
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElongated Fe–N–C containing trace atomic Co dopants for high power density PEMFCs†
Jiayao
Cui
ab,
Junyong
Min
a,
Hao
Wang
 *ac,
Jianglan
Shui
d,
Lishan
Peng
e,
Zhenye
Kang
f,
Jieyuan
Liu
d,
Qingjun
Chen
*e,
Shuo
Bai
a and
Yanrong
Liu
*abc
*ac,
Jianglan
Shui
d,
Lishan
Peng
e,
Zhenye
Kang
f,
Jieyuan
Liu
d,
Qingjun
Chen
*e,
Shuo
Bai
a and
Yanrong
Liu
*abc
aCAS Key Laboratory of Green Process and Engineering, State Key Laboratory of Mesoscience and Engineering, Beijing Key Laboratory of Ionic Liquids Clean Process, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: haowang@ipe.ac.cn; qjchen@ipe.ac.cn; yrliu@ipe.ac.cn
bSchool of Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cLongzihu New Energy Laboratory, Zhengzhou Institute of Emerging Industrial Technology, Henan University, Zhengzhou 450000, China
dSchool of Materials Science and Engineering, Beihang University, Beijing, China
eGanjiang Innovation Academy Chinese Academy of Sciences, Ganzhou 341000, China
fState Key Laboratory of Marine Resource Utilization in South China Sea, Hainan Provincial Key Lab of Fine Chemistry, School of Chemical Engineering and Technology, Hainan University, Haikou 570228, China
First published on 10th May 2024
Abstract
Developing single-atom Fe–N4/C catalysts is crucial for the large-scale implementation of proton exchange membrane fuel cells (PEMFCs). While Fe–N4/C catalysts are inherently active in accelerating the slow ORR process, their performance is still inferior to that of Pt/C. Herein, a trace Co-doped Fe single-atom catalyst (Fe(tCo)–N–C) containing more active Fe2N8 sites has been synthesized. Interestingly, compared with typical FeN4 sites in an Fe–N–C electrocatalyst, the Fe2N8 sites generate a larger Fe–N bond length due to Co-doping. The elongated Fe–N bond in Fe2N8 lowers the d-band center and charge density of iron sites, enhancing the ORR process by facilitating the formation of *OOH and generation and desorption of *OH. Fe(tCo)–N–C manifested excellent acidic and alkaline ORR activity, with a half-wave potential (E1/2) of 0.80 V in HClO4 solution and 0.89 V in KOH medium. More importantly, high peak power densities (Pmax) were realized by applying Fe(tCo)–N–C in PEMFCs, with the Pmax reaching 890 mW cm−2 in H2–O2 and 380 mW cm−2 in H2–air. Additionally, trace Co dopants in the catalyst improved carbon graphitization and provided high ORR catalytic stability. This research introduces an innovative approach to engineering highly active Fe2N8 sites, providing valuable insights for the sustainable progress of PEMFC technology.
Keywords: Proton exchange membrane fuel cells; Oxygen reduction reaction; Platinum-group-metal-free catalysts; Single-atom catalysts; Bimetallic active sites.
1 Introduction
The challenges induced by the shortage of fossil energy have triggered the development of renewable energy,1–7 among which hydrogen energy has the nature of being non-polluting and having high energy density.8 PEMFCs have aroused a lot of attention as hopeful power trains to replace internal combustion engines without generating emissions.9,10 The cathodic ORR occurs at the solid–gas–liquid three-phase boundary with multiple charge and proton transfer processes, leading to sluggish kinetics and thus hindering practical efficiency. The current catalyst of choice uses platinum (Pt) to accelerate the kinetics, but the use of this costly and scarce material is significantly impeding the large-scale implementation of PEMFCs.11 Thus, there is a very urgent need to explore alternative inexpensive earth-abundant PGM-free counterparts until their activity and stability are competitive or even superior to those of PGMs.12,13Ever since the first demonstration of metal–nitrogen–carbon (M–N–C) by R. Jasinski in 1964,14 this type of material has aroused great interest as PGM-free ORR catalysts because of their high efficiency for atomic utilization, high electronic conductivity, and superior ORR catalytic activity.15–21 M–N–C catalysts can be derived from various precursor materials. Metal–organic frameworks (MOFs) have gained widespread attention in this context due to their porous nature, high conductivity, expansive specific surface area (SSA), and versatile properties, making them extensively explored.22,23 Especially via thermal treatment of zeolitic imidazolate frameworks (e.g., ZIF-8),24,25 volatile Zn nodes could evaporate at elevated temperature to generate micropores, and imidazole linkers are transformed into a densely-populated N-doped carbon (N-C) matrix.26 The additional metal doping in N-C enables catalysts with an appropriate geometric structure and charge distribution, thereby efficiently facilitating oxygen adsorption and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation.27
O bond dissociation.27
Within different kinds of M–N–C materials, nitrogen-coordinated Fe samples (Fe–N–C) are hailed as the best prospective PGM-free ORR catalysts, because of their suitable binding strength between intermediates and catalytic centers, which follows the Sabatier principle.28,29 To enhance the performance of ORR and address the inherent constraints of the intrinsic activity of Fe–N–C, researchers often introduce secondary metals to adjust the electronic structure of the Fe center or to construct dual-metal active centers.30–32 Such regulated active sites can optimize the chemical environment encompassing the metal center, especially the electronic structure,33,34 thereby boosting the desorption of intermediate species and cleavage of OO bonds.35 Additionally, the addition of a secondary metal has the potential to effectively improve the degree of graphitization, thus efficiently enhancing the catalyst stability.36 Therefore, metal atom doping is a useful strategy for boosting the performance of Fe–N–C and shows great promise for possible applications in PEMFCs. However, M1M2 replacing Zn on the ZIF skeleton followed by pyrolysis is commonly used to construct metal active sites. However, this approach could generate inactive metal species with many active sites buried in a bulky matrix and inaccessible to O2 and protons in PEMFCs, so there is a call for better synthesis methods. Ammonia treatment has been demonstrated to produce catalysts with much higher ORR intrinsic activity;37,38e.g., pyrrole nitrogen can be transferred from pyridine nitrogen by removing additional carbon atoms to generate high-purity FeN4.39 However, metal nitride particles are generally produced during NH3 pyrolysis and decrease the utilization of FexNy species. Therefore, thermal treatment, impregnation, and subsequent NH3 treatment are expected to precisely control the metal content; thus, the isolated active site density and ORR activity are potentially enhanced.
This study presents a methodology for obtaining an enhanced Fe–N–C ORR catalyst, incorporating the trace atomic Co dopants, referred to as Fe(tCo)–N–C, which is highly effective in catalyzing the ORR in half cells and PEMFCs. To prepare Fe(tCo)–N–C, a Co-doped ZIF-8 precursor material was carbonized into a tCo–N–C support, which was then impregnated with Fe3+, followed by an NH3 treatment. Findings from this investigation highlight that the Fe2N8 sites within Fe(tCo)–N–C exhibit notably superior intrinsic ORR activity compared to conventional Fe–N4 sites found in typical Fe–N–C ORR electrocatalysts. This enhancement can be attributed to the regulated electric charge density and lowered d-band center of the iron atom, which facilitates *OOH formation and the generation and desorption of *OH, culminating in a significantly improved ORR performance. Optimized Fe(tCo)–N–C manifests superb ORR activity in a half cell (E1/2 of 0.80 V in HClO4, 0.89 V in KOH) and PEMFC (Pmax of 0.89 W cm−2 for H2–O2, 0.38 W cm−2 for H2–air). More importantly, in contrast to a typical Fe–N–C ORR electrocatalyst, Fe(tCo)–N–C, featuring the Fe2N8 sites and trace atomic Co dopants shows improved stability during fuel cell testing. It retains about 89% of its initial activity even after a 60 hour durability test. The developed catalysts provide great candidates for PGM-free catalysts for half cells and PEMFCs and represent a highly promising avenue for designing single-atom catalysts with bimetallic sites.
2 Results and discussion
The preparation scheme is depicted in Fig. 1a. In addition, Fig. S1† illustrates the synthesis of ZIF-8 and the Co-doped ZIF-8 precursor, denoted ZnCo-ZIF, in line with evidence from X-ray diffraction (XRD). XRD images show that the ZnCo-ZIF precursor maintained the ZIF-8 crystalline structure. We then utilized scanning electron microscopy (SEM) to inspect the ZnCo-ZIF precursor (Fig. S2†). The microstructure of Fe(tCo)–N–C was examined using SEM and a transmission electron microscope (TEM). According to Fig. 1b, target catalysts maintained a uniform rhombic dodecahedron morphology, similar to that of the ZnCo-ZIF precursor (Fig. S2b†), after two carbonization processes. After the last step of thermal treatment in NH3, the target catalysts exhibited a much smoother surface and smaller particle size of approximately 400 nm (Fig. 1b and S3†). Moreover, Fe(tCo)–N–C and its precursor were analyzed by XRD. According to Fig. S4 and S5a,† only two carbon peaks at 23° and 44° were manifested in the investigated samples, assigned to the amorphous carbon plane (002) and graphitized carbon plane (100),39,40 respectively. No metal-related crystalized species were detected, suggesting that all impregnated Fe3+ ions were translated into isolated sites and embedded in the carbon substrate. The disorderliness of the carbon morphology in Fe(tCo)–N–C was also verified via high-resolution (HR) TEM images, as shown in Fig. 1c, which indicates that no distinct particles were visible. Notably, abundant micropores can also be noticed in the HR-TEM pattern of the studied catalysts. Aberration-corrected (AC) HAADF scanning transmission electron microscopy (STEM) revealed uniformly distributed atomically dispersed metal sites (highlighted by bright spots) throughout the carbon matrix, as illustrated in Fig. 1d. Interestingly, in HAADF-STEM images, there are abundant dual bright spots that can be classified as diatomic active sites. Energy-dispersive spectrum (EDS) mapping images demonstrate the even dispersion of primary elements on the carbon substance (Fig. 1e and f). In Fig. 1f, it is evident that the Co content is extremely low. EDS results indicate that the atomic ratio of Fe to Co is as high as 25.9. | ||
| Fig. 1 (a) Graphical synthetic routes of the trace Co-doped Fe–N–C catalyst (Fe(tCo)–N–C). Electron microscopy images in (b) scanning and transmission (b insert); (c) high-resolution transmission; (d and e) AC HAADF scanning transmission; and (f) EDS mapping images of Fe(tCo)–N–C. | ||
To examine the effects of the trace atomic Co dopants, a reference sample without the addition of Co (Fe–N–C) was synthesized. To comprehend structural and compositional distinctions between reference Fe–N–C and corresponding Fe(tCo)–N–C, we conducted XRD, Brunauer–Emmett–Teller (BET) analysis, Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS). As indicated in Fig. S5a,† no distinct metal particles can be seen in either Fe(tCo)–N–C or Fe–N–C, showing the disordered carbon structure, in agreement with HR-TEM observed in Fig. 1c. BET results and details are shown in Fig. S5b and c and Tables S1 and S2,† in which Fe(tCo)–N–C exhibited a greater SSA (1486 m2 g−11) and microporous volume (Vmicro: 0.56 cm3 g−1) than Fe–N–C, showing more favorable structural features and surface areas for the ORR. Moreover, it was observed that typical I-type N2 adsorption isotherms with abundant micropores (<2 nm) are displayed in Fig. S5b and c,† further revealing structural advantages towards the ORR. Meanwhile, Raman analysis further disclosed the degree of defects and graphitization of Fe(tCo)–N–C and Fe–N–C. As depicted in Fig. S5d,† two distinct peaks were observed at 1590 cm−1 and 1350 cm−1, corresponding to G bands (representing graphitic sp2 C) and D bands (indicative of defects in the graphitic structure), respectively.41 A lower ID/IG value (intensity ratio) of Fe(tCo)–N–C compared to that of Fe–N–C revealed that a catalyst containing the trace Co-dopants would show a higher degree of graphitization, which can contribute to reinforcing the stability.
The surface composition was unveiled by an XPS test. XPS survey results in Fig. S6† showed that Fe(tCo)–N–C was different from Fe–N–C as it contained Fe, Co, O, C, and N. However, most Fe and Co atoms are located at the surface of the pore structure rather than at the outer surface of the material, leading to the lower intensity of XPS Co 2p and Fe 2p peaks. Hence, characteristic peaks are not observed in Co 2p and Fe 2p spectra. Table S3† summarizes the detailed C, N and O contents of the above two catalysts according to the XPS test. The ratio of Fe to Co content was also assessed via inductively coupled plasma optical emission spectrometry (ICP-OES) and mass spectrometry (ICP-MS). The Fe/Co ratio in Fe(tCo)–N–C was determined to be 23.7, consistent with the ratio of 25.9 obtained from the EDS mapping results of AC HAADF STEM. This consistency provided further verification of the trace doping of Co in the material. Furthermore, high-resolution N 1s spectroscopy was conducted to elucidate the types and proportions of various nitrogen species (Fig. S7a†).26 According to Fig. S7a and Tables S4 and S5,† both the catalysts prepared through pre-doping and impregnation strategies show abundant active Me–N species and pyridinic N, indicating that more active sites can be obtained through the pre-doping and impregnation strategy. Simultaneously, it becomes apparent that both the catalysts exhibit an abundance of pyrrolic and proper graphitic N species. Theoretical calculations provided significant inspiration by uncovering that pyrrolic N has a dual impact. First, the O2 adsorption energy of bonded Fe atoms is affected. In addition, adjacent C atoms are turned into active sites for the ORR.39,40 Pyridinic N can anchor the metal and lower the ORR overpotential, whereas graphitic N can enhance electrical conductivity.42,43 Moreover, C 1s and O 1s spectra are demonstrated in Fig. S8 and S9.† The C 1s spectra depicted in Fig. S8† indicate that in Fe(tCo)–N–C and Fe–N–C, the main coordinated forms are CC, C–N and CN bonds, in accordance with their ZnCo-ZIF precursor. Meanwhile, Fig. S9† reveals that oxygen is primarily adsorbed onto carbon and metal surfaces.
To further detect the active site coordination of the catalysts, X-ray absorption spectra (XAS) were performed with respect to extended-edge (EXAFS) and near-edge (XANES) measurements. As XANES results in Fig. 2a indicate, Fe(tCo)–N–C has an Fe absorption edge that is positioned in proximity to iron phthalocyanine (FePc), manifesting that the iron valency is approximately +2. In Fig. 2b, the FT-EXAFS spectra of FePc indicate the presence of an Fe–N structure, where the Fe–N signal is visible at around 1.5 Å. In contrast, the FT-EXAFS spectra of Fe2O3 demonstrate the existence of Fe–O bonds, with the Fe–O signal observed around 1.4 Å. Specifically, Fe foil spectra reveals the presence of Fe–Fe bonds, and the Fe–Fe signal is situated at around 2.2 Å. In contrast, our Fe(tCo)–N–C sample exhibits an Fe–N signal, with the notable absence of Fe–Fe and Fe–O signals. This structural configuration closely resembles that of FePc and differs from the Fe2O3 structure. These results suggest that Fe species are atomically distributed and coordinated with N, in conformity to HAADF-STEM and HR-TEM results. In addition, wavelet transform (WT)-EXAFS curves are provided in Fig. 2c to illustrate the atomic alignment and configuration. The maximum intensity was observed only in low k-space (∼3.8 Å−1) on Fe(tCo)–N–C, assigned to Fe–N(O) without Fe–Fe bonding (∼7.5 Å−1) or an Fe–Co path (∼7.8 Å−1),15 further demonstrating the atomic dispersion of Fe species without aggregation. In addition, to obtain coordination information about FeNx in Fe(tCo)–N–C, quantitative EXAFS fitting was implemented (Fig. 2d and e). Table S7† shows specific fitting parameters, in which the Fe–N coordination number is approximately 3.8, and the bond length is 2.01 Å. We conducted XAS analysis on the control sample of Fe–N–C to assess the influence of the introduction of Co on the active site. As depicted in Fig. 2a and b, similar to the Co-containing Fe(tCo)–N–C sample and FePc, the valence state of Fe remains approximately +2, with Fe also existing as isolated atoms. Significantly, the WT-EXAFS curves of Fe–N–C in Fig. 2f offered a slightly different arrangement and configuration, suggesting existing forms of active sites different from those of Fe(tCo)–N–C. By employing quantitative EXAFS fitting, the traditional pyridine-type FeN4 configuration in Fe–N–C was confirmed, and the precise details of its coordination number and bond length are outlined in Table S7.† In contrast with the typical FeN4 configuration (Fig. 2g and S11a†), the Fe(tCo)–N–C catalyst with a larger bond length (2.011 Å vs. 1.93 Å) suggests a different Fe–N configuration rather than typical Fe–N4. To confirm the possible configuration, four reasonable models (Fig. S11b–e†) were proposed, in which the most likely structure (Fe2N8) is identified by considering Fe–N bond distance and coordination number (Fig. 2i and S11e†).
 | ||
| Fig. 2 Fe k-edge (a) XANES spectrum; (b) Fourier transform (FT)-EXAFS spectrum; (c) k2-weighted χ(k) WT-EXAFS spectra; (d and e) EXAFS fitting analysis of Fe(tCo)–N–C. Fe k-edge (f) k2-weighted χ(k) WT-EXAFS spectra; (g and h) EXAFS fitting analysis of Fe–N–C.(i) The proposed configuration of the Fe2N8 active sites. | ||
In this work, the oxygen reduction performance of Fe(tCo)–N–C, Pt/C and other contrasting samples was disclosed by electrochemical results. In Ar- and O2-saturated acid electrolyte, cyclic voltammetry (CV) was conducted to initially assess the ORR activity. According to Fig. 3a, unlike under the Ar-saturated electrolyte where there was no ORR activity, a distinct reduction peak was witnessed in the O2-saturated electrolyte, exhibiting the oxygen reduction activity of the catalysts. Notably, unlike Fe–N–C and tCo–N–C, Fe(tCo)–N–C revealed a more positive peak, indicating its superior ORR activity. Furthermore, by means of linear sweep voltammetry (LSV), the investigated catalysts were further assessed for their catalytic activity. As displayed in Fig. 3b and c, Fe(tCo)–N–C manifests satisfactory performance for ORR with an E1/2 of 0.80 V. This is comparable to Pt/C and surpasses the performance of Fe–N–C, tCo–N–C, and N–C. As shown in Table S9,† Fe(tCo)–N–C is amongst the best PGM-free acidic ORR catalysts. Moreover, the reduced activity observed in tCo–N–C and N–C confirms that FeNx sites are the main place where the ORR occurs. The enhanced ORR catalysis of Fe(tCo)–N–C compared with that of Fe–N–C indicates that possible active sites induced by the trace Co dopants may demonstrate higher intrinsic ORR catalytic activity.
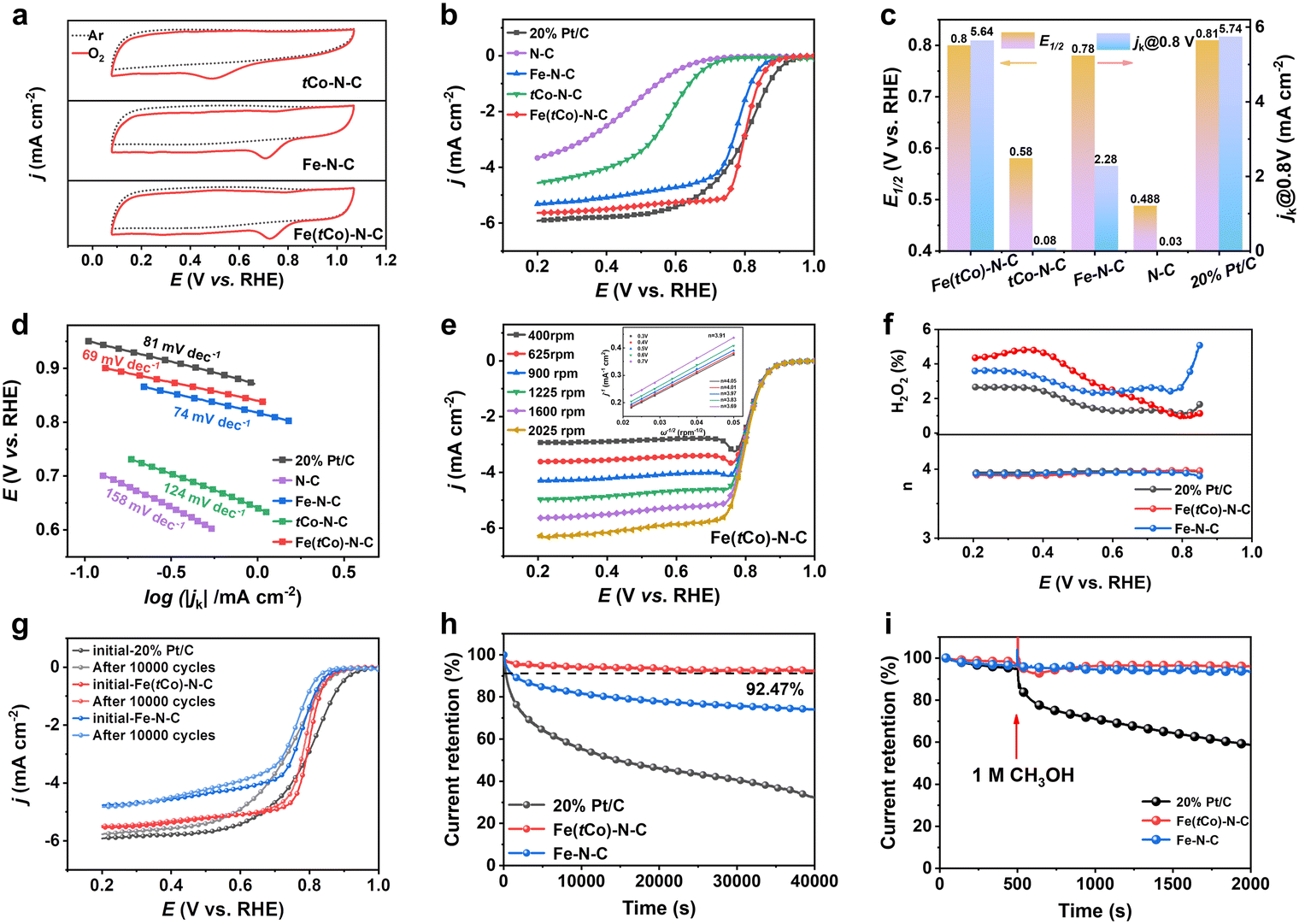 | ||
Fig. 3 (a) CV plots under Ar- and O2-saturated acidic electrolyte of Fe(tCo)–N–C, Fe–N–C, and tCo–N–C; (b) LSV plots (1600 rpm); (c) jk at 0.8 V and E1/2; (d) Tafel slopes for Fe(tCo)–N–C and other control samples; (e) LSV plots of Fe(tCo)–N–C at from 400 to 2025 rotating rates (inset: related K–L curves from 0.3–0.7 V); (f) H2O2 yield and corresponding electron transfer numbers; (g) LSV plots before and after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles within 0.6–1.0 V at 200 mV s−1; (h) CA at 0.7 V (1600 rpm); (i) Tolerance to methanol (1 M) for Pt/C, Fe(tCo)–N–C, and Fe–N–C (0.6 V, 1600 rpm) (O2-saturated 0.1 M HClO4 was applied unless otherwise stated). 000 cycles within 0.6–1.0 V at 200 mV s−1; (h) CA at 0.7 V (1600 rpm); (i) Tolerance to methanol (1 M) for Pt/C, Fe(tCo)–N–C, and Fe–N–C (0.6 V, 1600 rpm) (O2-saturated 0.1 M HClO4 was applied unless otherwise stated). | ||
In this study, the ORR kinetics of various catalysts were investigated to acquire an in-depth understanding. The kinetic current density (jk) and Tafel slopes were calculated and compared among different catalysts. As can be seen from Fig. 3c, Fe(tCo)–N–C exhibited a satisfactory jk at 0.8 V, which was equivalent to that of Pt/C and higher than that of tCo–N–C and Fe–N–C. A minimum Tafel slope was observed in Fe(tCo)–N–C in Fig. 3d, indicating an efficient ORR catalytic kinetic process. Furthermore, using the Koutecky–Levich (K–L) equation, the kinetic process and electron transfer numbers (n) were revealed and obtained, in which linear K–L plots derived from various rotating rate LSV curves and the calculated n verified a first-order ORR kinetic process and a highly selective 4e− transfer in Fe(tCo)–N–C; see Fig. 3e. The reaction pathway was further identified through the implementation of rotating ring disk electrode (RRDE) tests, wherein the acquired n value and H2O2% are depicted in Fig. 3f. Within the test potential range, Fe(tCo)–N–C and Fe–N–C showed satisfactory n values, close to 4 (∼3.94), and the H2O2 yield was below 4.8%, suggesting a desirable 4e− transfer pathway for the ORR. In contrast, Pt/C showed a value of 3.96 and an H2O2 yield below 2.7%. To eliminate the 2e− ORR activity of tCo–N–C, the H2O2 byproduct was measured in this work. It is undeniable that tCo–N–C exhibited significant 2e− activity, as illustrated in Fig. S12.† However, the H2O2 yield for Fe(tCo)–N–C significantly decreased to a level as low as that in Pt/C, suggesting that the 2e− activity was inhibited and nearly negligible in Fe(tCo)–N–C. Instead, its dominant activity was primarily associated with 4e− reactive processes.
To examine the stability of Fe(tCo)–N–C for ORR in an acidic medium, plots of chronoamperometric (CA) measurements at 1600 rpm were assessed for 40000 s. According to Fig. 3h, Fe(tCo)–N–C retained 92.47% of initial current density after 40000 s, whereas, in the same situation, Pt/C and Fe–N–C maintained only 32% and 74%, respectively. The results demonstrated the superiority of Fe(tCo)–N–C synthesized through a pre-doping and impregnation strategy, which was further confirmed by the Raman analysis, which showed a higher degree of graphitization and, thus, higher stability compared with Fe–N–C. Furthermore, an accelerated stability test (AST) was also completed by scanning 10000 cycles at 200 mV s−1 (0.6–1.0 V) to assess the stability. In the acidic electrolyte, after 10000 cycles, the level of E1/2 degradation of Fe(tCo)–N–C was significantly lower than that of Pt/C or Fe–N–C (Fig. 3g). After accelerated stability tests, the samples were subjected to post-reaction characterization, which encompassed TEM observations, XPS analysis, and ICP-MS tests for dissolved metals conducted in electrolytes. As depicted in Fig. S13 and S14,† even after undergoing 10000 CV cycles, the catalysts maintained their original rhombic dodecahedron morphology, showing no evidence of particle cracking, deformation, or aggregation of metal centers. Furthermore, the XPS analysis conducted after the CV cycles and nitrogen species percentage for Fe(tCo)–N–C and Fe–N–C are depicted in Fig. S7b and S15 and Tables S4 and S5.† Simultaneously, through a comparative analysis of nitrogen species content before and after the reaction, it was observed that the loss of pyridinic N and Me–N species in Fe(tCo)–N–C was less than that in Fe–N–C. This observation suggests that Fe–N–C underwent more pronounced electrochemical corrosion during the reaction. An ICP-MS test of the electrolyte after the stability tests was conducted to evaluate the dissolution of metallic sites during the ORR.44 Based on findings presented in Fig. S16 and Table S6,† the concentration of dissolved Fe in the electrolyte for Fe(tCo)–N–C is lower than that in Fe–N–C, indicating that the former is more stable. Apart from stability and activity, tolerance to methanol is also important when applying ORR catalysts in methanol fuel cells. According to Fig. 3i, introducing 1 M methanol had no obvious impact on Fe(tCo)–N–C, but significantly decreased the current on Pt/C. This finding suggests that Fe(tCo)–N–C can resist methanol poisoning, showing potential for application in the methanol fuel cells. Furthermore, the resistance of the tCo–N–C catalyst to methanol poisoning was also evaluated, as shown in Fig. S17.† The test results show that tCo–N–C can resist methanol poisoning, while the current of tCo–N–C in 0.1 M HClO4 is very low.
Additionally, in alkaline electrolytes, Fe(tCo)–N–C exhibits satisfactory ORR performance. Fe(tCo)–N–C demonstrated superior activity for ORR, with an E1/2 of 0.89 V in 0.1 M KOH, which was significantly superior to that of Pt/C (0.84 V) (Fig. S18†). By means of 10000 cycles AST and 40000 s CA measurements, the stability of Fe(tCo)–N–C was assessed. From Fig. S18,† there was almost no decay in activity on Fe(tCo)–N–C after 10000 CV cycles (0.6–1.0 V; 200 mV s−1), while Pt/C showed noticeable deterioration in performance. Besides, in comparison to Pt/C, the stability benefits of Fe(tCo)–N–C were also verified through CA tests (Fig. S19†). The low H2O2 yield below 4% and close-to-4 n also reveal a favorable 4e− reaction pathway (Fig. S20†). Meanwhile, in alkaline medium, Fe(tCo)–N–C demonstrated notable resistance to methanol poisoning, as evidenced by further confirmation in Fig. S21.†
To clarify reasons behind the superior ORR performance demonstrated by Fe(tCo)–N–C compared to Fe–N–C and verify the proposed configuration, DFT calculations were undertaken on the proposed Fe2N8 sites and typical Fe–N4 in Fe–N–C. The adsorbed states of O2 and oxygenated intermediate species (*OOH, *O, *OH) on the Fe–N4 and Fe2–N8 sites are depicted in Fig. S22 and S23.† As can be seen from Fig. 4a, the Gibbs free energy of the two configurations, FeN4 and Fe2N8, was calculated at 1.23 V, 0 V, and the corresponding applied potential. On both sites at U = 0 V, all elementary steps behaved with a consistent decrease in free energy, suggesting a spontaneous reaction process. Moreover, according to the DFT calculations at U = 1.23 V, the formation of *OH in Fe2–N8 and Fe–N4 was the rate-determining step (RDS). For the RDS, the free energy change on the Fe2–N8 structure was lower (0.51 eV) than that on Fe–N4 (0.63 eV), demonstrating that Fe2–N8 is more favorable than Fe–N4 in accelerating the ORR. Additionally, the free energy change on the Fe2–N8 structure was lower than that on Fe–N4 in view of first (*O2 → *OOH) and last (*OH → H2O) steps, indicating easier formation of *OOH and desorption of *OH intermediates on Fe2–N8, resulting in a favorable 4e− reaction process. To further explain the superiority of the Fe2N8 sites, the electronic structure of Fe centers on the Fe2–N8 and Fe–N4 sites was examined using Bader charge analysis. According to Fig. 4b and c, there was a slight increase in charge transport from the iron center to surrounding N atoms at the Fe2–N8 site in comparison to Fe–N4, which could be related to the elongation of the Fe–N bond.45 Based on the regulated charge density on Fe centers, the adsorption of intermediate species *O would be weakened and dissociation would be facilitated, further facilitating the ORR. Moreover, the PDOS (projected density of states) of the iron atoms in Fe2–N8 was also analyzed to further reveal the origin of the higher ORR activity. As indicated in Fig. 4d and e and Table S8,† a clear negative shift in the Fe-d orbital on Fe2–N8 can be observed compared to Fe–N4, which represents a d band center shift toward lower energies and an increase in filling of antibonding orbitals.46 This increased filling of Fe antibonding orbitals would weaken the interplay between Fe sites and adsorbate *O2/*O/*OH, thus boosting the oxygen reduction process. These results explain the higher intrinsic ORR activity observed on Fe2–N8 sites in the Fe(tCo)–N–C catalyst compared to that on the typical Fe–N4 sites, which is consistent with electrochemical data. Furthermore, the proposed ORR process on an Fe2–N8 site in Fe(tCo)–N–C is shown in Fig. 4f.
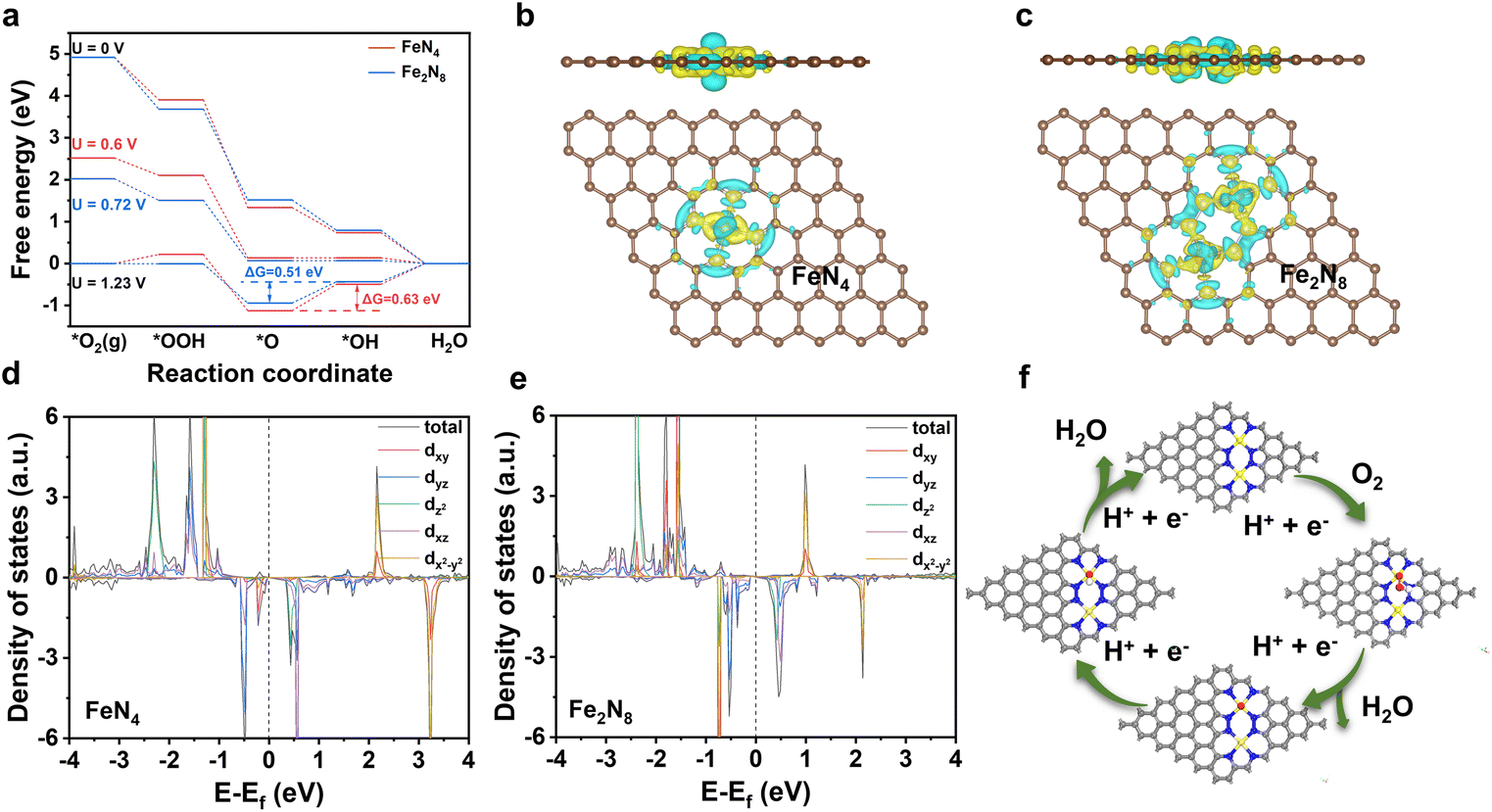 | ||
| Fig. 4 DFT calculations of the ORR mechanism and electronic structures of Fe2N8 and FeN4. (a) Free energy diagrams of Fe2N8 and FeN4 at 0 V, 1.23 V, and the applied potential (0.6 V for FeN4 and 0.72 V for Fe2N8). The difference in charge distribution on (b) FeN4 and (c) Fe2N8; yellow (blue) represents the charge accumulation (depletion). The PDOS curves of (d) FeN4 and (e) Fe2N8 (Fe-d orbitals). (f) The proposed ORR process on an Fe2–N8 site in Fe(tCo)–N–C. | ||
Herein, practical application of Fe(tCo)–N–C as a cathode catalyst was evaluated by assembling a PEMFC using Pt/C as an anode catalyst (Fig. 5a). Scribner 850e with O2 (or air) and H2 at 80 °C under 100% RH (relative humidity) was used to evaluate the PEMFC performance. Flow rates of 0.4 and 0.3 L min−1 for O2 (or air) and H2, respectively, were adopted. Fe–N–C without the trace Co dopants was also tested for comparison. Impressively, PEMFC with Fe(tCo)–N–C showed high activity and power densities (Pmax) in H2–O2 and H2–air atmospheres. As can be seen from Fig. 5b–e, the high OCV (open circuit voltage) for Fe(tCo)–N–C and contrast catalyst (0.98 V under H2–O2, 0.93 V under H2–air) indicated that they have high ORR intrinsic activity. Moreover, Fe(tCo)–N–C displayed much more favorable polarization curves with rising current density compared to Fe–N–C. Under 22 psi O2, 0.31 A cm−2 (@0.8 V) was achieved on Fe(tCo)–N–C, which was double that of Fe–N–C (0.16 A cm−2@0.8 V). Increasing the air pressure to 22 psi, 0.12 A cm−2@0.8 V was generated on Fe(tCo)–N–C, which was triple that of Fe–N–C (0.04 A cm−2@0.8 V). Furthermore, the corresponding Pmax for Fe(tCo)–N–C and Fe–N–C were also obtained. Based on data presented in Fig. 5b and c, Fe(tCo)–N–C demonstrated a significantly higher Pmax of approximately 890 mW cm−2 in H2–O2 and 380 mW cm−2 in H2–air, while Fe–N–C exhibited inferior values in H2–O2 and H2–air, as can clearly be seen from Fig. 5d and e. The high-intrinsic-activity Fe2N8 active centers, dominant microporous SSA, abundant porous structure, and good electroconductivity can assist in increasing the performance of Fe(tCo)–N–C in PEMFCs by efficiently decreasing the activation, ohmic, and concentration polarization. Furthermore, the abundant porosity can suppress micropore flooding; thus, the utilization rate of catalytic sites and catalyst deactivation can be increased and prevented, respectively. The results described above illustrate that Fe(tCo)–N–C could effectively facilitate oxygen reduction and show great prospects as a PGM-free catalyst for PEMFC applications. To assess the stability of the as-prepared catalyst for practical application in PEMFC and verify the effect of the addition of Co on PEMFC stability, we performed a 60 hour test of chronoamperometric current for Fe(tCo)–N–C and Fe–N–C. As shown in Fig. 5f, the stability of Fe(tCo)–N–C with Co dopants is higher than that of Fe–N–C without Co during fuel cell tests. The results indicate that Fe(tCo)–N–C maintained 89% of its original current density after continuous operation for 60 h.
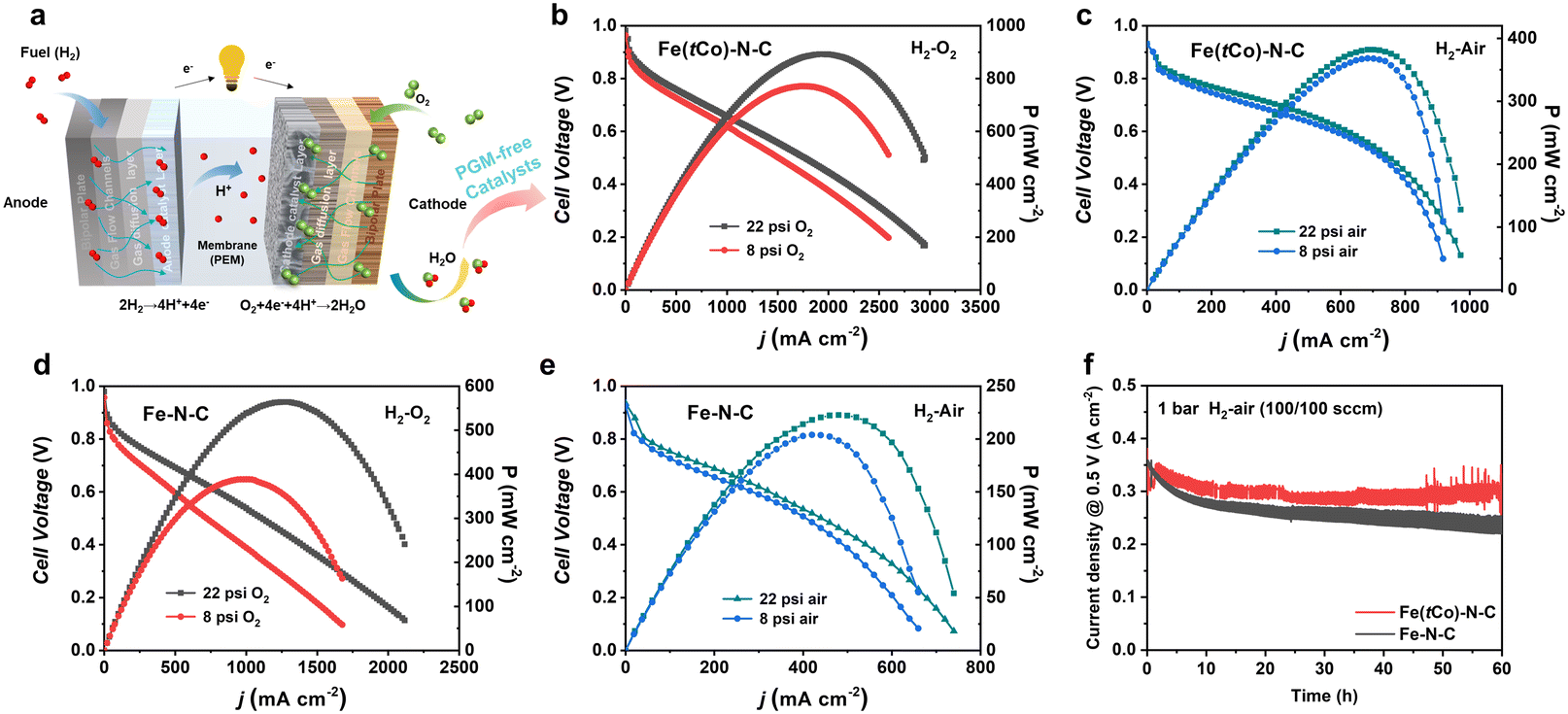 | ||
| Fig. 5 (a) Schematic plot of PEMFCs. Performance plots of Fe(tCo)–N–C in (b) H2–O2 and (c) H2–air. PEMFC results of Fe–N–C in (d) H2–O2 and (e) H2–air. (f) Current density–time function of a single cell, with a constant voltage of 0.5 V. Cathode loading: 3 mg cm−2; 80 °C; relative humidity: 100%. | ||
3 Conclusions
In this study, a superior trace atomic Co-doped Fe(tCo)–N–C electrocatalyst was successfully developed through a pre-doping, impregnation and ammonia activation strategy. Fe(tCo)–N–C had E1/2 of 0.89 V and 0.80 V in alkaline and acidic electrolytes for the ORR, respectively. The trace Co dopants effectively facilitated the formation of the Fe2N8 sites instead of the typical FeN4 structure in common Fe–N–C electrocatalysts and enhanced the degree of graphitization, making for high ORR stability. Fe(tCo)–N–C displayed a high Pmax of 890 mW cm−2 when used in PEMFCs with H2–O2 and 380 mW cm−2 in H2–air conditions. More importantly, unlike a typical Fe–N–C ORR electrocatalyst, Fe(tCo)–N–C with the Fe2N8 sites and trace atomic Co dopants shows improved stability during fuel cell testing. The results indicate that Fe(tCo)–N–C still retained 89% of its original current density after continuous operation for 60 h. Advanced synchrotron experiments confirmed the configuration of the Fe2N8 sites with elongated Fe–N bonds compared with FeN4 in Fe–N–C, while the DFT calculations elucidated the relation between the elongated Fe–N bond and catalytic activity for Fe(tCo)–N–C. Benefiting from the reduced d-band center and improved occupation of antibonding orbitals, the interplay between the Fe sites and adsorbates can be moderated, effectively facilitating the desorption of *OH and production of *OOH/*OH. Additionally, the decreased charge density of the Fe atoms caused by the elongated Fe–N bond effectively hindered the adsorption of *O species. This research offers an updated design approach to heighten PGM-free PEMFC performance by adjusting the electronic and geometric structures of a catalyst.4 Experimental section
4.1 Materials
All materials, including cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O) (99%) (Aladdin), ferric chloride (FeCl3) (90%) (Macklin), 2-methylimidazole (98%) (Macklin), zinc(II) nitrate hexahydrate (Zn(NO3)2·6H2O) (99%) (Aladdin), isopropanol (99.5%) (Macklin), and methanol (99.5%) (Sinopharm Chem. Reagent Co., Ltd.), were used without further purification. A Johnson Matthey 20% Pt/C catalyst was applied. Nafion™ 117 (∼5 wt%) was acquired from Sigma Aldrich. Deionized water (18.2 MΩ specific resistance) was employed in this study.4.2 Catalyst preparations
Fig. 1a describes the process for synthesizing Fe(tCo)–N–C. Initially, a Co-doped ZIF-8 precursor (ZnCo-ZIF) was obtained by thoroughly stirring a methanol solution of transition metal salts and dimethylimidazole. Then, the as-prepared ZnCo-ZIF precursor was carbonized to produce a tCo–N–C support with Co doping to adsorb Fe3+ ions. Subsequently, tCo–N–C powders adsorbed with a certain amount of Fe3+ (tCo–N–C + Fe3+) were subjected to thermal activation in an NH3 atmosphere for 15 min to make final Fe(tCo)–N–C. Reference samples of the N–C support stemming from ZIF-8 pyrolysis, and Fe–N–C without Co doping derived from N–C impregnation and thermal treatment were also prepared. Details on the catalyst preparation, characterization, electrochemical measurements, and theoretical calculations are available in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (No. 2202052 and Z200012), National Natural Science Foundation of China (No. 92061125), Hebei Natural Science Foundation (B2020103043), Science and Technology Program of Inner Mongolia (2021GG0237), and Key R&D Program of Henan Province (No. 231111241800). The authors truly appreciate Prof. Suojiang Zhang (IPE, CAS) for his great support and careful academic guidance.References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, eaad4998 CrossRef PubMed.
- C. F. Shih, T. Zhang, J. Li and C. Bai, Powering the future with liquid sunshine, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- M. Muhyuddin, E. Berretti, S. A. Mirshokraee, J. Orsilli, R. Lorenzi, L. Capozzoli, F. D'Acapito, E. Murphy, S. Guo, P. Atanassov, A. Lavacchi and C. Santoro, Formation of the active site structures during pyrolysis transformation of Fe-phthalocyanine into Fe-Nx-C electrocatalysts for the oxygen reduction reaction, Appl. Catal., B, 2024, 343, 123515 CrossRef CAS.
- Y. Persky, Y. Yurko, R. Z. Snitkoff-Sol, N. Zion and L. Elbaz, Tuning the performance of Fe-porphyrin aerogel-based PGM-free oxygen reduction reaction catalysts in proton exchange membrane fuel cells, Nanoscale, 2024, 16, 438–446 RSC.
- L. Peles-Strahl, H. C. Honig, Y. Persky, D. A. Cullen, A. Dahan and L. Elbaz, Modular Iron-bipyridine-based conjugated aerogels as catalysts for oxygen reduction reaction, ACS Catal., 2023, 13, 14377–14384 CrossRef CAS.
- S. A. Mirshokraee, M. Muhyuddin, J. Orsilli, E. Berretti, A. Lavacchi, C. Lo Vecchio, V. Baglio, R. Viscardi, A. Zaffora, F. Di Franco, M. Santamaria, L. Olivi, S. Pollastri and C. Santoro, Mono-, bi- and tri-metallic Fe-based platinum group metal-free electrocatalysts derived from phthalocyanine for oxygen reduction reaction in alkaline media, Nanoscale, 2024, 16, 6531–6547 RSC.
- S. A. Mirshokraee, M. Muhyuddin, J. Orsilli, E. Berretti, L. Capozzoli, A. Lavacchi, C. Lo Vecchio, V. Baglio, A. Galli, A. Zaffora, F. Di Franco, M. Santamaria, L. Olivi, S. Pollastri and C. Santoro, Mono-, bi- and tri-metallic platinum group metal-free electrocatalysts for hydrogen evolution reaction following a facile synthetic route, Ind. Chem. Mater., 2023, 1, 343–359 RSC.
- H. Zhang, L. Osmieri, J. H. Park, H. T. Chung, D. A. Cullen, K. C. Neyerlin, D. J. Myers and P. Zelenay, Standardized protocols for evaluating platinum group metal-free oxygen reduction reaction electrocatalysts in polymer electrolyte fuel cells, Nat. Catal., 2022, 5, 455–462 CrossRef CAS.
- J. Cui, Q. Chen, X. Li and S. Zhang, Recent advances in non-precious metal electrocatalysts for oxygen reduction in acidic media and PEMFCs: An activity, stability and mechanism study, Green Chem., 2021, 23, 6898–6925 RSC.
- S. T. Thompson and D. Papageorgopoulos, Platinum group metal-free catalysts boost cost competitiveness of fuel cell vehicles, Nat. Catal., 2019, 2, 558–561 CrossRef CAS.
- D. Banham and S. Ye, Current status and future development of catalyst materials and catalyst layers for proton exchange membrane fuel cells: An industrial perspective, ACS Energy Lett., 2017, 2, 629–638 CrossRef CAS.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- L. Xue, Y. Li, X. Liu, Q. Liu, J. Shang, H. Duan, L. Dai and J. Shui, Zigzag carbon as efficient and stable oxygen reduction electrocatalyst for proton exchange membrane fuel cells, Nat. Commun., 2018, 9, 3819 CrossRef PubMed.
- R. Jasinski, A new fuel cell cathode catalyst, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- M. Xiao, Y. Chen, J. Zhu, H. Zhang, X. Zhao, L. Gao, X. Wang, J. Zhao, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, Climbing the apex of the ORR volcano plot via binuclear site construction: Electronic and geometric engineering, J. Am. Chem. Soc., 2019, 141, 17763–17770 CrossRef CAS PubMed.
- H. Wang, L. Osmieri, H. Yu, M. J. Zachman, J. H. Park, N. N. Kariuki, F. C. Cetinbas, S. Khandavalli, S. Mauger, D. J. Myers, D. A. Cullen and K. C. Neyerlin, Elucidating the impact of the ionomer equivalent weight on a platinum group metal-free PEMFC cathode via oxygen limiting current, SusMat, 2023, 3, 72–90 CrossRef CAS.
- H. Zhang, H. Gu, G. Shi, K. Yu, C. Yang, H. Tong, S. Zhao, M. Chang, C. Zhu, C. Chen and L. Zhang, Two-dimensional covalent framework derived nonprecious transition metal single-atomic-site electrocatalyst toward high-efficiency oxygen reduction, Nano Lett., 2023, 23, 3803–3809 CrossRef CAS PubMed.
- C.-W. Ye and L. Xu, Recent advances in the design of a high performance metal-nitrogen-carbon catalyst for the oxygen reduction reaction, J. Mater. Chem. A, 2021, 9, 22218–22247 RSC.
- L. Ni, C. Gallenkamp, S. Wagner, E. Bill, V. Krewald and U. I. Kramm, Identification of the catalytically dominant iron environment in iron- and nitrogen-doped carbon catalysts for the oxygen reduction reaction, J. Am. Chem. Soc., 2022, 144, 16827–16840 CrossRef CAS PubMed.
- J. Tao, X. Wang, M. Xu, C. Liu, J. Ge and W. Xing, Non-noble metals as activity sites for ORR catalysts in proton exchange membrane fuel cells (PEMFCs), Ind. Chem. Mater., 2023, 1, 388–409 RSC.
- J. Luo, G. I. N. Waterhouse, L. Peng and Q. Chen, Recent progress in high-loading single-atom catalysts and their applications, Ind. Chem. Mater., 2023, 1, 486–500 RSC.
- H. Wang, B. H. Chen and D. J. Liu, Metal-organic frameworks and metal-organic gels for oxygen electrocatalysis: Structural and compositional considerations, Adv. Mater., 2021, 33, 2008023 CrossRef CAS PubMed.
- M. Gong, A. Mehmood, B. Ali, K.-W. Nam and A. Kucernak, Oxygen reduction reaction activity in non-precious single-atom (M–N/C) catalysts-contribution of metal and carbon/nitrogen framework-based sites, ACS Catal., 2023, 13, 6661–6674 CrossRef CAS PubMed.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M. T. Sougrati, M. Primbs, A. M. Bonastre, D. Fongalland, G. Drazic, P. Strasser and A. Kucernak, High loading of single atomic iron sites in Fe-NC oxygen reduction catalysts for proton exchange membrane fuel cells, Nat. Catal., 2022, 5, 311–323 CrossRef CAS.
- L. Jiao, J. Li, L. I. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Chemical vapour deposition of Fe-N-C oxygen reduction catalysts with full utilization of dense Fe-N4 sites, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Atomic-level modulation of electronic density at cobalt single-atom sites derived from metal-organic frameworks: Enhanced oxygen reduction performance, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, An isolated zinc-cobalt atomic pair for highly active and durable oxygen reduction, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Progress in the development of Fe-based PGM-free electrocatalysts for the oxygen reduction reaction, Adv. Mater., 2019, 31, 1806545 CrossRef PubMed.
- W. Wang, Q. Jia, S. Mukerjee and S. Chen, Recent insights into the oxygen-reduction electrocatalysis of Fe/N/C materials, ACS Catal., 2019, 9, 10126–10141 CrossRef CAS.
- D. Zhang, W. Chen, Z. Li, Y. Chen, L. Zheng, Y. Gong, Q. Li, R. Shen, Y. Han, W. C. Cheong, L. Gu and Y. Li, Isolated Fe and Co dual active sites on nitrogen-doped carbon for a highly efficient oxygen reduction reaction, Chem. Commun., 2018, 54, 4274–4277 RSC.
- S. H. Yin, J. Yang, Y. Han, G. Li, L. Y. Wan, Y. H. Chen, C. Chen, X. M. Qu, Y. X. Jiang and S. G. Sun, Construction of highly active metal-containing nanoparticles and FeCo-N4 composite sites for the acidic oxygen reduction reaction, Angew. Chem., Int. Ed., 2020, 59, 21976–21979 CrossRef CAS PubMed.
- B. Wang, J. Zou, X. Shen, Y. Yang, G. Hu, W. Li, Z. Peng, D. Banham, A. Dong and D. Zhao, Nanocrystal supracrystal-derived atomically dispersed Mn-Fe catalysts with enhanced oxygen reduction activity, Nano Energy, 2019, 63, 103851 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B. A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J. N. Zhang, Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- M. Liu, N. Li, S. Cao, X. Wang, X. Lu, L. Kong, Y. Xu and X. H. Bu, A “pre-constrained metal twins” strategy to prepare efficient dual-metal-atom catalysts for cooperative oxygen electrocatalysis, Adv. Mater., 2022, 34, 2107421 CrossRef CAS PubMed.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Synergistic effect of well-defined dual sites boosting the oxygen reduction reaction, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- Z. Qiao, S. Hwang, X. Li, C. Wang, W. Samarakoon, S. Karakalos, D. Li, M. Chen, Y. He, M. Wang, Z. Liu, G. Wang, H. Zhou, Z. Feng, D. Su, J. S. Spendelow and G. Wu, 3D porous graphitic nanocarbon for enhancing the performance and durability of Pt catalysts: A balance between graphitization and hierarchical porosity, Energy Environ. Sci., 2019, 12, 2830–2841 RSC.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells, Science, 2009, 324, 71–74 CrossRef PubMed.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- N. Zhang, T. Zhou, M. Chen, H. Feng, R. Yuan, C. a. Zhong, W. Yan, Y. Tian, X. Wu, W. Chu, C. Wu and Y. Xie, High-purity pyrrole-type FeN4 sites as a superior oxygen reduction electrocatalyst, Energy Environ. Sci., 2020, 13, 111–118 RSC.
- J. Guo, B. Li, Q. Zhang, Q. Liu, Z. Wang, Y. Zhao, J. Shui and Z. Xiang, Highly accessible atomically dispersed Fe-Nx sites electrocatalyst for proton-exchange membrane fuel cell, Adv. Sci., 2021, 8, 2002249 CrossRef CAS PubMed.
- X. F. Gong, J. B. Zhu, J. Z. Li, R. Gao, Q. Y. Zhou, Z. Zhang, H. Z. Dou, L. Zhao, X. L. Sui, J. J. Cai, Y. L. Zhang, B. Liu, Y. F. Hu, A. P. Yu, S. H. Sun, Z. B. Wang and Z. W. Chen, Self-templated hierarchically porous carbon nanorods embedded with atomic Fe-N4 active sites as efficient oxygen reduction electrocatalysts in Zn-Air batteries, Adv. Funct. Mater., 2021, 31, 2008085 CrossRef CAS.
- Y. J. Wu, Y. C. Wang, R. X. Wang, P. F. Zhang, X. D. Yang, H. J. Yang, J. T. Li, Y. Zhou, Z. Y. Zhou and S. G. Sun, Three-dimensional networks of S-doped Fe/N/C with hierarchical porosity for efficient oxygen reduction in polymer electrolyte membrane fuel cells, ACS Appl. Mater. Interfaces, 2018, 10, 14602–14613 CrossRef CAS PubMed.
- L. Liu, G. Zeng, J. Chen, L. Bi, L. Dai and Z. Wen, N-doped porous carbon nanosheets as pH-universal ORR electrocatalyst in various fuel cell devices, Nano Energy, 2018, 49, 393–402 CrossRef CAS.
- J. Xu, H. Jin, T. Lu, J. Li, Y. Liu, K. Davey, Y. Zheng and S. Z. Qiao, IrOxnH2O with lattice water-assisted oxygen exchange for high-performance proton exchange membrane water electrolyzers, Sci. Adv., 2023, 9, eadh1718 CrossRef CAS PubMed.
- Z. Miao, X. Wang, Z. Zhao, W. Zuo, S. Chen, Z. Li, Y. He, J. Liang, F. Ma, H.-L. Wang, G. Lu, Y. Huang, G. Wu and Q. Li, Improving the stability of non-noble-metal M-N-C catalysts for proton-exchange-membrane fuel cells through M-N bond length and coordination regulation, Adv. Mater., 2021, 33, 2006613 CrossRef CAS PubMed.
- Z. Wang, X. Jin, C. Zhu, Y. Liu, H. Tan, R. Ku, Y. Zhang, L. Zhou, Z. Liu, S.-J. Hwang and H. J. Fan, Atomically dispersed Co2-N6 and Fe-N4 costructures boost oxygen reduction reaction in both alkaline and acidic media, Adv. Mater., 2021, 33, 2104718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4im00043a |
| This journal is © Institute of Process Engineering of CAS 2024 |