
Robust interaction of cobalt phthalocyanine and nitrogen-doped ordered mesoporous carbon for CO2 reduction paired with the electro-oxidative synthesis of sulfonamide derivatives†
Samin
Barat-Abtahi
a,
Faranak
Jafari-Hafshejani
a,
Fahimeh
Varmaghani
 *ab,
Babak
Karimi
*ab and
Hamzeh H.
Veisi
a
*ab,
Babak
Karimi
*ab and
Hamzeh H.
Veisi
a
aDepartment of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan 45137-66731, Iran. E-mail: f.varmaghani@iasbs.ac.ir; Karimi@iasbs.ac.ir
bResearch Center for Basic Sciences and Modern Technologies (RBST), Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan 45137-66731, Iran
First published on 20th November 2023
Abstract
In this work, a high-performance hybrid electrocatalyst based on cobalt phthalocyanine combined with N-doped ordered mesoporous carbon (CoPc@GIOMC) was developed for CO2 reduction. To achieve this goal, we designed an innovative paired electrolyzer that integrates CO2 reduction to CO with the efficient oxidative coupling of certain phenylhydroxylamines to produce biologically important sulfonamide and organosulfone derivatives. The hybrid electrode exhibited a significant partial current density of 33 mA cm−2 in the selective reduction of CO2 to CO at an overpotential of 0.57 V vs. RHE (the onset potential was 0.3 V vs. RHE) and a faradaic efficiency of 90% in a near-natural aqueous solution (pH = 7.4). This excellent performance can be attributed to a high nitrogen content, numerous defect sites, and a large surface area of our N-doped mesoporous carbon material, which facilitated the strong and stable attachment of CoPc on GIOMC. Furthermore, in the context of coupling cathodic CO2 reduction with the synthesis of biologically important sulfonamide and organosulfone derivatives, the electrolysis system demonstrated a high anodic faradaic efficiency, reaching nearly 100% at 6 mA cm−2 after 135 minutes of electrolysis together with a satisfactory energy efficiency of up to 70%.
Introduction
The ever-increasing dependence on fossil fuel consumption has resulted in a continuous increase in carbon dioxide emissions in recent decades. Thus, to enhance the quality of life, protect the earth, and mitigate the effects of climate change, it is crucial to find solutions that either lower carbon dioxide emissions or develop innovative, sustainable methods for transforming carbon dioxide from fossil fuels into valuable and/or environment-friendly products. Among the wide range of products produced from CO2 conversion, CO is an important synthetic target, for it is utilized as an inexpensive C1 building block in chemical industries.1 In addition, a mixture of carbon monoxide and hydrogen in different proportions is mainly utilized for producing hydrocarbon-based fuels via the Fischer–Tropsch process.2Among the current methodologies, one of the most promising approaches to reduce the CO2 level is the electrochemical CO2 reduction reaction (eCO2RR), which can effectively transform waste CO2 into value-added chemicals and carbon-based fuels using electricity from renewable energy sources, environment-friendly reaction conditions, mild operations, and an atom efficient method.3–5 However, the electrolysis process in water is associated with certain challenges, such as the hydrogen evolution reaction (HER), a poor selectivity and activity for the desired products, and a high over-potential and current density. The use of catalysts and catalytic reactions can help to reduce energy consumption and enhance the selectivity and activity of the process.6,7
Numerous electrocatalysts have been extensively investigated for the electrochemical reduction of carbon dioxide (eCO2RR). Among them, molecular complexes, specifically cobalt phthalocyanine (CoPc), are of great interest owing to their highly active catalytic sites, ability to tune their electronic structure through ligand manipulation, well-defined structure-catalytic mechanism, and improved CO2 reduction for selective product formation.8–11 However, it has been observed that CoPc exhibits a significantly low electrical conductivity.12–14 Thus, to address this limitation, researchers have proposed the synthesis of hybrids consisting of CoPc and carbonaceous materials, which can serve as effective electrocatalysts. Indeed, carbonaceous materials have the potential to function as suitable substrates for anchoring CoPc, thereby presenting a significant opportunity for the development of highly efficient supported molecular catalysts. Among the various carbonaceous materials, ordered mesoporous carbons (OMCs) have attracted significant attention due to their abundant pores, regular structure, large and accessible surface area, and high electrical conductivity. These unique properties make OMCs highly promising for applications in energy storage devices and electrocatalysis.15–19 Furthermore, the introduction of nitrogen doping in OMCs enhances their performance, leading to exceptional characteristics. N-doped OMCs with a significant number of defect sites have been synthesized to increase their charge density and surface activity.20–22 N-doped OMCs have been increasingly applied in the eCO2RR in aqueous solution, given that they can possess a broad range of nitrogen contents and pore size distribution and potentially improve the electrode/electrolyte interface by increasing the wettability and conductivity.23–25 These materials can also serve as a platform or matrix to anchor molecular catalysts, offering great opportunities to build highly efficient supported molecular catalysts. In CoPc@N-OMCs, the high surface area of OMCs and abundant defect sites caused by the embedded nitrogen in the carbon framework facilitate the efficient loading of CoPc, whereas the strong π–π stacking interaction between CoPc and N-doped OMCs suppresses the problem of weak electrical conductivity.26 It is highly probable that the lone-pair electron associated with certain embedded nitrogen can also play a role in facilitating the efficient dispersion of CoPc centers on N-doped carbon platforms most likely through coordination with the vacant orbitals of cobalt centers.
Although substantial efforts have been dedicated to the development of efficient catalysts for eCO2RR by employing phthalocyanine-based carbon composites to date, there is still ample opportunity to enhance their efficiency. In recent years, our research efforts have been focused on the field of N-doped OMCs through the documentation of the synthesis process of various carbon materials derived from ionic liquids using the nanocasting technique. These materials have been utilized in diverse organic transformations as a support for various metal nanoparticles.27–32 Additionally, they have been employed in numerous significant electrochemical investigations owing to their exceptional electronic characteristics.33–37 In particular, Karimi et al. reported the application of guanine-rich ionic liquid-derived ordered mesoporous carbon (GIOMC)38 and studied the efficiency of this material as a catalyst for fructose dehydration38 and electrocatalytic oxygen reduction.36 GIOMC was synthesized by carbonizing a homogeneous mixture of 1-methyl-3-phenethyl-1H-imidazolium hydrogen sulfate and guanine as both carbon and nitrogen sources in the presence of SBA-15 mesoporous silica as a hard scaffold.38 Recently, we further showed the role of nitrogen in GIOMC for the spontaneous non-covalent attachment and electro-grafting of redox-active materials.37
Considering the high nitrogen content of GIOMC and its suitable electrocatalytic activity observed in our previous studies, we postulated that the utilization of this mechanically robust and electrically conductive N-doped carbon has the potential to significantly enhance the strong interaction between phthalocyanine and carbon, leading to the development of a stable electrode with improved electrochemical performance in eCO2RR. The initial idea behind this hypothesis is based on the fact that GIOMC with a high surface area and considerable number of nitrogen defect sites possesses at least two additional distinct characteristics, i.e., superior conductivity and hydrophobic framework. Altogether these characteristics may provide a means for strong interaction with the hydrophobic phthalocyanine aromatic rings and vacant Co orbitals, thereby greatly improving the electrochemical efficiency of the resulting phthalocyanine-based carbon composites.
Alternatively, one of the critical challenges in improving the efficiency of eCO2RR is the design of an appropriate electrolyzer. Generally, it is important to note that the oxidation of water, as the anodic reaction, occurs in a typical eCO2RR electrolyzer. This reaction is characterized by slow kinetics and necessitates high oxidation potentials. However, through the implementation of paired electrosynthesis, it is possible to replace the ineffective and useless water oxidation with a more advantageous anodic process that occurs at significantly lower oxidation potentials.39–41 Paired electrochemical systems can potentially lead to the production of valuable products at both the anode and cathode ends, resulting in up to a 50% reduction in energy consumption.42 Taking cues from these distinct advantages, several paired electrochemical methods have been introduced thus far to address this issue and increase the efficiency of eCO2RR simultaneously.42 In line with this, we hypothesized that a promising alternative to the kinetically slow formation of O2 during the undesirable oxidation of water may be accomplished by pairing the anodic production of medicinal-based products in aqueous and mild conditions with the cathodic eCO2RR, which is an innovative approach that has not been explored to date. Among the biological products, sulfonamides, also known as sulfa drugs, are an important class of chemical medicines that contain sulfonamide functional groups.43 Sulfonamides are inherently bioactive compounds that are commonly used as enzyme inhibitors and for correcting pathological conditions. Since the Second World War, sulfonamides have been widely used in the development of antimicrobial, antitumor, anti-inflammatory, and antiviral drugs, following their initial use in the treatment of severe injuries.44,45 Additionally, organosulfones have gained significance in the field of chemistry due to their biological activity and wide-ranging applications in medicinal chemistry.46 Consequently, it is important for the pharmaceutical industry to synthesize organosulfones, particularly various types of structurally diverse sulfonamides under environmentally friendly and mild conditions. Sulfonamides and organosulfones are typically synthesized through multiple steps and rigorous reactions. Nevertheless, there have been limited instances where green electrochemical methods have been utilized for the synthesis of these compounds.47–50
To test the above-mentioned hypotheses, we aimed to create a paired electrolysis setup for CO2RR. This setup involved using a hybrid of CoPc@GIOMC as the cathodic electrocatalyst, while simultaneously oxidizing phenylhydroxylamine derivatives and catechol in the presence of arylsulfinic acids in the anodic part. The goal was to produce five new and four known sulfonamide and organosulfone derivatives under environmentally friendly conditions. In this case, a hybrid material based on CoPc was prepared by impregnating CoPc in GIOMC. Subsequently, this material was used to modify the working electrode in a CO2 electrolyzer. In the second part of the current study, we developed a paired electrolysis system for the selective electro-reduction of CO2 to CO, which was coupled with the electrochemical synthesis of sulfonamides under environmentally friendly conditions. It was anticipated that the electrochemical oxidation of phenylhydroxylamine (or N-(p-tolyl)hydroxylamine, 4-(hydroxyamino)benzoyl chloride, or catechol) would occur in the anodic region in the presence of arylsulfinic acids. The lower oxidation potentials of these materials compared to water oxidation significantly reduced the applied voltage and increased the energy efficiency to a great extent.
Experimental
Chemicals
Chemicals and reagents including cobalt(II) phthalocyanine (CoPc), Nafion, P-123, tetraethyl orthosilicate, alkyl bromide, 1-methylimidazole, phosphoric acid, acetic acid, toluene, dichloromethane, sulfuric acid, hydrochloric acid, 4-chlorosulfinic acid, benzene sulfinic acid, 4-toluenesulfinic acid (TSA), nitrobenzene (NB), 4-nitrotoluene (NT), 4-nitrobenzoyl chloride (NBCl), ammonium chloride, zinc powder, dimethyl formamide, and sodium hydroxide were purchased from Merck and Sigma-Aldrich. Also, 2-bromo-1-phenylethane, guanine and cesium hydride were prepared from Across Systems. 5% Nafion (ethanolic solution) and Nafion 117 were purchased from Electrochemi Baspar Company. All chemicals were used as received with no further purification. Graphite paper (99%) was purchased from Nano-bazar.Material characterization
FTIR analysis was performed using a Bruker-Vector 22 instrument. A 200 kV Schottky field emitter transmission electron microscope (TEM) was used to determine the presence of pores and structures in the CoPc@GIOMC hybrid. The nitrogen adsorption–desorption analysis was performed using a BELSORP-MAX analyzer at 77 K. The materials were degassed for 6 h at 353 K before analysis. The BET method was used to determine the surface area. The Barrett–Joyner–Halena (BJH) and DH techniques were utilized to determine the pore size distribution. The surface morphology analysis and elemental mapping of the materials were performed using a scanning electron microscopy (TESCAN MIRA 2). The Raman spectra of GIOMC and CoPc@GIOMC hybrid were recorded using a Ram-532-004. Thermogravimetric analysis (TGA) was performed using a NETZSCH STA-404 in the temperature range of 25–700 °C under an N2 atmosphere. The possible liquid products during electrochemical CO2 reduction were assessed using a KNALIER D-14163 instrument with RI (K-2301) and UV (K-2600) detectors. XPS analysis was performed using a SPECS instrument, model: FlexPS. XRD data was obtained using an XRD Bruker D8 Advance. Gas chromatography was used for the analysis of the gas products on an Agilent 7890A. The GC was equipped with a Plot Q/molecular sieve column. Helium was used as the carrier gas with FID/TCD detectors. NMR analysis was performed using a Bruker-400 MHz instrument. The GC data for the determination of the anodic faradaic efficiency was recorded using a Varian CP-3800 gas chromatograph instrument (GC) equipped with a capillary column and flame-ionization detector (FID) using an internal standard method. UV-Vis spectra were obtained with a Cary 100 Bio. The pH values of the solutions were reported using a Metrohm pH meter. ICP-MS was performed on a PerkinElmer model: ELAN 9000.Electrochemical measurements
An Autolab PGSTAT 30 with a three-electrode system was used for cyclic voltammetry and chronoamperometry and a two-electrode setup used for the potential-time plots. The electrolysis in either non-paired or paired CO2 reductions and also electrochemical reduction of 4-nitrotoluene (NT) and 4-nitrobenzyl chloride (NBCl) were carried out using an Atomlab potentiostat with a digital Coulomb counter. Impedance (EIS) measurements were carried out using Zahner/Zennium instrument. All electrochemical experiments of eCO2RR were performed in a two-compartment H-type cell with Nafion 117 as the membrane separator. In the cyclic voltammetry experiments, the cell was equipped with a modified glassy carbon (GC) (surface area: 0.034 cm2) and a platinum wire as the working and counter electrode, respectively. A 0.7 cm2 piece of graphite paper modified with the CoPc@GIOMC hybrid was used as the working electrode in chronoamperometry and electrolysis under the controlled potential of CO2RR. A soft graphite plate (20 mm length, 5 mm width and 40 mm height) was utilized as the counter electrode. The constant current electrolysis was performed with CoPC@GIOMC/graphite paper as the cathode and graphite plate as the anode. The electrochemical reduction of NT and NBCl was performed in a divided home-made cell consisting of three soft graphite plates (surface area of each plate: 20 cm2) as the working electrode accompanied with a graphite plate (10 cm2) as the counter electrode. The working electrode for voltammetric studies of the anodic part during paired electrolysis was a bare glassy carbon electrode. The electrolyte in the cathodic part of the CO2 electrochemical cell was 0.5 M KHCO3 saturated with CO2 (pH = 7.4). The electrolyte in the anodic part of the CO2 electrochemical cell in the non-paired configuration was also 0.5 M KHCO3. In all electrochemical analyses, Ag/AgCl (3 M) was used as the reference electrode and converted to RHE according to eqn (1).| ERHE = EAg/AgCl + 0.059pH + 0.2 | (1) |
In the electrochemical data, iR corrections were exerted. The gas products were analyzed from the headspace of electrolysis cell by injection via a gas-tight syringe.
The faradaic efficiency (FE) was obtained according to eqn (2).
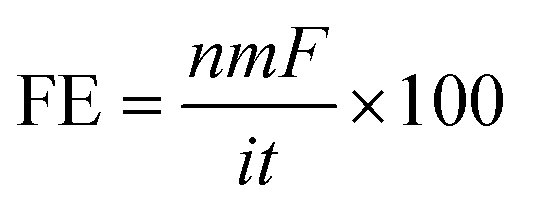 | (2) |
The partial current density of CO and H2 was calculated according to eqn (3).
| jCO or H2 = FECO or H2 × itotal | (3) |
Turnover Frequency (TOF) was calculated with eqn (4).
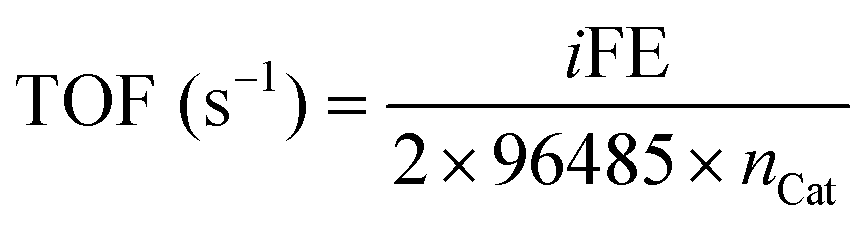 | (4) |
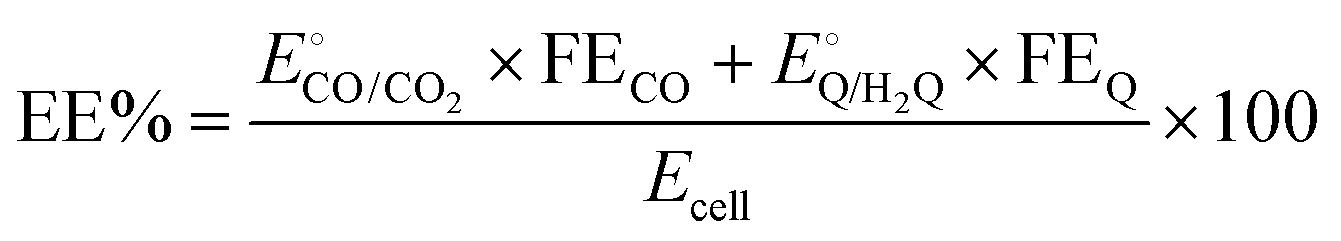 | (5) |
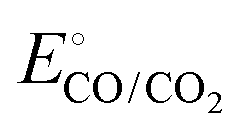 and
and 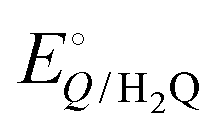 are −0.11 and 0.795 V (ref. 51) vs. SHE, respectively. The cathodic and anodic reactions were performed in a solution with pH values of 7.4 and 3.0, respectively. E° vs. RHE was determined to be −0.56 and +0.618 V for the reduction of CO2 to CO and oxidation of catechol (H2Q) to quinone (Q), respectively, according to the Nernst equation (eqn (6)).
are −0.11 and 0.795 V (ref. 51) vs. SHE, respectively. The cathodic and anodic reactions were performed in a solution with pH values of 7.4 and 3.0, respectively. E° vs. RHE was determined to be −0.56 and +0.618 V for the reduction of CO2 to CO and oxidation of catechol (H2Q) to quinone (Q), respectively, according to the Nernst equation (eqn (6)).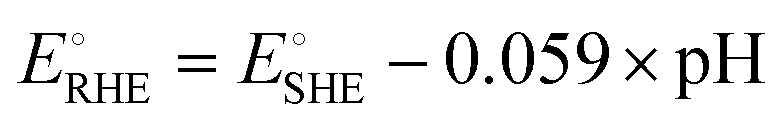 | (6) |
Preparation of CoPc@GIOMC hybrid
Firstly, 3 mg of GIOMC was dispersed in 2 mL of DMF under sonication for 30 min. Next, 2 mg of CoPc was added to 2 mL of DMF and stirred for 30 min. Then, 0.3 mL of CoPc in DMF was added to dispersed GIOMC, followed by 30 min sonication to obtain CoPC@GIOMC (0.9%). Different amounts of CoPc were used for other ratios of CoPC to GIOMC to prepare the other hybrids. In the next step, 20 μL of Nafion 5% (v/v) dissolved in ethanol was added to the suspension and further sonicated for 30 min. The hybrid was ready to use after 20 h stirring at room temperature.Modification of the GC electrode and graphite paper with CoPc@GIOMC hybrid
For the voltammetry studies, the GC electrode was polished with two pieces of felt cloth containing 0.3 and 0.05 μm alumina and thoroughly washed with water. Thereafter, 2 μL of ink was dropped on the surface of the GC electrode. In the chronoamperometry studies, the graphite paper was washed with acetone, and then modified by drop-drying 60 μL of the hybrid on 0.6 cm2 of the surface paper (loading: 1.37 × 10−8 mol CoPc).Chemical synthesis of hard template of SBA-15
Ordered mesoporous silica was synthesized based on the previous work.36,38 In brief, Pluronic acid (P-123) surfactant and tetraethyl orthosilicate as the silica source were mixed in acidic solution. Then, the mixture was placed in static conditions for 24 h to produce the desired silica mesoporous structure. Subsequently, the solid was filtered and washed several times with ethanol. The surfactant was removed after two days using Soxhlet apparatus and ethanol solvent.Synthesis of 1-methyl-3-phenethyl-1H-imidazolium hydrogen sulfate (MPIHS)
The preparation of MPIHS was described in the previous report.36,38 Briefly, 80 mmol 2-bromo-1-phenylethane and 73.1 mmol 1-methylimidazole were dissolved in 50 mL dry toluene. The solution was refluxed under an argon atmosphere for 24 h. At the end of the reaction, the contents of the flask were cooled to room temperature. The reaction mixture turned into two phases. The obtained ionic liquid was refluxed in 100 mL of dry toluene for 3 h, and then dried. To anion exchange Br− with HSO4−, 30 mmol of 1-methyl-3-phenylethyl imidazolium bromide (8 g) was added to a flask containing 100 mL of dry dichloromethane. Then 98% sulfuric acid (30 mmol, 1.6 mL) was slowly added to the flask in an ice bath and refluxed for two days. The solvent was evaporated under vacuum conditions and the obtained MPIHS was dried in a vacuum oven at 60 °C.Preparation of guanine-rich ionic liquid-derived ordered mesoporous carbon (GIOMC)
A solution of 0.5 g guanine and 5 g MPIHS at 100 °C was added to 1 g SBA-15. The resulting paste was carbonized at 800 °C for 3 h under argon gas. The silica template was removed by stirring with HF.36,38Chemical synthesis of phenylhydroxylamine (PHA)
Nitrobenzene (1 mL, 9.75 mmol) was dissolved in a mixture of ethanol/water (5 mL/10 mL) containing ammonium chloride (0.615 g, 11.5 mmol), and then vigorously stirred. While stirring, zinc dust (1.3 g, 20 mmol) was added over 20 min. Thereafter, the stirring was continued for another 20 min. The solution at the end of the reaction was filtered and washed with distilled hot water. Then, it was centrifuged until a clear and bright yellow solution of PHA was obtained.Electrochemical synthesis of N-(p-tolyl) hydroxylamine (THA) and 4-(hydroxyamino)benzoyl chloride (HABCl)
4-Nitrotoluene (1 mmol, 0.136 g) (or 4-nitrobenzoyl chloride (1 mmol, 0.184 g)) was added to an electrochemical cell containing ethanol/phosphate buffer (0.5 M, pH = 3.0) (20/80 v/v%). The electrolysis was carried out under a controlled potential condition at −0.38 V vs. Ag/AgCl until the substrate was completely converted to its reduced form at the surface of the cathode. The progress of the reaction was monitored by cyclic voltammetry.Electro-organic synthesis of sulfonamide and organosulfone derivatives in the anodic part of an H-type cell
PHA (or THA, HABCl or catechol) was added to the anodic part of an H-type cell containing 4-toluenesulfinic acid sodium salt (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) (benzensulfinic acid or 4-chlorosulfinic acid) in 100 mL ethanol/phosphate buffer (0.5 M, pH = 3.0) (20/80 v/v%) under stirring. At the end of the reaction, the solvent was evaporated until the white solid product appeared significantly. The obtained solid was washed with water and dried at room temperature. The product was characterized employing spectroscopic techniques.
:1 ratio) (benzensulfinic acid or 4-chlorosulfinic acid) in 100 mL ethanol/phosphate buffer (0.5 M, pH = 3.0) (20/80 v/v%) under stirring. At the end of the reaction, the solvent was evaporated until the white solid product appeared significantly. The obtained solid was washed with water and dried at room temperature. The product was characterized employing spectroscopic techniques.
Results and discussion
Synthesis and structural properties of CoPc@GIOMC hybrid
Firstly, GIOMC was prepared by carbonizing a homogeneous mixture of 1-methyl-3-phenethyl-1H-imidazolium hydrogen sulfate and guanine as both the carbon and nitrogen sources in the presence of SBA-15 mesoporous silica as a hard scaffold following our previous reported procedure. The XPS analysis of GIOMC showed the presence of carbon (75.6%), oxygen (10.6%) and high content of nitrogen (13.8%). Deconvolution of the nitrogen region showed four signals related to pyridinic, pyrrolic, graphitic and nitros-oxide nitrogens. The TEM analysis together with the BET and BJH results revealed well-ordered mesoporous structures with a regular arrangement of meso channels, reflecting the ideal replication of hexagonal structure of SBA-15 and narrow pore distribution.36,38The CoPc@GIOMC hybrid was obtained by adding an optimal volume (0.3 mL) of a DMF solution of CoPc to a pre-sonicated GIOMC suspension in DMF and sonicating the mixture, as described in the Experimental section. Among the solvents investigated, including DMF, acetone, and ethanol, DMF was determined to be a notably more appropriate solvent due to its effective carbon dispersion capabilities. Numerous reports have been published on the investigation and elucidation of the π–π stacking interaction between metal phthalocyanine and carbon structures.52–56 Therefore, the hybridization of CoPc and GIOMC was also expected to enhance eCO2RR because of the strong interaction between CoPc and the carbon structure, generating π–π stacking interaction between the phthalocyanine ligand and carbon plane in a similar way. It was also supposed that the direct interaction of the immobilized Co species and embedded nitrogen in the nanospaces of GIOMC (Co–N interactions) modulates the electronic properties of the Co atom. In addition, the high electrical conductivity of GIOMC was confirmed in our recent report.37 Therefore, it is reasonable to speculate that the high conductivity of GIOMC can reduce the electrical resistance. Consequently, any applied potential during electrolysis would facilitate the electron transfer process rather than having to overcome the resistance. With this hypothesis, a series of the hybrids was prepared with different weight ratios of CoPc and GIOMC.
Given that the Co atom plays a key role in electrocatalytic reduction, the obtained hybrids were labeled according to their theoretical Co percentage. The SEM images of GIOMC and CoPc@GIOMC (0.9%) are shown in Fig. 1I and II, respectively, indicating that the incorporation of CoPc in GIOMC did not significantly affect the carbon structure. The elemental mapping of the hybrid (Fig. 1III) confirmed the uniform distribution Co species throughout the surface of GIOMC, thus strongly highlighting the notion that the nitrogen is crucial for the effective dispersion of CoPc. This is most likely due to the acidic properties of the Co species and basic nature of the embedded nitrogen atoms. This strong affinity of Co to nitrogen with electron-donating properties ensures the diffusion of CoPc into the nanospaces of GIOMC, thus providing a means for the homogeneous distribution of CoPc on the CoPc@GIOMC hybrid. The amount of Co loaded on the electrode, which was calculated with ICP-MS, was 0.85%, in good agreement with the theoretical value of 0.9%. This indicates that the majority of the CoPc complexes were anchored in the hybrid structure. The structural pattern of the hybrid was investigated using TEM. Fig. 2 and Fig. S1† show the ordered arrays of carbon rods replicated from the pores of SBA-15. Selected area electron diffraction (SAED) of different parts of the hybrid did not show a crystalline structure related to CoPc in the hybrid, which is most likely due to its efficient distribution in the carbon structure. Fig. 3I displays the XRD pattern of CoPc, GIOMC and CoPc@GIOMC (0.9%). The diffraction peaks at 2θ = 7.58° and 9.82° for CoPc correspond to the (100) and (102) crystal planes of β-CoPc, respectively. The XRD pattern of the hybrid did not reveal any clear characteristic peaks related to CoPc, which can be attributed to either the low loading of CoPc on GIOMC or the strong interaction of each CoPc molecule with the graphitic planes of GIOMC. Fig. 3II shows a comparison of the Raman spectra of GIOMC and CoPc@GIOMC (0.9%). The Raman spectrum of GIOMC shows two distinct peaks corresponding to the D and G bands at 1334 and 1566 cm−1, respectively. It is well-known that the G band is due to the graphite nature of the carbon structure and the D band is related to structural defects and edge effects. The relative ratio of the D to G bands (ID/IG) is more than 1, indicating the presence of significant defect sites in the structure of GIOMC due to the presence of nitrogen in GIOMC. Generally, the presence of heteroatoms increases the number of defect sites in carbon structures. It is worth noting that the ratio of the D and G bands in the CoPc@GIOMC hybrid increased to 1.3, which is attributed to the increase in the number of defect sites in the hybrid by introducing CoPc in the structure. Moreover, the increase in ID/IG may be a result of the close interaction of CoPC with GIOMC via π–π stacking interaction and the change in the aromaticity of the carbon structures.52–56 A close look of the spectra revealed two additional signals located at 1540 and 1480 cm−1, corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–N bonds of CoPc, respectively. Furthermore, the slight shift in the G band (from 1566 to 1556 cm−1) confirms the robust interaction via π–π stacking interaction of CoPc to GIOMC.52–56 The structural properties of the CoPc@GIOMC hybrid were also analyzed by means of FTIR spectroscopy. Fig. S2† shows the vibration bands of CoPc at 733, 1286, 1333, 1462 and 3677 cm−1, which can be assigned to Co–N, C–N, CN, and CC of the macrocyclic ring and C–H, respectively. In addition, Fig. 3III shows a comparison of the FTIR spectra of GIOMC and CoPC@GIOMC (0.9%), revealing the appearance additional characteristic signals at 680, 1117, 1261 and 3149 cm−1, which are related to the vibration of Co–N, C–N bending, C–N stretching of isoindole and C–H stretching, respectively. The signals appearing at 2925 and 2958 cm−1 are possibly related to the stretching vibration of the C–H bonds in DMF, which is trapped in GIOMC. Additionally, the shift in the vibration bands of CoPc in the hybrid compared with that of free CoPc indicates the interaction of CoPc with surface functional groups of GIOMC. On the other hand, the signals of GIOMC also shifted, further confirming the strong interaction of CoPc with a carbon structure. Fig. 3IV displays the nitrogen adsorption–desorption diagram and Fig. 3V shows the pore size distribution for both GIOMC and CoPc@GIOMC (0.9%) in GIOMC. Both plots show the pattern of type IV isotherm in the pressure ratio (P/P0) range of 0.4 to 0.8, which reflects the characteristic of mesoporous structures. The total pore volume and BET surface of the pristine GIOMC decreased from 0.443 m3 g−1 and 545.8 m2 g−1 to 0.24 cm3 g−1 and 200.3 m2 g−1 for CoPc@GIOMC (0.9%), respectively. This clearly indicates the successful incorporation of CoPc in the mesochannels of GIOMC. TGA analysis under N2 atmosphere was comparatively performed for GIOMC, CoPc and the CoPc@GIOMC (0.9%) hybrid (Fig. S3†). GIOMC showed a small weight loss below 100 °C, which is most likely attributed to water or other surface adsorbed species. The weight loss gradually started and increased from 200 °C onward, indicating the decomposition of the carbon structure. On the contrary, CoPc@GIOMC (0.9%) tended to exhibit greater stability than GIOMC against weight loss under identical conditions, demonstrating a possible synergistic stabilizing effect between the supported CoPc species and GIOMC.
N and C–N bonds of CoPc, respectively. Furthermore, the slight shift in the G band (from 1566 to 1556 cm−1) confirms the robust interaction via π–π stacking interaction of CoPc to GIOMC.52–56 The structural properties of the CoPc@GIOMC hybrid were also analyzed by means of FTIR spectroscopy. Fig. S2† shows the vibration bands of CoPc at 733, 1286, 1333, 1462 and 3677 cm−1, which can be assigned to Co–N, C–N, CN, and CC of the macrocyclic ring and C–H, respectively. In addition, Fig. 3III shows a comparison of the FTIR spectra of GIOMC and CoPC@GIOMC (0.9%), revealing the appearance additional characteristic signals at 680, 1117, 1261 and 3149 cm−1, which are related to the vibration of Co–N, C–N bending, C–N stretching of isoindole and C–H stretching, respectively. The signals appearing at 2925 and 2958 cm−1 are possibly related to the stretching vibration of the C–H bonds in DMF, which is trapped in GIOMC. Additionally, the shift in the vibration bands of CoPc in the hybrid compared with that of free CoPc indicates the interaction of CoPc with surface functional groups of GIOMC. On the other hand, the signals of GIOMC also shifted, further confirming the strong interaction of CoPc with a carbon structure. Fig. 3IV displays the nitrogen adsorption–desorption diagram and Fig. 3V shows the pore size distribution for both GIOMC and CoPc@GIOMC (0.9%) in GIOMC. Both plots show the pattern of type IV isotherm in the pressure ratio (P/P0) range of 0.4 to 0.8, which reflects the characteristic of mesoporous structures. The total pore volume and BET surface of the pristine GIOMC decreased from 0.443 m3 g−1 and 545.8 m2 g−1 to 0.24 cm3 g−1 and 200.3 m2 g−1 for CoPc@GIOMC (0.9%), respectively. This clearly indicates the successful incorporation of CoPc in the mesochannels of GIOMC. TGA analysis under N2 atmosphere was comparatively performed for GIOMC, CoPc and the CoPc@GIOMC (0.9%) hybrid (Fig. S3†). GIOMC showed a small weight loss below 100 °C, which is most likely attributed to water or other surface adsorbed species. The weight loss gradually started and increased from 200 °C onward, indicating the decomposition of the carbon structure. On the contrary, CoPc@GIOMC (0.9%) tended to exhibit greater stability than GIOMC against weight loss under identical conditions, demonstrating a possible synergistic stabilizing effect between the supported CoPc species and GIOMC.
 | ||
| Fig. 1 The SEM images of (I) GIOMC, (II) CoPc@GIOMC (0.9%) hybrid, and (III) elemental mapping analysis of C, O, N, and Co. | ||
 | ||
| Fig. 2 The TEM image of the CoPc@GIOMC (0.9%) hybrid. | ||
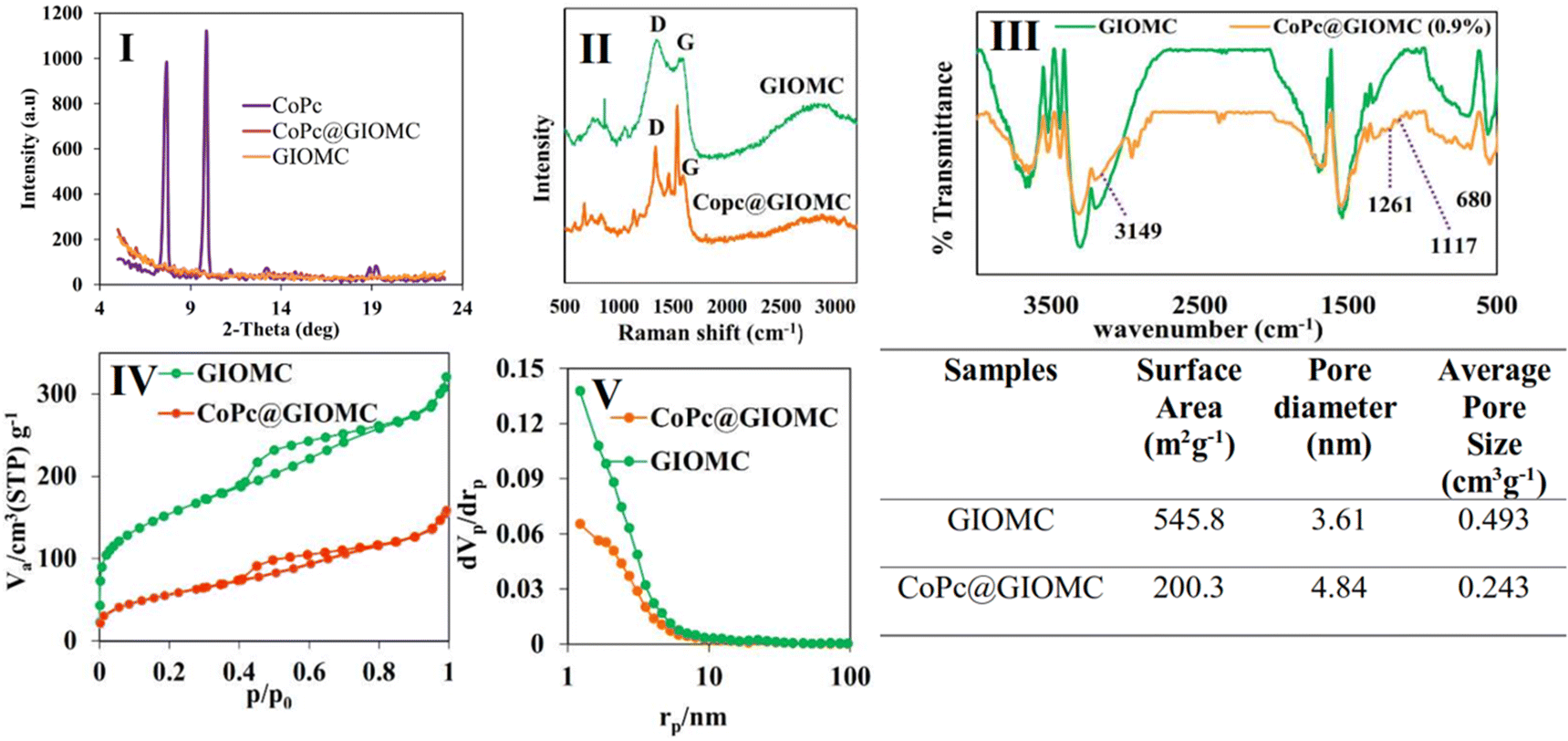 | ||
| Fig. 3 (I) The XRD pattern of pure CoPc, CoPc@GIOMC (0.9%) hybrid, and pure GIOMC. (II) The Raman spectra, (III) FTIR spectra, (IV) N2 adsorption–desorption isotherm, and (V) pore size distribution of GIOMC and the CoPc@GIOMC (0.9%) hybrid. Table: surface area, pore diameter, and average pore size of GIOMC and the CoPc@GIOMC (0.9%) hybrid. | ||
Electrocatalytic behavior of CoPc@GIOMC electrocatalyst toward eCO2RR in non-paired electrochemical cell
After the characterization of CoPc@GIOMC (0.9%), next we studied the electrocatalytic behavior of the electrode modified with CoPc@GIOMC (0.9%) using cyclic voltammetry with a three-electrode system in an H-type cell by drop-coating 2 μL of ink on the working electrode. As low as 4.5 × 10–10 mol of CoPc was loaded on the electrode surface and the voltammetric responses of the modified electrode in 0.5 M KHCO3 aqueous solution were measured. The potential was swept toward negative potentials in the range of +0.5 to −1.1 V vs. RHE in 5 mV s−1 in solution under two different conditions, i.e., N2-solution saturated and N2-plus CO2-saturated solution (Fig. 4I). The current density significantly increased in the CO2-saturated solution, clearly confirming the electrocatalytic reduction of CO2 with an onset potential of −0.3 V vs. RHE (overpotential of −0.21 V for CO in pH = 7.4). Voltammetry was further conducted in CO2-saturated 0.5 M KHCO3 solution with the electrode modified with different ratios of CoPc to GIOMC to optimize the amount of CoPc (Fig. 4II). As can be clearly seen, the modified electrode exhibited a low current density at high CoPc concentrations (6.3% and 7.8%), which is most probably due to CoPc aggregation on the electrode surface. The aggregated CoPc molecules have low conductivity, which can significantly reduce the catalytic efficiency, and consequently, the current density. It is noteworthy that the current density was smoothly increased by decreasing the amount of loaded CoPc on GIOMC, demonstrating the higher activity of the Co species due to the strong interaction of the individual Co centers with the embedded nitrogen atoms and robust anchoring of CoPc by π–π stacking interaction.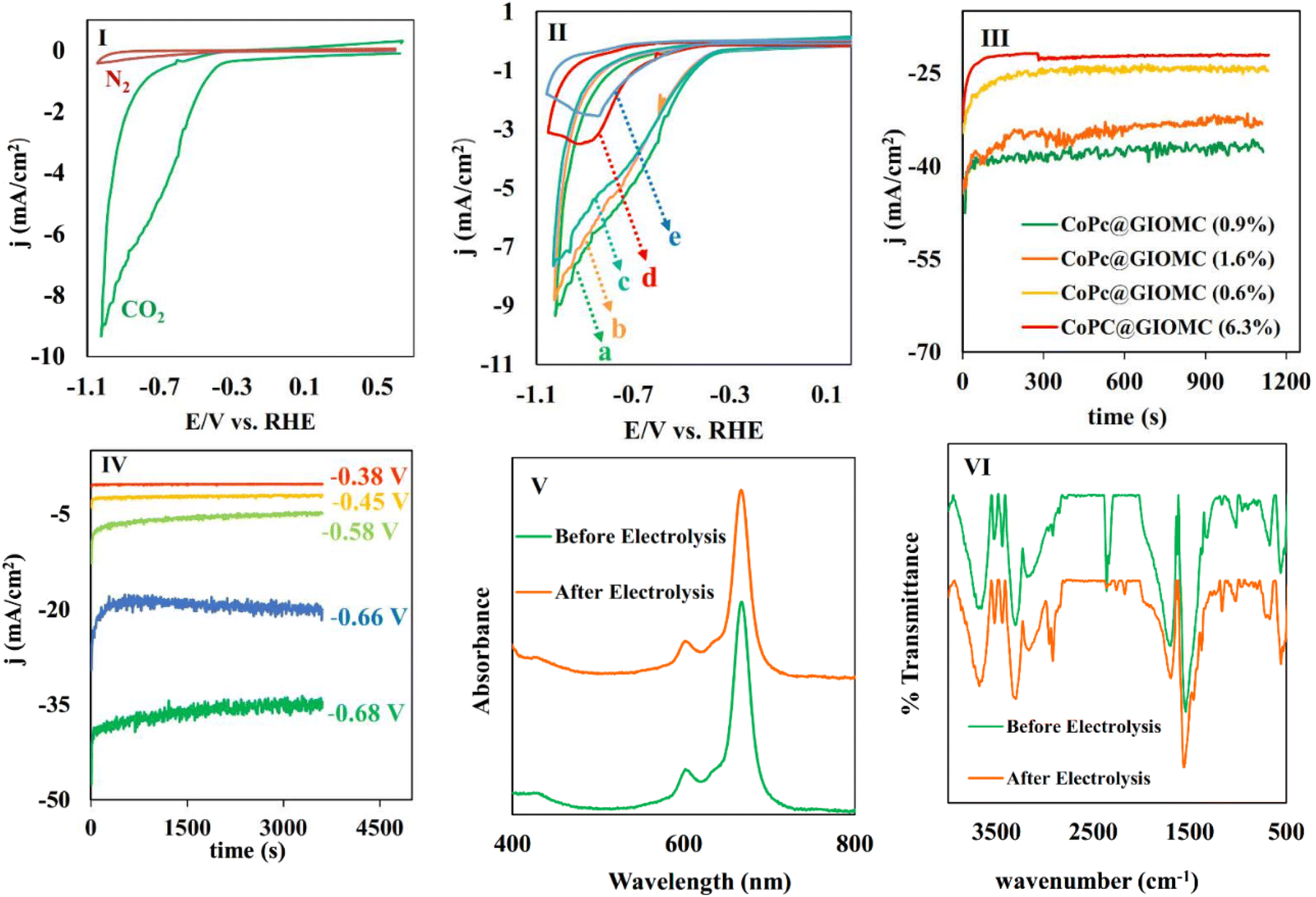 | ||
| Fig. 4 (I) The cyclic voltammograms of the CoPc@GIOMC (0.9%)-modified electrode in N2- and CO2-saturated 0.5 M KHCO3 solution at 5 mV s−1; (II) the cyclic voltammograms of the electrode modified with CoPc@GIOMC with various Co loadings of (a–e) 0.9%, 1.6%, 0.6%, 6.3%, and 7.8%; (III) the chronoamperograms of CoPc@GIOMC with various ratios of CoPc to GIOMC at −0.68 V vs. RHE; (IV) the chronoamperograms of CoPc@GIOMC (0.9%) at different potentials in CO2-saturated 0.5 M KHCO3, (V) the UV-vis spectra of CoPc loaded on the electrode before and after 1 h electrolysis at −0.66 V; and (VI) a comparison of the FTIR spectra of CoPc@GIOMC loaded on the electrode before and after 1 h electrolysis at −0.66 V. | ||
Linear sweep voltammetry (LSV) curves were also recorded for GC electrodes modified with CoPc@GIOMC with different percentages of Co, as well as for the electrodes modified with pure CoPc and pure GIOMC (Fig. S4†). To ensure a valid comparison, the quantities of pure CoPc and pure GIOMC deposited onto the electrode were adjusted to match the amount used for CoPc@GIOMC (0.9%). As clearly observed, the current density observed for the electrode modified with GIOMC was much lower than that of the hybrids with 0.9% to 1.6% Co. The low current density of the electrode modified with GIOMC indicated that it did not play a direct role in the reduction of CO2. The significant increase in current density observed in the hybrid with 0.9–1.6% of Co indicates that GIOMC is a suitable support for CoPc. The remarkable increase in the current density was due to the super electrical conductivity of GIOMC, the considerable nitrogen content, and the high surface area of the mesoporous carbon. In addition, although the amount of CoPc in the electrode modified with pure CoPc and CoPc@GIOMC (0.9%) was the same, the current density observed in the hybrid was significantly higher. This demonstrates the effectiveness of GIOMC in enhancing the conductivity, and also effectively isolating the Co centers on the electrode surface. The CoPc@GIOMC (0.9%)-modified electrode was finally chosen as the actual electrocatalyst system having the optimal loading of CoPc for the subsequent studies. Lower CoPc values than 0.9% were found to be less efficient because of the presence of insufficient electro-active sites.
In the next stage, we continued our studies by conducting a chronoamperometry experiment under the same conditions of cyclic voltammetry using a piece of graphite paper (0.7 cm2) as the working electrode. To ensure that the optimal CoPc/GIOMC ratio that resulted in the best output in the voltammetry studies also worked well for the chronoamperometry investigations, the same screening using different CoPc@GIOMC ratios was also conducted in the chronoamperogram measurement (Fig. 4III). Accordingly, it was found that the modified electrode comprised of CoPc@GIOMC samples with 6.3%, 1.6% and 0.6% of CoPc resulted in inferior activity compared to the material having 0.9% Co species. These results once again confirmed that the highest current density can be obtained in the chronoamperometric studies using modified electrode with CoPc@GIOMC (0.9%). The electrochemical performance of CoPc@GIOMC (0.9%) was evaluated at different applied potentials in a CO2 atmosphere and its chronoamperograms were recorded (Fig. 4IV). The current density sharply increased at −0.66 V vs. RHE, reaching the maximum of 37 mA cm−1 by exerting an overpotential of 0.57 V, which indicates its significant activity toward eCO2RR. The reduction of CO2 at a potential −0.66 V was easily performed without any kinetic restrictions. The presence of mesochannels and high porosity of the CoPc@GIOMC hybrid material increased the mass transfer of the electrolyte and CO2. Consequently, even with a slight increase in potential, the current density experienced a significant increase. For electrochemical catalysis using modified electrodes, the stability of the electrode system and steady-state and reproducible responses are important issues that should be resolved. Therefore, the chemical and physical stability of the CoPc-supported species were assessed by comparing the UV-Vis spectra of the CoPc@GIOMC (0.9%) modified electrode before and after 1 h of electrolysis. This study showed that CoPc remained largely stable on the surface of the electrode after electrolysis. Subsequently, to shed light on this observation, the UV-Vis spectra were also obtained by dissolving CoPc from the surface of the spent electrode in DMF with sonication and measuring the absorbance of the resulting solution (Fig. 4V). The same experiment was also applied to a solution obtained by dissolving CoPc from the fresh electrode surface before electrolysis employing essentially the same protocol. The absence of any remarkable difference between the UV spectra of two solutions of CoPc clearly confirmed the high stability of CoPc on the electrode (Fig. 4V). The stability of the hybrid materials was further confirmed by FTIR spectroscopy (Fig. 4VI). The FTIR spectra of CoPc@GIOMC (0.9%) on the electrode before and after 1 h electrolysis were recorded and compared. There was no significant difference in these spectra, indicating that CoPc did not apparently undergo any chemical or physical changes during electrolysis. X-ray photoelectron spectroscopy (XPS) was performed on the hybrid before electrolysis and after 1 h electrolysis at −0.66 V. Fig. 5 shows the cobalt photoemission region for the CoPc@GIOMC (0.9%) hybrid. It should be noted that the high-resolution XPS core spectrum for Co2p in the fresh CoPc@GIOMC hybrid could be easily fitted. In both cases, Co 2p3/2 and Co 2p1/2 were deconvoluted using a peak separation of 14.62 eV and the peaks constrained to a 3:2 area ratio at binding energies (BE) of 781.27 eV and 795.89 eV, respectively. These signals can be attributed to the presence of Co2+ oxidation state (Fig. 5I). It is very important to note that the XPS spectrum of the CoPc@GIOMC (0.9%) sample after 1 h electrolysis remained almost unchanged, confirming the high stability of the CoPc species under the described conditions (Fig. 5II). In addition, the absence of any additional detectable intensities in both samples also indicated that the presence of nitrogen in GIOMC did not apparently affect the oxidation state of the cobalt species. After ensuring the stability and high activity of the CoPc@GIOMC (0.9%)-modified electrode under the indicated electrochemical conditions, we turned our attention to studying the selectivity of the present electro-catalyst toward eCO2RR. Here, the products of electrolysis were analyzed by GC and HPLC for gaseous and liquid products, respectively (Fig. 6I).
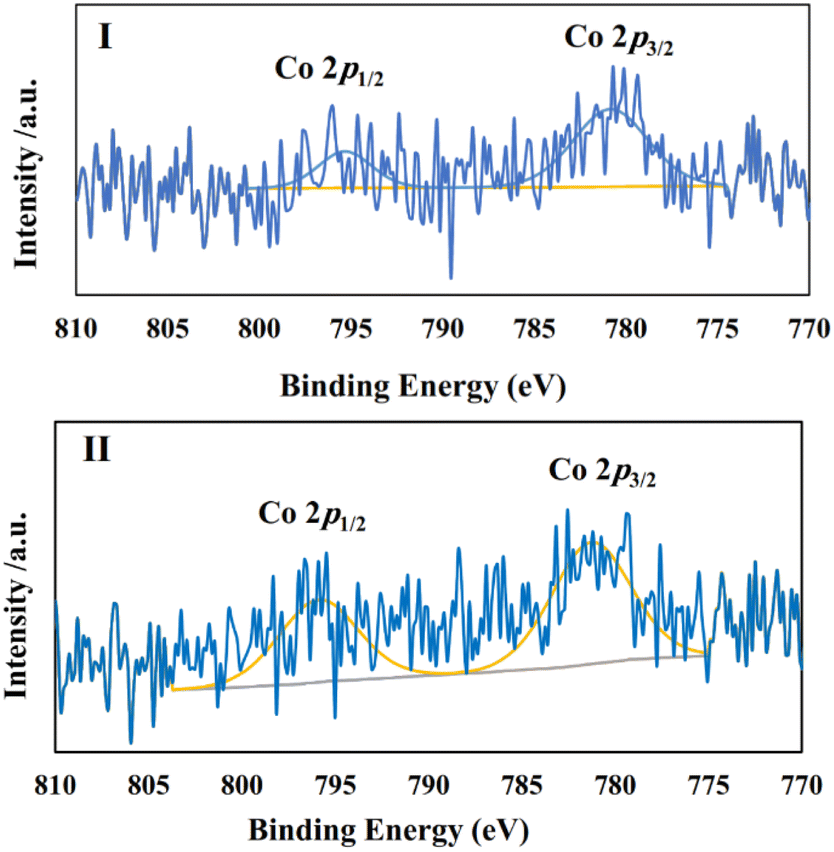 | ||
| Fig. 5 XPS spectra for the Co 2p region of CoPc@GIOMC (0.9%) (I) before electrolysis and (II) after 1 h electrolysis at −0.66 V vs. RHE. | ||
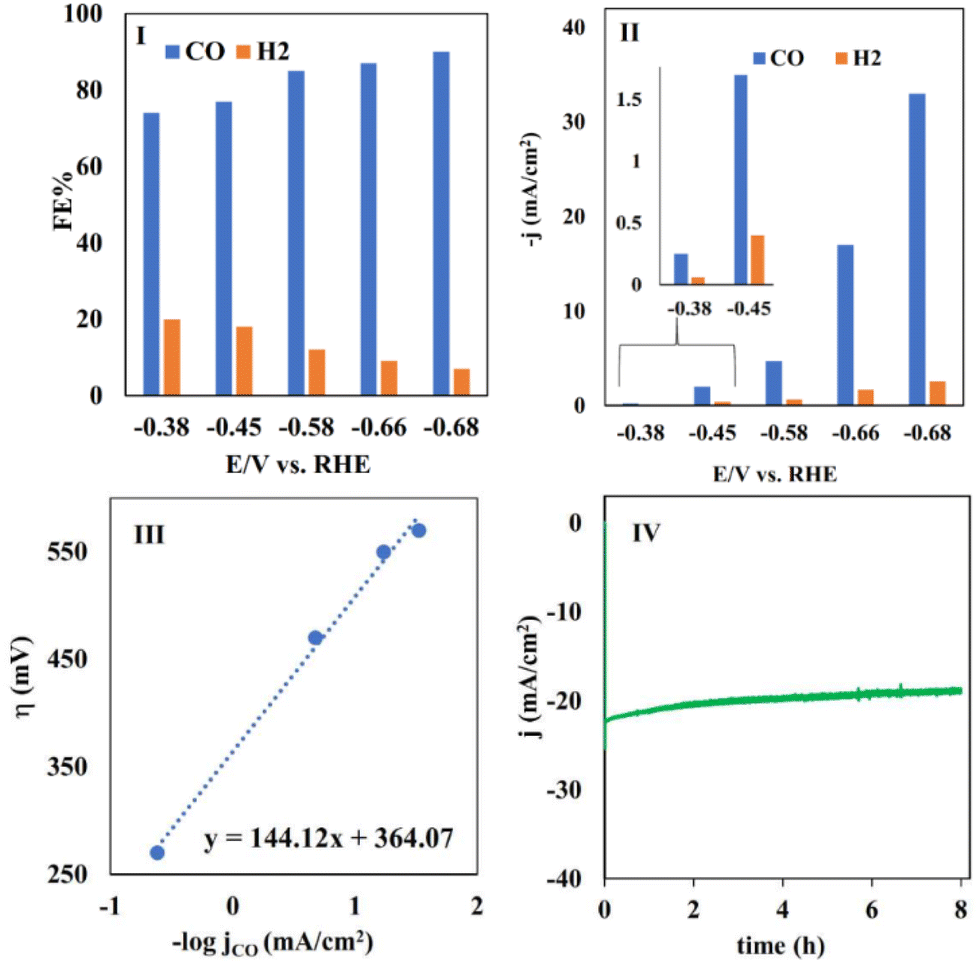 | ||
| Fig. 6 (I) The FE and (II) partial current density of CO and H2 measured in the electrolysis of CO2 at different potentials using CoPc@GIOMC (0.9%) in the same solution as mentioned in Fig. 4I. (III) A plot of η vs. −logjCO for CO production in the electrolysis of CO2 using CoPc@GIOMC (0.9%) at various overpotentials in 0.5 M KHCO3, and (IV) the recorded current density during 8 h electrolysis of CO2 at −0.66 V vs. RHE. | ||
No liquid product was detected, and carbon monoxide (CO) was produced as the main product, together with small quantities H2. This high selectivity towards the formation of carbon monoxide can undoubtedly be attributed to the significance of the presence of cobalt centers in our hybrid-based electrode system. The faradaic efficiency (FE) was calculated in the potential range of −0.38 to −0.68 V vs. RHE. The FECO reached the maximum of 90% at −0.68 V versus RHE, with a TOF of 11.2 s−1. The observed high FE (faradaic efficiency) and current density can be attributed to the efficient mass transfer and adsorption of carbon dioxide on the CoPc@GIOMC surface in close proximity to the cobalt centers. This is due to the high surface area and electron-donating properties of the embedded nitrogen in the GIOMC framework, thus finally facilitating the effective electroreduction of absorbed CO2. Fig. 6II shows the partial current density for CO and H2 at different exerted potentials. The current density for CO was much higher than that for H2 at almost all the applied potentials. The slope of the Tafel plot (logjCO–logη) was calculated to be 144 mV dec−1, reflecting the kinetics and mechanism of eCO2RR using CoPc@GIOMC (0.9%) (Fig. 6III). The slope is close to the theoretical value of 118 mV, verifying that the rate-determining step is most likely the electroreduction of CO2 to the *COOH intermediate.57 The operation of the bulk electrolysis was also studied over time at −0.66 V versus RHE. Accordingly, the stability of the CoPc@GIOMC (0.9%)-modified electrode was tested by performing continuous electrolysis for 8 h, which resulted in a stable and nearly constant current density of 20 mA cm−1, thus indicating the high stability and durability of our electrocatalyst system (Fig. 6IV). Table 1 compares the performance of the present catalyst with other CoPc based-electrocatalysts reported for eCO2RR, demonstrating the clear efficiency of CoPc@GIOMC. The kinetic performance of the catalyst toward eCO2RR was also examined with electrochemical impedance spectroscopy (EIS), which showed a low solid/electrode charge-transfer resistance (Rct), as evidenced by the Nyquist plot (Fig. S5†). This issue is most likely related to the significant electrical conductivity of GIOMC. Alternatively, the high concentration of the electrolyte increased the conductivity of the solution.
| Catalyst | Catalyst loading (mol cm−2) | Electrolyte | Main products (%) | TOF (s−1) | V vs. RHE | Partial j (mA cm−2) | Ref. |
|---|---|---|---|---|---|---|---|
| CoPc@GIOMC | 2.28 × 10−8 | 0.5 M KHCO3 | CO, 90 | 11.2 | −0.68 | 33.2 | This study |
| CoPc/MWCNT | 2.33 × 10−8 | 0.5 M KHCO3 | CO, 92 | 4.08 | −0.68 | 13.1 | 58 |
| CoPc-CN/CNT | 3.5 wt% | 0.5 M KHCO3 | CO, 88 | 1.4 | −0.46 | 5.6 | 12 |
| CoFPc | 1.3 × 10−8 | 0.5 M KHCO3 | CO, 93 | 1.6 | −0.8 | 4.4 | 59 |
| CoPc-1 | 3.78 × 10−8 | 0.5 M KHCO3 | CO, 94 | 0.29 | −0.54 | 2.2 | 60 |
| CoPc2@carbon powder | 1.44 × 10−8 | 0.5 M KHCO3 | CO, 93 | 6.8 | −0.68 | 18.1 | 58 |
| CoPc-P1-COF | 3.7 wt% | 0.5 M KHCO3 | CO, 95 | 4.9 | −0.9 | 21.2 | 61 |
| CoTNPc | 6.57 wt% | 0.5 M KHCO3 | CO, 94 | — | −0.88 | 12.6 | 10 |
| CoOCPc | 5.04 wt% | 0.5 M KHCO3 | CO, 91 | — | −0.78 | 6.8 | 10 |
| CoPPc-CNT | 5.6 wt% | 0.5 M KHCO3 | CO, 96 | 0.41 | −0.6 | 3.6 | 62 |
| CoPPc@g-C3N4-CNT | 6.5 wt% | 0.5 M KHCO3 | CO, 95 | 4.9 | −0.8 | 21.9 | 63 |
Design of electrolyzer for eCO2RR on CoPc@GIOMC electrocatalyst coupled with electro-oxidative synthesis of sulfonamide and organosulfone derivatives
Anodic part: In the anodic part, the process involved the electrochemical oxidation of some electroactive species with an oxidation potential significantly lower than water, including N-phenylhydroxylamine (PHA), N-(p-tosyl)hydroxylamine (THA), 4-(hydroxyamino)benzoyl chloride (HABCl) and catechol in the presence of arylsulfinic acids as nucleophiles. PHA derivatives were prepared through the chemical reduction of the corresponding substituted nitrobenzenes using a mixture of ammonium chloride and zinc powder as the reducing agent and catalyst, respectively. The electrochemical properties of the various PHAs are discussed in detail in the ESI.† Having the optimized electrochemical synthesis of THA by applying the controlled-potential electrolysis of NT at −0.4 V vs. RHE in a divided cell, as outlined in the ESI,† we set out to perform parallel paired electrolysis for the transformation of CO2 to CO in the cathodic part and simultaneous electro-oxidation of PHA in the presence of 4-toluenesulfinic acid (TSA) at the anodic section. A three-electrode configuration in an H-type cell equipped with a working electrode (catalyst) made of CoPc@GIOMC (0.9%)-modified graphite paper and a counter electrode made of graphite plate was employed. The working electrode was immersed in a CO2-saturated 0.5 M KHCO3 aqueous solution as the catholyte, while the counter electrode was immersed in a solution of PHA as the substrate and TSA as the nucleophile (1:1 ratio) in ethanol/water (aqueous phosphate buffer solution, 0.5 M, pH = 3) (20/80 v/v%) as the anolyte. The Ag/AgCl reference electrode was placed as near as possible to the working electrode. The electrolysis was conducted at −0.66 V vs. RHE and the progress of the electrolysis in the anodic part was monitored with cyclic voltammetry (Fig. 7I). The A1 and A2 signals correspond to the oxidation of PHA and TSA, respectively. These signals decreased gradually, while the A3 peak related to the corresponding product appeared. This clearly verifies the inhibition of the water oxidation and the progress of the desired reaction by the efficient oxidation of PHA. Neither the oxidized form of TSA nor the over-oxidized products were detected, verifying that only PHA molecules were oxidized at the surface of the counter electrode. Generally, the oxidation process with the lowest required potential occurs during electrolysis. The oxidation potential of the species in the electrolysis cell followed the order of water > TSA > product > PHA. The initial view is that concentration polarization occurs parallel to the decrease in PHA. Therefore, in addition to PHA, it is possible for other species to participate in the electro-oxidation process. However, the high level of purity observed in the products suggests efficient electron transfer and a high current density. The chronoamperograms were recorded at different applied potentials to further assess the electrolysis process using the same procedure as the non-paired electrolysis (Fig. 7II). The current density reached a maximum of 40 mA cm−2 at −0.68 V vs. RHE. The data clearly confirms the efficiency of the designed electrolyzer, which is due to the lower oxidation potential of PHA compared to water oxidation. Fig. S8† shows the representative chronoamperograms of the eCO2RR cell coupled with THA and catechol in the presence of TSA at different potentials. Similar outcomes were also achieved using PHA in comparison to that for THA. The current densities observed in the chronoamperograms of the electrolysis cell during the oxidation of THA and catechol were measured to be 39 and 30 mA cm−2 at a potential of −0.68 V, respectively. In the next stage, the electrolysis was tested by applying different constant currents to evaluate the cell voltage and its variation over the time. Fig. 7III shows the current densities in the chronoamperograms, demonstrating the voltage and its variations for the two-electrode eCO2RR cell coupled with electrochemical oxidation of THA in the presence of TSA at a current density of 2, 4 and 6 mA cm−2. Notably, a cell potential as low as 1.22 V was obtained at 2 mA cm−2. However, the cell voltage smoothly increased with an increase in the current density from 1.22 to 1.32. The cell voltage in the two-electrode system was determined by the potential difference between the anode and cathode. Due to the relatively low oxidation potential of THA, the potential difference between the anode and cathode was reduced in comparison to the non-paired system. By increasing the current from 2 to 4 and 6 mA cm−2, an elevated overvoltage was required to facilitate rapid electron transfer. Consequently, the anodic and cathodic potentials underwent a shift towards more positive and negative values, respectively. Therefore, the potential difference increased until the water underwent oxidation. However, the results indicate that when the current density increased from 2 to 6 mA cm−2, there was only a marginal increase of 0.1 V in the potential. This suggests that the electron transfer rate was sufficiently high and there was no occurrence of water oxidation with an increase in current.
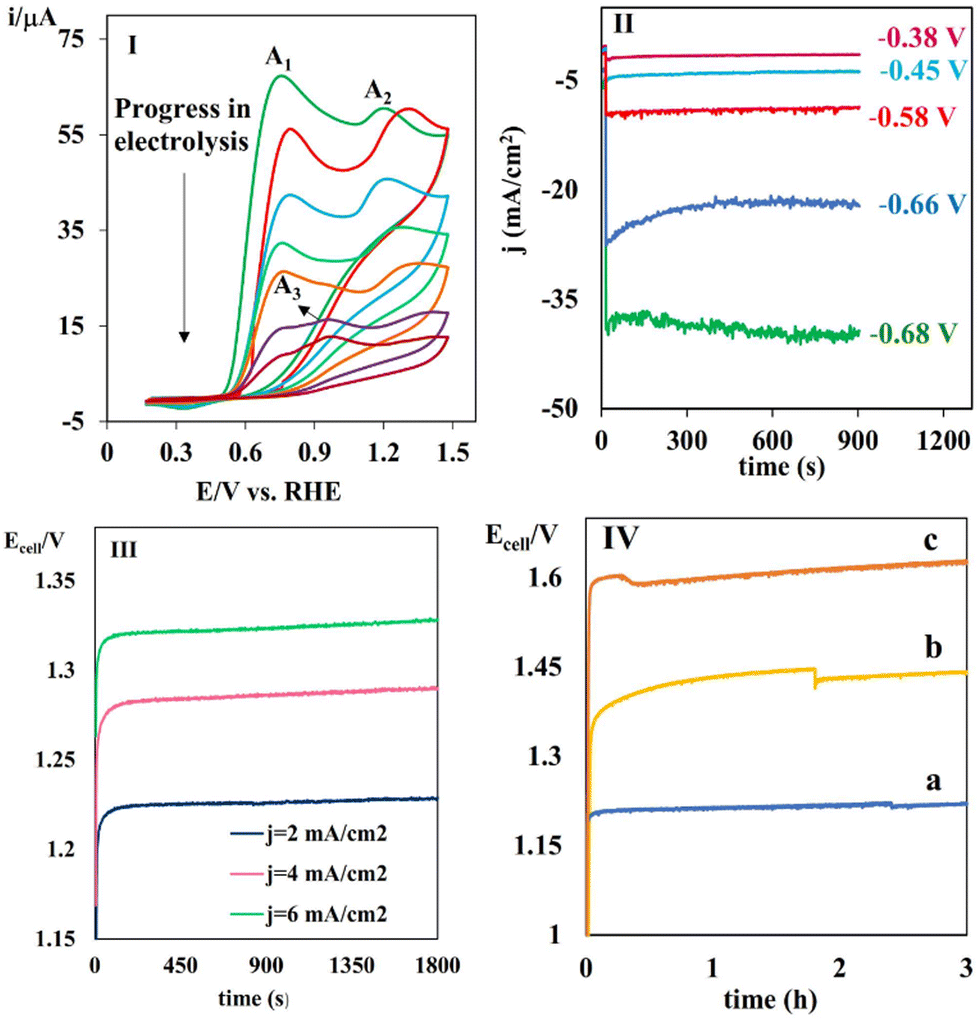 | ||
| Fig. 7 (I) The cyclic voltammograms of the anodic compartment of the paired electrolysis at −0.66 V vs. RHE containing 0.4 mmol 4-PHA and TSA at GCE at 100 mV s−1; (II) the chronoamperograms of the cathodic compartment of the paired electrolysis of eCO2RR coupled with the electrochemical oxidation of PHA in the presence of TSA at different potentials; (III) the E–t plot of the two-electrode setup of the paired electrolysis of eCO2RR coupled with the electrochemical oxidation of THA in the presence of TSA at various current densities; and (IV) the E–t plot for paired electrolysis in two-electrode setup containing (a) THA, (b) PHA, and (c) catechol in the presence of TSA in anodic part at 2 mA cm−2. | ||
Subsequently, the stability of the anodic half-reaction of the oxidation of THA in the presence of TSA was evaluated during 3 h electrolysis in a two-electrode cell configuration. Moreover, the electrolysis at a constant current of 2 mA cm−2 exhibited a stable voltage of 1.22 V for 3 h (Fig. 7IV). The consistent anodic and cathodic potential difference observed over time suggests that the anodic and cathodic processes remained unchanged. Furthermore, as the concentration of PHA decreased, it is evident that the water oxidation reaction did not substitute the PHA oxidation reaction. It is noteworthy that a product of high purity was obtained following a three-hour electrolysis process. Curve b and c in Fig. 7IV and Fig. S9† collectively demonstrate the same trend for the electrochemical oxidation of PHA and catechol in the presence of TSA. Table 2 lists the structures of five new (entries 1–5) and three known sulfonamides (entries 6–8) produced by the eCO2RR-paired electrolysis. Fig. S10† shows the overall reaction of the oxidation of catechol in the presence of TSA.
|
|
||
|---|---|---|
| Entry | R1 | R2 |
| 1 | CH3 | CH3 |
| 2 | CH3 | H |
| 3 | CH3 | Cl |
| 4 | COCl | CH3 |
| 5 | COCl | H |
| 6 | H | CH3 |
| 7 | H | H |
| 8 | H | Cl |
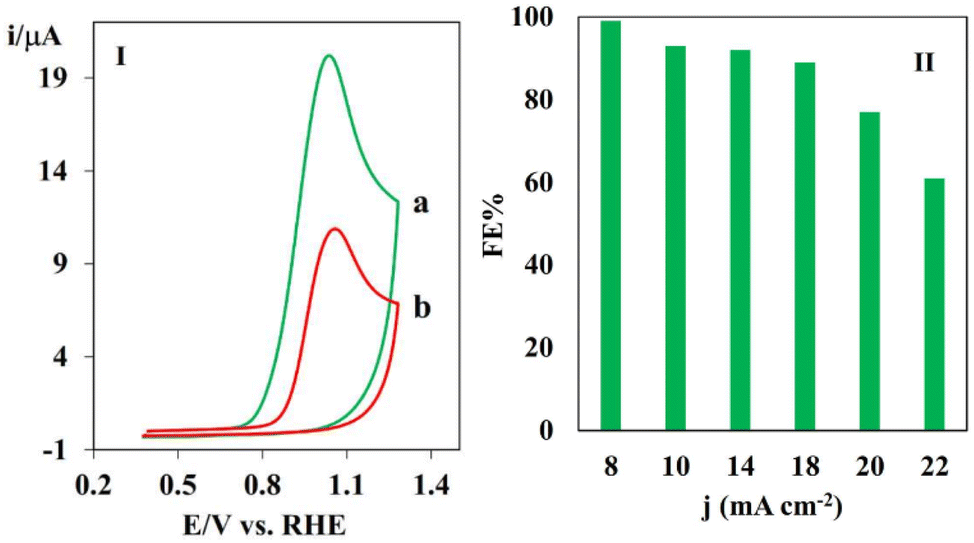
It should be noted that because of the slight oxidation of the PHA derivatives by oxygen or their slight instability in water, the anodic faradaic efficiency could not be determined accurately. Hence, the FE values for the anodic portion were determined by conducting the electrochemical oxidation of catechol, which is known to be more stable in air and water compared to PHA derivatives in the presence of TSA (Fig. 8).64 In this regard, a two-electrode electrolysis cell was created by adding 1 mmol catechol and 1 mmol TSA in ethanol/water (0.5 M phosphate buffer solution pH = 3) in the anodic compartment and CO2-saturated 0.5 M KHCO3 in the cathodic compartment. The resulting two-electrode cell was subjected to a current density of 14 mA cm−2 for 135 min of electrolysis. Fig. 8I shows the cyclic voltammograms at the beginning (curve a) and after 135 min of electrolysis (curve b). The consumed catechol was calculated by using the difference in the peak currents in curves a and b. The magnitude of the anodic peak observed at 0.9 V, which corresponds to the oxidation of catechol, exhibited a decrease as the electrolysis progressed, concomitant with the depletion of catechol. Therefore, the reduction in the magnitude of the anodic peak was directly proportional to the number of moles of catechol that underwent oxidation. The same procedure was repeated by applying current densities of 8, 10, 20 and 22 mA cm−2. The faradaic efficiencies reached 90–100% at current densities of 8 to 18 mA cm−2, respectively, indicating the excellent performance of the system (Fig. 8II). The high FE indicates that the electrical energy is primarily utilized for the oxidation of catechol. The FE experienced a decrease at higher current densities, which is primarily attributed to the presence of competitive background water oxidation. However, at higher current densities, water oxidation emerged as a significant side reaction, which is attributed to the concentration polarization and a potential shift towards more positive values. We also studied the oxidative electrochemistry of catechol by measuring the anodic FE using GC analysis (Fig. S11–13†). The chromatograms of the solutions were recorded before and after 135 min electrolysis. The calculated FE values were in good agreement with the results obtained from the voltammetric method. The energy efficiency (EE) was also calculated as a criterion of energy consumption. The theoretical cell voltage was calculated based on the theoretical electrode potential of both the anode and cathode processes. The energy efficiency for eCO2RR coupled with the oxidation of catechol in the presence of TSA at 2 mA cm−2 in the two-electrode setup was calculated to be 70%.
 | ||
| Fig. 8 (I) The cyclic voltammograms of the anodic part of the paired electrolysis (a) at the start and (b) after 135 minutes electrolysis in a two-electrode set-up at 14 mA cm−2 containing catechol and TSA in the anodic part, and (II) the plot of FE% vs. current density for the anodic part of the electrolysis mentioned in part I. | ||
Conclusion
In summary, we successfully prepared the CoPc@GIOMC hybrid material as a high-performance electrocatalyst toward eCO2RR. The electrocatalyst exhibited a high current density of 33 mA cm−2 at −0.68 V vs. RHE in 0.5 M KHCO3 with an FE of 90% for CO and TOF of 11.2 s−1, demonstrating the fast catalytic kinetics. In addition, we designed paired electrolysis for eCO2RR using a CoPC@GIOMC/graphite paper composite as the cathode coupled with the oxidation of PHA derivatives and catechol with a significantly lower oxidation potential than water on the graphite plate as an anode to synthesize sulfonamide and organosulfone derivatives. This study will open an avenue to develop novel paired eCO2RR with the synthesis of various types of value-added medicinally important materials under mild and green conditions.Author contributions
Samin Barat-Abtahi and Faranak Jafari-Hashejani: the role of both the authors is exactly the same. The paper was extracted from the master's thesis of these two authors. Investigation, conducting experiments, data curation, validation; Fahimeh Varmaghani: conceptualization, supervision, cell design, methodology, validation, writing – original draft, writing – review & editing; Babak Karimi: conceptualization, supervision, methodology, validation, writing – review & final editing; Hamzeh H. Veisi: conducting experiments, data curation, validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the IASBS Research Council for supporting this work. Fahimeh Varmaghani acknowledges Iran National Science Foundation: INSF (No: 4004120) to support the work. Babak Karimi also appreciates Alexander von Humboldt-Foundation for donation a prestigious Georg-Förster Award no. (Ref 3.4-1116632-IRN-GFPR) for partially supporting this work.References
- M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed.
- Y. Chen, J. Wei, M. S. Duyar, V. V. Ordomsky, A. Y. Khodakov and J. Liu, Chem. Soc. Rev., 2021, 50, 2337–2366 RSC.
- W. Choi and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 15341–15357 RSC.
- A. J. Martín, G. O. Larrazábal and J. Pérez-Ramírez, Green Chem., 2015, 17, 5114–5130 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS.
- C. Wang, Z. Lv, W. Yang, X. Feng and B. Wang, Chem. Soc. Rev., 2023, 52, 1382–1427 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS.
- Q. Feng, Y. Sun, X. Gu and Z. Dong, Electrocatalysis, 2022, 13, 675–690 CrossRef CAS.
- H. Tian, K. Wang, Z. Shui, M. A. Raza, H. Xiao, M. Que, L. Zhu and X. Chen, Mater. Lett., 2022, 310, 131482 CrossRef CAS.
- H. Gu, L. Zhong, G. Shi, J. Li, K. Yu, J. Li, S. Zhang, C. Zhu, S. Chen, C. Yang, Y. Kong, C. Chen, S. Li, J. Zhang and L. Zhang, J. Am. Chem. Soc., 2021, 143, 8679–8688 CrossRef CAS PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- Z. Jiang, Y. Wang, X. Zhang, H. Zheng, X. Wang and Y. Liang, Nano Res., 2019, 12, 2330–2334 CrossRef CAS.
- P. Tian, B. Zhang, J. Chen, J. Zhang, L. Huang, R. Ye, B. Bao and M. Zhu, Catal. Sci. Technol., 2021, 11, 2491–2496 RSC.
- R. Voo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef.
- T. Y. Ma, L. Liu and Z. Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC.
- J. C. Ndamanisha and L. P. Guo, Anal. Chim. Acta, 2012, 747, 19–28 CrossRef CAS.
- F. Varmaghani, B. Karimi and S. Mallakpour, Electrochim. Acta, 2018, 269, 312–320 CrossRef CAS.
- M. T. Ghafari, F. Varmaghani, B. Karimi and V. Khakyzadeh, Analyst, 2020, 145, 596 RSC.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- Y. Liang, L. Cai, L. Chen, X. Lin, R. Fu, M. Zhang and D. Wu, Nanoscale, 2021, 7, 3971–3975 RSC.
- C. Zhang, J. Liu, Y. Ye, Z. Aslam, R. Brydson and C. Liang, ACS Appl. Mater. Interfaces, 2018, 10, 2423–2429 CrossRef CAS.
- M. Koshy, S. Chen, D. U. Lee, M. B. Stevens, A. M. Abdellah, S. M. Dull, G. Chen, D. Nordlund, A. Gallo, C. Hahn, D. C. Higgins, Z. Bao and T. F. Jaramillo, Angew. Chem., Int. Ed., 2020, 59, 4043–4050 CrossRef.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv. Sci., 2021, 8, 2102886 CrossRef CAS.
- P. Lu, Y. Yang, J. Yao, M. Wang, S. Dipazir, M. Yuan, J. Zhang, X. Wang, Z. Xie and G. Zhang, Appl. Catal., B, 2019, 241, 113–119 CrossRef CAS.
- H. L. Zhu, Y. Q. Zheng and M. Shui, ACS Appl. Energy Mater., 2020, 3, 3893–3901 CrossRef CAS.
- B. Karimi, H. Barzegar and H. Vali, Chem. Commun., 2018, 54, 7155–7158 RSC.
- B. Karimi, H. Behzadnia, M. Bostina and H. Vali, Chem. – Eur. J., 2012, 18, 8634–8640 CrossRef.
- B. Karimi, H. Behzadnia and H. Vali, ChemCatChem, 2014, 6, 745–748 CrossRef CAS.
- S. Z. Alizadeh, B. Karimi and H. Vali, ChemCatChem, 2022, 14, e202101621 CrossRef.
- H. H. Veisi, M. Akbari, B. Karimi, H. Vali and R. Luque, Green Chem., 2023, 25, 4076–4089 RSC.
- H. H. Veisi, B. Karimi, M. Heydaria and R. Luque, Green Chem., 2023, 25, 7653 RSC.
- B. Karimi, H. Behzadnia, M. Rafiee and H. Vali, Chem. Commun., 2012, 48, 2776–2778 RSC.
- S. H. Kazemi, B. Karimi, S. A. Aghdam, H. Behzadnia and M. A. Kiani, RSC Adv., 2015, 5, 69032–69041 RSC.
- A. Korani, A. Salimi and B. Karimi, Electroanalysis, 2017, 29, 2646–2655 CrossRef CAS.
- M. Rafiee, B. Karimi and H. Shirmohammadi, Electrocatalysis, 2018, 9, 632–639 CrossRef CAS.
- N. Shamsvand, F. Varmaghani, B. Karimi and H. Hassanaki, Analyst, 2023, 148, 1309 RSC.
- B. Karimi, H. M. Mirzaei, H. Behzadnia and H. Vali, ACS Appl. Mater. Interfaces, 2015, 7, 19050 CrossRef CAS.
- Y. Wang, S. Gonell, U. R. Mathiyazhagan, Y. Liu, D. Wang, A. J. Miller and T. J. Meyer, ACS Appl. Energy Mater., 2018, 2, 97 CrossRef.
- S. Zhong, Z. Cao, X. Yang, S. M. Kozlov, K. W. Huang, V. Tung, L. Cavallo, L. J. Li and Y. Han, ACS Energy Lett., 2019, 4, 600–605 CrossRef CAS.
- M. A. Bajada, S. Roy, J. Warnan, K. Abdiaziz, A. Wagner, M. M. Roessler and E. Reisner, Angew. Chem., Int. Ed., 2020, 59, 15633–15641 CrossRef CAS PubMed.
- A. Vass, B. Endrődi and C. Janáky, Curr. Opin. Electrochem., 2021, 25, 100621–100700 CrossRef CAS.
- S. Apaydın and M. Török, Bioorg. Med. Chem. Lett., 2019, 29, 2042–2050 CrossRef.
- M. J. García-Galán, M. S. Díaz-Cruz and D. Barceló, TrAC, Trends Anal. Chem., 2008, 27, 1008–1022 CrossRef.
- C. T. Supuran, A. Casini and A. Scozzafava, Med. Res. Rev., 2003, 23, 535–558 CrossRef CAS PubMed.
- J. Meesin, P. Katrun, C. Pareseecharoen, M. Pohmakotr, V. Reutrakul, D. Soorukram and C. Kuhakarn, J. Org. Chem., 2016, 81, 2744–2752 CrossRef CAS PubMed.
- B. Mokhtari, D. Nematollahi and H. Salehzadeh, Green Chem., 2018, 20, 1499–1505 RSC.
- F. Varmaghani, D. Nematollahi, S. Mallakpour and R. Esmaili, Green Chem., 2012, 14, 963–967 RSC.
- F. Varmaghani, M. Hassan, D. Nematollahi and S. Mallakpour, New J. Chem., 2017, 41, 8279–8288 RSC.
- D. Nematollahi and F. Varmaghani, Electrochim. Acta, 2008, 53, 3350–3355 CrossRef CAS.
- https://en.wikipedia.org/wiki/Catechol .
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- J. H. Yang, Y. Gao, W. Zhang, P. Tang, J. Tan, A. H. Lu and D. Ma, J. Phys. Chem. C, 2013, 117, 3785–3788 CrossRef CAS.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- C. Zhang, R. Hao, H. Yin, F. Liu and Y. Hou, Nanoscale, 2012, 4, 7326–7329 RSC.
- J. P. Mensing, T. Kerdcharoen, C. Sriprachuabwong, A. Wisitsoraat, D. Phokharatkul, T. Lomas and A. Tuantranont, J. Mater. Chem., 2012, 17094–17099 RSC.
- H. Zhu, Y. Zheng and M. Shui, ACS Appl. Energy Mater., 2020, 3, 3893–3901 CrossRef CAS.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulie, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Isci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef.
- N. Morlanes, K. Takanabe and V. Rodionov, ACS Catal., 2016, 6, 3092–3095 CrossRef CAS.
- J. Luangchaiyaporn, D. Wielend, D. Solonenko, H. Seelajaroen, J. Gasiorowski, M. Monecke, G. Salvan, D. R. T. Zahn, N. S. Saricifci and P. Thamyongkit, Electrochim. Acta, 2021, 367 Search PubMed.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS PubMed.
- J. Chen, J. Li, W. Liu, X. Ma, J. Xu, M. Zhu and Y.-F. Han, Green Chem., 2019, 21, 6056–6061 RSC.
- T. T. Li, Y. Mei, H. Li, J. Qian, M. Wu and Y. Q. Zheng, Inorg. Chem., 2020, 59, 14184–14192 CrossRef CAS.
- D. Nematollahi and R. A. Rahchamani, J. Electroanal. Chem., 2002, 520, 145–149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02977h |
| This journal is © The Royal Society of Chemistry 2024 |