
Iridium-catalyzed asymmetric, complete hydrogenation of pyrimidinium salts under batch and flow†
Zhi
Yang‡
a,
Yu
Chen‡
a,
Linxi
Wan
a,
Yuxiao
Li
a,
Dan
Chen
a,
Jianlin
Tao
d,
Pei
Tang
*a and
Fen-Er
Chen
 *abc
*abc
aSichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: rfchen@fudan.edu.cn; peitang@scu.edu.cn
bEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai 200433, China
cShanghai Engineering Center of Industrial Asymmetric Catalysis for Chiral Drugs, Shanghai 200433, China
dCentral Nervous System Drug Key Laboratory of Sichuan Province, Luzhou, Sichuan 646000, China
First published on 5th December 2023
Abstract
A highly enantioselective method for the complete hydrogenation of pyrimidinium salts using Ir/(S,S)-f-Binaphane complex as the catalyst was presented in this study. This approach affords facile access to a range of fully saturated chiral hexahydropyrimidines, which are prevalent in many bioactive molecules. The reactions showcase high yields and enantioselectivities under mild reaction conditions without additives. Successful application of this methodology in a continuous flow fashion further broadened its practical utility.
Introduction
Hydropyrimidine moieties with chiral centers play pivotal roles as structural scaffolds in numerous natural products and drug-like molecules exhibiting remarkable biological activities. These include naturally occurring alkaloids lycojapomine A1 and hetiamacin C2,3 as well as highly selective α1a-adrenoceptor antagonist L-771688![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and tetrahydropyrimidinone HIV protease inhibitors5 (Fig. 1). From a sustainable chemistry perspective, innovating atom-economical synthetic strategies to access industrially and pharmaceutically important chiral N-heterocyclic building blocks is vital. Transition metal-catalyzed asymmetric hydrogenation (AH) of N-heteroaromatic compounds offers a direct and ecofriendly synthetic approach for the facile construction of valuable functionalized cyclic heterocycles, which offers excellent atom economy, with almost no waste or byproducts, and thus is a highly sustainable and “green” strategy. However, the AH reactions of pyrimidines have received comparatively less attention than those of other well-known prochiral substrates, such as indoles, quinolines, isoquinolines, and pyridines.6–10
4 and tetrahydropyrimidinone HIV protease inhibitors5 (Fig. 1). From a sustainable chemistry perspective, innovating atom-economical synthetic strategies to access industrially and pharmaceutically important chiral N-heterocyclic building blocks is vital. Transition metal-catalyzed asymmetric hydrogenation (AH) of N-heteroaromatic compounds offers a direct and ecofriendly synthetic approach for the facile construction of valuable functionalized cyclic heterocycles, which offers excellent atom economy, with almost no waste or byproducts, and thus is a highly sustainable and “green” strategy. However, the AH reactions of pyrimidines have received comparatively less attention than those of other well-known prochiral substrates, such as indoles, quinolines, isoquinolines, and pyridines.6–10
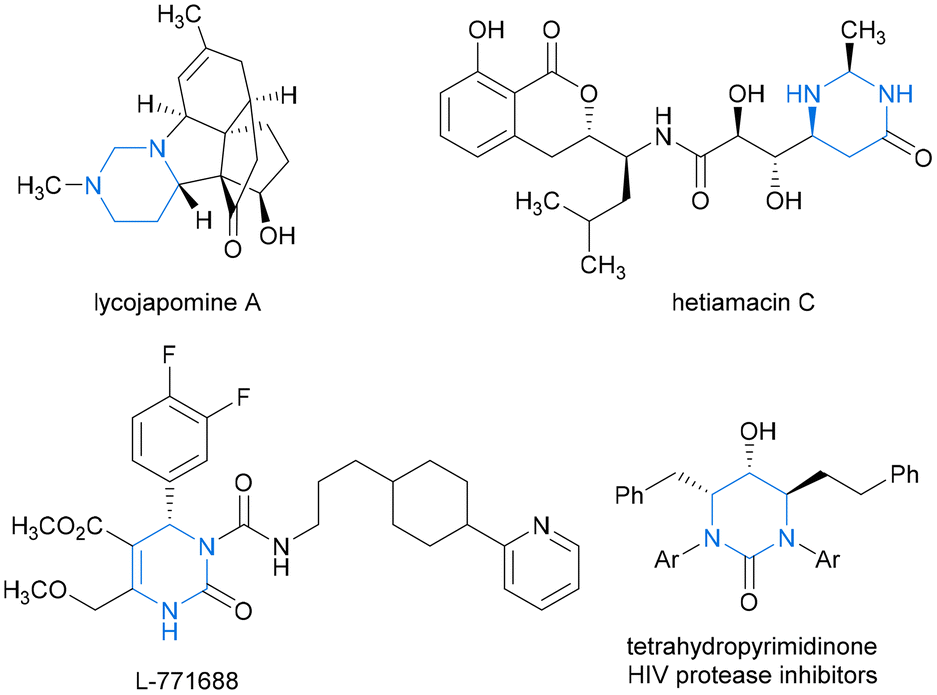 | ||
| Fig. 1 Representative examples of natural products and bioactive molecules containing hydropyrimidine moieties. | ||
The inherent problems are apparent: pyrimidine is one of the most stable aromatic structures that might impede reduction. Furthermore, both the pyrimidine and hydrogenated product, possessing strong coordination abilities, could deactivate chiral catalysts. Therefore, methods for AH of pyrimidine derivatives are notably scarce. In 2015, Kuwano et al. first documented the iridium-catalyzed AH of 4-substituted pyrimidines. This approach affords amidine products with high yields and enantioselectivities (Scheme 1a).11 Notably, lanthanide triflate is crucial for achieving high enantioselectivity and activating the heteroarene substrate. Subsequently, Zhou et al. reported an efficient palladium-catalyzed AH of 2-hydroxypyrimidines for the facile synthesis of chiral cyclic ureas.12 The key to success in this case lay in the hydroxyl-oxo tautomerism of 2-hydroxypyrimidine, which weakens aromaticity. They also reported an efficient iridium-catalyzed AH of 2-hydroxypyrimidines, which complements the earlier palladium-catalyzed AH of 2-hydroxypyrimidines (Scheme 1b).13 Furthermore, Zhou demonstrated the chiral phosphoric acid-catalyzed transfer hydrogenation of 2-hydroxypyrimidines, employing Hantzsch ester or dihydrophenanthridine as the hydrogen source (Scheme 1c).14 Despite these achievements, the employed approaches exhibit limitations such as the need for additives or low atom economy. Furthermore, there are barely any studies regarding the direct and highly stereoselective complete hydrogenation of pyrimidines for synthesizing diverse fully saturated chiral hexahydropyrimidines.
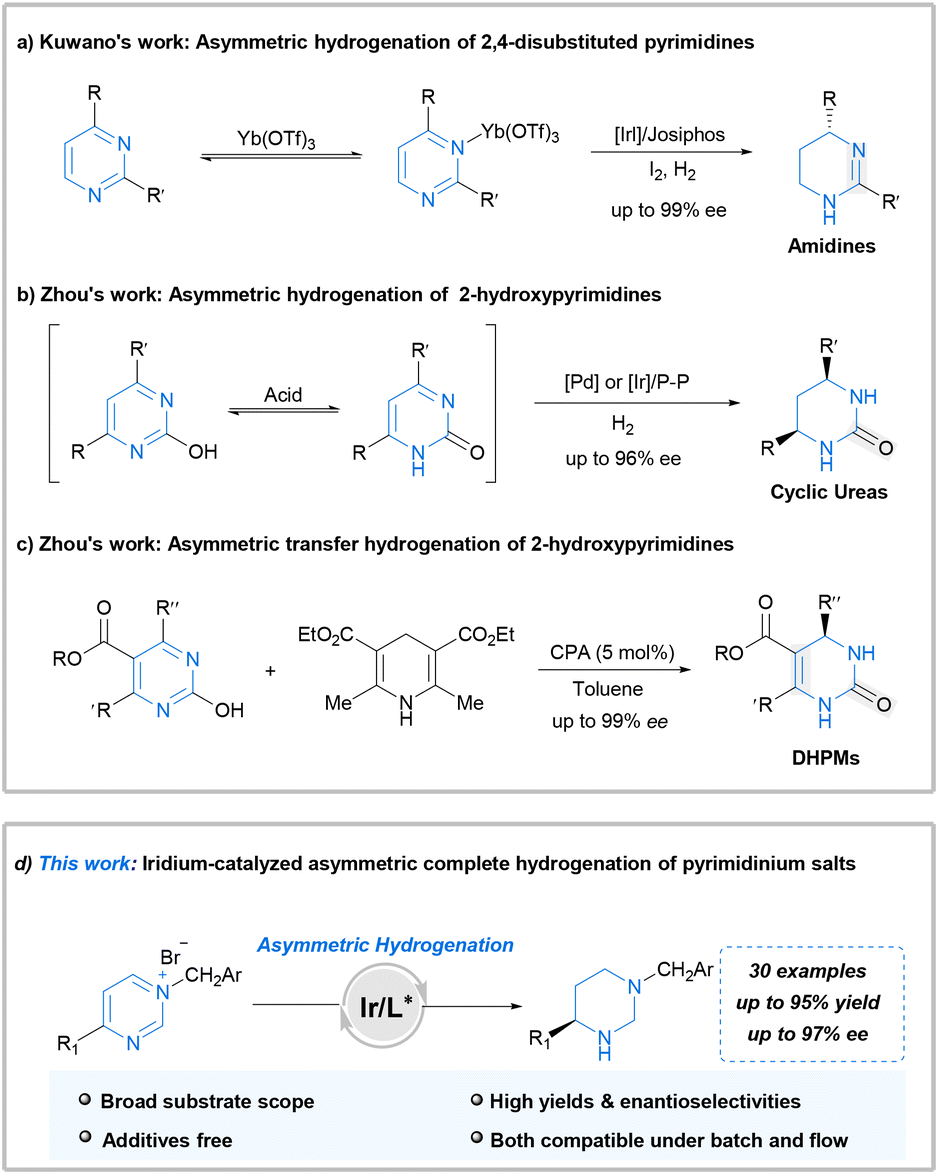 | ||
| Scheme 1 Asymmetric hydrogenation of pyrimidine derivatives. | ||
The hydrogenation activity of iminium salts is typically higher than that of the corresponding imines.15–18 Moreover, a successful approach for the AH of pyridines involves quaternization of the substrate.19 The structural analogy between pyridines and pyrimidines inspired us to attempt the quaternization of the pyrimidine nitrogen atom. This approach leads to the weakening of aromaticity and prevents substrate coordination to the catalyst during AH. Moreover, complete hydrogenation of arenes is one of the most effective methods for converting planar molecules into saturated three-dimensional structures, which are critical building blocks in many aspects of life.20,21 As part of our continuous interests in the synthesis of chiral amines via AH,22–27 herein, we disclose the first successful application of iridium-catalyzed complete AH for synthesizing a diverse range of fully saturated chiral hexahydropyrimidines from pyrimidinium salts without using any additional additives. Furthermore, application of this methodology was conducted successfully under continuous flow conditions (Scheme 1d). This flow transformation greatly reduced the hazard of hydrogen accumulation in batch with shorter reaction time and is more friendly to scale-up.
Results and discussion
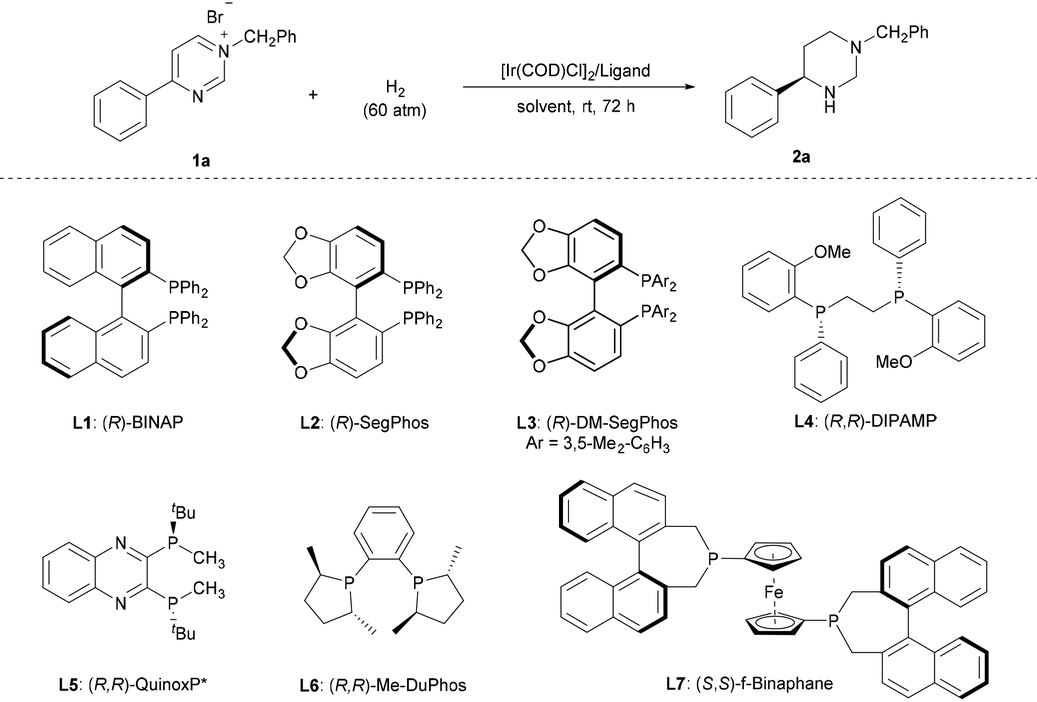
Numerous investigations have demonstrated the high efficacy of Ir complexes as catalysts in the enantioselective hydrogenation of N-heteroaromatic compounds.6–10 Thus, we commenced our study by employing N-benzyl-4-phenylpyrimidinium bromide (1a) as the model substrate and ethyl acetate as the solvent in the presence of 1.0 mol% [Ir(COD)Cl]2 at 25 °C. The assessment of ligands revealed that the classic axially chiral bisphosphine (R)-BINAP L1, (R)-SegPhos L2, and (R)-DM-SegPhos L3 afforded the product with poor enantioselectivities despite showing high activities (Table 1, entries 1–3). Notably, the ligands (R,R)-DIPAMP L4 and (R,R)-QuinoxP* L5, commonly used in AH, resulted in poor enantioselectivities (Table 1, entries 4 and 5). In the presence of (R,R)-Me-DuPhos L6, product 2a was produced in 85% yield but as a racemate (Table 1, entry 6). Moreover, product 2a could be obtained in 91% yield and a decent enantioselectivity (77% ee) using the ligand (S,S)-f-Binaphane L7 (Table 1, entry 7). Based on this result, we explored different reaction conditions with L7. A brief screening of the solvents revealed that although DCE, 1,4-dioxane, toluene, CHCl3, and dichloromethane (DCM) were suitable for completing this transformation (Table 1, entries 8–12); the reaction in DCM exhibited the best result (90% yield, 93% ee). The reaction at a lower reaction temperature (−20 °C) showed a comparable reactivity (91% yield) and an increased enantioselectivity (96% ee) (Table 1, entry 13). Notably, an enantioselective and complete hydrogenation of pyrimidinium salts has been successfully developed without additives.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Ligand | Yieldb (%) | eec (%) |
| a Reaction condition: 1a (0.20 mmol), [Ir(COD)Cl]2 (1.0 mol%), ligand (2.2 mol%), H2 (60 atm), solvent (3.0 mL), 25 °C, 72 h. b Isolated yields. c Determined by chiral HPLC. d The reaction was carried out at −20 °C. | ||||
| 1 | EtOAc | L1 | 88 | 54 |
| 2 | EtOAc | L2 | 90 | 47 |
| 3 | EtOAc | L3 | 90 | 56 |
| 4 | EtOAc | L4 | 85 | 12 |
| 5 | EtOAc | L5 | 87 | 46 |
| 6 | EtOAc | L6 | 85 | 0 |
| 7 | EtOAc | L7 | 91 | 77 |
| 8 | DCE | L7 | 89 | 80 |
| 9 | 1,4-Dioxane | L7 | 90 | 86 |
| 10 | PhMe | L7 | 89 | 91 |
| 11 | CHCl3 | L7 | 89 | 91 |
| 12 | DCM | L7 | 90 | 93 |
| 13d | DCM | L7 | 91 | 96 |
With the optimal reaction conditions established, we explored the substrate scope for the methodology; the results are summarized in Scheme 2. Expectedly, most of the substrates could be well-hydrogenated under the standard reaction conditions, affording the corresponding chiral hexahydropyrimidines in satisfactory yields and enantioselectivities. Initially, we explored the scope of pyrimidines bearing different substituents. We investigated the effect of benzene ring–substituent positions on the products (2b–2d). The substituents at para- and meta-positions in the benzene ring were efficiently hydrogenated, affording products with high yields (2b–2c, 88%–92% yields) and enantioselectivities (95%–96% ee). However, the enantioselectivity of the ortho-substituted product (2d) was adversely affected (63% ee), presumably as a result of steric effect. Furthermore, the presence of electron-donating substituents at the para-position of the phenyl group had no noticeable effect on the reaction, with all substrates yielding similar results (2b, 2e–2f, 92% yield, 96%–97% ee) to those obtained for 2a. Electron-withdrawing substituents at the para-position of the phenyl group were also well-tolerated to yield 2g–2j in uniformly good yields and excellent stereoselectivities (2g–2j, 88%–92% yields, 90%–93% ee). Moreover, substrates bearing halide substituents are also compatible with this catalytic system (2k–2m, 91–93% yields, 94–95% ee). Changing the phenyl group on the pyrimidine ring to 2-naphthyl or biphenyl also yielded excellent results (2n, 95% ee; 2o, 97% ee) compared with that obtained for 1a. In addition, disubstituted substrates were also successfully reacted under the optimized condition (2p, 94% yield, 93% ee). Substrates bearing alkyl groups were well-converted, although with lower enantioselectivity (2q–2r, 90%–91% yields, 49%–58% ee). Subsequently, we examined various substituents, including electron-donating and electron-withdrawing substitutents, on the N-benzyl group. All of these were well-tolerated, as were the halide substituents on the benzene ring at the para-, meta- and ortho-positions (4a–4j, 90%–94% yields, 93%–96% ee). Furthermore, disubstituted substrates also performed well (4k–4l, 92%–94% yields, 95–97% ee). The absolute configuration of 2o was unambiguously assigned using single-crystal X-ray analysis, and those of other products were assigned via analogy. To highlight the practical utility of our approach, a gram-scale AH of N-benzyl-4-phenylpyrimidinium bromide 1a was performed. The desired product 2a was obtained in 93% yield and 96% ee, and the catalyst loading could be reduced to 0.1 mol% (S/C = 500) at this scale (Scheme 3a). To broaden the application of our methodology, several transformations were performed on the product (Scheme 3b). First, the chiral hexahydropyrimidine 2a was easily converted to chiral cyclic urea 5 in 85% yield with 97% ee using triphosgene. Thereafter, late-stage modification of drugs using 2a was explored, for example, in the preparation of optically active stereoscopic compound 6 as a single diastereoisomer from (S)-Naproxen. Furthermore, hydroxylamine 7 was obtained in the presence of mCPBA in 91% yield without any compromise in the ee. Finally, the chiral 1-phenyl-1,3-diamine 8 was obtained in moderate yield (65%) and excellent ee (99%) by introducing a nitrogen-protecting group followed by ring opening. The chiral 1-phenyl-1,3-diamine structure is a key core of chiral ligands used for transition metal-based asymmetric catalysis28 and a core moiety in the FDA-approved HIV antiretroviral drug, Maraviroc.29–32 Notably, there was no substantial loss in the enantiomeric purity during the abovementioned transformations.
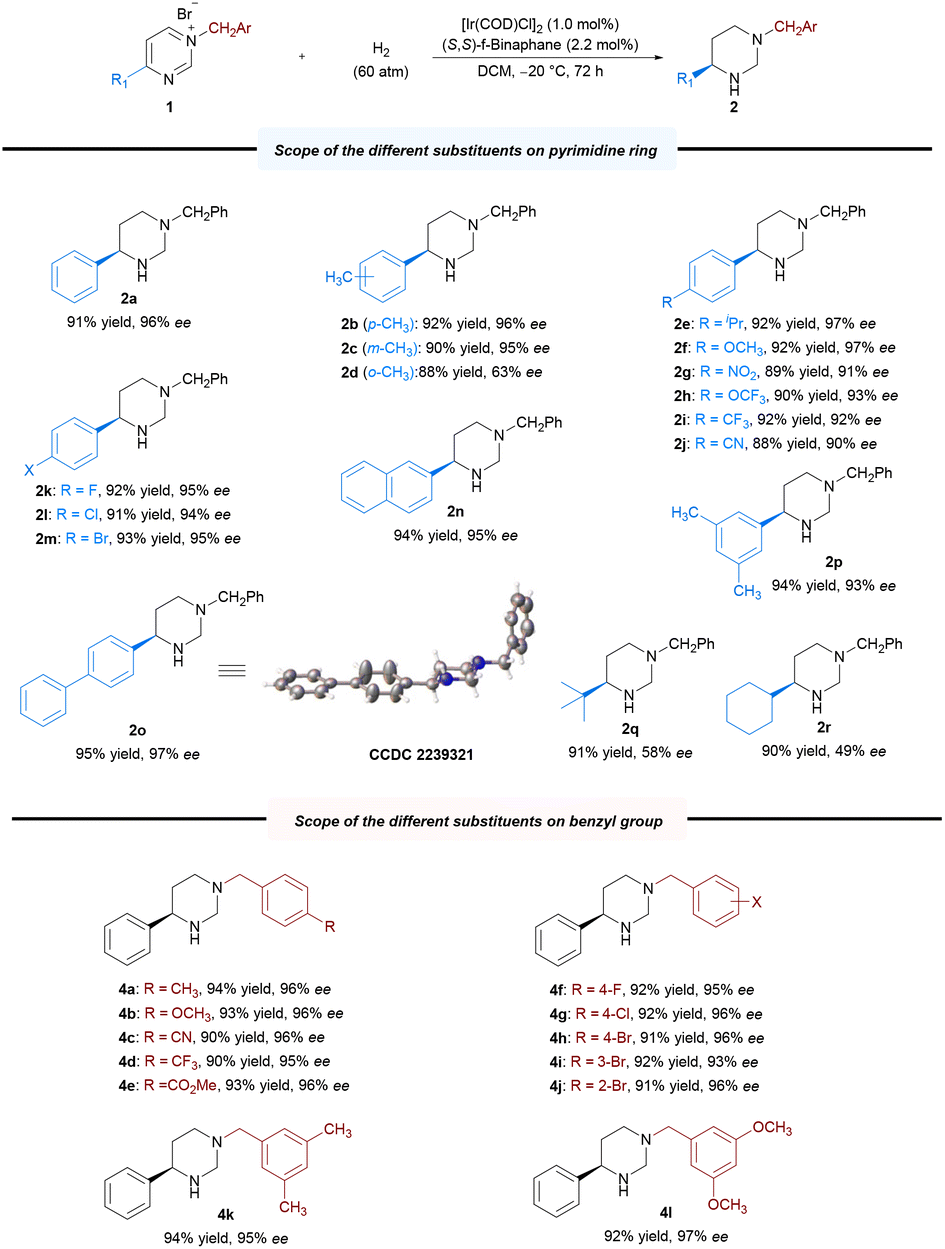 | ||
| Scheme 2 Iridium-catalyzed asymmetric hydrogenation of the N-benzyl-4-substituted pyrimidinium salts 1. Reaction conditions: 1 (0.2 mmol), [Ir(COD)Cl]2 (1.0 mol%), (S,S)-f-Binaphane (2.2 mol%), H2 (60 atm), DCM (3.0 mL), −20 °C, 72 h. | ||
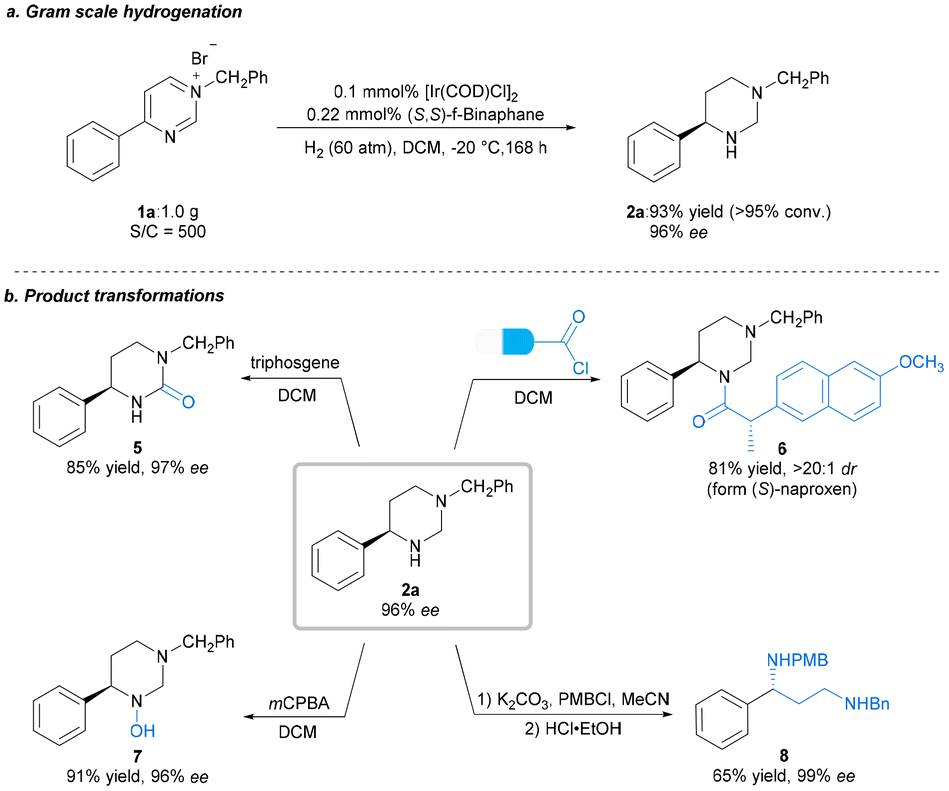 | ||
| Scheme 3 Gram-scale experiment and product transformations. | ||
To investigate the pathway, an isotopic labeling experiment was carried out by hydrogenation of 1a with D2 gas (Scheme 4a) and a plausible stepwise hydrogenation process was proposed (Scheme 4b). The salt 1a first undergoes a 1,2-hydride addition, the formed dihydropyrimidine is then go through a tautomerization; then, hydrogenation of imide gives the enamine; subsequent the enamine intermediate proceeds in a tautomerization of enamine to imine by the in situ-formed HBr, followed by asymmetric hydrogenation of imine to give the product 2a with high ee.
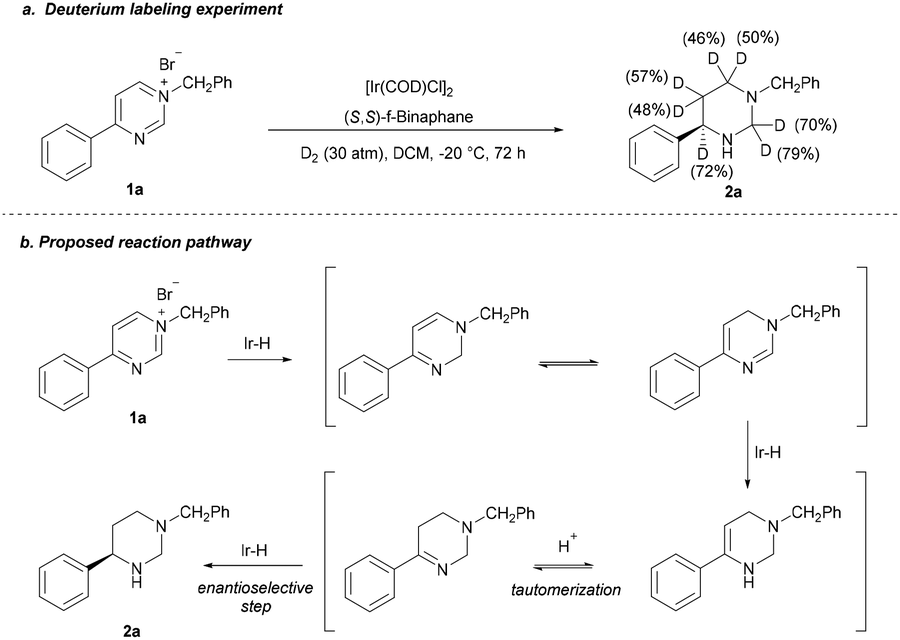 | ||
| Scheme 4 Deuterium labeling experiment and the proposed reaction pathway. | ||
Recently, flow chemistry gained significant attention due to its benefits, like short residence time, high surface area-to-volume ratio, excellent reproducibility and easy scale-up.33–36 Importantly, hydrogen gas is highly flammable and can easily form explosive mixtures with oxygen in the air. In a continuous flow system, hydrogen gas can be handled and controlled more safely, reducing potential hazards.37 Therefore, the iridium-catalyzed asymmetric and complete hydrogenation of pyrimidinium salt 1a was also applied to the continuous flow system (Table 2). Due to the solubility of the substrate, the exploration of solvent effects revealed that mixed solvent DCM/CHCl3 was the best choice with 87% ee (Table 2, entries 1–4), although a large amount of partially hydrogenated byproduct 9 was produced. Next, we increased back pressure and reaction temperature, also extended the reaction time. To our delight, higher selectivity for 2a was observed (Table 2, entries 5–7), where the ratio of 2a to 9 was up to 10:1 (Table 2, entry 7). However, only moderate enantioselectivity was obtained (Table 2, entries 5–7, 77%–84% ee). Surprisingly, when the temperature was reduced to 50 °C and the gas flow rate was increased to 120 sccm, 94% ee was obtained with almost no by-product (Table 2, entry 8, 2a:9 > 20:1). Furthermore, by increasing the concentration of the reaction solution to 0.05 M, the desired product 2a was obtained in high yield with excellent enantioselectivity at 50 °C and 8 MPa H2 within 25.0 min (Table 2, entry 9). Subsequently, a gram-scale continuous production was performed, resulting in the production of 3.5 g of product 2a with 92% yield and 95% ee within 10.0 h.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Conc. (M) | Gas (sccm) | Liquid (mL min−1) | Solvent | BPR (MPa) | T ( °C) | t (min) | Conv.b (%) |
2a:9 |
eec (%) |
|
a Reaction condition: DCM/CHCl3 = 4:1, [Ir(COD)Cl]2 (1.0 mol%), ligand (2.2 mol%).
b Determined by chiral 1H NMR.
c Determined by chiral HPLC.
|
||||||||||
| 1 | 0.03 | 32 | 0.5 | CHCl3 | 4 | 60 | 19.2 | >95 | 1:20 |
— |
| 2 | 0.03 | 32 | 0.5 | PhMe + MeOH | 4 | 60 | 19.2 | >95 | 2:7 |
80 |
| 3 | 0.03 | 32 | 0.5 | DCM + MeOH | 4 | 60 | 19.2 | >95 | 4:3 |
80 |
| 4 | 0.03 | 32 | 0.5 | DCM + CHCl3 | 4 | 60 | 19.2 | >95 | 1:7 |
87 |
| 5 | 0.03 | 48 | 0.5 | DCM + CHCl3 | 6 | 60 | 38.4 | >95 | 9:2 |
84 |
| 6 | 0.03 | 48 | 0.5 | DCM + CHCl3 | 6 | 80 | 38.4 | >95 | 7:1 |
78 |
| 7 | 0.03 | 64 | 0.5 | DCM + CHCl3 | 8 | 80 | 38.4 | >95 | 10:1 |
77 |
| 8 | 0.03 | 120 | 0.5 | DCM + CHCl3 | 8 | 50 | 25.0 | >95 | >20:1 |
94 |
| 9 | 0.05 | 120 | 0.5 | DCM + CHCl3 | 8 | 50 | 25.0 | >95 | >20:1 |
95 |
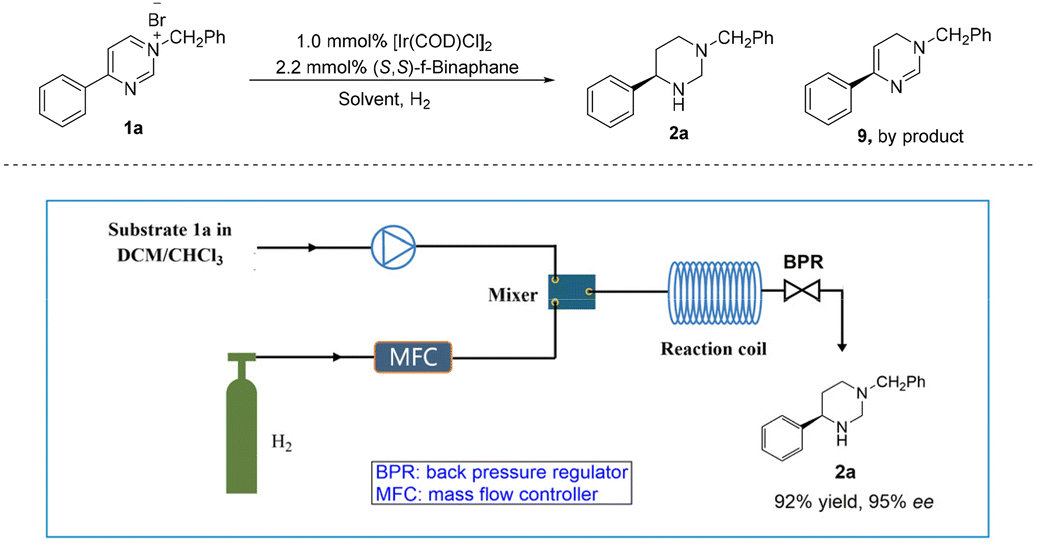
Conclusions
In summary, we have successfully developed a highly efficient iridium-catalyzed complete AH of 4-substituted pyrimidinium salts without using additives. This approach facilitates the synthesis of a diverse array of fully saturated chiral hexahydropyrimidines with significant yields (up to 95% yield) and high enantioselectivities (up to 97% ee). Additionally, AH at the gram scale under batch and flow were both conducted efficiently with satisfactory reactivity and enantioselectivity. Notably, the chiral hexahydropyrimidine can be converted into other functional groups. Certainly, the progress achieved herein extends the utility of AH in the context of aromatic heterocycles and enriches the methods for the synthesis of chiral amines. Further research regarding AH of heteroarenes and imines are currently underway in our laboratory, and the results will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (U21A20278).References
- D. Xia, Z.-H. Wang, J.-M. Jiang, X.-W. Yang, Y. Gao, Y.-Y. Xu, L.-Y. Chang, D. Zhu, B.-J. Zhao, X.-L. Zhu, J. Zhang, Z.-Q. Yin and K. Pan, Org. Lett., 2022, 24, 4684–4688 CrossRef CAS PubMed.
- S. Liu, X. Han, Z. Jiang, G. Wu, X. Hu, X. You, J. Jiang, Y. Zhang and C. Sun, J. Antibiot., 2016, 69, 769–772 CrossRef CAS PubMed.
- S. Tsukaguchi, M. Enomoto, R. Towada, Y. Ogura and S. Kuwahara, Eur. J. Org. Chem., 2019, 6110–6116 CrossRef CAS.
- J. C. Barrow, P. G. Nantermet, H. G. Selnick, K. L. Glass, K. E. Rittle, K. F. Gilbert, T. G. Steele, C. F. Homnick, R. M. Freidinger, R. W. Ransom, P. Kling, D. Reiss, T. P. Broten, T. W. Schorn, R. S. L. Chang, S. S. O'Malley, T. V. Olah, J. D. Ellis, A. Barrish, K. Kassahun, P. Leppert, D. Nagarathnam and C. Forray, J. Med. Chem., 2000, 43, 2703–2718 CrossRef CAS PubMed.
- G. V. De Lucca, J. Liang and I. De Lucca, J. Med. Chem., 1999, 42, 135–152 CrossRef CAS PubMed.
- D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- A. Cabré, X. Verdaguer and A. Riera, Chem. Rev., 2022, 122, 269–339 CrossRef PubMed.
- A. N. Kim and B. M. Stoltz, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS PubMed.
- P. Tang, H. Wang, W. Zhang and F. Chen, Green Synth. Catal., 2020, 1, 26–41 CrossRef.
- R. Gunasekar, R. L. Goodyear, I. P. Silvestri and J. Xiao, Org. Biomol. Chem., 2022, 20, 1794–1827 RSC.
- R. Kuwano, Y. Hashiguchi, R. Ikeda and K. Ishizuka, Angew. Chem., Int. Ed., 2015, 54, 2393–2396 CrossRef CAS PubMed.
- G.-S. Feng, M.-W. Chen, L. Shi and Y.-G. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5853–5857 CrossRef CAS PubMed.
- G.-S. Feng, L. Shi, F.-J. Meng, M.-W. Chen and Y.-G. Zhou, Org. Lett., 2018, 20, 6415–6419 CrossRef CAS PubMed.
- F.-J. Meng, L. Shi, G.-S. Feng, L. Sun and Y.-G. Zhou, J. Org. Chem., 2019, 84, 4435–4442 CrossRef CAS PubMed.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
- G. Hou, F. Gosselin, W. Li, C. McWilliams, Y. Sun, M. Weisel, P. D. O'Shea, C.-Y. Chen, I. W. Davies and X. Zhang, J. Am. Chem. Soc., 2009, 131, 9882–9883 CrossRef CAS PubMed.
- G. Hou, W. Li, M. Ma, X. W. Zhang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 12844–12846 CrossRef CAS PubMed.
- G. Hou, R. Tao, Y. Sun, X. Zhang and F. Gosselin, J. Am. Chem. Soc., 2010, 132, 2124–2125 CrossRef CAS PubMed.
- For a review on different substrate activation methods for the catalytic asymmetric hydrogenation of N-heteroarenes, see: B. Balakrishna, J. L. Núñez-Rico and A. Vidal-Ferran, Eur. J. Org. Chem., 2015, 5293–5303 CrossRef CAS.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. E. Chan, Chem. Biol. Drug Des., 2014, 83, 450–461 CrossRef CAS PubMed.
- W. Zhang, X. Chen, Y. An, J. Wang, C. Zhuang, P. Tang and F. Chen, Chem. – Eur. J., 2020, 26, 10439–10443 CrossRef CAS PubMed.
- W. Li, M. Jiang, W. Chen, Y. Chen, Z. Yang, P. Tang and F. Chen, J. Org. Chem., 2021, 86, 8143–8153 CrossRef CAS PubMed.
- W. Zhang, Z. Wang, G. Lin, Y. Xue, M. Wu, P. Tang and F. Chen, Org. Lett., 2022, 24, 2409–2413 CrossRef CAS PubMed.
- W. Chen, X. Yi, H. Qu, Y. Chen, P. Tang and F. Chen, Chin. Chem. Lett., 2022, 33, 5080–5083 CrossRef CAS.
- W. Zhang, Y. Xue, S. Konduri, G. Lin, M. Wu, P. Tang and F. Chen, Green Synth. Catal., 2022, 3, 291–293 CrossRef.
- Z. Yang, Y. Chen, L. Wan, X. Cen, P. Tang and F. Chen, Chem. Commun., 2022, 58, 10869–10872 RSC.
- G. Facchetti, R. Gandolfi, M. Fusè, D. Zerla, E. Cesarotti, M. Pellizzoni and I. Rimoldi, New J. Chem., 2015, 39, 3792–3800 RSC.
- W.-X. Huang, L.-J. Liu, B. Wu, G.-S. Feng, B. Wang and Y.-G. Zhou, Org. Lett., 2016, 18, 3082–3085 CrossRef CAS PubMed.
- B. Qu, H. P. R. Mangunuru, S. Tcyrulnikov, D. Rivalti, O. V. Zatolochnaya, D. Kurouski, S. Radomkit, S. Biswas, S. Karyakarte, K. R. Fandrick, J. D. Sieber, S. Rodriguez, J.-N. Desrosiers, N. Haddad, K. McKellop, S. Pennino, H. Lee, N. K. Yee, J. J. Song, M. C. Kozlowski and C. H. Senanayake, Org. Lett., 2018, 20, 1333–1337 CrossRef CAS PubMed.
- L.-S. Zheng, F. Wang, X.-Y. Ye, G.-Q. Chen and X. Zhang, Org. Lett., 2020, 22, 8882–8887 CrossRef CAS.
- Z.-S. Ye, R.-N. Guo, X.-F. Cai, M.-W. Chen, L. Shi and Y.-G. Zhou, Angew. Chem., Int. Ed., 2013, 52, 3685–3689 CrossRef CAS PubMed.
- C. Wiles and P. Watts, Green Chem., 2014, 16, 55–62 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- J. Liao, S. Zhang, Z. Wang, X. Song, D. Zhang, R. Kumar, J. Jin, P. Ren, H. You and F.-E. Chen, Green Synth. Catal., 2020, 1, 121–133 CrossRef.
- A. Xu, C. Li, J. Huang, P. Hang, C. Zhao, L. Song, H. You, X. Zhang and F.-E. Chen, Chem. Sci., 2023, 14, 9024–9032 RSC.
- R. Porcar, A. Mollar-Cuni, D. Ventura-Espinosa, S. V. Luis, E. García-Verdugo and J. A. Mata, Green Chem., 2022, 24, 2036–2043 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2239321. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc04364a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |