
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cycloaddition–dehydration continuous flow chemistry for renewable para-xylene production from 2,5-dimethylfuran and ethylene over phosphorous-decorated zeolite beta†
Zhaoxing
Wang
 a,
Tejas
Goculdas
ab,
Yung Wei
Hsiao
a,
Wei
Fan
bc and
Dionisios G.
Vlachos
*ab
a,
Tejas
Goculdas
ab,
Yung Wei
Hsiao
a,
Wei
Fan
bc and
Dionisios G.
Vlachos
*ab
aDepartment of Chemical and Biomolecular Engineering, 151 Academy St, University of Delaware, Newark, DE 19716, USA. E-mail: vlachos@udel.edu
bCatalysis Center for Energy Innovation, Delaware Energy Institute (DEI), 221 Academy St, University of Delaware, Newark, DE 19716, USA
cDepartment of Chemical Engineering, University of Massachusetts, Amherst, MA 01003, USA
First published on 3rd July 2024
Abstract
Continuous manufacturing of platform chemicals from lignocellulose is highly desirable for a fossil fuel independent future. We demonstrate highly selective production of para-xylene (pX) from ethylene and 2,5-dimethylfuran (DMF) in a packed bed microreactor using phosphorous-decorated zeolite beta (P-BEA), with pX selectivity up to 97% at 80% DMF conversion. We map the effect of reactor temperature, space velocity, concentration, gas-to-liquid ratio, and process pressure. Time-on-stream (TOS) and in situ regeneration studies show minimal productivity degradation over ∼5 h TOS and full productivity restoration upon regeneration for multiple cycles. Most non-selective Brønsted acidity occurs at low TOS and is attributed to the remaining trace Al bridge site. External mass transfer limitations are implicated at low space velocities. We combine the TOS data with NMR, XRD, and Raman to develop structure–performance insights into the catalyst behavior. A comparison with mesoporous P-supported materials illustrates that P-BEA is an excellent catalyst for size selectivity and long-term stability.
Introduction
Lignocellulosic biomass upgrading has the potential to produce low carbon emissions fuels and chemicals and enhance energy security.1 Among the pathways in valorizing biomass, lignocellulose-derived furans, such as furfural and 5-hydroxymethylfurfural (HMF) from the C5 and C6 sugars, have been studied intensively. The tandem cycloaddition and dehydration of 2,5-dimethylfuran (DMF), a key platform chemical derived from HMF, can produce para-xylene (pX), as shown in Fig. 1, an essential fossil fuel-dependent polymer precursor.2,3 The catalytic conversion of DMF and ethylene to pX forms an oxanorbornene cycloadduct via a Diels–Alder reaction, which is dehydrated to water and pX. The Diels–Alder step with ethylene is difficult but can still proceed homogeneously or over Lewis acid site (LAS) catalysts.4–10 At low Brønsted acid site concentrations, the second step typically controls the rate; conversely, the Diels–Alder cycloaddition controls the reaction at higher Brønsted acid site (BAS) concentrations.8 Moreover, the cycloaddition reaction is equilibrium controlled, and the retro Diels–Alder is favored at high reaction temperatures.11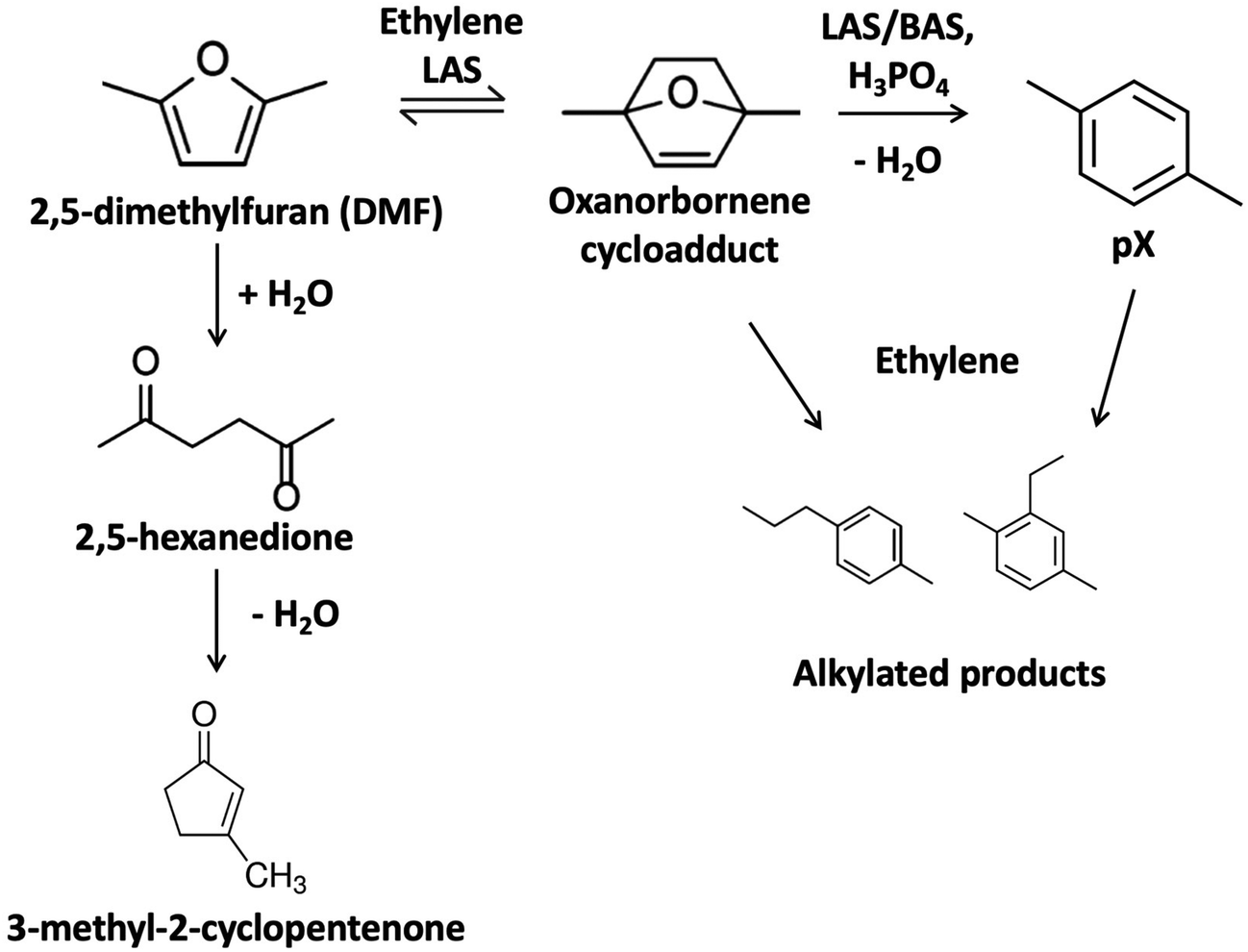 | ||
| Fig. 1 Cycloaddition of 5-dimethylfuran (DMF) and ethylene and dehydration of the oxanorbornene cycloadduct to form pX. | ||
Cho et al. demonstrated the highest pX yield of 97% in batch mode using the P-BEA catalyst prepared by dealumination of the CP814E commercial zeolite beta and incorporation of phosphoric acid.12 Gulbinski et al. further showed the interplay between phosphoric acid and the silica support for high pX yields in batch.13 Kim et al. investigated mesoporous titanium phosphate catalysts for converting biomass-derived furans to pX and toluene via Diels–Alder cycloaddition. Hydrothermally synthesized MTiP catalysts, particularly MTiP-E, showed superior performance in converting DMF and ethylene to pX, with over 90% selectivity and conversion. The same catalysts also efficiently converted 2-MF to toluene.16 Feng et al. demonstrated DMF and ethylene to pX chemistry over tin phosphate catalysts. The SnPO catalysts, featuring tetrahedrally coordinated Sn(IV) active sites, achieved a high pX yield of 93% after 18 h in batch. The superior catalytic performance of these SnPO catalysts was attributed to the enhanced main reaction via adjustable acid sites upon varying the P/Sn molar ratio, improved mass transfer efficiency, and carbon tolerance stemming from the mesoporous support structure.17 Wu et al. investigated the same reaction using zirconium phosphate (ZrP-x) catalysts. ZrP-2.5 achieved a pX yield of 89.2% at 72.3 mmol pX per g per h at 250 °C after 12 h. This high performance was attributed to a balance of Brønsted and Lewis acid sites and the catalyst's mesoporous structure.18
Large-scale implementation of biomass conversion requires continuous flow reactors for the manufacturing of chemicals. From a process development perspective, flow reactors allow TOS data collection, a better understanding of catalyst deactivation, and higher temperatures with shorter residence times, mostly unattainable by batch reactors. The distributed valorization of lignocellulosic biomass can also benefit from continuous operation of flow reactors, reducing the demand for cycling batch reactors. Prior works exploring the feasibility of flow chemistry have been reported. Yu et al. demonstrated for the first time this tandem reaction in a continuous flow reactor using H-, Sn-, and Zr-BEA catalysts. They found that both Brønsted and Lewis acid sites can catalyze the oxanorbornene dehydration reaction at similar rates, with Sn-BEA achieving the highest pX selectivity of 94% at 7.5% DMF conversion and the highest DMF conversion of 35% with 74% selectivity at 423 °C.14 The Brønsted acid sites in H-BEA were unstable, primarily due to oligomerizing DMF and 2,5-hexanedione.14 In another continuous flow reactor effort, Mendoza Mesa et al.15 proposed using liquid acrylic acid as the dienophile instead of ethylene and achieved over 80% pX selectivity over H-BEA (Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al = 150) using 0.3 M DMF in n-heptane for over 8 h time-on-stream (TOS). These works show the potential to further enhance pX chemistry productivity via catalyst optimization and rational reactor design. In this work, we explore pX production from ethylene and DMF in a gas/liquid three phase packed bed microreactor using phosphorous-decorated zeolite beta (P-BEA). We demonstrate high performance compared to previous flow reactor experiments with pX selectivity up to 97% at 80% DMF conversion. We map the effect of reactor temperature, space velocity, concentration, gas-to-liquid ratio, process pressure, and TOS. The near complete dealumination of zeolite beta in this work, coupled with flow reactor experiments, reveals transient catalyst behavior not typically seen in batch reactors due to prolonged startup and heating times typical of bench-scale batch reactors. The remnant aluminum bridge sites are strong Brønsted acids that lead to nonspecific reactions seen in the flow reactor, as the early collectible liquid outlet stream experiences all possible active sites.
:Al = 150) using 0.3 M DMF in n-heptane for over 8 h time-on-stream (TOS). These works show the potential to further enhance pX chemistry productivity via catalyst optimization and rational reactor design. In this work, we explore pX production from ethylene and DMF in a gas/liquid three phase packed bed microreactor using phosphorous-decorated zeolite beta (P-BEA). We demonstrate high performance compared to previous flow reactor experiments with pX selectivity up to 97% at 80% DMF conversion. We map the effect of reactor temperature, space velocity, concentration, gas-to-liquid ratio, process pressure, and TOS. The near complete dealumination of zeolite beta in this work, coupled with flow reactor experiments, reveals transient catalyst behavior not typically seen in batch reactors due to prolonged startup and heating times typical of bench-scale batch reactors. The remnant aluminum bridge sites are strong Brønsted acids that lead to nonspecific reactions seen in the flow reactor, as the early collectible liquid outlet stream experiences all possible active sites.
Experimental methods
Catalyst performance evaluation
A typical reaction used a 152 mm × 4.6 mm ID stainless steel tube as the reactor. P-BEA and P-SBA-15 catalysts were pelletized into 150–250 µm pellets after pressing in an ½ inch I.D. hydraulic press die under 3 metric tons. Glass beads (150–212 µm, Sigma Aldrich) and quartz wool were used to pack the reactor. A syringe pump (Chemyx Fusion 6000-X) and a 20 mm stainless steel syringe (Chemyx) provided liquid feed at pressure. A mass flow controller (Brooks Instrument 5850) regulated the ethylene gas flow (99.999%, Matheson). The gas and liquid sides of the flow path consist of 1/16″ OD, 0.02″ ID tubing that join in a 1/16″ tee (Swagelok SS-100-3) from orthogonal directions. An HP 5890 series II gas chromatograph was used to heat the preheating coil (SS316, 1/16″ OD, 1.00 m × 0.02″ ID, Restek), and an Equilibar ZF back pressure regulator (BPR) was used to maintain the pressure. The liquid outflow was collected with 300 µL gas chromatograph autosampler vials immediately downstream of the BPR upon cooling to room temperature within the sampling vial.During a typical flow reaction (Fig. 2), the reactor was first heated to the reaction temperature under slow N2 flow at 1 atm, then pressurized with ethylene to the desired process pressure before starting the gas and liquid flows immediately after. The first liquid samples exit the outlet after ∼40 minutes. The gas is automatically separated from the liquid upon cooling and depressurization. The liquid product was characterized on an Agilent 7890B gas chromatograph (GC) equipped with an HP-INNOWAX column (30 m length, 0.25 mm film thickness, 0.25 µm ID). An Agilent GCMS with an identical HP-INNOWAX column was used to identify product species (Fig. S1†). In all samples with high pX yield, meta-xylene (mX) was also observed in the gas chromatogram at approximately 2% of the pX yield. In samples with >1% yield of alkylated products, 2-ethyl-p-xylene was the dominant product and was considered a model compound of alkylation reactions beyond the primary reaction pathway. DMF (Acros Organics, 99%) and pX (Sigma-Aldrich, 99%) in n-heptane (Fisher Scientific, 99%) external calibration curves were created for the concentration ranges relevant to this work. The pX calibration curves were used for the alkylated products. For in situ catalyst regeneration, the reactor column was placed in a tube furnace; its inlet was connected to flowing air at 15 sccm at 1 atm, heated to 550 °C at 10 °C min−1, and held for 5 h. In these experiments, the conversion, yield, selectivity, and carbon balance are evaluated as follows:
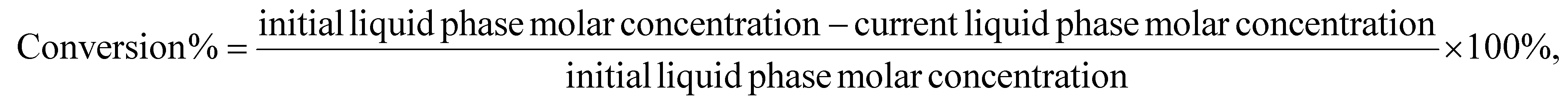

and
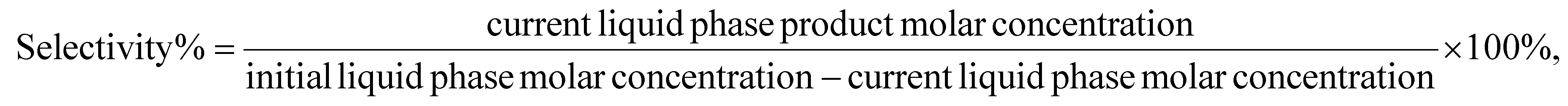

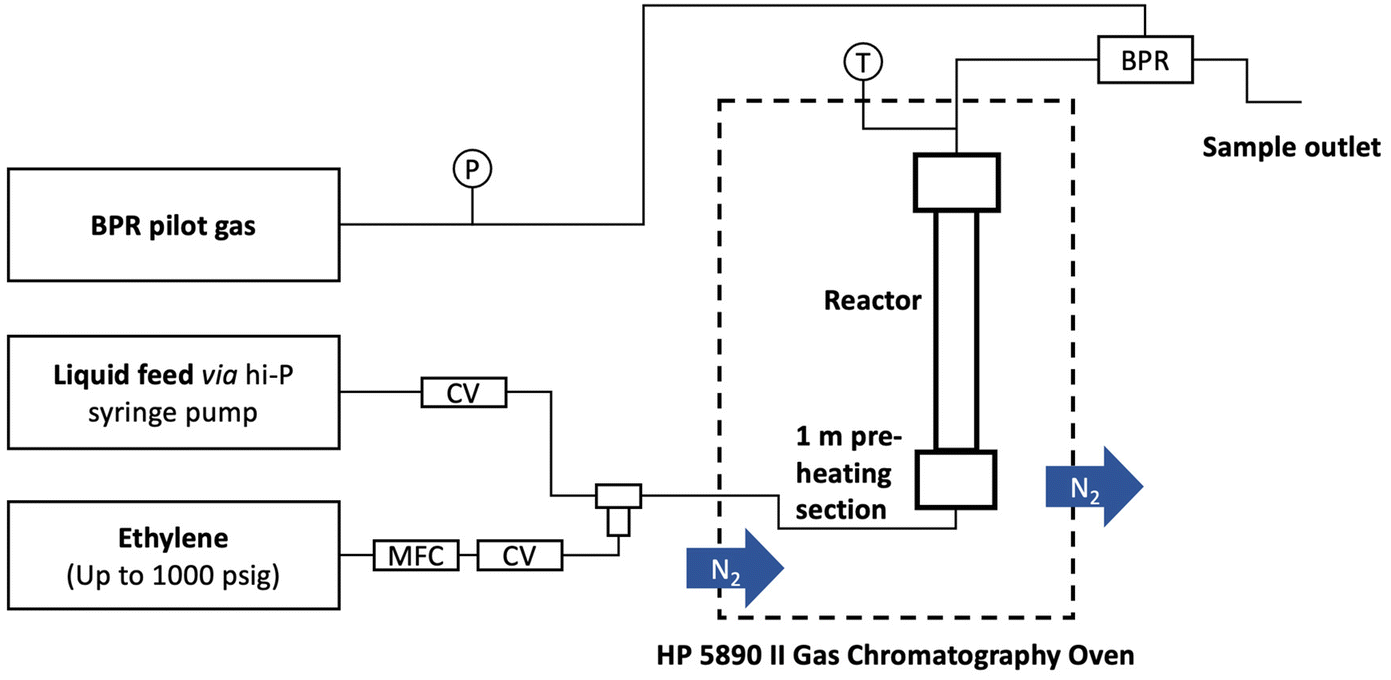 | ||
| Fig. 2 Flow chemistry setup. | ||
Catalyst preparation
Briefly, phosphoric acid-decorated, dealuminated zeolite beta (P-deAlBEA, Si/P = 25) was prepared by dealuminating commercial zeolite beta (Zeolyst CP814E, Si/Al = 12.5) using nitric acid at 95 °C for 24 h and wetness impregnation of phosphoric acid followed by calcination at 600 °C under airflow, similar to the works by Cho et al. and Gulbinski et al.12,13 ICP analysis suggests near-complete dealumination, with Si:Al ratios exceeding 900 for all catalyst samples. P-SiO2 and P-SBA-15 were prepared via incipient wetness impregnation on the respective parent support with 6 nm pore size, followed by calcination in static air at 500 °C for 3 h, as reported elsewhere.19 For flow reactions, all catalysts except P-SiO2 (150–212 µm silica gel) were pelletized to 150–250 µm after pressing under 30000 psi.
Catalyst characterization
N2 adsorption–desorption was performed at 77.359 K on a Micromeritics ASAP 2000 Brunauer Emmett Teller (BET) analyzer. X-ray diffraction (XRD) of the catalysts was acquired on a Bruker D8 powder diffractometer equipped with monochromatic Cu-Kα radiation at λ = 1.54 Å with a 45 kV acceleration voltage and 40 mA current. Samples were scanned between 4 and 40° (2θ) at a 0.05° s−1 scan rate using a 1.2 mm tall, 7 mm wide slit. Scanning electron microscopy (SEM) was performed using a Zeiss Auriga 60 high resolution focused ion beam & scanning electron microscope. For elemental analysis, ICP-OES was performed by Galbraith Laboratories using the GLI Procedure ME-70. Thermal analysis data was recorded with a thermogravimetric analyzer (Discovery TGA, TA Instruments). Approximately 10 mg of powdered sample was equilibrated at 30 °C on a platinum wafer before heated to 700 °C at a heating rate of 10 °C min−1 in air at 20 mL min−1. Fresh and spent catalyst pellets were pressed onto glass slides for Raman analysis, and a Horiba LabRAM HR Evolution Raman spectrometer with a Syncerity OE detector was used with λ0 = 325 nm. An acquisition time of 30 s was used over 3 accumulations. All solid-state (SS)-NMR spectroscopy measurements were carried out on a Bruker AVANCE III HD spectrometer operating at a proton resonance frequency of 600.13 MHz. Samples were prepared by mixing the catalyst sample in a 1:10 ratio with quartz pellets. The mixture was then spun at the magic angle of 54.74° in a 3.2 mm zirconia rotor with a K-FEL cap at 14 kHz. A direct polarization pulse sequence was used with a 3.65 μs pulse and a recycle delay of 2 seconds. For each spectrum, 512 scans were collected and averaged at room temperature.
Results and discussion
Catalyst evaluation and optimization
First, we employed P-BEA (Si:P = 25, denoted as P-BEA(25) below) to reproduce the highest batch performance reported in the literature of 94% pX yield at 98% DMF conversion after 24 h in batch at 250 °C.12,13 A typical chromatogram is shown in Fig. S1.† ICP, catalyst performance (ESI, Fig. S2†), XRD (below), and N2 adsorption–desorption (Fig. S3 and Table S1†) results corroborate with the catalyst similarities. The differences between batch and flow mode and headspace pose a challenge in comparing performance. Differences in flow and phase behavior, and inevitably mass transfer, necessitate re-optimizing the operating conditions of the flow reactor.
Our optimization efforts have led to optimum conditions near 750 psig and 375 °C. Increasing the pressure from 300 and 750 psig (Fig. 3a) led to monotonic pX yield and selectivity improvement. We attribute this to increased cycloadduct concentration at higher ethylene partial pressures, as the retro Diels–Alder reaction is heavily favored at higher temperatures.11 When changing the gas-to-liquid (G:L) flow ratio (Fig. 3b), a volcano-type behavior is seen as the pX yield and DMF conversion peak near a G:L ratio of 400 v/v. The increase in DMF conversion with increasing gas flow rate is probably due to enhanced external mass transfer and the decline at high flow rates to reduced residence time, while the pX selectivity increases monotonically due to the higher ethylene availability from enhanced external mass transfer. We use a G:L volume ratio of 400 for operational ease for subsequent experiments. We further illustrate this behavior by analyzing the Thiele modulus and Mears criteria in the ESI.†
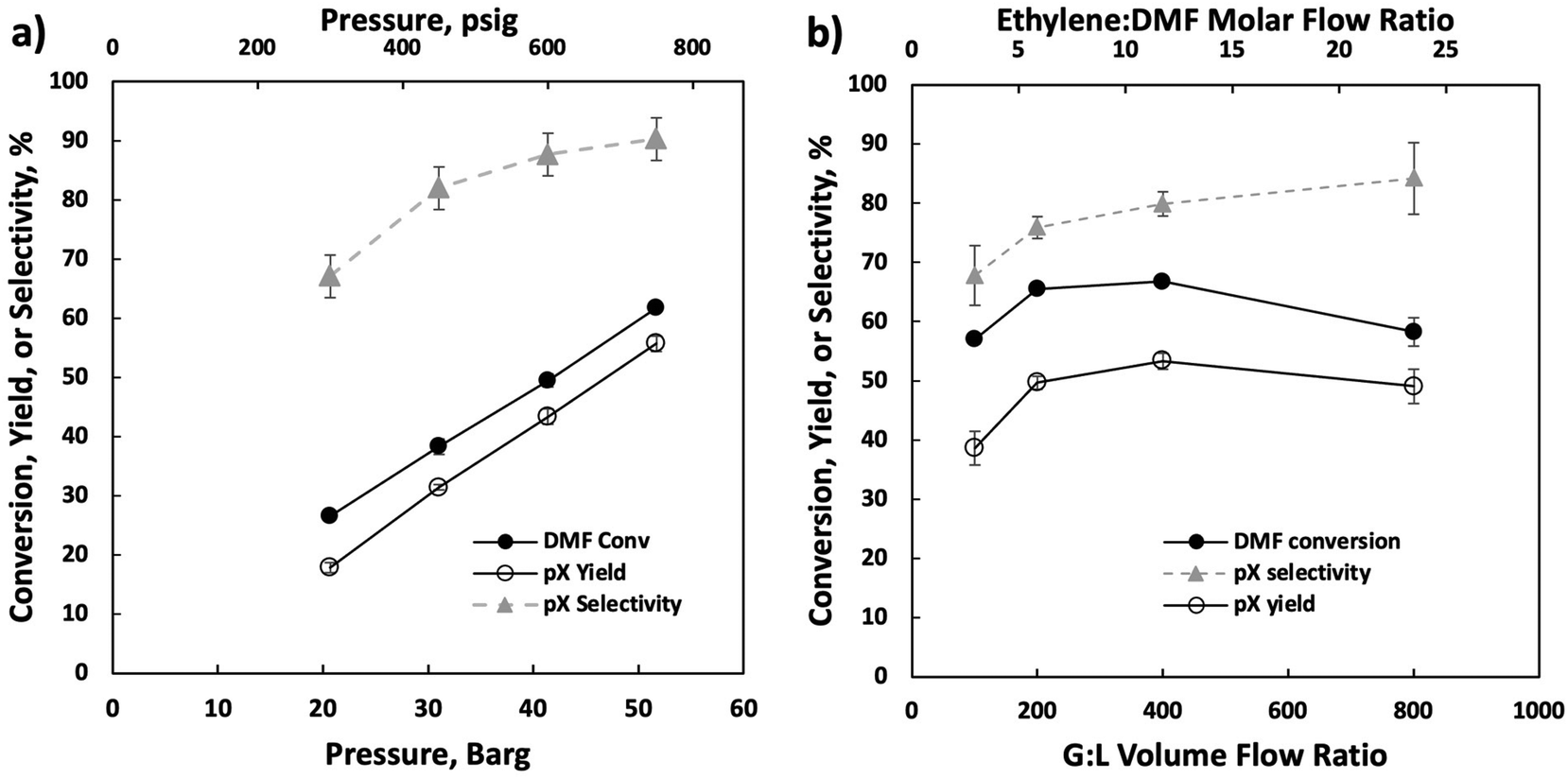 | ||
| Fig. 3 Performance vs. flow reactor pressure (a) and gas:liquid feed ratio (b). Conditions: 375 °C, 20 wt% DMF in n-heptane feed, 0.025 mL min−1 liquid feed. Only the gas flow rate was varied in changing the gas-to-liquid (G:L) ratio. | ||
We vary the weight hourly space velocity (WHSV) to optimize productivity further (Fig. 4). Specifically, 1/WHSV was increased to 9.3 h at varied DMF feed concentrations and temperatures. Interestingly, the DMF conversion, pX yield, and pX selectivity showed non-monotonic behavior, except for DMF conversion at 20 wt%, where DMF conversion increases monotonically with inverse WHSV, potentially due to non-selective reactions.
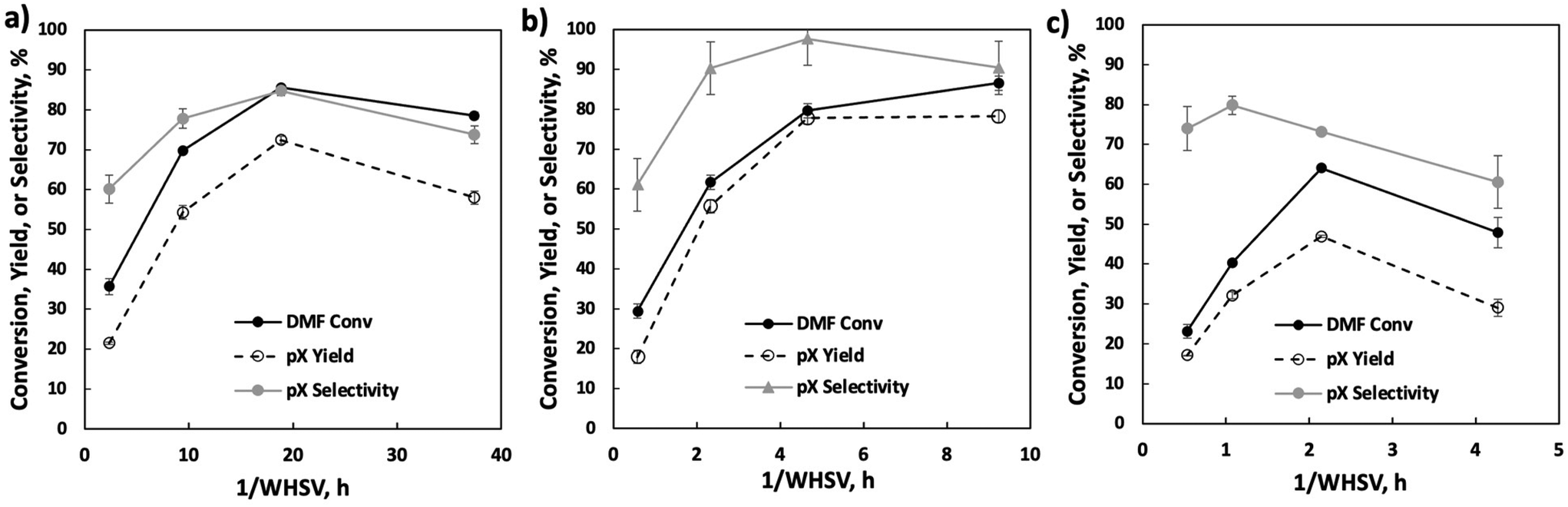 | ||
| Fig. 4 Reactor performance at various 1/WHSVs using 5 wt% (2.36 h, 9.43 h, 18.86 h, and 37.4 h, panel a), 20 wt% (0.57 h, 2.33 h, 4.66 h, and 9.24 h, panel b), and 40 wt% (0.54 h, 1.07 h, 2.15 h, and 4.26 h, panel c) DMF in n-heptane liquid feed. Conditions: 375 °C, 750 psig back pressure, 0.5 g P-BEA(25). | ||
To understand the process stream phase behavior under the experimental conditions in the flow reactor, COSMO-RS (Amsterdam Modeling Suite) and NRTL (ASPEN Plus) models were employed. Detailed simulation setup and results are discussed in the ESI Note 1.† The process streams are either gas phase or a gas-like supercritical phase. Short residence times at fast flows lead to high external mass transfer and limited contact time of the cycloadduct over the active site. Conversely, long residence times come with slow flow and mass transfer, and thus external mass transfer limitations. The concave curve shapes in Fig. 4 demarcate a clear transition from external mass transfer to potentially internal diffusion or kinetically limited with increasing space velocity.
Further productivity optimization included a 1/WHSV study with 20 wt% DMF feed at various temperatures (Fig. 5). A temperature of 375 °C achieves the highest pX selectivity. DMF appears particularly prone to non-selective condensation reactions at 400 °C, as indicated by the early drop in selectivity at 1/WHSV above 2 h, lack of quantifiable liquid phase byproducts, and a product mixture turning brown (inset in Fig. 5).
 | ||
| Fig. 5 Reactor performance at various 1/WHSVs at 350 °C (1.15 h, 2.31 h, 4.61 h, and 9.15 h, panel a), 375 °C (0.57 h, 2.33 h, 4.66 h, and 9.24 h, panel b), and 400 °C (1.18 h, 2.35 h, 4.70 h, and 9.33 h, panel c). Conditions: 750 psig back pressure, 0.5 g P-BEA(25), 20 wt% DMF feed; 0.025 mL min−1 liquid flow rate. | ||
Phosphorous loading and support effects
Under fast gas flows, internal mass transfer limitation could be relevant, especially at higher temperatures. In this work, the P-BEA(25) catalyst shows a disordered distribution of zeolite micro aggregates with nano-crystallites 20–40 nm in size, as confirmed by SEM (Fig. S4†). Various works have been attempted to combat internal mass transfer, using for example hierarchical zeolite beta.15,20–23 We thus compare the stable-regime performance (>1 h TOS) of P-BEA(25) against phosphoric acid-impregnated mesoporous silica gel (6 nm mesopores) and SBA-15 (6 nm mesopores), P-BEA(12.5) equivalent to the initial Al content (Fig. 6).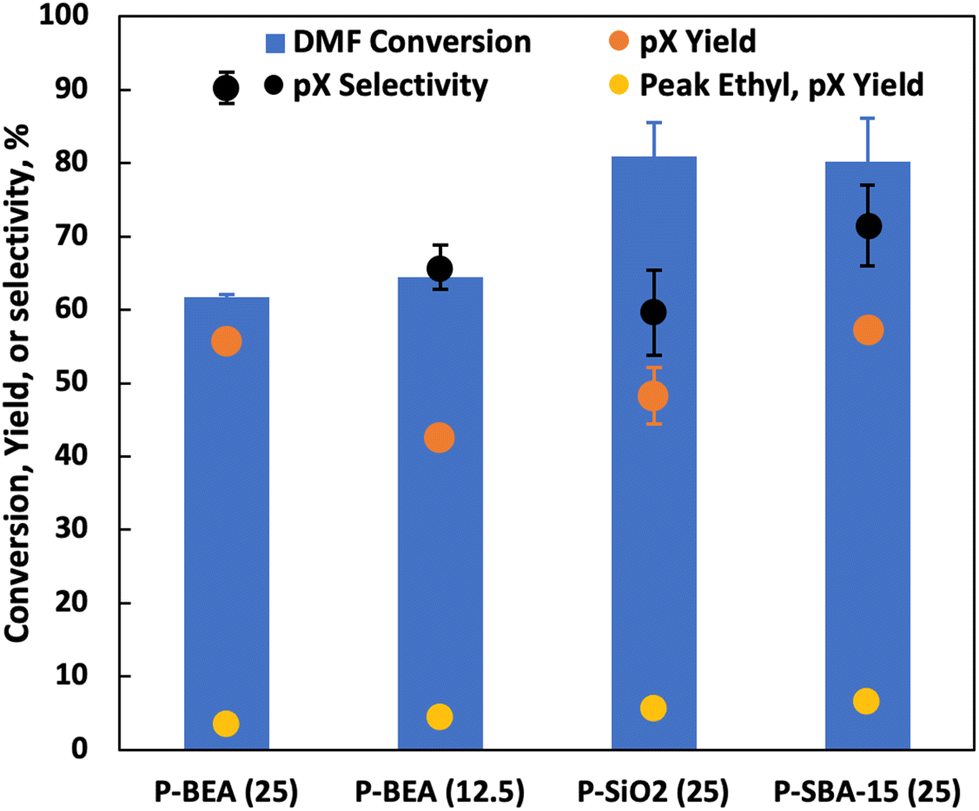 | ||
| Fig. 6 Performance of various P-catalysts at 375 °C, 20 wt% DMF in n-heptane, 750 psig, 0.025 mL min−1, and 10 sscm gas feed. P-BEA at two phosphoric acid loadings, 0.5 g of 150–250 µm pellets of P-SBA-15 (6 nm mesopores) at Si/P = 25 (same as P-BEA(25)), 150–212 µm SiO2 (6 nm mesopores as per Sigma Aldrich); this is not the most selective space velocity for P-BEA(25) (1/WHSV = 2.5 h vs. ∼5 h with 97% selectivity). All data taken after the 1st h of stabilization since the start of gas and liquid flows, except for the peak ethyl pX yield. | ||
Other phosphorous-decorated mesopore supports, P-SiO2 and P-SBA-15, show enhanced productivity of ethyl-p-xylene, with slightly reduced pX yield for P-SiO2(25) at 50% and P-SBA-15(25) show comparable pX yield (57% vs. 56%) to P-BEA (25). However, the reduced pX selectivity due to a higher occurrence of alkylated products results in lower pX selectivity for both P-SiO2(25) and P-SBA-15(25). Compared to P-BEA(25), P-BEA(12.5) exhibits comparable ethyl-p-xylene yield, a reduced pX yield, and a slightly higher DMF conversion. The GC-FID shows an abundance of other alkylated products. This could result from aggregated P sites giving rise to non-selective chemistry. High phosphoric acid concentrations in batch reactions have also reduced pX selectivity.13
Time-on-stream behavior of P-BEA
We further explore the productivity of the P-BEA(25) catalyst in extended TOS experiments (Fig. 7). As the pX yield stabilizes after 1 h, the pX selectivity improves with long TOS and resets with regeneration. Over longer times across two regeneration cycles, the DMF conversion reduces slightly, but the pX yield is unaffected after two regeneration cycles. After taking out small portions of the spent catalyst before regeneration, the pellets retain their shape while dark in appearance. Significant coking is indicated by TGA (Fig. 8), with 22.5 wt% loss at 700 °C after ∼10 h TOS. The spent catalyst shows minimal mass loss until 500 °C, where up to 22.5 wt% loss is seen after being heated to up to 700 °C, indicative of heavy/polyaromatic coke dominance. The catalyst stability is further demonstrated with TOS up to ∼60 h before the pressure drop across the column exceeds 30 psi, leading to liquid-only reactor outflow at significantly reduced DMF conversion. One cycle of in situ regeneration led to near-full restoration of productivity (Fig. S7†).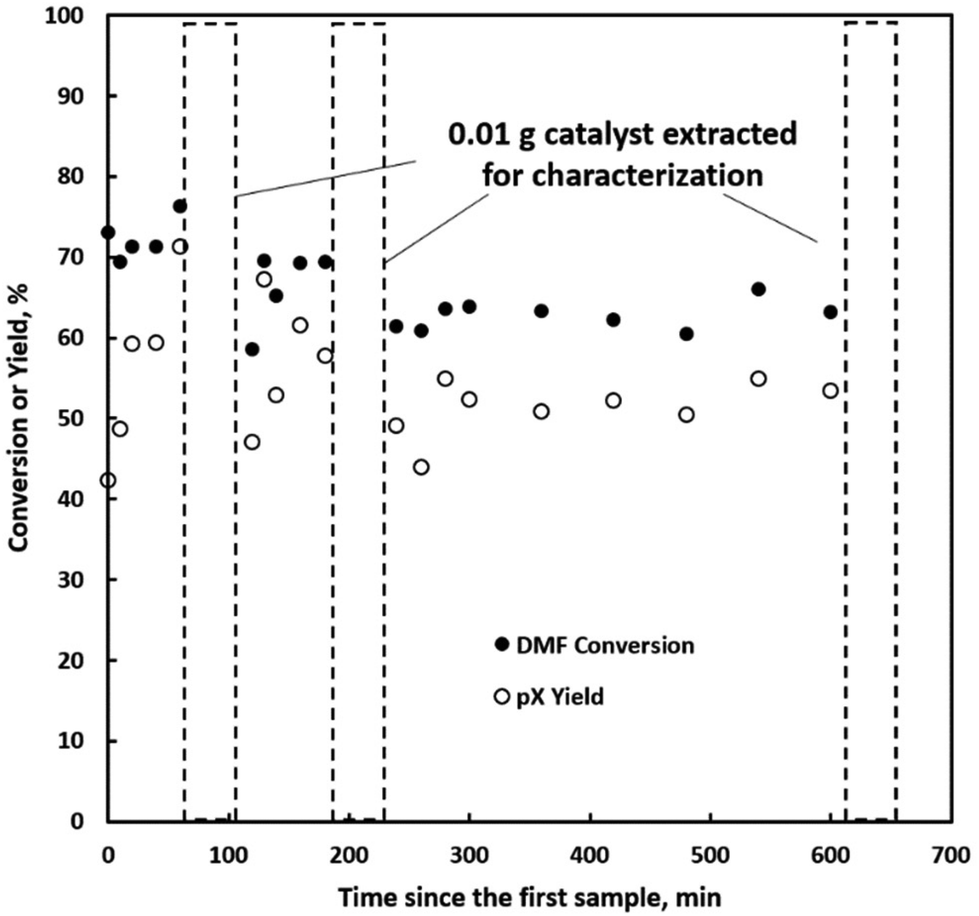 | ||
| Fig. 7 Time-on-stream (TOS) performance of P-BEA(25) with small portions of the catalyst taken out at 1, 2, and 8 h. The time after catalyst sampling was set to 50 min for easy visualization. Conditions: 375 °C, 20 wt% DMF in n-heptane, 750 psig, 0.025 mL min−1, 10 sccm gas feed, 0.5 g 150–250 µm pellets, 0.01 g catalyst taken out at each instant. | ||
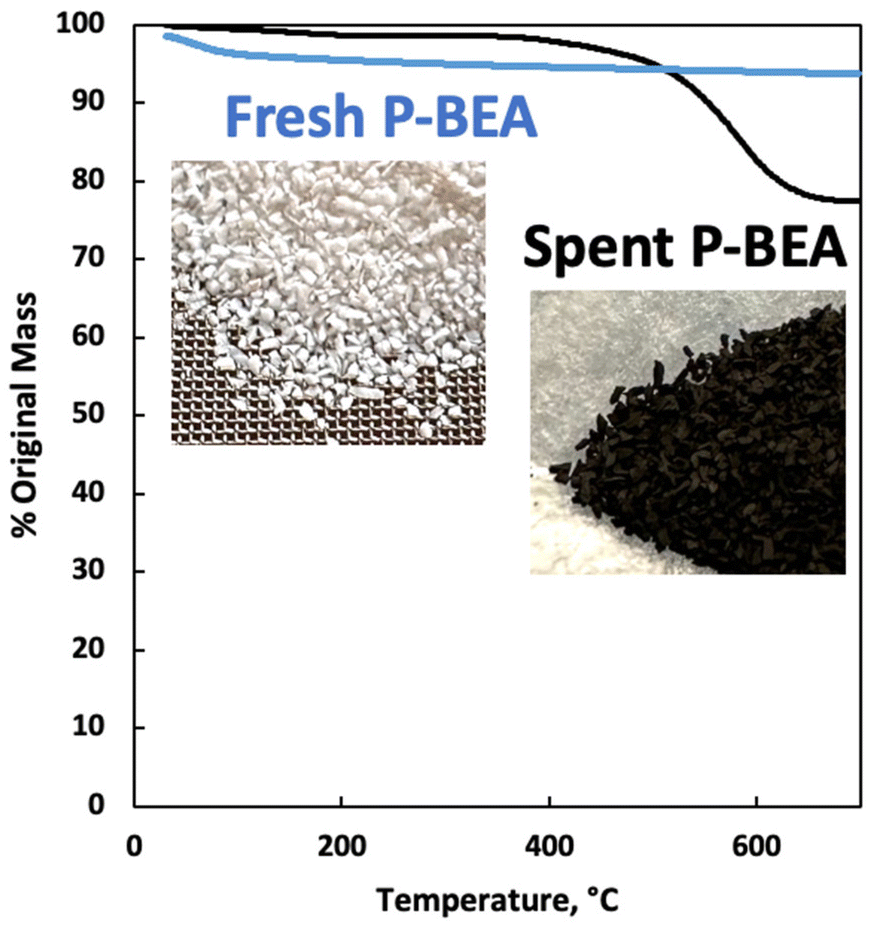 | ||
| Fig. 8 Thermogravimetric analysis (TGA) of fresh and spent P-BEA(25). 20 wt% DMF in feed. Catalyst after10 h overall TOS exposed between 250 °C and 425 °C. | ||
Continuous flow reactors provided ease of catalyst regeneration without the need to recover the catalysts after each batch reaction. In situ regeneration of the catalyst performed for multiple cycles shows little degradation in pX yield despite a slight decrease in DMF conversion (Fig. S5 & S6†). Motivated by this, we characterized small catalyst quantities (∼10 mg) near the end of the reactor at short TOS before resealing the reactor and resuming the reaction. A slight reduction in productivity over time is seen in Fig. 7, attributed to catalyst loss from sampling for characterization.
Raman spectroscopy on the fresh samples shows an utterly flat baseline between 600 and 1850 cm−1 (Fig. 9). Raman spectra on the spent catalysts show typical coking signatures. No significant change in coking between 1 and 8 h TOS and on different supports is seen. A strong signature of G (1620 cm−1) and D (broad peak at 1350 cm−1) bands without apparent differences in sub-signals from the D1–D4 bands, indicative of identical coking pathways with TOS, are seen.24 The same analysis on fresh and spent (2 h TOS) P-SiO2 and P-SBA15 shows that the spent spectra are nearly identical to P-BEA (Table 1), indicating a similar coking mechanism.25
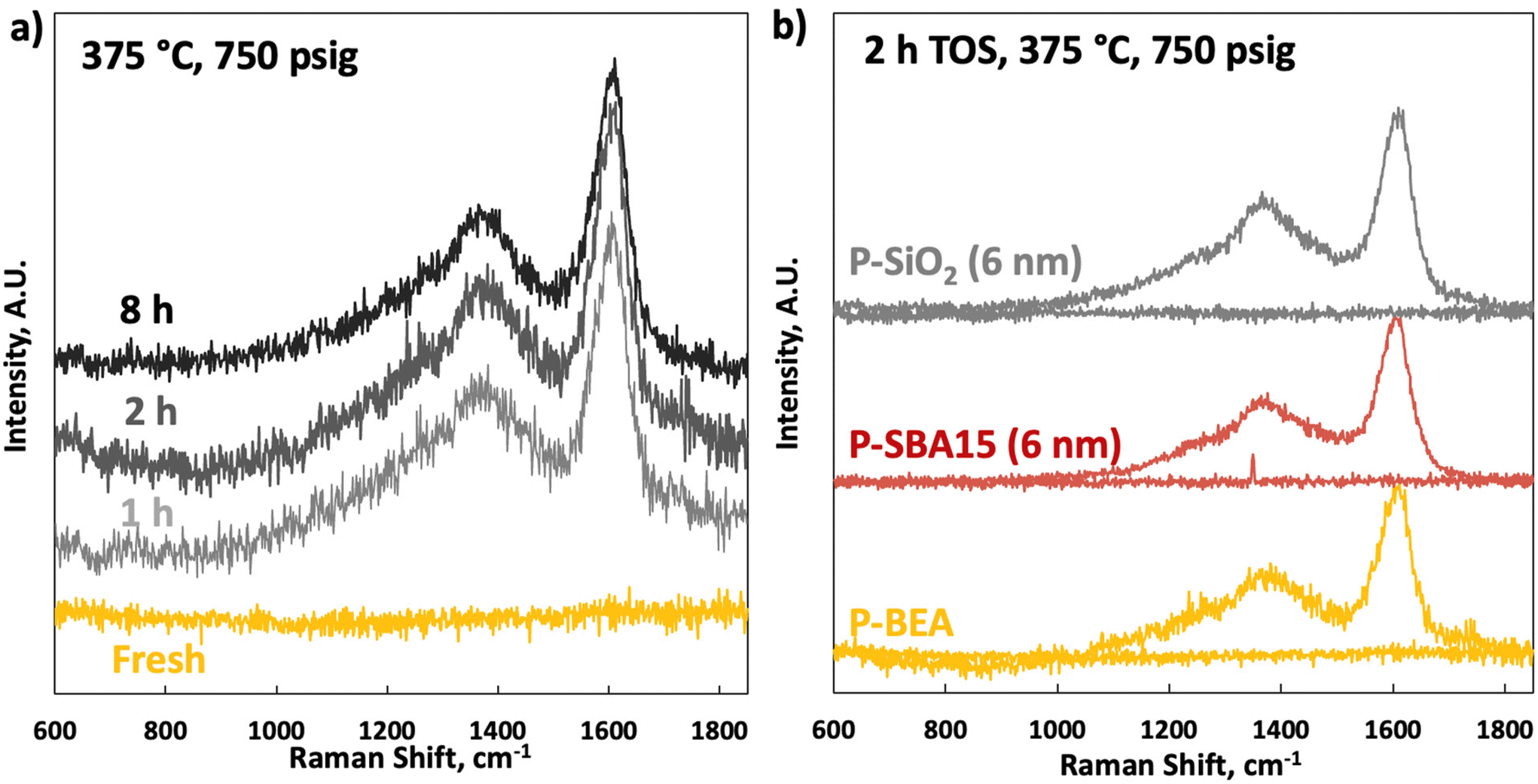 | ||
| Fig. 9 Raman spectra of (a) fresh P-BEA(25) and after 1, 2, and 8 h time on stream; and (b) fresh P-BEA, P-SBA-15 (6 nm mesopores) and P-SiO2 (6 nm mesopores) at Si/P = 25 loading, and after 2 h time on stream. Conditions: 375 °C, 20 wt% DMF in n-heptane, 750 psig, 0.025 mL min−1, 10 sccm gas feed, 0.5 g of 150–250 µm pellets, 1/WHSV of 2.5 h, 0.01 g catalyst taken out at each sampling point. | ||
| Sample | q0 (0 shift), % | q1 (−11 ppm), % | Q2, Q3, Q4 |
|---|---|---|---|
| Fresh | 73.4 | 26.6 | Not detected |
| 1 h TOS | 75.3 | 24.7 | |
| 2 h TOS | 72.9 | 27.1 | |
| 70 h TOS + regeneration | 57.8 | 42.2 |
We conducted solid-state 31P NMR on the P-BEA(25) catalysts to investigate the P speciation (Fig. 10). The intense signal at 0 ppm (q0) is from H3PO4, likely trapped in the micropores without chemical bonding. The −11 ppm q1 signal is from O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(OSi or OP)(OH)2. Signals indicating higher degree of P agglomeration, such OP(OSi or OP)2(OH) at approximately −24 ppm and OP(OP)m(OSi)n between −30 and −35 ppm, (m + n = 3), are absent.25,26 Comparison of fresh and TOS samples shows negligible change in P speciation over short TOS, except a slight reduction in peak sharpness at longer TOS. As minimal reactivity loss is seen and portions of the catalyst were removed from the reactor, the signal reduction is unlikely an effect of significant P loss. This indicates a low degree of P aggregation within the P-BEA(25) zeolite, and while the overall signal intensity decreases with increasing TOS, the relative q0/q1 ratio remains constant. The combination of modest P loading and low mesoporosity of P-BEA(25) catalyst gives high activity and stability compared to other supports and the higher loading of P-BEA(12.5). After approximately 70 h TOS and in situ catalyst regeneration, the q0/q1 ratio slightly decreases, reflecting potential P loss in the form of H3PO4 even though the catalyst activity is largely sustained post-regeneration.
P(OSi or OP)(OH)2. Signals indicating higher degree of P agglomeration, such OP(OSi or OP)2(OH) at approximately −24 ppm and OP(OP)m(OSi)n between −30 and −35 ppm, (m + n = 3), are absent.25,26 Comparison of fresh and TOS samples shows negligible change in P speciation over short TOS, except a slight reduction in peak sharpness at longer TOS. As minimal reactivity loss is seen and portions of the catalyst were removed from the reactor, the signal reduction is unlikely an effect of significant P loss. This indicates a low degree of P aggregation within the P-BEA(25) zeolite, and while the overall signal intensity decreases with increasing TOS, the relative q0/q1 ratio remains constant. The combination of modest P loading and low mesoporosity of P-BEA(25) catalyst gives high activity and stability compared to other supports and the higher loading of P-BEA(12.5). After approximately 70 h TOS and in situ catalyst regeneration, the q0/q1 ratio slightly decreases, reflecting potential P loss in the form of H3PO4 even though the catalyst activity is largely sustained post-regeneration.
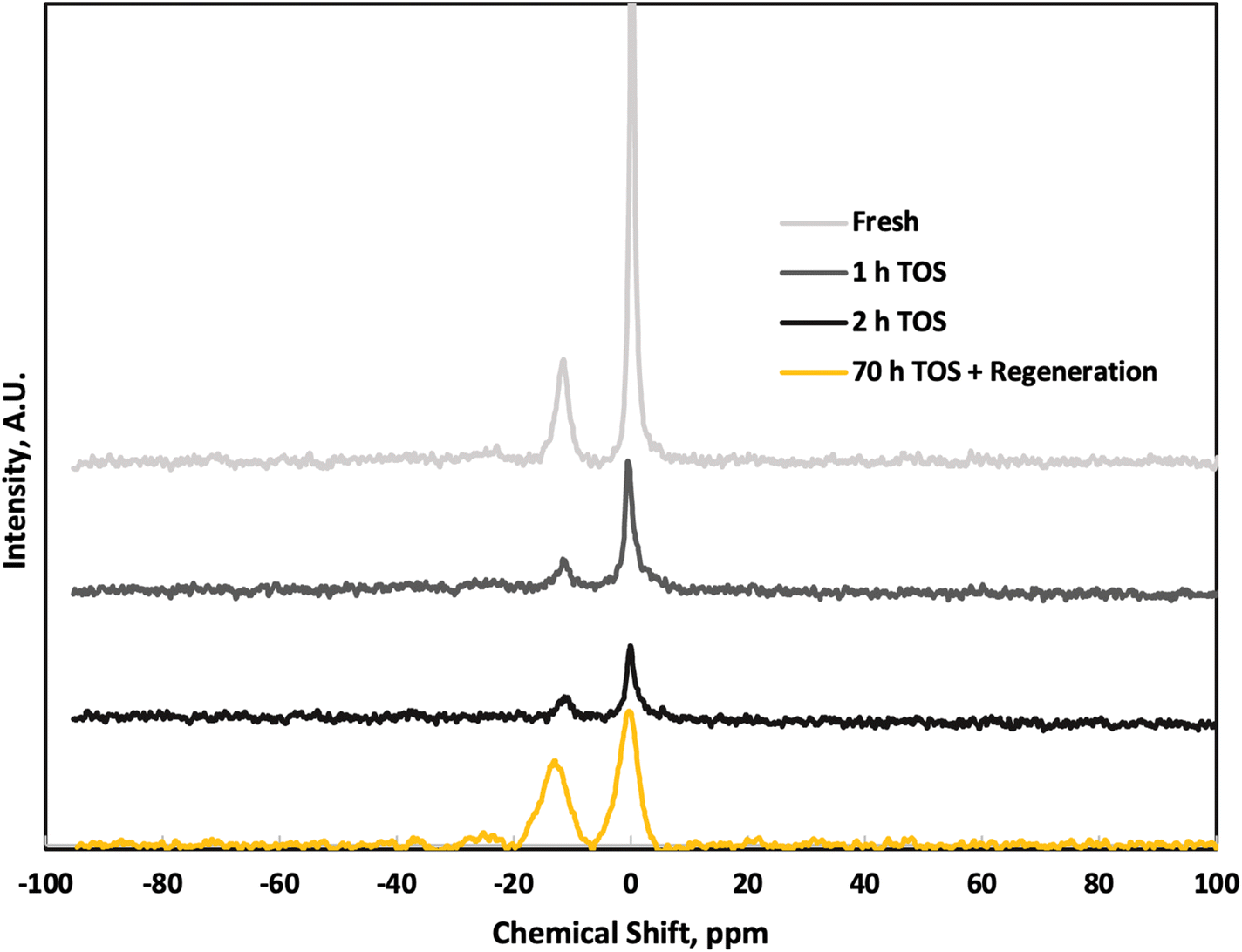 | ||
| Fig. 10 31P NMR spectra of fresh P-BEA (25) and post-reaction at 1 and 2 h TOS, and post-reaction after in situ regeneration following ∼70 hours of continuous operation. 375 °C, 20 wt% DMF in n-heptane, 750 psig, 0.025 mL min−1, 10 sccm gas feed, 0.5 g 150–250 µm pellets, 0.01 g catalyst sampled at each TOS. | ||
The XRD patterns of the P-BEA(25) show the typical zeolite beta characteristics after synthesis (Fig. 11).27 The peak widths at 2θ = 8° and 22° of the fresh and 8 h TOS samples are the same despite a decrease in peak intensities, potentially due to coke. Slight crystallinity degradation upon long TOS and in situ regeneration (from pressure, DMF concentration, temperature effects, and WHSV experiments) without an impact on productivity was also observed in our prior batch experiments.
 | ||
| Fig. 11 X-ray diffraction patterns of fresh P-BEA(25) and spent catalysts (1, 2, and 8 h time on stream), and after approx. 70 h time on stream and one regeneration cycle. | ||
Conclusions
We report high selectivity and high yield of para-xylene (pX) from 2,5-dimethylfural (DMF) and ethylene in a micro-packed bed flow reactor over phosphorous-decorated zeolite beta, with peak pX selectivity of 97% at 1/WHSV of 4.7 h at 80% DMF conversion. We identify optimal process pressure, gas-to-liquid feed ratio, DMF concentration, and space velocity and revealed external mass transfer limitations at slow flow rates (1/WHSV > 5 h at 375 °C) and kinetics/internal mass transfer limitations at faster flow rates. Compared to mesoporous supports, like silica gel and SBA-15, P-BEA provides size selectivity and suppresses non-specific reactions, by incorporating phosphorous within the micropores achieving catalyst stability over long operation. In situ regeneration of the catalyst performed for multiple cycles shows slight pX yield and DMF conversion reduction. Future implementation of flow chemistry to enable the production of renewable pX will benefit from a comprehensive techno-economic analysis and life cycle assessment.Data availability
All key data is provided in the manuscript or the ESI.† Any additional information can be requested from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the National Science Foundation under grant no. 2134471. We thank Dr Sean Najmi for his insightful contribution to the 31P NMR characterization used in this work.References
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520–540 RSC.
- W. Partenheimer, Catal. Today, 1995, 23, 69–158 CrossRef CAS.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- C.-C. Chang, H. Je Cho, J. Yu, R. J. Gorte, J. Gulbinski, P. Dauenhauer and W. Fan, Green Chem., 2016, 18, 1368–1376 RSC.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- J. J. Pacheco, J. A. Labinger, A. L. Sessions and M. E. Davis, ACS Catal., 2015, 5, 5904–5913 CrossRef CAS.
- C.-C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2014, 16, 585–588 RSC.
- R. E. Patet, N. Nikbin, C. L. Williams, S. K. Green, C.-C. Chang, W. Fan, S. Caratzoulas, P. J. Dauenhauer and D. G. Vlachos, ACS Catal., 2015, 5, 2367–2375 CrossRef CAS.
- R. E. Patet, W. Fan, D. G. Vlachos and S. Caratzoulas, ChemCatChem, 2017, 9, 2523–2535 CrossRef CAS.
- N. Nikbin, S. Feng, S. Caratzoulas and D. G. Vlachos, J. Phys. Chem. C, 2014, 118, 24415–24424 CrossRef CAS.
- R. C. Cioc, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Angew. Chem., 2022, 134, 1–35 CrossRef.
- H. J. Cho, L. Ren, V. Vattipalli, Y.-H. Yeh, N. Gould, B. Xu, R. J. Gorte, R. Lobo, P. J. Dauenhauer, M. Tsapatsis and W. Fan, ChemCatChem, 2017, 9, 398–402 CrossRef CAS.
- J. Gulbinski, L. Ren, V. Vattipalli, H. Chen, J. Delaney, P. Bai, P. Dauenhauer, M. Tsapatsis, O. A. Abdelrahman and W. Fan, Ind. Eng. Chem. Res., 2020, 59, 22049–22056 CrossRef CAS.
- J. Yu, S. Zhu, P. J. Dauenhauer, H. J. Cho, W. Fan and R. J. Gorte, Catal. Sci. Technol., 2016, 6, 5729–5736 RSC.
- J. A. Mendoza Mesa, F. Brandi, I. Shekova, M. Antonietti and M. Al-Naji, Green Chem., 2020, 22, 7398–7405 RSC.
- H. Kim, J. R. Kim, Y.-K. Park, J.-M. Ha and J. Jae, Catal. Today, 2024, 425, 1–13 Search PubMed.
- X. Feng, Z. Cui, K. Ji, C. Shen and T. Tan, Appl. Catal., B, 2019, 259, 1–13 CrossRef.
- C. Wu, T. Wu, J. Li, C.-L. Liu and W.-S. Dong, Appl. Catal., A, 2023, 663, 119323 CrossRef CAS.
- S. Liu, T. R. Josephson, A. Athaley, Q. P. Chen, A. Norton, M. Ierapetritou, J. I. Siepmann, B. Saha and D. G. Vlachos, Sci. Adv., 2019, 5, eaav5487 CrossRef PubMed.
- A. Feliczak-Guzik, Microporous Mesoporous Mater., 2018, 259, 33–45 CrossRef CAS.
- R. Barakov, N. Shcherban, P. Mäki-Arvela, P. Yaremov, I. Bezverkhyy, J. Wärnå and D. Y. Murzin, ACS Sustainable Chem. Eng., 2022, 10, 6642–6656 CrossRef CAS.
- M. Hartmann, A. G. Machoke and W. Schwieger, Chem. Soc. Rev., 2016, 45, 3313–3330 RSC.
- Z. Guo, X. Li, S. Hu, G. Ye, X. Zhou and M. O. Coppens, Angew. Chem., Int. Ed., 2020, 59, 1548–1551 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- S. K. Das, M. K. Bhunia, D. Chakraborty, A. R. Khuda-Bukhsh and A. Bhaumik, Chem. Commun., 2012, 48, 2891–2893 RSC.
- S. P. Szu, L. C. Klein and M. Greenblatt, J. Non-Cryst. Solids, 1992, 143, 21–30 CrossRef.
- T. O. Bok, E. P. Andriako, E. E. Knyazeva and I. I. Ivanova, RSC Adv., 2020, 10, 38505–38514 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01904k |
| This journal is © The Royal Society of Chemistry 2024 |