
Direct air-induced arylphosphinoyl radicals for the synthesis of benzo[b]phosphole oxides†
Mingqing
Huang
,
Haiyang
Huang
 *,
Mengyao
You
,
Xinxin
Zhang
,
Longgen
Sun
,
Chao
Chen
,
Zhichao
Mei
,
Ruchun
Yang
and
Qiang
Xiao
*
*,
Mengyao
You
,
Xinxin
Zhang
,
Longgen
Sun
,
Chao
Chen
,
Zhichao
Mei
,
Ruchun
Yang
and
Qiang
Xiao
*
Institute of Organic Chemistry, Jiangxi Science & Technology Normal University, Key Laboratory of Organic Chemistry, Jiangxi Province, Nanchang 330013, China. E-mail: huanghaiyang1209@163.com; xiaoqiang@tsinghua.org.cn
First published on 4th December 2023
Abstract
Benzo[b]phosphole oxides are valuable and significant organic functional molecules. Therefore, extensive efforts have been dedicated to the development of an environmentally friendly and convenient synthetic strategy for benzo[b]phosphole oxides. However, several critical issues still persist in the currently available protocols. In this study, we present a direct air-oxidized strategy enabling the transformation of arylphosphine oxides into phosphinoyl radicals, which were further utilized in the synthesis of benzo[b]phosphole oxides by combining with various alkynes. In addition, the results of DFT calculations show that phosphinoyl radical formation could involve an O2-mediated O–H bond homolysis instead of the commonly recognized P–H bond homolysis mechanism.
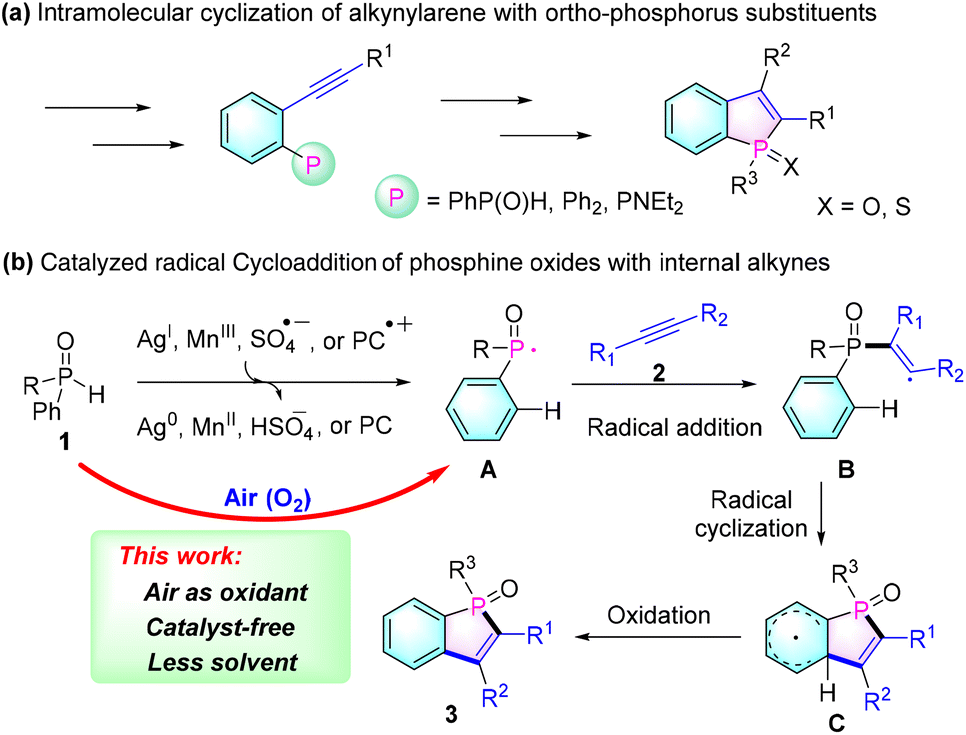
Due to their unique photoelectronic and physical properties, π-conjugated phospholes have attracted significant attention from synthetic and materials chemists.1 In particular, benzo[b]phospholes have emerged as promising scaffolds for organic functional molecules, attracting increasing interest in the fields of organic functional materials, such as organic light-emitting diodes (OLEDs),2 photovoltaic cells3 and bioimaging probes.4 However, synthetic approaches for these scaffolds are relatively deficient. The common methods for preparing these types of molecules involve the cyclization of ortho-alkynyl phosphorus substrates by treatment with strong bases or expensive transition metal catalysts (Scheme 1a).5 Yet, this synthetic strategy requires complicated and multistep reactions for preparing their precursors.
 | ||
| Scheme 1 Synthetic methods of benzo[b]phospholes. | ||
Recently, an efficient intermolecular radical cycloaddition of secondary phosphine oxides 1 with internal alkynes 2 has been developed to enable a convenient synthesis of benzo[b]phosphole oxides (Scheme 1b). These methods involve the use of stoichiometric amounts of Ag2O (2 equiv.),6 AgOAc (2 equiv.),7 Mn(III),8 Cu(II) catalyzed TBHP (2 equiv.),9 or a persulphate (5 equiv.),10 which oxidize the diarylphosphine oxide 1 to generate the necessary phosphinoyl radical A. Then, the resulting radical intermediate A combines with alkynes 2 to form the phosphole ring through an alkenyl radical intermediate B and the subsequent cyclohexadienyl intermediate C. Although this approach has proven useful in synthesizing numerous benzo[b]phosphole oxides 3, it still requires the use of stoichiometric amounts of expensive metals or potentially explosive peroxides as oxidants.
Very recently, in order to avoid the use of expensive transition metal catalysts or dangerous reagents, Lakhdar's group demonstrated a visible light-photocatalyzed synthetic methodology.11 In this approach, the phosphinoyl radical A is generated through the oxidation of diarylphosphine oxide 1 using the oxidized photocatalyst (PC˙+). While this represents a significant advancement in terms of safety and environmental friendliness, an excess of organic oxidant (N-ethoxy-2-methylpyridinium tetrafluoroborate) is still required for the formation of phosphinoyl radicals. Therefore, there remains a strong demand for the development of simpler, more cost-effective, environmentally friendly, and safe methods for the preparation of benzo[b]phosphole oxides.
On the basis of these works on the radical cycloaddition of phosphine oxides with alkynes, we hypothesize that the key step of this protocol is the formation of phosphinoyl radical intermediate A, which would efficiently undergo addition with an alkyne and then spontaneously undergo the subsequent steps to afford benzo[b]phosphole oxides 3. On the other hand, the P–H bonds of phosphine oxides are known to have relatively low bond dissociation energies (317 kJ mol−1 for diphenylphosphine oxide);12 therefore, we anticipated developing a cheap, green, and mild oxidant system (for example, oxygen in air) for synthesizing benzo[b]phosphole oxides without requiring metal catalysts or potentially explosive reagents.13
To keep the process as green as possible, we examined the reaction of diphenylphosphine oxide 1a with diphenylacetylene 2a only under an air atmosphere and heating conditions without any additional oxidants, bases, or photocatalysts (Table 1). In line with our working hypothesis, we were pleased to find that a handful of O2 existing in the air can oxidize 1a to the essential phosphinoyl radical species, followed by its cycloaddition with 2a to afford the desired product 3a. From the results of solvent screening (including DCE, DMF, DMSO, Py, toluene and 1,4-dioxane), pyridine (Py) and dimethylformamide (DMF) were confirmed to be the optimal solvents for the task (entries 1–6). Then, 2.8 equivalents of diphenylphosphine oxide were determined to be necessary to deplete alkynes (entries 6–11). Subsequently, we checked the effect of the amount of DMF solvent and the results showed that an increase in solvent usage caused a slower reaction rate (entries 12 and 13) and a highly concentrated system led to a significant decrease in the reaction yield (entries 14 and 15). When the solvent was reduced to 0.1 mL, an alkenyl by-product 3a′ was observed, which was possibly formed due to the alkenyl radical intermediate capture of hydrogen from 1a (see the ESI†).14 In addition, gradually decreasing the temperature to 60 °C resulted in a required longer reaction time and a relatively low yield (entries 16–18). Also, we conducted the reaction in an oxygen-poor environment and found that the reaction still worked but the yield was significantly reduced (entry 19).
|
|
||||
|---|---|---|---|---|
| Entry | 1a (equiv.) | Solvent (mL) | Temp. (°C)/time (h) | Yield (%) |
| a Reaction conditions: 1a (2–3 equiv.), 2a (0.5 mmol), in solvent (0.1–0.5 mL) at temperatures ranging from 60 to 90 °C in air. b Under an argon atmosphere. | ||||
| 1 | 3.0 | DCE (0.5) | 90/24 | 51 |
| 2 | 3.0 | 1,4-Dioxane (0.5) | 90/24 | 13 |
| 3 | 3.0 | DMSO (0.5) | 90/24 | Trace |
| 4 | 3.0 | Toluene (0.5) | 90/24 | 72 |
| 5 | 3.0 | Py (0.5) | 90/24 | 79 |
| 6 | 3.0 | DMF (0.5) | 90/24 | 81 |
| 7 | 2.8 | DMF (0.5) | 90/24 | 82 |
| 8 | 2.6 | DMF (0.5) | 90/24 | 80 |
| 9 | 2.4 | DMF (0.5) | 90/24 | 65 |
| 10 | 2.0 | DMF (0.5) | 90/24 | 57 |
| 11 | 1.5 | DMF (0.5) | 90/24 | 42 |
| 12 | 2.8 | DMF (1.0) | 90/24 | 75 |
| 13 | 2.8 | DMF (0.8) | 90/24 | 79 |
| 14 | 2.8 | DMF (0.2) | 90/24 | 64 |
| 15 | 2.8 | DMF (0.1) | 90/24 | 57 |
| 16 | 2.8 | DMF (0.5) | 80/24 | 72 |
| 17 | 2.8 | DMF (0.5) | 70/24 | 65 |
| 18 | 2.8 | DMF (0.5) | 60/48 | 66 |
| 19b | 2.8 | DMF (0.5) | 90/48 | 21 |
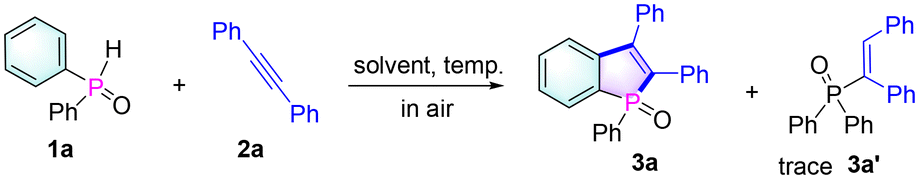
With the optimized conditions in hand, a large variety of benzophosphole oxides were synthesized from different alkynes and phosphine oxides as shown in Table 2. At first, diphenylphosphine oxide 1a reacts smoothly with various diphenylacetylene derivatives 2 under standard conditions, affording benzophosphole oxides (3a–3j) in good to excellent yields (48–85%). Next, the reactions between 1a and dialkylalkynes were also investigated and the corresponding products 3 (3k–3n) were obtained in medium yields (57–67%). Remarkably, with the tested unsymmetrical alkynes, complete control of selectivity was achieved (3p–3r). This regioselectivity can be attributed to the ability of aryl groups to stabilize the formed alkenyl radical (B) or the steric hindrance effect of the substituent (3p). In addition, good to excellent yields were also obtained when different phosphine oxides were combined with diphenylacetylene 2a to give benzo[b]phosphole oxides 3s–3aa. Notably, some phosphole oxides with alkyl substituents on the phosphorus center were also achieved in good yields.
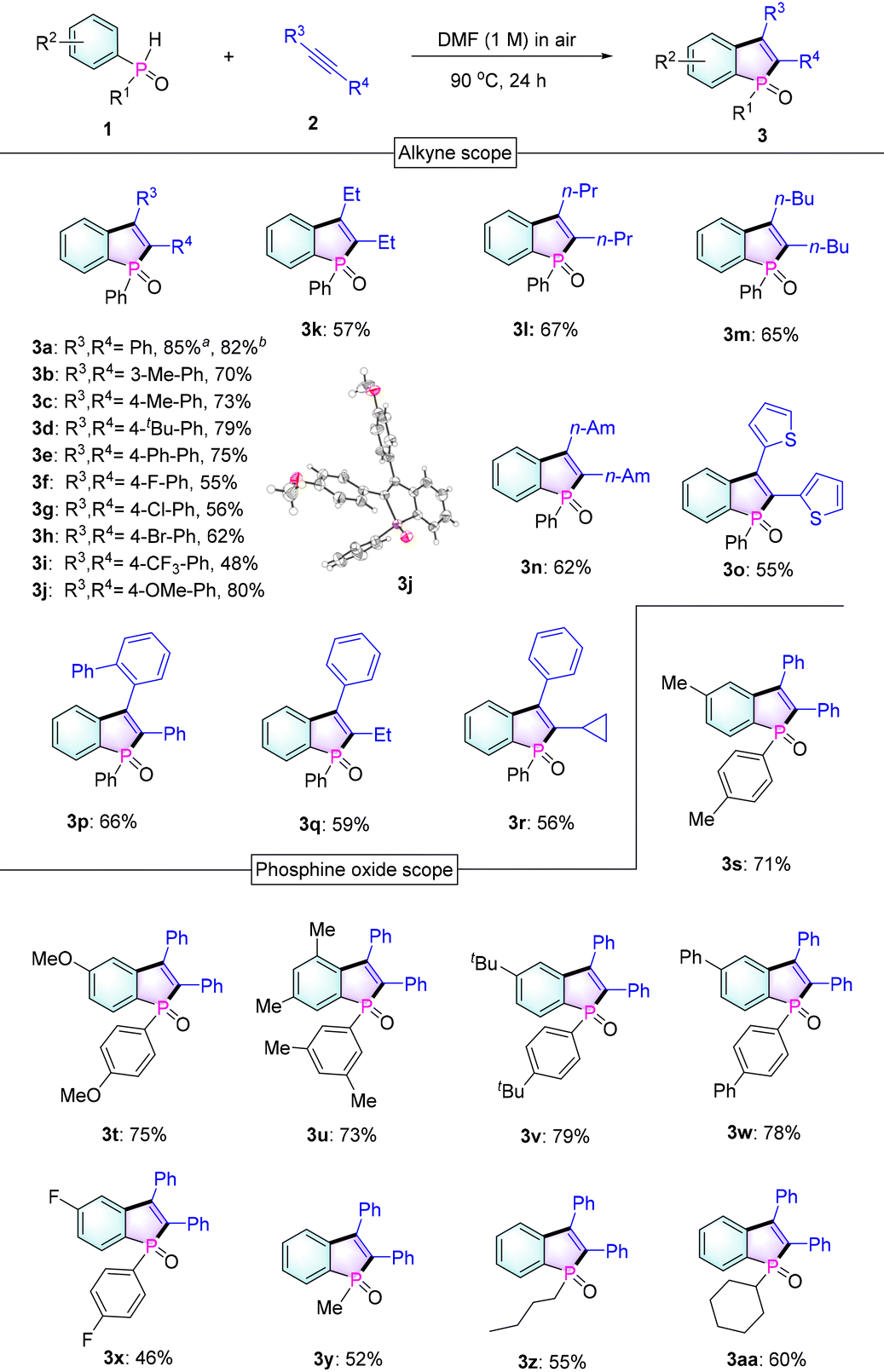
Next, to evaluate the scalability of the reaction, a gram-scale reaction was carried out employing 1a and 2a (2 g scale) as reactants under standard conditions. To our delight, the reaction could be executed without any compromise in the yield (82%, Table 2), thus proving the effectiveness of our protocol. In addition, as expected, 1a could react with diynes 2ab and 2ac to give bis-benzophosphole oxides 3ab and 3ac (Scheme 2), which would be of interest for applications in thin-film photovoltaics and organic light-emitting diodes.3a,b,5a
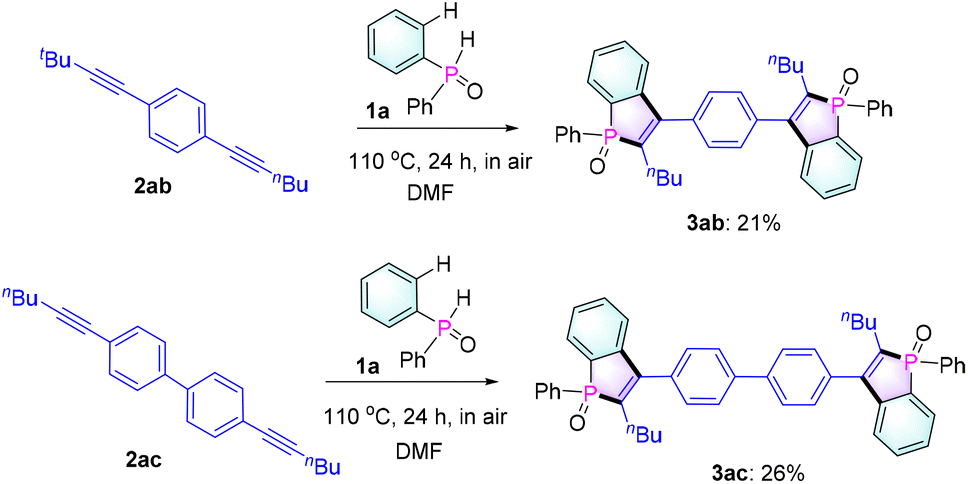 | ||
| Scheme 2 Application studies on diynes. | ||
To elucidate the mechanistic aspects of the current reaction, we investigated the radical scavenging effect by adding either 2,2,6,6- tetramethylpiperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT) to the reaction system. Remarkably, when two equivalents of radical scavengers were added, this reaction was significantly suppressed (Scheme 3). ESI-HRMS analysis confirmed the formation of a TEMPO adduct. These findings strongly suggest that the reaction likely proceeds through a free radical pathway. Moreover, we also observed that the stirring rate had a considerable effect on the reaction rate. In fact, no reaction occurred if the stirring stopped, which further supported that the O2 in the air was the trigger for the radical reaction.
 | ||
| Scheme 3 Free radical capture with BHT and TEMPO. | ||
To investigate the details of the reaction mechanism and clarify the experimental observations, density functional theory (DFT) calculations were performed with 1a and 2a as model substrates for the intermolecular phosphacyclization between arylphosphine oxides and alkynes (Fig. 1).15 Typically, the generation of a phosphinoyl radical, which is a critical rate-determining step for radical cyclization, benefits from the low bond energy of the P–H bond. This leads to an easy homolytic cleavage upon exposure to activated oxidizing agents. However, the calculation results indicated that the P–H bond homolysis of diphenylphosphine oxide 1a has a much higher barrier of 35.3 kcal mol−1 than the O–H bond homolysis of its λ3-isomer (24.7 kcal mol−1) using O2 (triplet state) as an oxidizing agent, implying that in previous works, the formation of the phosphinoyl radical could also involve O–H bond cleavage instead of the usually recognised P–H bond cleavage.6–10 In addition, according to the electron spin density of int-1, 48.05% of the radical electrons are located at the P-center and 29.78% at the O-centre. Thus a more radical feature on the P atom is responsible for the subsequent intermolecular radical addition with alkynes to afford int-2 through the transition state ts-2 with a low barrier of 8.5 kcal mol−1. Then, the expected radical cyclization and oxidative dehydrogenation were successively conducted to obtain the desired benzo[b]phosphole oxides.
 | ||
| Fig. 1 Computed energy profile (kcal mol−1) for the reaction at the M06-2X-D3/def-TZVP/SMD(DMF)//B3LYP-D3/6-31G(d,p)/SMD(DMF) level. | ||
Conclusions
In conclusion, we have successfully developed a natural air(O2)-induced radical addition and cyclization of arylphosphine oxides with various unactivated alkynes. This methodology allows for the synthesis of benzo[b]phosphole oxides without the need for additional oxidants or metal catalysts in a small amount of solvent under simple air conditions, making it a safer and more environmentally friendly strategy (the green metrics analysis is summarized in Table S1†). Furthermore, on the basis of experimental observations and theoretical calculations, we have proposed a plausible reaction mechanism for this transformation. The mechanism involves the formation of a phosphinoyl radical followed by radical addition and cyclization with alkynes. Notably, our results indicate that phosphinoyl radical formation can be achieved through the O–H bond homolysis of isomerized λ3-arylphosphine rather than the previously recognized P–H bond cleavage of λ5-arylphosphine oxide.Author contributions
H. Huang and Q. Xiao directed the project and co-wrote the manuscript; H. Huang conceived the work, designed the experiments, conducted the DFT calculations and analysed the data; M. Huang, M. You, X. Zhang, L. Sun, C. Chen, Z. Mei, and R. Yang performed the experiments and collected the experimental data; all authors have given approval regarding the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22066011), the Jiangxi Provincial Natural Science Foundation (No. 20224BAB203006), the Education Department of Jiangxi Province (No. GJJ2201315 and GJJ2201340), and the Jiangxi Science & Technology Normal University (No. 2018BSQD025 and 2022KFJJ003).References
- (a) J. Guo, J. Li, T. Wu, X. Peng, S. Wang, Z. Zhao and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 7837–7844 CrossRef CAS PubMed; (b) N. Yoshikai, M. Santra and B. Wu, Organometallics, 2017, 36, 2637–2645 CrossRef CAS; (c) F. Bu, E. Wang, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Tang, Chem. – Eur. J., 2015, 21, 4440–4449 CrossRef CAS PubMed; (d) S. Furukawa, S. Haga, J. Kobayashi and T. Kawashima, Org. Lett., 2014, 16, 3228–3231 CrossRef CAS PubMed; (e) Z. Zhuang, F. Bu, W. Luo, H. Peng, S. Chen, R. Hu and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 1836–1842 RSC; (f) Y. Matano, A. Saito, T. Fukushima, Y. Tokudome, F. Suzuki, D. Sakamaki and H. Imahori, Angew. Chem., 2011, 123, 8166–8170 CrossRef; (g) T. Delouche, A. Mocanu, T. Roisnel, R. Szucs, E. Jacques, Z. Benko and M. Hissler, Org. Lett., 2019, 21, 802–806 CrossRef CAS PubMed.
- (a) J. Lee, N. Aizawa and T. Yasuda, J. Mater. Chem. C, 2018, 6, 3578–3583 RSC; (b) C. H. Lin, C. W. Hsu, J. L. Liao, Y. M. Cheng, Y. Chi, T. Y. Lin and C. L. Ho, J. Mater. Chem., 2012, 22, 10684 RSC; (c) C. Fave, T. Y. Cho, M. Hissler, C. W. Chen, T. Y. Luh, C. C. Wu and R. Réau, J. Am. Chem. Soc., 2003, 125, 9254–9255 CrossRef CAS PubMed; (d) H. C. Su, O. Fadhel, C. J. Yang, T. Y. Cho, C. Fave, M. Hissler and R. Réau, J. Am. Chem. Soc., 2006, 128, 983–995 CrossRef CAS PubMed; (e) R. F. Chen, R. Zhu, Q. L. Fan and W. Huang, Org. Lett., 2008, 10, 2913–2916 CrossRef CAS PubMed; (f) O. Fadhel, M. Gras, N. Lemaitre, V. Deborde, M. Hissler, B. Geffroy and R. Réau, Adv. Mater., 2009, 21, 1261–1265 CrossRef CAS; (g) D. Joly, D. Tondelier, V. Deborde, W. Delaunay, A. Thomas, K. Bhanuprakash and R. Réau, Adv. Funct. Mater., 2012, 22, 567–576 CrossRef CAS; (h) H. Chen, W. Delaunay, L. Yu, D. Joly, Z. Wang, J. Li and R. Réau, Angew. Chem., Int. Ed., 2012, 51, 214–217 CrossRef CAS PubMed; (i) F. Riobé, R. Szűcs, P. A. Bouit, D. Tondelier, B. Geffroy, F. Aparicio and M. Hissler, Chem. – Eur. J., 2015, 21, 6547–6556 CrossRef PubMed.
- (a) H. Tsuji, K. Sato, Y. Sato and E. Nakamura, Chem. – Asian J., 2010, 5, 1294–1297 CrossRef CAS PubMed; (b) H. Tsuji, K. Sato, Y. Sato and E. Nakamura, J. Mater. Chem., 2009, 19, 3364 RSC; (c) Y. Matano, A. Saito, Y. Suzuki, T. Miyajima, S. Akiyama, S. Otsubo and H. Imahori, Chem. – Asian J., 2012, 7, 2305–2312 CrossRef CAS PubMed; (d) K. H. Park, Y. J. Kim, G. B. Lee, T. K. An, C. E. Park, S. K. Kwon and Y. H. Kim, Adv. Funct. Mater., 2015, 25, 3991–3997 CrossRef CAS; (e) Y. H. Cheng, H. L. Wong, E. Y. H. Hong, S. L. Lai, M. Y. Chan and V. W. W. Yam, ACS Appl. Energy Mater., 2020, 3, 3059–3070 CrossRef CAS.
- (a) C. Wang, M. Taki, Y. Sato, A. Fukazawa, T. Higashiyama and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 10374–10381 CrossRef CAS PubMed; (b) C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 15213–15217 CrossRef CAS PubMed; (c) E. Yamaguchi, C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539–4543 CrossRef CAS PubMed; (d) C. Wang, M. Taki, K. Kajiwara, J. Wang and S. Yamaguchi, ACS Mater. Lett., 2020, 2, 705–711 CrossRef CAS; (e) C. Reus, M. Stolar, J. Vanderkley, J. Nebauer and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 11710–11717 CrossRef CAS PubMed.
- (a) H. Tsuji, K. Sato, L. Ilies, Y. Itoh, Y. Sato and E. Nakamura, Org. Lett., 2008, 10, 2263 CrossRef CAS PubMed; (b) T. Sanji, K. Shiraishi, T. Kashiwabara and M. Tanaka, Org. Lett., 2008, 10, 2689 CrossRef CAS PubMed; (c) A. Fukazawa, Y. Ichihashi, Y. Kosaka and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1729 CrossRef CAS PubMed; (d) A. Fukazawa, M. Hara, T. Okamoto, E.-C. Son, C. Xu, K. Tamao and S. Yamaguchi, Org. Lett., 2008, 10, 913 CrossRef CAS PubMed; (e) A. Fukazawa, H. Yamada and S. Yamaguchi, Angew. Chem., Int. Ed., 2008, 47, 5582 CrossRef CAS PubMed; (f) Y. Zhou, Z. Gan, B. Su, J. Li, Z. Duan and F. Mathey, Org. Lett., 2015, 17, 5722 CrossRef CAS PubMed; (g) B. Wu and N. Yoshikai, Org. Biomol. Chem., 2016, 14, 5402–5416 RSC.
- Y. R. Chen and W. L. Duan, J. Am. Chem. Soc., 2013, 135, 16754–16757 CrossRef CAS PubMed.
- (a) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 12975–12979 CrossRef CAS PubMed; (b) W. Ma and L. Ackermann, Synthesis, 2014, 2297–2304 CAS.
- (a) H. C. Fisher, O. Berger, F. Gelat and J. L. Montchamp, Adv. Synth. Catal., 2014, 356, 1199–1204 CrossRef CAS; (b) O. Berger and J. L. Montchamp, J. Org. Chem., 2019, 84, 9239–9256 CrossRef CAS PubMed.
- P. Zhang, Y. Gao, L. Zhang, Z. Li, Y. Liu, G. Tang and Y. Zhao, Adv. Synth. Catal., 2016, 358, 138–142 CrossRef CAS.
- D. Ma, W. Chen, G. Hu, Y. Zhang, Y. Gao, Y. Yin and Y. Zhao, Green Chem., 2016, 18, 3522–3526 RSC.
- V. Quint, F. Morlet-Savary, J. F. Lohier, J. Lalevee, A. C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436–7441 CrossRef CAS PubMed.
- X.-Q. Pan, J.-P. Zou, W.-B. Yi and W. Zhang, Tetrahedron, 2015, 71, 7481–7529 CrossRef CAS and references therein.
- (a) H. Guo, A. Yoshimura, T. Chen, Y. Saga and L. B. Han, Green Chem., 2017, 19, 1502–1506 RSC; (b) T. Hirai and L. B. Han, Org. Lett., 2007, 9, 53–55 CrossRef CAS PubMed.
- H. Wang, Y. Li, Z. Tang, S. Wang, H. Zhang, H. Cong and A. Lei, ACS Catal., 2018, 8, 10599–10605 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2300782 and 2300783. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc03947a |
| This journal is © The Royal Society of Chemistry 2024 |