
Electrochemical chlorination of least hindered tertiary and benzylic C(sp3)–H bonds†
Jianyou
Zhao
,
Jiatai
Zhang
,
Pengkai
Fang
,
Jintao
Wu
,
Fan
Wang
and
Zhong-Quan
Liu
 *
*
Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization, College of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210023, China. E-mail: liuzhq@lzu.edu.cn
First published on 1st December 2023
Abstract
Functionalization of C(sp3)–H bonds should greatly benefit the synthesis of natural products and pharmaceuticals. One of the greatest challenges is to develop new synthetic strategies for industrial applicability. Herein we report an electrochemical method for the chlorination/bromination of tertiary and secondary benzylic C(sp3)–H bonds. This method tolerates many functional groups. Moreover, this reaction can be easily scaled up to 100 grams without losing its efficiency.
C(sp3)–Cl bonds are found in a wide range of natural products and pharmaceutical molecules.1 In addition, diverse functional group transformation can be conveniently achieved by the substitution of C(sp3)–Cl bonds.2 Therefore, the exploration of efficient methods for the incorporation of chlorine atoms into organic molecules has received great attention.3 Selective chlorination of inert C(sp3)–H bonds represents one of the most attractive strategies.4 As demonstrated in Scheme 1, considerable advances in direct chlorination of C(sp3)–H bonds have been made in the past few decades. Traditional chlorination of simple alkanes usually uses toxic chlorine as the Cl source but it suffers from poor selectivity (Scheme 1a).5 Wohl–Ziegler halogenation and related radical C–H bond halogenation are usually limited to the relatively active benzylic and allylic C(sp3)–H bonds (Scheme 1b).6 The typical Hoffmann–Löffler–Freytag (HLF) reaction enables remote C(sp3)–H bond chlorination (Scheme 1c).7 However, this strategy generally relies on preformed substrates and a 1,5 (or 6)-H shift to N- or O-centered radicals. Recently, several attractive protocols have been developed to realize site-selective chlorination of the C(sp3)–H bond in complex molecules (Scheme 1d).8 For example, Alexanian et al. reported a series of visible-light-promoted site-selective aliphatic C–H halogenation reactions using N-haloamides.9 In 2016, Chen and co-workers accomplished the halogenation and azidation of tertiary C(sp3)–H bonds using Zhdankin's azidoiodinane as the H-atom abstractor.10 Very recently, selective chlorination of the C(sp3)–H bond suitable for remote functionalization using the same hypervalent iodine reagent was achieved by Hartwig et al.11 At the same time, Leonori and co-workers discovered an aminium radical-promoted C(sp3)–H bond chlorination with high site selectivity.12 However, most of these systems require preforming Cl source and/or using unstable radical precursors, which are inappropriate for large-scale industrial production.
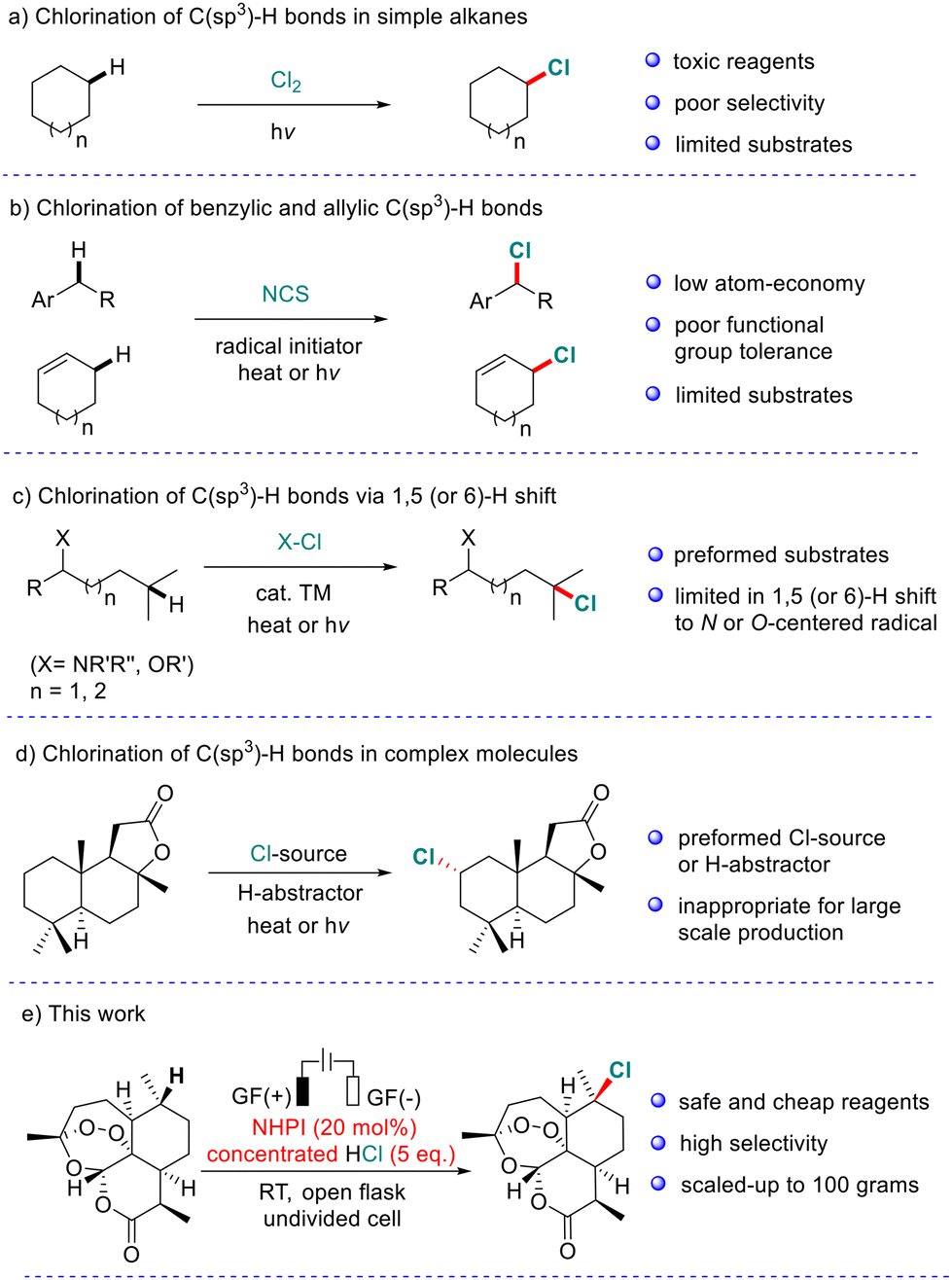 | ||
| Scheme 1 Chlorination of C(sp3)–H bonds. | ||
Therefore, the development of new strategies for remote functionalization including halogenation of the inert C(sp3)–H bond that is suitable for both late-stage functionalization of complex molecules and large-scale synthesis remains a great challenge.13 Herein, we wish to report a regio-selective and scalable chlorination/bromination of tertiary and benzylic C(sp3)–H bonds catalyzed by N-hydroxyphthalimide (NHPI) in an undivided cell (Scheme 1e).
NHPI is a cheap and safe reagent that is widely used as an efficient catalyst for C(sp3)–H bond functionalization.14 Previous studies showed that the 1 e−/1 H+ NHPI/phthalimido-N-oxyl (PINO) couple exhibits a redox potential of 1.44 V vs. SCE in CH3CN and is quasi-reversible.15 In addition, the rate of chlorine generation in situ could be regulated by electrochemical parameters.16 Inspired by these pioneering studies and our continuous efforts on free-radical C(sp3)–H bond functionalization,17 we began to explore an electrochemistry-driven remote C(sp3)–H bond halogenation catalyzed by NHPI.
We started to optimize the reaction conditions using isoamyl benzoate 1a as a model substrate (Table 1). To our delight, the desired product 1b was obtained in a yield of 65% when graphite felt (GF) was used as the anode and Pt sheet as the cathode with tetraethylammonium tetrafluoroborate (Et4NBF4) as the electrolyte under constant current in an undivided cell (entry 1). The yields of 1b decreased under application of higher or lower currents (entries 2 and 3). Next, NaCl was used instead of hydrochloric acid as the chlorine source; however, no product was observed (entry 4). Graphite felt as an electrode afforded the product in 49% yield (entry 5). The yield did not improve when Cl4-NHPI was used as the catalyst instead of NHPI (entry 6). When the amount of HCl was adjusted to 5 equiv., we obtained 1b in 79% yield (entry 7). Further examination revealed that the yield of 1b decreased upon increasing the equivalents of HCl (entry 8). Only 14% yield of 1b was obtained without NHPI (entry 9). Air bubbling or nitrogen atmosphere gave low yields of the desired product (entries 10 and 11). With graphite felt as the electrode, we found that the substrate concentration and graphite felt size have large influences on current efficiency (entry 12 vs. entry 13). As expected, no reaction occurred in the absence of an electric current (entry 14).
|
|
|||
|---|---|---|---|
| Entry | Catalyst/Cl source (eq.)/current(mA)/electrode/atmosphere/time (h) | RSMb (%) | Yield of 1bc (%) |
| a Reaction conditions: 0.5 mmol of substrate, 20 mol% of NHPI, 5.0 equiv. of HCl (concentrated hydrochloric acid, 12.0 mol L−1), 2.0 equiv. of Et4NBF4, MeCN (8 mL), graphite felt anode (1.0 cm × 1.0 cm × 0.5 cm), Pt plate cathode (1.0 cm × 1.0 cm × 0.2 mm), 10 mA, open air, room temperature, 24 h, 8.95 F mol−1. b RSM is short for the recovery of starting material. c Isolated yield. d ND = not detected. e MeCN (5 mL), 8.95 F mol−1, η = 8%. f 1.0 mmol of substrate, MeCN (5 mL), graphite felt anode (1.5 cm × 1.5 cm × 0.5 cm), 2.80 F mol−1, η = 26%. | |||
| 1 | NHPI (20 mol%)/HCl (10)/10/GF(+)|Pt(−)/open air/24 | <10 | 65 |
| 2 | NHPI (20 mol%)/HCl (10)/5/GF(+)|Pt(−)/open air/48 | <10 | 62 |
| 3 | NHPI (20 mol%)/HCl (10)/15/GF(+)|Pt(−)/open air/24 | <10 | 54 |
| 4 | NHPI (20 mol%)/NaCl (10)/10/GF(+)|Pt(−)/open air/48 | 100 | NDd |
| 5 | NHPI (20 mol%)/HCl (10)/10/GF(+)|GF(−)/open air/48 | 17 | 49 |
| 6 | Cl4-NHPI (20 mol%)/HCl (10)/10/GF(+)|Pt(−)/open air/24 | 15 | 63 |
| 7 | NHPI (20 mol%)/HCl (5)/10/GF(+)|Pt(−)/open air/24 | <10 | 79 |
| 8 | NHPI (20 mol%)/HCl (20)/10/GF(+)|Pt(−)/open air/24 | <10 | 39 |
| 9 | NHPI (0)/HCl (5)/10/GF(+)|Pt(−)/open air/24 | 75 | 14 |
| 10 | NHPI (0)/HCl (5)/10/GF(+)|Pt(−)/air bubbles/24 | 18 | 34 |
| 11 | NHPI (20 mol%)/HCl (5)/10/GF(+)|Pt(−)/N2/24 | 54 | 27 |
| 12e | NHPI (20 mol%)/HCl (5)/10/GF(+)|GF(−)/open air/24 | 14 | 70 |
| 13f | NHPI (20 mol%)/HCl (5)/10/GF(+)|GF(−)/open air/15 | <10 | 73 |
| 14 | NHPI (20 mol%)/HCl (5)/0/GF(+)|Pt(−)/open air/24 | 100 | NDd |
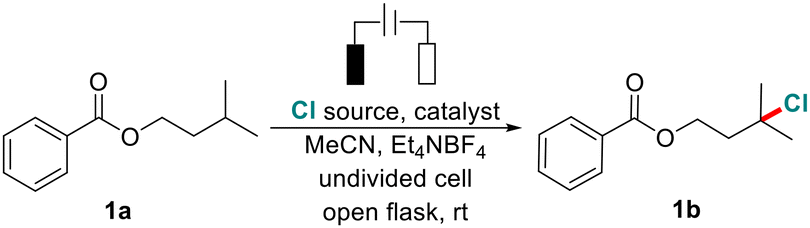
With the optimized conditions in hand, we then investigated the substrate scope as shown in Scheme 2. A variety of molecules that have multiple inert C(sp3)–H bonds were smoothly transformed into the desired alkyl chlorides in 46%–88% isolated yields (over 50 examples). Substrates containing aromatic (1a–15a), naphthenic (16a), heterocyclic aromatic (17a–20a), benzylic (21a), alkyl (22a) hydrocarbon carboxylates, benzenesulfonates (23a), and N-alkyl phthalimide (24a), all underwent chlorination of the 3° C(sp3)–H bonds in good yields and site selectivity. It should be noted that the chlorination proceeds simultaneously on the electron-rich aromatic ring (5a). Cyclic 3° C(sp3)–H bonds can also be smoothly chlorinated selectively (25a and 26a). Furthermore, chlorination preferentially occurs at more electron-rich and sterically less hindered sites when multiple 3° C(sp3)–H bonds are present in one molecule (27a–32a).
 | ||
Scheme 2 Substrate scope. Reaction conditions: 0.5 mmol substrate, 2.0 equiv. of Et4NBF4, 20 mol% of NHPI, 5.0 equiv. of HCl (concentrated hydrochloric acid, 12.0 mol L−1), 8 mL MeCN, graphite felt anode (1.0 cm × 1.0 cm × 0.5 cm), Pt plate cathode (1.0 cm × 1.0 cm × 0.2 mm), 10 mA, open air, room temperature, 24 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1.0 mmol substrate, 5 mL MeCN, graphite felt (1.5 cm × 1.5 cm × 0.5 cm) as the anode and cathode, 15 h. bTime = 36 h. c15 mA. dYields are based on 1H NMR analysis. eTime = 48 h. fMeCN/DCM (8 mL, 3/1) instead of MeCN. gN2. h5 mA, time = 12 h. 1.0 mmol substrate, 5 mL MeCN, graphite felt (1.5 cm × 1.5 cm × 0.5 cm) as the anode and cathode, 15 h. bTime = 36 h. c15 mA. dYields are based on 1H NMR analysis. eTime = 48 h. fMeCN/DCM (8 mL, 3/1) instead of MeCN. gN2. h5 mA, time = 12 h. | ||
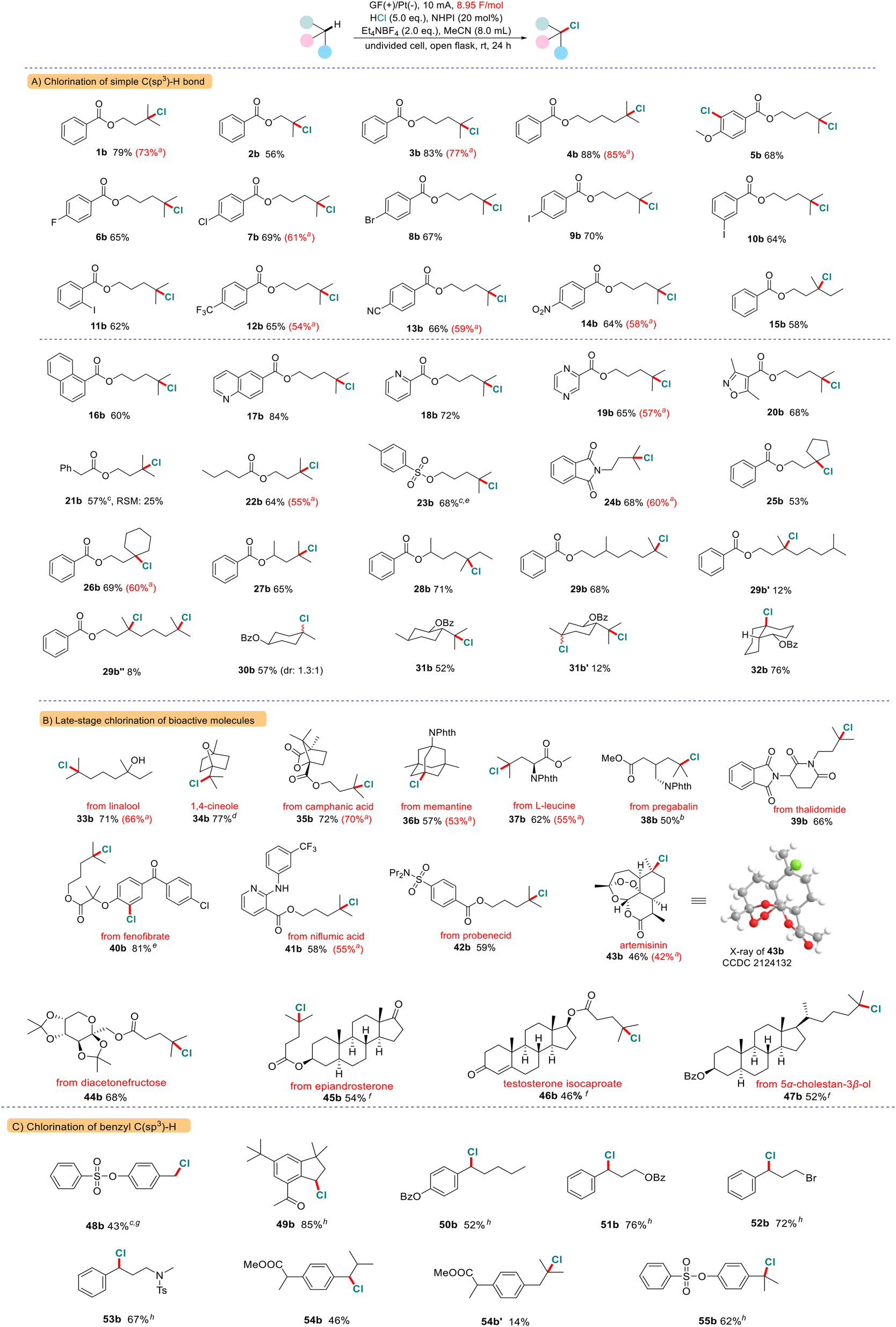
Next, we conducted the chlorination of various bioactive molecules that contain multiple C(sp3)–H bonds and diverse functional groups to evaluate its potential for late-stage functionalization of complex molecules (Scheme 2B). We are pleased to find that the tertiary C(sp3)–H bond of a series of natural perfume molecules and their derivatives such as linalool (33a), eucalyptol (34a) and camphanic acid (35a) were smoothly converted to the corresponding C(sp3)–Cl bonds in high yields. Furthermore, this chlorination is also amenable to a wide range of pharmaceutical ingredients (36a–42a). For example, an analogue of fenofibrate, a drug used to treat abnormal blood lipid levels, gave the bis-chlorinated product 40b in 81% yield. To our delight, even more complex molecules like artemisinin 43a, which contains a peroxide bridge and five 3° C(sp3)–H bonds, underwent the reaction smoothly to afford the chlorinated product 43b in 46% isolated yield (with 38% recovery of starting material). The chlorination occurred selectively at the C6 methine position as the major product, which was confirmed by X-ray analysis (CCDC No. 2124132†). Furthermore, we found that the carbohydrate and steroid derivatives were compatible with this system (44a–47a). For instance, testosterone isocaproate 46a is a class of anti-inflammatory and anti-allergic adrenocortical hormone drugs, which can be selectively chlorinated at the methine position of the side chain. The C(25)–H bond in the cholesterol derivative (47a) was smoothly converted to the corresponding C(25)–Cl bonds with high selectivity. Next, we examined an array of molecules containing benzyl C(sp3)–H bonds (Scheme 2C). We found that selective chlorination preferentially happened at the benzylic position (48a–55a). The 1°, 2° and 3° benzylic C(sp3)–H bonds can be converted into the expected C(sp3)–Cl bonds in moderate to high yields. In the case of ibuprofen methyl ester 54a, two competitively chlorinated products were obtained (54b and 54b′). The electrochemical chlorination mainly occurred at the more electron-rich benzylic site and was less at the tertiary C–H bond.
In addition, we replaced hydrochloric acid and Et4NBF4 with nBu4NBr to test whether bromination of the C(sp3)–H bond would occur under similar conditions (Scheme 3). We found that both tertiary and benzylic C(sp3)–H bonds can be selectively converted into the desired C(sp3)–Br bonds (56c–59c).
 | ||
| Scheme 3 Electrochemical bromination of the C(sp3)–H bond. Reaction conditions: 0.5 mmol substrate, 20 mol% of NHPI, 3.0 equiv. of nBu4NBr, 8 mL of MeCN, graphite felt anode (1.0 cm × 1.0 cm × 0.5 cm), Pt plate cathode (1.0 cm × 1.0 cm × 0.2 mm), 15 mA, open air, room temperature, 24 h. a5 mA, 12 h. | ||
In order to verify the potential applications of this method in synthetic organic chemistry, we carried out the following scaled-up experiments and transformations of the product (Scheme 4). As demonstrated in Scheme 4A, we treated 11.3 g of artemisinin under our protocol and obtained 43b in 49% yield (η = 36%), while also recovering the starting material and NHPI in 30% and 85% yields, respectively. Furthermore, a scaled-up experiment to 100 g was carried out (Scheme 4B). We successfully isolated 68% yield (η = 27%) of the expected chloride 1b and recovered 83% yield of NHPI. It is noteworthy that very cheap and easily recycled graphite felt electrodes are used in these large-scale experiments, which make this process highly appropriate for industrial production. As shown in Scheme 4C, we used chloride 1b as the representative product to exhibit its value in diverse transformations (see the ESI† for details). The chloride can be conveniently converted into azide, alcohol, alkene, fluoride, iodide and deuterated alkane (60d–65d).
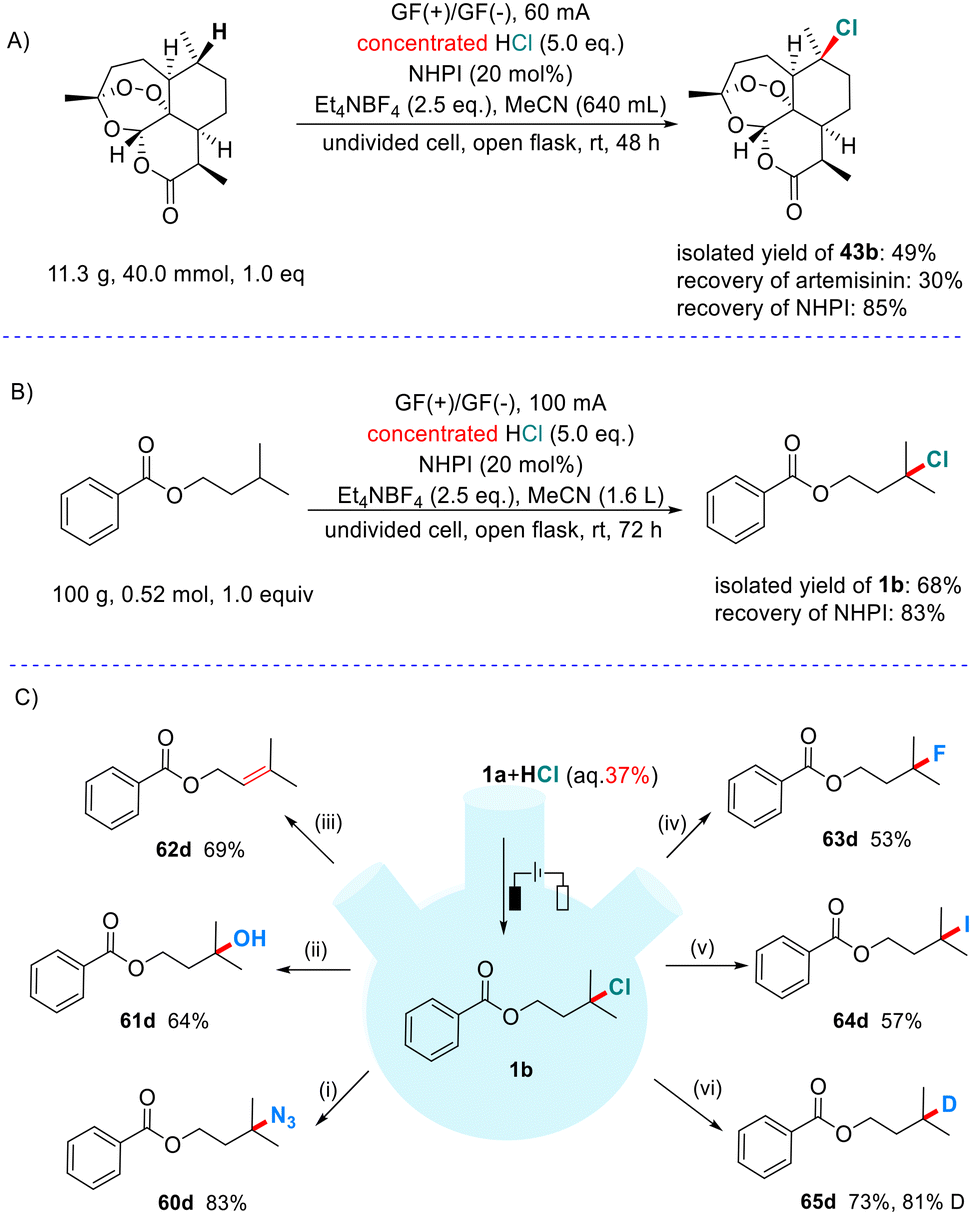 | ||
| Scheme 4 Scaled-up experiments and transformations of the product. Reaction conditions for 1b conversion: iNaN3, DMF, 55 °C. iipyridine, NaOH, acetone/H2O, 70 °C. iiipyridine, acetone/H2O, 70 °C. ivPyridine, NaOH, acetone/H2O, 70 °C, 24 h; then DAST, MeCN, −78 to 40 °C, 6 h. vNaI, acetone, 60 °C. vi20 mol% of N-ethyl-3,6-bis(dimethylamino)carbazole, 30 mol% of (nPrS)2, HCO2Na, DMSO/H2O, blue LEDs. | ||
Next, cyclic voltammetry studies of this system were carried out (Scheme 5A see the ESI† for details). We found that the oxidation potential of NHPI decreased from 1.94 V to 1.77 V under acidic conditions. Although this is higher than that under basic conditions (about 1.44 V with pyridine),15 it becomes easier to oxidize under either basic or acidic conditions rather than a neutral system.
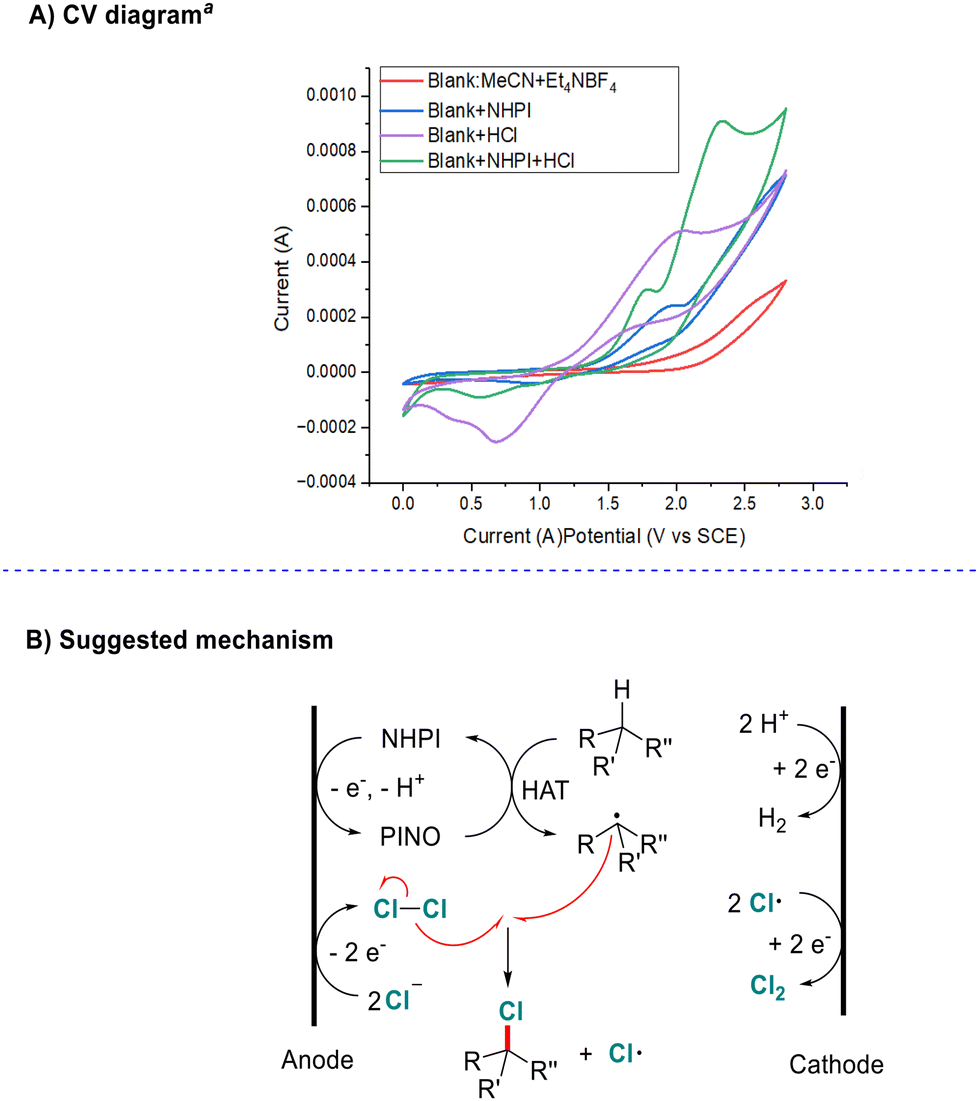 | ||
| Scheme 5 Mechanistic studies. (A) Cyclic voltammograms. aConditions: a glassy carbon working electrode, Ag/AgCl (3 M KCl) reference electrode, and a platinum wire counter electrode, Et4NBF4 (0.1 mmol in 10.0 mL of MeCN), 0.1 V s−1 scan rate with blank (red line); blank + 0.01 M NHPI (blue line); blank + 0.01 M concentrated HCl (purple line); blank + 0.01 M NHPI + 0.01 M concentrated HCl (green line). (B) Suggested reaction mechanism. | ||
On the basis of previous research studies and the results described above, we proposed a possible mechanism, as shown in Scheme 5B. Initially, the reaction begins with the anodic oxidation of chlorine ions to generate the chlorine radical and then Cl2. Meanwhile, single-electron oxidation of NHPI followed by deprotonation affords radical PINO. Then hydrogen-atom-transfer (HAT) from alkane to PINO generates an alkyl radical and regenerates NHPI. Finally, chlorine-atom-transfer from Cl2 to the alkyl radical affords the product and chlorine radical. In solution, HAT from alkane to a chlorine atom might be another pathway to generate alkyl radicals. The cathodic half-reaction should be hydrogen (H2) and Cl2 evolution via reduction of protons and chlorine atoms.
In conclusion, we discovered a practical and site-selective method for least hindered tertiary and benzylic C(sp3)–H bond halogenation. Through this protocol, a wide range of molecules, including complex natural products and pharmaceuticals, can easily be converted into the corresponding chlorides on a large scale. The present strategy features simple operation (an undivided cell), low cost (cheap NHPI as the catalyst, hydrochloric acid as the chlorine source, and graphite felt as the electrode), safe reactants (chemical oxidant and sensitive initiator free), good functional group tolerance and excellent site selectivity, scalability (scaled-up to 100 g), and mild conditions (open flask, room temperature). We hope it will open a door for the promotion of laboratory C(sp3)–H bond functionalization to industrial application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is supported by the National Natural Science Foundation of China (No. 21971116).References
- (a) B. Gál, C. Bucher and N. Z. Burns, Mar. Drugs, 2016, 14, 206–216 CrossRef PubMed; (b) W.-J. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396–4434 CrossRef PubMed; (c) W.-Y. Fang, L. Ravindar, K. P. Rakesh, H. M. Manukumar, C. S. Shantharam, N. S. Alharbi and H.-L. Qin, Eur. J. Med. Chem., 2019, 173, 117–153 CrossRef PubMed; (d) J. Zeng and J. Zhan, Isr. J. Chem., 2019, 59, 387–402 CrossRef; (e) D. Chiodi and Y. Ishihara, J. Med. Chem., 2023, 66, 5305–5331 CrossRef PubMed.
- (a) T. C. Atack, R. M. Lecker and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 9521–9523 CrossRef PubMed; (b) S. K. Bose, S. Brand, H. O. Omoregie, M. Haehnel, J. Maier, G. Bringmann and T. B. Marder, ACS Catal., 2016, 6, 8332–8335 CrossRef; (c) Z. Lin, Y. Lan and C. Wang, Org. Lett., 2019, 21, 8316–8322 CrossRef CAS PubMed; (d) H. A. Sakai, W. Liu, C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 11691–11697 CrossRef CAS PubMed; (e) M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47 CrossRef CAS; (f) S. Wang, M. Sun, H. Zhang, J. Zhang, Y. He and Z. Feng, CCS Chem., 2021, 3, 2164–2173 CrossRef CAS; (g) P. Li, C. Guo, S. Wang, D. Ma, T. Feng, Y. Wang and Y. Qiu, Nat. Commun., 2022, 13, 3774–3781 CrossRef CAS PubMed; (h) D. Wood and S. Lin, Angew. Chem., Int. Ed., 2023, 62, e202218858 CrossRef CAS PubMed; (i) M. Zhang, Z. Ye and W. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202306248 CrossRef PubMed.
- (a) U. Tilstam and H. Weinmann, Org. Process Res. Dev., 2002, 6, 384–393 CrossRef CAS; (b) W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847–12849 CrossRef CAS; (c) X. Yang, Y. Sun, T.-Y. Sun and Y. Rao, Chem. Commun., 2016, 52, 6423–6426 RSC; (d) S.-S. Lv, X.-P. Yan, C.-K. Li, S.-F. Zhou, A. Shoberu and J.-P. Zou, ChemistrySelect, 2020, 5, 5670–5674 CrossRef CAS; (e) A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412–484 CrossRef CAS; (f) M. A. Lopez, J. A. Buss and S. S. Stahl, Org. Lett., 2022, 24, 597–601 CrossRef CAS; (g) Q. Zhang, S. Liu, X. Tian, Y. Liu, S. Fan, B. Huang and A. Whiting, Green Chem., 2022, 24, 384–393 RSC; (h) J. Ye, D. Zhang, S. Salli, Y. Li, F. Han, Y. Mai, F. Rosei, Y. Li, Y. Yang, F. Besenbacher, H. Niemantsverdriet, E. Richards and R. Su, Angew. Chem., Int. Ed., 2023, 62, e202302994 CrossRef CAS.
- For selected recent reviews on C(sp3)–H bond functionalization, see: (a) X. Huang and J. T. Groves, ACS Catal., 2016, 6, 751–759 CrossRef CAS; (b) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS; (c) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS PubMed; (d) Y. Liu, T. You, H.-X. Wang, Z. Tang, C.-Y. Zhou and C.-M. Che, Chem. Soc. Rev., 2020, 49, 5310–5358 RSC; (e) T. G. Luu, Y. Jung and H.-K. Kim, Molecules, 2021, 26, 7380–7423 CrossRef CAS PubMed; (f) M. Galeotti, M. Salamone and M. Bietti, Chem. Soc. Rev., 2022, 51, 2171–2223 RSC; (g) D. L. Golden, S.-E. Suh and S. S. Stahl, Nat. Rev. Chem., 2022, 6, 405–427 CrossRef CAS; (h) Z. Zhang, P. Chen and G. Liu, Chem. Soc. Rev., 2022, 51, 1640–1658 RSC.
- (a) A. A. Fokin and P. R. Schreiner, Adv. Synth. Catal., 2003, 345, 1035–1052 CrossRef; (b) W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847–12849 CrossRef PubMed.
- (a) C. Djerassi, Chem. Rev., 1948, 43, 271–317 CrossRef; (b) L. Han, J.-B. Xia, L. You and C. Chen, Tetrahedron, 2017, 73, 3696–3701 CrossRef PubMed; (c) H. Tu, S. Zhu, F.-L. Qing and L. Chu, Tetrahedron Lett., 2018, 59, 173–179 CrossRef; (d) Z.-H. Li, B. Fiser, B.-L. Jiang, J.-W. Li, B.-H. Xu and S.-J. Zhang, Org. Biomol. Chem., 2019, 17, 3403–3408 RSC; (e) M. Xiang, C. Zhou, X.-L. Yang, B. Chen, C.-H. Tung and L.-Z. Wu, J. Org. Chem., 2020, 85, 9080–9087 CrossRef.
- For a recent review, see: (a) W. Guo, Q. Wang and J. Zhu, Chem. Soc. Rev., 2021, 50, 7359–7377 RSC. For selected recent examples, see: (b) Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894–1897 CrossRef PubMed; (c) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef PubMed; (d) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed; (e) T. Liu, M. C. Myers and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 306–309 CrossRef PubMed; (f) M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2018, 57, 296–299 CrossRef; (g) H. Guan, S. Sun, Y. Mao, L. Chen, R. Lu, J. Huang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 11413–11417 CrossRef; (h) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940–12944 CrossRef; (i) R.-Z. Liu, J. Li, J. Sun, X.-G. Liu, S. Qu, P. Li and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4428–4433 CrossRef PubMed; (j) M. A. Short, M. F. Shehata, M. A. Sanders and J. L. Roizen, Chem. Sci., 2020, 11, 217–223 RSC; (k) Y. Zhu, J. Shi and W. Yu, Org. Lett., 2020, 22, 8899–8903 CrossRef PubMed; (l) A. N. Herron, C.-P. Hsu and J.-Q. Yu, Org. Lett., 2022, 24, 3652–3656 CrossRef.
- (a) J. Ozawa and M. Kanai, Org. Lett., 2017, 19, 1430–1433 CrossRef PubMed; (b) G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Angew. Chem., Int. Ed., 2018, 57, 1251–1255 CrossRef; (c) S. Duewel, L. Schmermund, T. Faber, K. Harms, V. Srinivasan, E. Meggers and S. Hoebenreich, ACS Catal., 2020, 10, 1272–1277 CrossRef; (d) J. Jin, Y. Zhao, S. H. Kyne, K. Farshadfar, A. Ariafard and P. W. H. Chan, Nat. Commun., 2021, 12, 4065–4076 CrossRef PubMed; (e) Y. He, C. Tian, G. An and G. Li, Chin. Chem. Lett., 2024, 35, 108546 CrossRef; (f) Q. He, Z. Cao, Y. Zhang, G. Chen and Y. Wang, Adv. Synth. Catal., 2023, 365, 2711–2717 CrossRef; (g) T. Nanjo, A. Matsumoto, T. Oshita and Y. Takemoto, J. Am. Chem. Soc., 2023, 145, 19067–19075 CrossRef.
- (a) V. A. Schmidt, R. K. Quinn, A. T. Brusoe and E. J. Alexanian, J. Am. Chem. Soc., 2014, 136, 14389–14392 CrossRef PubMed; (b) R. K. Quinn, Z. A. Könst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 696–702 CrossRef; (c) A. M. Carestia, D. Ravelli and E. J. Alexanian, Chem. Sci., 2018, 9, 5360–5365 RSC.
- Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679–2683 RSC.
- A. Fawcett, M. J. Keller, Z. Herrera and J. F. Hartwig, Angew. Chem., Int. Ed., 2021, 60, 8276–8828 CrossRef.
- A. J. McMillan, M. Sienkowska, P. D. Lorenzo, G. K. Gransbury, N. F. Chilton, M. Salamone, A. Ruffoni, M. Bietti and D. Leonori, Angew. Chem., Int. Ed., 2021, 60, 7132–7139 CrossRef.
- For selected very recent reviews on late-stage C–H functionalization, see: (a) J. Börgel and T. Ritter, Chem, 2020, 6, 1877–1887 CrossRef; (b) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS; (c) P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS.
- (a) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef CAS PubMed; (b) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS; (c) M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22–25 CrossRef CAS PubMed; (d) T. Kato and K. Maruoka, Angew. Chem., Int. Ed., 2020, 59, 14261–14264 CrossRef CAS; (e) C. Yang, L. A. Farmer, D. A. Pratt, S. Maldonado and C. R. J. Stephenson, J. Am. Chem. Soc., 2021, 143, 10324–10332 CrossRef CAS PubMed; (f) M. A. Hoque, J. Twilton, J. Zhu, M. D. Graaf, K. C. Harper, E. Tuca, G. A. Dilabio and S. S. Stahl, J. Am. Chem. Soc., 2022, 144, 15295–15302 CrossRef CAS; (g) S. Zhang and M. Findlater, ACS Catal., 2023, 13, 8731–8751 CrossRef CAS PubMed; (h) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS; (i) I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed; (j) J. Ke, W. Liu, X. Zhu, X. Tan and C. He, Angew. Chem., Int. Ed., 2021, 60, 8744–8749 CrossRef CAS; (k) C. Jiang, Y. Liao, H. Li, S. Zhang, P. Liu and P. Sun, Adv. Synth. Catal., 2023, 365, 1205–1210 CrossRef CAS; (l) X. Chen, Y.-G. Huang, W.-Q. Zhong and J.-M. Huang, Org. Lett., 2023, 25, 4562–4566 CrossRef CAS PubMed.
- M. Masui, T. Ueshima and S. Ozaki, N-Hydroxyphthalimide, as an Effective Mediator for the Oxidation of Alcohols by Electrolysis, J. Chem. Soc., Chem. Commun., 1983, 479–480 RSC.
- (a) B. D. W. Allen, M. D. Hareram, A. C. Seastram, T. McBride, T. Wirth, D. L. Browne and L. C. Morrill, Org. Lett., 2019, 21, 9241–9246 CrossRef CAS; (b) F. Liu, N. Wu and X. Cheng, Org. Lett., 2021, 23, 3015–3020 CrossRef CAS.
- (a) Z.-Q. Liu, L. Sun, J.-G. Wang, J. Han, Y.-K. Zhao and B. Zhou, Org. Lett., 2009, 11, 1437–1439 CrossRef CAS PubMed; (b) Z. Cui, X. Shang, X.-F. Shao and Z.-Q. Liu, Chem. Sci., 2012, 3, 2853–2858 RSC; (c) Y. Tian and Z.-Q. Liu, Green Chem., 2017, 19, 5230–5235 RSC; (d) Y. Tian, C. Sun, R. Tan and Z.-Q. Liu, Green Chem., 2018, 20, 588–592 RSC; (e) L. Yang, Z. Qiu, J. Wu, J. Zhao, T. Shen, X. Huang and Z.-Q. Liu, Org. Lett., 2021, 23, 3207–3210 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2124132. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc03849a |
| This journal is © The Royal Society of Chemistry 2024 |