
Chemical recycling of polycarbonate and polyester without solvent and catalyst: mechanochemical methanolysis†
Hyo Won
Lee
ab,
Kwangho
Yoo
b,
Lars
Borchardt
*b and
Jeung Gon
Kim
 *ac
*ac
aDepartment of Chemistry and Research Institute of Physics and Chemistry Jeonbuk National University, Jeonju, 54896, Republic of Korea. E-mail: jeunggonkim@jbnu.ac.kr
bInorganic Chemistry I, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: lars.borchardt@rub.de
cDepartment of JBNU-KIST Industry-Academia Convergence Research, Jeonbuk National University, Jeonju 54896, Republic of Korea
First published on 4th January 2024
Abstract
In this study, we present a green and economical approach to chemical recycling of commercial polycarbonates and poly-esters, specifically poly(bisphenol A carbonate), poly(ethylene terephthalate), and poly(lactic acid). Our method involves mechanochemical ball-milling of a heterogeneous mixture of plastic and methanol, resulting in quantitative depolymerization to yield monomers or useful chemical units that already have high demands. We found that the energy-intensive step is forming physical contact between the reactants, rather than the chemical methanolysis itself. Mechanochemical ball-milling facilitates sufficient physical contact and energy transfer between plastics and methanol, eliminating the need for solvents and catalysts. Our study demonstrates a practical and sustainable process with minimal chemical input and simple output for the chemical recycling of these plastics.
Introduction
Due to their low cost, ease of production, and desirable properties, plastics have become ubiquitous. However, the resulting plastic waste has become a significant environmental issue, requiring immediate attention.1,2 While reducing plastic usage and mechanical recycling are temporary solutions, chemical recycling offers a long term solution.3,4 By breaking down plastic waste into its monomers through clean depolymerization, it can be repolymerized, preserving the benefits of plastic while minimizing the environmental footprint. Or polymer degradation to value-added chemicals, called chemical upcycling, is another way to have waste plastics as a chemical resource. As a result, significant investments have been made in chemical recycling research and development.This report centers on the chemical recycling of carbonate and ester polymers, including poly(bisphenol-A carbonate) (BPA-PC), poly(ethylene terephthalate) (PET), and poly(lactic acid) (PLA), which are significant constituents of common plastics.5–7 BPA-PC waste is concerning because of its bisphenol A content, which is a xenoestrogen.8,9 PET is the plastic targeted for efficient mechanical recycling.10,11 However, the market for post-PET products is insufficient to handle current PET waste levels. PLA is a representative bio-based green plastic that is quickly replacing other petro-based polymers. Nonetheless, its degradation in nature is slow, leading to persistent plastic issues such as microplastics.7,12 All of these materials require practical and fully circular chemical recycling processes to become sustainable plastics.
Polycarbonates, such as BPA-PC, and polyesters, such as PET and PLA, contain chemically active ester and carbonate groups, making their chemical recycling an area of interest.13–15 Previous reports on chemical recycling using alcohols and amines have claimed high efficiency and mild conditions, but they require homogeneous conditions and activating reagents, resulting in complex processes and high costs. In addition, PET presents a particular challenge due to its poor solubility in common solvents.16,17 Recently, Wang and colleagues reported the successful catalyst- and solvent-free degradation of BPA-PC, PET, and PLA using amino alcohols.18 The direct amidation between reagent and polymer was feasible under proper conditions and reagents. However, PET and PLA depolymerization resulted in diol products that do not have market need at this moment. Therefore, there is a need to develop a catalyst- and solvent-free chemical recycling process that produces monomers or chemical products with existing bulk usage, which would be highly desirable for sustainable plastic recycling.
The present study proposes the use of mechanochemistry as a means of enhancing the sustainability and cost-effectiveness of the chemical recycling of BPA-PC, PET, and PLA.19–21 Mechanochemistry employs physical processes like collision, shearing, and grinding to initiate chemical reactions, leading to efficient mixing and energy transfer, solvent elimination, and maximum green chemistry. This technique has been shown to be effective in degrading even highly insoluble plastics, such as BPA-PC, PET, and PLA, using methanol, into their constituent raw materials through high-speed ball-milling transesterifications (Fig. 1), without the need for solvents or catalysts.22–24 The mechanical energy and contact provided by mechanochemistry promote the depolymerization of the ester- and carbonate-based polymers, even with highly immiscible methanol, resulting in a simple and efficient process with minimal chemical input and low energy consumption. The kinetic study of model system proved that high thermal energy input is necessary to overcome physical immiscibility and catalyst is not necessary for transesterification. The mechanochemical methanolysis of BPA-PC, PET, and PLA with methanol quantitatively produced BPA, dimethyl carbonate (DMC), dimethyl terephthalate (DMT), and methyl lactate. These products have high demand in the current market and can be reused in the production of BPA-PC and PET or other applications.
 | ||
| Fig. 1 Concept of mechanochemical recycling methanolysis of carbonate & ester plastics: no catalyst, no solvent, and high efficiency. | ||
Experimental
Materials
All reagents were purchased from Alfa Aesar and used without further purification. The PC pellet had the shape of an elliptical column, with an average width of 3.9 mm, length of 2.8 mm, and height of 3.0 mm from Lotte Chemical in South Korea [SC-1190P, number average molecular weight (Mn) = 16.4 kg mol−1, dispersity (Đ) = 2.15]. The PLA cup was made of 100% PLA and was purchased from e·SOL company in South Korea (Mn = 80.6 kg mol−1, Đ = 2.51). The ball-milling experiments were conducted using a Retsch Planetary Mill PM 100 instrument with either a 25 mL or 125 mL stainless-steel vessel and stainless-steel balls with diameters of 5 mm. To obtain PET powder, PET cup pieces were placed in a SUS 25 mL mixer mill container, frozen in liquid nitrogen, and milled twice for 3 min using a Retsch Mixer Mill MM 400. The general ball-milling experiments were performed using a Retsch Planetary Mill PM 100 instrument with either a 25 mL or 125 mL stainless-steel vessel and stainless-steel balls with diameters of 5 mm.Measurement methods
The 1H NMR spectra were measured by a Bruker AVANCE III HD-400 MHz Fourier transform NMR spectrometer. Mn and Đ were estimated using a size exclusion chromatography (SEC) equipped with a refractive index (RI) detector. The equipment consisted of a Waters 1515 isocratic pump, a Waters 2414 differential refractive index detector, and a column-heating module with Shodex HK-0403 and HK-404L columns placed in series. The samples were eluted with HPLC-grade tetrahydrofuran (THF) at 40 degrees and 1.0 mL min−1. A calibration curve was obtained with 16 monodispersed polystyrene standards (purchased from Alfa Aesar).Representative procedure for BPA-PC methanolysis
PC pellet (0.25 g, 1.0 mmol of carbonate functionality) and dimethyl carbonate (0.17 mL, 2.0 mmol) were added to a stainless-steel container (25 mL) having 50 stainless steel balls with a diameter of 5 mm. The vessel was placed in a planetary ball-milling machine and milled at 600 rpm for 30 min. After milling, methanol (1.2 mL, 30 mmol) was added, and the mixture was further milled at 600 rpm for 6 h. The container was opened and an aliquot of the reaction mixture was taken, and filtered through a syringe filter (45 mM) for 1H NMR (CDCl3) and SEC analysis.Chemical recycling of compact disc
CD (5.0 g, 20 mmol of repeat unit) and dimethyl carbonate (3.4 mL, 40 mmol) were added to a stainless-steel container (125 mL) containing 500 stainless steel balls with a diameter of 5 mm. The vessel was placed in a planetary ball-milling machine and milled at 600 rpm for 30 min. After milling, methanol (24 mL, 600 mmol) was added, and the mixture was further milled at 600 rpm for 6 h. The resulting mixture was transferred with methanol to a round-bottom flask, and the volatiles were removed. The flask was subjected to acid work-up using 1 M HCl and then separated with dichloromethane. The product was purified by silica filter to afford BPA (4.1 g, 90%).Chemical recycling of PLA cup
PLA cup (2.2 g, 30 mmol of repeat unit) was added to a stainless-steel container (125 mL) containing 250 stainless steel balls with a diameter of 5 mm, along with methanol (24 mL, 600 mmol). The vessel was placed in a planetary ball-milling machine and milled at 600 rpm for 6 h. The resulting mixture was transferred to a round-bottom flask with methanol, and the volatiles were removed. The flask was subjected to acid work-up using 1 M HCl and then separated with dichloromethane. A solvent removal afforded methyl lactate (3.1 g, 98%).Chemical recycling of PET cup
PET powder (2.8 g, 15 mmol of repeat unit) and methanol (43 mL, 1.05 mol) were added to stainless steel container (125 mL) having 300 stainless steel balls with a diameter of 5 mm. The vessel was placed in a planetary ball-milling machine and milled at 650 rpm for 6 h. The resulting mixture was transferred with methanol in a round bottom flask, and the volatiles were removed. The flask was subjected to acid work-up using 1 M HCl and then separated with dichloromethane. The product was purified by silica filter to afford DMT (2.4 g, 82%).Results and discussion
Model methanolysis with diphenyl carbonate
We first evaluated a model methanolysis of carbonate using diphenyl carbonate (DPC), an aromatic carbonate molecule that is chemically relevant to BPA-PC.25 We observed that a non-catalytic methanolysis reaction pathway is available, which produces methyl phenyl carbonate (MPC), DMC, and phenol (Fig. 2 and Table S1†). DPC esterification with various alcohols generates valuable organic carbonates. However, all previous reports utilized a catalyst, and no study has proposed non-catalytic alcoholysis. Only one example showcased catalyst-free aminolysis.18 | ||
| Fig. 2 Catalyst-free DPC methanolysis to DMC. (A) DPC and methanol mixture at 100 °C. (B) Planetary ball milling (SUS 12 mL, 5 mm balls 50 ea, 600 rpm). | ||
To the vial, we added DPC (1.0 mmol) and MeOH (70 mmol). DPC was slightly soluble in MeOH at ambient conditions, and no chemical reaction occurred. However, upon heating the mixture to 100 °C in a closed vessel, it became homogeneous and promoted non-catalytic methanolysis to MPC, DMC, and phenol (Fig. 2A). Within an hour, 57% of DPC had turned into 51% MPC and 6% DMC. The consumption of DPC continued, but the second methanolysis from MPC to DMC was slow. After a near quantitative DPC conversion, a significant amount of MPC (55%) remained.
The same combination was tested under mechanochemical conditions, whereby a heterogeneous mixture of DPC and MeOH was subjected to ball-milling in a 12 mL stainless steel (SS) jar containing 50 × 5 mm SS balls, using planetary ball milling (PM100, Restch) at 600 rpm (Fig. 2B). The results indicated that DPC conversion to MPC and DMC occurred more rapidly than in solution reactions at 100 °C. Within 1 hour, more than half of DPC (75%) had transformed to MPC (60%) and DMC (15%), while milling for 3 hours completely consumed DPC, producing MPC (27%) and DMC (73%). Full conversion to DMC was achieved after 5 hours. The reaction profile over time showed a temporary accumulation of MPC before the second step, similar to that observed in solution reactions. These findings suggest that mechanochemical ball milling methanolysis followed the same reaction profile. The mechanochemical condition exhibited superior performance to the conventional one. However, there is a possibility that the SS media used in the milling process may have catalyzed the methanolysis. There have been reports of milling media participating in the reaction.26–28 To investigate this, we conducted a solution reaction at 100 °C with a fine powder of SS (SUS304, 100 mesh) to elucidate the media effect. However, there was no significant rate acceleration by metal powder, ruling out the possibility of direct catalysis by the milling media (see ESI, Table S2 and Fig. S2†).
Mechanochemical methanolysis of BPA-PC
The conditions used for non-catalytic direct methanolysis of DPC were tested on BPA-PC (Table 1). A commercial BPA-PC powder with a number average molecular weight (Mn) of 16.4 kg mol−1 and a dispersity (Đ) of 2.15, measured using size exclusion chromatography (SEC) in tetrahydrofuran with polystyrene standard, was reacted with 70 mmol of MeOH to 1 mmol of carbonate unit. However, unlike DPC, BPA-PC was insoluble in MeOH even at 100 °C and failed to promote depolymerization (entry 1). There was only a marginal reduction in molecular weight to Mn = 14.3 kg mol−1. If methanolysis had occurred, random chain scission would have happened throughout the polymer, resulting in multimodal molecular weight distribution.29,30 However, the BPA-PC after MeOH treatment maintained an unimodal dispersity (Đ = 2.04) similar to the starting BPA-PC. And no trace of oligomeric or monomeric species was detected. Thus, we ruled out non-catalytic solution-phase methanolysis as the depolymerization mechanism. In the model reaction of DPC methanolysis, methanol served as both solvent and reactant. However, when it did not act as a solvent with a polymeric carbonate, no reaction occurred.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Type | MeOH (mmol) | Conv. (%) | BPA (%) | BPAMC (%) | BPADC (%) | Comments |
| a Reaction time – 6 hours. b Conversions and yields were determined by 1H NMR spectroscopy after methanol removal. | |||||||
| Solution reaction, 100 °C | |||||||
| 1 | Powder | 70 | 0 | — | — | ||
| Planetary ball-milling, SUS 25 mL, 5 mm × 50 ea, 600 rpm | |||||||
| 2 | Powder | 70 | 97 | 78 | 18 | 1 | 12 ml jar |
| 3 | 70 | 99 | 99 | — | — | ||
| 4 | 30 | 99 | 99 | — | — | ||
| 5 | 10 | 55 | 14 | 10 | 31 | ||
| 6 | Pellet | 30 | 0 | — | — | — | |
| 7 | 30 | 99 | 99 | — | — | DMC 2 mmol | |
Subsequently, mechanochemical conditions were employed for the same mixture. Planetary ball milling of the heterogeneous mixture of BPA-PC (powder, 1 mmol) and MeOH (70 mmol) with 50 SS balls (5 mm diameter) in a 12 mL SS jar resulted in near-quantitative depolymerization after 6 hours (entry 2). However, the resulting mixture consisted of monomeric species, including BPA (78%), BPA-dimethylcarbonates (BPADC, 1%), and BPA-monomethylcarbonate (BPAMC, 18%). Switching to a 25 mL jar led to full conversion to BPA and DMC (entry 3), likely due to better ball movement and mixing facilitated by the increased volume. Reducing the amount of MeOH to 30 equiv. retained the depolymerization efficiency with quantitative production of BPA and DMC (entry 4). However, further reduction below 30 equiv. compromised the reactivity (entry 5).
The time-resolved reaction tracking allowed us to understand how the depolymerization process progressed (Fig. 3). After an hour, the mixture remained heterogeneous. The 1H NMR spectra contained a mixture of polymeric BPA-PC (yellow), monomeric BPAMC (blue), and BPA (green). Although a significant amount of monomeric species was produced, the BPA-PC portion maintained a high molecular weight (9.7 kg mol−1). BPA-PC on the surface reacted with methanol during collisions and generated low molecular weight fragment`s, while the core BPA-PC remained unreacted. After 3 hours, a homogeneous mixture was formed, as shown by both the 1H NMR and SEC results, with no traces of polymeric BPA-PC. The reaction continued homogeneously, and after 5 hours of ball milling, only BPA was detected.
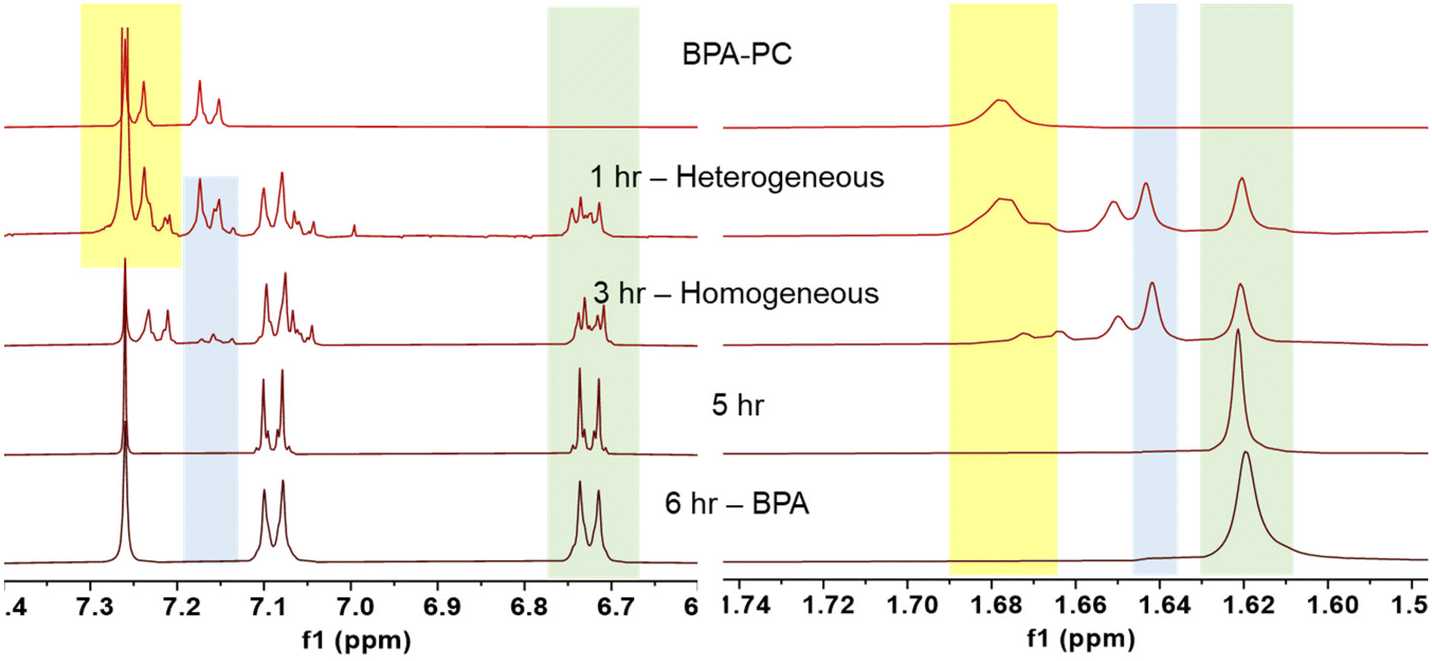 | ||
| Fig. 3 1H NMR spectra changes of aromatic and dimethyl protons during methanolysis of BPA-PC. | ||
1H NMR measurements were obtained in CDCl3 after filtration and methanol removal to enhance the visibility of the BPA peaks. However, since DMC has a boiling point of 90 °C, it evaporated during the process. To confirm the production of DMC, a separate measurement using non-deuterated proton NMR spectroscopy was performed on a crude aliquot, and the presence of DMC was confirmed (Fig. S4†).31
To confirm the catalytic effect of the metallic components in the SS jar and balls, the reaction was conducted using a zirconia jar and balls. A mixture of BPA-PC (powder, 1 mmol) and MeOH (30 mmol) in a 50 mL zirconia jar with 26 g of 5 mm balls resulted in partial depolymerization. The resulting mixture contained 37% BPA, 26% BPAMC, and 5% BPADC. This supports the notion that PC methanolysis can occur without metal catalysis, as demonstrated in Fig. S5.† The reduced reactivity could be attributed to the weaker mechanical energy resulting from the lower density of zirconia. However, we cannot completely rule out the potential catalytic effect of the metallic components in the SS jar and balls.
We also evaluated the effect of mechanical fragmentation. Mechanical force on polymer chains could result in chain fragmentation, which might help the BPA-PC depolymerization process.32,33 But, the same milling process without MeOH did not show a notable effect on PC. A molecular weight change of less than 2% from 16.4 kg mol−1 to 16.1 kg mol−1 proved that representative engineering plastic BPA-PC with excellent impact resistance endured a milling force (Fig. S6†). The depolymerization proceeded solely by methanolysis.
In the following study, the shape of BPA-PC was investigated. A chunk shape was more prevalent than powder in waste plastics. Therefore, pellet PC was depolymerized to simulate waste-PC under identical conditions. After six hours, the pellet shape PC retained its shape and could not be broken down to powder through milling due to its mechanical robustness. Furthermore, depolymerization was not detected through 1H NMR (entry 6). To address this, a liquid-assisted grinding system was devised, where a small amount of a good solvent, such as DMC, was used to swell the pellet and penetrate its polymer domain. The final mixture contained the same three components, making the postseparation process less complicated. The BPA-PC pellets (1 mmol) and DMC (2 mmol) were milled for 30 minutes, and the resulting powder had a larger surface area. Further milling with MeOH (30 mmol) allowed for complete depolymerization to BPA and DMC (entry 7).
In a previous study by Kim et al., BPA-PC methanolysis was analyzed step-by-step at high temperatures.34 They used pellet BPA-PC and methanol in a tightly closed tube for depolymerization. At high temperatures above 160 °C, depolymerization to BPA and DMC occurred. To evaluate the effect of temperature on high-speed ball-milling, we monitored the temperature change of the reactor and the reaction mixture. At the end of milling, the reaction jar was opened, and an infrared thermometer recorded the temperature of the mixture. The reaction of Table 1, entry 4 was repeated, and the reactor reached 54 °C, where the thermal reaction would not proceed (Fig. S7†). We speculated that high thermal energy input to near 200 °C was necessary to locate MeOH inside the BPA-PC domain under heterogeneous conditions, rather than chemical transesterification. Mechanochemical conditions successfully provided both sufficient contact and activation energy.
The conditions developed at a 1 mmol (255 mg) scale were scaled up to grams (Fig. 4). BPA-PC (5.0 g, 20 mmol) was treated with DMC (3.4 mL, 40 mmol) in a 125 mL SS jar containing 500 ∅ 5 mm balls. Crushed BPA-PC was mixed with MeOH (24 mL, 600 mmol). Milling at 600 rpm for 6 hours resulted in quantitative depolymerization to BPA and DMC. Pure BPA (3.9 g) was isolated after silica filtration.
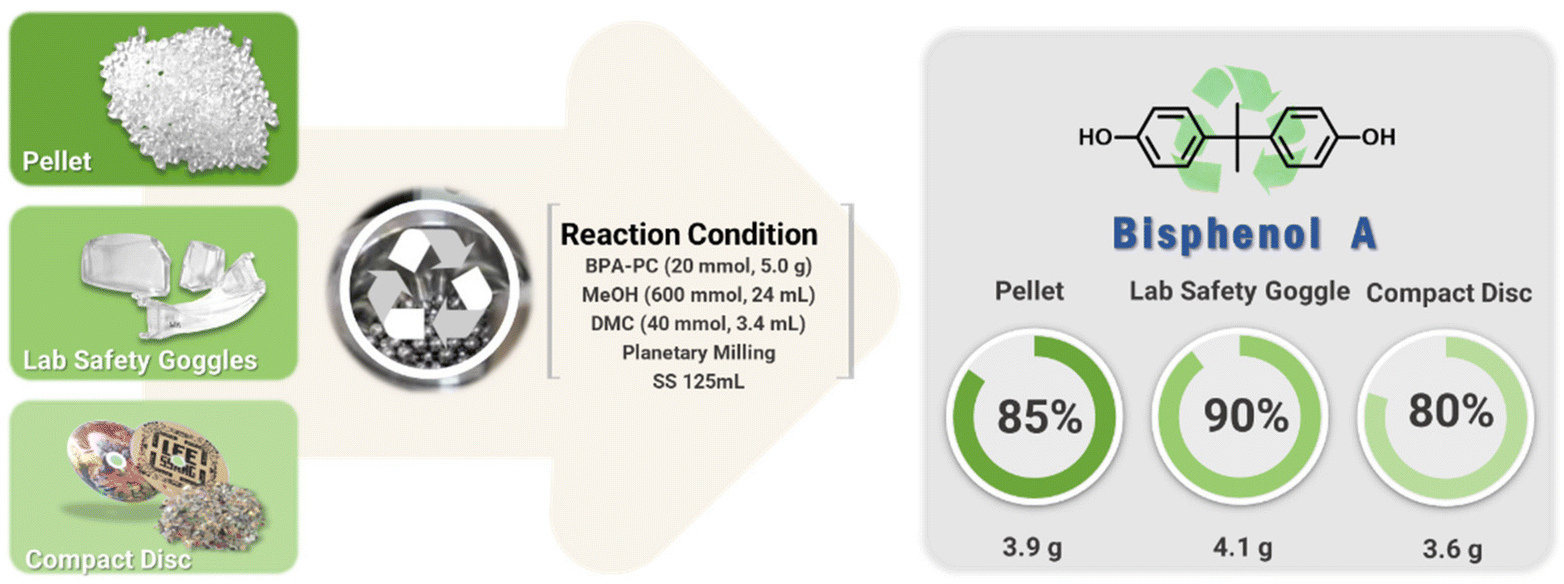 | ||
| Fig. 4 Mechanochemical methanolysis of BPA-PC from various sources. | ||
The chemical recycling of waste polycarbonates continued using compact discs (CD, Mn = 16.4 kg mol−1, Đ = 2.15) and safety glasses (Mn = 21.1 kg mol−1, Đ = 2.55), which contained additives such as UV stabilizers and mold release agents that could interfere with the depolymerization process. The mechanochemical process proceeded regardless of the molecular weight and additives. The full conversions to BPA and DMC showed promise for the mechanochemical process. BPA were isolated of 4.1 g (90%) from CD and 3.6 g (80%) from safety glasses, respectively.
Mechanochemical methanolysis of polyesters, PET and PLA
The chemical depolymerization of PET and PLA was then investigated (Fig. 5). As the ester and carbonate groups in these polymers are chemically similar, particularly in terms of transesterification reactions, it was expected that the methanolysis conditions used for BPA-PC would also be effective for PET and PLA. The mechanochemical hydrolysis of PET has been reported previously.35–38 The examples used activation additives such as enzymes and sodium hydroxide, transferring similar conventional combinations to mechanochemical conditions. Thus, additional purification, neutralization, and waste generation are inevitable. Our catalyst- and solvent-free methanolysis of polyesters expects simple mechanochemistry exclusive processes as seen in BPA-PC. To test this hypothesis, PET and PLA obtained from used cold beverage containers were chopped and added to a ball-milling jar containing MeOH. The chunk-type PLA (2.2 g, 30 mmol) and MeOH (20 equiv.) underwent quantitative methanolysis under otherwise identical conditions (3.1 g methyl lactate, 98% isolation yield). However, the mechanically more robust PET required additional treatment to achieve complete methanolysis. While chunk-type PET did not undergo methanolysis, powder PET (2.8 g, 15 mmol) with methanol (70 equiv.) resulted in a mixture of MeOH, DMT, and ethylene glycol (Fig. S11†). After silica filtration and removal of volatiles, pure DMT was obtained (2.4 g, 82%).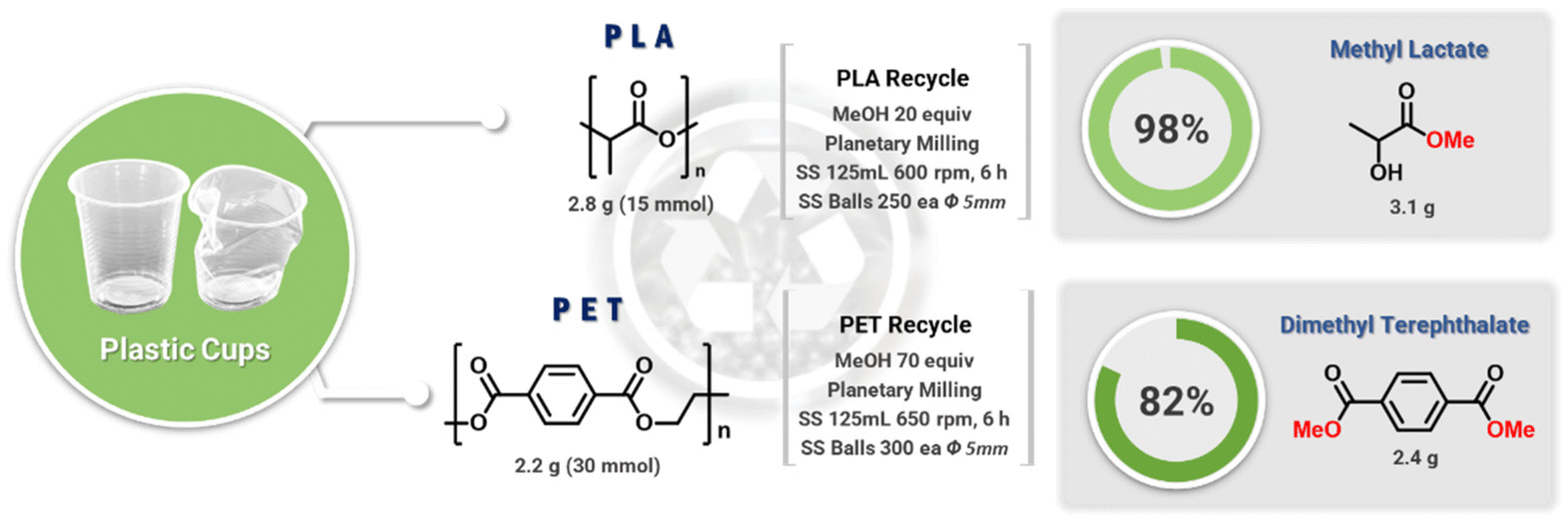 | ||
| Fig. 5 Mechanochemical depolymerization of after-use PET and PLA containers. | ||
Conclusions
Our findings suggest that mechanochemistry has the potential to enhance the sustainability and cost-effectiveness of chemical recycling. The technique employs physical processes such as collision, shearing, and grinding to induce chemical reactions, leading to efficient mixing and energy transfer, elimination of auxiliary chemicals, and maximum green chemistry. This approach was shown to be effective in degrading even highly insoluble conditions, such as BPA-PC, PET, and PLA, into their constituent raw materials through high-speed ball-milling transesterifications, without the need for solvents or catalysts. The final products, BPA, DMC, DMT, and methyl lactate, can be resubjectable to BPA-PC and PET production or have existing high demands in the market. We currently investigate scale-up options for further economic evaluations.Author contributions
Conceptualization: HWL, JGK, LB; investigation: HWL, KY; funding acquisition: JGK, LB; supervision: JGK, LB; writing – original draft: JGK; writing – review & editing: HWL, YK, JGK, LB.Conflicts of interest
The authors declare the following competing financial interest(s): a patent applications KR10-2023-0103879 and PCT/KR2023/018625 submitted by the Jeonbuk National University.Acknowledgements
This research was supported Samsung Science & Technology Foundation SRFC-MA1902-05 (HWL, JGK), National Research Foundation of Korea (NRF-2019R1A2C1087769) (HWL, JGK), and European Union's Horizon 2020 research and innovation program of European Research Council (grant agreement no. 948521) (KY, LB).References
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- A. Rahimi and J. M. García, Nat. Rev., 2017, 1, 0046 Search PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- D. J. Brunelle, ACS Symp. Ser., 2005, 898, 1–5 CAS.
- K. Ravindranath and R. A. Mashelkar, Chem. Eng. J., 1986, 41, 2197–2214 CrossRef CAS.
- E. Castro-Aguirre, F. Iñiguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS PubMed.
- S. A. Vogel, Am. J. Public Health, 2009, 99, S559–S566 CrossRef PubMed.
- C. M. Metz, Workplace Health Saf., 2016, 64, 28–36 CrossRef PubMed.
- F. Awaja and D. Pavel, Eur. Polym. J., 2005, 41, 1453–1477 CrossRef CAS.
- F. Welle, Resour., Conserv. Recycl., 2011, 55, 865–875 CrossRef.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- J. G. Kim, Polym. Chem., 2020, 11, 4830–4849 RSC.
- E. Barnard, J. J. R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- B.-K. Kim, G.-C. Hwang, S.-Y. Bae, S.-C. Yi and H. Kumazawa, J. Appl. Polym. Sci., 2001, 81, 2102–2108 CrossRef CAS.
- T. Sako, I. Okajima, T. Sugeta, K. Otake, S. Yoda, Y. Takebayashi and C. Kamizawa, Polym. J., 2000, 32, 178–181 CrossRef CAS.
- X. Zhou, M. Chai, G. Xu, R. Yang, H. Sun and Q. Wang, Green Chem., 2023, 25, 952–959 RSC.
- S. J. James and T. Friščić, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed.
- G. S. Lee, H. W. Lee, H. S. Lee, T. Do, J.-L. Do, J. Lim, G. I. Peterson and T. Friščić, Chem. Sci., 2022, 13, 11496–11505 RSC.
- T. Yong, G. Báti, F. García and M. C. Stuparu, Nat. Commun., 2021, 12, 5187 CrossRef CAS PubMed.
- E. R. Baral, J. H. Lee and J. G. Kim, J. Org. Chem., 2018, 83, 11768–11776 CrossRef CAS PubMed.
- Y. Sawama, N. Yasukawa, K. Ban, R. Goto, M. Niikawa, Y. Monguchi, M. Itoh and H. Sajiki, Org. Lett., 2018, 20, 2892–2896 CrossRef CAS PubMed.
- S. Hwang, S. Grätz and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC.
- M. Wohlgemuth, M. Mayer, M. Rappen, F. Schmidt, R. Saure, S. Grätz and L. Borchardt, Angew. Chem., Int. Ed., 2022, 61, e202212694 CrossRef CAS PubMed.
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- H. J. Jung, S. Park, H. S. Lee, H. G. Shin, Y. Yoo, E. R. Baral, J. H. Lee, J. Kwak and J. G. Kim, ChemSusChem, 2021, 14, 4301–4306 CrossRef CAS PubMed.
- T. R. Hoye, B. M. Eklov, T. D. Ryba, M. Voloshin and L. J. Yao, Org. Lett., 2004, 6, 953–956 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- G. I. Peterson, W. Ko, Y.-J. Hwang and T.-L. Choi, Macromolecules, 2020, 53, 7795–7802 CrossRef CAS.
- D. Kim, B.-K. Kim, Y. Cho, M. Han and B.-S. Kim, Ind. Eng. Chem. Res., 2009, 48, 6591–6599 CrossRef CAS.
- V. Štrukil, ChemSusChem, 2021, 14, 330–338 CrossRef PubMed.
- A. W. Tricker, A. A. Osibo, Y. Chang, J. X. Kang, A. Ganesan, E. Anglou, F. Boukouvala, S. Nair, C. W. Jones and C. Sievers, ACS Sustainable Chem. Eng., 2022, 10, 11338–11347 CrossRef CAS.
- E. Ambrose-Dempster, L. Leipold, D. Dobrijevic, M. Bawn, E. M. Carter, G. Stojanovski, T. D. Sheppard, J. W. E. Jeffries, J. M. Ward and H. C. Hailes, RSC Adv., 2023, 13, 9954–9962 RSC.
- S. Kaabel, J. P. D. Therien, C. E. Deschênes, D. Duncan, T. Friščic and K. Auclair, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026452118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03643j |
| This journal is © The Royal Society of Chemistry 2024 |