
Cu-Catalyzed, electron-relayed three-component synthesis of 2-alkenylbenzothiazoles with cathodic ammonia evolution†
Chengxian
Hu
,
Dan
Wang
,
Lu
Wang
,
Ying
Fu
 * and
Zhengyin
Du
*
* and
Zhengyin
Du
*
Key Laboratory of Eco-functional Polymer Materials of the Ministry of Education, College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, P. R. China. E-mail: clinton_du@126.com; fu_yingmail@126.com
First published on 5th December 2023
Abstract
A novel one-pot, three-component reaction of 2-aminothiophenols, aldehydes and malononitrile was studied under electrochemical conditions. The reaction was conducted using TBABF4 as an electrolyte and CuI as a catalyst in an undivided cell at room temperature. The proposed reaction mechanism showed that CuI acted as an “electron relay”.
2-Substituted benzothiazoles are important nitrogen-containing heterocyclic compounds with a wide range of biological activities. The thiazole ring and condensed benzene ring of 2-substituted benzothiazoles have received much attention due to their special “double loop” system. The 2-substituted benzothiazoles ring system prevalent in natural products and drug molecules is present in oceans and on land, and the value of pharmaceutical application has gained increased attention.1–4 Consequently, the search for new strategies to synthesize 2-substituted benzothiazoles is important. A one-pot multicomponent reaction is an efficient and economical synthetic method because it avoids the separation and purification of intermediates, and complicated steps. It can also be used to rapidly assemble complex structures that are molecularly diverse, and is used widely in the design and synthesis of new drugs, combinatorial chemistry, and synthesis of natural products.5 The research and development of a malononitrile-participated multicomponent reaction have attracted great attention in recent years, especially with regard to the synthesis of heterocyclic compounds.6–8 In 2016, Wang and colleagues uncovered the catalysis of piperidine for the annulation reaction of 4H-chromenes, aromatic aldehydes and malononitrile to prepare a series of oxacyclic compounds under room temperature (Scheme 1a).9 Recently, Jonnalagadda and co-workers demonstrated a simple and efficient method for the synthesis of 2-(benzothiazol-2-yl)-3-(substituted-phenyl) acrylonitriles (Scheme 1b).10 Then, the research teams of Evdokimov, Guo and Shingare independently studied the one-pot reaction of aldehydes, malononitrile and o-aminothiols in different reaction media, and finally 2-amino-4-phenyl-6-(phenylsulfanyl)-3,5-dicyanopyridines were obtained via cyclization and dehydrogenation reactions (Scheme 1c).11
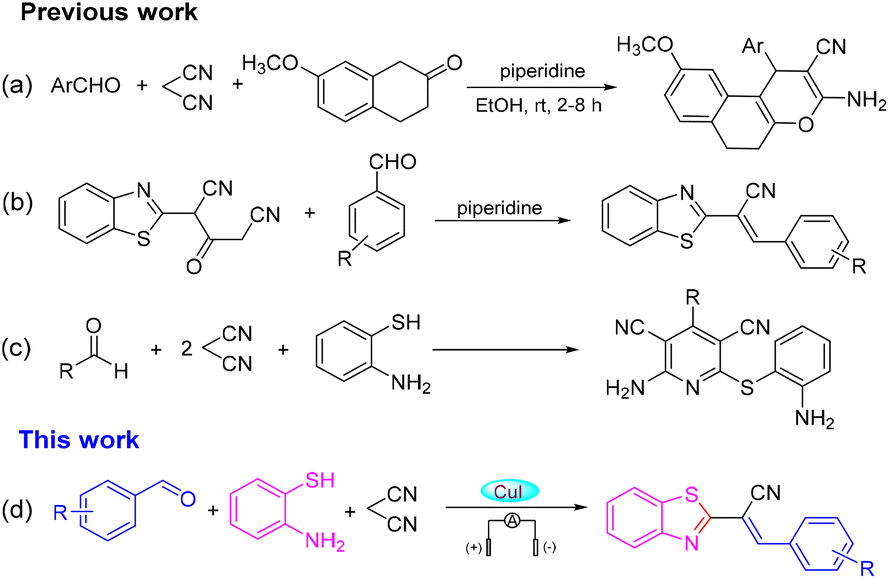 | ||
| Scheme 1 Previous and proposed work. | ||
Nevertheless, finding a method for synthesizing 2-substituted benzothiazoles at high yields under mild reaction conditions and simple operation has been the goal of organic chemists.12 Organo-electro synthesis that uses electric current as a redox reagent has attracted increasing attention due to its great potential in the development of “green” and sustainable methods. The electro-synthetic method has five important advantages: (i) avoids the use of toxic or dangerous oxidants and reducing agents; (ii) electrons are clean reaction reagents; (iii) the reaction system (in addition to raw materials and products) usually does not contain other reagents; (iv) reduced consumption of materials; (v) the product is easy to separate, of high purity, and causes low environmental pollution.13 The synthesis of 2-substituted benzothiazoles by electrochemical methods can offer new avenues to access natural products and drug molecules. With our persistent interest in green organic synthesis and considering the green characteristics of electrochemical synthesis,14,15 we report herein a highly atom-economical, multicomponent reaction of 2-aminothiophenols, aldehydes and malononitrile to prepare 2-alkenyl-substituted benzothiazoles through dual electrochemical synthesis and catalysis of transition metals (Scheme 1d).
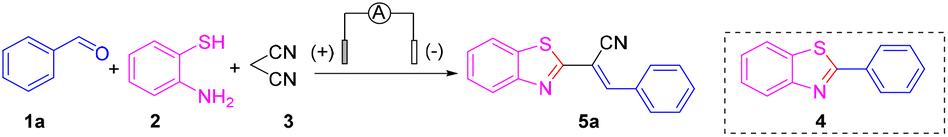
First, benzaldehyde (1a), 2-aminothiophenol (2) and malononitrile (3) were selected as model substrates for optimization of the electrosynthesis conditions. The electro-chemical reaction was performed in an undivided cell equipped with a carbon anode and a platinum cathode at a constant current of 12 mA (Table 1). To screen electrolytes, we did the template reaction using different electrolytes in the absence of any catalyst. Unfortunately, only a trace amount of the desired product 5a was observed when using NaClO4, LiClO4, NaOAc, KI, tetrabutylammonium hexafluorophosphate (TBAPF6), and tetrabutylammonium tetrafluoroborate (TBABF4) as electrolytes (Table 1, entries 1–6). Instead, 2-phenylbenzothiazole 4, via a Knoevenagel condensation between malononitrile 3 and benzaldehyde 1a, was obtained in a moderate yield. When several imidazonium ionic liquids were utilized instead of electrolytes, the target product, (E)-2-(benzothiazol-2-yl)-3-phenylacrylonitrile (5a), was obtained in ≤30% yield with almost 100% E-selectivity (Table 1, entries 7–10). To further improve the product yield, we chose to add copper salts as transition-metal catalysts. Encouragingly, under constant-current electrolysis conditions, by adding various copper salts (CuI, CuBr, CuCl, and Cu(OAc)2), the yield of product 5a was increased significantly to 82% (Table 1, entries 11–14). When CuI was chosen as a catalyst, the reaction was fulfilled smoothly after 2 h and a yield of up to 84% for 5a was obtained (Table 1, entry 15). Under these conditions, neither 4 nor other byproducts were observed. Therefore, we chose to add CuI (10 mol%) as a catalyst to ensure a better reaction effect.
|
|
||||
|---|---|---|---|---|
| Entry | Electrolyte | Catalyst | Time (h) | 5a yieldb (%) |
| a Reaction conditions: Two-electrode undivided cell: C anode, Pt cathode; benzaldehyde 1a (0.5 mmol), 2-aminothiophenol 2 (0.5 mmol), malononitrile 3 (0.5 mmol), electrolytes (20 mol%), catalysts (10 mol%), EtOH (5 mL), rt, I = 12 mA. b Isolated yield. | ||||
| 1 | NaClO4 | — | 4 | Trace |
| 2 | LiClO4 | — | 4 | n.r. |
| 3 | NaOAc | — | 4 | Trace |
| 4 | KI | — | 4 | n.r. |
| 5 | TBAPF6 | — | 4 | Trace |
| 6 | TBABF4 | — | 4 | Trace |
| 7 | C8MImBF4 | — | 6 | 26 |
| 8 | BMImBF4 | — | 6 | 22 |
| 9 | HMImBF4 | — | 6 | 17 |
| 10 | C6MImBF4 | — | 6 | 13 |
| 11 | TBABF4 | CuI | 4 | 82 |
| 12 | TBABF4 | CuBr | 4 | 77 |
| 13 | TBABF4 | CuCl | 4 | 73 |
| 14 | TBABF4 | Cu(OAc)2 | 4 | 52 |
| 15 | TBABF4 | CuI | 2 | 84 |
To improve the reaction yield further, we continued to screen the reaction solvent (Table 2). EtOH was the best solvent for this reaction, whereas the yield of 5a was reduced significantly when CH3OH, DCM, EA, acetonitrile (ACN), DMF, ethyl lactate (EL), and THF were used as solvents (Table 2, entries 1–9). This may have been because polar solvents can promote dissolution of substrates and electrolytes to transfer electrons efficiently. However, when water was used as a solvent, a trace amount of 5a was obtained (Table 2, entry 5). This showed that water had a potent inhibitory effect on the reaction. Then, we screened the current intensity (Table 2, entries 2, 10 and 11). Decreasing the operating current to 10 mA and increasing it to 14 mA furnished the product in a slightly reduced yield of 79% and 75%, respectively (Table 2, entry entries 10 and 11). Next, we used a carbon rod and platinum plate in pairs and recombined them into different electrolytic cells to screen electrode materials (Table 2, entries 2 and 12–14). Under these electrode pairs, the product yield was lower than that obtained using carbon as an anode and platinum plate as the cathode (Table 2, entry 2).
|
|
||||
|---|---|---|---|---|
| Entry | Solvents | Current (mA) | Anode/cathode | 5a yieldb (%) |
| a Reaction conditions: Two-electrode undivided cell: C anode, Pt cathode; benzaldehyde 1a (0.5 mmol), 2-aminothiophenol 2 (0.5 mmol), malononitrile 3 (0.5 mmol), solvents (5 mL), TBABF4 (32.2 mg, 20 mol%), CuI (9.5 mg, 10 mol%), rt, 2 h. b Isolated yield. | ||||
| 1 | MeOH | 12 | C/Pt | 56 |
| 2 | EtOH | 12 | C/Pt | 84 |
| 3 | DCM | 12 | C/Pt | 54 |
| 4 | EA | 12 | C/Pt | 48 |
| 5 | H2O | 12 | C/Pt | Trace |
| 6 | ACN | 12 | C/Pt | 63 |
| 7 | DMF | 12 | C/Pt | 34 |
| 8 | EL | 12 | C/Pt | 32 |
| 9 | THF | 12 | C/Pt | 10 |
| 10 | EtOH | 10 | C/Pt | 79 |
| 11 | EtOH | 14 | C/Pt | 75 |
| 12 | EtOH | 12 | Pt/Pt | 52 |
| 13 | EtOH | 12 | C/C | 55 |
| 14 | EtOH | 12 | Pt/C | Trace |
Having the optimized reaction conditions in hand, we set out to evaluate the scope of the substrate in this reaction (Scheme 2). The reaction was compatible with a series of electron-donating groups and electron-withdrawing groups, providing a moderate-to-excellent yield of coupling products in most cases (54% to 91%) (5a–5r). The reactions of benzaldehydes with an electron-donating group such as methoxy methyl on para-, ortho-, and meta-positions proceeded as smoothly as those for 5a to produce 5b–5c, 5e–5f and 5h–5i in high-to-excellent yields (82–91%).
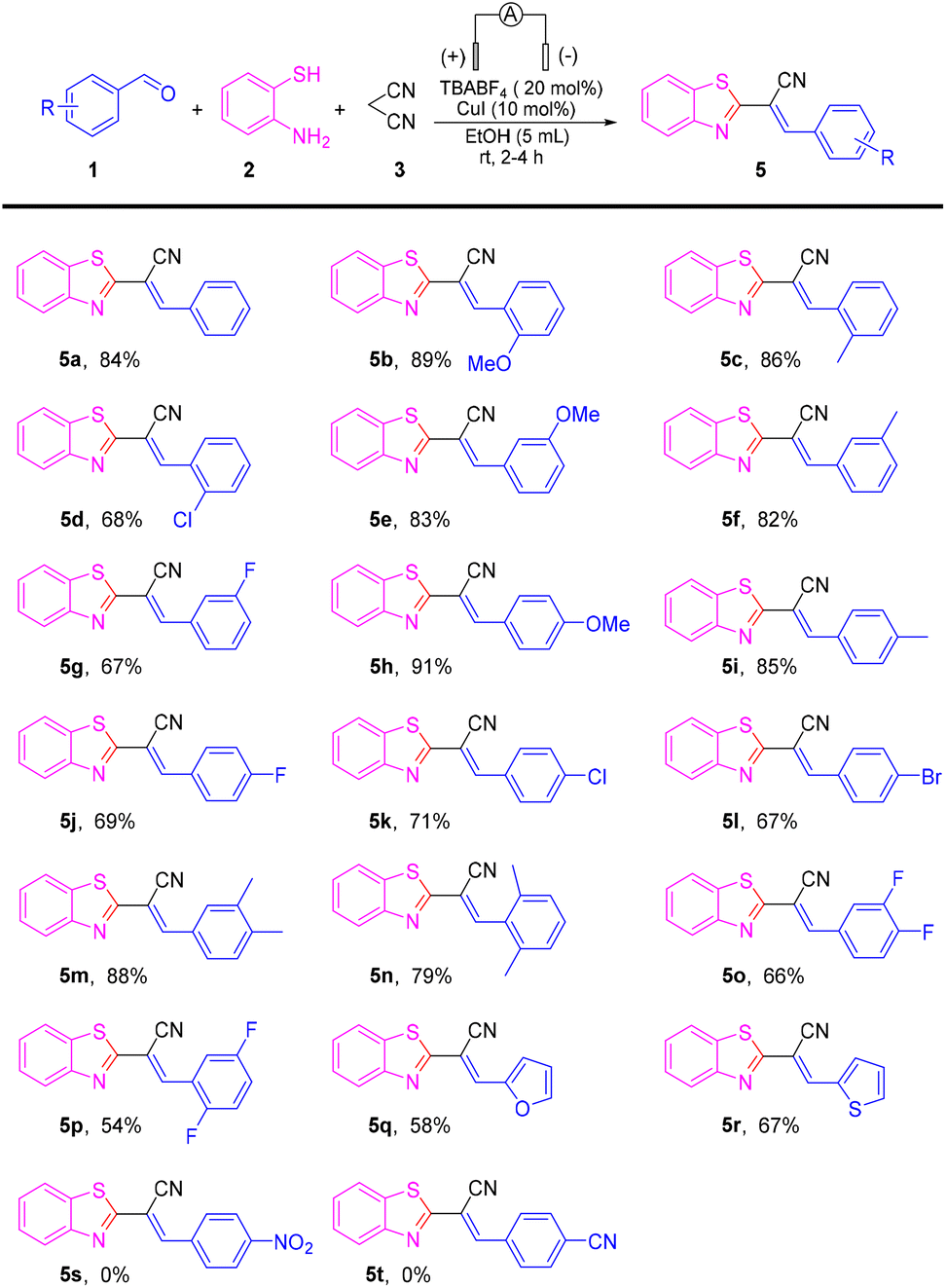 | ||
Scheme 2 Substrate scope. (a) Reaction conditions: Two-electrode undivided cell: C anode, Pt cathode; benzaldehydes 1 (0.5 mmol), 2-aminothiophenol 2 (0.5 mmol), malononitrile 3 (0.5 mmol), solvent (5 mL), rt, TBABF4 (32.2 mg, 20 mol%), CuI (9.5 mg,10 mol%), rt, I ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) 12 mA, 2 h, 1.8 F mol−1; (b) isolated yield. 12 mA, 2 h, 1.8 F mol−1; (b) isolated yield. | ||
Halogen atoms (F, Cl, and Br) were well-tolerated to deliver the corresponding products 5d, 5g, 5j–5l in 67–71% yields. Benzaldehydes with two electron-donating substituents were good substrates, affording desired coupling products 5m–5n in moderate-to-high yields. However, when two halogen groups were attached to the aromatic ring, the yield was reduced (e.g., 5p had only 54% yield). On the other hand, aldehydes with heterocycles could also produce the target product, such as 5q and 5r, but at a lower yield than aromatic aldehydes. Unfortunately, when the strong electron-withdrawing groups –NO2 and –CN were connected to benzaldehydes, the reaction was stagnant and no product was obtained. To gain further insight into the mechanism of this transformation, a series of control experiments were carried out (Scheme 3). The reaction could proceed smoothly in air and argon atmospheres. Therefore, it could be described as an electrochemical anodic oxidation rather than the oxidation of oxygen in air (Scheme 3a). The condensation product 6a and 2-aminothiophenol underwent a reaction under a standard condition to obtain a yield of 79% for the target product. In the process of this reaction, benzaldehyde and malononitrile first reacted to obtain 6a (Scheme 3b). Later, upon the addition of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), 2,6-di-tert-butyl-4-methylphenol (BHT), and 1,1-diphenylethylene to the reaction mixture, only a trace amount of the desired product 5a was obtained, thereby suggesting that the reaction involved a free radical (Scheme 3c). To verify the applicability of our method, the reaction was then amplified to a scale of 7 mmol, and 5a was formed in 62% yield (1.18 g) (Scheme 3d), which showed the feasibility and practicability of this protocol.
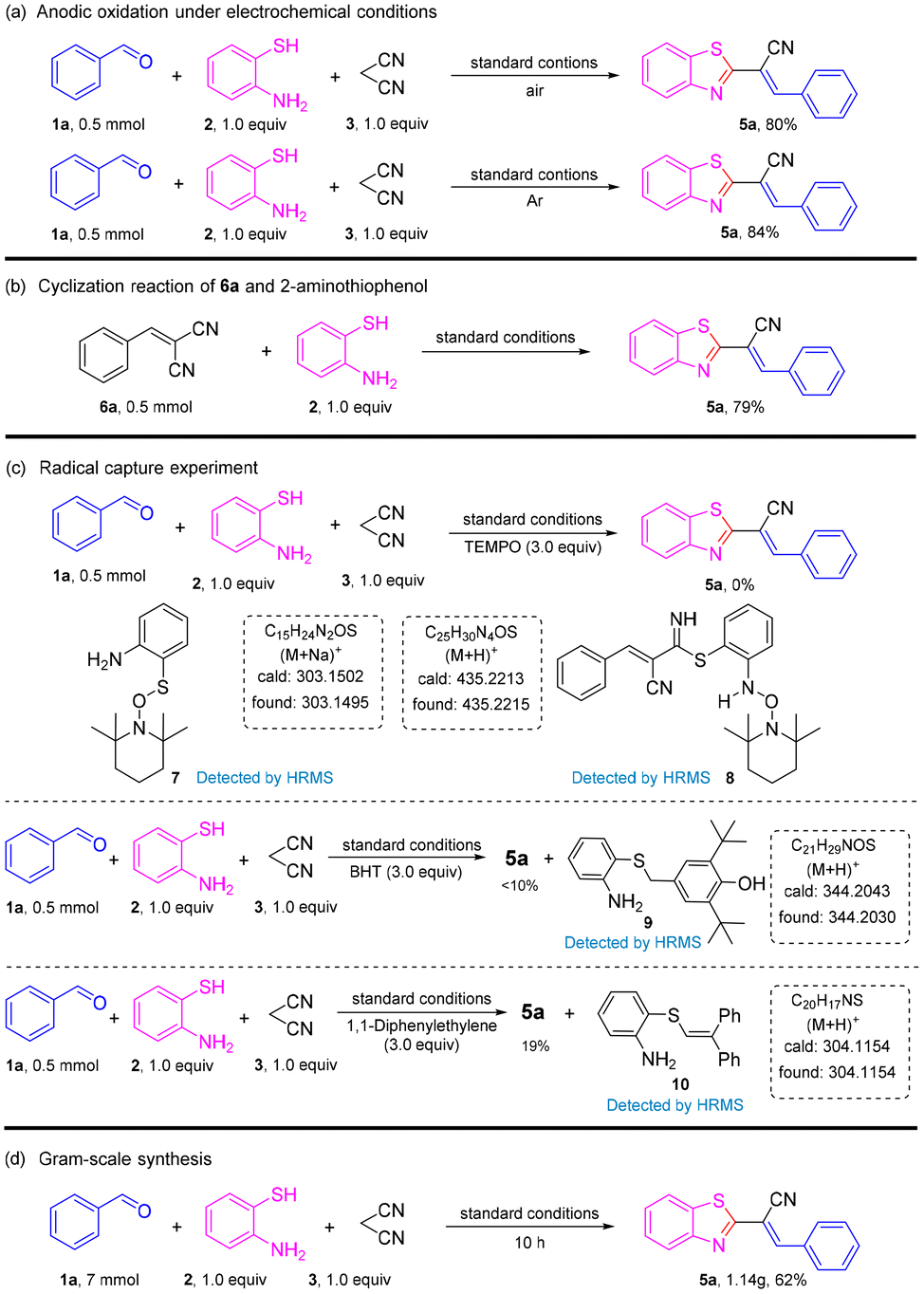 | ||
| Scheme 3 Control experiments. | ||
To delve deeper into the mechanism, cyclic voltammetry (CV) was conducted. As shown in Scheme 4, obvious oxidation peaks of 2-aminothiophenol were observed at 0.73 and 1.06 V. In addition, one oxidation peak of benzaldehyde was found at 1.25 V. The lower oxidation peak of 2-aminothiophenol than that of benzaldehyde indicated that 2-aminothiophenol may be oxidized before 1a. Considering the literature,16 and combining the results of Scheme 3 and CV experiments, a possible mechanism was proposed (Scheme 5). Initially, a Knoevenagel condensation between malononitrile 3 and benzaldehyde 1a leads to the formation of intermediate A. Cu(I) is oxidized to Cu(II) at the anode and reacts with 2-aminothiophenol 2 to form B. Then, radical B adds to the CN bond to afford the intermediate C, which undergoes 1,6-HAT to give the radical intermediate D, followed by an intramolecular cyclization of D to afford intermediate E. Finally, E releases radical NH2 to obtain the target product 5a. Meanwhile, the cathodic reduction of protons generates the byproduct NH3, which excludes the demand for sacrificial external chemical oxidants.
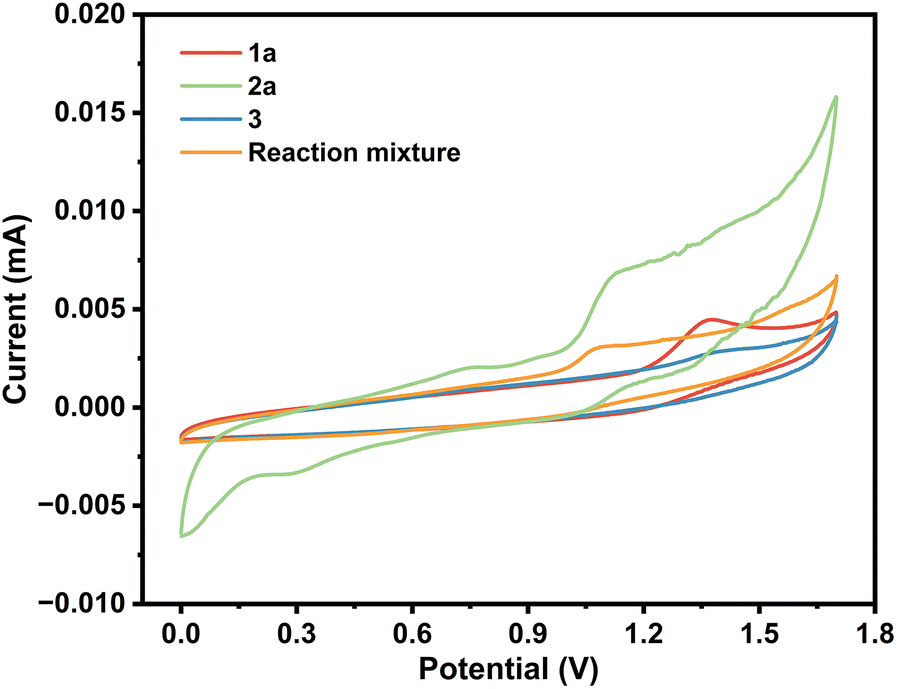 | ||
| Scheme 4 Experiments using cyclic voltammetry. | ||
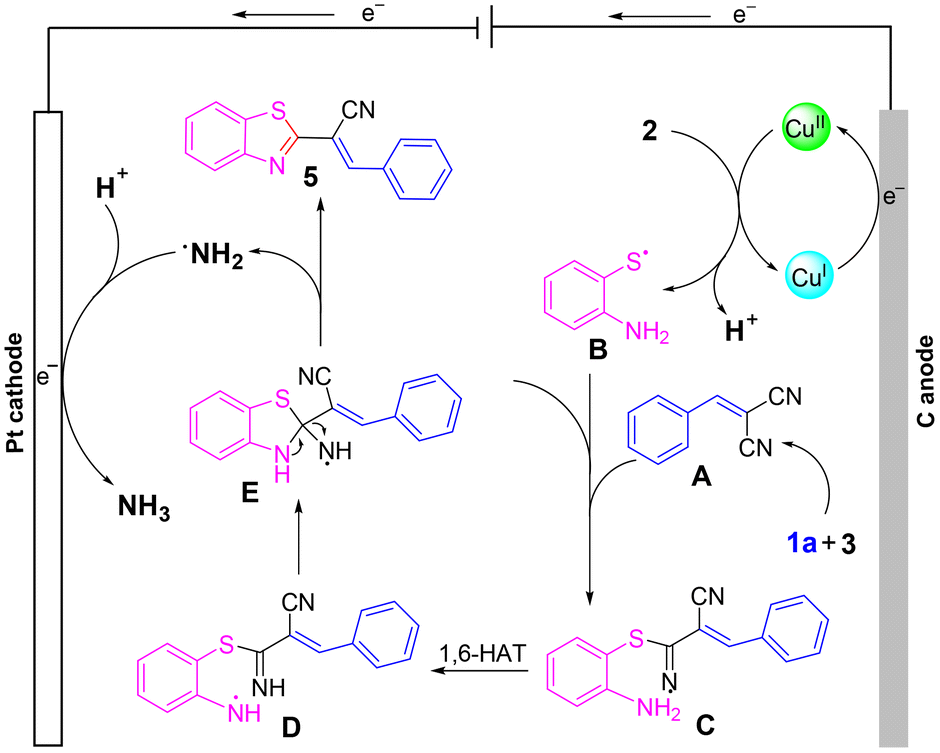 | ||
| Scheme 5 Postulated mechanism. | ||
Conclusions
We found a CuI-catalyzed, one-pot, three-component reaction of benzaldehydes, 2-aminobenziphenol and malononitrile under electrochemical conditions, wherein CuI underwent an “electron relay”. A series of 2-alkenyl-substituted benzothiazoles were obtained in high yields in a short reaction time without the need for external heating or oxidants. Ammonia and water were byproducts of this reaction. Our method had a wide substrate scope and good tolerance of functional groups. The one-step reaction avoids the separation of intermediates, and shows good practicable value.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21762039, 22161041) and the Natural Science Foundation of Gansu Province (20JR5RA521) is acknowledged.References
- (a) G. P. Gunawardana, S. Kohmoto, S. P. Gunasekera, O. J. McConnell and F. E. Koehn, J. Am. Chem. Soc., 1988, 110, 4856–4858 CrossRef CAS; (b) A. K. Chakraborti, S. Rudrawar, K. B. Jadhav and G. Kaur, Green Chem., 2007, 9, 1335–1340 RSC; (c) P. Natarajan, N. K. Brara and J. J. Kaura, Org. Chem. Front., 2018, 5, 1527–1531 RSC.
- (a) R. H. Tale, Org. Lett., 2002, 4(10), 1641–1642 CrossRef CAS PubMed; (b) X. M. Zhu, F. G. Zhou, Y. Yang, G. B. Deng and Y. Liang, ACS Omega, 2020, 5, 13136–13147 CrossRef CAS PubMed; (c) A. Hasan and R. Ali, Results Chem., 2022, 4, 100338 CrossRef.
- M. B. Harriet, F. Bret and B. Paul, Drugs, 1996, 52, 549–563 CrossRef PubMed.
- (a) M. L. Morningstar, T. Roth and D. W. Farnsworth, J. Med. Chem., 2007, 50, 4003–4015 CrossRef CAS PubMed; (b) G. Sreenivasa, E. Jayachandran, B. Shivakumar, K. Jayaraj and M. K. Vijay, Arch. Pharm. Sci. Res., 2009, 1, 150–157 CAS; (c) J. Das, R. V. Moquin, J. Lin, C. Liu, A. M. Doweyko and H. F. DeFex, Bioorg. Med. Chem. Lett., 2003, 13, 2587–2590 CrossRef CAS PubMed; (d) M. Singh, S. K. Singh, M. Gangwar, G. Nath and S. K. Singh, RSC Adv., 2014, 4, 19013–19023 RSC; (e) Y. Liu, Y. Wang, G. Dong, Y. Zhang, W. Zhang and C. Sheng, Med. Chem. Commun., 2013, 4, 1551–1561 RSC.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS; (c) B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS PubMed; (d) K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef PubMed.
- (a) S. Ambethkar, V. Padmini and N. Bhuvanesh, Adv. Res., 2015, 6, 975–985 CrossRef CAS; (b) C. E. Hsieh, C. Y. Tsao and C. H. Chuang, J. Org. Chem., 2021, 86, 12168–12180 CrossRef CAS; (c) K. Kriis, A. Miller, K. Erkman and T. Kanger, J. Org. Chem., 2022, 87, 7422–7435 CrossRef CAS.
- F. Freeman, Chem. Rev., 1969, 69, 591–624 CrossRef CAS PubMed.
- (a) R. Mallepalli and R. S. Perali, Tetrahedron Lett., 2016, 57, 4541–4543 CrossRef CAS; (b) I. Yavari, R. Solgi and K. K. Aliyeh, Heterocycl. Chem., 2019, 56, 3396–3402 CrossRef CAS; (c) S. O. Hourieh and S. G. Javad, Res. Chem. Intermed., 2022, 48, 2069–2085 CrossRef.
- X. S. Wang, D. Q. Shi, X. Y. Wei and Z. M. Zong, Chin. J. Org. Chem., 2004, 24, 1454–1457 CAS.
- S. Maddila and S. B. Jonnalagadda, Bull. Chem. Soc. Ethiop., 2012, 26, 467–471 CAS.
- (a) N. M. Evdokimov, A. S. Kireev, A. A. Yakovenko, M. Y. Antipin, I. V. Magedov and A. Kornienko, J. Org. Chem., 2007, 72, 3443–3453 CrossRef CAS; (b) J. J. Tian and H. Y. Guo, Chin. J. Org. Chem., 2012, 32, 193–196 CrossRef CAS; (c) P. V. Shinde, B. B. Shingate and M. S. Shingare, Bull. Korean Chem. Soc., 2011, 32, 459–462 CrossRef CAS.
- (a) N. Fu, G. S. Sauer, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (b) Q. L. Yang, Y. K. Xing, X. Y. Wang and T. S. Mei, J. Am. Chem. Soc., 2019, 141, 18970–18976 CrossRef CAS PubMed; (c) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (d) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (e) Y. Liu, H. Yi and A. Lei, Chin. J. Chem., 2018, 36, 692–697 CrossRef CAS.
- (a) Y. Y. Jiang, K. Xu and C. C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (b) K. D. Moeller, Tetrahedron, 2000, 56, 9527–9554 CrossRef CAS; (c) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC.
- (a) F. Gang, G. Xu, T. Dong, L. Yang and Z. Y. Du, Chin. J. Org. Chem., 2015, 35, 1428–1440 CrossRef CAS; (b) H. Wu, J. Wu and Z. Y. Du, Chin. J. Org. Chem., 2017, 37, 1127–1138 CrossRef CAS; (c) H. Li, P. Yang, Z. Xu, Z. Y. Du and Y. Fu, Chin. J. Org. Chem., 2020, 40, 2476–2482 CrossRef CAS; (d) D. Wang, Q. Li, M. T. Li, Y. Fu and Z. Y. Du, Curr. Org. Chem., 2021, 25, 1298–1320 CrossRef CAS; (e) C. X. Hu, D. Mou, X. Zhang, Y. Fu, C. D. Huo and Z. Y. Du, Org. Biomol. Chem., 2022, 20, 8120–8124 RSC; (f) C. X. Hu, R. K. Liu, Z. T. Ning, D. Mou, Y. Fu and Z. Y. Du, Org. Biomol. Chem., 2022, 20, 8280–8284 RSC; (g) L. Y. Wang, M. T. Li, Z. T. Ning, X. Zhang, Y. Fu and Z. Y. Du, J. Org. Chem., 2023, 88, 6374–6381 CrossRef CAS PubMed.
- (a) Q. L. Yang, P. Fang and T. S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS; (b) T. H. Meyer and L. Ackermann, Chem., 2020, 6, 2484–2496 CrossRef CAS; (c) X. Fan, X. He, X. Ji, L. Zhang, J. Li, B. Ying and X. Sun, Inorg. Chem. Front., 2023, 10, 1431–1435 RSC; (d) J. Liang, Z. Li, L. Zhang, X. He, Y. Luo, B. Tang and X. Sun, Chem., 2023, 9, 1768–1827 CrossRef CAS; (e) W. Song, L. Yue, X. Fan, S. Hamdye and X. Sun, Inorg. Chem. Front., 2023, 10, 3489–3514 RSC; (f) Z. Zhang, Y. Liu, X. Su, Z. Zhao, Y. Chen and S. Gao, Nano Res., 2023, 16, 6632–6641 CrossRef CAS; (g) X. Xu, L. Hu, Z. Li, L. Xie, S. Sun, Q. Kong, X. Sun and Q. Liu, Sustainable Energy Fuels, 2022, 6, 4130–4136 RSC; (h) G. Ma, F. Sun, L. Qiao, Q. Shen, L. Wang, Q. Tang and Z. Tang, Nano Res., 2023, 16, 10867–10872 CrossRef CAS; (i) X. He, Z. Li, J. Yao, K. Dong, X. Li, L. Hu, Y. Luo, M. S. Hamdy, L. Xie, Q. Liu and X. Sun, iScience, 2023, 26, 107100 CrossRef CAS PubMed.
- (a) Q. L. Yang, X. Y. Wang, P. Fang and T. S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS PubMed; (b) Z. Z. Ning, Z. C. Zhang, Y. Tang and P. J. Feng, Sci. China: Chem., 2022, 65, 1962–1967 CrossRef CAS; (c) X. Cheng, A. W. Lei, T. S. Mei, H. C. Xu, K. Xu and C. C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS; (d) N. Fu, L. Song, Y. Shen, J. C. Siu and S. Lin, J. Am. Chem. Soc., 2019, 141, 14480–14485 CrossRef CAS PubMed; (e) Q. Q. Shi, D. H. Wei, L. C. Michelle and L. Yu, J. Am. Chem. Soc., 2022, 144, 3137–3145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03884j |
| This journal is © The Royal Society of Chemistry 2024 |