
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteractions between liquid ammonia and graphitic materials†
Cheng-Wei
Lin
 a,
Zhiyin
Yang
a,
Ailun
Huang
a,
Xueying
Chang
a,
Chenxiang
Wang
a,
Fan
Yang
b,
Chen
Wei
c,
Markus
Thiel
a,
Yuto
Katsuyama
a,
Lihua
Jin
c,
David
Jassby
bd and
Richard B.
Kaner
*ae
a,
Zhiyin
Yang
a,
Ailun
Huang
a,
Xueying
Chang
a,
Chenxiang
Wang
a,
Fan
Yang
b,
Chen
Wei
c,
Markus
Thiel
a,
Yuto
Katsuyama
a,
Lihua
Jin
c,
David
Jassby
bd and
Richard B.
Kaner
*ae
aDepartment of Chemistry and Biochemistry and California NanoSystems Institute, University of California, Los Angeles, Los Angeles, California 90095, USA. E-mail: kaner@chem.ucla.edu
bDepartment of Civil and Environmental Engineering, University of California, Los Angeles, Los Angeles, California 90095, USA
cDepartment of Mechanical and Aerospace Engineering, University of California, Los Angeles, Los Angeles, California 90095, USA
dCalifornia NanoSystem Institute, University of California, Los Angeles, Los Angeles, California 90095, USA
eDepartment of Materials Science and Engineering and California NanoSystems Institute, University of California, Los Angeles, Los Angeles, California 90095, USA
First published on 18th December 2023
Abstract
Interactions between ammonia gas and graphitic materials, including adsorptions and N-doping at elevated temperatures, are known. However, liquid ammonia has rarely been studied other than for the Birch reduction reaction. Here, we report influences of liquid ammonia on graphite oxide (GO) and reduced graphite oxide (rGO), at both the atomic and macroscopic scales. It is demonstrated that GO and rGO do not disperse in liquid ammonia regardless of charge transfer due to the incompatible surface energy and polarity. X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), and Raman spectroscopy studies of GO sheets treated with liquid ammonia, and liquid nitrogen as a control, for different time periods demonstrate ∼0.6 at% pyridinic N-doping and non-restoration of conjugation in the absence of thermal energy. Macroscopically, liquid ammonia is incapable of altering the shape or size of assembled GO and single walled carbon nanotube (SWCNT) freestanding membranes. However, loud popping sounds along with the generation of pockets, which leads to lower stiffness within the GO membranes, were observed after treating with liquid ammonia. In addition, shrinkage of porous rGO aerogels and counter-intuitively an increase in electrical resistance of SWCNT/cellulose composite membranes after treatment with liquid ammonia, are reported.
1. Introduction
Liquid ammonia is a non-aqueous ionizing solvent that is often used for organic synthesis, especially for Birch reduction reactions, where alkali metals can be dissolved in liquid ammonia to form electrical conducting solutions of solvated electrons possessing a blue color due to transmission of light at low concentrations and a gold color due to reflection of light at high concentrations.1 The solution comprised of solvated electrons that are caged by ammonia molecules, serves as one of the most powerful chemical reducing agents, while the dissolved metals act as counter-cations.2 For example, conducting polyacetylene was n-doped with the multi-valent cations, Al3+, Mg2+, Ca2+ and Eu2+, in liquid ammonia, as reported by Kaufman et al.3 Howard and co-workers demonstrated ionic solutions of 2D transition dichalcogenides with liquid ammonia, in the presence of lithium and potassium.4 Highly reduced graphene oxide and hydrogenated graphene were prepared by reacting graphene oxide with a solution of liquid ammonia and sodium.5,6Liquid ammonia is also known to be able to solvate cellulose and alter the chemical structural arrangement of cellulose upon evaporation, as confirmed by X-ray diffraction.7,8 This phenomenon has been used to twist the shape of wood and to shrink U.S. dollar bills.9,10 As graphite oxide (GO) possesses hydroxyl, epoxide, phenol, carboxyl, and carbonyl groups at its edges and basal planes, a hypothesis was made as to whether liquid ammonia, which can readily form hydrogen bonding with these functional groups, is capable of solvating graphite oxides, thus leading to a shrinkage or deformations of macroscopic GO assemblies. On the other hand, reduced graphene oxide (rGO) is ubiquitously used for sensing a trace amount of ammonia gas.11,12 The lowering of resistivity in rGO when exposed to ammonia gas is due to charge transfer between ammonia gas molecules and rGO.13,14 As Chiou et al. recently revealed, charge transfer is able to stabilize carbon nanotube dispersions in m-cresol,15 suggesting an analogous question: is charge transfer powerful enough to form stabilized colloidal suspensions of rGO in liquid ammonia?
Previous studies regarding the interactions between GO and ammonia are mostly associated with the reduction and N-doping of GO by ammonia gas at an elevated temperature. The synthesis of substitutional N-doped graphene was reported by Liu et al. by introducing ammonia gas in the chemical vapor deposition (CVD) growth of graphene at 800 °C.16 Dai and co-workers developed the simultaneous reduction and N-doping of GO by annealing GO in the presence of ammonia gas at temperatures higher than 300 °C.17 Supercritical ammonia (200 °C, 15 MPa) was used to exfoliate graphite along with the simultaneous N-doping of graphene.18 However, with the insight that GO can be thermally reduced to rGO,19 the effect of ammonia therefore can only be fairly evaluated once the thermal energy is separated from the experiment. It is noteworthy that Lee and co-workers demonstrated the partial reduction and functionalization of GO after 24 hours of treatment with ethylenediamine at room temperature.20 Since ethylenediamine and liquid ammonia possess similar chemical properties including strong electron donation and serving as effective ligands for powerful reduction reactions,21,22 it is of great interest to investigate the interactions between GO and liquid ammonia.
In this report, we first investigate the stability of colloidal suspensions of rGO and GO in liquid ammonia and hydroxide aqueous solutions. Then, N-doping and reduction effects of GO in liquid ammonia are probed by XPS, Raman spectroscopy, and XRD, with comparisons to GO treated with liquid nitrogen. In addition, materials properties of macroscopic assemblies of GO, rGO, and SWCNT after soaking in liquid ammonia are reported. Our results give insights into the surface chemistry, N-doping and reduction mechanisms of GO at low temperatures, and the interactions between liquid ammonia and graphitic materials at both the microscopic and macroscopic scale, which can enrich the knowledge base of interactions between liquid ammonia and carbon materials.
2. Experimental methods
2.1 Materials
Graphite oxide powder was synthesized through a modified Hummer's method,23 freeze dried and collected as a brown powder. Reduced graphite oxide powder (Ultra Graphene), was purchased from Nanotech Energy Inc. The partially oxidized graphite oxide was synthesized through a modified Hummer's method, and stored in plastic jars as a 4.86% paste. The partially oxidized graphite oxide paste was specifically used for fabricating membranes. The single-walled carbon nanotubes (SWCNTs) used for dispersion were acquired from Carbon Solutions, Inc. (P2-SWNT, CO#1815). The SWCNTs (SWCNT-COOH, 99%) used to make freestanding films were purchased from Cheap Tubes Inc. (USA). Chlorosulfonic acid (99%) was purchased from Sigma-Aldrich. The ammonia gas tank (AM 5.5SP-G, ammonia >99.9995%, moisture <1.0 ppm) was purchased from Praxair. All chemicals were used as received.2.2 Characterization
X-ray diffraction patterns were collected on a powder X-ray diffractometer (PANalytical, X'Pertpro) with Cu Kα radiation (λ = 1.54184 Å). The morphologies of materials were examined using a JEOL-6700F FE-SEM except for the SWCNT/cellulose composite membranes which were imaged using a JEOL JSM 6610. FT-IR spectroscopy by attenuated total reflectance was performed on a Cary 600 Series FTIR Microscope (Agilent Technologies). XPS measurements were obtained using a Kratos Axis Ultra DLD spectrometer with a monochromatic Al Kα X-ray source (λ = 1486.6 eV). UV-vis spectra were taken on a Shimadzu UV-3101PC UV-vis-NIR scanning spectrophotometer with quartz cuvettes. Raman spectra were collected with a Renishaw InVia Raman Microscope using 633 nm laser excitation. Thermogravimetric analyses were carried out in alumina crucibles using a Pyris Diamond TG/DTA Analyzer (PerkinElmer) under a flow of argon. The heating profile used was from 30 °C to 300 °C with a ramp rate of 5 °C min−1. The tensile tests were obtained by using an Instron model 5944 with a loading rate of 0.1% s−1. The electrical conductance was measured by a multimeter with a probe distance of 0.5 inch.2.3 Graphite exfoliation
5 mg of graphite powders (Carbon Graphite Materials, Inc., M102) were each placed in 6 mL scintillation vials with 5 mL aliquots of base solutions, NaOH, KOH, and NH4OH. The vials then were bath sonicated (Ultrasonic Bath 2.8 L, Fisher Scientific) for one hour at room temperature, followed by centrifugation (Allegra X-15R Centrifuge, SX4750, Beckman Coulter) at 500 r.p.m. for one hour. The supernatant was then carefully transferred out via a pipette for UV-vis measurements.2.4 Carbon materials dispersions in liquid ammonia
To demonstrate the stability of dispersions of carbon materials in liquid ammonia, 3–5 mg of each desired material was first placed in a 7 mL scintillation vial. Liquid ammonia was then collected in an Erlenmeyer flask by condensing ammonia gas with an acetone/dry ice cooling bath (−78 °C). The vials with the carbon materials were pre-chilled in the cooling bath before ∼5 mL of liquid ammonia was transferred in through disposable Pasteur glass pipets. Those solutions were then probe sonicated for 2 min (2 s/2 s) at 90% intensity (Ultrasonic Processor, 450 W), while the vials were sitting in the acetone/dry ice cooling bath.2.5 Fabrication of GO membranes
200 mg of partially oxidized GO paste (4.86%) and 4 mL of deionized water were placed in a 20 mL scintillation vial for a few minutes of bath sonication. The solution was then vacuum filtered through filter paper (Millipore, AAWP, 0.8 μm) to obtain a freestanding membrane after being air dried in a fumehood for several days.2.6 Fabrication of rGO aerogels
In a typical synthesis, graphite oxide (GO) was prepared by a modified Hummers' method.23 0.5 g of urea and 25 mg of the resulting GO was dispersed in 10 mL of DI water. After ultrasonication for 10 min at room temperature, a homogeneous dispersion (2.5 mg mL−1) was obtained. The dispersion was heated at 180 °C for 8 h in a 30 mL sealed Teflon-lined stainless-steel autoclave. The reaction was cooled down naturally. A cylindrical reduced graphite oxide aerogel monolith was produced in a clear and colorless solution. The resulting rGO wet gels were washed with D.I. water three times and freeze dried at −40 °C and preserved for further experiments.2.7 Fabrication of SWCNT membranes
The densified SWCNT films were prepared via a thermally-induced evaporation process. Briefly, the SWCNT powders (SWNT-COOH) were first completely dissolved in chlorosulfonic acid (CSA).24 Then, 5 mL of SWCNT in CSA was slowly evaporated in a glass Petri dish which was continually heated on a heating plate. The densified SWCNT films formed on the bottom of the Petri dish after the complete evaporation of the CSA. The as-produced SWCNT films could be readily peeled off in the water bath.2.8 Synthesis of polyaniline emeraldine base
As described in a previous report,25 in a typical synthesis, 3.2 mmol of aniline was first dissolved in 10 mL of 1.0 M HCl solution. In another vial, ammonium persulfate (0.8 mmol) was dissolved in 10 mL of 1.0 M HCl solution. These two solutions were mixed quickly and then left overnight. The solution was then centrifuged and washed with 0.1 M HCl to remove ions and residues. The product was stirred in a round bottom flask with 2.0 M NH4OH for over three hours, followed by centrifugation and washing with copious amount of D.I. water. The final emeraldine base powder was obtained after air-drying the product for several days.2.9 Synthesis of aniline tetramer emeraldine base
In a modified procedure,26 0.92 (g) of aniline dimer was dissolved in a solution comprised of 10 mL of ethanol and 50 mL of 0.1 M HCl, and stirred for one hour. In another beaker, 0.81 (g) of ferric chloride was dissolved in 125 mL of 0.1 M HCl assisted by gentle sonication. The ferric chloride solution was then poured into the aniline dimer solution and then stirred for 2 h. The product was then centrifuged and washed with 0.1 M HCl at least 6 times. Afterwards, the product was reacted with 2.0 M NH4OH for 2 h, followed by centrifugation and washing with D.I. water several times. The final powder in the emeraldine base form was collected after air-drying for days. The final product was confirmed by NMR.2.10 Fabrication of 10 wt% SWCNT/cellulose composite membranes
0.15 g of paper (Boise Aspen 30) was shredded and then probe sonicated for 1 h (2 s/2 s) in 100 mL of D.I. with 80% intensity. 15 mg of SWCNT were added followed by another 15 min of probe sonication. The dispersion was then vacuum filtered and air-dried for several days.3. Results and discussion
3.1 Dispersions of GO and rGO
Our previous study revealed that the stability of rGO colloidal suspensions in NH4OH solutions are dependent on the pH, up to pH 11, due to surface charge (zeta potential) in accord with the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory.27 The classical DLVO force is comprised of electrostatic forces and van der Waals forces. The electrostatic force is enhanced as the pH increases, reaching an equilibrium against the van der Waals force at a certain pH, that then leads to destabilization and precipitation at an even higher pH. Thereafter, several articles addressed the exfoliation of graphite in the presence of diluted NH4OH;28,29 however, the use of high concentrations of NH4OH and other basic hydroxide solutions have rarely been addressed.30 In Fig. 1a–c, rGO sheets were probe sonicated in serial dilutions of NaOH, KOH, and NH4OH, and left undisturbed overnight. The vials on the most right-hand side are 1.0 M for NaOH and KOH, and 28–30 wt% for NH4OH, with pH values of 14. Vials to the left were diluted ten times from the right ones. It is observed that NaOH and KOH solutions follow classical DLVO theory, where negative zeta potential builds up as the pH increases. The electrostatic repulsions make rGO stable and well-dispersed colloids within the pH range between 10 to 12. As seen, the dispersion concentrations decay at an even higher pH environments for NaOH and KOH solutions as the increasing electrostatic repulsions overpower the van der Waals attractions, resulting in precipitation.27 However, the DLVO theory seems not to be the only governing factor for NH4OH solutions. The most direct evidence can be found in Fig. 1d, in which the NH4OH dispersion (right) shows a much stronger Tyndall effect compared to NaOH (left) and KOH (middle) dispersions at a pH of 14, indicating a comparatively superior dispersibility of NH4OH. Such variations in the dispersion concentration are evident even after 3 days left undisturbed (Fig. S1†).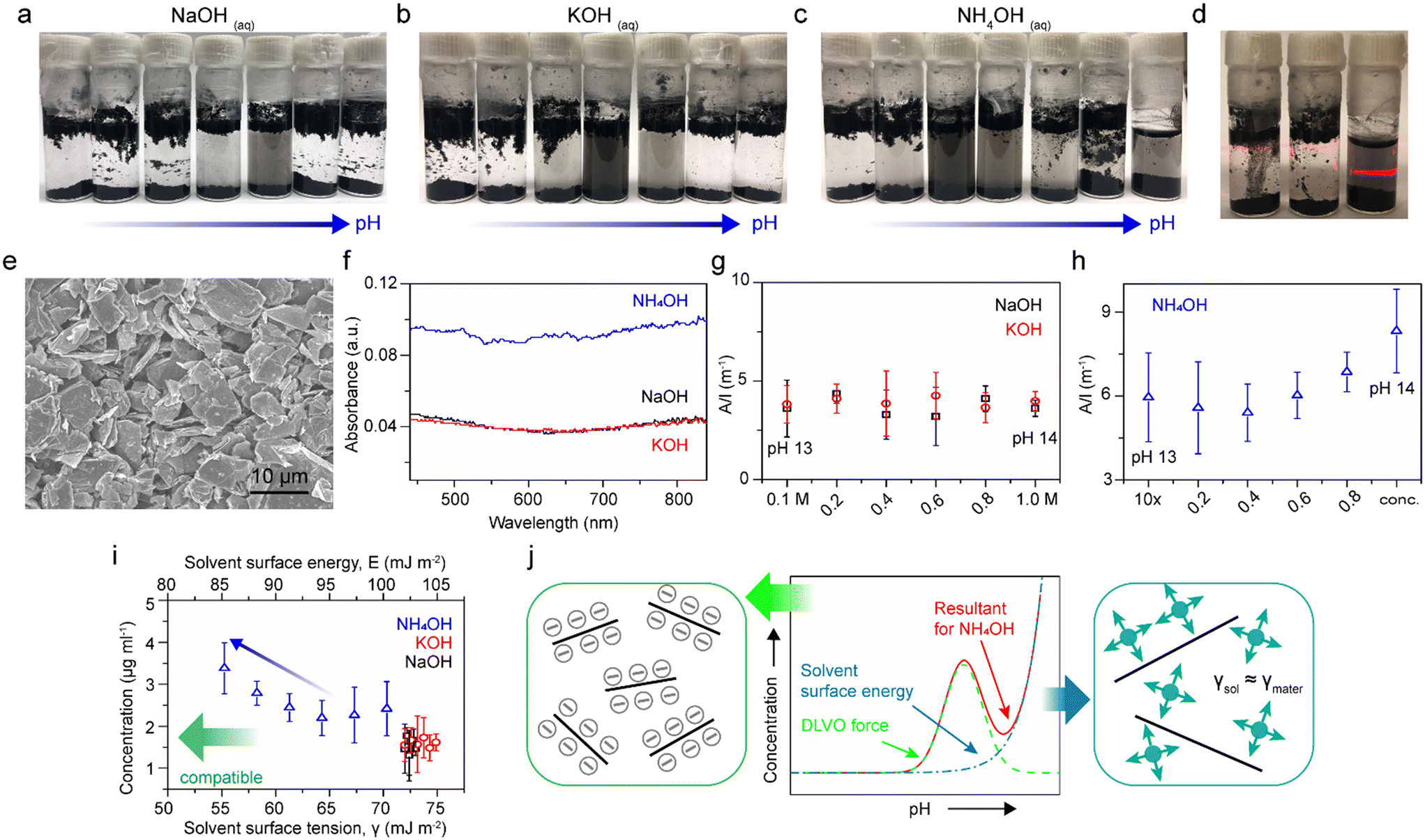 | ||
| Fig. 1 Dispersions of rGO sheets in serial dilutions of (a) NaOH, (b) KOH, and (c) NH4OH aqueous solutions. (d) At pH 14, a NH4OH solution (right) shows a strong Tyndall effect compared to solutions of NaOH (left) and KOH (middle). (e) SEM image of pristine graphite. (f) Absorption spectra of graphite dispersed in KOH, NaOH, and NH4OH after exfoliation and centrifugation. Optical absorbance at λ = 660 nm divided by cell length (A/l) versus the fraction of pH 14 solutions in their ten-time-diluted counterparts for graphite dispersions in solutions of (g) NaOH and KOH, and (h) NH4OH. (i) Concentration of graphite dispersions in solutions of NaOH, KOH, and NH4OH versus solvent surface tension. The concentration was calculated based on the absorbance value at λ = 660 nm, with the absorption coefficient equal to 2460 L g−1 m−1. (j) A scheme showing the stability of graphite dispersions in NH4OH versus pH (from neutral to basic regime) is due to following factors: the DLVO force (green) and the compatibility (blue) between the solvent surface energy (γsol) and the surface energy of the materials (γmater). | ||
In order to investigate this phenomenon more thoroughly, graphite powders (Fig. 1e) were added to a series of NaOH, KOH, and NH4OH solutions, from 0.1 M to 1.0 M for NaOH and KOH solutions, and from 2.8–3.0 wt% to 28–30 wt% for NH4OH solutions. The solutions in glass vials were bath ultrasonicated for 1 h, followed by centrifuge at 500 r.p.m. for 1 h. The dispersions were collected carefully, and then characterized by UV-vis-IR absorption spectroscopy. The absorption spectra of dispersions at pH 14 were recorded in Fig. 1f. Since a correlation between the concentration of dispersed graphite with the absorbance at a wavelength of 660 nm has been reported, the absorbance at 660 nm divided by the cell length (A/l) versus the fractions of pH 14 solutions in their ten-time-diluted counterparts are plotted (Fig. 1g and h).31 It is demonstrated that the value of A/l has no dependency on pH for NaOH and KOH dispersions but an upward inclination for NH4OH from pH 13 to pH 14.
By considering energetics, it is known that surface energies of materials and solvents have to be compatible in order to minimize their mixing enthalpy.32 Coleman and co-workers have concluded that the most compatible solvent surface tension value for graphite dispersions should be in the region of 40–50 mJ m−2.31 Therefore, the concentrations of graphite dispersions, assuming that the Lambert–Beer's law applies to all dispersions with the absorption coefficient equals to 2460 L g−1 m−1, were plotted as functions of solvent surface energy and solvent surface tension (Fig. 1i). The top y-axis, solvent surface energy, was calculated using the equation γ = E − TS, with a universal value for surface entropy of S = 0.1 mJ K−1 m−2. The surface tensions of 1.0 M NaOH, 1.0 M KOH, and concentrated (28–30 wt%) NH4OH are found to be 73, 74.9, and 55.25 mJ m−2, respectively; while the surface tensions of their diluted solutions are calculated based on the assumption of 72 mJ m−2 for the surface tension of water and linear correlations with the component fractions (Fig. S2†).33,34 The apparent increase in the graphite dispersion concentration as the NH4OH concentration is increased ten-fold (from 2.8–3.0 wt% to 28–30 wt% blue arrow), in conjunction with dispersion concentrations of NaOH (black) and KOH (red), corroborates that the surface tension of NH4OH does play as significant of a role in these dispersions.
In this regard, the stability of graphite dispersions versus pH of NH4OH solutions (from neutral to the basic regime) are a resultant (red) of two factors, the DLVO force (green) and the solvent surface energy (blue), as illustrated in Fig. 1j. The DLVO theory well describes the scenario of graphite dispersed in NaOH and KOH solutions as density changes of these two solutions among the span of pH are minute. However, in the case of NH4OH, the compatibility of the solvent surface energy becomes dominant at the high pH regime where the dispersion concentration surges. As a result, the concentration profile of the depicted scheme (Fig. 1j) is in a good agreement with Fig. 1c.
Fig. 2a shows that rGO does not form a stable dispersion in liquid ammonia even after probe sonication, which can be attributed to the incompatible surface tensions between liquid ammonia (21.97 mJ m−2) and graphite (30–40 mJ m−2).35,36 Therefore, the aforementioned NH4OH solutions are considered as co-solvents comprised of liquid ammonia and water, with surface tensions of 21.97 and 72 mJ m−2, respectively. Predictably, NH4OH at 0 degree Celsius, which is able to take up 50 wt% of ammonia, may serve as a good solvent for graphite dispersions due to the compatibility of surface tension (44.55 mJ m−2).33 Besides, the poor dispersion of rGO in liquid ammonia can also be forecast by the Hansen solubility parameters. As Ruoff and co-workers observed, for colloidal suspensions of rGO in various organic solvents, the sum of the polarity cohesion parameter (δp) and hydrogen bonding cohesion parameter (δh) has to fall within the range of 13 to 29, in order to create a good dispersion of rGO.37 Since the polarity cohesion parameter and hydrogen bonding cohesion parameter of ammonia are 16.7 and 18.8, respectively, the instability of graphite dispersion in liquid ammonia is then validated.38 Furthermore, by re-examining the m-cresol-carbon nanotube charge-transfer complex reported by Chiou et al., we found that the surface tension of m-cresol (35.69 mJ m−2) is not far from that of carbon nanotubes (∼40 mJ m−2),15,39 thus the charge transfer successfully helped stabilize SWCNTs in m-cresol. However, in the case of liquid ammonia and graphite or rGO, the massive difference in surface tensions results in unstable suspensions regardless of charge transfer (∼0.03 electrons per molecule).29,40
 | ||
| Fig. 2 Images of (a) rGO, (b) GO, and (c) SWCNT being probe sonicated in liquid ammonia (left) and left undisturbed for one hour (right). The emeraldine base form of (d) the aniline tetramer and (e) polyaniline in liquid ammonia (left) and 28–30 wt% NH4OH aqueous solutions (right). | ||
GO, with abundant functional groups such as hydroxyl, phenol, carboxyl, quinone, and epoxide, is known to disperse in solvents with high dielectric constants, such as water, dimethyl sulfoxide (DMSO), and N-methyl-2-pyrrolidone (NMP).41 Liquid ammonia, with a low dielectric constant (22.7) and a low polarity (∼0.4), is found not be able to solvate GO despite the formation of hydrogen bonding between the liquid ammonia and functional groups of GO (Fig. 2b). Additionally, dispersibilities of other materials were tested. SWCNTs are shown to precipitate in liquid ammonia after probe sonication (Fig. 2c), likely due to the aforementioned incompatible surface energies. Aniline tetramer in the emeraldine base form is found to be well dissolved in liquid ammonia (left), but not in concentrated NH4OH (right) (Fig. 2d). With a similar dielectric constant to ethanol, liquid ammonia serves as a good solvent for the aniline tetramer, showing a bluish color.42 Polyaniline, with a higher dielectric constant compared to the tetramer counterpart, does not disperse in liquid ammonia nor concentrated NH4OH (Fig. 2e), confirming that de-doping is indeed the mechanism for polyaniline-based ammonia sensors instead of swelling of polymer chains.43
3.2 Surface characterizations
Chemical interactions between GO and liquid ammonia were investigated by XPS after soaking GO in liquid ammonia and liquid nitrogen as a control, for 1 min, 30 min, and 7 h. To prevent gas adsorption, XPS measurements were taken after treated samples were air-dried for more than one week. The XPS survey spectra are presented in Fig. 3a, where a trace amount of nitrogen is found in pristine GO (0.9 at%) as ascribed to the fabrication process for GO.23 Unlike GO being annealed in ammonia gas and hydrogen gas at elevated temperatures (>300 °C), the peaks at 286.66 eV and 287.8 eV, corresponding to C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups, respectively, in the high resolution C(1s) XPS spectra do not show significant changes (Fig. 3b–h),17 indicating that no reduction nor oxidation has occurred. Hence, the reduction of GO in the presence of ammonia gas at elevated temperatures can be attributed solely to the thermal energy applied. Detailed analysis of the area-under-the-curve integrations of deconvolutions of C(1s) are shown in Fig. 3i. The percentages of graphitic, C–O, CO, C–N, and CN for pristine GO are 43.40%, 42.84%, 8.78%, 1.98%, and 2.99%, respectively. As seen, the gradual increase in percentages of C–N and CN bonding were observed for GO treated with liquid ammonia, 3.24% and 3.98% for GO treated with liquid ammonia for 7 h, indicating potential N-doping.
O functional groups, respectively, in the high resolution C(1s) XPS spectra do not show significant changes (Fig. 3b–h),17 indicating that no reduction nor oxidation has occurred. Hence, the reduction of GO in the presence of ammonia gas at elevated temperatures can be attributed solely to the thermal energy applied. Detailed analysis of the area-under-the-curve integrations of deconvolutions of C(1s) are shown in Fig. 3i. The percentages of graphitic, C–O, CO, C–N, and CN for pristine GO are 43.40%, 42.84%, 8.78%, 1.98%, and 2.99%, respectively. As seen, the gradual increase in percentages of C–N and CN bonding were observed for GO treated with liquid ammonia, 3.24% and 3.98% for GO treated with liquid ammonia for 7 h, indicating potential N-doping.
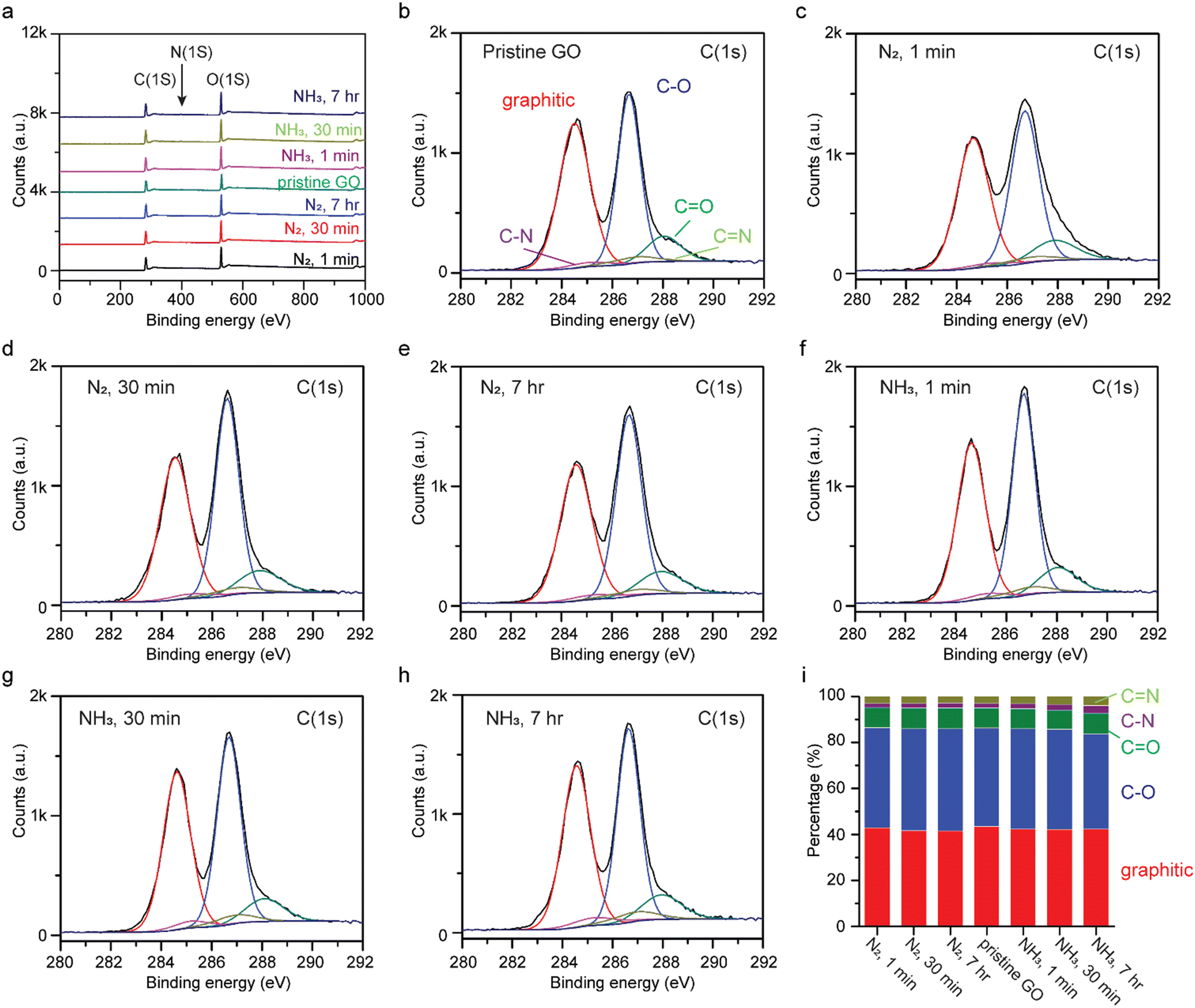 | ||
| Fig. 3 (a) XPS surveys, (b–h) high resolution C(1s) spectra, and their (i) area-under-the-curve ratios of peaks for pristine GO, and GO samples treated with liquid nitrogen and liquid ammonia for 1 min, 30 min, and 7 h. | ||
XPS reveals that N-doping can still occur in the absence of thermal energy for GO immersed in liquid ammonia (Fig. 4a). The samples soaked in liquid nitrogen show similar nitrogen contents to pristine GO, as expected. The nitrogen content increases from 0.9 at% for pristine GO to 1.46, 1.61, and 1.51 at% for GO soaked in liquid ammonia for 1 min, 30 min, and 7 h, respectively. High resolution N(1s) XPS spectra were carried out in order to clarify the bonding configurations. Since GO is considered a graphitic acid, the formation of ammonium carboxylate between carboxylic functional groups at GO and liquid ammonia is plausible, but its binding energy does not fall into the detected regime. The formation of amides, however, requires heat to undergo the dehydration process, and is not feasible in this case. The deconvolution of the N(1s) spectra were fit into two peaks, peak A at a lower energy and peak B at a higher energy. Peak A, at around 398.4 eV, corresponds to pyridinic nitrogen; while peak B, at around 400.5 eV, represents pyrrolic nitrogen, tertiary and quaternary amine, or amide.16,17,44–47 The results indicate that pristine GO and GO treated with liquid nitrogen exhibit stronger signals for peak B, while enhancements in peak A were observed for samples of GO soaked in liquid ammonia (Fig. 4b–f and S4†). The much stronger signal ratio of peak A to peak B for samples treated with liquid ammonia indicates N-doping. This N-doping trend is in line with GO annealed in ammonia gas at temperatures of 300–700 °C, where pyridinic N-doping was found. The peak positions, in terms of binding energy, of fit to peak A and peak B for all samples are fairly consistent, which rules out the formation of quaternary nitrogen replacing carbon atoms in the rGO sp2 network.17
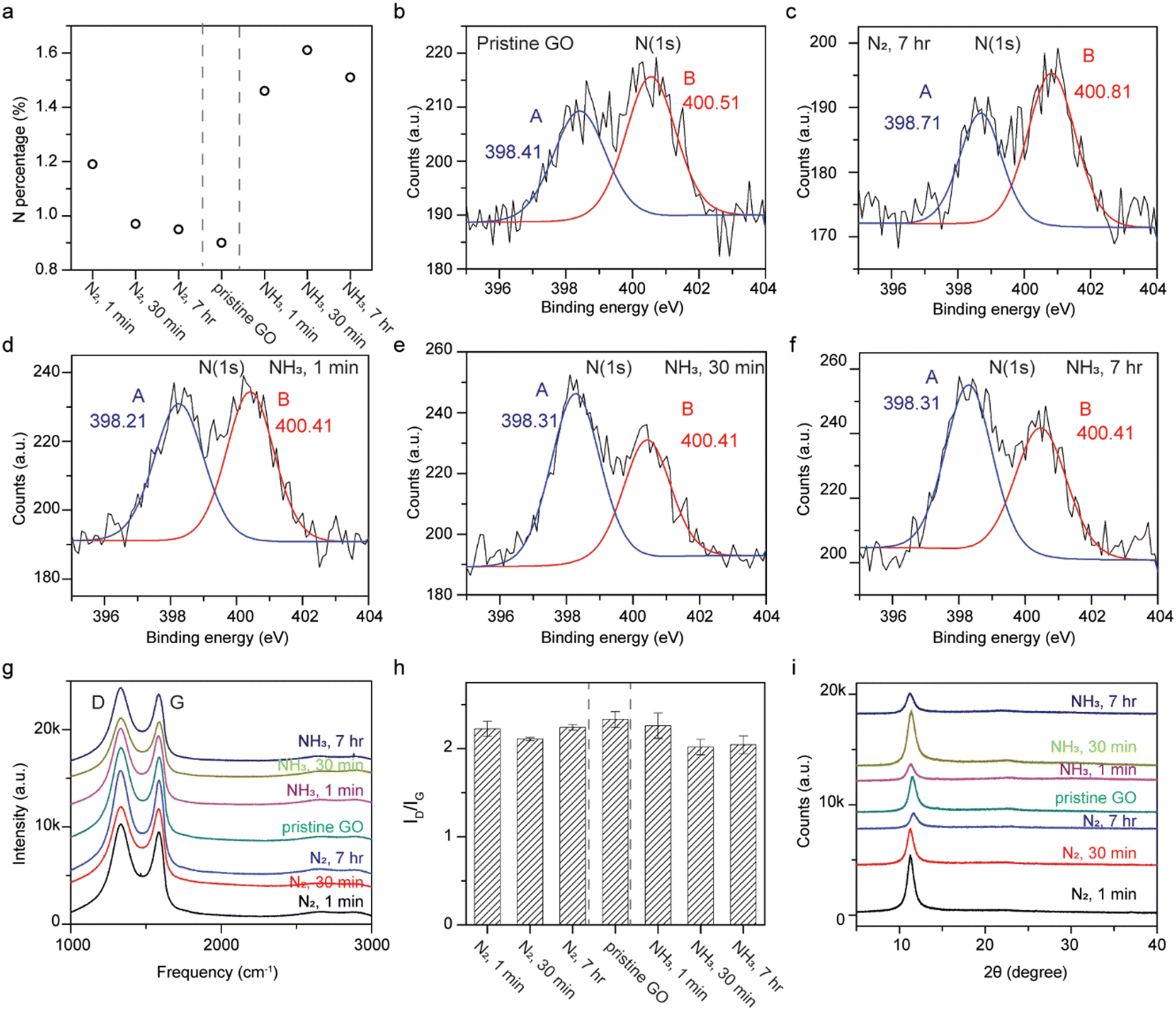 | ||
| Fig. 4 (a) Nitrogen contents of pristine GO, and GO treated with liquid nitrogen and liquid ammonia for 1 min, 30 min, and 7 h. High resolution N(1s) spectra of (b) pristine GO, (c) GO treated with liquid nitrogen for 7 h, and GO treated with liquid ammonia for (d) 1 min, (e) 30 min, and (f) 7 h. The peaks are fit at both low and high energies, A (blue) and B (red) components, with their centered positions. (g) Raman spectra, (h) D/G intensity ratios, and (i) XRD spectra of pristine GO, and GO treated with liquid nitrogen and liquid ammonia for 1 min, 30 min, and 7 h. | ||
Raman spectra show no obvious 2D peak at ∼2700 cm−1, indicating the non-reduction of GO and the non-restoration of conjugation (Fig. 4g).5,20 The consistency in the G peak positions of all samples verifies the non-incorporation of quaternary nitrogen into the carbon network.17,45,48 The marginally decreased D/G intensity ratio for GO treated with liquid nitrogen with respect to pristine GO are likely due to the error inherent in integrations of area-under-the-curve (Fig. 4h). On the other hand, the ID/IG for samples treated with liquid ammonia compared to pristine GO exhibit discernible decreases, which is opposite from that found for the Birch reduction reaction of GO, but consistent with GO reduced by ethylenediamine and ethylamine at room temperature.5,20 The prominently lower D/G ratios for GO treated with liquid ammonia suggest that the diminished functional groups reacting with ammonia may be responsible for the formation of pyridinic nitrogen and less out-of-place vibrations (D peak).
X-ray diffraction shows no graphitic peaks at 2θ = 26.3° verifying the absence of reduction of GO to rGO (Fig. 4i). A peak at 2θ = 11.572° corresponding to a lattice spacing of 7.6 Å was observed for pristine GO. For all treated GO samples, slight downshifts of the (002) peaks (2θ ∼ 11.3°) with a slightly greater d-spacing (7.8 Å) were detected, which should not be ascribed to intercalations as the sizes of ammonia and nitrogen are 3.26 Å and 3.05 Å, respectively. Instead, water may be the clue for this puzzle. GO, with its hygroscopic nature, picks up water from the environment, leading to expansions of the d-spacing with higher humidity.49 Thus, the low temperature treatment of GO by either liquid ammonia (∼244 K) or liquid nitrogen (∼77 K) led to undesired depositions of water from the ambient, which later caused downshifts of the (002) peaks.
3.3 GO membranes treated with liquid ammonia
To elucidate whether GO macroscopic assemblies shrink or deform upon the evaporation of liquid ammonia, GO freestanding membranes were fabricated via a typical vacuum filtration set-up. In Fig. 5a, an as-made GO membrane is shown on top of a filter membrane. GO membranes soaked in liquid nitrogen for 1 min and 30 min, and another one soaked in liquid ammonia for 30 min show no apparent differences from the as-made one, all exhibit smooth and semi-reflective surfaces. No deformation nor size alterations were observed. However, the GO membranes soaked in liquid ammonia for 1 min generated continuous loud popping sounds as ammonia gas was expelled, creating pockets in the membrane. When the GO membranes were removed from the liquid ammonia, these pockets were especially prevalent at the periphery of the membranes (Fig. 5b). The cross-sectional areas and top surfaces of all the membranes appear alike (Fig. 5c, S5 and S6†), except for the GO membrane soaked in liquid ammonia for 1 min which possessed ubiquitous hollow pockets on the size of hundreds of micrometers (Fig. 5d). We hypothesize that the short soaking period (1 min) of liquid ammonia generates thermal shock (∼100 K) when the GO membrane was exposed to the room temperature environment. The strong affinities, owing to the hydrogen bonding between liquid ammonia and functional groups of GO, and the possible formation of ammonium carboxylate result in trapped and longer retention of ammonia molecules.50,51 Thus, the instantaneous vaporization of ammonia due to the thermal shock generated loud popping sounds and created hollow pockets in the near-edge areas; however, such a phenomenon was not observed in the case of graphite paper and highly oriented pyrolytic graphite (HOPG).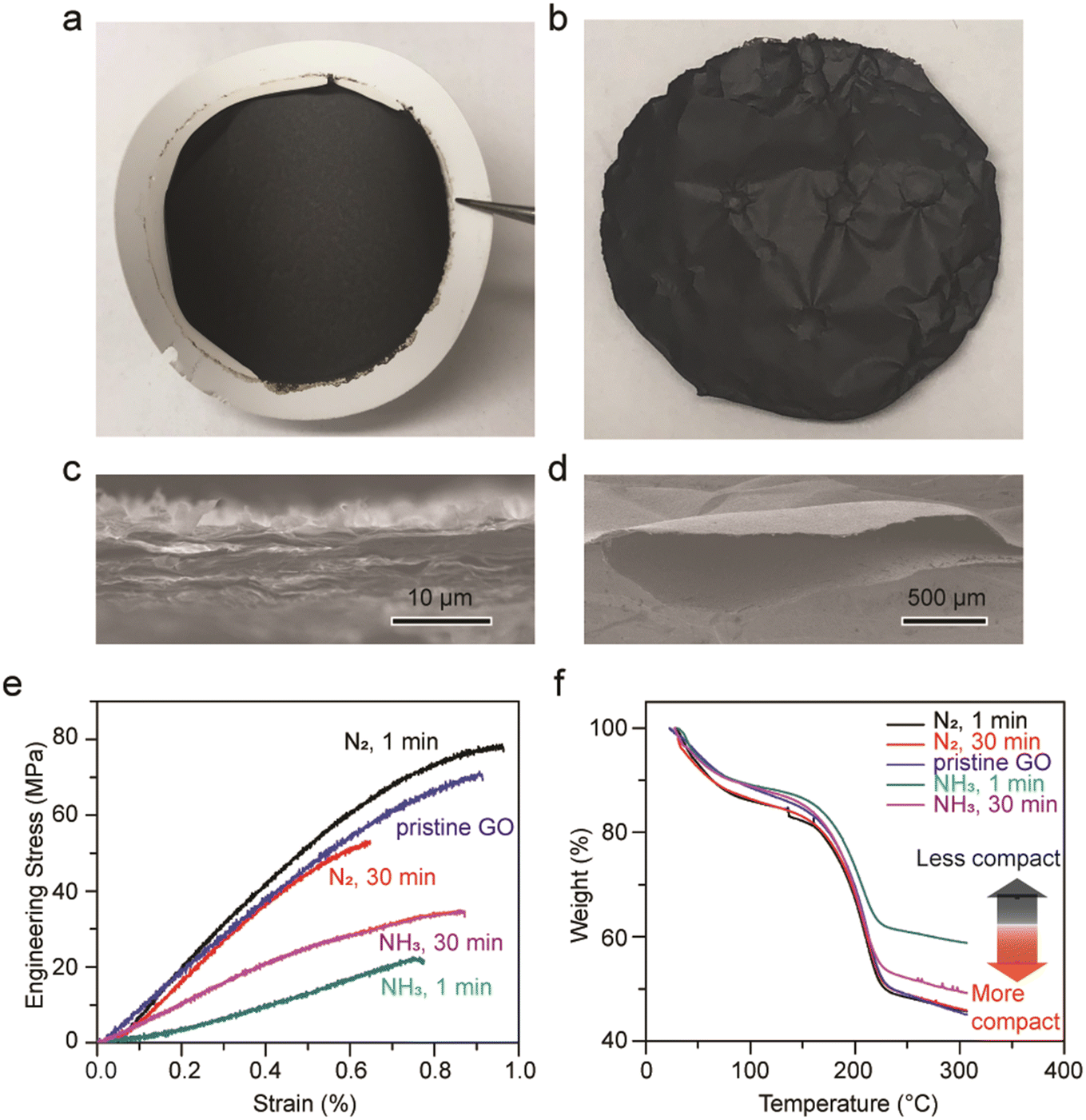 | ||
| Fig. 5 Optical images and cross-sectional SEM images of (a and c) as-made GO film and (b and d) GO film treated with liquid ammonia for 1 min. (e) Stress–strain curves and (f) TGA curves of pristine GO, and GO samples treated with liquid ammonia and liquid nitrogen for 1 min and 30 min. | ||
The impacts on the microstructure and interlayer forces of the membranes after treatment were also investigated. As no apparent differentiation in the cross-sectional images were observed, GO membranes were subjected to the tensile test where strips of GO membranes were clamped with tensile forces applied at a constant loading rate. As shown in Fig. 5e, the Young's modulus of pristine GO is about 10 GPa, similar to reported values.52 GO membranes soaked in liquid nitrogen for 1 min and 30 min exhibit similar Young's modulus as pristine GO, indicating that the van der Waals attractions and hydrogen bonding between GO sheets stay unaffected. In contrast, the Young's modulus of GO membranes soaked in liquid ammonia for 1 min and 30 min were found to be 3 GPa and 5 GPa, respectively. Since GO membranes soaked in liquid ammonia and liquid nitrogen share similar d-spacings based on XRD, the weakening in the van der Waals forces between GO sheets should not be detrimental. Therefore, the lowering in stiffness for GO membranes soaked in liquid ammonia may be ascribed to the structural alterations on the mesoscale, i.e., pores or pockets that are not within the XRD detectable regime.
Fig. 5f shows TGA curves for all the GO samples. The weight percentages remaining at 300 °C for pristine GO and GO membranes soaked in liquid nitrogen are approximately 46%, which are lower than that for GO membranes soaked in liquid ammonia for 1 min (59.0%) and 30 min (49.5%), meaning that the GO membranes treated with liquid ammonia exhibit less weight loss. It is observed that the trend of the TGA curves is in accordance with the trend of the stress–strain curves, where samples with lower Young's modulus retain higher percentages of weight. These results can be understood by the geometry of GO, where a dense GO solid was found to release more gas than a less compact GO foam. More carbon dioxide gas was generated from dense GO solids because of the closer GO sheets allowing just released carbon monoxide to react sufficiently with epoxide and hydroxyl groups on adjacent GO sheets to generate carbon dioxide.53 In this regard, GO membranes soaked in liquid ammonia are considered to possess comparatively porous structures, thus leading to a weaker stiffness.
3.4 rGO aerogels treated with liquid ammonia
In addition to compact GO membranes, extremely porous rGO aerogels made of thermal reduction of GO with urea followed by freeze-drying were examined. The as-made porous rGO aerogels possess a density around 13.3 mg cm−3 and a diameter around 0.5 inch. Profound and rapid shrinkages in the size of the rGO aerogels after soaking in liquid ammonia was observed. However, this phenomenon is intrinsically different from the shrinkage of dollar bills upon the evaporation of liquid ammonia as different levels of shrinkage of rGO aerogels after soaking in liquid nitrogen and water were obtained, as shown in Fig. 6a. The changes in diameter for rGO aerogels treated with liquid nitrogen, liquid ammonia, and water were found to be 0%, 21.0%, and 44.8%, respectively. Densities were measured to be 13.9, 29.8, and 88.0 mg cm−3 for rGO treated with liquid nitrogen, liquid ammonia, and water, respectively (Fig. 6b). For the rGO aerogel treated with water, the density can increase by almost 7 fold. The volume change percentage is found to be related to the surface tension of the solvent used (Fig. 6c). This relationship can be reasoned by the capillary pressures generated upon evaporation of solvents.54 According to the Young–Laplace equation, the capillary pressure, P, is directly proportional to the surface tension, γ, and inversely proportional to the radius of the pores, R (inset). Therefore, the capillary pressure of shrinkage increases as the surface tension of the solvent increases. Similar phenomenon was observed earlier in the case of GO doughs,55 and was later reported with rGO hydrogel monoliths treated by various organic solvents.54 It is noteworthy that much faster shrinkage of the rGO aerogels treated with liquid ammonia compared to water were observed, which can be attributed to the much higher vapor pressure of ammonia (1.07 MPa) compared to water (2.3 kPa).56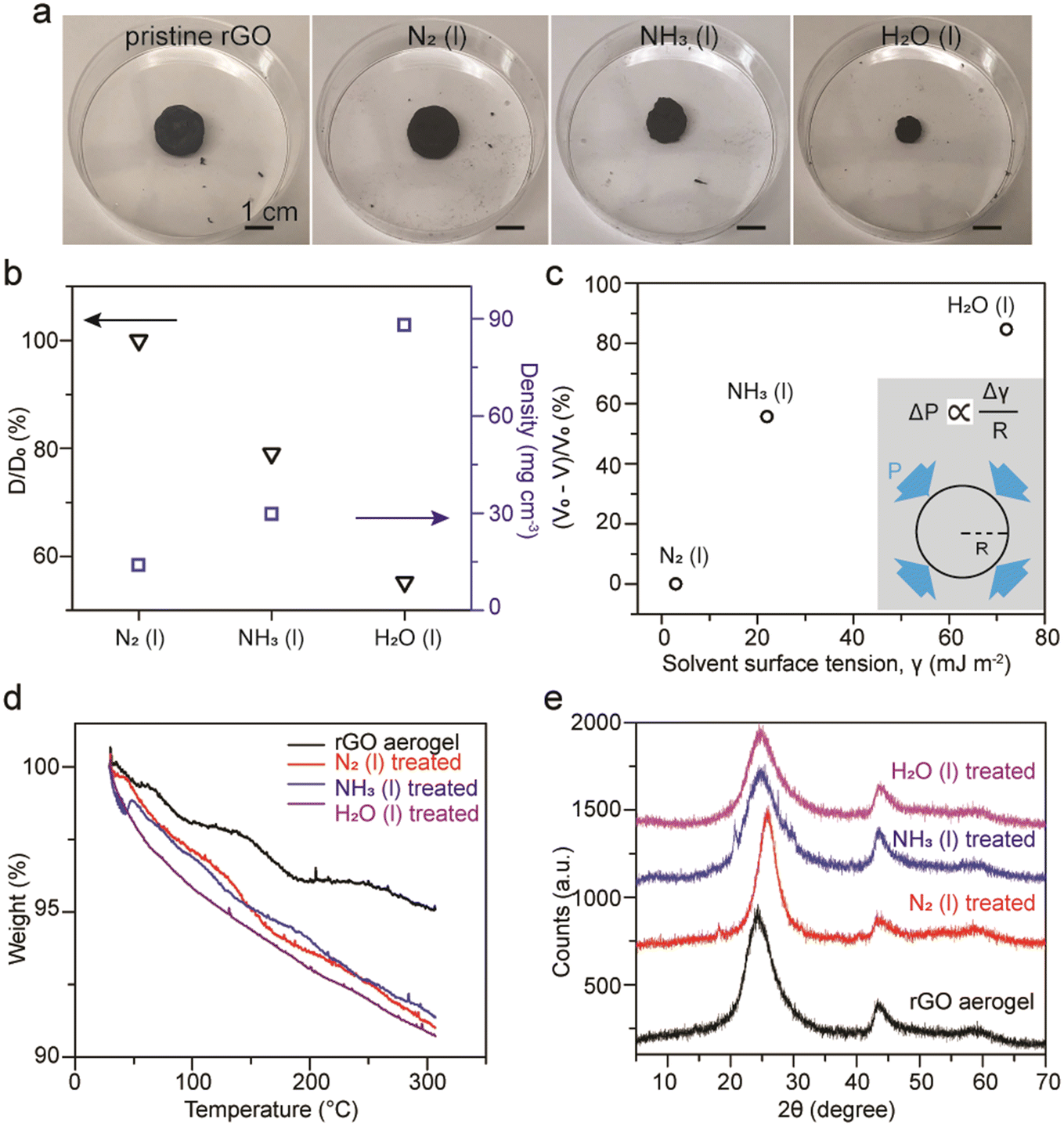 | ||
| Fig. 6 (a) Images and (b) changes in diameter and densities of a pristine rGO aerogel, and rGO aerogels soaked in liquid nitrogen, liquid ammonia, and water followed by two days of air-drying. (c) The percentage of the volume shrinkage of the rGO aerogels correlates with the surface tension of the solvent it is soaked in, which can be explained by the Young–Laplace equation (see inset). (d) TGA curves and (e) XRD spectra of a pristine rGO aerogel, and rGO aerogels treated with liquid nitrogen, liquid ammonia, and water, after air-drying. | ||
For TGA analysis under N2 gas flow, pristine rGO aerogels retain 95% of their initial weight, while rGO aerogels treated with liquid nitrogen, liquid ammonia, and water retain 91–92% at 300 °C (Fig. 6d). We have observed that the weight retentions for treated rGO aerogels at 300 °C can vary from 90% to 95%, likely due to regional anisotropic compressions for the rGO aerogel monoliths. The somewhat inconsistent trend between the weight retention percentage and the shrinkage percentage of the aerogels indicates that the evaluation of porosity of carbon materials based on weight loss may not be applicable for rGO due to its sparse amount of oxygen functional groups. XRD patterns exhibit typical rGO signals of (002) at 25° and (102) at 43° (Fig. 6e). The broadening in diffraction peaks with weak intensities indicate that the rGO sheets do not restack in a highly ordered fashion upon capillary compression.
3.5 Cellulose composite membranes
As SWCNTs were demonstrated to not disperse in liquid ammonia (Fig. 2e), no changes in appearance nor electrical resistance of the SWCNT freestanding membranes treated with liquid ammonia were observed (Fig. S8†). The 10 wt% SWCNT/cellulose composite membranes, on the other hand, do exhibit about 30% shrinkage in diameter after five treatments with liquid ammonia (Fig. 7a and b). Both the composite membranes and pure paper membranes showed a “doughy status”, i.e., elongations along the directions of applied tensile forces, right after being removed from liquid ammonia. The transition from rigid to wiggly cellulose fibers in the SEM images is likely the main cause for the macroscopic shrinkage of the membrane (Fig. 7c and d).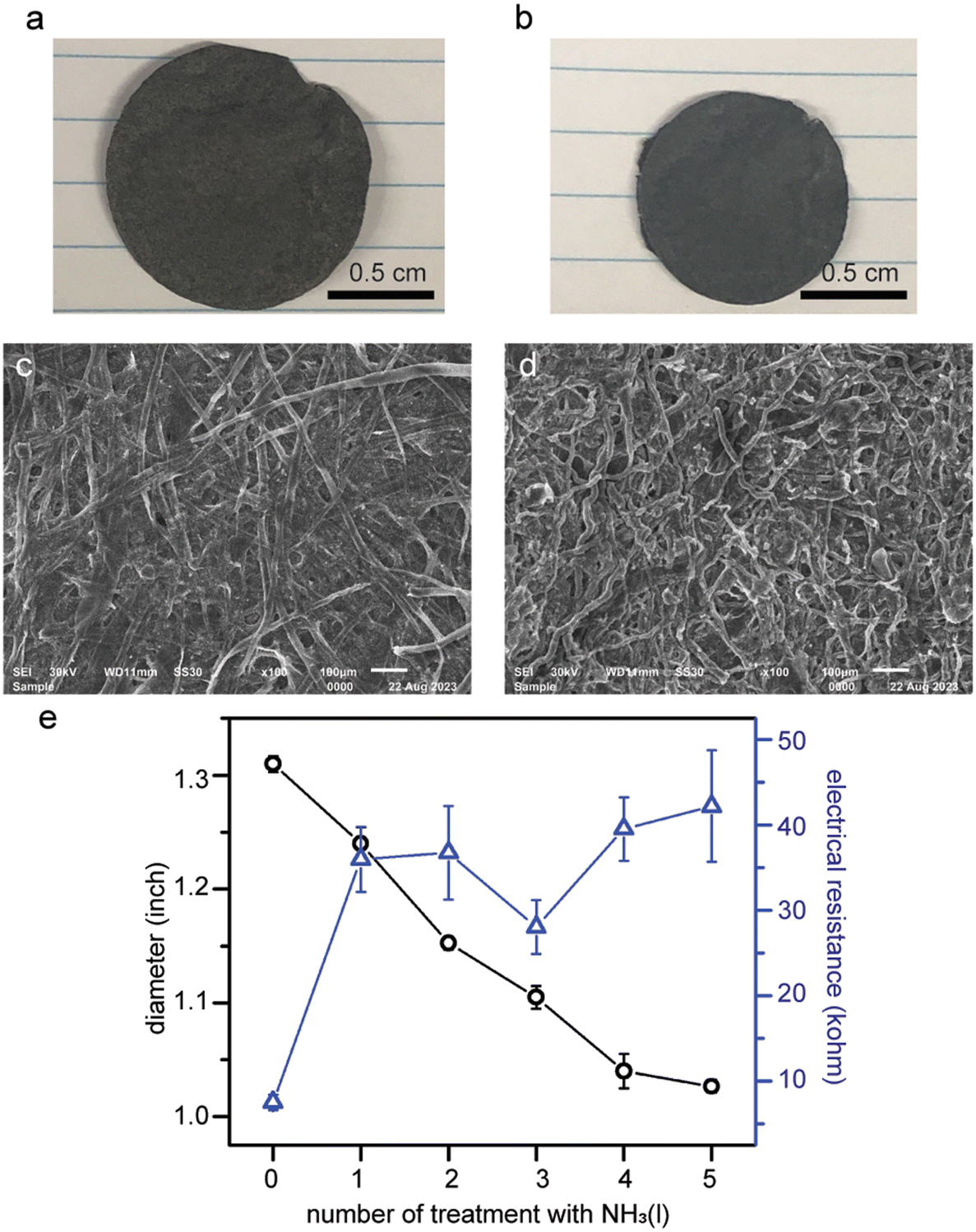 | ||
| Fig. 7 Optical images and SEM images of (a and c) an as-made SWCNT/cellulose composite membrane and (b and d) the composite membrane treated with liquid ammonia five times. (e) Changes in both diameter and electrical resistance of the composite membrane were observed. | ||
Unexpectedly, we found out that the electrical resistance of the composite membrane increased by 3 fold despite the appreciable shrinkage in diameter (Fig. 7e). The same phenomenon was also observed for pencil-traces-on-paper. We speculate that the decreases in the electrical conductance can be attributed to the creases and buckling of the composite membranes upon the evaporation of ammonia.
Conclusions
GO, rGO, and SWCNTs do not form stable dispersions in liquid ammonia. Precipitation results from the incompatible surface tensions regardless of charge transfer, and differences in polarity between liquid ammonia and functional groups of GO. The immersion of GO in liquid ammonia renders ∼0.6 at% pyridinic N-doping and no restoration in conjugation without the assistance of thermal energy, as confirmed by XPS, XRD, and Raman spectroscopy. Unlike cellulous-based materials, no shrinkage nor deformations of GO and SWCNT membranes were observed upon the evaporation of liquid ammonia. However, assembled GO membranes showed lowered stiffness after treatments with liquid ammonia, which can be ascribed to the generation of meso- and macropores due to instantaneous vaporization of trapped ammonia molecules. The different levels of compactness in the membrane integrity were validated by TGA analysis. On the other hand, rGO aerogels were found to shrink by 55% in volume upon the evaporation of liquid ammonia. The unexpected increase in the electrical resistance of SWCNT/cellulose composite membranes was observed despite the shrinkage in size. These insights presented in this report should lead to an improved understanding of liquid ammonia interactions with carbon based materials from both a chemistry and materials science perspective.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Dr. Myung Ki Hong Endowed Chair in Materials Innovation at UCLA for financial support (R. B. K.), the Funai Foundation for Information Technology (Y. K.), and the Honjo International Scholarship Foundation (Y. K.).References
- A. J. Birch, J. Chem. Soc., 1944, 430–436 RSC.
- C. B. Wooster and K. L. Godfrey, J. Am. Chem. Soc., 1937, 59, 596–597 CrossRef.
- R. B. Kaner, S.-M. Huang, C.-H. Lin and J. H. Kaufman, Synth. Met., 1989, 28, D115–D125 CrossRef.
- P. L. Cullen, K. M. Cox, M. K. Bin Subhan, L. Picco, O. D. Payton, D. J. Buckley, T. S. Miller, S. A. Hodge, N. T. Skipper, V. Tileli and C. A. Howard, Nat. Chem., 2017, 9, 244–249 CrossRef.
- H. Feng, R. Cheng, X. Zhao, X. Duan and J. Li, Nat. Commun., 2013, 4, 1539 CrossRef.
- A. Y. S. Eng, H. L. Poh, F. Šaněk, M. Maryško, S. Matějková, Z. Sofer and M. Pumera, ACS Nano, 2013, 7, 5930–5939 CrossRef PubMed.
- A. J. Barry, F. C. Peterson and A. J. King, J. Am. Chem. Soc., 1936, 58, 333–337 CrossRef.
- M. Wada, H. Chanzy, Y. Nishiyama and P. Langan, Macromolecules, 2004, 37, 8548–8555 CrossRef.
- C. Schuerch, Ind. Eng. Chem., 1963, 55, 39 CrossRef.
- C. K. F. Hermann, J. Chem. Educ., 1997, 74, 1357 CrossRef.
- Y. Dan, Y. Lu, N. J. Kybert, Z. Luo and A. T. C. Johnson, Nano Lett., 2009, 9, 1472–1475 CrossRef.
- J. D. Fowler, M. J. Allen, V. C. Tung, Y. Yang, R. B. Kaner and B. H. Weiller, ACS Nano, 2009, 3, 301–306 CrossRef CAS.
- H. E. Romero, P. Joshi, A. K. Gupta, H. R. Gutierrez, M. W. Cole, S. A. Tadigadapa and P. C. Eklund, Nanotechnology, 2009, 20, 245501 CrossRef.
- G. Lu, L. E. Ocola and J. Chen, Nanotechnology, 2009, 20, 445502 CrossRef PubMed.
- K. Chiou and J. Huang, Matter, 2020, 3, 302–319 CrossRef.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS.
- S. P. Sasikala, K. Huang, B. Giroire, P. Prabhakaran, L. Henry, A. Penicaud, P. Poulin and C. Aymonier, ACS Appl. Mater. Interfaces, 2016, 8, 30964–30971 CrossRef CAS.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS.
- N. H. Kim, T. Kuila and J. H. Lee, J. Mater. Chem. A, 2013, 1, 1349–1358 RSC.
- J. Burrows, S. Kamo and K. Koide, Science, 2021, 374, 741–746 CrossRef CAS.
- R. Raymond, L. Reggel, W. A. Steiner, S. Ergun and I. Wender, Nature, 1960, 185, 379–380 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- V. A. Davis, A. N. G. Parra-Vasquez, M. J. Green, P. K. Rai, N. Behabtu, V. Prieto, R. D. Booker, J. Schmidt, E. Kesselman, W. Zhou, H. Fan, W. W. Adams, R. H. Hauge, J. E. Fischer, Y. Cohen, Y. Talmon, R. E. Smalley and M. Pasquali, Nat. Nanotechnol., 2009, 4, 830–834 CrossRef CAS PubMed.
- J. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851–855 CrossRef CAS.
- Z. Shao, P. Rannou, S. Sadki, N. Fey, D. M. Lindsay and C. F. J. Faul, Chem. – Eur. J., 2011, 17, 12512–12521 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- I. Janowska, K. Chizari, O. Ersen, S. Zafeiratos, D. Soubane, V. D. Costa, V. Speisser, C. Boeglin, M. Houllé, D. Bégin, D. Plee, M.-J. Ledoux and C. Pham-Huu, Nano Res., 2010, 3, 126–137 CrossRef CAS.
- H. Ma, Z. Shen, M. Yi, S. Ben, S. Liang, L. Liu, Y. Zhang, X. Zhang and S. Ma, J. Colloid Interface Sci., 2017, 503, 68–75 CrossRef CAS.
- W. W. Liu and J. N. Wang, Chem. Commun., 2011, 47, 6888 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- J. H. Hildebrand, J. M. Prausnitz and R. L. Scott, Regular and Related Solutions: The Solubility of Gases, Liquids, and Solids, Van Nostrand Reinhold Company, 1970 Search PubMed.
- G. C. Ware, MSci thesis, Kansas State University, 1928 Search PubMed.
- P. M. Dunlap and S. R. Faris, Nature, 1962, 196, 1312–1313 CrossRef CAS.
- L. X. Benedict, N. G. Chopra, M. L. Cohen, A. Zettl, S. G. Louie and V. H. Crespi, Chem. Phys. Lett., 1998, 286, 490–496 CrossRef CAS.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155406 CrossRef.
- S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, Hoboken, 2nd edn, 2007 Search PubMed.
- S. D. Bergin, V. Nicolosi, P. V. Streich, S. Giordani, Z. Sun, A. H. Windle, P. Ryan, P. P. Niraj, Z.-T. T. Wang, L. Carpenter, W. J. Blau, J. J. Boland, J. P. Hamilton and J. N. Coleman, Adv. Mater., 2008, 20, 1876–1881 CrossRef CAS.
- Z. Zhang, X. Zhang, W. Luo, H. Yang, Y. He, Y. Liu, X. Zhang and G. Peng, Nanoscale Res. Lett., 2015, 10, 359 CrossRef.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- C.-W. Lin, R. L. Li, S. Robbennolt, M. T. Yeung, G. Akopov and R. B. Kaner, Macromolecules, 2017, 50, 5892–5897 CrossRef CAS.
- S. Virji, J. Huang, R. B. Kaner and B. H. Weiller, Nano Lett., 2004, 4, 491–496 CrossRef CAS.
- K. Kinoshita, Carbon: Electrochemical and Physicochemical Properties, Wiley, New York, 1988 Search PubMed.
- T. Kato, Y. Yamada, Y. Nishikawa, T. Otomo, H. Sato and S. Sato, J. Mater. Sci., 2021, 56, 15798–15811 CrossRef CAS.
- Y. Yamada, H. Tanaka, S. Kubo and S. Sato, Carbon, 2021, 185, 342–367 CrossRef CAS.
- Y. Yamada, J. Kim, S. Matsuo and S. Sato, Carbon, 2014, 70, 59–74 CrossRef CAS.
- B. Das, R. Voggu, C. S. Rout and C. N. R. Rao, Chem. Commun., 2008, 5155 RSC.
- J. Abraham, K. S. Vasu, C. D. Williams, K. Gopinadhan, Y. Su, C. T. Cherian, J. Dix, E. Prestat, S. J. Haigh, I. V. Grigorieva, P. Carbone, A. K. Geim and R. R. Nair, Nat. Nanotechnol., 2017, 12, 546–550 CrossRef CAS PubMed.
- S. Tang and Z. Cao, J. Phys. Chem. C, 2012, 116, 8778–8791 CrossRef CAS.
- M. Seredych and T. J. Bandosz, J. Phys. Chem. C, 2007, 111, 15596–15604 CrossRef CAS.
- C.-N. Yeh, K. Raidongia, J. Shao, Q.-H. Yang and J. Huang, Nat. Chem., 2015, 7, 166–170 CrossRef CAS PubMed.
- L. Klemeyer, H. Park and J. Huang, ACS Mater. Lett., 2021, 3, 511–515 CrossRef CAS.
- C. Qi, C. Luo, Y. Tao, W. Lv, C. Zhang, Y. Deng, H. Li, J. Han, G. Ling and Q.-H. Yang, Sci. China Mater., 2020, 63, 1870–1877 CrossRef CAS.
- C.-N. Yeh, H. Huang, A. T. O. Lim, R.-H. Jhang, C.-H. Chen and J. Huang, Nat. Commun., 2019, 10, 422 CrossRef CAS PubMed.
- C. S. Cragoe, C. H. Meyers and C. S. Taylor, Vapor Pressure of ammonia, Gov. Pr. Off., Washington, 1920 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI includes deconvolutions of XPS spectra, SEM images of GO and SWCNT membranes, and FT-IR spectra. See DOI: https://doi.org/10.1039/d3lf00194f |
| This journal is © The Royal Society of Chemistry 2024 |