
Sustainable electrocatalytic oxidation of N-alkylamides to acyclic imides using H2O†
Jing
Qi
a,
Xiyan
Wang
a,
Gan
Wang
a,
Srinivas Reddy
Dubbaka
b,
Patrick
O'Neill
c,
Hwee Ting
Ang
 *a and
Jie
Wu
*a
*a and
Jie
Wu
*a
aDepartment of Chemistry, National University of Singapore, 117545, Singapore. E-mail: hweeting@nus.edu.sg; chmjie@nus.edu.sg
bPfizer Manufacturing Pte Ltd, Manufacturing Technology Development Centre (MTDC), 138623, Singapore
cPfizer Ireland Pharmaceuticals, Process Development Centre, Ringaskiddy (PDC), Co-Cork, 637578, Ireland
First published on 13th December 2023
Abstract
The synthesis of imides holds significant importance in various scientific disciplines and industrial applications due to their extensive use in biomolecules, inorganic compounds, and organic compounds. Traditional methods for imide synthesis often rely on the use of toxic or expensive oxidizing agents, limiting their sustainability and practicality. Herein, we present a green and environmentally friendly approach for the synthesis of imides from N-alkylamides using electrocatalytic oxidation with H2O as the green oxygen source. This sustainable and atom-efficient approach outperforms traditional methods by eliminating the need for toxic or expensive oxidants while simultaneously achieving high yields under mild reaction conditions. The protocol exhibits broad substrate compatibility, enabling the transformation of a wide range of N-alkyl (methyl, ethyl and cyclopropyl) amides into imides. The resulting imide products can serve as valuable building blocks for the synthesis of biologically relevant 1,2,4-triazole compounds. Furthermore, the practicality of this method is demonstrated through a gram-scale reaction, affirming its efficiency and potential for industrial applications. Our work presents a sustainable and versatile strategy for imide synthesis, aligning with green chemistry principles and sustainable manufacturing practices.
Introduction
Given its abundance on Earth, nitrogen is an essential element in a myriad of biomolecules, inorganic compounds, and organic compounds, offering diverse properties and functions. Imides, a class of nitrogen-containing organic compounds, have gained significant attention due to their extensive applications across various disciplines, including medicinal chemistry, polymer science, and synthetic chemistry.1–6 Moreover, imides are commonly found in natural products and bioactive compounds, further underscoring their significance. Notable examples of bioactive imide-containing compounds (Scheme 1A) include formothion (an effective organophosphate insecticide),7 aniracetam (a cognitive-enhancing brain supplement),8 tetraacetylethylene-diamine (TAED, a bleach activator with an estimated annual consumption of 75 kt in detergents)9 and dermagen (a skincare product that stimulates the body's circulatory system and revitalizes the skin).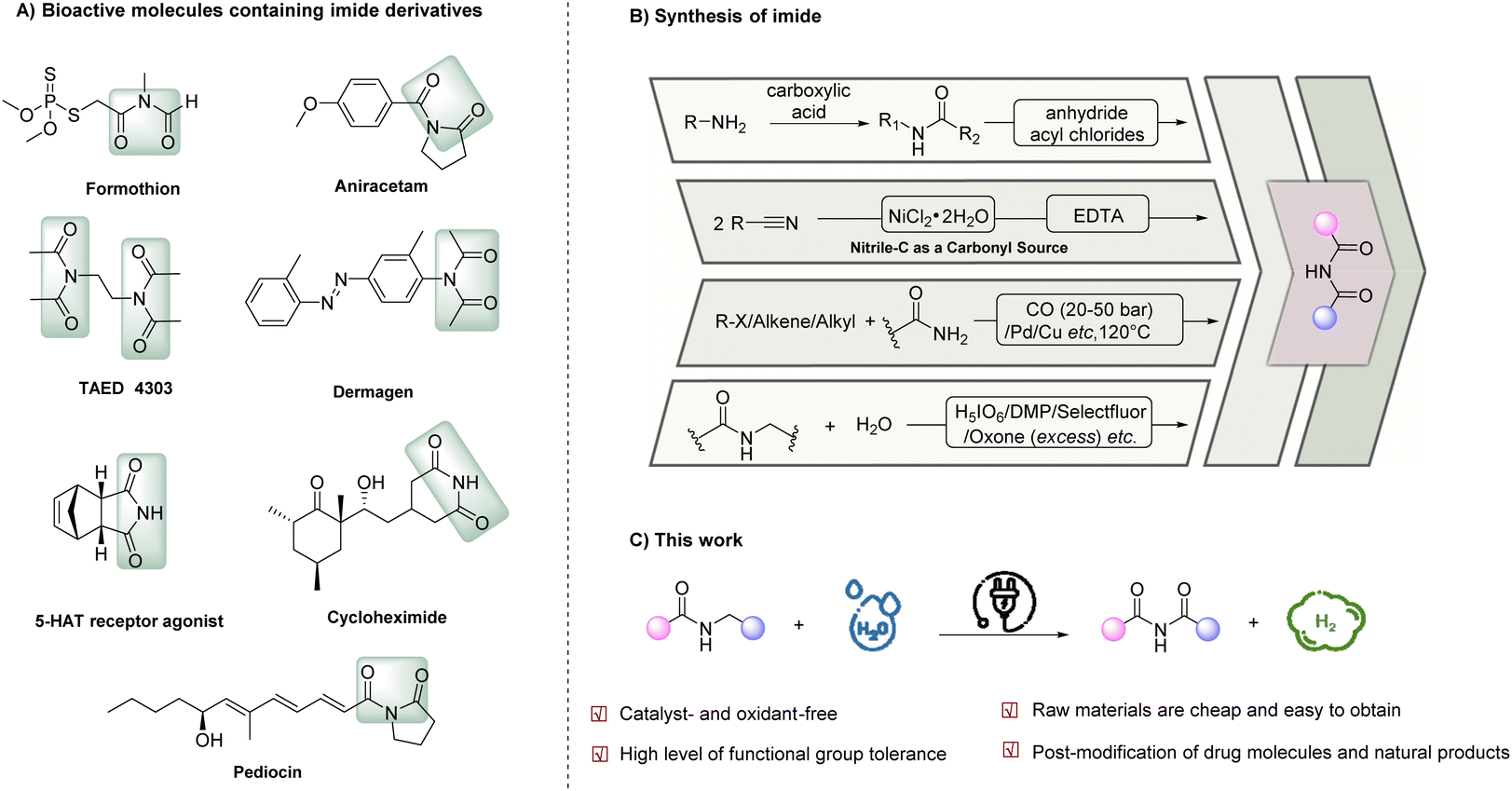 | ||
| Scheme 1 Electrochemical oxidation of amides to imines. (A) Selected examples of imine motifs in pharmaceutical compounds. (B) Reported methods of the synthesis of imides. (C) This work: the development of electrochemical α-oxidation of N-alkyl amides using water as the oxygen source. | ||
Conventional methods to prepare imides typically involve nucleophilic substitution reactions between amides and activated carboxylic acid derivatives, such as acyl chlorides and anhydrides, under harsh conditions.10–12 However, these methods suffer from a limited substrate scope owing to their poor functional group tolerability and constraints in substrate availability. Additionally, the use of activating reagents in these methods generates stoichiometric harmful wastes, underscoring the need for milder and more sustainable methods that align with the principles of green chemistry and sustainable synthesis. In recent years, various atom-economical catalytic strategies have emerged for imide synthesis (Scheme 1B), which include: (i) utilization of nitriles through the addition of nitriles onto the electrophilic carbonyl carbons of other nitriles, followed by hydration,13–15 (ii) carbonylative coupling of amides with carbon monoxide and aryl or alkyl precursors16–18 and (iii) selective α-oxidation of N-alkylamides. Among these strategies, the selective α-oxidation of N-alkylamides stands out as the most straightforward approach. However, this method remains challenging due to competing N-dealkylation reactions that result in the formation of imides as minor products. Although the discovery of ruthenium oxide (RuO4) as an oxidant has shown promise, its high oxidation potential can degrade aromatic rings and cleave C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, limiting its applicability.19–22 Recently, significant breakthroughs have been achieved by several research groups. For instance, the Lu group developed a transition-metal- and halogen-free oxidation of N-benzylamides using K2S2O8/O2 as an oxidant and pyridine as a phase-transfer catalyst in MeCN, but it is not applicable to N-alkylamides.23 Furthermore, in contrast to the extensive literature on the oxidation of α-C(sp3)–H bonds to carbonyl groups, only a limited number of oxidants (e.g., H5IO6,22 Dess–Martin periodinane (DMP),24 Selectfluor,25 or Oxone26) have been found effective for the oxidation of such bonds in N-alkylamides to produce the corresponding imides. Moreover, these protocols generally require excessive amounts of oxidants and high temperatures, resulting in poor atom economy, waste generation, and limited functional group compatibility. In 2022, Cai and colleagues reported an efficient metal-free, photoinduced strategy for the α-oxidation of N-alkylamides through a hydrogen atom transfer (HAT) process.27 However, this method required an excess of N-bromosuccinimide (NBS) as the HAT reagent. Hence, there remains a pressing need for more sustainable and efficient methods for the α-oxidation of N-alkylamides in imide synthesis that exhibit high atom-efficiency and chemoselectivity.
C bonds, limiting its applicability.19–22 Recently, significant breakthroughs have been achieved by several research groups. For instance, the Lu group developed a transition-metal- and halogen-free oxidation of N-benzylamides using K2S2O8/O2 as an oxidant and pyridine as a phase-transfer catalyst in MeCN, but it is not applicable to N-alkylamides.23 Furthermore, in contrast to the extensive literature on the oxidation of α-C(sp3)–H bonds to carbonyl groups, only a limited number of oxidants (e.g., H5IO6,22 Dess–Martin periodinane (DMP),24 Selectfluor,25 or Oxone26) have been found effective for the oxidation of such bonds in N-alkylamides to produce the corresponding imides. Moreover, these protocols generally require excessive amounts of oxidants and high temperatures, resulting in poor atom economy, waste generation, and limited functional group compatibility. In 2022, Cai and colleagues reported an efficient metal-free, photoinduced strategy for the α-oxidation of N-alkylamides through a hydrogen atom transfer (HAT) process.27 However, this method required an excess of N-bromosuccinimide (NBS) as the HAT reagent. Hence, there remains a pressing need for more sustainable and efficient methods for the α-oxidation of N-alkylamides in imide synthesis that exhibit high atom-efficiency and chemoselectivity.
In light of the aforementioned obstacles and our ongoing interest in electrocatalysis as an environmentally friendly and sustainable synthetic approach, this research paper presents a straightforward electrocatalytic oxidation of N-alkyl (methyl, ethyl, and cyclopropyl) amides to imides using water as a green and atom-efficient oxygen source, eliminating the need for an external oxidant (Scheme 1C). Significantly, our protocol enables the direct conversion of a broad scope of low-cost and simple bulk chemicals into valuable imides in a single step, with hydrogen gas (H2) as the only by-product.
Results and discussion
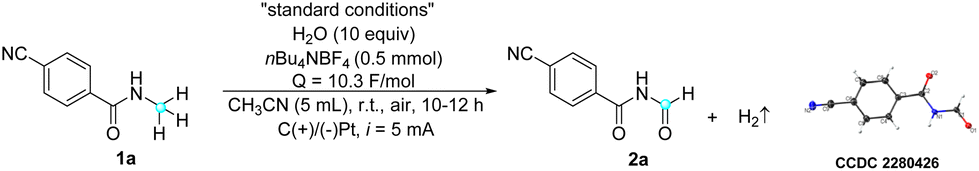
The feasibility of electrocatalytic selective α-oxidation of N-alkyl amides was initially investigated using amide 1a as a model substrate with a simple two-electrode configuration in an undivided cell (Table 1). The optimal conditions were determined to be constant current electrolysis in CH3CN at room temperature, with water (10 equiv.) as the oxygen source. Under these conditions (entry 1), the desired imide product 2a was obtained in a 73% yield, along with an inevitable dealkylation reaction that resulted in a 20% yield of the dealkylated product. Control experiments revealed that no reaction occurred in the absence of electricity, and starting material 1a was recovered (entry 2). Furthermore, the necessity of water in this reaction was substantiated through the following experiments. In the absence of water, only 20% of the product was obtained, likely due to the presence of adventitious water in the solvent and atmosphere (entry 3). Substituting water with TFA (entry 4) resulted in a comparable product yield to that obtained under standard conditions,28 whereas the use of concentrated HCl (12 M) as an additive led to a low product yield of 13% (entry 5). In an additional experiment conducted under an oxygen atmosphere without additional water, a low yield of only 13% was obtained, ruling out the possibility of oxygen gas in the air serving as the oxygen source (entry 6). Moreover, a control experiment conducted under an argon atmosphere (entry 7) showed no difference compared to that conducted under air (entry 1), further indicating the non-participation of oxygen in the reaction. These results strongly suggest that water and TFA serve not only as proton sources but also play a crucial role as oxygen sources in the reaction, which requires further investigation in the mechanistic study. Notably, although TFA appears to be slightly superior to water, possibly due to its stronger nucleophilicity and acidity, thereby promoting the reaction with higher efficiency, we chose water for its greener profile and better atom economy in line with green chemistry principles. Current intensity experiments indicated that both excessively low and high current intensities led to a decrease in product yield (entries 8 and 9). Changing the electrolyte from nBu4NBF4 to nBu4NPF6 (entry 10) slightly decreased the reaction efficiency. Similarly, changing the cathode from platinum to graphite was found to be less effective (entry 11). It is worth noting that the structure of product 2a was further confirmed by X-ray crystallography.|
|
||
|---|---|---|
| Entry | Variation from “standard conditions” | Yieldb (%) |
| a Conditions: amide (0.2 mmol, 1 equiv.), H2O (10 equiv.), nBu4NBF4 (0.5 mmol), CH3CN (5 mL), C anode, P cathode (current intensity = 3.3 mA cm−2), r.t. and i = 5 mA, undivided cell. b Isolated yield. c No water added. | ||
| 1 | None | 73 |
| 2 | Without electricity | 0 |
| 3 | Without H2O | 20 |
| 4 | TFA instead of H2O | 75 |
| 5 | HCl (12 M) instead of H2O | 13 |
| 6c | Under an oxygen atmosphere | 15 |
| 7 | Under an argon atmosphere | 71 |
| 8 | i = 3 mA | 64 |
| 9 | i = 10 mA | 70 |
| 10 | nBu4NPF6 instead of nBu4NBF4 | 68 |
| 11 | C(+)/C(−) instead of C(+)/P(−) | 59 |
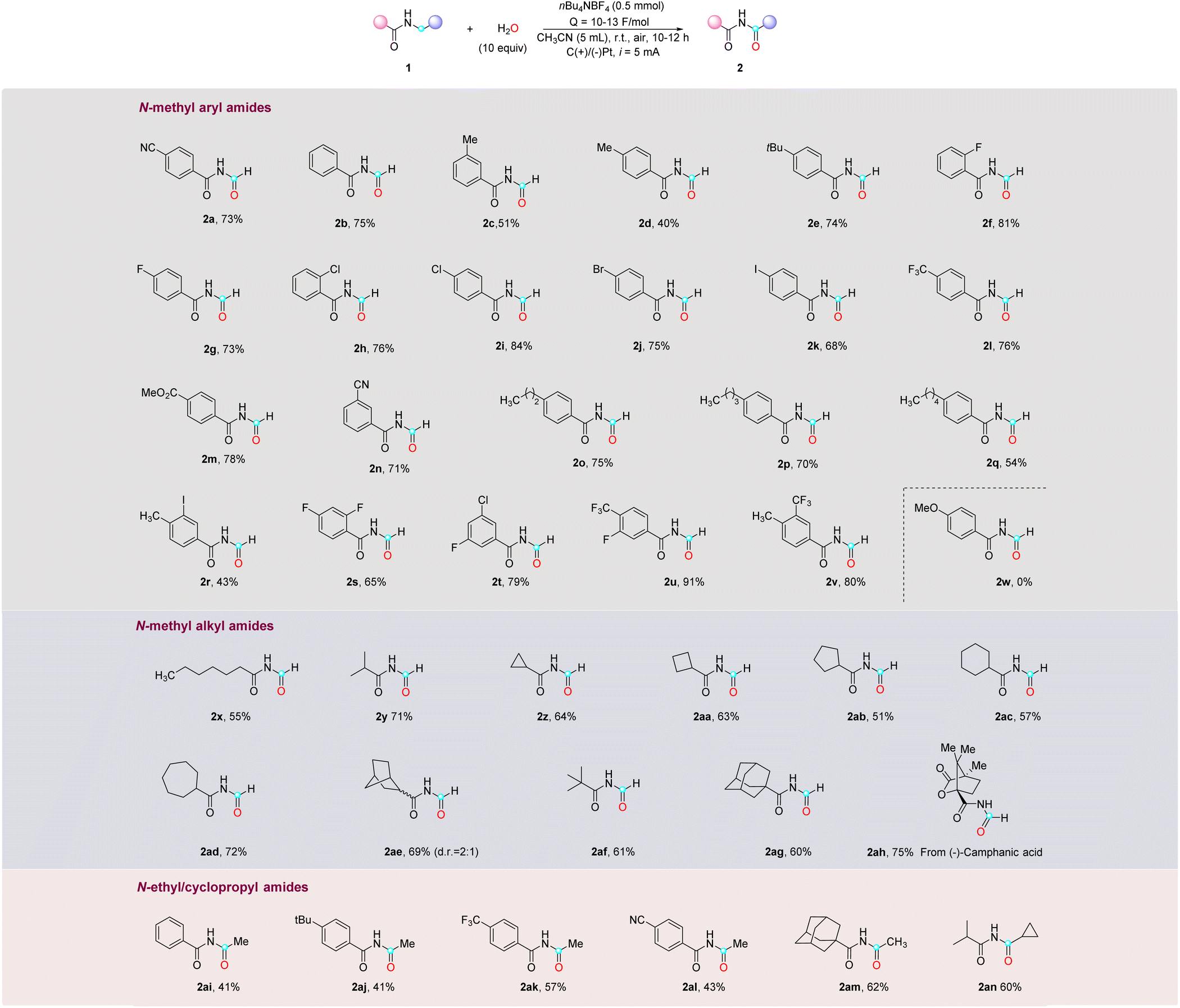
Using the optimized conditions, a series of N-methyl as well as N-ethyl aryl- and alkyl-amides were explored (Table 2). In general, the reaction exhibited broad substrate compatibility, allowing for the incorporation of various substituents on the aromatic rings, regardless of their electronic nature and substitution pattern. Alkyl mono-substituted phenyl rings, including methyl-, tert-butyl-, and long-chain alkyl groups, all reacted smoothly to provide the desired imide products (2b–2e, 2o–2q) in moderate to high yields (40–75%). Halide substituents such as F, Cl, Br, and I were well tolerated, enabling the synthesis of the corresponding imides (2f–2k) with good yields (68–81%). Amides bearing electron-withdrawing groups, such as cyano, trifluoromethyl, and ester, in either the para- or meta-position, also underwent smooth reactions to afford the desired products (2a, 2l–2n) in excellent yields (>70%). Furthermore, disubstituted aryl groups, such as methyl and iodo (2r), difluoro (2s), fluoro and chloro (2t), trifluoro and fluoro (2u), and methyl and trifluoro (2v), exhibited successful transformations. It is noteworthy that, despite a slightly lower yield observed in the case of 2r, the result is deemed satisfactory considering the labile nature and inherent instability of the C–I bond. When the aromatic ring was substituted with an electron-donating group like methoxy (2w), the starting material was consumed without yielding the desired imide product, which is expected because the electron-rich aromatic ring is more susceptible to oxidation and subsequent decomposition on the electrode surface compared to the amides. Additionally, attempts to expand the scope to include heteroaryl substrates were unsuccessful, yielding no desired products (see the ESI†). Aside from N-methyl arylamides, N-methyl alkylamides were also viable substrates in this reaction, demonstrating successful transformations with primary, secondary, and tertiary alkyl groups (2x–2y, 2af) in good yields (55–71%). Additionally, cyclic structures including cyclopropane, cyclobutane, cyclopentane, cyclohexane, cycloheptane, bicycloheptane, and adamantane rings were found to be compatible, furnishing the desired imide products (2z–2ae, 2ag) in good yields (51–72%). Lastly, attempts were made to perform late-stage modifications on the derivatives of bioactive molecules bearing N-methyl amide. The N-methyl amide derived from (−)-camphanic acid (1ah) was successfully converted into the corresponding imide product 2ah with a yield of 75%. However, N-methyl amides derived from other bioactive molecules, such as abietic acid, stearic acid, and oleanolic acid, were unsuccessful primarily in undergoing conversion into the corresponding imide products due to their limited solubility in CH3CN (see the ESI†). The scope of this reaction can be extended to N-ethyl and cyclopropyl amides as well, leading to moderate yields of imides for both aryl and alkyl amides (2ai–2an), with an unreacted starting material that was not fully consumed even with a prolonged reaction time. It should be noted that other N-alkyl amides with longer chain lengths were not successful in this reaction (see the ESI†).
| a Conditions: amide (0.2 mmol, 1 equiv.), H2O (10 equiv.), nBu4NBF4 (0.5 mmol), CH3CN (5 mL), C anode, Pt cathode (current intensity = 3.3 mA cm−2), r.t. and i = 5 mA, undivided cell. Isolated yields are reported, unless noted otherwise. |
|---|
|
To further validate the role of water as the oxygen source in this reaction, an isotope labeling study using 18O-water (H218O, 85 atom %18O) and amide 1v was conducted (Scheme 2a). High-resolution mass spectrometry (HRMS) analysis of the resulting product showed 59% incorporation of the 18O isotope (labeled product 2v′) and 27% of a non-labeled product, which can be attributed to residual water present in the solvent. This result strongly reinforces the notion that the oxygen atom in the product originates from water. In order to elucidate the mechanism by which water contributes an oxygen atom to the substrates, a plausible explanation involves the oxidation of amide substrates on the anode to generate a cationic intermediate, which subsequently undergoes nucleophilic attack by water, leading to the formation of an alcohol moiety. To evaluate this hypothesis, commercially available N-(hydroxymethyl)-benzamide (II), a potential intermediate in the reaction, was subjected to the optimized reaction conditions (Scheme 2b), and the desired product 2b was obtained with a high yield of 82%. This finding strongly supports the assumption that N-(hydroxymethyl)benzamide (II) acts as a plausible reaction intermediate which supports the mechanism.
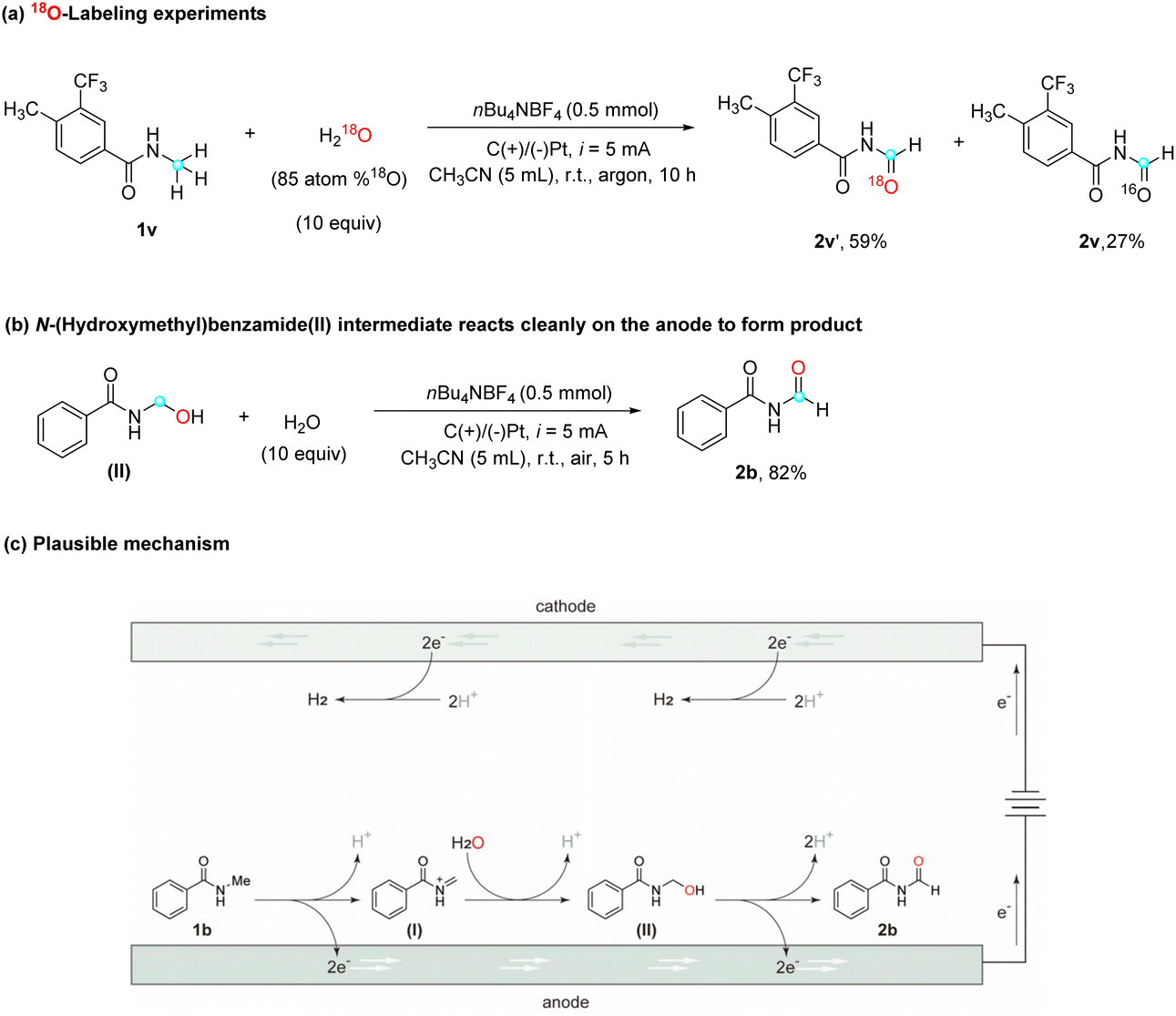 | ||
| Scheme 2 Proposed mechanisms with supporting evidence. (a) 18O-Labeling experiment. (b) N-(Hydroxymethyl)benzamide(II) intermediate reacts cleanly on the anode to form a product. (c) Plausible reaction mechanisms. | ||
Based on the findings from the above studies and existing literature, a plausible mechanism is proposed for this electrocatalytic selective α-oxidation of N-alkyl amides to imides, using N-methyl amide 1b as a representative substrate (Scheme 2c). The mechanism begins with the anodic oxidation of N-methyl amide 1b (EP/2 = 2.35 V vs. Ag/AgCl, determined experimentally with cyclic voltammetry), which involves the transfer of two electrons, giving rise to the formation of cationic intermediate I. Subsequently, intermediate I undergoes nucleophilic addition with water, resulting in the generation of intermediate II. Following the anodic oxidation of intermediate II and the subsequent release of a proton, the desired imide product 2b is formed.29 It is worth noting that, during this electrochemical reaction, protons are simultaneously reduced at the cathode, resulting in the generation of H2 as a byproduct.
This metal- and oxidant-free direct electrochemical oxidation of N-alkyl amides to imides using water as the oxygen source represents a highly green and sustainable approach (Scheme 3). To explore its potential for industrial applications, a gram-scale reaction was performed using substrate 1v (10 mmol) under optimal reaction conditions. Encouragingly, the desired imide 2v was obtained in a yield of 65%, affirming the optimal reaction conditions and the feasibility of scaling up the electrocatalytic oxidation of amides to imides. Additionally, imides find significant utility as precursors for the synthesis of 1,2,4-triazoles through the Einhorn–Brunner reaction,30 which involves the condensation of imides and hydrazines. Triazole is a nitrogen heterocyclic compound, consisting of three nitrogen atoms and two carbon atoms, with diverse applications in various fields and holds significant importance. Extensive research on 1,2,4-triazoles by Pattan et al. has revealed their antibacterial, antifungal, antitubercular, and anti-inflammatory activities.31 Notably, many FDA-approved drug molecules, such as itraconazole, fluconazole, and voriconazole, contain the 1,2,4-triazole scaffold, further highlighting its significance in medicinal applications. By utilizing imide 2b and phenylhydrazine, for instance, a remarkable 96% yield of 1,5-diphenyl-1H-1,2,4-triazole 3 can be obtained, showcasing the potential of imides as precursors for the synthesis of biologically relevant 1,2,4-triazole compounds.
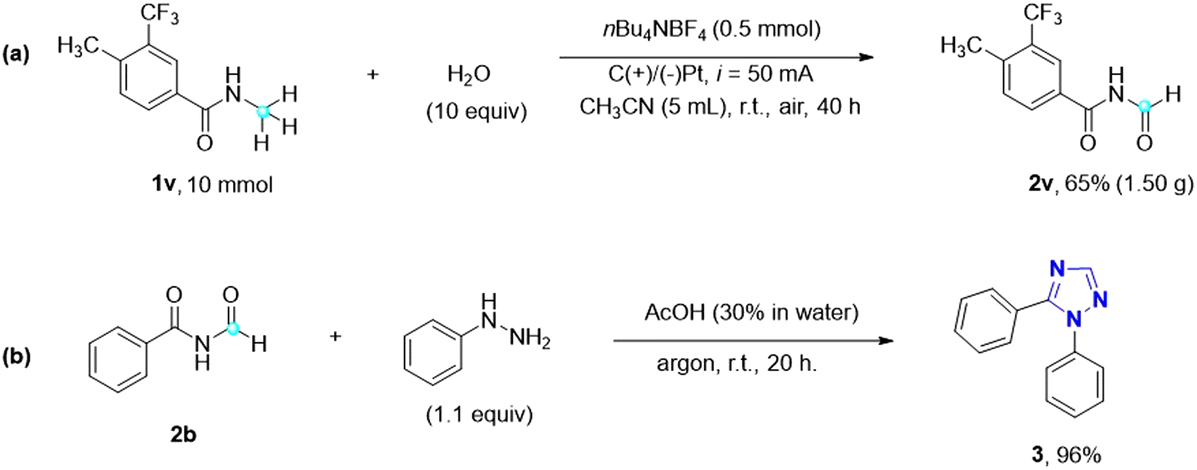 | ||
| Scheme 3 Practicality and application of the imide products. (a) Gram-scale reaction of 1v. (b) Application: synthesis of 1,5-disubstituted 1,2,4-triazole. | ||
Conclusions
In conclusion, our study presents a straightforward and environmentally friendly method for synthesizing imides from N-alkyl amides. Although our protocol is currently limited to N-methyl, ethyl, and cyclopropyl amides, it offers several notable advantages: (1) our electrocatalytic oxidation using water as the oxygen source offers a sustainable alternative to conventional methods that rely on toxic or expensive oxidants; (2) this method showcases an atom- and step-economical synthesis, providing a sustainable approach for the synthesis of imides; (3) it demonstrates a remarkably broad scope, allowing for the transformation of a wide range of N-methyl and ethyl amides into imides, thereby expanding the synthetic scope for imide derivatives; and (4) this protocol exhibits high reactivity, delivering good yields of the desired imide products under mild reaction conditions. Furthermore, the ability to achieve high yields on a gram-scale affirms the feasibility and practicality of our method, making it suitable for broader adoption in various synthetic processes. Additionally, the imide products can serve as valuable building blocks for the synthesis of 1,2,4-triazoles, which are important compounds with diverse applications particularly in drug molecules. We envision that further development and optimization of this method will lead to its widespread use in medicinal chemistry, contributing to the advancement of green chemistry principles and sustainable manufacturing practices.Author contributions
J. Q. conducted all experiments and characterized the novel compounds. J. W. designed the experiments. H. T. A. and J. W. wrote the manuscript. S. D. and P. N. were responsible for the funding application and oversaw the strategic direction of the project. X. W. and G. W. contributed to the analysis and interpretation of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by Pfizer (A-0008361-00-00), the Agency for Science, and the Prime Minister's Office of Singapore under its NRF-CRP Programme (NRF-CRP27-2021-0001).References
- A. M. Higgins and R. A. L. Jones, Nature, 2000, 404, 476–478 CrossRef CAS PubMed.
- M. Klinge, H. Cheng, T. M. Zabriskie and J. C. Vederas, J. Chem. Soc., Chem. Commun., 1994, 1379–1380 RSC.
- S. M. Capitosti, T. P. Hansen and M. L. Brown, Org. Lett., 2003, 5, 2865–2867 CrossRef CAS PubMed.
- L. Ross and L. A. Guarino, Virology, 1997, 232, 105–113 CrossRef CAS PubMed.
- K. Krohn, C. Franke, P. G. Jones, H.-J. Aust, S. Draeger and B. Schulz, Liebigs Ann. Chem., 1992, 1992, 789–798 CrossRef.
- Q. T. Zhang and J. M. Tour, J. Am. Chem. Soc., 1997, 119, 5065–5066 CrossRef CAS.
- N. N. Singh and A. K. Srivastava, Environ. Res., 1982, 28, 335–339 CrossRef CAS PubMed.
- K. Nakamura, CNS Drug Rev., 2002, 8, 70–89 CrossRef CAS PubMed.
- E.-S. Kim, K. Y. Park, J.-M. Heo, B. J. Kim, K. D. Ahn and J.-G. Lee, Ind. Eng. Chem. Res., 2010, 49, 11250–11253 CrossRef CAS.
- W. Kantlehner, P. Fischer, W. Kugel, E. Möhring and H. Bredereck, Justus Liebigs Ann. Chem., 1978, 1978, 512–527 CrossRef.
- N. A. Al-Awadi, J. Chem. Soc., Perkin Trans. 2, 1990, 2187–2189 RSC.
- P. Y. Reddy, S. Kondo, T. Toru and Y. Ueno, J. Org. Chem., 1997, 62, 2652–2654 CrossRef CAS PubMed.
- K. Kikukawa, K. Kono, F. Wada and T. Matsuda, Bull. Chem. Soc. Jpn., 1982, 55, 3671–3672 CrossRef CAS.
- M. N. Kopylovich, A. J. L. Pombeiro, A. Fischer, L. Kloo and V. Y. Kukushkin, Inorg. Chem., 2003, 42, 7239–7248 CrossRef CAS PubMed.
- A. O. Gálvez, C. P. Schaack, H. Noda and J. W. Bode, J. Am. Chem. Soc., 2017, 139, 1826–1829 CrossRef PubMed.
- A. Schnyder and A. F. Indolese, J. Org. Chem., 2002, 67, 594–597 CrossRef CAS PubMed.
- A. Umehara, S. Shimizu and M. Sasaki, ChemCatChem, 2023, 15, e202201596 CrossRef CAS.
- Y. Li, K. Dong, F. Zhu, Z. Wang and X.-F. Wu, Angew. Chem., Int. Ed., 2016, 55, 7227–7230 CrossRef CAS PubMed.
- J. P. N. Papillon and R. J. K. Taylor, Org. Lett., 2000, 2, 1987–1990 CrossRef CAS PubMed.
- Y. Takeuchi, T. Shiragami, K. Kimura, E. Suzuki and N. Shibata, Org. Lett., 1999, 1, 1571–1573 CrossRef CAS.
- C. Altomare, A. Carotti, G. Casini, S. Cellamare, M. Ferappi, E. Gavuzzo, F. Mazza, G. Pantaleoni and R. Giorgi, J. Med. Chem., 1988, 31, 2153–2158 CrossRef CAS.
- L. Xu, S. Zhang and M. L. Trudell, Chem. Commun., 2004, 1668–1669 RSC.
- Y. Hu, L. Zhou and W. Lu, Synthesis, 2017, 4007–4016 CAS.
- K. C. Nicolaou and C. J. N. Mathison, Angew. Chem., Int. Ed., 2005, 44, 5992–5997 CrossRef CAS PubMed.
- Z. Jin, B. Xu and G. B. Hammond, Tetrahedron Lett., 2011, 52, 1956–1959 CrossRef CAS.
- C. Mei, Y. Hu and W. Lu, Synthesis, 2018, 2999–3005 CAS.
- H. H. Chang, X. X. He, Z. L. Zang, C. H. Zhou and G.-X. Cai, Asian J. Org. Chem., 2022, 11, e202200500 CrossRef CAS.
- H. Long, T.-S. Chen, J. Song, S. Zhu and H.-C. Xu, Nat. Commun., 2022, 13, 3945 CrossRef CAS PubMed.
- D. Wang, P. Wang, S. Wang, Y.-H. Chen, H. Zhang and A. Lei, Nat. Commun., 2019, 10, 2796 CrossRef PubMed.
- Y.-I. Lin, J. J. Hlavka, P. Bitha and S. A. Lang Jr., J. Heterocycl. Chem., 1983, 20, 1693–1695 CrossRef CAS.
- P. P. Gadhave, N. S. Dighe, S. R. Pattan, D. Pravin, D. S. Musmade and R. V. Shete, Ann. Biol. Res., 2010, 1, 82–89 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2280426. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc04010k |
| This journal is © The Royal Society of Chemistry 2024 |