
Digital microfluidics with distance-based detection – a new approach for nucleic acid diagnostics†
Man
Ho
ab,
N.
Sathishkumar
 ab,
Alexandros A.
Sklavounos
ab,
Jianxian
Sun
a,
Ivy
Yang
c,
Kevin P.
Nichols
d and
Aaron R.
Wheeler
*abe
ab,
Alexandros A.
Sklavounos
ab,
Jianxian
Sun
a,
Ivy
Yang
c,
Kevin P.
Nichols
d and
Aaron R.
Wheeler
*abe
aDepartment of Chemistry, University of Toronto, 80. St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: aaron.wheeler@utoronto.ca
bDonnelly Centre for Cellular and Biomolecular Research, University of Toronto, 160 College Street, Toronto, Ontario M5S 3E1, Canada
cDepartment of Chemical Engineering & Applied Chemistry, University of Toronto, 200 College Street, Toronto, Ontario M5S 3E5, Canada
dGlobal Health Labs, Bellevue, WA 98007, USA
eInstitute of Biomedical Engineering, University of Toronto, 164 College Street, Toronto, Ontario M5S 3G9, Canada
First published on 21st November 2023
Abstract
There is great enthusiasm for using loop-mediated isothermal amplification (LAMP) in point-of-care nucleic acid amplification tests (POC NAATs), as an alternative to PCR. While isothermal amplification techniques like LAMP eliminate the need for rapid temperature cycling in a portable format, these systems are still plagued by requirements for dedicated optical detection apparatus for analysis and manual off-chip sample processing. Here, we developed a new microfluidic system for LAMP-based POC NAATs to address these limitations. The new system combines digital microfluidics (DMF) with distance-based detection (DBD) for direct signal readout. This is the first report of the use of (i) LAMP or (ii) DMF with DBD – thus, we describe a number of characterization steps taken to determine optimal combinations of reagents, materials, and processes for reliable operation. For example, DBD was found to be quite sensitive to background signals from low molecular weight LAMP products; thus, a Capto™ adhere bead-based clean-up procedure was developed to isolate the desirable high-molecular-weight products for analysis. The new method was validated by application to detection of SARS-CoV-2 in saliva. The method was able to distinguish between saliva containing no virus, saliva containing a low viral load (104 genome copies per mL), and saliva containing a high viral load (108 copies per mL), all in an automated system that does not require detection apparatus for analysis. We propose that the combination of DMF with distance-based detection may be a powerful one for implementing a variety of POC NAATs or for other applications in the future.
Introduction
The global SARS-CoV-2 pandemic has highlighted a need for point-of-care (POC) disease diagnostics that can be operated outside of the laboratory. Among many technologies that can be used for this application, nucleic acid amplification tests (NAATs) are particularly attractive because of their high analytical and clinical sensitivities.1,2 The most popular format for POC NAATs are methods that rely on the polymerase chain reaction (PCR), which are advantageous because they are directly comparable to the gold-standard laboratory diagnostics that also rely on PCR.3 A disadvantage of PCR is the inherent requirement for temperature cycling, which can be challenging to package into a miniaturized device footprint. Thus, there is great interest in the development of isothermal techniques relying on methods that do not require temperature cycling, which can be more amenable to miniaturization.The most common isothermal technique used in NAATs is loop-mediated isothermal amplification (LAMP).4 In addition to not requiring temperature cycling, LAMP combines high specificity (as it employs multiple primers specific to the target sequence5) with rapid reaction kinetics, requiring only ∼30 minutes to complete a given amplification.6–8 For RNA viruses such as SARS-CoV-2, the reverse transcribed variant of this technique, RT-LAMP,6,8 is preferred, as it first converts viral RNA to DNA and then amplifies the DNA sequences for detection. There are several recent reports of miniaturized systems that use RT-LAMP to detect SARS-CoV-2 (ref. 1, 6, 7 and 9–14) and other diseases,15–19 and there is a growing interest in the development of alternate LAMP reagents designed for field applications.20 These are important advances for the field; however, to date, all systems reported have required (I) the use of a dedicated detection apparatus for analysis, and/or (II) off-chip sample processing prior to loading onto the device.
Here, we have developed an RT-LAMP-based microfluidic system for SARS-CoV-2 that overcomes limitations (I) and (II). To tackle limitation (I) (the requirement of dedicated detectors), we have adopted a technique known as distance-based detection (DBD), that was developed and popularized by the Henry group at Colorado State University.21 In distance-based detection, the analyte is wicked into a paper matrix, and its quantity is revealed (by eye, with no requirement of a “detector”) by a coloured or fluorescent trail left on the surface. With proper attention to concentrations and kinetics, the length of the trail can “semi-quantitatively” reveal the amount of analyte being investigated. The method was initially developed and applied to environmental applications,21–26 while variations of this technique have recently been applied to detect PCR-amplified DNA in NAAT applications.27,28 To our knowledge, distance-based detection has never been used with LAMP for post-amplification analysis.
Limitation (II) (the requirement of off-chip sample processing prior to loading onto the device) is a pernicious one in the microfluidics community, often “hidden” or not emphasized in papers describing the latest results. However, in practice, the requirement for the user to perform manual processing steps prior to use is a substantial limitation that makes many potential POC techniques impractical for use in the field.2 Here we used digital microfluidics (DMF) to address this limitation, providing automated sample processing, and reducing the burden on the user. DMF has been used to automate PCR,29–32 LAMP33,34 and other NAAT techniques,35,36 but has never before been combined with distance-based detection.
The process introduced here is summarized in Fig. 1. A DMF device designed for isothermal amplification was adapted from previous work35 and an automated sample processing protocol was developed and implemented to amplify the target nucleic acid (SARS-CoV-2 RNA). Following the amplification, the DMF system facilitated the purification of the produced amplicon solution using Capto™ adhere microbeads and the introduction of intercalating dye and the final delivery to the DBD substrate. We propose that the innovations introduced here may represent a general approach that can be useful for a wide range of POC diagnostic applications in the future.
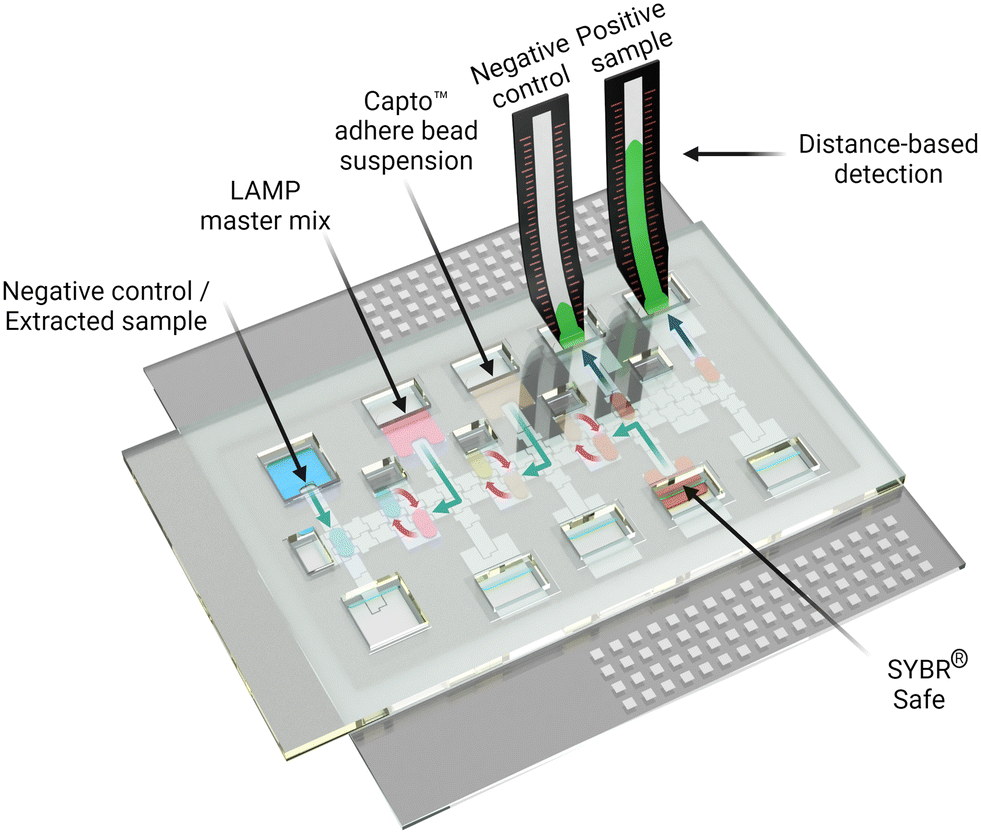 | ||
| Fig. 1 Digital microfluidics for LAMP-based diagnostics with distance-based detection. In the rendering, extracted viral RNA (blue) is loaded into a DMF cartridge. LAMP master mix (pink) is also introduced, and the cartridge is heated to 65 °C. LAMP amplicons (yellow) are then cleaned up using Capto™ adhere beads (Lt. brown). Processed LAMP amplicons are introduced into a DBD substrate after mixing with intercalating dye (SYBR® Safe, Orange). The fluorescent signal (green) on the DBD allows for semi-quantitative measurement of viral load in the saliva sample. | ||
Experimental
Reagents and materials
Unless specified otherwise, reagents were purchased from Invitrogen™ ThermoFisher Scientific (Mississauga, Canada). Tetronic 90R4 (BASF Corp., Germany) was generously donated by BASF Corporation (Wyandotte, USA). Parylene-C dimer was from Specialty Coating System (Indianapolis, USA). Chromium and photoresist-coated glass slides (2 in × 3 in) used to fabricate DMF devices were purchased from Telic Company (Valencia, USA). ITO-coated glass slides (25 mm × 75 mm × 0.7 mm) were purchased from Riley Supplies (Richmond Hill, Canada). FluoroPel 1101V and PFC110 solvent were purchased from Cytonix, LLC (Beltsville, USA). Acrylic sheets and ITO-coated ITO-coated polyethylene terephthalate film (PET-ITO) were purchased from McMaster-Carr (Princeton, NJ) and Memcon (Stevensville, MI) respectively. Medical grade, pressure-sensitive adhesive tapes (AR Care® 90106NB and 7761-19) were graciously donated by Adhesives Research (Glen Rock, PA). Whatman™ Grade 1 chromatography paper (200 × 200 cm) and grade 4 filter paper (240 mm diameter) were purchased from GE Healthcare (Mississauga, Canada). Clear laminating plastic sheets were purchased from Amazon (Amazon, Canada). PCR plate sealing film (Microseal ‘B’) and Hard-shell PCR plates (96 wells, thin wall) were purchased from Bio-Rad (Mississauga, Canada). SARS-CoV-2 rapid colorimetric LAMP assay kits (E2019S, Lot #10112519) were purchased from New England Biolabs (Ipswich, USA). Inactivated cultured SARS-CoV-2 viral samples at 1.2 × 106 plaque forming units per milliliter (PFU mL−1) were generously provided by the National Microbiology Laboratory (NML) of the Public Health Agency of Canada. Capto™ adhere anion exchangers agarose beads (25 mL, Part# GE17-5444-10) were purchased from Sigma Aldrich (Oakville, Canada). A pooled human saliva sample (pre-pandemic, COVID-19 negative, #MBS170697) was purchased from MyBioSource (San Diego, USA).Fabrication of distance-based detection substrates
Distance-based detection substrates, each comprising a loading zone (circle, 5 mm diameter) and a liquid flowing zone (35 mm length with 1 mm tick markings along the length, and 3 mm width), were formed using techniques adapted from those described by Wang et al.27 Briefly, the designs were formed using Inkscape and then printed on Whatman™ grade 1 and grade 4 paper substrates using a Xerox ColorQube 8580 Wax Printer (North York, Canada) with “photo” mode activated. The patterned paper substrates were then transferred to a hot plate and baked at 120 °C for 1 minute. After cooling, two printed sheets were placed with the top sides facing each other and were placed in plastic laminating sheets and laminated using a laminator (TCC-330, Tamerica, USA) at 120 °C. The sheets were then separated, yielding two DBD substrates (each with a layer of laminate on its back).Digital microfluidic device/cartridge fabrication and operation
Digital microfluidic devices (two plates separated by a spacer) and cartridges (devices featuring a plastic enclosure and integrated reservoirs) were manufactured in-house. Briefly, devices were formed from two plates: an ITO-coated top plate and a bottom plate with patterned electrodes on a glass substrate. The bottom plates were fabricated from 2 in × 3 in chromium-coated glass substrates at the Toronto Nanofabrication Center (TNFC) as described previously35via UV photolithography and wet etching. Patterned bottom plates were then coated with a ∼6 μm layer of parylene-C via chemical vapor deposition at the TNFC, and then spin-coated with 1% w/w Fluoropel 1101V dissolved in PFC110 at 2000 rpm for 30 seconds. The coated bottom plates were post-baked in a dry oven at 120 °C for 15 minutes. Each bottom plate featured an array of 80 actuation electrodes (2.2 × 2.2 mm each) connected to 8 reservoir electrodes (16.4 × 6.7 mm each). The actuation electrodes were square with interdigitated borders (140 μm peak-to-peak sinusoid), with inter-electrode gaps of 30–80 μm. The top plates were prepared by spin-coating ITO-coated glass slides with Fluoropel as described above for the bottom plates and then were heated on a hot plate at 120 °C for 15 minutes. Devices were assembled by joining the top and bottom plates with spacers formed from two layers of double-sided tape (3M Company, Maplewood, USA) (∼180 μm). The volume of a unit droplet (i.e., one that covers the actuation electrode) was ∼1.3 μL.Digital microfluidic cartridges were fabricated and assembled as described previously.35 Briefly, 3 in × 3 in bottom plates were formed as above. The top plates were formed from rigid and transparent acrylic (1.5 mm thick) interfaced to PET-ITO (MITO-60-125, 60 Ω sq.−1, 125 μm thick). Both acrylic and PET-ITO substrates were diced into 8 cm × 5 cm pieces and punctuated with through-holes to form reservoirs35 using a 40 W CO2 benchtop laser cutter (Full Spectrum Laser). The acrylic and PET-ITO substrates were bound together using a bio-compatible tape (ARcare® 90106 NB), dip-coated in 1% w/w Fluoropel 1101V in PFC110, spun at 2000 rpm for 30 s to remove excess solution, and then dried at room temperature for 30 minutes. Each DMF cartridge was assembled by joining an acrylic/PET-ITO top plate with a bottom plate using a double-stacked adhesive gasket (AR-Care® 7761-19 medical grade tape, ∼220 μm).
After the formation of devices or cartridges, a DBD substrate was trimmed to expose the edge of the loading zone and then was inserted between the top and bottom plates on DMF devices (through one of the through-holes for cartridges) and positioned such that the loading zone was positioned on one of the reservoirs. Each device or cartridge was then interfaced through pogo-pin connectors to DropBot, a digital microfluidic control system which is described in detail elsewhere37 (specifically, a custom/modified version of DropBot with integrated PWM-driven heating for on-chip amplification35 was used here). Droplets were actuated by applying a force of 25 μN mm−1 using the open-source MicroDrop 2.0 software, conditions that were determined to be below the saturation force38 for all liquids that were used.
Characterization of distance-based detection performance
UltraPure™ Salmon sperm DNA (10 mg mL−1, lot# 1998818, Invitrogen) was diluted in nuclease-free distilled water to yield a series of standard solutions at concentrations of 0, 25, 50, 75, 100, 125, 150, 175, 200, 225 and 250 μg mL−1. 100× SYBR® Safe (Lot# 2265984, Invitrogen) was prepared by diluting SYBR® Safe concentrate (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×) in distilled water. In each experiment, 5 μL of DNA standard solution was mixed with 5 μL 100× SYBR® Safe solution in a centrifuge tube. The resulting mixture, with a total volume of 10 μL, was then pipetted on the loading zone of a DBD substrate formed from a Whatman™ grade 1 or 4 substrate and allowed to elute through capillary action. The liquid front on the reader was marked by a pencil once the front appeared to be no longer moving (typically within ∼5 min), and each DBD substrate was analyzed as described below. All assays were repeated 3 times.
000×) in distilled water. In each experiment, 5 μL of DNA standard solution was mixed with 5 μL 100× SYBR® Safe solution in a centrifuge tube. The resulting mixture, with a total volume of 10 μL, was then pipetted on the loading zone of a DBD substrate formed from a Whatman™ grade 1 or 4 substrate and allowed to elute through capillary action. The liquid front on the reader was marked by a pencil once the front appeared to be no longer moving (typically within ∼5 min), and each DBD substrate was analyzed as described below. All assays were repeated 3 times.
Optimization of intercalator dye for distance-based detection
Four standard salmon DNA solutions at concentrations of 0, 10, 25, and 50 μg mL−1, were prepared as described above, for analysis using DBD substrates formed from grade 4 Whatman™ paper. Six concentrations of SYBR® Safe and SYBR® Green I (Lot# 2311857, Invitrogen) intercalating dyes (diluted to 1, 5, 10, 25, 50, 100× each in nuclease-free water) were assessed by mixing 5 μL of DNA standard with 5 μL of intercalating dye and then pipetting the mixture onto the loading zone of a distance-based detection substrate, for analysis as described above.SARS-CoV-2 RNA extraction from viral samples and quantification by qPCR
Mock clinical samples were formed by spiking pre-pandemic human saliva with 0, 10 or 4.6 × 104 PFU mL−1 cultured, inactivated SARS-CoV-2. These samples were processed by QIAamp viral RNA kits (Qiagen, Germantown, USA, including wash and elution buffers) following the instructions given by the manufacturer without any modification. Briefly, a mini spin column was pre-conditioned with lysis buffer and carrier RNA. 140 μL of the viral sample was then transferred to the mini spin column, followed by pulse-vortexing for 15 seconds and incubation at room temperature for 10 minutes. The spin column was then washed with ethanol and wash buffer, and the RNA was eluted in the elution buffer. The final volume was ∼140 μL and the RNA concentrations were determined by qPCR using methods described elsewhere.39Processing and analysis off-chip by RT-LAMP and qRT-LAMP
In positive (+ve) RT-LAMP experiments, extracted RNA from viral samples (see above) was amplified using a WarmStart® reverse transcribed LAMP (RT-LAMP) colorimetric assay kit with primers specific to SARS-CoV-2 RNA, following instructions given by the manufacturer. Briefly, (i) 12.5 μL of 2× master mix, (ii) 2.5 μL of primer mix, (iii) 2.5 μL of guanidine hydrochloride, (iv) 2.5 μL of nuclease-free water, and (v) 5.0 μL of RNA sample were combined in a well in a semi-skirted 96-well PCR plate. The sample was then heated at 65 °C in a C1000 touch thermal cycler (BioRad, Mississauga, Canada) for 17 minutes or 40 minutes before cooling to 4 °C. In negative (−ve) RT-LAMP experiments, (i), (ii), (iii), and 7.5 μL of nuclease-free water were combined and then exposed to the same amplification protocol. All amplification reactions in different conditions were repeated 3 times.For real-time qRT-LAMP experiments, the amplification mixtures were prepared by combining (i), (ii), (iii), 2.5 μL of 10× SYBR® Green I, and 5 μL of the RNA sample. These samples were heated in a CFX 96 touch real-time PCR detection system (BioRad, Mississauga, Canada) for 40 minutes at 65 °C, with a fluorescent measurement performed every 30 s. The lid temperature of the instrument was set at 95 °C, and there was a 5-second pre-heat step at 65 °C. All amplification reactions in different conditions were repeated 3 times.
Optimization of Capto™ adhere agarose beads for LAMP mixture clean-up
A 1 mL aliquot of Capto™ adhere agarose bead suspension was transferred to a 2 mL centrifuge tube. After centrifugation at 5000 rpm for 1 min, the supernatant was removed, 1 mL of nuclease-free water was added, and the beads were resuspended by pipetting. The centrifuge/wash process was repeated three times, resulting in a 1 mL final suspension. Five ratios of sample to Capto™ bead suspensions were assessed, by combining 10 μL aliquots of RT-LAMP −ve control (after amplification) with different volumes of the washed Capto™ adhere bead suspension (0, 4, 6, 8, 10 μL). These samples were mixed thoroughly by pipetting for 1 min, followed by incubation at room temperature for 1 min. A 5 μL aliquot of this mixture was then mixed with 5 μL 100× SYBR® SAFE and pipetted onto the loading zone of a distance-based detector, for analysis as described above. Control experiments using five volumes of nuclease-free water (0, 4, 6, 8, 10 μL) (in place of the bead suspension) were evaluated using the same procedure.Sample processing off-chip with distance-based detection
Samples were processed in five steps (1–5) as follows. (1) Extracted SARS-CoV-2 RNA (9.84 × 108 copies per mL) was amplified in the PCR instrument as described above (for qRT-LAMP). (2) Amplified products were then diluted to 75%, 50%, 25%, or 10% (v/v) in amplified −ve control solution with 0.1% w/w T90R4. (3) Aliquots (20 μL) of diluted products were mixed with 20 μL Capto™ adhere beads and incubated at room temperature for 1 min. (4) Each bead suspension was mixed thoroughly with a 20 μL aliquot of 100× SYBR® Safe by pipetting. (5) After the beads settled at the bottom of the tube, a 10 μL aliquot of supernatant (amplified and cleaned-up product with SYBR® Safe) was collected and analyzed by distance-based detection (as described below). All experiments were repeated 3 times.Sample processing on-chip with distance-based detection
In DMF experiments, all reagents contained 0.1% w/w T90R4. In most DMF experiments, samples were amplified and diluted off-chip, through steps (1) and (2) above, and then cleaned up and analyzed on DMF devices in five steps (A–E), as described here. (A) 4 μL aliquots each of amplified/diluted sample and centrifuged/washed Capto™ bead suspension were loaded into separate reservoirs and dispensed onto the array of electrodes. (B) Each pair of droplets was merged and actively mixed by rotating in a circular pattern for five minutes. (C) An 8 μL aliquot of 100× SYBR® Safe solution was loaded into a reservoir, and a triple-unit droplet (∼3.9 μL) was dispensed onto the array of electrodes. (D) The dye droplet was merged with the sample/bead droplet, and the merged droplet was then mixed by rotating in a circular pattern for 2 minutes. (E) The resultant droplet was driven to the distance-based detector, where it touched the loading zone and wicked into the liquid flowing zone, for analysis.In some experiments, extracted RNA samples were amplified on-chip in DMF cartridges (Fig. S1†) prior to carrying out steps (A)–(E). Briefly, the modified DropBot system (with integrated, PWM-controlled resistive heater) and evaporation mitigation strategy (sealed cartridges) described in Narahari et al.35 was used to load, dispense, and merge unit-droplets of solutions (i–v) above (with concentrations scaled to maintain reagent ratios). The merged droplet (total volume 8 μL) was heated at 65 °C for 40 minutes and allowed to cool, before using it as the amplified sample droplet in step (A) above.
Distance-based detection
The total distance travelled by the liquid front on the distance-based detectors dt was determined by eye in comparison to the printed scale on the devices (often marked with a pencil). The length travelled by the reporter in the presence of the DNA sample ds was qualitatively observed by imaging the DBD with a Pixel 6 smartphone with filters [Brightline® single-band-pass filter (561/14 nm) and fluorescence filter (485 nm)] positioned in front of the flashlight and camera, respectively. The normalized sample travel distance was dn = ds/dt, which was related to the chromatographic retention factor as k = (1 − dn)/dn.In semi-quantitative experiments, ds were determined by imaging the length-based detector in a Lonza FlashGel® Dock coupled to a personal computer, using the Camera application on Windows 11; RGB images were saved at the maximum resolution of the camera (1920 × 1080) as JPG files. A script in ImageJ was used to isolate the blue channel which exhibited the highest contrast of the fluorescent traces. The background pixel intensity was determined for each image by averaging the pixel intensities outside of the fluorescent trace. In moving through the front (from high to low intensity), the first band of pixels (40 width × 1 length) with an average intensity less than or equal to that of the background was designated as the front. The distance travelled ds was then interpolated using the position of the front relative to the nearest indicator line as the total distance from the first line (near the loading zone).
Results and discussion
Distance-based detection
Here, we describe a new NAAT for viral diagnostics that may someday be useful for portable applications outside of the laboratory. This is an area of intense interest in the field; we focused on mitigating two key limitations, (1) the requirement of systems to be equipped with dedicated detection apparatus, and (2) the requirement of off-chip sample processing. We approached the first challenge using distance-based detection21–26 (DBD), a technique in which analyte amounts are reported based on the length travelled of a visual signal on a patterned paper reporter. The signal can simply be identified by the eye (using a simple illumination system), such that it effectively replaces the detection apparatus (bulky equipment) with a strip of paper.The application of DBD substrates for nucleic acid analytes was first reported by Wang and coworkers in 2018.27 The system is quite clever – by analogy to chromatography, an intercalating fluorescent dye, SYBR® Green I (SG I), serves as the “chromatographic analyte”. The analyte is strongly retained by the “stationary phase” (cellulose fibers in the DBD substrate), and in most “mobile phases” (buffer solutions), the chromatographic analyte remains bound to the cellulosic surface (the “stationary phase”) and does not move when the mobile phase wicks through the system. But because SG I forms complexes with double-stranded DNA (dsDNA) when dsDNA is included in the mobile phase, the chromatographic analyte migrates through the substrate. The distance that the chromatographic analyte travels is determined by the equilibrium between association with stationary and mobile phases, which can be tuned by changing the concentration of dsDNA in the solution. [Note: in actual practice, the DNA in solution is the “analyte” that is being measured. We use the term “chromatographic analyte” to refer to the behavior of the fluorescent reporter as it moves through the DBD.] The system has been well characterized,28 and seems to be quite versatile, being compatible with a variety of different types of samples and solvents. Here, we sought to develop and apply similar methods to serve as part of a miniaturized detection system for viral diagnostics.
As a first step, DBD substrates were formed from two types of Whatman™ paper, grade 1 and grade 4. DBD performance was evaluated with a series of DNA standards (0–250 μg mL−1) mixed with an SYBR® dye reporter. As shown in Fig. 2A, the DNA standard solutions illustrated the expected trend, with SYBR® dye travel distance correlated to dsDNA concentration. As shown in Fig. 2B, when viewed semi-quantitatively, the normalized travel distance (dn) as a function of dsDNA concentration was reproducible for both paper types (Fig. 2A), with an average relative standard deviation of 5.7% (grade 1) and 3.8% (grade 4) across all DNA concentrations tested. The relationship between travel distance and DNA concentration was strongest for both types of substrates between 0–100 μg mL−1 DNA. The travel distances observed for this concentration range (approximately 0.1 ≤ dn ≤ 0.5) correspond to chromatographic retention factors in the (typically desirable) range of approximately 9 ≥ k ≥ 1.
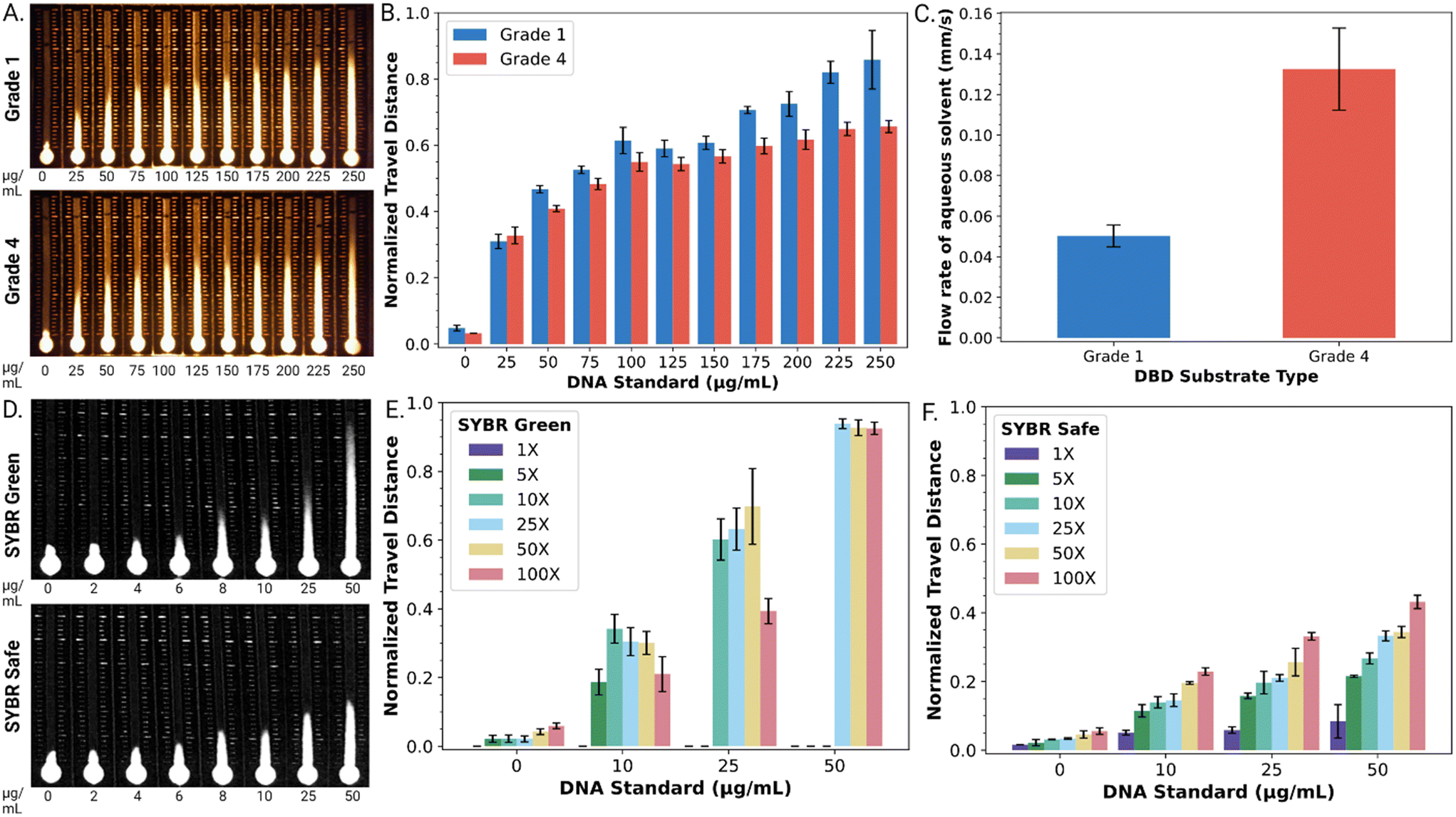 | ||
| Fig. 2 Optimization of distance-based detection of dsDNA. A. Representative fluorescent images of dsDNA standards (0 to 250 μg mL−1) in grade 1 (top) and grade 4 (bottom) Whatman™ paper DBD substrates using 100× SYBR® Safe intercalating dye. B. Graph of normalized travel distance as a function of concentration of dsDNA in grade 1 (blue) and 4 (red) substrates. C. Graph of flow rates observed in grade 1 (blue) and grade 4 (red) substrates for 10 μL 0.1% w/w T90R4 in distilled water. D. Representative fluorescent images (blue channel only) of dsDNA standards (0 to 50 μg mL−1) in DBD substrates after exposure to 100× SYBR® Green I (top) and SYBR® Safe (bottom) dyes. E and F. Plots of normalized travel distances as a function of dsDNA concentration for varying concentrations of SYBR® Green I (Fig. 2E) or SYBR® Safe (Fig. 2F) [1× – purple, 5× – dark green, 10× – light green, 25× – blue, 50× – yellow, and 100× – red]. Error bars represent 1 std. deviation for n = 3 replicates per condition. | ||
We further evaluated the flow rate of DBD substrates formed from both types of paper by measuring the time required for a fixed volume of aqueous solvent to travel a selected distance (Fig. 2C). The flow rate in DBDs formed from grade 4 paper (0.13 ± 0.02 mm s−1) was found to be 2.6 times faster than that observed in DBDs formed from grade 1 paper (0.05 ± 0.01 mm s−1). This agreed with our expectations (noting that velocity in a porous medium scales with pore size), given that grade 4 and grade 1 papers have pore diameters of ∼20–25 μm and ∼11 μm, respectively. Based on this result and the lower level of standard error, we used DBD substrates formed from grade 4 paper for all subsequent experiments.
As a next step, the fluorescent reporter was optimized for the application described here. The fluorescent reporter used in the original studies,27,28 SYBR® Green I (SG I) dye, works well for this application, but it is toxic and expensive (approx. $0.83 per μL). We were thus interested in exploring alternative reporters, such as the less toxic and less expensive (approx. $0.23 per μL) SYBR® Safe (SS) dye. As shown in Fig. 2D, both SG I and SS were found to produce a strong travel distance/DNA concentration trend.
SYBR® dyes are sold by their manufacturer as aqueous solutions with their quantities reported as concentration factors for their uses in gel electrophoresis and quantitative PCR (i.e., 1×, 50×, 100×, etc.). The effects of reporter concentration for the two dyes were probed for DBD performance. As shown in Fig. 2E, at high SG I dye concentrations (≥25×), the normalized distance travelled approaches unity (and the retention factor in the stationary phase approaches zero) at high [dsDNA], implying more dye interactions with the dsDNA. On the other hand, at low dye concentrations (≤10×), SG I did not adequately resolve the range of DNA concentrations tested here. In contrast, as shown in Fig. 2F, the retention factor for SS in the stationary phase is more modest (implying weaker dye interactions with dsDNA), but the entire range of SS concentrations (1–100×) provided adequate distance-based detection resolution of the DNA concentrations tested here.
The most useful comparison of the two reporters (SG 1 and SS) was found upon close inspection of the geometries of the “fronts” of the fluorescent traces on the DBD substrates. The SG I trace exhibited a characteristic ‘fading front’ effect that obscured the ability to reliably determine the precise travel distance. For example, a linear plot of fluorescence intensity for a front formed by 100× SG I in the presence of 25 μg mL−1 DNA on a DBD is shown in Fig. S2.† As shown, the intensity gradually diminishes over a substantial distance (∼136 pixels or ∼5.4 mm). In contrast, the SS trace has a much more modest ‘fading front’ effect. For example, as shown in Fig. S2,† the distance between maximum intensity and background level for 100× SS in the presence of 25 μg mL−1 DNA is only ∼35 pixels (or ∼1.5 mm). Thus, with the desire for a rapid, reliable visual readout, SS (at the 100× level) was selected as the reporter for the remainder of the work described here.
Finally, the most important aspect of DBD is the potential for analysis by “eye”, without requiring a dedicated detection apparatus. Thus, while some data were acquired for semi-quantitative analysis using a scanner, in all qualitative experiments, we used a modified cell phone camera/flashlight system, illustrated in Fig. S3.†
Distance-based detection of LAMP products
Armed with an optimized DBD system for evaluating dsDNA, we turned our attention to its ability to detect LAMP products, noting that all previous reports of this type of system (i.e., DBD for NAAT applications) have been applied exclusively to detecting PCR products.27,28 After amplification, the LAMP reaction mixture contains multiple short base-pair components, including primer–dimers and mixtures of oligonucleotides and concatemers,5,7–9 forming a much more complex background than what is typically found in PCR. We hypothesized that some of these LAMP-specific components might interact with the dye reporter and thus interfere with DBD performance. To probe this phenomenon, a series of negative LAMP experiments making use of an amplified negative (−ve) control solution (containing all the reagents but lacking RNA template and dsDNA analytes) was devised. As shown in Fig. S4A,† after amplification, this mixture results in an elongated response of dn > 0.5 (∼15 mm travel distance), limiting greatly the available range of the DBD, and suggesting that unmodified LAMP products may be incompatible with distance-based detection for the conditions reported here.To mitigate this issue, a sample clean-up approach was adapted from a protocol reported previously by Soares and coworkers9 relying on Capto™ adhere agarose (CAA) beads. Briefly, the CAA approach employs a solid phase that selectively binds low molecular-weight nucleotides (<100 bp), leaving the desired LAMP products (concatemers of the target sequence, typically with large molecular weight) in solution. In practice, CAA beads are added to the mixture after amplification, and after a short incubation step (1 min), they are removed to allow the analysis of the supernatant. This approach was found to be useful for distance-based detection – for example, Fig. S4B† shows distance-based detection signals for amplified −ve controls after exposure to increasing volumes of CAA bead suspension. Semi-quantitative DBD results are shown in Fig. S4C,† indicating that exposure to CAA beads causes the dn of the mixture to decrease from 0.55 to below 0.30, reducing the baseline signal. Thus, this clean-up process was employed for all the remaining experiments described here.
As indicated above, the CAA approach makes LAMP compatible with DBD – to our knowledge, this is the first report of this (LAMP–DBD) combination. But the extra sample processing introduced by this procedure (including mixing with bead suspension, incubating, and recovery of supernatant) is an additional hurdle for portable, point-of-care applications. Thus, we turned to digital microfluidics (DMF) to automate the new method. DMF is a technique in which droplets of reagents and samples are programmed and controlled on-demand, in a format that is convenient for point-of-care use, even in remote and resource-limited settings.40,41 DMF has been used for a wide range of applications involving rigid magnetic particles,40–47 but to our knowledge has never been applied to handling suspensions of large (∼75 μm diameter), malleable particles such as the CAA beads described here.
In practice, suspensions of CAA beads were found to be compatible with DMF operations, including dispensing, splitting, and merging droplets. As shown in Fig. 3A, an automated program was developed in which a droplet of LAMP-amplified product was mixed with a droplet of CAA bead suspension and SYBR® reporter, and then driven to an integrated DBD. The solution phase (containing the desirable high molecular weight analytes) wets the DBD spontaneously, allowing for distance-based detection, while the beads (containing the undesirable low molecular weight constituents) remain behind. Typical results generated off-chip (manually) and on-chip (by DMF) are shown in Fig. 3B. In sum, the (DMF) system appears to be a useful solution to limitations related to manual processing steps for the method described here.
 | ||
| Fig. 3 Automated sample cleanup. A. Pictures (i–viii) illustrating sample processing steps in a DMF cartridge. (i) A DMF cartridge has three reagents: white – Capto™ Adhere bead suspension (bottom left reservoir), red – LAMP products (bottom, second-from-left reservoir), and blue – SYBR® Safe (top, middle reservoir). CAA bead suspension and LAMP products are dispensed (ii), moved and mixed (iii) and then split (iv). A droplet of SYBR® Safe is dispensed (v) and mixed (vi) with the processed LAMP product. The droplet (purple) is then delivered (vii) to the DBD and wicked onto it (viii) by capillary action. B. A plot of normalized travel distance as a function of percentage composition (0–75% v/v) of LAMP-amplified +ve sample diluted in LAMP-amplified −ve mix, processed with CAA bead clean-up off-chip (blue markers) and on DMF (red markers). Error bars represent ±1 std. deviation of normalized travel distance for n = 3 replicates per composition. | ||
To our knowledge, DMF has never before been combined with DBD. In fact, there are only a few reports describing the interface of DMF with lateral flow/wicking substrates for any application, either for sample delivery48 or sample analysis.49 In its current implementation, DBD substrates were interfaced with DMF cartridges by hand. In the future, automated processes (appropriate for scalable manufacture) might be developed in which integrated DBD substrates are formed directly in inkjet-printed DMF devices50–52 or are assembled together via roll-to-roll or pick-and-place techniques. However these devices are ultimately manufactured, we believe this to be a powerful combination that is likely to be useful for a wide range of potential applications in the future.
DMF–LAMP–DBD for automated NAATs
Armed with an automated DMF–LAMP–DBD system, we turned to evaluating its suitability for use with a diagnostic NAAT application – detection of SARS-CoV-2 RNA in saliva. As a first step toward this goal, we characterized the performance of a commercial qRT-LAMP kit53 combined with highly specific primer sets10 for the envelope (E) gene and nucleocapsid (N) genes in the virus. A suspension of cultured, inactivated SARS-CoV-2 at a density of 1.2 × 106 PFU mL−1 was acquired and determined by qPCR to contain 9.84 × 108 copies per mL of the viral genome. The qRT-LAMP method was then applied to a dilution series of this sample, and as shown in Fig. S5,† it was found to be appropriate for detecting viral RNA at all concentrations tested (9.84 × 103–9.84 × 108 copies per mL) after 30 minutes of amplification. This range is (in principle) compatible with the detection of SARS-CoV-2 in the saliva of infected patients, which ranges between 104–108 copies per mL.54Finally, as a proxy for real samples, cultured, inactive SARS-CoV-2 was spiked into pre-pandemic human saliva. (Note that the sample pre-processing employed here ensures that solutions loaded onto the DBD substrates do not suffer from differences in wicking velocity caused by variations in saliva viscosity.55) Specifically, saliva samples spiked with SARS-CoV-2 viral loads at the low (10 PFU mL−1) and high (4.6 × 104 PFU mL−1) end of the clinical range were prepared and then confirmed by qPCR to contain (4.88 ± 0.78) × 104 and (2.20 ± 0.29) × 108 copies per mL (Table S1†). From the kinetic data in Fig. S5,† we hypothesized that an amplification time of 17 minutes would be useful for distinguishing between high and low viral loads (noting that the conventional 40-minute amplification would likely yield indistinguishable signals). This hypothesis was borne out qualitatively (Fig. 4A) and semi-quantitatively (Fig. 4B), where a 17 min amplification yielded statistically significant differences between the three samples (two-sided t-test, pblank-low = 0.02863, plow-high = 0.00077). Finally, as illustrated in Fig. S6,† the entire process including amplification, cleanup, and distance-based detection can be automated on DMF, with a run time of 50 minutes. Note that there are two key pre-processing steps that are performed manually in this method – viral lysis and extraction – but we propose that in the future, they might be automated by DMF as well for end-to-end automation, similar to what was reported previously35 for a Zika virus diagnostic (that did not use LAMP or DBD).
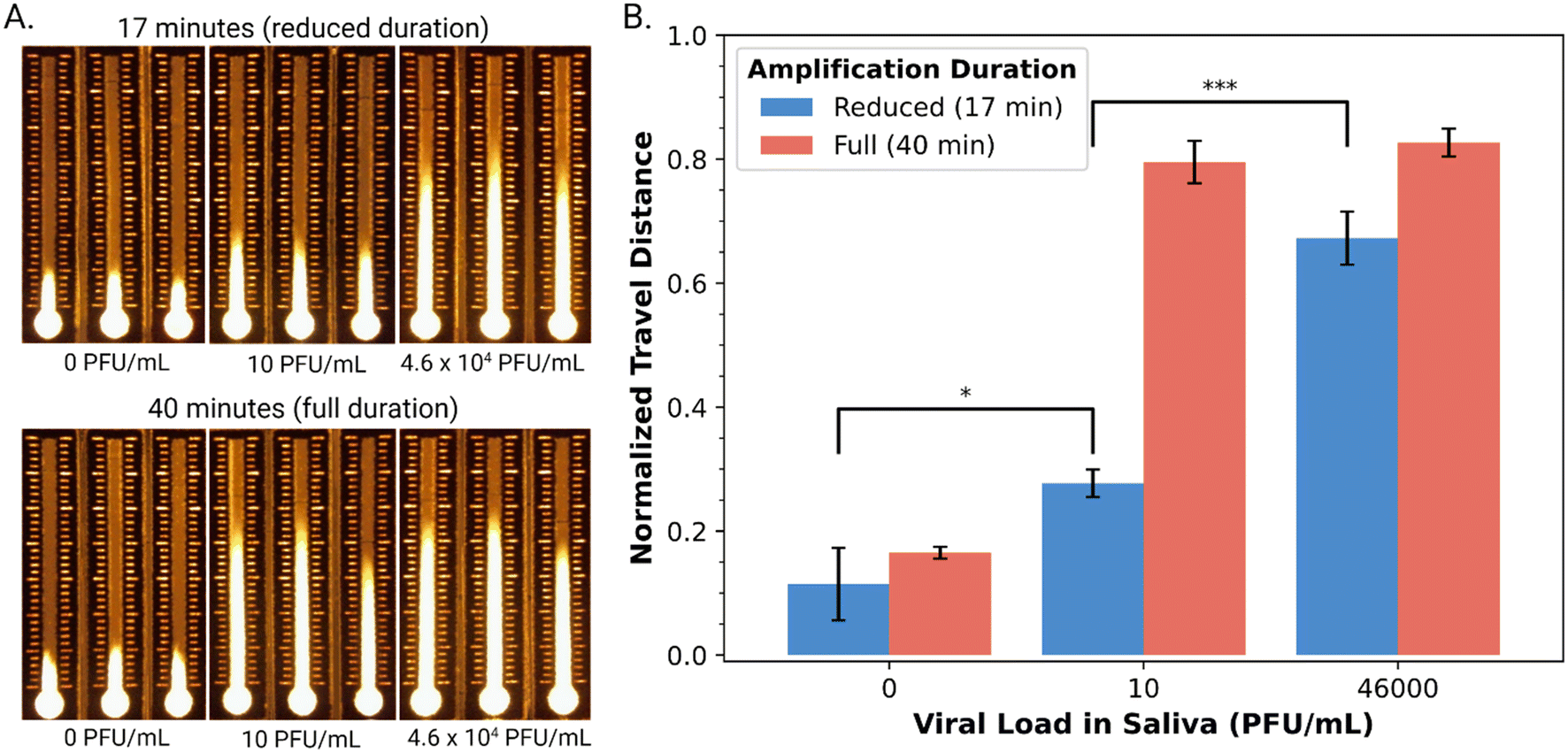 | ||
| Fig. 4 DBD analysis of amplified viral RNA extracted from human saliva spiked with SARS-CoV-2. A. Representative fluorescent images of DBD substrates after exposure to amplified viral RNA extracted from three (spiked) viral loads in saliva (0-blank, 10-low, 4.6 × 104-high PFU mL−1) after reduced (17 min, top) and full (40 min, bottom) duration of RT-LAMP amplification. B. Plot of normalized travel distance as a function of viral load in saliva (0, 10, 4.6 × 104 PFU mL−1) for reduced (17 min-blue) and full (40 min-red) amplification. Error bars represent ±1 std. deviation of normalized travel distance for n = 3 replicates per condition; a single asterisk (*) indicates statistical significance with p < 0.05 and three asterisks (***) indicate statistical significance with p < 0.001. | ||
Conclusions
In this work, we introduce the combination of distance-based detection (DBD) and digital microfluidics (DMF) to address two limitations of POC NAATs. First, DBD allows for analysis without a dedicated detection apparatus, to provide signal output and (semi-) quantitative data about the analyte by eye. Second, DMF allows for automated sample handling and processing, reducing user burden and error while maintaining assay performance. We propose that the combination of DMF with distance-based detection may be a powerful one for future generations of NAATs that are intended for use at the point of care.In addition to introducing DMF–DBD, another key innovation reported here is the adaptation of LAMP to be compatible with DBD. LAMP and other isothermal techniques are popular for developers of portable diagnostic tests, as instrumentation can be simpler than what is needed to implement the more common PCR (which requires temperature cycling). But in initial tests, LAMP proved to have undesirably high background for DBD, limiting its effective dynamic range. A new procedure was developed in which LAMP products were automatically cleaned-up by a solid-phase depletion procedure (using Capto™ adhere beads) in the DMF chip prior to analysis. This innovation allowed the technique to differentiate between positive and negative saliva samples, and between positive samples spiked with SARS-CoV-2 at “high” (108 genome copies per mL) and “low” (104 copies per mL) viral loads.
In sum, the new system allows for a hands-free automated test for the presence of a viral target. Critically, very little user intervention is required, and no dedicated detection apparatus is needed – the result can be determined by eye. To date, this system has not yet been used outside of the laboratory, but we propose that it is a good bet for future demonstrations as a “portable” instrument that can be operated in many different environments. In sum, this system has the potential to contribute to redefining the landscape of diagnostic testing, rendering the experience more accessible and precise while simultaneously reducing the demands placed on healthcare professionals and patients alike.
Author contributions
K. P. N. and A. R. W. conceived of the idea of coupling digital microfluidic sample processing with lateral flow strip-like detectors; M. H. and A. A. S. adapted the idea to include distance-based detection. A. A. S. designed and fabricated the distance-based detectors. M. H. and A. A. S. characterized DBD methods and materials. M. H. conducted qRT-LAMP amplification assays and all DMF experiments. M. H. conceived the idea and developed the sample clean-up procedure based on Capto™ adhere beads. M. H. and N. S. conducted amplification assays and DBD analysis on viral samples. M. H., N. S. and A. A. S. conducted image analysis and DBD data analysis. J. S. and I. Y. conducted qPCR experiments and data analysis. M. H., A. R. W., and N. S. interpreted the results and wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Aaron Au and Professor Chris Yip (University of Toronto) for access to optical components and advice on cell phone imaging. We thank Professor Elizabeth Edwards and Professor Hui Peng for access to and advice regarding the use of the qPCR system used here (University of Toronto). We thank Dr. Teodor Veres (National Research Council of Canada, NRC) for illuminating discussions about microfluidic diagnostics. Finally, we acknowledge the NRC for the Pandemic Response Challenge Program (PR-015.1), the Centre for Research and Applications in Fluidic Technologies (Project Award), the Natural Sciences and Engineering Research Council of Canada (ALLRP 554378-20), and the University of Toronto (Temerty KT Grant and Connaught COVID-19 Fund) for funding.References
- T. Kang, J. Lu, T. Yu, Y. Long and G. Liu, Biosens. Bioelectron., 2022, 206, 114109 CrossRef CAS.
- E. M. Euliano, A. A. Sklavounos, A. R. Wheeler and K. J. McHugh, Sci. Transl. Med., 2023, 14, eabm1732 CrossRef PubMed.
- B. Shu, C. Zhang and D. Xing, Biosens. Bioelectron., 2017, 97, 360–368 CrossRef CAS.
- S. Fu, G. Qu, S. Guo, L. Ma, N. Zhang, S. Zhang, S. Gao and Z. Shen, Appl. Biochem. Biotechnol., 2010, 163, 845–850 CrossRef.
- T. Notomi, Y. Mori, N. Tomita and H. Kanda, J. Microbiol., 2015, 53, 1–5 CrossRef CAS.
- L. Mautner, C.-K. Baillie, H. M. Herold, W. Volkwein, P. Guertler, U. Eberle, N. Ackermann, A. Sing, M. Pavlovic, O. Goerlich, U. Busch, L. Wassill, I. Huber and A. Baiker, Virol. J., 2020, 17, 160 CrossRef CAS PubMed.
- B. A. Rabe and C. Cepko, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 24450–24458 CrossRef CAS PubMed.
- M. N. Aoki, B. de Oliveira Coelho, L. G. B. Góes, P. Minoprio, E. L. Durigon, L. G. Morello, F. K. Marchini, I. N. Riediger, M. do Carmo Debur, H. I. Nakaya and L. Blanes, Sci. Rep., 2021, 11, 9026 CrossRef CAS.
- R. R. G. Soares, A. S. Akhtar, I. F. Pinto, N. Lapins, D. Barrett, G. Sandh, X. Yin, V. Pelechano and A. Russom, Lab Chip, 2021, 21, 2932–2944 RSC.
- K. J. M. Moore, J. Cahill, G. Aidelberg, R. Aronoff, A. Bektaş, D. Bezdan, D. J. Butler, S. V. Chittur, M. Codyre, F. Federici, N. A. Tanner, S. W. Tighe, R. True, S. B. Ware, A. L. Wyllie, E. E. Afshin, A. Bendesky, C. B. Chang, R. Dela Rosa, E. Elhaik, D. Erickson, A. S. Goldsborough, G. Grills, K. Hadasch, A. Hayden, S.-Y. Her, J. A. Karl, C. H. Kim, A. J. Kriegel, T. Kunstman, Z. Landau, K. Land, B. W. Langhorst, A. B. Lindner, B. E. Mayer, L. A. McLaughlin, M. T. McLaughlin, J. Molloy, C. Mozsary, J. L. Nadler, M. DSilva, D. Ng, D. H. OConnor, J. E. Ongerth, O. Osuolale, A. Pinharanda, D. Plenker, R. Ranjan, M. Rosbash, A. Rotem, J. Segarra, S. Schürer, S. Sherrill-Mix, H. Solo-Gabriele, S. To, M. C. Vogt, A. D. Yu and C. E. Mason, J. Biomol. Tech., 2021, 32, 228–275 CrossRef.
- Y.-S. Tsai, C.-H. Wang, H.-P. Tsai, Y.-S. Shan and G.-B. Lee, Anal. Chim. Acta, 2022, 1219, 340036 CrossRef CAS PubMed.
- Y.-R. Jhou, C.-H. Wang, H.-P. Tsai, Y.-S. Shan and G.-B. Lee, Sens. Actuators, B, 2022, 358, 131447 CrossRef CAS PubMed.
- L. Malic, D. Brassard, D. Da Fonte, C. Nassif, M. Mounier, A. Ponton, M. Geissler, M. Shiu, K. J. Morton and T. Veres, Lab Chip, 2022, 22, 3157–3171 RSC.
- T. AbdElFatah, M. Jalali, S. G. Yedire, I. I. Hosseini, C. del Real Mata, H. Khan, S. V. Hamidi, O. Jeanne, R. Siavash Moakhar, M. McLean, D. Patel, Z. Wang, G. McKay, M. Yousefi, D. Nguyen, S. M. Vidal, C. Liang and S. Mahshid, Nat. Nanotechnol., 2023, 18, 922–932 CrossRef CAS.
- R. Bester and H. J. Maree, J. Virol. Methods, 2022, 306, 114543 CrossRef CAS.
- M. Jang, S. Kim, J. Song and S. Kim, Anal. Bioanal. Chem., 2022, 414, 4685–4696 CrossRef CAS.
- S. Sharma, E. Thomas, M. Caputi and W. Asghar, Biosensors, 2022, 12, 298 CrossRef CAS.
- M. Gaber, A. A. Ahmad, A. M. El-Kady, M. Tolba, Y. Suzuki, S. M. Mohammed and N. A. Elossily, PLoS One, 2022, 17, e0265760 CrossRef CAS PubMed.
- J. S. Kumar, P. D. Yadav, A. M. Shete, T. Majumdar, S. Patil and P. K. Dash, Int. J. Infect. Dis., 2021, 112, 346–351 CrossRef CAS PubMed.
- D. Seevaratnam, F. Ansah, Y. Aniweh, G. A. Awandare and E. A. H. Hall, Anal. Bioanal. Chem., 2022, 414, 6309–6326 CrossRef CAS.
- D. M. Cate, W. Dungchai, J. C. Cunningham, J. Volckens and C. S. Henry, Lab Chip, 2013, 13, 2397 RSC.
- C. T. Gerold, E. Bakker and C. S. Henry, Anal. Chem., 2018, 90, 4894–4900 CrossRef CAS PubMed.
- D. M. Cate, S. D. Noblitt, J. Volckens and C. S. Henry, Lab Chip, 2015, 15, 2808–2818 RSC.
- L. Cai, Y. Fang, Y. Mo, Y. Huang, C. Xu, Z. Zhang and M. Wang, AIP Adv., 2017, 7, 85214 CrossRef.
- M. Rahbar, A. R. Wheeler, B. Paull and M. Macka, Anal. Chem., 2019, 91, 8756–8761 CrossRef CAS PubMed.
- L. G. A. Dias, L. C. Duarte, K. M. P. Pinheiro, N. S. Moreira and W. K. T. Coltro, Talanta, 2023, 7, 100216 CrossRef.
- A. G. Wang, T. Dong, H. Mansour, G. Matamoros, A. L. Sanchez and F. Li, ACS Sens., 2018, 3, 205–210 CrossRef CAS PubMed.
- T. Dong, G. A. Wang, M. W. Li and F. Li, Anal. Methods, 2019, 11, 5376–5380 RSC.
- Z. Hua, J. L. Rouse, A. E. Eckhardt, V. Srinivasan, V. K. Pamula, W. A. Schell, J. L. Benton, T. G. Mitchell and M. G. Pollack, Anal. Chem., 2010, 82, 2310–2316 CrossRef CAS.
- W. A. Schell, J. L. Benton, P. B. Smith, M. Poore, J. L. Rouse, D. J. Boles, M. D. Johnson, B. D. Alexander, V. K. Pamula, A. E. Eckhardt, M. G. Pollack, D. K. Benjamin, J. R. Perfect and T. G. Mitchell, Eur. J. Clin. Microbiol. Infect. Dis., 2012, 31, 2237–2245 CrossRef CAS.
- R. Sista, Z. Hua, P. Thwar, A. Sudarsan, V. Srinivasan, A. Eckhardt, M. Pollack and V. Pamula, Lab Chip, 2008, 8, 2091 RSC.
- M. J. Jebrail, R. F. Renzi, A. Sinha, J. Van De Vreugde, C. Gondhalekar, C. Ambriz, R. J. Meagher and S. S. Branda, Lab Chip, 2015, 15, 151–158 RSC.
- L. Wan, T. Chen, J. Gao, C. Dong, A. H.-H. Wong, Y. Jia, P.-I. Mak, C.-X. Deng and R. P. Martins, Sci. Rep., 2017, 7, 14586 CrossRef.
- T. Hoang, B.-H. Ly, T.-X. Le, T.-T. Huynh, H.-T. Nguyen, T. Van Vo, T. T. H. Pham and K. Huynh, Microsyst. Technol., 2020, 26, 1863–1873 CrossRef CAS.
- T. Narahari, J. Dahmer, A. Sklavounos, T. Kim, M. Satkauskas, I. Clotea, M. Ho, J. Lamanna, C. Dixon, D. G. Rackus, S. J. R. da Silva, L. Pena, K. Pardee and A. R. Wheeler, Lab Chip, 2022, 22, 1748–1763 RSC.
- M. Kühnemund, D. Witters, M. Nilsson and J. Lammertyn, Lab Chip, 2014, 14, 2983–2992 RSC.
- R. Fobel, C. Fobel and A. R. Wheeler, Appl. Phys. Lett., 2013, 102, 193513 CrossRef.
- I. Swyer, R. Fobel and A. R. Wheeler, Langmuir, 2019, 35, 5342–5352 CrossRef CAS PubMed.
- M. Fuzzen, N. B. J. Harper, H. A. Dhiyebi, N. Srikanthan, S. Hayat, L. M. Bragg, S. W. Peterson, I. Yang, J. X. Sun, E. A. Edwards, J. P. Giesy, C. S. Mangat, T. E. Graber, R. Delatolla and M. R. Servos, Sci. Total Environ., 2023, 881, 163292 CrossRef CAS PubMed.
- A. H. C. Ng, R. Fobel, C. Fobel, J. Lamanna, D. G. Rackus, A. Summers, C. Dixon, M. D. M. Dryden, C. Lam, M. Ho, N. S. Mufti, V. Lee, M. A. M. Asri, E. A. Sykes, M. D. Chamberlain, R. Joseph, M. Ope, H. M. Scobie, A. Knipes, P. A. Rota, N. Marano, P. M. Chege, M. Njuguna, R. Nzunza, N. Kisangau, J. Kiogora, M. Karuingi, J. W. Burton, P. Borus, E. Lam and A. R. Wheeler, Sci. Transl. Med., 2018, 10, eaar6076 CrossRef.
- A. K. Knipes, A. Summers, A. A. Sklavounos, J. Lamanna, R. P. S. de Campos, T. Narahari, C. Dixon, R. Fobel, Y. D. Ndjakani, L. Lubula, A. Magazani, J. J. Muyembe, Y. Lay, E. Pukuta, D. Waku-Kouomou, L. Hao, J. K. Kayembe, C. Fobel, J. Dahmer, A. Lee, M. Ho, J. G. C. Valenzuela, D. G. Rackus, R. Shih, B. Seale, A. Chang, G. Paluku, P. A. Rota, A. R. Wheeler and H. M. Scobie, PLoS One, 2022, 17, e0278749 CrossRef CAS PubMed.
- T. Kokalj, E. Pérez-Ruiz and J. Lammertyn, New Biotechnol., 2015, 32, 485–503 CrossRef CAS.
- K. Choi, A. H. C. Ng, R. Fobel, D. A. Chang-Yen, L. E. Yarnell, E. L. Pearson, C. M. Oleksak, A. T. Fischer, R. P. Luoma, J. M. Robinson, J. Audet and A. R. Wheeler, Anal. Chem., 2013, 85, 9638–9646 CrossRef CAS.
- L. Coudron, M. B. McDonnell, I. Munro, D. K. McCluskey, I. D. Johnston, C. K. L. Tan and M. C. Tracey, Biosens. Bioelectron., 2019, 128, 52–60 CrossRef CAS.
- Y. Wang, Q. Ruan, Z.-C. Lei, S.-C. Lin, Z. Zhu, L. Zhou and C. Yang, Anal. Chem., 2018, 90, 5224–5231 CrossRef CAS PubMed.
- M. Kühnemund, D. Witters, M. Nilsson and J. Lammertyn, Lab Chip, 2014, 14, 2983–2992 RSC.
- J. Leipert and A. Tholey, Lab Chip, 2019, 19, 3490–3498 RSC.
- C. Dixon, J. Lamanna and A. R. Wheeler, Lab Chip, 2020, 20, 1845–1855 RSC.
- A. Abadian, S. S. Manesh and S. J. Ashtiani, Microfluid. Nanofluid., 2017, 21, 65 CrossRef.
- R. Fobel, A. E. Kirby, A. H. C. Ng, R. R. Farnood and A. R. Wheeler, Adv. Mater., 2014, 26, 2838–2843 CrossRef CAS PubMed.
- C. Dixon, A. H. C. Ng, R. Fobel, M. B. Miltenburg and A. R. Wheeler, Lab Chip, 2016, 16, 4560–4568 RSC.
- H. Ko, J. Lee, Y. Kim, B. Lee, C.-H. Jung, J.-H. Choi, O.-S. Kwon and K. Shin, Adv. Mater., 2014, 26, 2335–2340 CrossRef CAS PubMed.
- Sars-Cov-2 Rapid Colorimetric Lamp Assay Kit, https://international.neb.com/products/e2019-sars-cov-2-rapid-colorimetric-lamp-assay-kit#Product%20Information, (accessed 28 March 2023).
- Y. M. Bar-On, A. Flamholz, R. Phillips and R. Milo, eLife, 2020, 9, e57309 CrossRef PubMed.
- J. Noiphung, M. P. Nguyen, C. Punyadeera, Y. Wan, W. Laiwattanapaisal and C. S. Henry, Theranostics, 2018, 8, 3797–3807 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00683b |
| This journal is © The Royal Society of Chemistry 2024 |