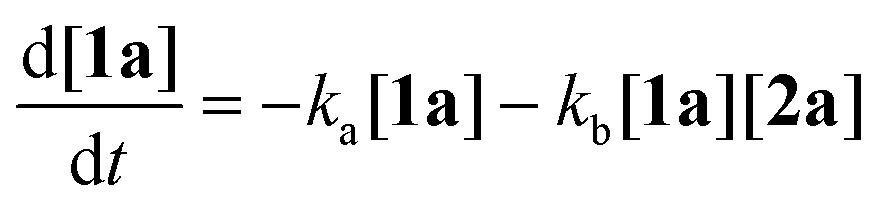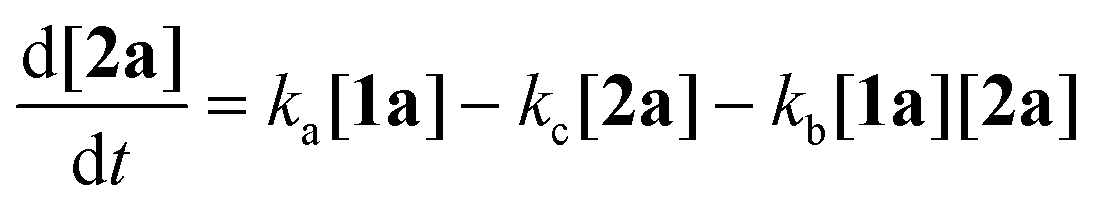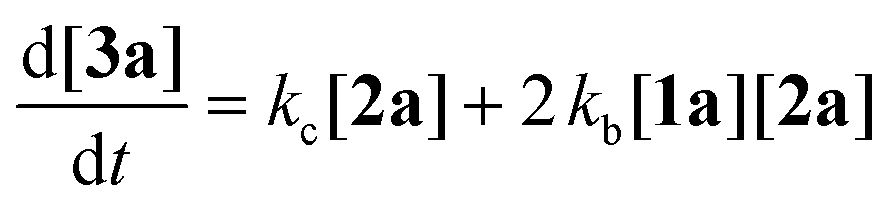DOI:
10.1039/D3GC02951D
(Paper)
Green Chem., 2024,
26, 375-383
Light-induced autoxidation of aldehydes to peracids and carboxylic acids†
Received
8th August 2023
, Accepted 21st November 2023
First published on 29th November 2023
Abstract
Autoxidation of aldehydes to peracids and carboxylic acids holds a significant impact in both academia and industry due to their wide applications in organic synthesis and environmental remediation. However, the multiple pathways involved in this reaction have hindered the development of sustainable methods for peracid synthesis. Herein, we conduct a comprehensive kinetic and mechanistic investigation to elucidate the interplay between these pathways. Subsequently, we introduce an efficient eco-friendly method to oxidize aldehydes to their corresponding peracids under sunlight or UV irradiation using oxygen as the sole oxidant without any additives or photocatalysts. Additionally, we demonstrate a simple method for the autoxidation of aldehydes to carboxylic acids via controlling key parameters such as the wavelength and solvent. These methods exhibit broad applicability to aromatic and aliphatic aldehydes, successful scaling up to the gram scale, and utilizing renewable solar energy making them good alternatives for traditional methods.
Introduction
Organic peracids are a widely recognized class of organic compounds with diverse applications in organic synthesis, materials science, and the environmental and medicinal fields.1 Owing to their distinctive peroxy groups (–O–O–) that readily donate oxygen atoms and compatibility with diverse organic solvents,2 some organic peracids such as meta-chloroperoxybenzoic acid (mCPBA) have been considered privileged oxidizing agents for several established reactions (Scheme 1A).1d Organic peracids are commonly synthesized by the perhydrolysis of carboxylic acids, acid chlorides, or anhydrides with hydrogen peroxide (H2O2).3 Other approaches, such as the photoinduced peroxidation of alkyl arenes (e.g., toluene), have recently been introduced to afford aromatic peracids.4 Autoxidation of aldehydes to carboxylic acids and peracids is one of the most appealing methods, primarily because of the readily available and cost-effective nature of aldehydes (Scheme 1B).5 Early reports used superstoichiometric amounts of hazardous oxidants with low atom efficiency, such as permanganate, chromate, perborate, percarbonate, and oxone.6 To address this problem, various strategies have been implemented using molecular oxygen as a greener alternative for the oxidation of aldehydes via organo-[N-hydroxyphthalimide (NHPI)]7 or metal-catalyzed (Cu, Ag, Ni, Mn, Fe, and Pt) approaches.8 Recently, Wang et al. disclosed an aerobic pH-adjusted method,9 and He et al. introduced a thermo-induced approach for the oxidation of aldehydes.10
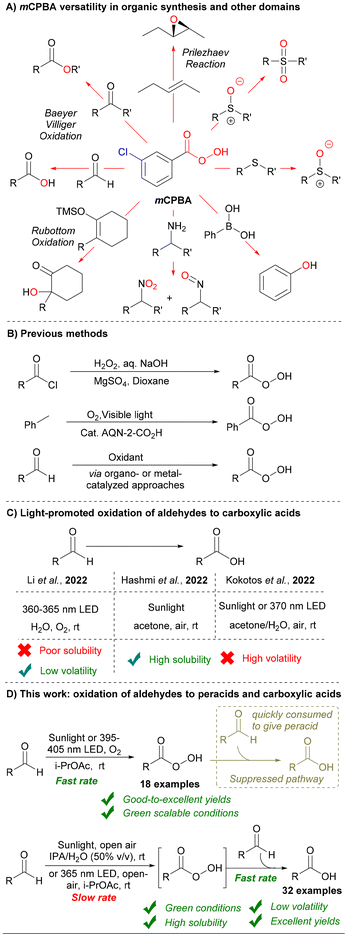 |
| | Scheme 1 Applications and synthetic approaches for peracids: previous methods and this work. | |
The concept of the photoexcitation of aldehydes to form a triplet radical pair, which initiates a radical-mediated pathway towards the corresponding peracids and carboxylic acids, has a long history.11 Apart from a few early reports that studied the photoinduced ozone-initiated oxidation of benzaldehyde to perbenzoic acid,12 most recent efforts have been dedicated to investigating the photoinduced oxidation of aldehydes to carboxylic acids.13 In the pursuit of aldehyde autoxidation, Safari et al. used porphyrin as a photocatalyst under visible light conditions,13b Cho et al. applied Ru/Ir-based photocatalysts under blue light,13c and Favre-Réguillon et al. used camphorquinone under white light in a flow photochemical system.13d Despite recent significant advancements, most of these approaches cannot avoid the use of organo- and metal catalysts, other additives, hazardous solvents, buffers, or strong bases, and many of them require complex setups, high pressures of molecular oxygen, or elevated temperatures, which hinder their classification as green and environmentally friendly processes.
In 2022, three groups independently reported highly atom-efficient and green photoinduced protocols for the aerobic oxidation of aldehydes to their corresponding carboxylic acids (Scheme 1C).13e–g Li et al. used molecular oxygen and water as the main solvent to afford carboxylic acids without catalysts or additives.13e Hashmi et al. demonstrated a sunlight-induced protocol using air, rather than pure oxygen, and acetone as the solvent.13f Kokotos et al. applied two conditions, sunlight, and 370 nm irradiation, to afford carboxylic acids (Scheme 1C).13g However, the photoinduced oxidation of aldehydes to their corresponding organic peracids has been underexplored, and the few explored protocols have either required high concentrations of organocatalysts (NHPI)14 or encountered challenges such as generalizing their findings or effectively separating the peracids.13f These limitations arose mainly from the multiple reaction pathways for the liquid-phase aerobic oxidation of aldehydes and how these pathways interfere with the conversion of peracids to the corresponding carboxylic acids.15 Herein, we further explore the competition between these interfering reaction pathways through mechanistic and kinetic studies. Building on our understanding and fine-tuning of all the reaction parameters, we established efficient, scalable, and eco-friendly conditions for the oxidation of aldehydes to their corresponding peracids under sunlight and UV irradiation. Varying key parameters (wavelength and solvent) provided alternative efficient conditions for synthesizing the corresponding carboxylic acids using sunlight or UV light (Scheme 1D).
Results and discussion
Exploring the reaction and condition optimizations
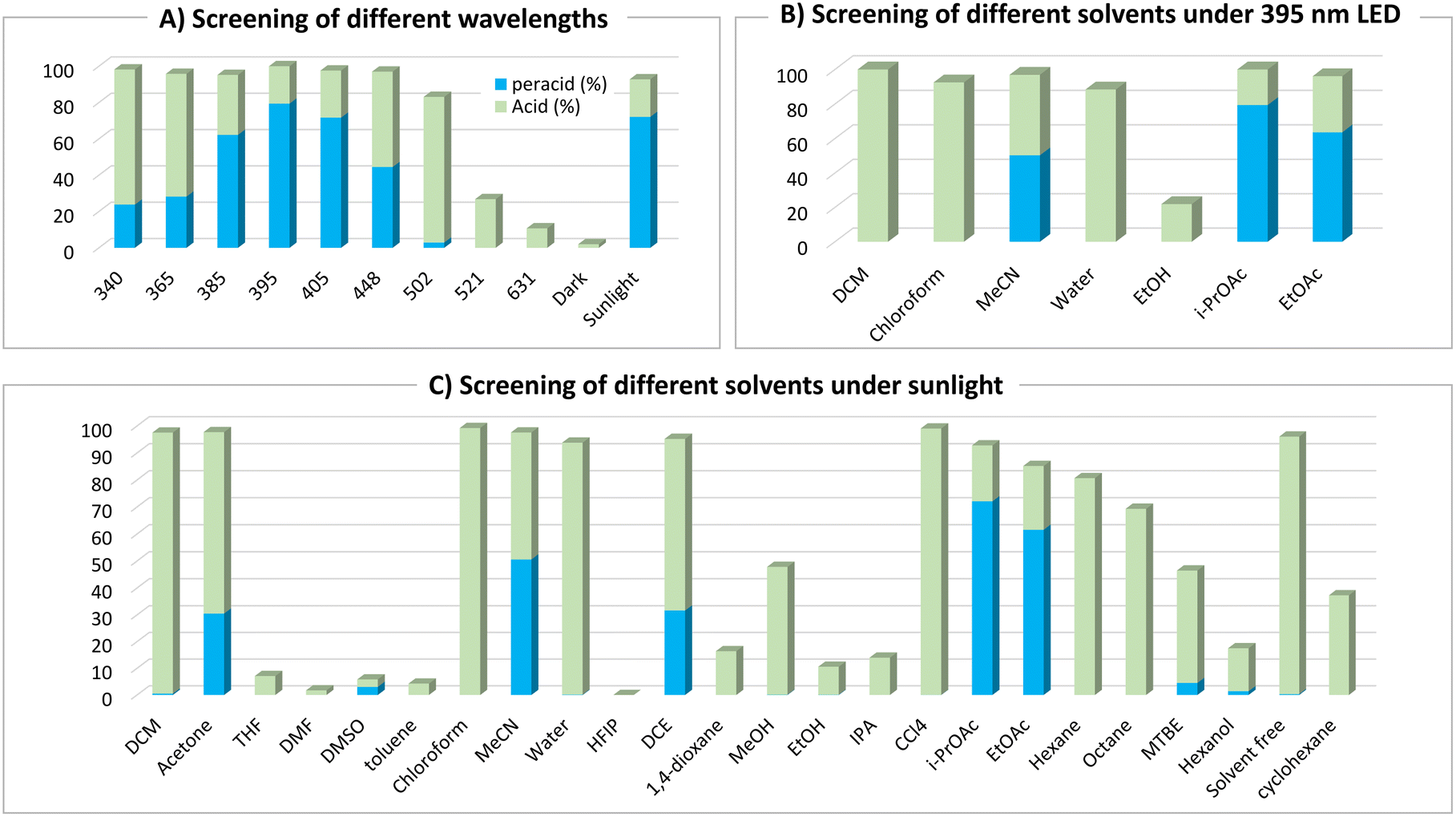
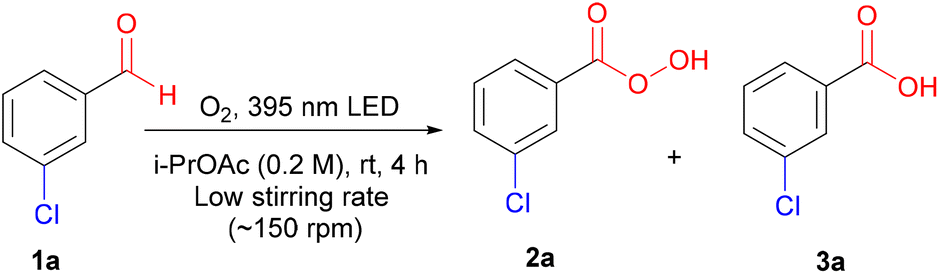
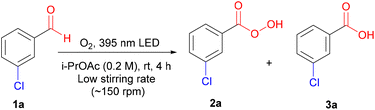
To investigate the photoinduced autoxidation of aldehydes to the corresponding peracids, meta-chlorobenzaldehyde 1a was chosen as the model starting material because of the favorable properties of mCPBA 2a, ensuring high safety and ease of handling.1d With iso-propyl acetate (i-PrOAc) as the solvent and 395 nm irradiation under an oxygen atmosphere for 4 h (see ESI† for details), 2a was obtained in a 79% yield (Table 1, entry 1). Light played a critical role in this reaction, because no product was observed under dark conditions (Table 1, entry 2). Shorter wavelengths (340–365 nm) afforded higher 3a/2a ratios, whereas longer wavelengths (521–631 nm) resulted in lower conversions (Table 1, entries 3 and 4, and Fig. 1A). Advantageously, sunlight irradiation gave a comparable 72% yield (Table 1, entry 5), which did not occur with indoor light because of its different power and wavelength spectra (Table 1, entry 6). In pursuit of a simpler setup, we replaced pure oxygen with air; however, the yield was reduced because of the activation of the side pathways at the expense of the main pathway (Table 1, entry 7). Under a nitrogen atmosphere, the reaction was suppressed, highlighting the crucial role of oxygen (Table 1, entry 8). Owing to the vulnerability of peracids to mechanical and thermal decomposition,16 we optimized our conditions at a low stirring rate (<200 rpm) to avoid any negative physical effects caused by vigorous stirring (see ESI† for details), which agrees with the observed lower yields at higher stirring rates (Table 1, entry 9).
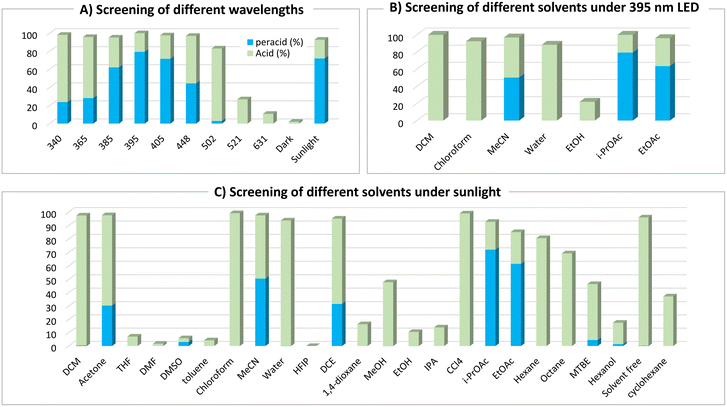 |
| | Fig. 1 Effect of different wavelengths and solvents on the reaction progress and peracid 2a/carboxylic acid 3a ratio. Reaction conditions: 1a (0.25 mmol), solvent (1.25 mL), under an oxygen atmosphere at room temperature (stirring rate = 150 rpm) for 4 h. | |
Table 1 Screening of reaction conditions
|
|
| Entry |
Variation from standard conditions |
2a (%) |
3a (%) |
| Reaction conditions: 1a (0.25 mmol), i-PrOAc (1.25 mL), under oxygen atmosphere at room temperature. Yields were determined by 1H-NMR. |
| 1 |
None |
79 |
20 |
| 2 |
No light |
— |
Trace |
| 3 |
365 nm LED |
28 |
68 |
| 4 |
521 nm LED |
— |
27 |
| 5 |
Outdoor sunlight |
72 |
21 |
| 6 |
Indoor artificial light |
— |
44 |
| 7 |
Open air |
54 |
45 |
| 8 |
Under nitrogen |
— |
Trace |
| 9 |
Stirring rate = 400 rpm |
68 |
30 |
| 10 |
Ethanol (0.2 M) |
— |
21 |
Considering the critical role of the solvent,17 we conducted a comprehensive screening of approximately 24 solvents under 405 nm (see ESI† for details), 395 nm, and sunlight irradiation (Fig. 1B and C). With alcoholic solvents, lower conversions were observed owing to the suppressed reaction rates, which could be attributed to the strong intermolecular forces (H-bonding) between these solvents and the aldehydes.17b Ethereal solvents such as tetrahydrofuran (THF) and 1,4-dioxane afforded only carboxylic acid 3a in modest yields (<20%), whereas methyl tert-butyl ether (MTBE) provided 42% of 3a and 4.5% of 2a, respectively. In contrast, chlorinated solvents afforded significantly higher yields of carboxylic acid 3a (>95%) than those achieved with other solvents, primarily because of their higher rates. However, dichloroethane (DCE) yielded a mixture of 3a and 2a in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Some nonpolar solvents (toluene and cyclohexane) and polar aprotic solvents [dimethylsulfoxide (DMSO) and dimethylformamide (DMF)] displayed low conversion rates, yielding only carboxylic acid 3a as the over-oxidation product. The use of water or neat conditions led to high yields of carboxylic acid 3a with no observation of peracid 2a. Some solvents showed promising results for the formation of 2a, such as acetone (31%) and acetonitrile (51%), while the best results were achieved with ester solvents [ethyl acetate (EtOAc), 62% and i-PrOAc, 72%]. In recent years, i-PrOAc has demonstrated significant value across numerous industrial and academic applications as a green and cost-efficient solvent,18 with some early studies highlighting its advantage in decreasing the decomposition rate of benzoyl peroxide.19
:1 ratio. Some nonpolar solvents (toluene and cyclohexane) and polar aprotic solvents [dimethylsulfoxide (DMSO) and dimethylformamide (DMF)] displayed low conversion rates, yielding only carboxylic acid 3a as the over-oxidation product. The use of water or neat conditions led to high yields of carboxylic acid 3a with no observation of peracid 2a. Some solvents showed promising results for the formation of 2a, such as acetone (31%) and acetonitrile (51%), while the best results were achieved with ester solvents [ethyl acetate (EtOAc), 62% and i-PrOAc, 72%]. In recent years, i-PrOAc has demonstrated significant value across numerous industrial and academic applications as a green and cost-efficient solvent,18 with some early studies highlighting its advantage in decreasing the decomposition rate of benzoyl peroxide.19
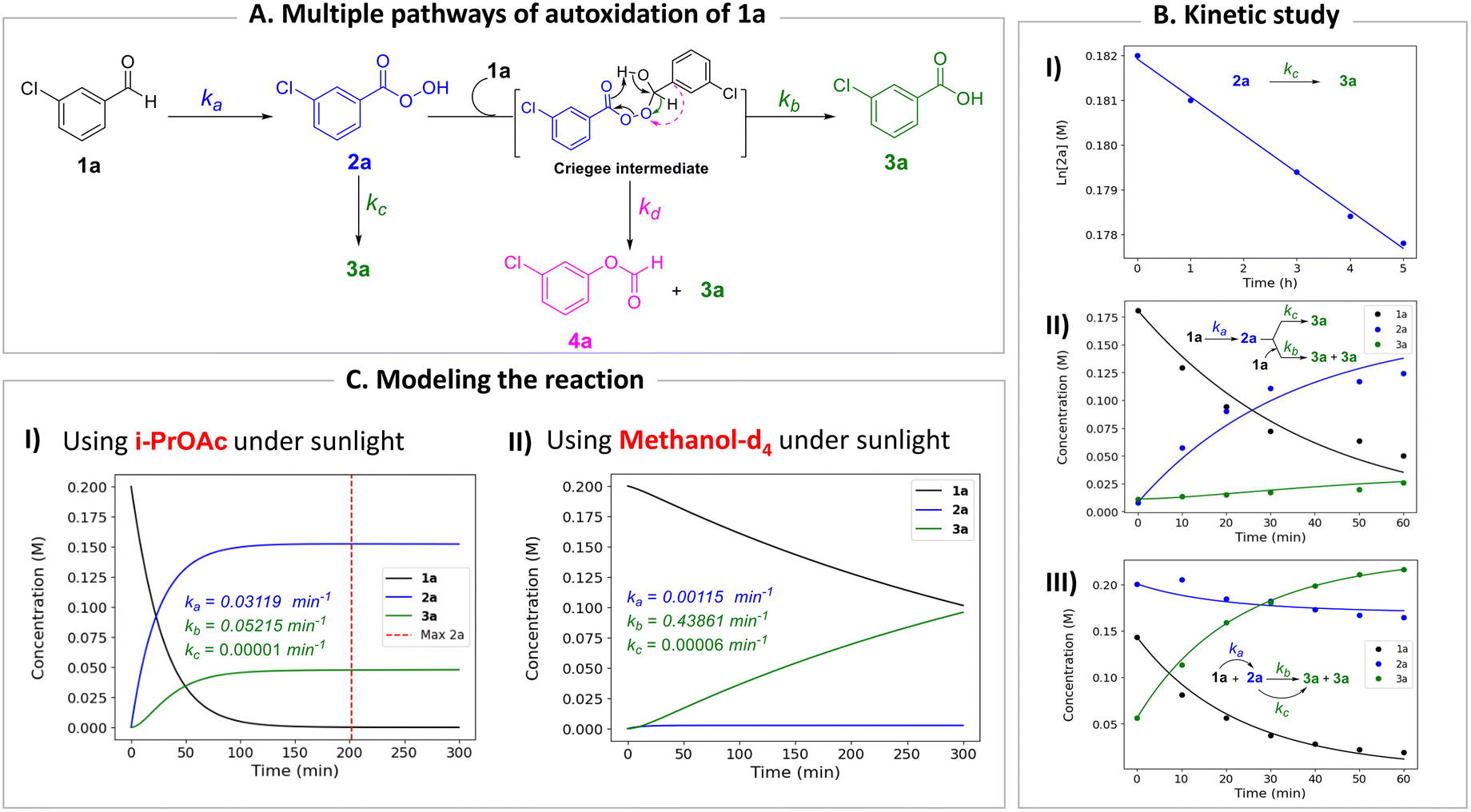
Mechanistic and kinetic investigations
To gain deeper insight into the critical role of solvents in suppressing or encouraging interfering reaction pathways, we conducted a detailed kinetic analysis under various conditions (see ESI† for details). Autoxidation of aldehydes begins with photoexcitation and reaction with oxygen through a radical-mediated pathway, generating the corresponding peracid 2a at rate ka (Fig. 2A). Following a mechanism similar to Baeyer–Villiger oxidation,20 the corresponding carboxylic acid 3a can be formed via a Criegee intermediate at rate kb. Another pathway for the rearrangement of the Criegee intermediate has been reported,17,21 which can afford a mixture of formate 4a and carboxylic acid 3a. Luckily, formate byproduct 4a was not observed under our conditions for substrate 1a.17a Additionally, carboxylic acid 3a was formed by the direct decomposition of 2a at rate kc.14 Favre-Réguillon et al. investigated these pathways and the key factors that favored one pathway over another.14,21,22 Lehtinen et al. and Jin, Peng, Zhao et al. have demonstrated the effect of the solvent on the rearrangement of peracid-aldehyde adducts during the aerobic autoxidation of aldehydes via experimental and computational efforts.17 However, most of these mechanistic investigations have focused on the competition between the two pathways governing the rearrangement of the Criegee intermediate (kbvs. kd) and used an aliphatic aldehyde (2-ethylhexanal) to conduct their investigation. Herein, our primary focus is to analyze the interplay between the pathways involved in the production of the peracid and its subsequent degradation (kavs. kb and kc) to determine the optimal conditions for peracid synthesis and to ascertain the most suitable point to terminate the reaction. Our system can be defined using three ordinary differential equations (ODEs) as follows:| | 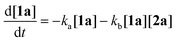 | (1) |
| | 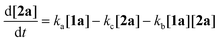 | (2) |
| | 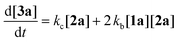 | (3) |
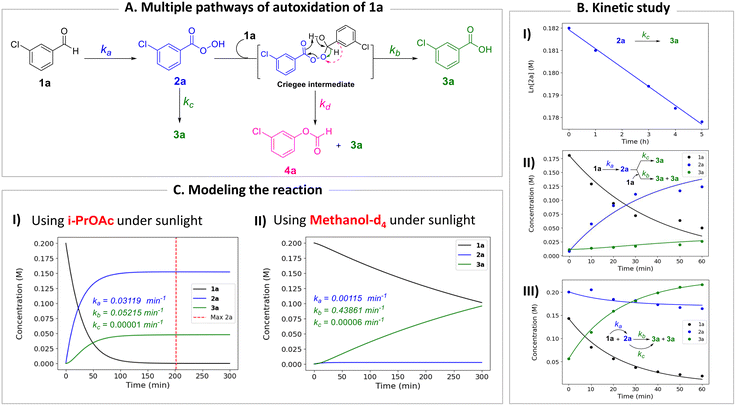 |
| | Fig. 2 Kinetic study. (A) Multiple pathways for the autoxidation of meta-chlorobenzaldehyde (1a). (B) Kinetic analysis: (I) calculation of the rate of peracid decomposition kc; (II) and (III) calculation of the rates ka and kb. (C) Modeling the autoxidation of aldehyde 1a under different conditions: (I) using i-PrOAc as a solvent under sunlight and (II) using MeOH-d4 as a solvent under sunlight. | |
Tracking the changes in the concentrations of 1a, 2a, and 3a as a function of time under different initial conditions (Fig. 2B) enabled the calculation of ka, kb, and kc values through nonlinear regression analysis (via Python's SciPy library) and fitting our experimental data to the ODEs (eqn (1)–(3)) (see ESI† for details). By solving these equations, we modeled the concentration profiles of each species over time to visualize their generation and consumption under different conditions (Fig. 2C). Kinetic analysis revealed that the direct decomposition of 2a into 3a occurred at an extremely low rate compared to the other pathways, suggesting a negligible effect, especially with shorter reaction times. The primary competing pathways involved the generation of 2a from 1a and the subsequent reaction of 1a and 2a producing 3a. Under sunlight irradiation with i-PrOAc as the solvent, the rate constant (kb = 0.05215 min−1) for the reaction between 1a and 2a was 1.67 times faster than the rate constant (ka = 0.03119 min−1) for the generation of 2a; however, the significance of this side reaction (kb) depends on the concentrations of both 1a and 2a. Owing to the fast conversion of 1a to 2a under these conditions, the time frame when both concentrations of 1a and 2a are relatively high is very short, minimizing the effect of this side pathway (Fig. 2C-I). In contrast, other solvents such as methanol-d4 showed a kb rate approximately 400 times faster than ka, which explains why peracid 2a was not observed during the time-course study. The reason for the low conversion of 1a to 3a in methanol-d4 (Fig. 1C), despite the smooth progression of the reaction between 1a and 2a to form 3a, can be attributed to the slow rate-limiting step of 1a to 2a conversion (Fig. 2C-II). The kinetic study was extended to other light sources and wavelengths (see ESI† for details).
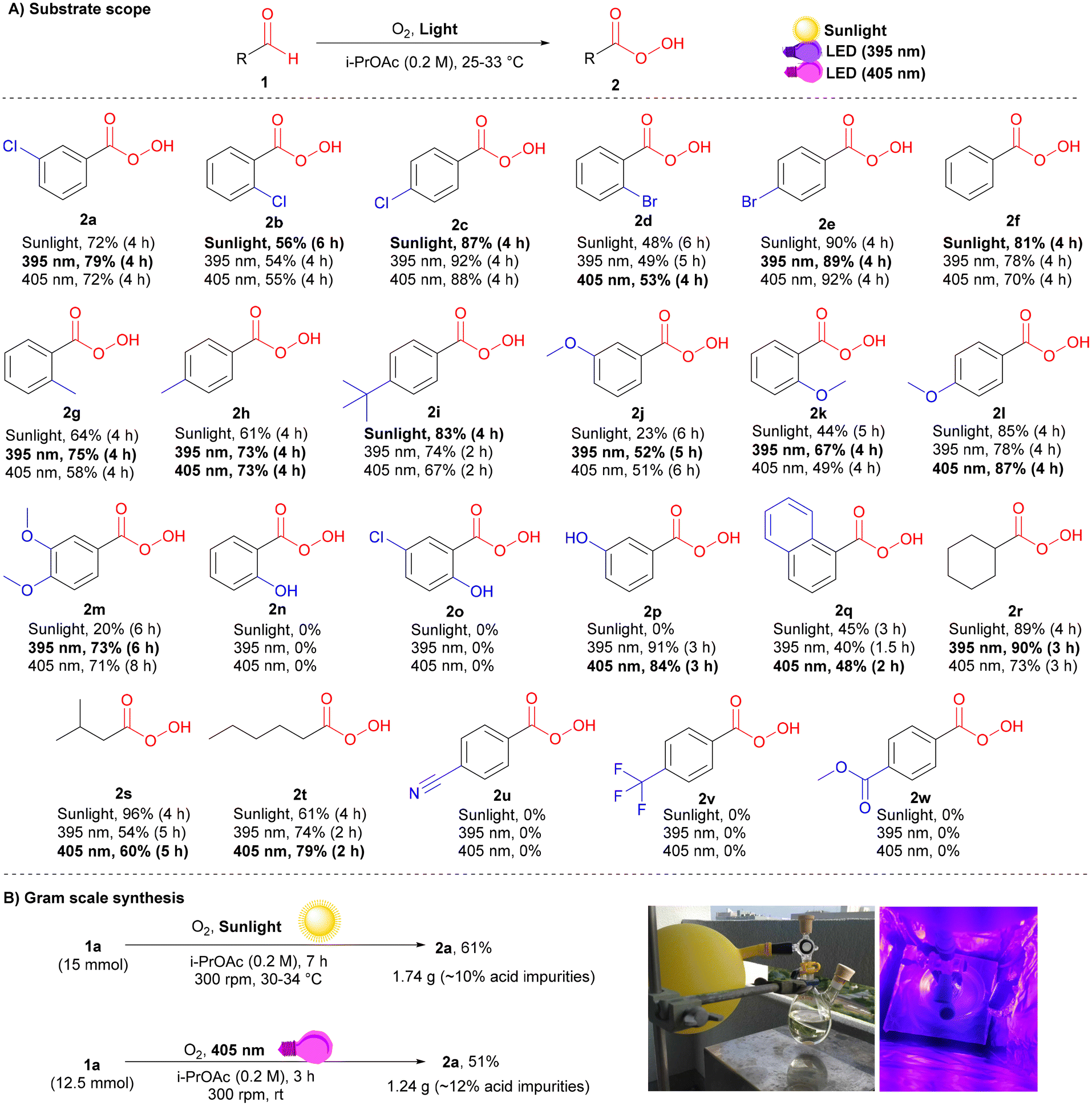
Exploring the scope of peracids
With the three high-yielding conditions in hand (see ESI† for details), the substrate scope of peracid 2 was investigated (Scheme 2A). Generally, unsubstituted (2f) and para-substituted aromatic aldehydes bearing electron-donating (EDGs; 2h, 2i, and 2l) or halogens (2c and 2e) afforded higher yields than their ortho- and meta-substituted analogs (2a, 2b, 2d, 2g, 2j, and 2k). This result highlights how both electronic and steric effects play key roles in the reactivities of different substrates in the multiple pathways. Accordingly, some substrates required either shorter or longer reaction times to obtain the maximum possible peracid yield. Dimethoxy-substituted benzaldehyde afforded good yields under LED irradiation (395 and 405 nm), whereas lower yields were observed under sunlight owing to the slower reaction rate (2m).
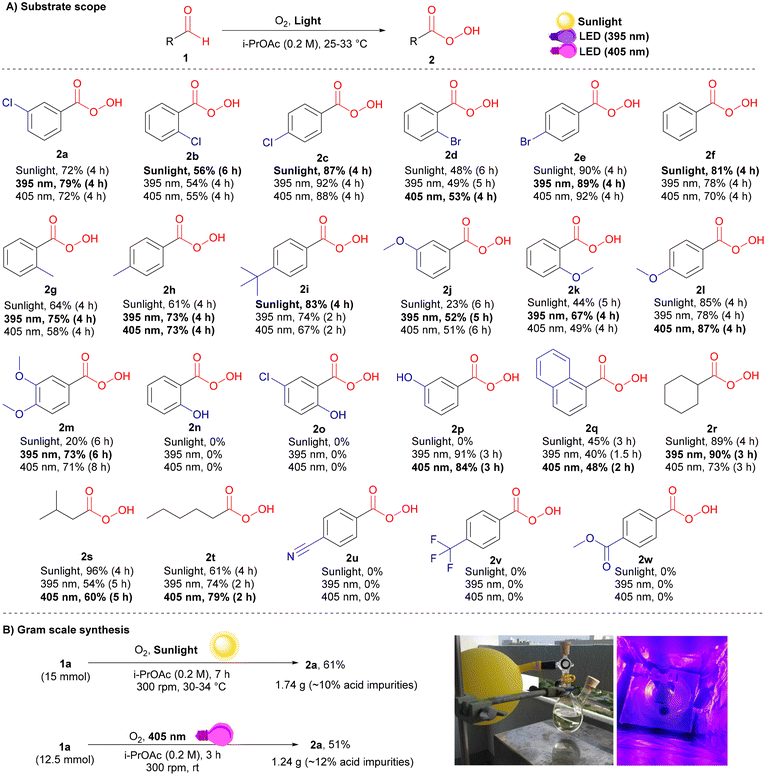 |
| | Scheme 2 Substrate scope and scalability for the photoinduced oxidation of aldehydes to peracids. (A) Reaction conditions 1 (0.25 mmol), i-PrOAc (1.25 mL), under an oxygen atmosphere (stirring rate = 150 rpm) at 25–33 °C. The yields were determined by 1H-NMR. Bold conditions are recommended for a good balance between high yield and safety (keeping the purity ∼70–75%, with ∼10% carboxylic acid impurities, is favourable in terms of stability and safety). (B) Gram scale synthesis of mCPBA under sunlight and 405 nm LED irradiation: reaction conditions 1a (12.5–15 mmol), i-PrOAc (0.2 M), under an oxygen atmosphere (stirring rate = 300 rpm) at 25–34 °C. The isolated yields are reported. | |
Stable ortho-substituted hydroxybenzaldehydes were resistant to photoinduced oxidation under additive-free conditions (2n and 2o),23 while their meta-substituted analogs (2p) afforded high yields of 91% and 84% under 395 nm and 405 nm irradiation, respectively. Aliphatic aldehydes were highly compatible with our conditions, giving the corresponding peracids in good yields (2r–2t). Benzaldehydes bearing electron-withdrawing groups (EWGs) showed very slow reaction rates, which terminated the possibility of peracid accumulation, as all generated peracids were directly converted to their corresponding carboxylic acids at a faster rate (3u–2w). Then, a gram scale synthesis was performed to give approximately 1.74 g (90% peracid) and 1.24 g (88% peracid) of 2a under sunlight and 405 nm irradiation, respectively (Scheme 2B).
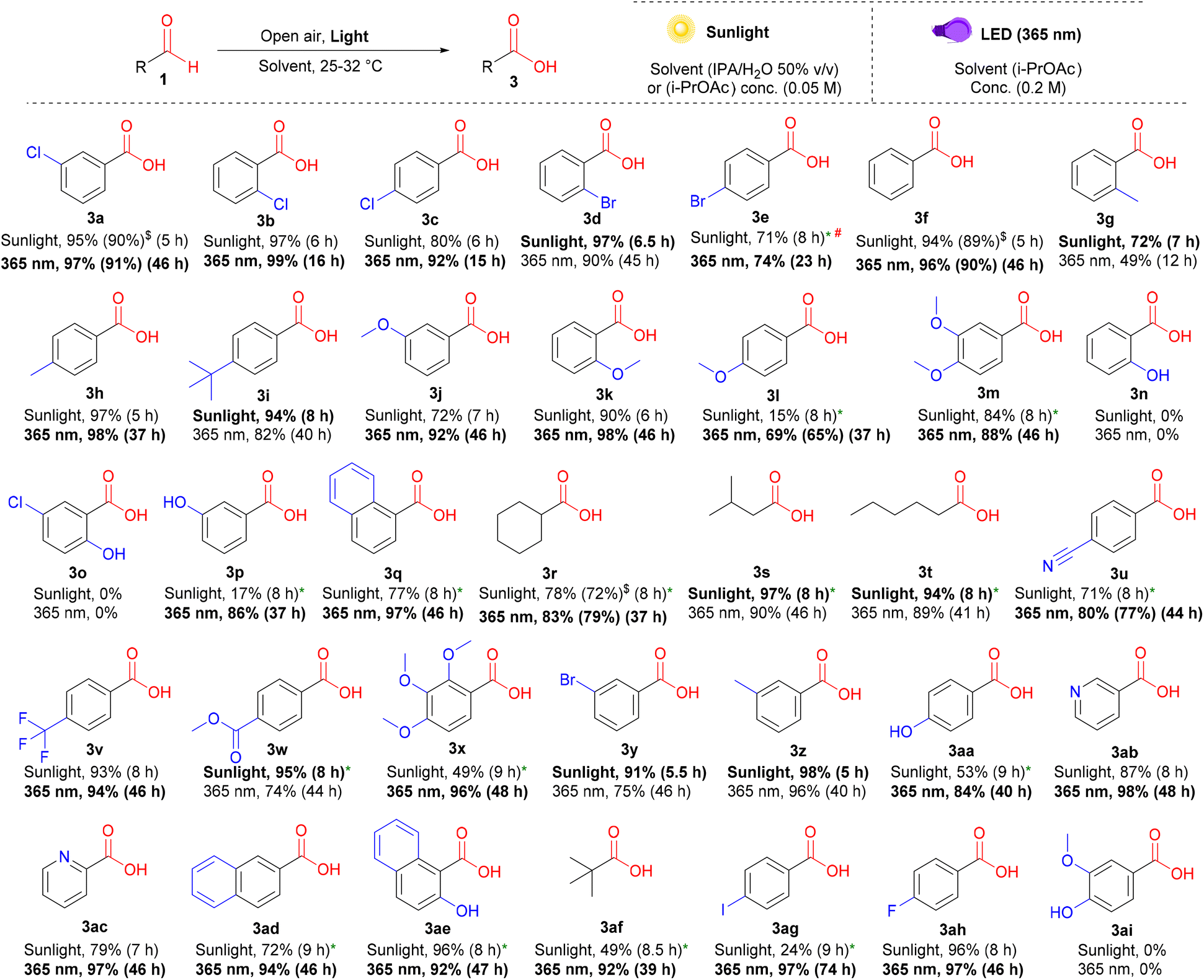
Exploring the scope of carboxylic acids
After the details of the reaction mechanism were clarified through our kinetic studies, further green and efficient methods for converting the peracids produced in situ into carboxylic acids were also explored. Based on our understanding of the role of different wavelengths and solvents in activating and suppressing competing pathways, we further optimized alternative conditions using a different wavelength (365 nm LED) or solvent system [iso-propyl alcohol (IPA)/water, 50% v/v] for the production of carboxylic acids 3 from aldehydes 1 using air as a safer and cheaper alternative to pure oxygen. We employed a mixed solvent system under sunlight irradiation to effectively address the most controversial aspects encountered in prior works13e–g concerning solubility and volatility.24 A broad spectrum of aliphatic and aromatic aldehydes with various EWGs and EDGs at different positions afforded carboxylic acids in high yields under the optimized conditions (Scheme 3). Some aldehydes displayed slower reaction rates (ka) in water-containing mixed solvent systems under sunlight. Hence, we utilized i-PrOAc as an alternative solvent, taking advantage of their slower (ka) rates, which would prevent the unwanted accumulation of peracids during the reaction (3l, 3m, 3p–3u, 3w, 3x, 3aa, and 3ad–3ag). Multi-methoxy-substituted benzaldehydes (3m and 3x), heterocyclic aldehydes (3ab and 3ac), and naphthaldehydes (3q, 3ad, and 3ae) gave good yields, especially under 365 nm irradiation. Although para-fluoro- (3ah) and para-chlorobenzaldehydes (3c) exhibited excellent yields under sunlight, their bromo- (3e) and iodo- (3ag) counterparts were less efficient under these conditions. Even with high dilution and scaling down, the yield of 3e could not be enhanced beyond 71%; however, all these substrates showed excellent yields when irradiated at 365 nm for longer times. Similar to peracids, ortho-substituted hydroxybenzaldehydes (3n and 3o) and vanillin (3ai) resisted photoinduced oxidation under additive-free conditions.
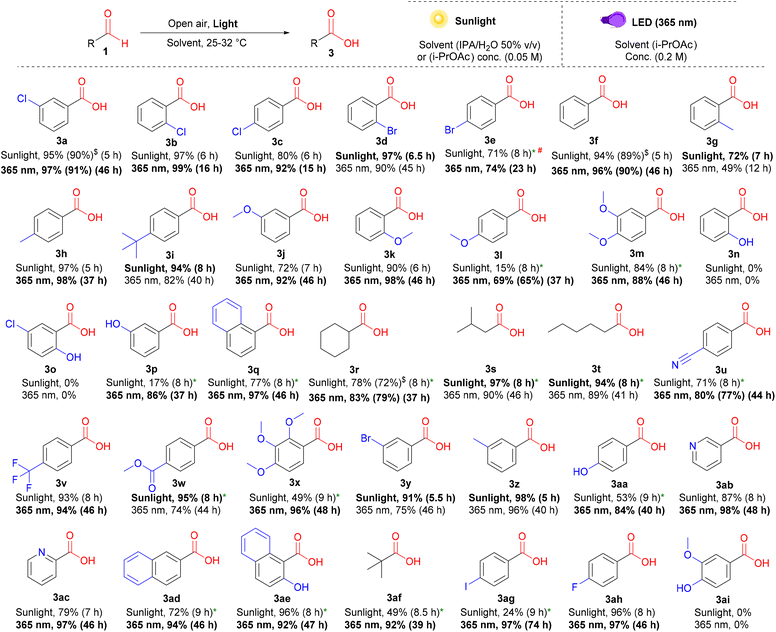 |
| | Scheme 3 Substrate scope for the photoinduced oxidation of aldehydes to carboxylic acids. Reaction conditions (under sunlight): 1 (0.0625 mmol), IPA/water 50% v/v (1.25 mL), under open air (stirring rate = 500 rpm) at 25–32 °C. The yields were determined by 1H-NMR (isolated yields in parentheses). # (conc. = 0.025 M). * (solvent = i-PrOAc). $ (0.25 mmol scale). Reaction conditions (under 365 nm LED): 1 (0.25 mmol), i-PrOAc (1.25 mL), open air (stirring rate = 500 rpm) at room temperature. The yields were determined by 1H-NMR (isolated yields in parentheses). | |
Control experiments
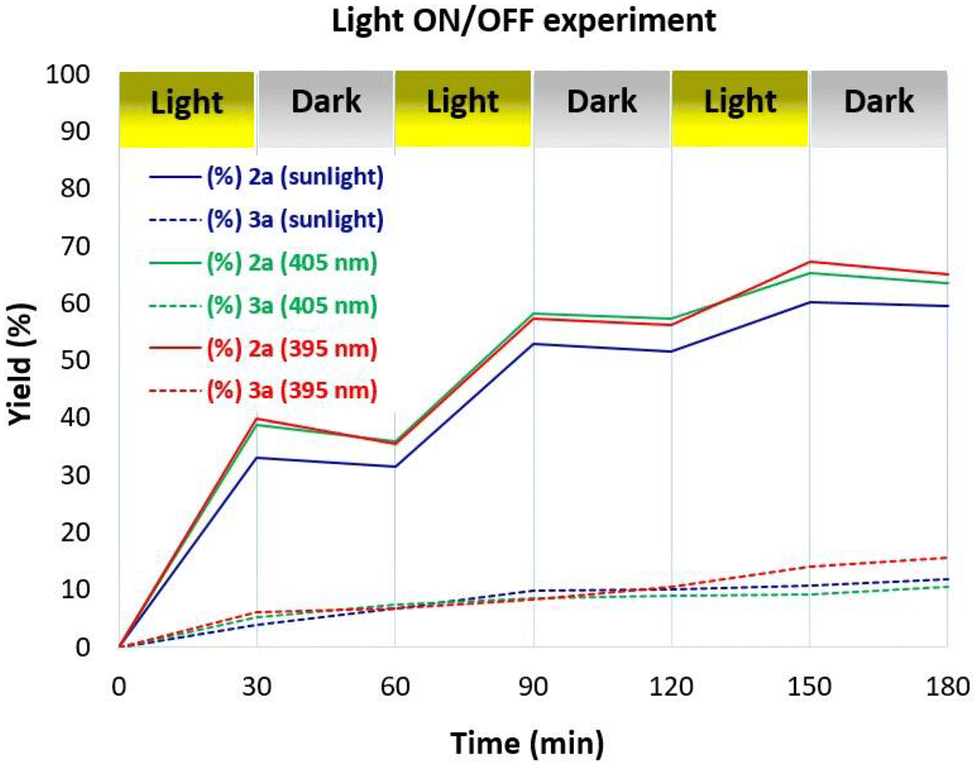
To further support the aldehyde autoxidation mechanism, some control experiments were performed. Light on/off experiments revealed the indispensability of continuous irradiation for the generation of peracid 2a from aldehyde 1a (Fig. 3). In the absence of light, the primary pathway leading from 1a to 2a was entirely suppressed, whereas the secondary pathway involving the Baeyer–Villiger rearrangement remained partially active, demonstrating the partial consumption of 2a and the concurrent generation of 3a.
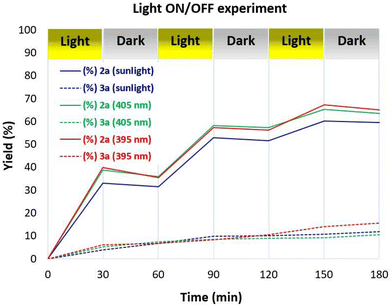 |
| | Fig. 3 Light-irradiation on/off experiments. | |
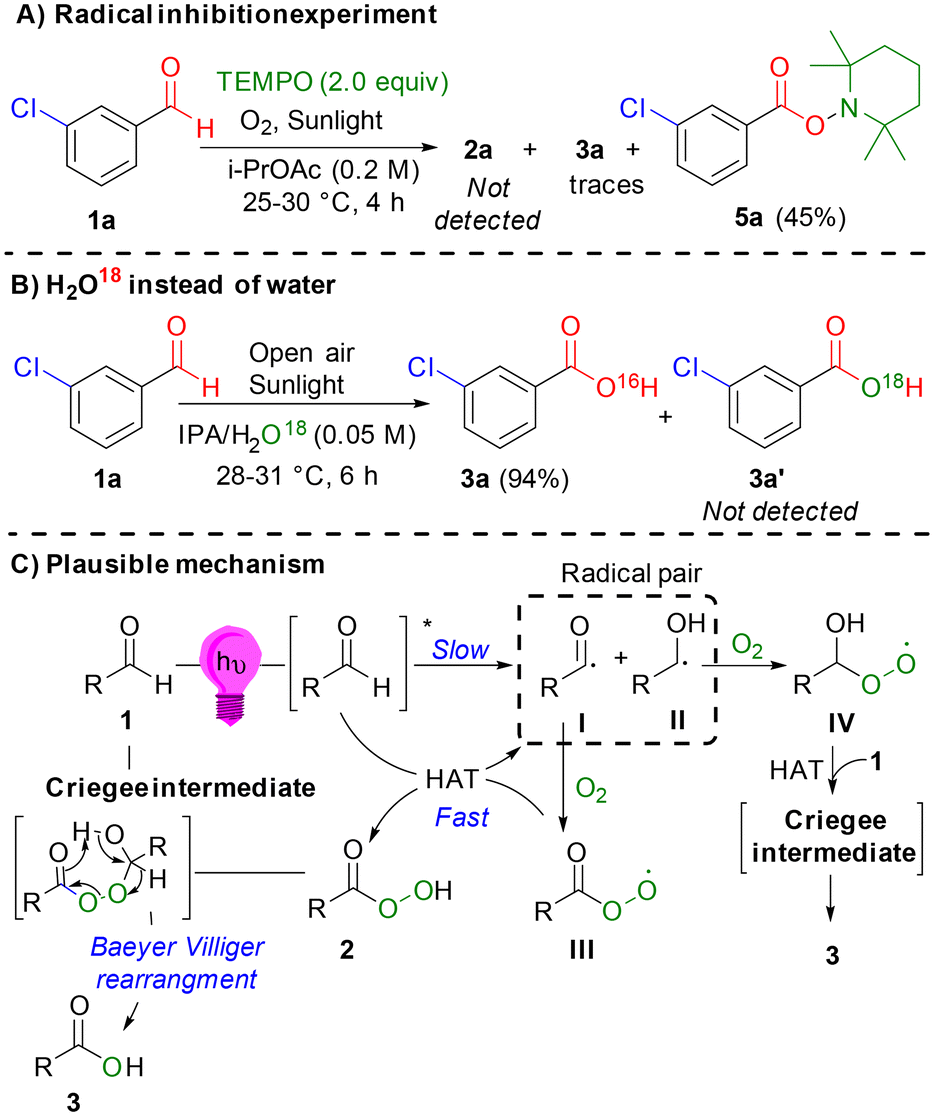
In the presence of the radical scavenger tetramethylpiperidine-1-oxyl (TEMPO), both carboxylic acid 3a and peracid 2a were not observed, which is consistent with the accepted radical-mediated pathway (Scheme 4A). When 18O-isotope labeled water was used in place of water under the optimized conditions for carboxylic acids, no 18O-labeled carboxylic acid 3a′ was observed, confirming that the origin of the oxygen atom in the product was the oxygen atmosphere rather than water (Scheme 4B). A plausible mechanism (Scheme 4C) can be proposed based on our control experiments (Fig. 3 and Scheme 4), kinetic analysis (Fig. 2), and related reports.11e,13e–g,21,22b Under light irradiation, aldehyde 1 is excited to form a highly reactive triplet radical pair (I and II), which undergoes a radical-mediated pathway with molecular oxygen to generate the corresponding peroxy radicals (III and IV). Peroxy radical III subsequently engages in a hydrogen atom transfer (HAT) reaction to form peracid 2 and benzoyl radical I. Through a pathway similar to Baeyer–Villiger oxidation,20 the corresponding carboxylic acid 3 can be formed via a Criegee intermediate. This intermediate is produced by the nucleophilic addition of peracid 2 or radical IV to aldehyde 1.
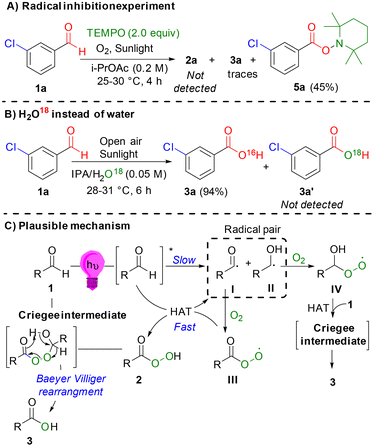 |
| | Scheme 4 Control experiments and plausible mechanism. | |
Conclusions
To explore the liquid-phase autoxidation of aldehydes, we conducted detailed kinetic and mechanistic studies to elucidate the key factors affecting the different rates of each step. We then developed an eco-friendly, practical, and simple photoinduction method to afford peracids in high-to-excellent yields using oxygen as the sole oxidant under sunlight or UV light irradiation. The substrate scope and limitations were carefully examined, and the scalability of this protocol was validated. An alternative green and efficient method for the production of various carboxylic acids in open air was introduced after adjusting the wavelength and solvent system. Further investigations for the nature of interactions between the solvents and reaction intermediates is currently under progress.
Experimental section
Gram-scale synthesis of mCPBA 2a
To a dry 100 mL two-neck round-bottom flask, meta-chlorobenzaldehyde (1a, 15 mmol) and i-PrOAc (60 mL) were added under an oxygen atmosphere (using the Schlenk line). The mixture was stirred (using a 20 mm magnet at 300 rpm) under sunlight at 30–34 °C. The progress of the reaction was monitored by No-D 1H-NMR spectroscopy. After 7 h, the reaction was stopped after reaching a plateau, and the solution was directly concentrated in vacuo (avoiding heating or high-speed rotation during concentration). The obtained compounds were washed with phosphate buffer (pH = 7.2 × 25 mL), extracted with ethyl acetate, dried over anhydrous sodium sulfate, and evaporated to obtain the final mCPBA 2a (1.74 g) containing approximately 10% acid impurities. Further purification was avoided, primarily because of safety concerns. Pure peracids are highly shock-sensitive and may explode or decompose under thermal or mechanical stress. Maintaining approximately 10% acid impurities in the sample is advisable, which aligns with commercially available mCPBA, ensuring both a reasonable level of purity and high yields.
Additional reaction information: place: (Osaka, Japan); date (04/07/2023); weather (sunny); outdoor temperature (30–34 °C); time (11:00 am–18:00 pm); humidity (41–48%); wind (3.2–14.5 km h−1).
Author contributions
M. S. H. S.: Designing the experiments, writing the paper, performing experiments and analysing the data. C. D., Y. T., A. K., and T. K.: Performing experiments and analysing the data. S. T. and M. K.: Supervising the project and editing the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Prof. Makoto Yasuda and Dr Yoshihiro Nishimoto for generously affording us high-power LED light for the large-scale synthesis. We are very appreciative to Amr Shaalan and Rashed Almasri for their insightful discussions and support during conducting the kinetic study. We also acknowledge the technical staff of the Comprehensive Analysis Center of SANKEN, Osaka University (Japan). The computational part was performed at the Research Center for Computational Science, Okazaki (IMS-RCCS-A-ja). This work was supported by JSPS KAKENHI Grant Numbers 21A204, 21H05217 and 22K06502, 21A204 from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), the Japan Society for the Promotion of Science (JSPS), JST CREST (No. JPMJCR20R1), and Hoansha Foundation.
References
-
(a) D. Kiejza, U. Kotowska, W. Polińska and J. Karpińska, Sci. Total Environ., 2021, 790, 148195 CrossRef CAS PubMed
 ;
(b) C. Shi, C. Li, Y. Wang, J. Guo, S. Barry, Y. Zhang and N. Marmier, Water, 2022, 14, 2309 CrossRef CAS ;
(c) N. Kaur and D. Kishore, Synth. Commun., 2014, 44, 721 CrossRef CAS ;
(d) H. Hussain, A. Al-Harrasi, I. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882 RSC ;
(e) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS PubMed ;
(f) T. Luukkonen and S. O. Pehkonen, Crit. Rev. Environ. Sci. Technol., 2017, 47, 1 CrossRef CAS .
;
(b) C. Shi, C. Li, Y. Wang, J. Guo, S. Barry, Y. Zhang and N. Marmier, Water, 2022, 14, 2309 CrossRef CAS ;
(c) N. Kaur and D. Kishore, Synth. Commun., 2014, 44, 721 CrossRef CAS ;
(d) H. Hussain, A. Al-Harrasi, I. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882 RSC ;
(e) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS PubMed ;
(f) T. Luukkonen and S. O. Pehkonen, Crit. Rev. Environ. Sci. Technol., 2017, 47, 1 CrossRef CAS .
- D. Swern, Chem. Rev., 1949, 45, 1 CrossRef CAS .
-
(a) B. T. Brooks and W. B. Brooks, J. Am. Chem. Soc., 1933, 55, 4309 CrossRef CAS ;
(b) L. S. Silbert, E. Siegel and D. Swern, J. Org. Chem., 1962, 27, 1336 CrossRef CAS ;
(c) J. Moyer and N. Manley, J. Org. Chem., 1964, 29, 2099 CrossRef CAS ;
(d) Y. Ogata and Y. Sawaki, Tetrahedron, 1967, 23, 3327 CrossRef CAS .
- M. Taguchi, Y. Nagasawa, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, Tetrahedron Lett., 2016, 57, 230 CrossRef CAS .
-
(a) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943 CrossRef CAS ;
(b)
J.-E. Bäckvall, Modern oxidation methods, John Wiley & Sons, 2011 Search PubMed .
-
(a) B. Ganem, R. P. Heggs, A. J. Biloski and D. R. Schwartz, Tetrahedron Lett., 1980, 21, 685 CrossRef CAS ;
(b) B. S. Bal, W. E. Childers Jr. and H. W. Pinnick, Tetrahedron, 1981, 37, 2091 CrossRef CAS ;
(c) S. O. Nwaukwa and P. M. Keehn, Tetrahedron Lett., 1982, 23, 3131 CrossRef CAS ;
(d) A. McKillop and D. Kemp, Tetrahedron, 1989, 45, 3299 CrossRef CAS ;
(e) A. McKillop and W. R. Sanderson, Tetrahedron, 1995, 51, 6145 CrossRef CAS ;
(f) B. R. Travis, M. Sivakumar, G. O. Hollist and B. Borhan, Org. Lett., 2003, 5, 1031 CrossRef CAS ;
(g) M. Hunsen, Synthesis, 2005, 2487 CrossRef CAS ;
(h) R. Bernini, A. Coratti, G. Provenzano, G. Fabrizi and D. Tofani, Tetrahedron, 2005, 61, 1821 CrossRef CAS ;
(i) M. Shibuya, T. Sato, M. Tomizawa and Y. Iwabuchi, Chem. Commun., 2009, 2009, 1739 RSC ;
(j) J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 2010, 12, 3618 CrossRef CAS ;
(k) A. B. Leduc and T. F. Jamison, Org. Process Res. Dev., 2012, 16, 1082 CrossRef CAS .
-
(a) A. K. Khatana, V. Singh, M. K. Gupta and B. Tiwari, Synthesis, 2018, 4290 CAS ;
(b) P.-F. Dai, J.-P. Qu and Y.-B. Kang, Org. Lett., 2019, 21, 1393 CrossRef CAS ;
(c) K. Takamatsu, M. Kasai, H. Nishizawa, R. Suzuki and H. Konno, Tetrahedron Lett., 2021, 81, 153320 CrossRef CAS ;
(d) B. S. Nagy, C. O. Kappe and S. B. Ötvös, Adv. Synth. Catal., 2022, 364, 1998 CrossRef CAS .
-
(a) D. R. Larkin, J. Org. Chem., 1990, 55, 1563 CrossRef CAS ;
(b) M. Liu, H. Wang, H. Zeng and C.-J. Li, Sci. Adv., 2015, 1, e1500020 CrossRef PubMed ;
(c) M. Liu and C. J. Li, Angew. Chem., Int. Ed., 2016, 55, 10806 CrossRef CAS ;
(d) H. Yu, S. Ru, G. Dai, Y. Zhai, H. Lin, S. Han and Y. Wei, Angew. Chem., Int. Ed., 2017, 56, 3867 CrossRef CAS ;
(e) X. Yan, Y.-H. Lai and R. N. Zare, Chem. Sci., 2018, 9, 5207 RSC ;
(f) L. J. Durndell, C. Cucuzzella, C. M. Parlett, M. A. Isaacs, K. Wilson and A. F. Lee, Catal. Today, 2019, 333, 161 CrossRef CAS .
- Y. Zhang, Y. Cheng, H. Cai, S. He, Q. Shan, H. Zhao, Y. Chen and B. Wang, Green Chem., 2017, 19, 5708 RSC .
- K.-J. Liu, Y.-L. Fu, L.-Y. Xie, C. Wu, W.-B. He, S. Peng, Z. Wang, W.-H. Bao, Z. Cao and X. Xu, ACS Sustainable Chem. Eng., 2018, 6, 4916 CrossRef CAS .
-
(a) H. L. Bäckström, J. Am. Chem. Soc., 1927, 49, 1460 CrossRef ;
(b) H. L. Bäckström and Ü. Riiner, Acta Chem. Scand., 1966, 20, 630 CrossRef ;
(c) R. Cundall and A. Davies, Trans. Faraday Soc., 1966, 62, 2444 RSC ;
(d) I. V. Khudyakov, P. F. McGarry and N. J. Turro, J. Phys. Chem., 1993, 97, 13234 CrossRef CAS ;
(e) M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833 CrossRef CAS PubMed .
-
(a) W. Jorissen and P. van der Beek, Recl. Trav. Chim. Pays-Bas, 1926, 45, 245 CrossRef CAS ;
(b) P. Van der Beek, Recl. Trav. Chim. Pays-Bas, 1932, 51, 411 CrossRef CAS ;
(c) T. W. Findley, D. Swern and J. T. Scanlan, J. Am. Chem. Soc., 1945, 67, 412 CrossRef CAS ;
(d) C. R. Dick and R. F. Hanna, J. Org. Chem., 1964, 29, 1218 CrossRef CAS .
-
(a) S.-I. Hirashima and A. Itoh, Chem. Pharm. Bull., 2007, 55, 156 CrossRef CAS ;
(b) M. Hajimohammadi, N. Safari, H. Mofakham and A. Shaabani, Tetrahedron Lett., 2010, 51, 4061 CrossRef CAS ;
(c) N. Iqbal, S. Choi, Y. You and E. J. Cho, Tetrahedron Lett., 2013, 54, 6222 CrossRef CAS ;
(d) Z. E. Hamami, L. Vanoye, P. Fongarland, C. de Bellefon and A. Favre-Réguillon, J. Flow Chem., 2016, 6, 206 CrossRef CAS ;
(e) J. Xu, X. Yue, L. He, J. Shen, Y. Ouyang, C. Liang and W. Li, ACS Sustainable Chem. Eng., 2022, 10, 14119 CrossRef CAS ;
(f) H. Shi, J. Li, T. Wang, M. Rudolph and A. S. K. Hashmi, Green Chem., 2022, 24, 5835 RSC ;
(g) C. S. Batsika, C. Koutsilieris, G. S. Koutoulogenis, M. G. Kokotou, C. G. Kokotos and G. Kokotos, Green Chem., 2022, 24, 6224 RSC .
- L. Vanoye, M. Abdelaal, K. Grundhauser, B. Guicheret, P. Fongarland, C. De Bellefon and A. Favre-Réguillon, Org. Lett., 2019, 21, 10134 CrossRef CAS PubMed .
- Y. Ogata and Y. Sawaki, J. Am. Chem. Soc., 1972, 94, 4189 CrossRef CAS .
- J. Yang, J. Jiang, J. Jiang, X. Pan, Y. Pan and L. Ni, J. Therm. Anal. Calorim., 2019, 135, 2309 CrossRef CAS .
-
(a) C. Lehtinen, V. Nevalainen and G. Brunow, Tetrahedron, 2001, 57, 4741 CrossRef CAS ;
(b) Z. Wang, Y. Qin, H. Huang, G. Li, Y. Xu, P. Jin, B. Peng and Y. Zhao, Front. Chem., 2022, 10, 855843 CrossRef CAS .
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC .
- S. Kato and F. Mashio, J. Soc. Chem. Ind., Jpn., 1957, 60, 1515 CAS .
-
(a) A. Baeyer and V. Villiger, Ber. Dtsch. Chem. Ges., 1899, 32, 3625 CrossRef ;
(b) M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737 CrossRef CAS .
- L. Vanoye and A. Favre-Réguillon, React. Chem. Eng., 2023, 8, 1043 RSC .
-
(a) L. Vanoye, A. Favre-Réguillon, A. Aloui, R. Philippe and C. de Bellefon, RSC Adv., 2013, 3, 18931 RSC ;
(b) L. Vanoye and A. Favre-Réguillon, Org. Process Res. Dev., 2022, 26, 335 CrossRef CAS .
- D. Rusinska-Roszak, Molecules, 2017, 22, 481 CrossRef PubMed .
-
(a) A. Favre-Réguillon, ACS Sustainable Chem. Eng., 2023, 11, 1619 CrossRef ;
(b) A. Favre-Réguillon and L. Vanoye, Green Chem., 2023, 25, 5756 RSC ;
(c) J. Xu, X. Yue, L. He, J. Shen, Y. Ouyang, C. Liang and W. Li, ACS Sustainable Chem. Eng., 2023, 11, 1622 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Carla
Dubois
ab,
Carla
Dubois