-based eCO2R system, Chu et al. demonstrated that attenuating the water content in an organic medium and using a nonnucleophilic buffer with a matched pKa simultaneously mitigates carbonation and the HER.64
Although progress has been made to address the salt precipitation and water flooding to extend the durability of acidic eCO2R MEAs, there is still a long way ahead to meet the requirements for stability (5 years), energy efficiency (∼50%), and SPCCO2 (30% and 15% for C1 and C2 products) according to the techno-economic assessment.17 To push acidic eCO2R MEAs toward large-scale implementation, research efforts also should be devoted to the following fields:
(1) Fabricating selective and stable eCO2R electrocatalysts for acidic MEAs. Some heterogeneous molecular electrocatalysts are intrinsically active and selective for acidic eCO2R in a pure water-fed BPM electrolyzer, which completely eliminates the salt precipitation issue. For example, the acid-tolerant [Ni(1,4,8,11-tetraazacyclotetradecane)]2+ catalyst achieves >30% CO selectivity at 100 mA cm−2 in a zero-gap r-BPM device fed with pure water and CO2.65 Cobalt phthalocyanine (CoPc) achieved 53% CO selectivity at 100 mA cm−2 and an SPC up to 51%.66 Cobalt tetraaminophthalocyanine covalently anchored on the positively charged polyfluorene backbone (PF-CoTAPc) achieves 82.6% FECO at 100 mA cm−2 and 87.8% CO2 utilization.67 More efforts are needed to develop electrocatalysts with intrinsically high eCO2R selectivity and stability in pure acid or water.
Nanostructured electrocatalysts have the benefit of reducing the current/area and modulating the concentration of reactants/intermediates/products. But the coexistence of H+, adsorbates such as *CO and negative bias in acidic eCO2R MEAs generates highly corrosive conditions for the popular M–N–C, Cu and Ag based electrocatalysts, causing demetallation in M–N–C and migration of surface-active metal atoms, and deactivating these electrocatalysts.68,69 We think the activity, selectivity, stability, and cost of the nanostructured electrocatalysts should all be taken into account and well-balanced. Constructing highly stable electrocatalysts, which are resilient to harsh conditions through strong metal–support interactions or surface polymer/carbon coating, may be able to alleviate the corrosion of electrocatalysts in acidic MEAs.
(2) Designing highly conductive and stable CPLs. CPLs play crucial roles in manipulating the CO2, H2O, CO32−/HCO3− and K+/Cs+ transportation in the triphasic gas–electrode–electrolyte interface.46 CPLs with high ionic conductivity and microphase separation structure are expected to decouple the CO2 and ion transfer pathway and boost the eCO2R kinetics.45 Moreover, more chemically and mechanically stable CPLs are highly desired to enhance the durability of acidic MEAs.
(3) Systematically engineering the cathode CL/membrane interface. For the conventional catalyst-coated substrate (CCS) method, the cathode CL/membrane ionic interface contributes the major voltage loss,70 which hinders the energy efficiency and current density. Constructing novel micro-/nano-structures such as three-dimensional (3D) ordered CLs with interlocked catalyst/membrane interface should boost the ion transfer and elevate the energy efficiency.71
(4) Quantitatively understanding and optimizing acidic eCO2R MEAs by numerical simulations. In contrast to the conventional try-and-error experimental approach, numerical simulations play a key role in the development of eCO2R electrolyzers by providing a deep and quantitative understanding of the transportation behavior of reactants/intermediates/products and their local concentration at the electrode/electrolyte interface with less cost.72–77 The insight gained from numerical simulation will provide useful guidance for developing selective and stable acidic eCO2R MEAs.
(5) Operando characterizations and monitoring the chemical microenvironment and interface in acidic eCO2R MEAs. Monitoring the chemical microenvironment and the interface during operando conditions is significant for unraveling the failure mechanism of acidic eCO2R MEAs, but is still at the early stage. Developing more in situ and operando techniques such as synchrotron radiation X-ray absorption spectroscopy (XAS), Raman spectroscopy, operando X-ray diffraction (XRD), and electrochemical impedance spectroscopy (EIS) with distribution of relaxation times (DRT) analysis is highly suggested to reveal the electronic structure of catalysts, evolution of the intermediates and local pH, salt precipitation and water flooding in GDEs, and changes in the catalyst/membrane interface.78–81
Author contributions
Q. B., L. X., Y. Z., M. M. and Z. J.: writing – original draft and editing, investigation, visualization, data curation. Y. P., Z. D. and F. L.: conceptualization, resources, supervision, methodology, writing – review and editing, funding acquisition.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 22309125, 22072101, 22075193, 22109099), the Natural Science Foundation of Jiangsu Province (No. BK20220483, BK20211306, BK20220027, BK20221239), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (22KJB150010), Six Talent Peaks Project in Jiangsu Province (No. TD-XCL-006), Chen Guang project from Shanghai Municipal Education Commission and Shanghai Education Development Foundation, and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.Notes and references
- S. M. Jordaan and C. Wang, Nat. Catal., 2021, 4, 915–920 CrossRef.
- C. Chen, H. Jin, P. Wang, X. Sun, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Chem. Soc. Rev., 2024, 53, 2022–2055 RSC.
- C. Tang, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- Z. Zhang, X. Huang, Z. Chen, J. Zhu, B. Endrődi, C. Janáky and D. Deng, Angew. Chem., Int. Ed., 2023, 62, e202302789 CrossRef CAS PubMed.
- C. Yan, D. Gao, J.-J. Velasco-Vélez and G. Wang, EES Catal., 2024, 2, 220–230 RSC.
- C. A. Giron Rodriguez, N. C. Kani, A. B. Moss, B. O. Joensen, S. Garg, W. Deng, T. Wilson, J. R. Varcoe, I. Chorkendorff and B. Seger, EES Catal., 2024, 2, 850–861 RSC.
- E. F. Johnson, E. Boutin, S. Liu and S. Haussener, EES Catal., 2023, 1, 704–719 RSC.
- S. Kwon, T.-H. Kong, N. Park, P. Thangavel, H. Lee, S. Shin, J. Cha and Y. Kwon, EES Catal., 2024, 2, 911–922 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455–2462 RSC.
- J. Han, B. Tu, P. An, J. Zhang, Z. Yan, X. Zhang, C. Long, Y. Zhu, Y. Yuan, X. Qiu, Z. Yang, X. Huang, S. Yan and Z. Tang, Adv. Mater., 2024, e2313926 CrossRef PubMed.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. D. Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- N. McQueen, K. V. Gomes, C. McCormick, K. Blumanthal, M. Pisciotta and J. Wilcox, Prog. Energy, 2021, 3, 032001 CrossRef.
- K. V. Petrov, C. I. Koopman, S. Subramanian, M. T. M. Koper, T. Burdyny and D. A. Vermaas, Nat. Energy, 2024, 9, 932–938 CrossRef CAS.
- C. P. O’Brien, R. K. Miao, S. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C.-T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustainability, 2021, 4, 911–919 CrossRef.
- B. Pan, J. Fan, J. Zhang, Y. Luo, C. Shen, C. Wang, Y. Wang and Y. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS.
- S. C. da Cunha and J. Resasco, Nat. Commun., 2023, 14, 5513 CrossRef CAS PubMed.
- A. Ozden, F. P. García de Arquer, J. E. Huang, J. Wicks, J. Sisler, R. K. Miao, C. P. O’Brien, G. Lee, X. Wang, A. H. Ip, E. H. Sargent and D. Sinton, Nat. Sustainability, 2022, 5, 563–573 CrossRef.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 1808066 CrossRef PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- S. Feng, X. Wang, D. Cheng, Y. Luo, M. Shen, J. Wang, W. Zhao, S. Fang, H. Zheng, L. Ji, X. Zhang, W. Xu, Y. Liang, P. Sautet and J. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202317942 CrossRef CAS PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- Y. Dong, M. Ma, Z. Jiao, S. Han, L. Xiong, Z. Deng and Y. Peng, Chin. Chem. Lett., 2024, 35, 109049 CrossRef CAS.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- C. J. Bondue, M. Graf, A. Goyal and M. T. M. Koper, J. Am. Chem. Soc., 2021, 143, 279–285 CrossRef CAS PubMed.
- S.-J. Shin, H. Choi, S. Ringe, D. H. Won, H.-S. Oh, D. H. Kim, T. Lee, D.-H. Nam, H. Kim and C. H. Choi, Nat. Commun., 2022, 13, 5482 CrossRef CAS PubMed.
- Z. Zhang, H. Li, Y. Shao, L. Gan, F. Kang, W. Duan, H. A. Hansen and J. Li, Nat. Commun., 2024, 15, 612 CrossRef CAS PubMed.
- G. P. Heim, M. A. Bruening, C. B. Musgrave, W. A. Goddard, J. C. Peters and T. Agapie, Joule, 2024, 8, 1312–1321 CrossRef CAS.
- X. Yang, H. Ding, S. Li, S. Zheng, J.-F. Li and F. Pan, J. Am. Chem. Soc., 2024, 146, 5532–5542 CrossRef CAS PubMed.
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed.
- A. Reyes, R. P. Jansonius, B. A. W. Mowbray, Y. Cao, D. G. Wheeler, J. Chau, D. J. Dvorak and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 1612–1618 CrossRef CAS.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- H.-G. Qin, F.-Z. Li, Y.-F. Du, L.-F. Yang, H. Wang, Y.-Y. Bai, M. Lin and J. Gu, ACS Catal., 2023, 13, 916–926 CrossRef CAS.
- S. Kato, S. Ito, S. Nakahata, R. Kurihara, T. Harada, S. Nakanishi and K. Kamiya, ChemSusChem, 2024, e202401013 CrossRef PubMed.
- J. Disch, S. Ingenhoven and S. Vierrath, Adv. Energy Mater., 2023, 13, 2301614 CrossRef CAS.
- S. Weng, W. L. Toh and Y. Surendranath, J. Am. Chem. Soc., 2023, 145, 16787–16795 CrossRef CAS PubMed.
- M. Fan, J. E. Huang, R. K. Miao, Y. Mao, P. Ou, F. Li, X.-Y. Li, Y. Cao, Z. Zhang, J. Zhang, Y. Yan, A. Ozden, W. Ni, Y. Wang, Y. Zhao, Z. Chen, B. Khatir, C. P. O’Brien, Y. Xu, Y. C. Xiao, G. I. N. Waterhouse, K. Golovin, Z. Wang, E. H. Sargent and D. Sinton, Nat. Catal., 2023, 6, 763–772 CrossRef CAS.
- D. Song, Y. Lian, M. Wang, Y. Su, F. Lyu, Z. Deng and Y. Peng, eScience, 2023, 3, 100097 CrossRef.
- J. Fan, B. Pan, J. Wu, C. Shao, Z. Wen, Y. Yan, Y. Wang and Y. Li, Angew. Chem., Int. Ed., 2024, 63, e202317828 CrossRef CAS PubMed.
- S. Favero, I. E. L. Stephens and M.-M. Titirci, Adv. Mater., 2024, 36, 2308238 CrossRef CAS PubMed.
- S. Garg, C. A. Giron Rodriguez, T. E. Rufford, J. R. Varcoe and B. Seger, Energy Environ. Sci., 2022, 15, 4440–4469 RSC.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O’Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339–348 CrossRef CAS.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- H.-G. Qin, Y.-F. Du, Y.-Y. Bai, F.-Z. Li, X. Yue, H. Wang, J.-Z. Peng and J. Gu, Nat. Commun., 2023, 14, 5640 CrossRef CAS PubMed.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Nat. Energy, 2024, 9, 81–91 CrossRef CAS.
- M. Heßelmann, J. K. Lee, S. Chae, A. Tricker, R. G. Keller, M. Wessling, J. Su, D. Kushner, A. Z. Weber and X. Peng, ACS Appl. Mater. Interfaces, 2024, 16, 24649–24659 CrossRef PubMed.
- L. Xue, Z. Gao, T. Ning, W. Li, J. Li, J. Yin, L. Xiao, G. Wang and L. Zhuang, Angew. Chem., Int. Ed., 2023, 62, e202309519 CrossRef CAS PubMed.
- M. Zhuansun, Y. Liu, R. Lu, F. Zeng, Z. Xu, Y. Wang, Y. Yang, Z. Wang, G. Zheng and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309875 CrossRef CAS PubMed.
- W. Li, Z. Yin, Z. Gao, G. Wang, Z. Li, F. Wei, X. Wei, H. Peng, X. Hu, L. Xiao, J. Lu and L. Zhuang, Nat. Energy, 2022, 7, 835–843 CrossRef CAS.
- Q. Xu, A. Xu, S. Garg, A. B. Moss, I. Chorkendorff, T. Bligaard and B. Seger, Angew. Chem., Int. Ed., 2023, 62, e202214383 CrossRef CAS PubMed.
- J. C. Bui, E. W. Lees, L. M. Pant, I. V. Zenyuk, A. T. Bell and A. Z. Weber, Chem. Rev., 2022, 122, 11022–11084 CrossRef CAS PubMed.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- H. Li, H. Li, P. Wei, Y. Wang, Y. Zang, D. Gao, G. Wang and X. Bao, Energy Environ. Sci., 2023, 16, 1502–1510 RSC.
- T. Alkayyali, A. S. Zeraati, H. Mar, F. Arabyarmohammadi, S. Saber, R. K. Miao, C. P. O’Brien, H. Liu, Z. Xie, G. Wang, E. H. Sargent, N. Zhao and D. Sinton, ACS Energy Lett., 2023, 8, 4674–4683 CrossRef CAS.
- M. Wang, L. Lin, Z. Zheng, Z. Jiao, W. Hua, G. Wang, X. Ke, Y. Lian, F. Lyu, J. Zhong, Z. Deng and Y. Peng, Energy Environ. Sci., 2023, 16, 4423–4431 RSC.
- Q. Wen, S. Pan, Y. Li, C. Bai, M. Shen, H. Jin, F. Ning, X. Fu and X. Zhou, ACS Energy Lett., 2022, 7, 3900–3909 CrossRef CAS.
- M. Sun, J. Cheng and M. Yamauchi, Nat. Commun., 2024, 15, 491 CrossRef CAS PubMed.
- B. Endrődi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef PubMed.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- A. T. Chu, O. Jung, W. L. Toh and Y. Surendranath, J. Am. Chem. Soc., 2023, 145, 9617–9623 CrossRef CAS PubMed.
- B. Siritanaratkul, M. Förster, F. Greenwell, P. K. Sharma, E. H. Yu and A. J. Cowan, J. Am. Chem. Soc., 2022, 144, 7551–7556 CrossRef CAS PubMed.
- B. Siritanaratkul, P. K. Sharma, E. H. Yu and A. J. Cowan, Adv. Mater. Interfaces, 2023, 10, 2300203 CrossRef CAS.
- G. Li, L. Huang, C. Wei, H. Shen, Y. Liu, Q. Zhang, J. Su, Y. Song, W. Guo, X. Cao, B. Z. Tang, M. Robert and R. Ye, Angew. Chem., Int. Ed., 2024, 63, e202400414 CrossRef CAS PubMed.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P.-C. Chen, H. Wang, C. J. Pollock, X. Huang, Y.-T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- X. Li, P. Zhang, L. Zhang, G. Zhang, H. Gao, Z. Pang, J. Yu, C. Pei, T. Wang and J. Gong, Chem. Sci., 2023, 14, 5602–5607 RSC.
- K. U. Hansen, L. H. Cherniack and F. Jiao, ACS Energy Lett., 2022, 7, 4504–4511 CrossRef CAS.
- L. Wan, J. Liu, D. Lin, Z. Xu, Y. Zhen, M. Pang, Q. Xu and B. Wang, Energy Environ. Sci., 2024, 17, 3396–3408 RSC.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2020, 13, 3592–3606 RSC.
- N. Gupta, M. Gattrell and B. MacDougall, J. Appl. Electrochem., 2006, 36, 161–172 CrossRef CAS.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. D. Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- S. Brückner, Q. Feng, W. Ju, D. Galliani, A. Testolin, M. Klingenhof, S. Ott and P. Strasser, Nat. Chem. Eng., 2024, 1, 229–239 CrossRef.
- B. O. Joensen, J. A. Zamora Zeledon, L. Trotochaud, A. Sartori, M. Mirolo, A. B. Moss, S. Garg, I. Chorkendorff, J. Drnec, B. Seger and Q. Xu, Joule, 2024, 8, 1754–1771 CrossRef CAS.
- A. B. Moss, S. Garg, M. Mirolo, C. A. Giron Rodriguez, R. Ilvonen, I. Chorkendorff, J. Drnec and B. Seger, Joule, 2023, 7, 350–365 CrossRef CAS.
- Y. Qu, K. Yang, W. Li, G. Wang, L. Xiao, G. Wang and L. Zhuang, ACS Energy Lett., 2024, 9, 3042–3048 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |