
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-polymerization using quantum dots for stable epoxy coatings†
Keroles B.
Riad
 a,
M. Reza
Kholghy
a and
Paula M.
Wood-Adams
*b
a,
M. Reza
Kholghy
a and
Paula M.
Wood-Adams
*b
aEnergy and Particle Technology Laboratory, Department of Mechanical and Aerospace Engineering, Carleton University, 1125 Colonel By Dr, Ottawa, Ontario K1S 5B6, Canada
bFaculty of Science and Engineering, University of Northern British Columbia, 3333 University Way, Prince George, BC. V2N 4Z9. Canada. E-mail: Paula.Wood-Adams@unbc.ca
First published on 18th April 2024
Abstract
Photo-polymerization is at the foundation of many industries such as dentistry, coatings, adhesives, and stereolithography 3D printing. However, the organic cationic photo-initiators currently used are toxic, expensive, and difficult to tune with respect to the wavelength of light required to initiate polymerization reactions. For example, current stereolithography 3D printing resins are unstable under sunlight. Here, we demonstrate that less expensive and non-toxic titania quantum dots made via the scalable flame spray pyrolysis technology can photo-polymerize epoxy when exposed to UVC (not present in sunlight on Earth), while being insensitive to UVA (present in natural sunlight on Earth) leading to resins that are photo-stable during end use. We use NMR and FTIR to demonstrate that photo-polymerization is catalyzed under UVC but not UVA, and nanoindentation to monitor the mechanical stability of epoxy films during post-polymerization UVA exposure. This approach allows precise control over the wavelengths of light under which photo-polymerization can and cannot occur, and is also transferable to other photo-catalytic reactions.
Keywords: Polymers; Photo-polymerization; Catalysis; Quantum dots; Nanotechnology.
1 Introduction
Photo-polymerization is preferable to heat-polymerization because light is less unpleasant relative to heat in dentistry applications, easier to apply to precise locations to achieve high printing resolution in circuit board manufacturing and stereolithography, and requires lower energy.1 However, there remains a need to control the critical wavelength required to initiate the photo-polymerization reaction and circumvent undesired reactions from taking place outside of controlled conditions.For example, the instability of stereolithography epoxy resins and aircraft coatings under sunlight originates from the fact that the absorption wavelength range of commercial photo-initiators overlaps with the solar spectrum on Earth.2 Existing photo-initiators have some sensitivity to UVA (315–400 nm) and UVB (295–315 nm) which represent 6% (ref. 3) of the solar intensity on Earth.4–7 These photo-initiators absorb sunlight and further polymerize the material, releasing carcinogenic volatile organic components, and continuously changing the material properties to the point of brittle fracture within weeks.2 UV absorbers, light stabilizers8 and inorganic surface coatings9 are often used to mitigate this instability. However, they only slow down degradation, and like sunscreen on the beach, coatings must be regularly reapplied. On the other hand, the ozone layer blocks light of wavelengths lower than 300 nm, or arguably 310 nm,10 from the solar spectrum on Earth.2 Thus, cheap and non-toxic photocatalysts that are sensitive exclusively to light with lower wavelength than the 300 nm lower limit of the solar spectrum on Earth are needed.
The findings presented here culminate multiple previously reported steps. First, we have demonstrated that metal oxide semiconducting nanoparticles can photo-polymerize epoxy via a cationic polymerization mechanism (shown in Fig. S1†).11 Semiconducting nanoparticles oxidize hydroxyl groups when excited by light with an energy that matches the particle bandgap energy, producing protons that initiate the cationic photo-polymerization reaction of epoxy. We note that in that previous study, we used P25 titania, crystal size of 25 nm, which is well above titania's 2 nm Bohr diameter and is therefore not a quantum dot. Notably, the use of quantum dots as initiators for photo-polymerization is a growing, yet immature, field as per the recent review by Waiskopf et al.12 Further, quantum dot/polymer nanocomposites facilitate multiple applications such as pollutant photodegradation,13 antimicrobial14 and antibacterial15 films, and full color displays.16 Then, we have shown that the flame spray pyrolysis (FSP) process, a mass-production nanoparticle synthesis technology, can synthesize metal oxide quantum dots with any bandgap energy from the UVC to the dark red region of the light spectrum.17 Finally, we have shown that flame-made titania contains a significant TiO2(B) crystalline fraction and is 2–3 times more efficient in catalyzing epoxy photo-polymerization than P25 titania,18 the gold standard19 for photocatalysis.
Titania is an inexpensive and abundant material with over 2 million tons per year produced, representing an $8.5 billion year industry.20 It is typically used in white paint, and coatings of medical pills as it is non-toxic. Our previous demonstration17 of the use of industrial flame synthesis process to synthesize titania quantum dots extends the affordability advantages of flame-made titania to applications of its quantum dots. For example, the size-dependent bandgap energy of titania quantum dots allows us to select the maximum wavelength of light that can excite the particle and initiate the catalysis of the epoxy photo-polymerization reaction.21
Finally, we have previously22 reported a methodical study assessing the impacts of various parameters on the kinetics of the epoxy photo-polymerization reaction catalyzed by titania nanoparticles. These parameters include the presence and concentration of an electron scavenger, the kind and concentration of alcohol used, and the concentration, crystal composition and size of the titania particles used.22
Here, we dive into the quantum regime and explore the advantages that arise from the increase in bandgap energy stemming from the quantum effect, and how they can be exploited to address the costly instability problems of photo-polymers when exposed to sunlight during end use. We demonstrate that flame-made titania quantum dots can catalyze the photo-polymerization of epoxy when exposed to UVC (not present in sunlight on Earth), while also being insensitive to UVA (present in sunlight on Earth) leading to photo-stable resins. That means that resins can be photo-polymerized using a UVC light source and remain stable when a coated or 3D-printed product is used outdoors where it is exposed to sunlight. In turn, the findings presented here could eliminate the need for expensive short-term fixes currently used to mitigate photo-instability such as regularly applying UV-blocking coatings. This approach can not only be used to mitigate the instability challenges of any photocatalytic material having to operate in sunlight, but it is also transferable to any photocatalytic application requiring strict control on the energy of light capable of initiating a specific reaction.
2 Results and discussion
Metal oxide quantum dots can be synthesized in flame by adding silica, which hinders the growth of the metal oxide crystal allowing for the very small crystal sizes necessary to achieve the quantum effect.17 In this study, we use particles composed of 10% TiO2 and 90% silica. We have previously17 reported the flame synthesis and full characterization (XRD, UV-vis spectroscopy, nitrogen adsorption BET) of the titania quantum dots used here. For the readers' convenience, those quantum dots exhibit the following properties: a bandgap energy of 4.3 eV (UVC region), which is higher than the 4.1 eV upper limit of sunlight on Earth; d(UV-vis) of 0.9 nm, lower than the 2 nm Bohr diameter of TiO2; a surface area of 282 m2 g−1; a d(BET) of 9 nm; and a mix of anatase and monoclinic crystal structures.17,18Fig. 1 shows a TEM image, not previously reported, of the TiO2 quantum dots used in this study. The TEM image shows two titania crystals embedded in an amorphous silica matrix. The two crystals appear to have sizes in the range between 2 and 4 nm. It is important to note the TEM imaging is biased towards larger crystals as they are much easier to locate, especially in this case where the titania content constitutes only 10 wt% with the balance being an amorphous silica matrix. This, and other previously17 reported TEM images, show that the morphology of the flame-made titania particles is a mixed phase where the quantum-sized titania crystals are embedded in a silica matrix, not a core–shell structure. This mixed phase structure has been found to improve dispersibility and catalytic performance, while a core–shell structure eliminates surface catalytic reactions.23
 | ||
| Fig. 1 TEM image of the flame-made silica-embedded TiO2 quantum dots used in this study. | ||
Fig. 2a shows an SEM image of the photo-polymerized films containing TiO2 quantum dots, demonstrating much better dispersibility relative to P25 TiO2 (Fig. 2b) that we investigated previously.11 The improved dispersibility can be assessed by how the absence of large titania agglomerates in Fig. 2a, how well spread out the quantum dots are, and how much smaller the are relative the P25 TiO2 ones shown in Fig. 2b. The improved dispersibility is due to the silica matrix in which the flame-made TiO2 quantum dots are embedded, consistent with literature.23
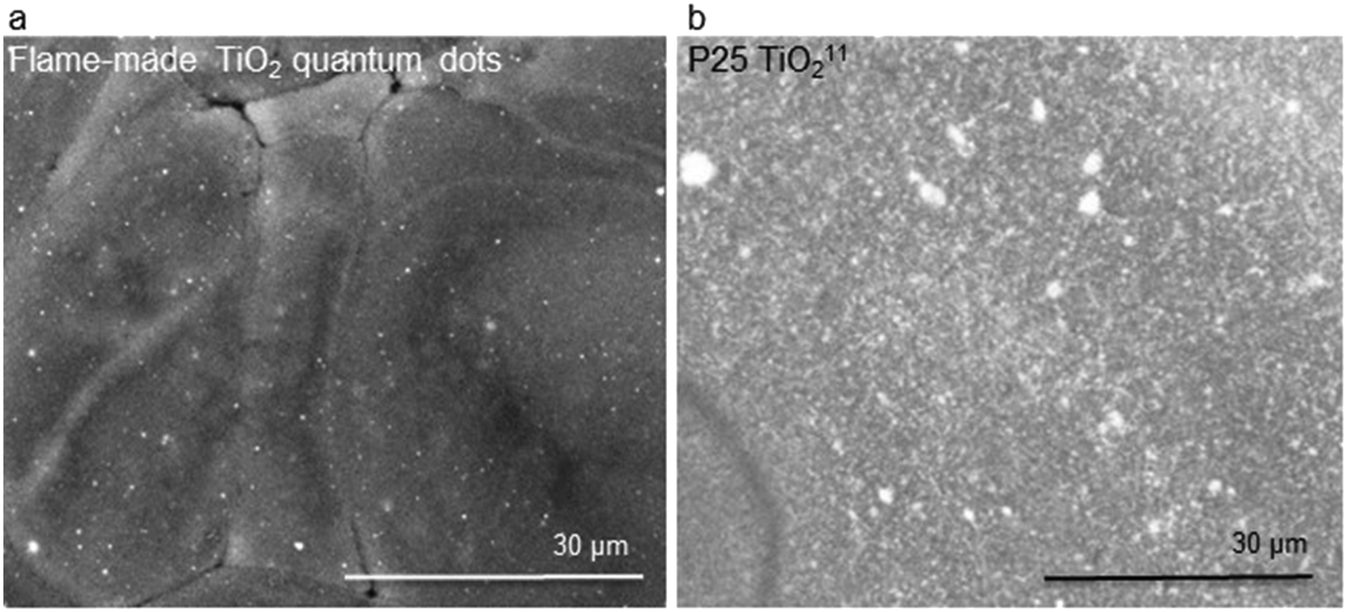 | ||
| Fig. 2 SEM images of a photo-polymerized film of a mixture containing epoxy, isopropanol (5 wt%), and TiO2: (a) quantum dots and (b) P25. The brightness and contrast in (a) were modified for clarity. The SEM image in (b) has been previously reported,11 and shown here for reference and convenience. | ||
Fig. 3 shows 13C NMR results. The shift at 51.7 ppm seen in Fig. 3a corresponds to epoxide rings in the unpolymerized monomer.11 The methylene carbons at 20–33 ppm, and methine carbons at 68–90 ppm seen in Fig. 3b and c expected for polymerized epoxy,24 overlap with shifts present in the initial monomer in the same region as shown in Fig. 3a. As such, the broadening of those shifts due to the increase in molecular weight that takes place seen in Fig. 3b and c should be used as the clearest indication of epoxy photo-polymerization. Fig. 3d shows the liquid-state 13C NMR spectra of a control system with neither photo-initiators nor photo-catalysts, radiated by UVC light. No broadening is observed in the spectrum shown in Fig. 3d, which is also almost identical to that of the unpolymerized epoxy monomers shown in Fig. 3a, demonstrating that UVC radiation does not photo-polymerize epoxy in the absence of a photo-initiator or a photo-catalyst. Fig. 3e and f show the solid-state 13C NMR spectra of epoxy systems containing the TiO2 quantum dots and radiated by UVA and UVC, respectively. The spectrum shown in Fig. 3e for the epoxy system containing TiO2 quantum dots and radiated by UVA is also identical to that of the unpolymerized epoxy monomers shown in Fig. 3a and no peak broadening is observed. This demonstrates that UVA radiation does not initiate the quantum dots to catalyze the epoxy photo-polymerization reaction. The shift at 51.7 ppm, corresponding to unpolymerized epoxide rings, still appears in the spectrum shown in Fig. 3f suggesting that the film is not fully polymerized. Nonetheless, the shape of the spectrum shown in Fig. 3f of the epoxy system with the TiO2 quantum dots radiated by UVC is very similar to that of the epoxy system photo-polymerized by P25 titania in Fig. 3c, with significant broadening in the observed shifts. This demonstrates that UVC radiation does indeed initiate the quantum dots to catalyze the epoxy photo-polymerization reaction.
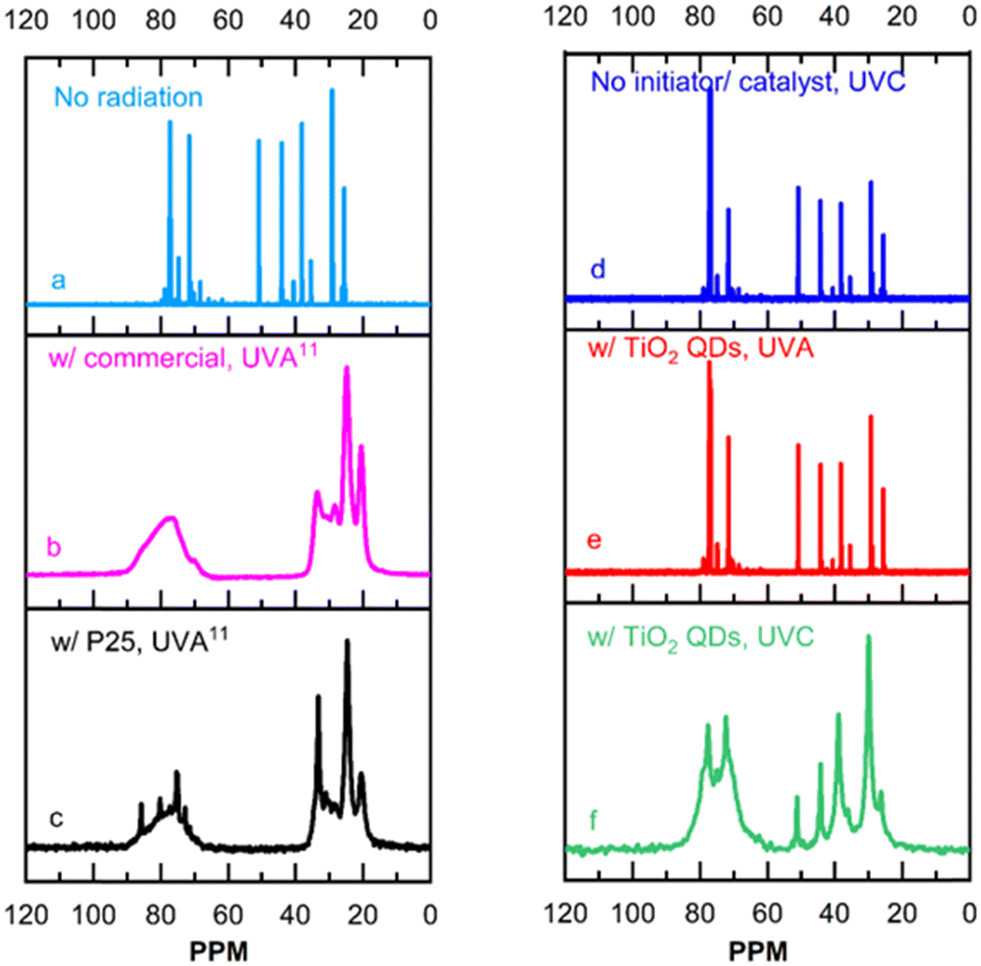 | ||
| Fig. 3 13C NMR of systems of cyclohexene oxide and isopropanol: (a) initiator/catalyst free, with no radiation; (b) with commercial cationic organic initiator and (c) with P25 TiO2 radiated by UVA light; (d) initiator/catalyst free, radiated by UVC light; (e) with TiO2 quantum dots, radiated by UVA light, and (f) with TiO2 quantum dots (QDs) radiated by UVC light. The 13C NMR spectra in (a)–(c) have been previously reported,11 and shown here for reference and convenience. Compositions and radiation times are presented in the experimental section. | ||
Fig. 4 shows reaction kinetics by following the consumption of epoxide groups via the FTIR peak at 910 cm−1, normalized relative to the aliphatic peak area (2800–3000 cm−1). We have previously11 validated the use of FTIR to quantify the degree of epoxy polymerization using chemical titration by HBr according to ASTM-D1652-97. No epoxy consuming reaction is detected in the control sample with neither a photo-initiator nor a photo-catalyst after prolonged exposure to UVC light (dashed line with circles). Similarly, no epoxy consuming reaction is detected in the control sample with the TiO2 quantum dots after prolonged exposure to UVA light (solid line with squares). However, we do observe a typical photo-polymerization pattern in the samples with TiO2 quantum dots when exposed to UVC light (dotted line with triangles). The FTIR findings in Fig. 4 that demonstrate that the TiO2 quantum dots are excited to catalyze the photo-polymerization of epoxy with UVC but not UVA light are consistent with 13C NMR data shown in Fig. 3.
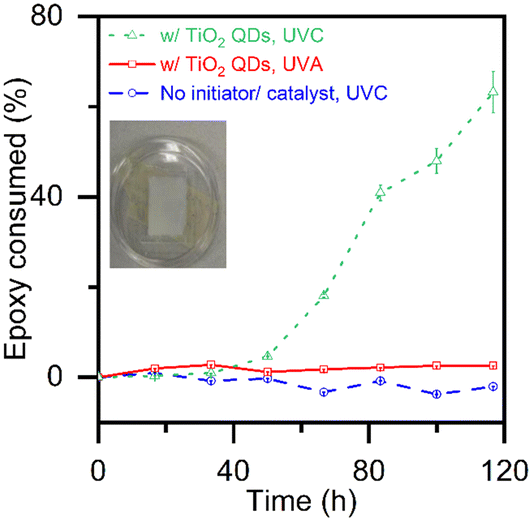 | ||
| Fig. 4 Epoxy consumption calculated based on the area of the FTIR peak at ∼910 cm−1 for epoxy and isopropanol systems: radiated by UVC light (dashed line with circles), with TiO2 quantum dots radiated by UVA light (solid line with squares), and with TiO2 quantum dots radiated by UVC light (dotted line with triangles). All peak areas are normalized relative to the aliphatic peak area (2800–3000 cm−1). Error bars indicate 90% confidence limits on the mean of 6 measurements for all t > 0. The inset shows a sample blade-coated film used for FTIR. | ||
Further, the epoxy films photo-polymerized with titania quantum dots (only 10 wt% of the TiO2 active catalyst) reach maximum epoxy consumption after 116 hours, 20% faster than the 150 hours it took those photo-polymerized with P25 titania11 (100 wt% of the TiO2 active catalyst), the gold standard for photocatalytic activity. This improved kinetic performance is likely due to the higher surface area of the titania quantum dots (282 m2 g−1)17 as compared to that of P25 (50 m2 g−1),18 as well as interactions between the anatase and the monoclinic crystal structures present18 in flame-made titania.
Finally, the epoxy films photo-polymerized with titania quantum dots show a substantial degree of polymerization. The maximum possible degree of polymerization in cross-linked thermosets depend on many factors including steric hindrance, degree of side reactions and monomer geometry.25 Nonetheless, the degree of polymerization of the epoxy films photo-polymerized with TiO2 quantum dots shown in Fig. 4 exceed the 33% gelation threshold estimated based on Flory's theory.26 Note that in the case of photo-polymerization reactions, the epoxy monomer used here is considered to have four functional groups.
Fig. 5 shows the mechanical properties of the photo-polymerized epoxy films as measured by nanoindentation: reduced modulus (Fig. 5a), hardness (Fig. 5b) and plastic deformation (Fig. 5c). The epoxy films photo-polymerized with titania quantum dots are shown as dotted lines with triangles, while those photo-polymerized with a commercial initiator are shown as solid lines with diamonds.
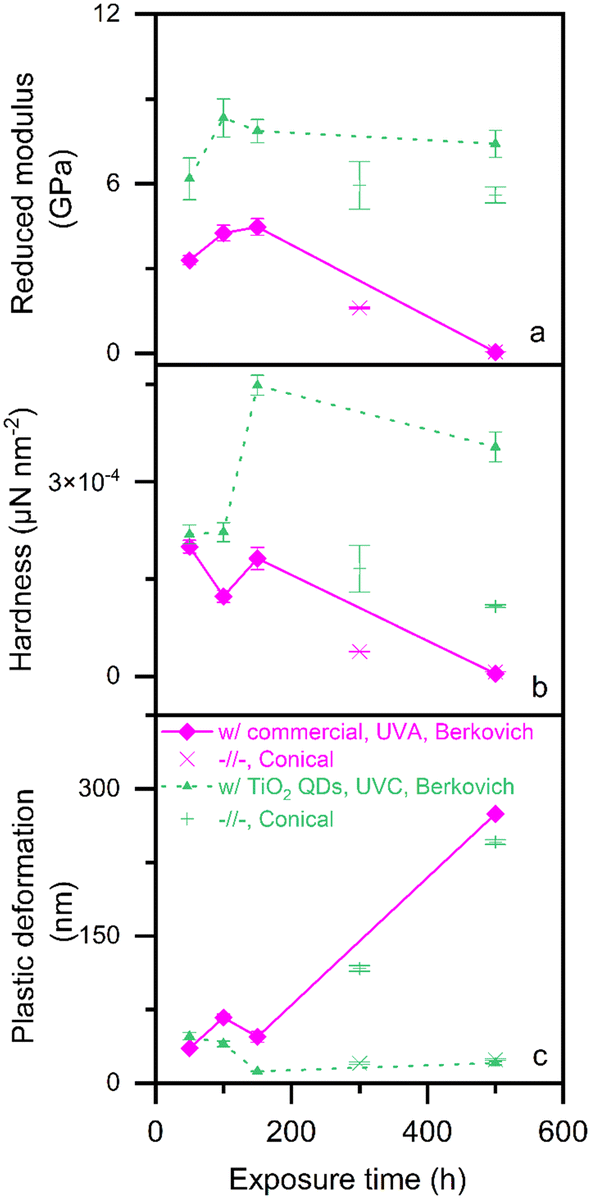 | ||
| Fig. 5 Nanoindentation measurements of sample films photo-polymerized with a commercial photo-initiator (solid lines with diamonds) and TiO2 quantum dots (dotted lines rectangles) showing (a) reduced modulus, (b) hardness, and (c) plastic deformation. The lines represent data obtained with a Berkovich tip, while the scattered points represent ones obtained with a conical tip. | ||
The data depicted using lines are obtained using a Berkovich tip, as is best practice.27 However, we also conducted spot checks on two specimens, depicted using points (× for commercial, + for TiO2 QDs), with a conical tip to assess the effect of tip geometry. In all the measured mechanical properties, the trend is the same with both tips. However, the values obtained using a conical tip are always lower in the cases of reduced modulus and hardness, and higher in the case of plastic deformation, relative to those obtained using the Berkovich tip.
We observe slight changes in local mechanical properties (Fig. 5) of both films over the first 150 hours of post-polymerization UVA exposure. The only substantial change in this period is the hardness (Fig. 5b) of the epoxy films photo-polymerized with titania quantum dots.
In all three cases, we observe that the epoxy films photo-polymerized with the titania quantum dots exhibit significantly better mechanical properties (higher reduced modulus and hardness, and less plastic deformation), than those of the films photo-polymerized with a commercial organic photo-initiator. In fact, at the 150 hours of UVA exposure mark, the epoxy films photo-polymerized with the titania quantum dots showed double the reduced modulus and hardness, and 75% less plastic deformation relative to those of the films photo-polymerized with a commercial organic photo-initiator. This mechanical property improvement is consistent with literature finding that the incorporation of titania increases the glass transition temperature and modulus of epoxy nanocomposites.28 Further, metal oxide quantum dots used as both photo-polymerization initiators as well as fillers have been shown to lead to nanocomposites with 85% better mechanical properties and provide antimicrobial functionality compared to when the quantum dots are used only as fillers.14
The mechanical properties of the epoxy films photo-polymerized with a commercial organic photo-initiator drastically deteriorated after 300 hours of exposure to UVA. Indeed, in the case of the epoxy films photo-polymerized, both the reduced modulus (Fig. 5a) and hardness (Fig. 5b) dropped to near zero, and plastic deformation (Fig. 5c) increased by an order of magnitude.
On the other hand, and most importantly, the mechanical properties of the epoxy films photo-polymerized with titania quantum dots remained stable for the 500 hours of UVA exposure that we tested. This indicates that UVA light, and in turn, sunlight, has no effect on the mechanical properties of the epoxy films photo-polymerized with titania quantum dots having a bandgap energy higher than the 4.1 eV threshold of the Solar spectrum on Earth.
3 Conclusions
We demonstrate that titania quantum dots with a 4.3 eV bandgap energy can catalyze epoxy photo-polymerization when radiated with UVC, while being insensitive to UVA, leading to products that are both stronger and photo-stable during end-use. These findings are the culmination of previous steps where we have 1) introduced metal oxide semiconducting as a new class of photocatalysts for epoxy photo-polymerization,11 2) demonstrated the ability of FSP to mass-produce metal oxide quantum dots with any bandgap energy from the UVC to the dark red region of light,17 and 3) demonstrated that the monoclinic crystal structure in flame-made titania significantly improves catalytic performance.18 The findings in this study, combined with the UVC to dark red range of quantum dot bandgap energies accessible by FSP,17 allow for the precise selection of the maximum light wavelength that can initiate catalytic reactions such as that of epoxy photo-polymerization.4 Experimental section
4.1 Materials
All materials used in this study are obtained from Sigma Aldrich and used as received. The epoxy monomer used is 1,4-cyclohexane dimethanol diglycidyl ether (mixture of cis and trans, technical grade). Other materials used are isopropanol (99.5%), xylene (reagent grade), commercial organic cationic initiator (bis(4-methylphenyl) iodonium hexafluorophosphate, 98%), titanium(IV) tetraisopropoxide (97%), and hexamethyldisiloxane (98.5%).4.2 Titania quantum dot particle flame synthesis
We have previously17 reported the flame synthesis procedure and full particle characterization of the titania quantum dots used in this study. Briefly, a 0.25 M solution (10 mol% titania with the balance being silica) is prepared by diluting the metal precursors with xylenes, and fed at 1 mL min−1 through the capillary of the FSP reactor and atomized by 3.75 L min−1 of oxygen. The titania and silica precursors used are titanium(IV) tetraisopropoxide and hexamethyldisiloxane, respectively. A pilot flame (1.25 L min−1 of CH4 premixed with 2.5 L min−1 of O2) served as an ignition source. The produced titania quantum dots are collected on glass fiber filters (Albet-Hahnemühle, GF 6, 25.7 cm diameter) placed in a water-cooled stainless-steel holder with the help of a vacuum pump (Busch, Seco SV 1040 C). Scheme 1 depicts the flame synthesis process.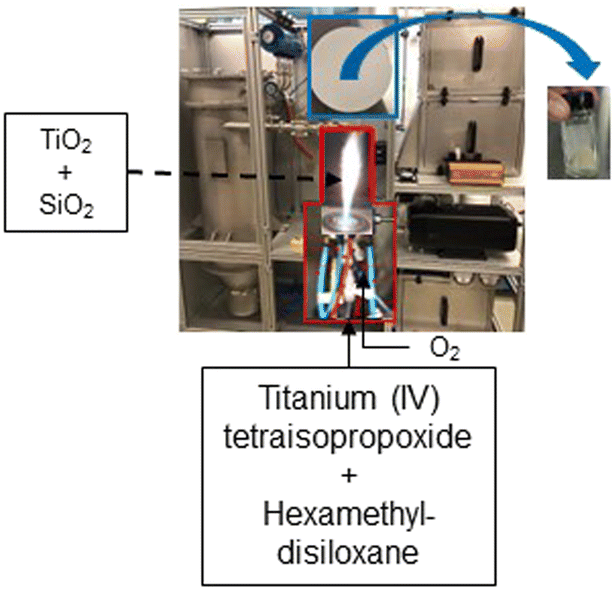 | ||
| Scheme 1 FSP synthesis process of titania quantum dots. | ||
The TiO2 quantum dots were further characterized using transmission electron microscopy (TEM) via a FEI Tecnai F2 G20 field emission transmission electron microscope, at a 120 kV acceleration voltage. The TiO2 quantum dots are dispersed in ethanol, and then drop-casted on a carbon coated TEM grid that is left to dry for 30 minutes before imaging.
4.3 Epoxy mixture preparation and photo-polymerization
We have previously11 reported the procedures for the preparation and photo-polymerization of epoxy film. Briefly, mixtures (1.5 g) consisting of epoxy monomer, 5 wt% of isopropanol, and a 5 wt% of either the titania quantum dot catalyst or commercial photo-initiator are stirred overnight. The mixtures with the titania quantum dots are further sonicated for 30 minutes with a Misonix Sonicator 3000 (5 seconds on, 20 seconds off). In some control samples, the titania quantum dots and the commercial photo-initiators are excluded.Samples are radiated under ambient conditions, no inert gas used, with UVA and UVC using a UVP (CL-1000L) crosslinker with a peak emission at 365 nm, and a UVP (CL-1000) crosslinker with a peak emission at 254 nm, respectively. Both crosslinkers had a light intensity of 4 mW cm−2 at the surface of the samples. The UVA crosslinker has a broad emission spectrum, shown in Fig. S2,† that spans from ∼310 nm to ∼435 nm. That broad emission spectrum of the UVA crosslinker overlaps with a portion of the UVB region, and almost perfectly matches the UV range present in sunlight on Earth.2,3 As such, the two UV crosslinkers selected in this work properly support the objective of this study: developing photocatalysts that are stable under sunlight during end use. Finally, note that the reaction kinetics of epoxy photo-polymerization are directly related to the light intensity used as shown in Fig. S3.†
Mixtures prepared for 13C NMR are photo-polymerized by being radiated for 50 hours in all cases. Mixtures are blade coated on microscope slides (example shown in the inset of Fig. 4) with 0.0635 cm thick Kapton tape (McMaster-Carr) for FTIR spectroscopy, and on AFM metal desks for nanoindentation measurements. Nanoindentation films with titania quantum dots and commercial photo-initiators are photo-polymerized by being radiated for 3 hours in the UVA crosslinker, and 133 hours in the UVC cross linkers respectively.
Scanning electron microscopy (SEM) imaging is conducted on the same films used for nanoindentation that experienced 500 hours of UVA exposure. A Hitachi S-3400N microscope with used at 30 Pa variable pressure and 15 kV.
4.4 Monitoring of the epoxy photo-polymerization chemical reaction
We monitor the epoxy photo-polymerization chemical reaction via13C nanomagnetic resonance spectroscopy (NMR) and Fourier transform infrared (FTIR) spectroscopy using the same procedures we previously reported.11Liquid-state 13C NMR experiments are conducted on samples showing no polymerization (Fig. 3a, d and e), while solid-state NMR experiments are conducted on the other mixtures that have polymerized, as assessed by a typical physical change from liquid to a “paste-like” state. Liquid-state spectra are recorded on a Bruker Avance III HD spectrometer operating at a field of 14.1 T with corresponding 13C and 1H Larmor frequencies of 150.87 and 599.99 MHz respectively, using a double resonance BBFO probe. 13C spectra of the soluble samples are acquired with NOE enhancement during the recycle delays. 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 384 transients are added with 1 s acquisition time a spectral width of 234 ppm and a recycle delay of 2 s. The 13C spectra of the solid samples are obtained on a Bruker Avance III HD spectrometer operating at a field of 9.4 T with corresponding 13C and 1H Larmor frequencies of 100.6 and 400.1 MHz respectively. A 4 mm double resonance probe was used operating at room temperature and a magic-angle spinning frequency of 12.5 kHz using a 2 ms long 30% ramped 1H to 13C cross polarization. A 50 kHz spectral width is used and 4096 transients are added with 15 ms acquisition time and a recycle delay of 5 s. High-power 1H decoupling was applied during the acquisition using SPINAL-64. The applied radio-frequency fields were 75 kHz and 85 kHz for 13C (cross-polarization) and 1H (cross-polarization and decoupling). All spectra are externally referenced to TMS (0 ppm) by setting the unshielded CH2 resonance of adamantane to 38.48 ppm.
384 transients are added with 1 s acquisition time a spectral width of 234 ppm and a recycle delay of 2 s. The 13C spectra of the solid samples are obtained on a Bruker Avance III HD spectrometer operating at a field of 9.4 T with corresponding 13C and 1H Larmor frequencies of 100.6 and 400.1 MHz respectively. A 4 mm double resonance probe was used operating at room temperature and a magic-angle spinning frequency of 12.5 kHz using a 2 ms long 30% ramped 1H to 13C cross polarization. A 50 kHz spectral width is used and 4096 transients are added with 15 ms acquisition time and a recycle delay of 5 s. High-power 1H decoupling was applied during the acquisition using SPINAL-64. The applied radio-frequency fields were 75 kHz and 85 kHz for 13C (cross-polarization) and 1H (cross-polarization and decoupling). All spectra are externally referenced to TMS (0 ppm) by setting the unshielded CH2 resonance of adamantane to 38.48 ppm.
FTIR spectroscopy is conducted using ThermoFisher's Nicolet IS10 spectrometer in ATR mode with 64 scans at 1 cm−1 resolution between 600 cm−1 and 4000 cm−1.
4.5 Monitoring of the mechanical properties of the photo-polymerized epoxy films
Nanoindentation is used to provide thin film mechanical properties,29 using the same procedure we previously reported.30 Briefly, we used a Hysitron Triboindenter TI 950 (Bruker) to conduct indents using a 25 μN load which was held for 5 seconds with loading and unloading times also being set to 5 seconds each. This process is repeated for 25 indents in a 5 × 5 matrix for each sample. We follow Cheng and Cheng31 to calculate the reduced modulus. Hardness is calculated as the load over the projected area at the beginning of the hold period. Plastic deformation is measured as the residual depth at the end of the unloading cycle.Author contributions
All authors contributed to the research and manuscript preparation. All authors have given approval to the final version of the manuscript.Conflicts of interest
Dr. Keroles Riad has recently incorporated his own startup on FSP-made nanoparticles. The startup is only a few months old, and this study predates the conceptualization of that startup.Acknowledgements
From Université du Québec à Montréal, we thank Dr. Alexandre Arnold for his help with NMR. From Polytechnique Montréal, we thank Stephen Brown and Dr. Jincheng Qian for their help with nanoindentation. From Concordia University, we thank Dr. Dmytro Kevorkov for his help with SEM. From Carleton University, we thank Dr. Jianqun Wang for his help with TEM, as well as Dr. Jeremy Laliberte for his help reading this manuscript and providing feedback. Further, we gratefully acknowledge the following funding programs: Canada Research Chair Program (# CRC-2019-232527), the Natural Sciences and Engineering Research Council of Canada (Discovery # RGPIN-2019-06330, Early Career Supplemental Award # DGECR-2019-00220, and Banting Postdoctoral Fellowship # FRN192340), and the MITACS Accelerate Entrepreneur program (IT39555), as well as Concordia University.References
- R. Peiffer, in Photopolymerization: Fundamentals and Applications, American Chemical Society, Washington, DC, 1997, ch. 1, pp. 1–14 Search PubMed.
- C. H. Hare, The degradation of coatings by ultraviolet light and electromagnetic radiation, J. Prot. Coat. Linings, 1992, 5, 58–61 Search PubMed.
- J. Crivello, The discovery and development of onium salt cationic photoinitiators, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- Y. Yagci, S. Jockusch and S. Turro, Photoinitiated polymerization: Advances, challenges, and opportunities, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- C. Jubsilp, T. Takeichi and S. Rimdusit, in Handbook of Benzoxazine Resins, ed. H. Ishida and T. Agag, Elsevier, Amsterdam, 2011, ch. 7, pp. 157–174 Search PubMed.
- J. Zhang, Y. Huang, X. Jin, A. Nazartchouk, M. Liu, X. Tong, Y. Jiang, L. Ni, S. Sun, Y. Sang, H. Liu, L. Razzari, F. Vetrone and J. Claverie, Plasmon enhanced upconverting core@trible-shell nanoparticles as recyclable panchromatic initiators (blue to infrared) for radical polymerization, Nanoscale Horiz., 2019, 4, 907–917 RSC.
- J. Crivello and S. Liu, Photoinitiated cationic polymerization of epoxy alcohol monomers, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 389–401 CrossRef CAS.
- G. P. Bierwagen and D. E. Tallman, Choice and measurement of crucial aircraft coatings system properties, Prog. Org. Coat., 2001, 41, 201–216 CrossRef CAS.
- B. A. Driel, P. J. Kooyman, K. J. Berg, A. Schmidt-Ott and J. Dik, A quick assessment of photocatalytic activity of TiO2 pigments – From lab to conservation studio!, Microchem. J., 2016, 126, 162–171 CrossRef.
- Y. Matsumi and M. Kawasaki, Photolysis of atmospheric ozone in the ultraviolet region, Chem. Rev., 2003, 103, 4767–4782 CrossRef CAS PubMed.
- K. B. Riad, A. A. Arnold, J. P. Claverie, S. V. Hoa and P. M. Wood-Adams, Photopolymerization using metal oxide semiconducting nanoparticles for epoxy-based coatings and patterned films, ACS Appl. Nano Mater., 2020, 3, 2875–2880 CrossRef CAS.
- N. Waiskopf, S. Magdassi and U. Banin, Quantum photoinitiators: Toward emerging photocuring applications, J. Am. Chem. Soc., 2021, 143, 577–587 CrossRef CAS PubMed.
- S. Abu-Melha, Distinguisable photocatalytic activity of nano polyaniline with quantum dots metal oxide as photocatalysts for photodegradation of Dianix blue dye and different industrial pollutants, Polyhedron, 2024, 252, 116781 CrossRef CAS.
- T. Naor, S. Gigi, N. Waiskopf, G. Jacobi, S. Shoshani, D. Kam, S. Magdassi, E. Banin and U. Banin, ZnO quantum photoinitiators as an all-in-one solution for multifunctional photopolymer nanocomposites, ACS Nano, 2023, 17, 20366–20375 CrossRef CAS PubMed.
- W. J. Chong, S. Shen, Y. Li, A. Trinchi, D. Pejak, I. Kyratzis, A. Sola and C. Wen, Additive manufacturing of antibacterial PLA-ZnO nanocomposites: Benefits, limitatons and open challenges, J. Mater. Sci. Technol., 2022, 111, 120–151 CrossRef CAS.
- W. Guo, J. Chen, T. Ma, Z. Chen, M. Li, H. Zeng and J. Lu, Direct photolithography pattering of quantum dot-polymer, Adv. Funct. Mater., 2024, 34, 2310338 CrossRef CAS.
- K. B. Riad, K. B. Hoa and P. M. Wood-Adams, Metal oxide quantum dots embedded in silica matrices made by flame spray pyrolysis, ACS Omega, 2021, 6, 11411–11417 CrossRef CAS PubMed.
- K. B. Riad, P. M. Wood-Adams and K. Wegner, Flame-made TiO2(B), Mater. Res. Bull., 2018, 106, 276–281 CrossRef CAS.
- B. Ohtani, B. Prieto-Mahaney, D. Li and R. Abe, What is Degussa (Evonik) P25? Crystalline composition analysis, reconstruction from isolated pure particles and photocatalytic activity test, J. Photochem. Photobiol., A, 2010, 216, 179–182 CrossRef CAS.
- A. Balakrishnan, J. D. Groeneveld, S. Pokhrel and L. Mädler, Frontispiece: Metal sulfide nanoparticles: Precursor chemistry, Chem. – Eur. J., 2021, 27, 6390–6406 CrossRef CAS PubMed.
- A. I. Ekimov and A. A. Onuschenko, Quantum size effect in three-dimensional microscopic semiconductor crystals, JETP Lett., 1981, 34, 345–349 Search PubMed.
- K. B. Riad, Photocuring epoxy with quantum dot for 3D printing, M. Sc. dissertation, Concordia University, 2016 Search PubMed.
- A. Teleki, M. C. Heine, F. Krumeich, M. K. Akhtar and S. E. Pratsinis, In situ coating of flame-made TiO2 particles with nanothin SiO2 films, Langmuir, 2008, 24, 12553–12558 CrossRef CAS PubMed.
- A. Yahiaoui, M. Belbachir, J. Soutif and L. Fontaine, Synthesis and structural analyses of poly (1,2-cyclohexene oxide) over solid acid catalyst, Mater. Lett., 2005, 48, 759–767 CrossRef.
- K. B. Riad, R. Schmidt, A. A. Arnold, R. Wuthrich and P. M. Wood-Adams, Characterizing the structural formation of epoxy-amine networks: The effect of monomer geometry, Polymer, 2016, 104, 83–90 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell Univ. Press, New York, 1992 Search PubMed.
- J. C. Hay, E. Y. Sun, G. M. Pharr, P. F. Becher and K. B. Alexander, Elastic anisotropy of β-silicon nitride whiskers, J. Am. Ceram. Soc., 1998, 81, 2661 CrossRef CAS.
- D. Morselli, F. Bondioli, M. Sangermano and M. Messori, Photo-cured epoxy networks reinforced with TiO2 in-situ generated by means of non-hydrolytic sol-gel process, Polymer, 2012, 53, 283–290 CrossRef CAS.
- R. Saha and W. D. Nix, Effects of the substrate on the determination of thin film mechanical properties by nanoindentation, Acta Mater., 2002, 50, 23–38 CrossRef CAS.
- K. B. Riad, S. V. Hoa and P. M. Wood-Adams, Photocuring graphene oxide liquid crystals for high-strength structural materials, ACS Omega, 2022, 7, 21192–21198 CrossRef CAS PubMed.
- Y. T. Cheng and C. M. Cheng, General relationship between contact stiffness, contact depth and mechanical properties for indentation in linear viscoelastic solids using axisymmetric indenters of arbitrary profiles, Appl. Phys. Lett., 2005, 87, 111914 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4im00026a |
| This journal is © Institute of Process Engineering of CAS 2024 |