
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic fast pyrolysis of cellulose to oxygenates: roles of homogeneous and heterogeneous catalysts
Yingchuan
Zhang
 *ab,
Zijing
Li
a,
Tao
Zhou
a and
Guangri
Jia
*a
*ab,
Zijing
Li
a,
Tao
Zhou
a and
Guangri
Jia
*a
aDepartment of Chemistry, The University of Hong Kong, Hong Kong SAR, China. E-mail: jiagr23@hku.hk
bDepartment of Chemical and Biological Engineering, University of Wisconsin–Madison, WI 53706, USA. E-mail: zhang2985@wisc.edu
First published on 6th September 2024
Abstract
Catalytic fast pyrolysis (CFP) of biomass is an efficient approach that can overcome the structural recalcitrance of solid biomass (e.g., crystalline cellulose) to produce sugar monomers and their derivatives within seconds. The composition of the product mixture, which is accumulated in a liquid called bio-oil, is highly tuneable through the use of in situ/ex situ catalysts for the downstream production of sustainable fuels and fine chemicals. This minireview summarises the recent advances in homogeneous and heterogeneous catalysts in the CFP production of versatile oxygenates as fuel precursors or bulk chemicals. First, a brief overview of primary CFP pathways, including cellulose-to-levoglucosan (LGA) conversion and the production of three important derivative anhydrosugars, is provided. Particular attention is paid to the roles of homogeneous and heterogeneous catalysts in promoting secondary reforming of LGA by dehydration and to alternative pathways via C3–C6 cyclisation or benzylic rearrangement over versatile catalysts (e.g., aqueous acids, zeolites, metal oxides) with Brønsted/Lewis acidity to produce a variety of oxygenates in bio-oil. This minireview may provoke more CFP technologies by clarifying the opportunities and challenges in the selective production of different reformed oxygenates, complementing CFP-based production of aromatics from biomass.
Broader contextBiomass is a renewable resource for the production of fuels and fine chemicals that can provide alternatives to petroleum commodities and therefore mitigate energy and environmental issues. Catalytic fast pyrolysis can efficiently depolymerise macromolecular components of native biomass in seconds, releasing small-molecule oxygenates or aromatics that are highly concentrated in bio-oil and can be directly used in downstream manufacturing. Depending on the catalyst properties and heating temperature, catalytic fast pyrolysis can provide a large variety of oxygenates, including anhydrosugars, furans and C2–C4 alcohols and aldehydes. This review summarises the recent discoveries on homogeneous and heterogeneous catalysts in the fast pyrolysis of cellulose. Particular attention is focused on the reaction chemistry for secondary dehydration, cyclisation and benzylic arrangement over different catalysts, leading to three anhydrosugars with unique chiral structures. The opportunities and challenges in catalyst site design, mechanism study and reactor engineering may provoke the development of precise and sustainable biorefineries based on catalytic fast pyrolysis. |
1. Introduction
Fast pyrolysis is a promising technology for the conversion of solid biomass into bio-oil. This process involves direct loading of the solid biomass into a reactor, followed by anaerobic heating to temperatures ranging from 300 °C to 800 °C at high rates of 100–300 °C s−1.1 Compared to other conversion methods such as acid hydrolysis, fermentation and catalytic fractionation, pyrolysis technology offers unique benefits in terms of process efficiency, feedstock feasibility and product selectivity.2,3 During the rapid heating stage, the covalent bonding network of the major components in the biomass, including cellulose, hemicellulose and lignin, is effectively disrupted. These macromolecules are fractionised and depolymerised via reactive-end fragment intermediates to ultimately release small molecules, such as anhydrosugars, furans, alcohols, aldehydes, carbon monoxide, carbon dioxide and hydrogen.4 The liquid product of this process can be collected using one-pot or multiple cold traps to yield bio-oil, which is a promising feedstock for the downstream production of fuels and fine chemicals. Moreover, fast pyrolysis enables high selectivity toward desirable products in bio-oil due to the rapid transfer of heat energy from the reactor to the biomass feedstock without undergoing multiple transition states, making it an economically and environmentally viable technology (Fig. 1a).5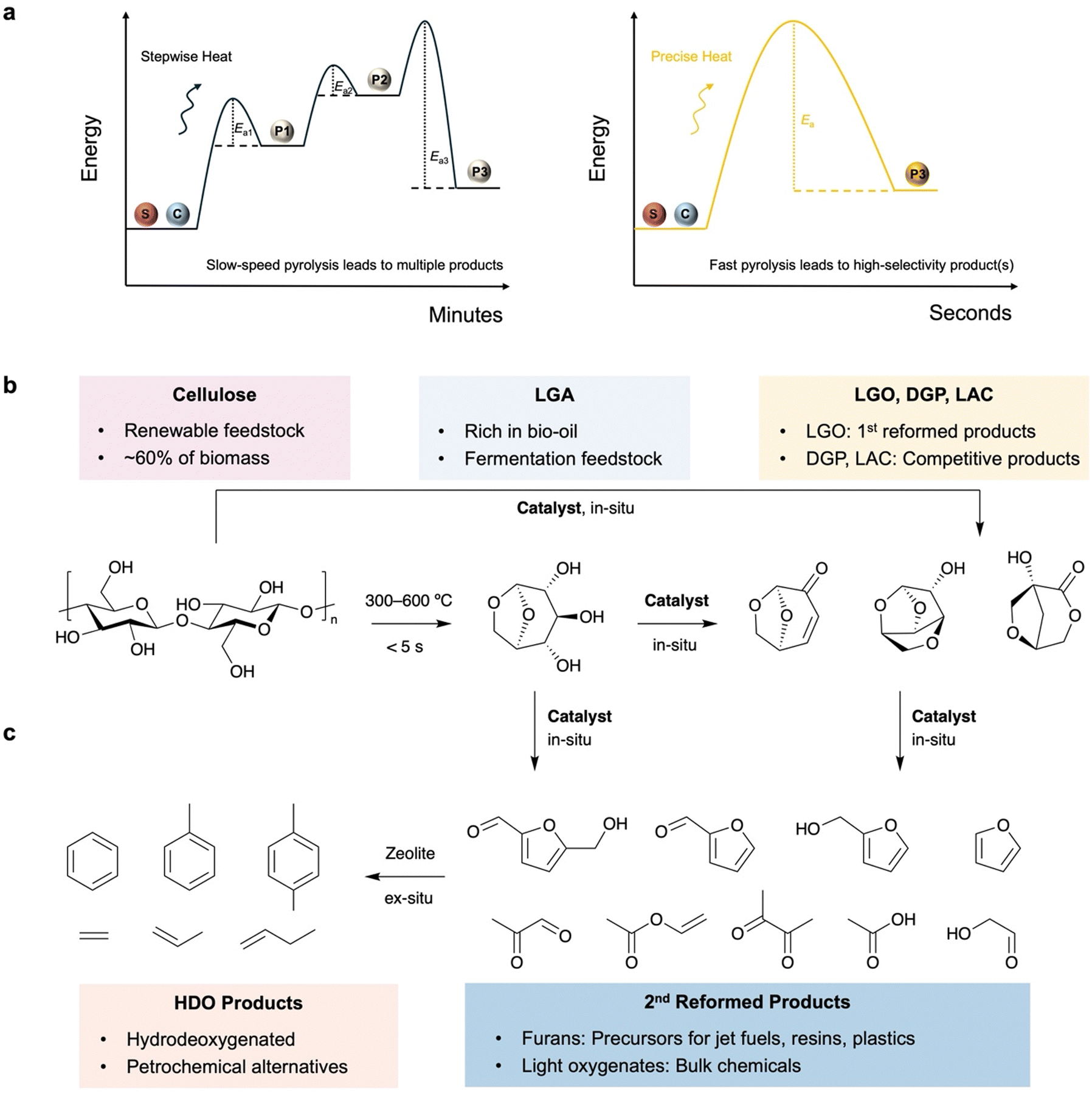 | ||
| Fig. 1 (a) Schematic illustration of reaction progress in slow pyrolysis and fast pyrolysis. Reaction scheme of cellulose CFP to produce (b) anhydrous sugars and (c) furans, light oxygenates and aromatics (catalysts to be discussed are in bold font). | ||
To further extend the product scope and improve product selectivity, versatile catalysts can be loaded in situ/ex situ to promote desirable reaction pathways and/or reform the initial pyrolysis products. This technology, known as catalytic fast pyrolysis (CFP), has been developed over the last few decades and has benchtop and industrial applications.6 Homogeneous catalysis using aqueous acids, ammonia hydroxide and ionic liquids, which are simply mixed with biomass feedstocks, is extensively employed in in situ CFP to boost the production of levoglucosan (LGA) and its derivatives such as levoglucosenone (LGO), 1,4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3,6-dianhydro-α-D-glucopyranose (DGP) and 1-hydroxy-3,6-dioxabicyclo[3.2.1]octan-2-one (LAC) (Fig. 1b). These compounds have unique stereochemical structures suitable for the synthesis of chiral pharmaceuticals and polymers, in addition to their direct use in fermentation towards bioalcohols.7 Heterogeneous catalysis provides opportunities for recyclability and tuneability in CFP to promote in situ depolymerisation and/or ex situ secondary reforming via different reaction pathways.8 There are numerous reports on zeolite-based CFP for the production of aromatics from cellulose through a cascade of depolymerisation, dehydration and hydrodeoxygenation (Fig. 1c).9,10 In contrast, the use of metal oxides in heterogeneous CFP can provide a variety of value-added oxygenates with high tuneability of the product scope and energy content, but this has rarely been summarised before.
:3,6-dianhydro-α-D-glucopyranose (DGP) and 1-hydroxy-3,6-dioxabicyclo[3.2.1]octan-2-one (LAC) (Fig. 1b). These compounds have unique stereochemical structures suitable for the synthesis of chiral pharmaceuticals and polymers, in addition to their direct use in fermentation towards bioalcohols.7 Heterogeneous catalysis provides opportunities for recyclability and tuneability in CFP to promote in situ depolymerisation and/or ex situ secondary reforming via different reaction pathways.8 There are numerous reports on zeolite-based CFP for the production of aromatics from cellulose through a cascade of depolymerisation, dehydration and hydrodeoxygenation (Fig. 1c).9,10 In contrast, the use of metal oxides in heterogeneous CFP can provide a variety of value-added oxygenates with high tuneability of the product scope and energy content, but this has rarely been summarised before.
Despite recent similar work on reaction engineering towards the production of common oxygenates and aromatics, to the best of our knowledge, the catalyst design and underlying chemistry of the production of specific anhydrosugars (LGO, DGP, LAC) via cellulose CFP have rarely been summarised before.11 In this minireview, recent discoveries on oxygenate production via CFP are summarised by comparing the roles of homogeneous and heterogeneous catalysts in (1) tuning the competitive pathways in the initial depolymerisation of cellulose and (2) promoting secondary reforming reactions including dehydration, isomerisation, cyclisation, retro-aldol condensation and hydrodeoxygenation. Challenges and future perspectives are further discussed in terms of catalyst design, in situ/operando catalyst characterisation and reaction engineering.
2. Reaction pathways of cellulose pyrolysis
Fast pyrolysis enables fractionation of lignin–carbohydrate complexes and depolymerisation of three components in biomass.12 Compared to cellulose and hemicellulose, lignin depolymerisation requires higher energy and re-condensation may occur after cracking of C–C and C–O bonds.13 In contrast, cellulose depolymerisation starts from the middle or ends of the chains, with the β-1,4-glycosidic bond undergoing homolytic cleavage involving radical intermediates to form an LGA or LGA-end anhydro-oligosaccharide and a glucose-end anhydro-oligosaccharide, along with the release of a water molecule (Fig. 2a).4 The glucose-end anhydro-oligosaccharide rapidly undergoes further dehydration to form another LGA end, ultimately releasing LGA. Overall, anhydro-oligosaccharides predominantly release LGA as a dehydrated product at C1–O and O–C4, leading to up to 80% selectivity in bio-oil.14,15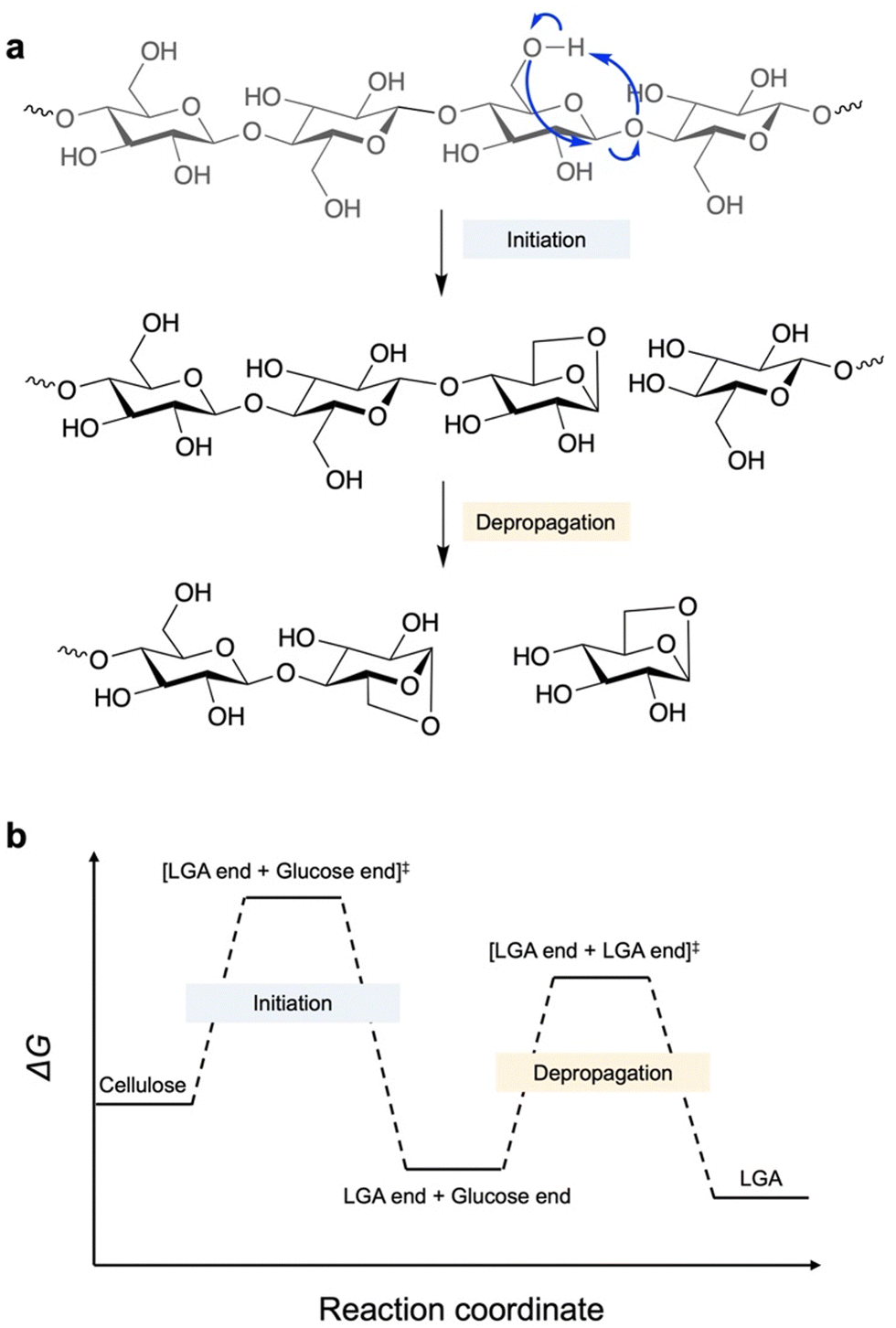 | ||
| Fig. 2 (a) Reaction scheme of cellulose depolymerisation to release LGA in pyrolysis. (b) Free energy change of key steps in cellulose depolymerization.15 | ||
While LGA can be directly utilised in fermentation to produce bioethanol, CFP provides opportunities for the in situ reformation of nascent LGA or the promotion of the competitive pathways towards other anhydrosugars, including LGO, DGP and LAC, which have unique stereochemistry.16 LGO maintains the intramolecular ester bond but loses two hydroxyl groups to provide an unsaturated bond and an aldehyde group. This makes LGO an important platform for the stereoselective synthesis of chiral compounds used as anticancer drugs (e.g., RAS inhibitors) and antibiotics (e.g., chloriolide).17 Its hydrogenated product, Cyrene, is considered to be a next-generation green aprotic solvent. Moreover, a cascade of reduction and hydrogenolysis enables production of tetrahydrofurandimethanol (THFDM) and further 1,6-hexanediol (HDO) for the green synthesis of chiral polymers, coatings and adhesives with potential stereoselectivity.18 DGP and LAC are deoxygenated products of LGA with multi-cyclic chiral structures that are higher in energy content and can be used to synthesize complex non-natural compounds.19 Moreover, these anhydrosugars can further undergo isomerisation, dehydration and/or decarboxylation to produce furan compounds including 5-hydroxymethylfurfural (HMF), furfural, furfuryl alcohol and furan, or retro-aldol reaction to generate light oxygenates (C2–C4) such as glycolaldehyde and 3-heptanol.20
3. Roles of catalysts in cellulose CFP
3.1 Strong Brønsted acid driven dehydration of LGA to LGO
In the presence of Brønsted acids, LGA can undergo in situ dehydration to form a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond between C3 and C4 and a ketone group at C5 in LGO.6,21 The reactive ketone group makes it a promising platform for subsequent transformations, such as coupling, amination, halogenation and ring-opening.22 If CFP catalysts have additional Lewis acid sites, LGO will be isomerised to HMF and will further undergo retro-aldol condensation to form C2–C4 light oxygenates (e.g., levulinic acid and acetaldehyde).23 Until recently, highly selective production of LGO had only been achieved in milligram-scale pyroprobe systems. Huber et al. reported large-scale production of LGO from cellulose by solvolysis (in a mixture of polar aprotic solvent and water) under harsh conditions (230 °C, catalysed by aqueous H2SO4 or propylsulfonic acid functionalized silica as a solid acid); however, the inevitable production of HMF in ∼30% yield makes separation difficult, and the large amount of waste increases the environmental impact.17,24,25 In comparison, if the previously developed CFP catalysts could be compatible with large-scale pyrolysis reactors, the CFP-based production of high-purity LGO should be more efficient and cost-effective compared to solvolysis.
C bond between C3 and C4 and a ketone group at C5 in LGO.6,21 The reactive ketone group makes it a promising platform for subsequent transformations, such as coupling, amination, halogenation and ring-opening.22 If CFP catalysts have additional Lewis acid sites, LGO will be isomerised to HMF and will further undergo retro-aldol condensation to form C2–C4 light oxygenates (e.g., levulinic acid and acetaldehyde).23 Until recently, highly selective production of LGO had only been achieved in milligram-scale pyroprobe systems. Huber et al. reported large-scale production of LGO from cellulose by solvolysis (in a mixture of polar aprotic solvent and water) under harsh conditions (230 °C, catalysed by aqueous H2SO4 or propylsulfonic acid functionalized silica as a solid acid); however, the inevitable production of HMF in ∼30% yield makes separation difficult, and the large amount of waste increases the environmental impact.17,24,25 In comparison, if the previously developed CFP catalysts could be compatible with large-scale pyrolysis reactors, the CFP-based production of high-purity LGO should be more efficient and cost-effective compared to solvolysis.
Within several seconds, LGO can be produced in CFP using homogeneous and heterogeneous catalysts at a temperature of 300–500 °C. As shown in Fig. 3b, a variety of aqueous acids (H3PO4, HCl, H2SO4) can catalyse cellulose depolymerisation and LGA dehydration to afford high-yield LGO as the major product in bio-oil, with a maximum selectivity of 50%.21,26,27 In these cases, the biomass solids are impregnated with 3–10 wt% acid prior to pyrolysis, which inevitably induces reactor corrosion and increases the environmental impact. As green alternatives, the sulfonated ionic liquid (IL) [bmmim][CF3SO3] (50 wt%, bmmim = 1-butyl-2,3-dimethylimidazol-3-ium) and deep eutectic solvents (DESs) containing p-toluenesulfonic acid (p-TSA) as a hydrogen bond donor have been employed in cellulose pyrolysis (5 °C min−1) and effectively cleave the β-1,4-glycosidic bond by protonating the bridging O atom.28–30 Despite numerous effects in this series of homogeneous catalysts, such as increased thermal stability towards recyclable use, the poor recovery rate and reactor corrosion make these approaches less beneficial to industrial applications. Nevertheless, these studies suggest that CFP production of LGO can effectively surpass the production of side products such as HMF, char and retro-aldol condensation compounds, providing a higher selectivity for LGO compared to solvolysis and aligning with the purpose of directly using bio-oil as a sustainable feedstock.
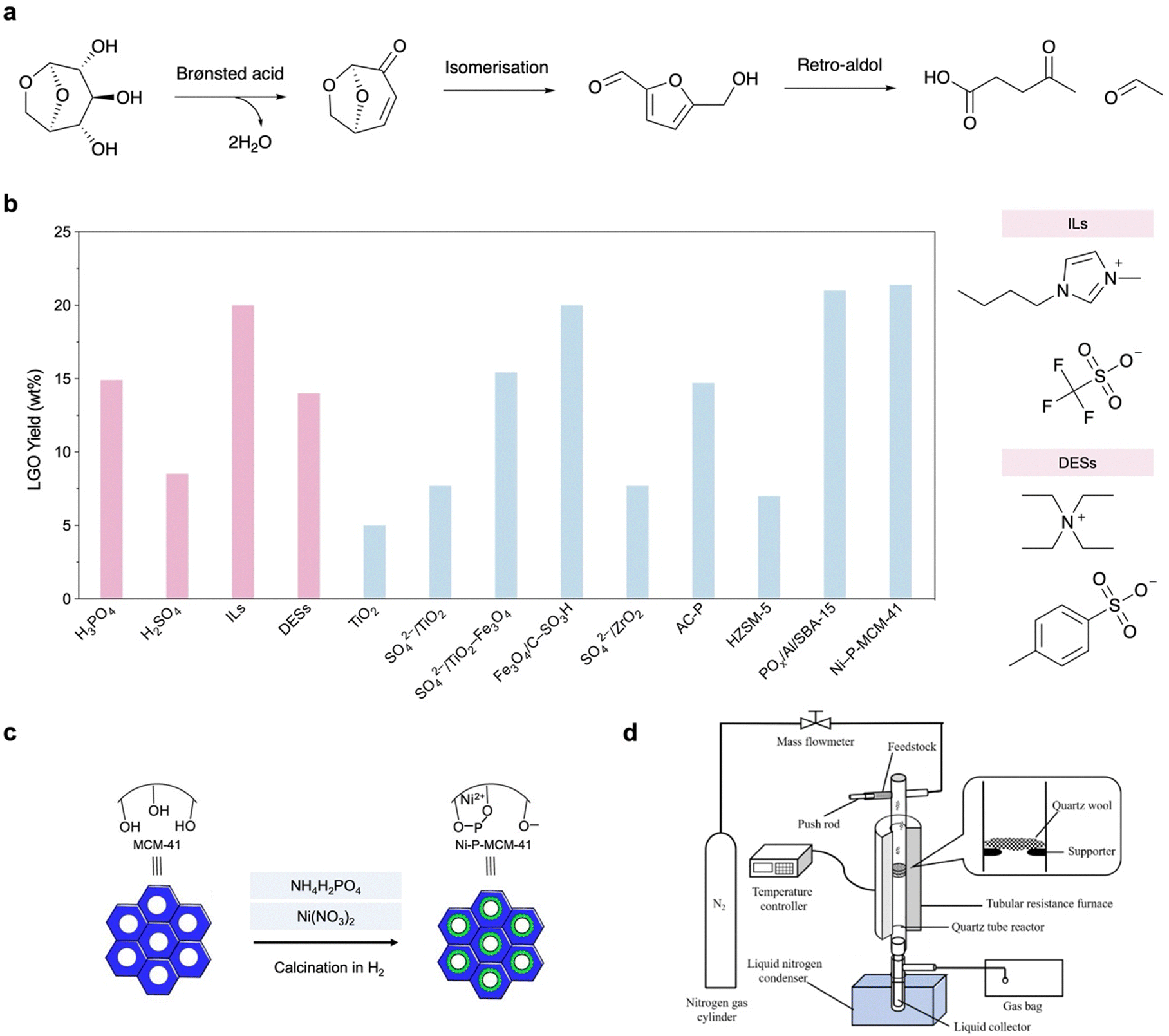 | ||
| Fig. 3 (a) Reaction scheme of LGA-to-LGO process and production of side-products. (b) Performance comparison of previously reported catalysts for LGO production in cellulose pyrolysis and molecular structures of ILs and DESs as representative of homogeneous catalysts (selected from the highest yield achieved under ambient conditions).21 (c) Modification of MCM-41 for cellulose CFP.31 (d) Reaction set-up for cellulose CFP to produce LGO.32 | ||
The application of heterogeneous catalysts in CFP holds great promise due to their durability and recyclability. Until recently, only solid acids, all of which were modified with acidic groups, had been developed in CFP for LGO production. Metal oxides, zeolites and activated carbon (AC) are used as supports. For example, TiO2 itself shows a very limited effect on LGO production. By sol–gel mixing of Ti(OH)4/ferric hydrates and sulfuric acid, solid acids SO42−/TiO2 and SO42−/TiO2–Fe3O4 (further endowed with magnetism for recyclable collection) have been developed and exhibit higher LGO yields from cellulose than sulfuric acid.33 SO42−/TiO2–Fe3O4 further exhibits its potential in CFP of native biomass, showing a LGO yield of 7 wt% when poplar wood is used as the feedstock. A similar magnetic solid acid, Fe3O4/C–SO3H, exhibits 20 wt% LGO yield with enhanced pyrolysis kinetics, which enables a lowered activation energy (from 187.4 to 168.1 kJ mol−1) and a smaller deviation factor representing concentrated heating in TG analysis.34 SO42−/ZrO2 exhibits long-span LGO production with an 8 wt% yield in a continuous reactor.35 H3PO4-impregnated activated carbon (P-AC) also shows an LGO yield of 18 wt%.36
However, the recyclability of all the sulfonated/phosphated catalysts is five cycles or less due to a loss in the acidic groups. Therefore, the development of alternative Brønsted acid sites with higher durability is of great significance for CFP-based LGO production in the future.
Zeolites are ideal Brønsted/Lewis acid catalysts but can be endowed with stronger acid properties by bridging O–H groups with adsorbed water or further decorated with acidic groups in a porous Al/Si/O framework, which enables a variety of applications for biomass upgrading reactions such as dehydration, decarbonylation and decarboxylation.37 Lewis-acidic HZSM-5 shows a limited LGO yield, suggesting the key role of Brønsted acid sites in LGA dehydration.38 To improve the catalytic performance in CFP towards LGO production, SBA-15 and MCM-41 are modified with phosphate groups and grafted with Al3+ and Ni2+ species in the framework, respectively (Fig. 3c).32,39 The resulting POx/Al/SBA-15 and Ni–P-MCM-41 exhibit LGO yields of 21 wt% and 10 wt%, respectively, and more importantly, comparative yields in CFP of native biomass. It is found that the loaded Al3+ and Ni2+ species can accelerate water deprotonation and increase the overall Brønsted acidity. Notably, XPS/XRD analyses suggest that they maintain structural durability even after 300 °C in a fixed bad, demonstrating the feasibility of acidic group modified zeolites with cooperation of Lewis and Brønsted acid sites for large-scale LGO production.
Despite the achievements of these heterogeneous CFP processes, acidic groups (sulfonate and phosphate) are still the crucial species to promote LGA dehydration, with inevitable loss after each cycle. Moreover, when the acidity is too high, the nascent LGO will further undergo undesirable reactions such as isomerisation to HMF, re-condensation to coke, and/or retro-aldol to light oxygenates and ultimately to CO2.21 Nevertheless, heterogeneous CFP is the most promising approach for LGO production, provided that an alternative Brønsted acid without sulfonate and phosphate groups and with tuneable acidity is used.
3.2 Oxalic acid mediated C3–C6 cyclisation to produce DGP
In comparison, the production of DGP, a promising chiral planform with a rigid and multi-cyclic structure but without CO and CC bonds, through the CFP of cellulose is rarely reported. DGP is a common by-product in acid-based CFP for LGO production, with a selectivity less than 10%. Until recently, only Lu et al. had reported an oxalic acid–catalysed approach to producing DGP in a 14 wt% yield with in situ loading (oxalic acid:cellulose = 9:1) (Fig. 4a).19 Compared to aqueous acids such as H3PO4 and H2SO4, the moderate acidity of oxalic acid without introduction of water plays a key role in promoting competitive reactions against LGA formation, leading to the alternative production of DGP. This process starts from the formation of an unsaturated CC bond that is preferably formed between C1 and C2 of a glucose unit, followed by the esterification of the hydroxyls at C3 and C6, resulting in a 3,6-acetal ring in DGP instead of a ring between C1 and C6 of LGA. A higher temperature (400–450 °C) than that used in LGO production is required due to higher energy barriers, but this is lowered by oxalic acid, which is directly involved in transition states instead of decomposition into formic acid for protonation (Fig. 4b). For the first time, this work suggests that a specific structure enabling hydrogen-bond-based transition state formation can promote the alternative pathway in cellulose CFP. However, oxalic acid plays a “catalyst-like” role that is not similar to other heterogeneous catalysis in DGP production, since it is ultimately decomposed into formic acid and CO2. It is of great significance to develop alternative heterogeneous catalysts that can induce similar interactions to stabilise the transition state before intermediate cyclisation to produce this valuable but less-exploited chiral platform using sustainable CFP approaches.
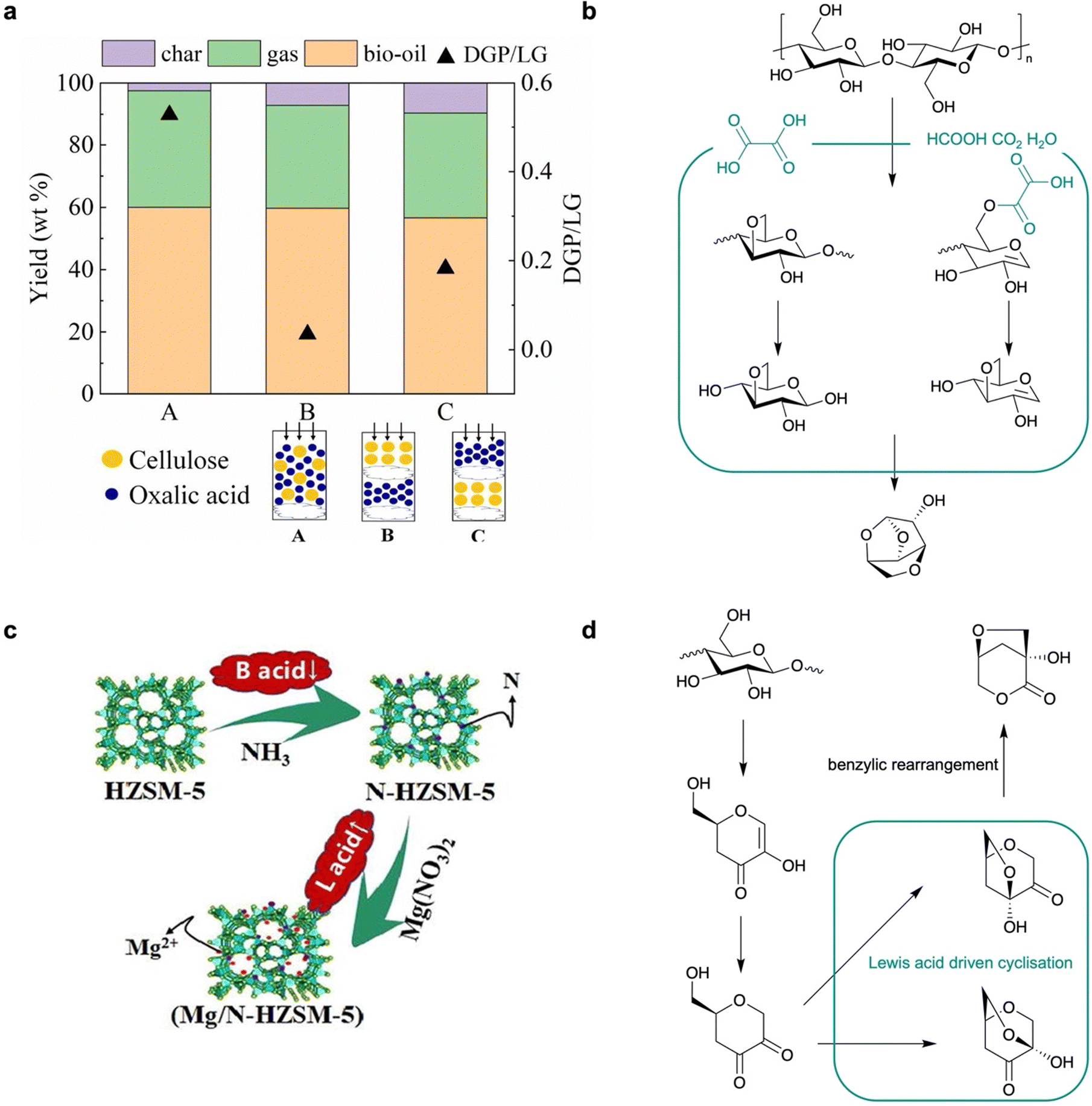 | ||
| Fig. 4 (a) Performance comparison of cellulose CFP with oxalic acid to produce DGP in different loading set-ups.18 (b) Reaction pathway of cellulose CFP to produce DGP. (c) Modification of HZSM-5 for cellulose CFP.40 (d) Reaction pathway of cellulose CFP to produce LAC. | ||
3.3 Lewis acid assisted benzylic rearrangement to produce LAC
In the presence of Lewis acid sites, cellulose CFP over metal oxides or metal-loaded zeolites can produce LAC with co-production of LGA or LGO. Fabbri et al. first reported an Al–Ti nanopowder catalysed slow pyrolysis of cellulose to produce LAC in a 6% yield, in which the separation and upgrading approaches to chiral amines further support the usefulness of LAC in the green synthesis of bioproducts.40 It was found that high temperatures (∼500 °C) can supress the generation of LGO to less than 1% in the bio-oil; the Lewis acid sites in the Al–Ti nanopowder can effectively coordinate with glucopyranoside intermediates to promote its benzylic rearrangement towards LAC, instead of Brønsted acid driven dehydration to produce LGP. Moreover, Lu et al. reported the CFP of cellulose to produce LAC in 33% selectivity using the Mg-modified nitride HZSM-5.41 To mitigate the deoxygenation capacity of HZSM-5, it is first nitridated by calcination in an NH3 atmosphere to block the Brønsted acid sites (Fig. 4c).4 Mg2+ species are further loaded into the mesoporous structure, which achieved a higher LAC yield than Fe-, Co-, Cu-, Ni- and Zn-loaded N-HZSM-5. The authors further developed a Ni–Sn layered double oxide (Ni–Ti LDO) with a 9% yield in cellulose CFP at lowered temperatures (320 °C).42 The LDO synthesized from layered double hydroxide (LDH) exhibits a special crystal structure of the NiSnO3 phase, providing enriched and active Lewis acid sites for cyclization, isomerization and benzyl rearrangement reactions towards LAC production (Fig. 4d). These works suggest that switching from Brønsted acid to Lewis acid catalysts in CFP can produce LAC as the main product instead of LGO or DGP, demonstrating the compatibility of versatile acid catalysts with CFP towards anhydrosugar production.3.4 Zeolite mediated bond cleavage to produce furans, light oxygenates and aromatics
With longer durations and/or ex situ catalyst loading, CFP can alternatively produce furans/light oxygenates or aromatics, in a complicated composition. In the presence of moderate Brønsted acid sites, all the initial products, including LGA and LGO, from cellulose depolymerisation will further undergo isomerisation and/or decarboxylation to produce furan compounds, or retro-aldol condensation to generate C2–C4 light oxygenates.20 Depending on the type of zeolite, the composition of bio-oil is tuneable, but it remains a mixture of these volatile compounds. Interestingly, Wang et al. demonstrated that in the different phases of the NaY zeolite, cellulose pyrolysis will lead to a different product scope.43 Furans are the dominant products on the surface of NaY, along with a small amount of light oxygenates from enhanced dehydration, acetal reaction and cyclisation of LGA. Anhydrous intermediates diffusing into the NaY pores will undergo hydrodeoxygenation (including decarbonylation and decarboxylation) to produce aromatics and phenols as the main products, likely due to enriched and stronger acid sites. Most zeolites promote secondary hydrodeoxygenation in cellulose CFP to produce aromatics or hydrocarbons, but NaY, H-form zeolite (CBV 400), and SAPO-series zeolites can alternatively produce furans.44,45 However, the underlying chemistry related to porosity, channel distance and/or acidity is still unclear. In general, a higher temperature (500–600 °C) is required compared to CFP for anhydrosugar production (300–400 °C), as C–C/C–O bond cleavage has a higher activation energy than dehydration, cyclisation and rearrangement (Fig. 5).46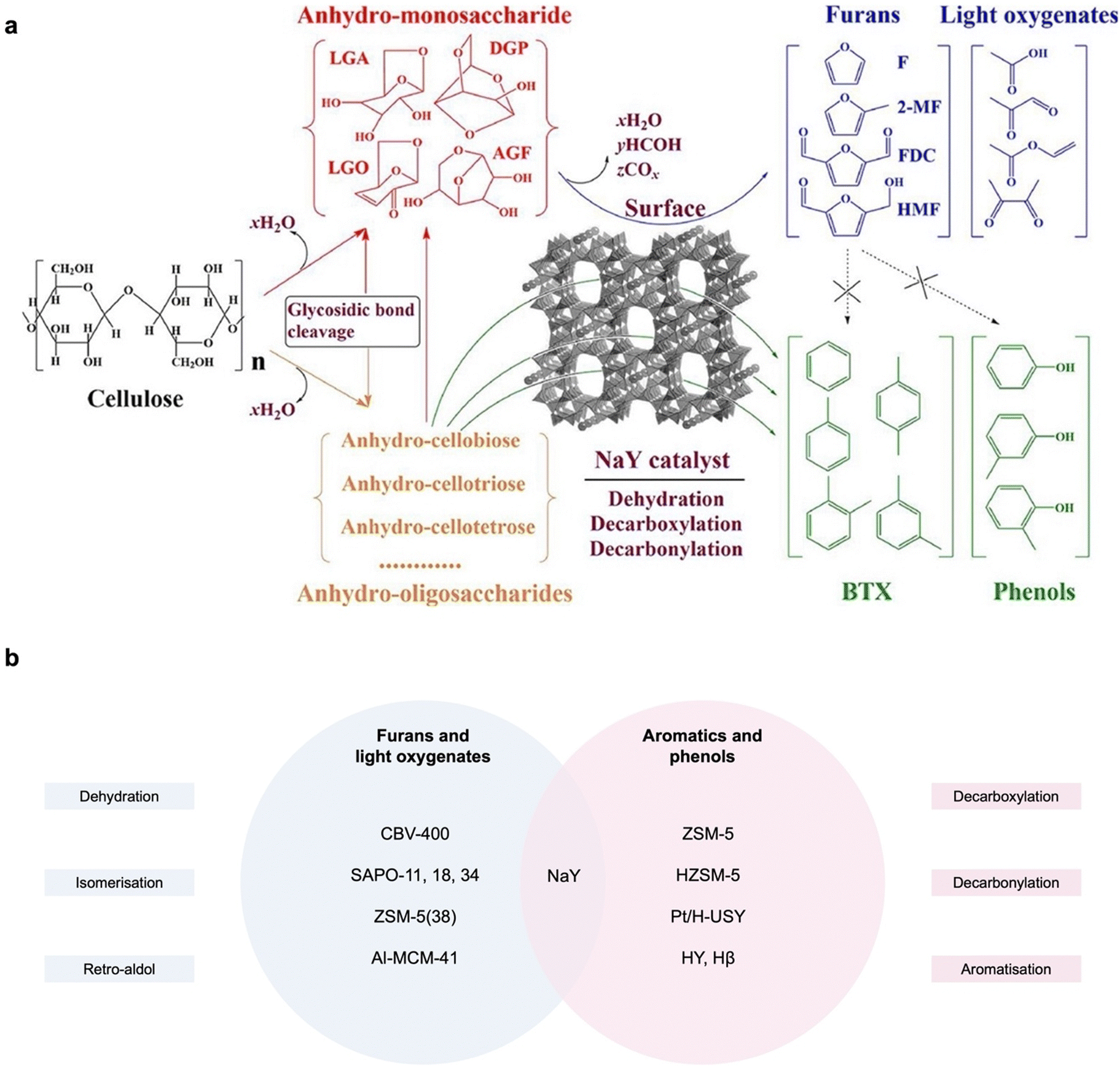 | ||
| Fig. 5 (a) Product scope of cellulose CFP on the surface or in the pores of NaY. (b) Reaction types of cellulose pyrolysis over zeolites to produce furan/light oxygenates or aromatics/phenols. | ||
4. Conclusion and future perspectives
Cellulose CFP is a highly efficient approach to directly converting native biomass into versatile precursors of fuels and fine chemicals. The ultrafast heating provides precise energy to specifically activate macromolecules in the biomass, thereby enabling high selectivity toward desirable products in the bio-oil. By employing different catalysts (Brønsted acids, Lewis acids, catalyst-like activators, metal active sites) in situ or ex situ, cellulose can undergo depolymerisation and subsequently a series of programmable reactions including dehydration, alternative cyclisation, rearrangement, isomerisation and retro-aldol condensation to produce LGA, LGO, DGP, LAC, furans and C2–C4 light oxygenates in several seconds. Notably, LGO, DGP, and LAC are promising building blocks for chiral pharmaceuticals, antibacterial agents and polymers, and can be produced in high-concentrated bio-oils with facile separation and cost effectiveness. The utilisation of heterogeneous catalysts with post-modifications (e.g., sulfonation, phosphorylation, metal loading, amination) further expands the reaction chemistry and process sustainability of CFP towards a bio-based circular economy. In CFP, homogeneous and heterogeneous catalysts each have unique strengths and weaknesses in terms of catalyst recyclability, product scope, reactor maintenance and mass/energy transfer (Fig. 6). | ||
| Fig. 6 Strengths and weaknesses of homogeneous and heterogeneous catalysts in CFP. | ||
However, there are still many uncertain aspects in terms of the reaction pathway, mass/species transfer and reactor engineering. The toolbox for studying biomass pyrolysis consisting of multiple reactions is lacking, as most of the previous work has only focused on reaction products and analyses such as thermogravimetric analysis (TGA) and pyrolysis-GC/MS.47In situ spectroscopy, especially the recently emerging photoelectron photo-ion coincidence spectroscopy (PEPICO), can directly detect intermediate species and facilitate a deeper understanding of the activation and transformation of functional groups in biomass molecules to confirm the existing mechanism from theoretical calculations.48–50 Moreover, operando characterisations of the catalyst surface and structure, such as microscopy, X-ray diffraction (XRD) and synchrony techniques, could effectively elucidate the role of active sites in CFP.51 In association with the previously developed thermodynamics and kinetics tools, molecular dynamics (MD) can facilitate a better understanding of mass and species transfer after their activation on the catalyst surface, as there will be a dynamic network derived from multiple covalent/noncovalent interactions within different components in biomass.52 As most of the volatile oxygenates produced in CFP are highly reactive, the rational design of reaction engineering with setting up coolers, condensers and phase separators is necessary for scalable CFP,53 by which an integrated valorisation of biomass and other waste resources (e.g., CO2, nitrate ions) is also possible in tandem CFP processes.54 Overall, combining catalyst design, thermochemistry and reaction engineering, there is a still long journey towards net-zero CFP biorefineries.
Data availability
There is no data availability in this review paper. All the data used to create figures are reproduced with permissions from previous publications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the HKPFS (PF20-48638) from Research Grants Council and the Presidential Scholarship from the University of Hong Kong.References
- M. S. Mettler, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 7797–7809 RSC.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- X. Wu, H. Jiang, H. Wang, F. Xu, H. Li, H. Zhang and S. Yang, Coord. Chem. Rev., 2024, 502, 215622 CrossRef.
- S. Wang, G. Dai, H. Yang and Z. Luo, Prog. Energy Combust. Sci., 2017, 62, 33–86 CrossRef.
- D. Neves, H. Thunman, A. Matos, L. Tarelho and A. Gómez-Barea, Prog. Energy Combust. Sci., 2011, 37, 611–630 CrossRef CAS.
- R. Liu, M. Sarker, M. M. Rahman, C. Li, M. C. Nishu, R. Cotillon and N. R. Scott, Prog. Energy Combust. Sci., 2020, 80, 100852 CrossRef.
- L.-Q. Jiang, Z. Fang, Z.-L. Zhao, A.-Q. Zheng, X.-B. Wang and H.-B. Li, Renewable Sustainable Energy Rev., 2019, 105, 215–229 CrossRef CAS.
- H. Zhang, Y.-T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 RSC.
- J. Liang, G. Shan and Y. Sun, Renewable Sustainable Energy Rev., 2021, 139, 110707 CrossRef CAS.
- L. Y. Jia, M. Raad, S. Hamieh, J. Toufaily, T. Hamieh, M. M. Bettahar, G. Mauviel, M. Tarrighi, L. Pinard and A. Dufour, Green Chem., 2017, 19, 5442–5459 RSC.
- X. Huang, J. Ren, J.-Y. Ran, C.-L. Qin, Z.-Q. Yang and J.-P. Cao, Fuel Process. Technol., 2022, 229, 107175 CrossRef CAS.
- Y. Zhang, W. Xu, N. Ma, Y. Shen, F. Xu, Y. Wang, N. Wu, Z. Guo and L. Jiang, Bioresour. Technol., 2022, 361, 127714 CrossRef CAS.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 1629–1636 CrossRef CAS.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS.
- J. D. Guthrie, C. E. R. Rowell, R. O. Anyaeche, K. Z. Alzarieni and H. I. Kenttämaa, Mass Spectrom. Rev., 2024, 43, 369–408 CrossRef CAS.
- M. S. Mettler, A. D. Paulsen, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 7864–7868 RSC.
- F. Cao, T. J. Schwartz, D. J. McClelland, S. H. Krishna, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
- S. H. Krishna, D. J. McClelland, Q. A. Rashke, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1278–1285 RSC.
- B. Hu, A. Cheng, Y. Li, Y. Huang, J. Liu, B. Zhang, K. Li, L. Zhao, T. Wang and Q. Lu, Chem. Eng. J., 2022, 436, 135200 CrossRef CAS.
- M. S. Mettler, S. H. Mushrif, A. D. Paulsen, A. D. Javadekar, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 5414–5424 RSC.
- S. Kudo, X. Huang, S. Asano and J. Hayashi, Energy Fuels, 2021, 35, 9809–9824 CrossRef CAS.
- M. B. Comba, Y. Tsai, A. M. Sarotti, M. I. Mangione, A. G. Suárez and R. A. Spanevello, Eur. J. Org. Chem., 2018, 590–604 CrossRef CAS.
- S. H. Krishna, T. W. Walker, J. A. Dumesic and G. W. Huber, ChemSusChem, 2017, 10, 129–138 CrossRef CAS.
- J. He, M. Liu, K. Huang, T. W. Walker, C. T. Maravelias, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 3642–3653 RSC.
- O. Oyola-Rivera, J. He, G. W. Huber, J. A. Dumesic and N. Cardona-Martínez, Green Chem., 2019, 21, 4988–4999 RSC.
- X. Sui, Z. Wang, B. Liao, Y. Zhang and Q. Guo, Bioresour. Technol., 2012, 103, 466–469 CrossRef CAS.
- B. Hu, Q. Lu, Y. Wu, W. Xie, M. Cui, J. Liu, C. Dong and Y. Yang, J. Energy Chem., 2020, 43, 78–89 CrossRef.
- S. Kudo, N. Goto, J. Sperry, K. Norinaga and J. Hayashi, ACS Sustainable Chem. Eng., 2017, 5, 1132–1140 CrossRef CAS.
- S. Kudo, Z. Zhou, K. Norinaga and J. Hayashi, Green Chem., 2011, 13, 3306 RSC.
- S. Saragai, S. Kudo, J. Sperry, U. P. M. Ashik, S. Asano and J. Hayashi, Bioresour. Technol., 2022, 344, 126323 CrossRef CAS PubMed.
- S. Sharma and U. P. Singh, Inorg. Chem. Commun., 2022, 139, 109359 CrossRef CAS.
- K. Li, B. Wang, D. Bolatibieke, D.-H. Nan, Z.-X. Zhang, M.-S. Cui and Q. Lu, J. Anal. Appl. Pyrolysis, 2020, 148, 104824 CrossRef CAS.
- Q. Lu, X. Ye, Z. Zhang, C. Dong and Y. Zhang, Bioresour. Technol., 2014, 171, 10–15 CrossRef CAS.
- L. Qian, F. Xu, S. Liu, G. Lv, L. Jiang, T. Su, Y. Wang and Z. Zhao, Cellulose, 2021, 28, 7579–7592 CrossRef CAS.
- Z. Wang, Q. Lu, X.-F. Zhu and Y. Zhang, ChemSusChem, 2011, 4, 79–84 CrossRef CAS.
- X. Ye, Q. Lu, X. Wang, H. Guo, M. Cui, C. Dong and Y. Yang, ACS Sustainable Chem. Eng., 2017, 5, 10815–10825 CrossRef CAS.
- L. Lin, A. M. Sheveleva, I. Da Silva, C. M. A. Parlett, Z. Tang, Y. Liu, M. Fan, X. Han, J. H. Carter, F. Tuna, E. J. L. McInnes, Y. Cheng, L. L. Daemen, S. Rudić, A. J. Ramirez-Cuesta, C. C. Tang and S. Yang, Nat. Mater., 2020, 19, 86–93 CrossRef CAS.
- X. Wei, Z. Wang, Y. Wu, Z. Yu, J. Jin and K. Wu, J. Anal. Appl. Pyrolysis, 2014, 107, 150–154 CrossRef CAS.
- J. A. Santander, M. Alvarez, V. Gutierrez and M. A. Volpe, J. Chem. Technol. Biotechnol., 2019, 94, 484–493 CrossRef CAS.
- D. Fabbri, C. Torri and I. Mancini, Green Chem., 2007, 9, 1374 RSC.
- Z. Zhang, B. Hu, Y. Li, K. Li and Q. Lu, Bioresour. Technol., 2020, 309, 123370 CrossRef CAS.
- Y. Li, K. Li, B. Hu, Z. Zhang, G. Zhang, S. Feng, T. Wang and Q. Lu, Ind. Crops Prod., 2021, 162, 113269 CrossRef CAS.
- W. Wang, Y. Shi, Y. Cui and X. Li, J. Anal. Appl. Pyrolysis, 2018, 131, 93–100 CrossRef CAS.
- H. Abou-Yousef and E. B. Hassan, J. Ind. Eng. Chem., 2014, 20, 1952–1957 CrossRef CAS.
- X. Chen, Y. Chen, Z. Chen, D. Zhu, H. Yang, P. Liu, T. Li and H. Chen, J. Anal. Appl. Pyrolysis, 2018, 129, 53–60 CrossRef CAS.
- K. B. Ansari, B. Kamal, S. Beg, Md. A. Wakeel Khan, M. S. Khan, M. K. Al Mesfer and M. Danish, Renewable Sustainable Energy Rev., 2021, 150, 111454 CrossRef CAS.
- S. Kelkar, C. M. Saffron, K. Andreassi, Z. Li, A. Murkute, D. J. Miller, T. J. Pinnavaia and R. M. Kriegel, Appl. Catal., B, 2015, 174, 85–95 CrossRef.
- Y. Zhang, W. Zhang, F. Zhang, Y. Xiao, G. Jia, W. Wang, F.-S. Ke and Z. Guo, Angew. Chem., Int. Ed., 2024, 63, 202402694 CrossRef.
- P. Hemberger, V. B. F. Custodis, A. Bodi, T. Gerber and J. A. van Bokhoven, Nat. Commun., 2017, 8, 15946 CrossRef.
- Z. Pan, A. Puente-Urbina, S. R. Batool, A. Bodi, X. Wu, Z. Zhang, J. A. van Bokhoven and P. Hemberger, Nat. Commun., 2023, 14, 4512 CrossRef CAS.
- J. Dou, Z. Sun, A. A. Opalade, N. Wang, W. Fu and F. Tao, Chem. Soc. Rev., 2017, 46, 2001–2027 RSC.
- G. Piccini, M.-S. Lee, S. F. Yuk, D. Zhang, G. Collinge, L. Kollias, M.-T. Nguyen, V.-A. Glezakou and R. Rousseau, Catal. Sci. Technol., 2022, 12, 12–37 RSC.
- A. Sharma, V. Pareek and D. Zhang, Renewable Sustainable Energy Rev., 2015, 50, 1081–1096 CrossRef CAS.
- H. Jiang, X. Wu, H. Zhang, Q. Yan, H. Li, T. Ma and S. Yang, SusMat, 2023, 3, 781–820 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |