
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of co-reactants on surface passivation by nanoscale hafnium oxide layers grown by atomic layer deposition on silicon†
Sophie L.
Pain
 *a,
Edris
Khorani
a,
Anup
Yadav
a,
Tim
Niewelt
abc,
Antonio
Leimenstoll
b,
Brendan F. M.
Healy
a,
Marc
Walker
d,
David
Walker
d,
Nicholas E.
Grant
a and
John D.
Murphy
a
*a,
Edris
Khorani
a,
Anup
Yadav
a,
Tim
Niewelt
abc,
Antonio
Leimenstoll
b,
Brendan F. M.
Healy
a,
Marc
Walker
d,
David
Walker
d,
Nicholas E.
Grant
a and
John D.
Murphy
a
aSchool of Engineering, University of Warwick, Coventry, CV4 7AL, UK. E-mail: sophie.l.pain@warwick.ac.uk
bFraunhofer Institute for Solar Energy Systems ISE, Heidenhofstraße 2, 79110 Freiburg, Germany
cChair for Photovoltaic Energy Conversion, Institute for Sustainable Systems Engineering, University of Freiburg, Emmy-Noether-Straße 2, 79110 Freiburg, Germany
dDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
First published on 7th December 2023
Abstract
Hafnium oxide thin films have attracted considerable interest for passivation layers, protective barriers, and anti-reflection coatings. Atomic layer deposition offers a route to produce conformal films at the nanometre scale, but there is a lack of clarity over how the growth conditions affect the film properties. Here we present a study into the role of different atomic layer deposition co-reactants (O2 plasma, O3 and H2O) in the growth of HfOx on n-type silicon from a tetrakis(dimethylamido)hafnium (TDMAH) precursor followed by post-deposition annealing (up to 500 °C). Through X-ray diffraction and X-ray photoelectron spectroscopy, we demonstrate variations in film composition, stoichiometry and crystallinity with co-reactant. Depth profiling conducted with X-ray photoelectron spectroscopy reveals differences in composition between the HfOx surface and the HfOx/Si interface. We also determine differences in fixed charge density and chemical passivation through photoconductance decay measurements and Kelvin probe analysis. We find surface recombination velocities (SRVs) <10 cm s−1 are possible for HfOx films, with the best passivation achieved for H2O-based HfOx (SRVs as low as ∼5 cm s−1). With TDMAH as a hafnium precursor, we show that neither co-reactant choice nor annealing environment influence the resulting charge polarity.
Introduction
Surface passivation aims to minimise unwanted recombination of charge carriers in semiconductors which, in the case of silicon, is key to improving solar cell efficiency. Passivation arises from both chemical and field effects, with the former involving termination of dangling bonds at the semiconductor surface, and the latter involving surface charges which can repel charge carriers. Deposition of dielectrics is an established passivation strategy,1 with atomic layer deposition (ALD) being commonly used. ALD is based on sequential self-terminating reactions, and offers thickness control with atomic layer precision, as well as uniform and conformal deposition.2–5 Deposition process temperature, pressure, duration, and other parameters play a key role in achieving the desired film properties.5 ALD-grown films are usually annealed ex situ to “activate” the passivation, with the passivation level strongly affected by the post-deposition annealing temperature due to variations in chemical and field effects.6,7The chemical reaction step of ALD can be conducted either thermally or enhanced by the presence of a plasma.4 In thermal ALD, one of the precursors is typically a gas such as O2, or a vapour such as H2O or O3. During plasma-enhanced ALD (PE-ALD), a plasma is generated from a gas (such as O2, N2 or H2), and the substrate surface is exposed to the species generated.
Recent studies have identified HfOx as a promising passivating layer.8–14 ALD-grown HfOx passivation studies have achieved surface recombination velocities (SRVs) <10 cm s−1 (compared to <2 cm s−1 for thermal SiO2 and <1 cm s−1 for ALD Al2O3)1,15,16 for films 10–20 nm thick.8–14 In addition to its potential as an insulating silicon passivation layer, the electronic properties of HfOx make it a candidate for inclusion within passivating contact structures, either as a hole-selective contact17,18 or as an interlayer.19 Beyond silicon passivation, HfOx has been extensively researched by the (micro)electronics industry for applications in transistors and capacitors20,21 due to its high dielectric constant, and as a protective barrier layer, due to the impressive chemical resistance of HfOx to etchants such as HF.22
Although there has been considerable interest in ALD-grown HfOx, there is a lack of consistency in film properties reported in the literature, as demonstrated in Table 1 of ref. 18. This is particularly evident when considering HfOx charge polarity, which has been reported as being both positive and negative,8–14 even for films processed under very similar conditions. This inconsistency could arise from several process parameters – such as hafnium precursor, co-reactant, deposition temperature and pressure, and post-deposition processing. To date, investigations have been conducted into the impact of varying co-reactant (mainly H2O and O3) with tetrakis(ethylmethylamido)hafnium (TEMAH), hafnium tetrachloride (HfCl4) and tetrakis(dimethylamido)hafnium (TDMAH) precursors,11,23,24 and into the effect of changing precursor while maintaining co-reactant.25 Multiple parameters were varied within these studies, making it difficult to ascribe changes in film properties and performance to any specific effect. Additionally, the inter- and intra-precursor variability in film properties suggests that conclusions drawn from one study may not necessarily be applicable to another investigation with different parameters.
Here we present a systematic study into the impact of co-reactants on film properties of HfOx grown with a TDMAH precursor. Oxidant choice can affect electrical properties, impurities, refractive indices, and morphology26 and hence we investigate three co-reactants: remote oxygen plasma (with resultant films hereafter denoted O2–HfOx), ozone (O3–HfOx) and water (H2O–HfOx). Post-deposition annealing conditions, such as annealing environment, are another key parameter varied in the literature,27 and we assess the impact of four annealing environments (air, forming gas, O2 and N2) on resulting passivation quality.
Results and discussion
Structural properties
To allow comparison between films grown from different co-reactants, a constant number of ALD cycles (200) was used for all samples discussed in this study. We have previously determined that 200 ALD cycles of O2–HfOx results in a 26 nm thick film,22 and we find similar film thicknesses with H2O–HfOx and O3–HfOx of 25 and 23 nm, respectively. Film thickness can play a significant role in HfOx film properties, notably with passivation when films are very thin,19 although the difference is lesser at greater thicknesses. These films are of a very similar thickness at a thickness level where passivation is not strongly thickness dependent, ensuring that comparison between film types is meaningful. Thickness measurements for each film type, and derived growth rates per ALD cycle are shown in Fig. S1(a) in the ESI.†Much of the behaviour of hafnium oxide, such as its chemical resistance22 and optical properties (shown in Fig. S1(b)†),28 has been attributed to crystallisation. We have previously investigated extensively the crystallisation behaviour of O2–HfOx as a function of annealing temperature,6,22 and have determined an amorphous-to-crystalline phase transition at 275–325 °C. Our prior work found that good passivation quality was achieved following annealing after this crystallisation point, whether this is as a result of crystallisation is as yet unclear. To assess whether there is any difference in crystallinity and this phase transition, we performed grazing-incidence X-ray diffraction (GI-XRD) on all film types, both in the as-deposited state and after annealing in air at ∼450 °C. The resulting GI-XRD patterns are presented in Fig. 1(a)–(c) for O2–HfOx, H2O–HfOx and O3–HfOx, respectively. Irrespective of co-reactant, all three HfOx film types are amorphous on deposition. The low intensity features seen in the 2θ range of 50–60° of each XRD pattern can be attributed to the (311) plane of the underlying c-Si (100) substrate.29 Following annealing at ∼450 °C, O2–HfOx and H2O–HfOx undergo a clear phase transition to monoclinic, although O3–HfOx does not, remaining amorphous. It is as yet unclear why O3–HfOx appears to have different crystallisation behaviour than its O2– and H2O–HfOx counterparts.
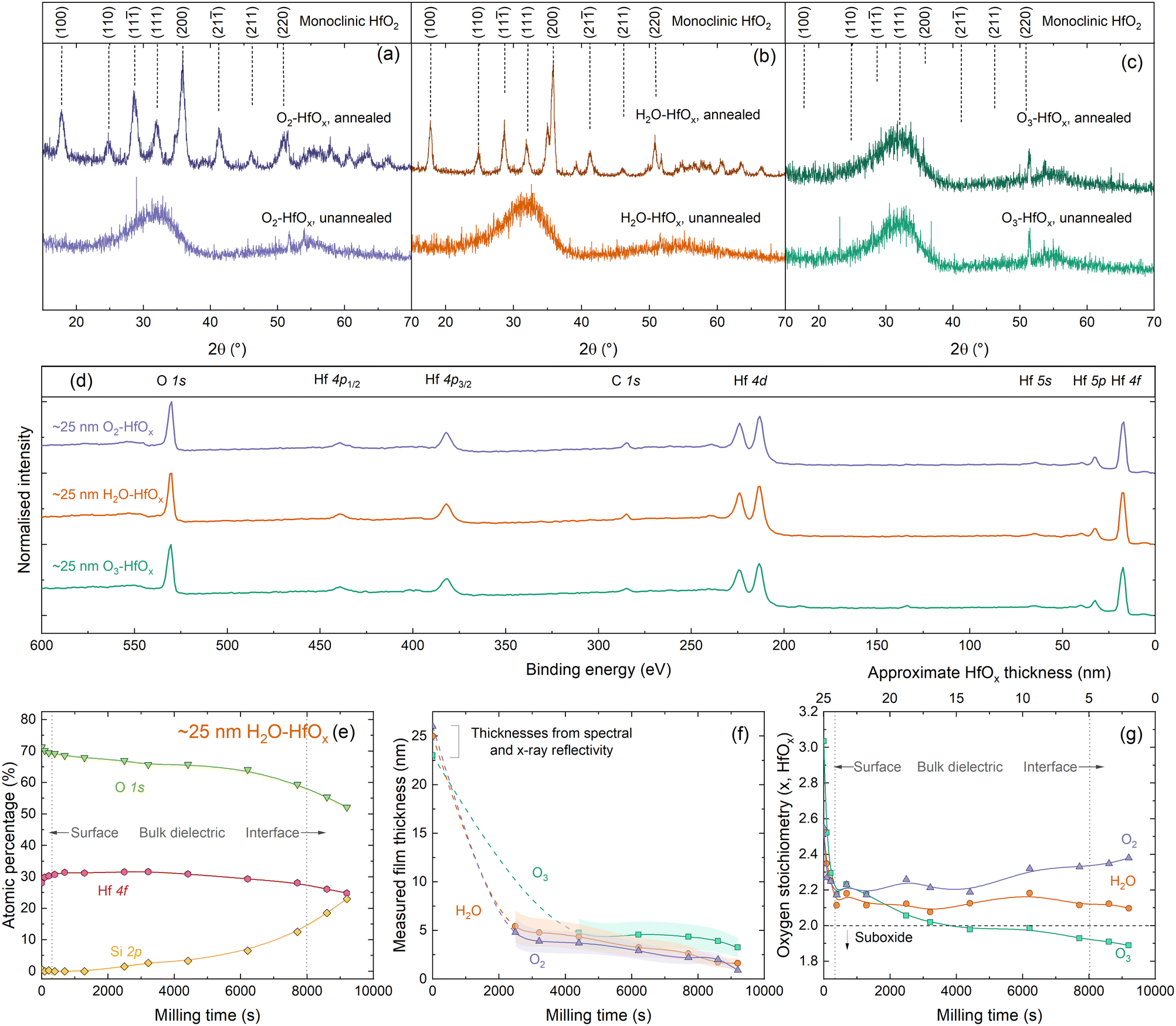 | ||
| Fig. 1 (a) GI-XRD patterns, using Cu Kα1/2, taken from polished silicon wafers coated with (a) O2–HfOx, (b) H2O–HfOx and (c) O3–HfOx as deposited (unannealed) or following annealing at 450–475 °C for 30 min in air. The sharp signals visible at 2θ = ∼27° for unannealed O2–HfOx and 2θ = ∼23° and ∼36° for unannealed O3–HfOx are measurement artefacts. Patterns are vertically offset for clarity. O2–HfOx GI-XRD data (purple lines) were previously published in ref. 6. Main crystallographic planes, corresponding to monoclinic HfO2, are labelled above.33 (d) XPS Survey scans for ∼25 nm O2–HfOx (purple), O3–HfOx (green) and H2O–HfOx (orange) after annealing at 450 °C. All XPS spectra were calibrated to the C 1s core level. Core levels are labelled. Survey scan was collected prior to milling with Ar+. (e) Atomic concentrations (%) determined via XPS for Hf (pink circles), O (green triangles) and Si (yellow diamonds, determined from intensity of signal at a binding energy of ∼99 eV) as a function of milling time for H2O–HfOx, shown for demonstration purposes. (f) Determined film thickness via the thickogram method32 for O2–HfOx, H2O–HfOx and O3–HfOx (purple triangles, orange circles and green squares, respectively) as a function of XPS milling time. Initial thicknesses determined via X-ray or spectral reflectivity. Connections between points serve as a guide to the eye. Shaded region corresponds to the relative uncertainty of determined thickness. (g) HfOx stoichiometry as a function of milling time. | ||
Chemical properties
To explore the origins of this differing behaviour, ∼25 nm films were subsequently characterised via X-ray photoelectron spectroscopy (XPS). Survey scans for each film are shown in Fig. 1(d), with core levels labelled. All XPS spectra were calibrated to the C 1s core level. The survey scans are dominated by Hf and O signals. No evidence of the underlying Si substrate is detectable, as the film thickness far exceeds the photoelectron sampling depth (∼10 nm).30,31There are no significant differences in the survey scans for each material, however closer inspection of the electronic core levels via high resolution scans reveals some variations in composition, both between film types and at each stage of depth profiling. Relative atomic concentrations of each species present were determined as a function of depth into the material. The Hf 4f and O 1s core levels were used to determine the stoichiometry of the HfOx films as a function of depth, and Si 2p was used for silicon (i.e., identifying when we have milled to the HfOx/Si interface). Three clear regions are evident from this depth profiling – surface/near-surface, bulk dielectric, and the HfOx/Si interface, as annotated in Fig. 1(e). The Hf 4f and Si 2p signals allow determination of an approximate milling rate. Once sufficient HfOx overlayer had been milled away to allow detection of the underlying Si substrate, film thicknesses could be determined via the thickogram method.32 These thickness are presented in Fig. 1(f).
For O2–HfOx and H2O–HfOx, this required cumulative milling of ∼2500 s, at which point hafnium oxide thicknesses of 4.8 and 5.4 nm were determined, respectively. O3–HfOx required ∼4400 s milling to thin the hafnium oxide layer sufficiently (to 4.8 nm) to detect the Si substrate. Once the substrate was detectable, milling rates of ∼7 × 10−4, 6 × 10−4 and 5 × 10−4 nm s−1 were determined from the measured dataset for O2–HfOx, H2O–HfOx and O3–HfOx, respectively. Slight variations in milling rates are not unexpected, and can arise from compositional variations and differential sputtering, although the differences in milling rate observed here are relatively small, within the same order of magnitude.
From determined atomic percentages, the oxygen stoichiometry x for HfOx can be inferred from 1/([Hf]/[O]). The whole of the O 1s and Hf 4f7/2 signal are used in this calculation, rather than any deconvoluted contribution, to facilitate comparison between different film types.
We note that there can be additional contributions to [O] which are not related to Hf–O bonds. Near the sample surface, some of the O 1s signal will be related to organic hydrocarbon contamination, hence at in this region [O] is likely overestimated. Furthermore, near the dielectric–silicon interface, SiOx interlayers are known to form under these conditions12 and could also skew determined [O]. Characterisation of this interfacial oxide by XPS is challenging, as the O 1s signal will comprise overlapping contributions from both Si–O and Hf–O bonding, in addition to the potential effects of sputter damage and differential sputtering. Hence, the whole [O] signal is considered, rather than any sub-component. Oxygen stoichiometries for all three films as a function of milling time/approximate film thickness are plotted in Fig. 1(g).
XPS analysis of the initial (un-milled) film shows an oxygen-rich film in each case, with x = 2.3 for O2–HfOx and H2O–HfOx and x = 2.5 for O3–HfOx. Following ∼400 s of milling, x decreases for O2–HfOx and H2O–HfOx to 2.1–2.2 and remains in this range with increased milling time. Following 9200 s of milling, x increases once more to ∼2.4 and ∼2.1 for O2–HfOx and H2O–HfOx, respectively. The initial decrease in x corresponds to a reduction in [O], likely related to removal of sample surface hydrocarbon contamination.
With milling time, there is a change in determined stoichiometry for O3–HfOx, which appears to become less oxygen-rich with milling time (depth). It is important to highlight that this change could arise from the milling process itself—the considerable difference in atomic mass between O and Hf would result in differential sputtering, whereby O is removed more readily than Hf, hence affecting the [Hf]/[O] ratio. The observed x reduces to ∼2.2 in the first ∼500 s, but continues to decrease monotonically with increased milling, eventually forming hafnium sub-oxide with x = 1.9.
The Hf 4f peak observed here can be deconvoluted into several contributing signals. Fig. 2 presents fitted spectra for both unmilled and milled O2–HfOx, O3–HfOx, and H2O–HfOx, respectively. The unmilled spectra are largely similar, although two small contributions are visible at ∼18 and ∼19 eV in the spectra of O3–HfOx. These peaks, corresponding to Hf 4f7/2 and Hf 4f5/2 respectively, may arise from surface contamination, as Zrinski et al. attribute signals at this binding energy to hafnium phosphate.34 The definitive source of this impurity is unclear, but it may have arisen from annealing in a tube furnace which had previously been used for phosphorous diffusions. Importantly, these signals are lost after three milling steps, indicating that this is localised to the surface, rather than distributed throughout the film, we do not consider it influential for film performance.
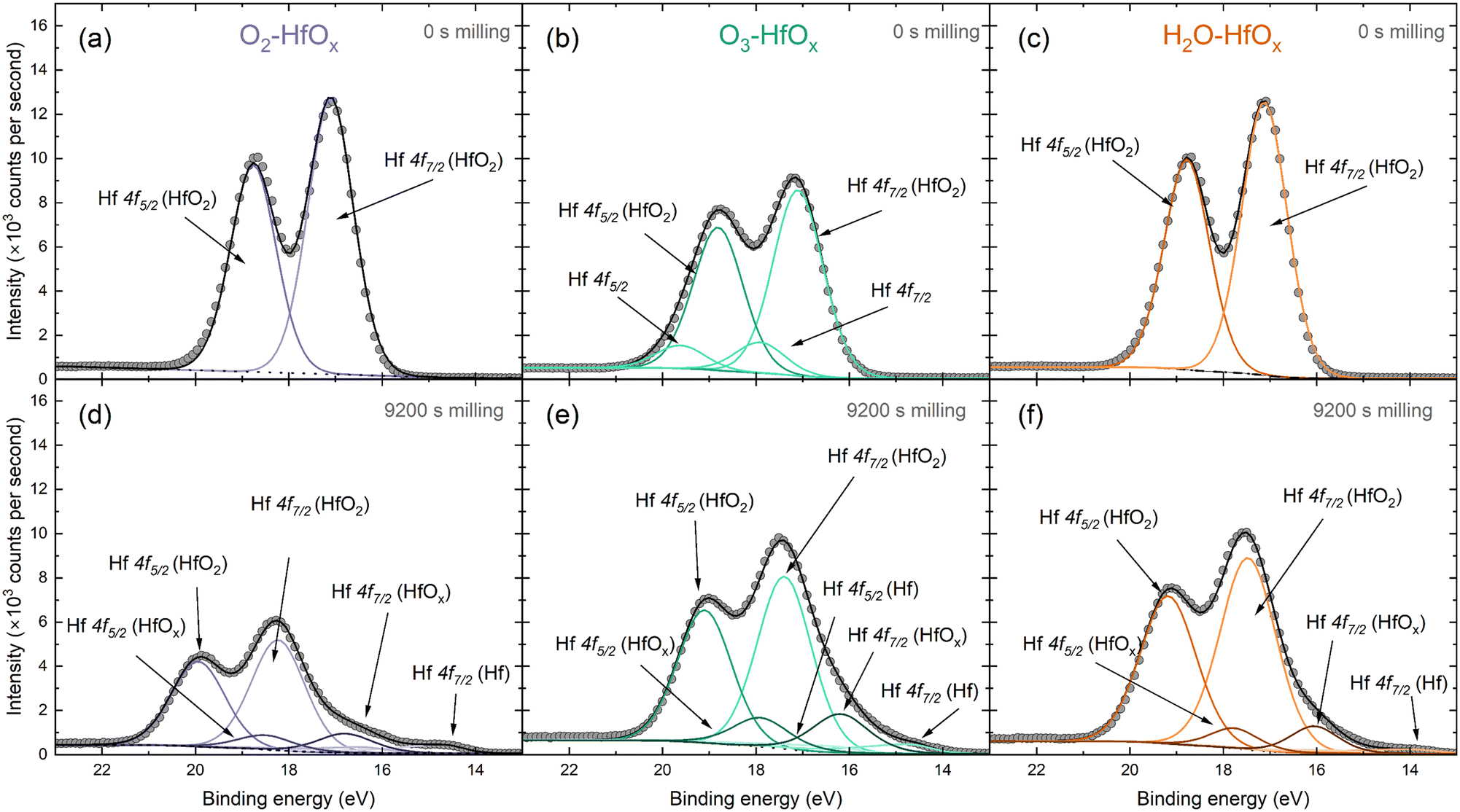 | ||
| Fig. 2 Peak deconvolution of HfOx 4f XPS signal, with Hf 4f5/2 and Hf 4f7/2 contributions identified from O2–HfOx, O3–HfOx and H2O–HfOx before milling (a–c) and following 9200 s of Ar+ milling (d–f). The grey circles are recorded data and dotted/dashed/solid lines correspond to fitted data. Fitting and chemical state analysis were supported by the NIST XPS database.35 | ||
Within the Hf 4f signal, the most prominent contributions are the 4f5/2 and 4f7/2 oxide signals, which also dominate the corresponding region of the milled XPS spectra. After extended milling, hafnium suboxide peaks (at ∼18 and ∼16 eV for Hf 4f5/2 and Hf 4f7/2, respectively) and metallic Hf peaks (at ∼17 and ∼14.6 eV for Hf 4f5/2 and Hf 4f7/2, respectively) are visible. The appearance of a metallic Hf signal as we near the interface could be linked to a change in interfacial composition, a product of differential sputtering or sputter damage.
Fig. S2 in the ESI† allows comparison of all three film types at each milling depth. In all cases, there is a shift in the position of both the Hf 4f5/2 and the Hf 4f7/2 oxide contributions to higher binding energies. This shift in Hf 4f position suggests a change in bonding configuration, with positive shifts generally associated with oxidation. Of these shifts, the most prominent are that of O2–HfOx and H2O–HfOx, with a shift towards a binding energy of ∼20 eV for the Hf 4f5/2 peak, whereas O3–HfOx the Hf 4f5/2 peak only shifts to ∼19 eV.
Passivation properties
A key metric in silicon passivation is the charge carrier lifetime in a passivated substrate. From this we can derive more interface-specific parameters such as surface recombination velocity (SRV) and dark surface saturation current J0s. Following film deposition, and after annealing in air for 30 min at temperatures 300–500 °C, effective lifetimes were recorded via photoconductance decay. Effective lifetimes were extracted at an excess carrier density of 1 × 1015 cm−3 and are plotted in Fig. 3(a). Corresponding injection dependent lifetime curves can be found in Fig. 3(b)–(d) for O2–HfOx, H2O–HfOx and O3–HfOx, respectively.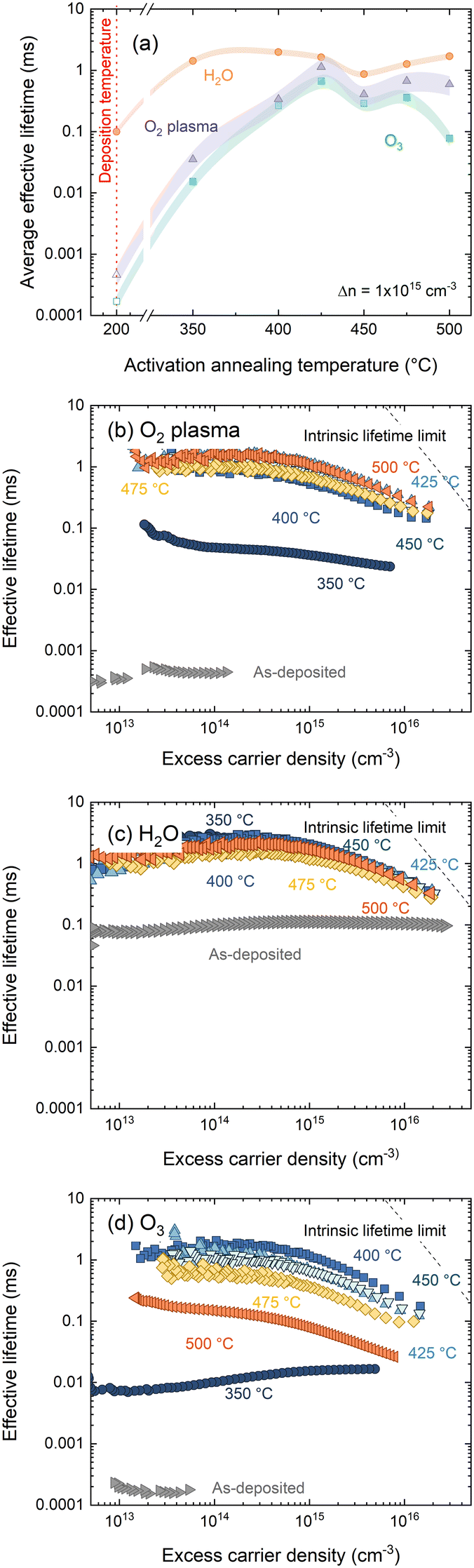 | ||
| Fig. 3 (a) Effective lifetimes extracted at Δn = 1 × 1015 cm−3 for O2–HfOx (purple triangles), O3–HfOx (green squares) and H2O–HfOx (orange circles), as deposited and after annealing in air at temperatures 350–600 °C. Samples were ∼150 μm, (100), 5 Ω cm, n-type Cz-silicon. Multiple samples were characterised following annealing at 400 °C (O2–HfOx, O3–HfOx), 450 °C (O2–HfOx, H2O–HfOx, O3–HfOx) and 500 °C (O2–HfOx), with the lifetime values presented herein an average of those samples. The shaded regions correspond to the experienced variation between samples. The as-deposited values at Δn = 1 × 1015 cm−3 for O2–HfOx and O3–HfOx, indicated with open triangles and squares, respectively, are estimated by the Sinton software. Injection dependent lifetime curves for (b) O2–HfOx, (c) H2O–HfOx and (d) O3–HfOx annealed in air at temperatures 350–600 °C and as-deposited. Also shown in each case is the effective lifetime limit.40 | ||
At all annealing temperatures studied, H2O–HfOx exhibits the most promising passivation quality, with single-side J0s values <20 fA cm−2 possible at all annealing temperatures, consistent with studies of thermal deposition of Al2O3 on n-type silicon.16 O2–HfOx and O3–HfOx initially passivate poorly and require an ‘activation’ anneal at around 400 °C for reasonable passivation (minimum J0s achieved of 33 and 71 fA cm−2 for O2–HfOx and O3–HfOx, respectively) to be realised.
The best passivation quality for O3–HfOx (albeit less competitive than the other films) occurs following annealing at a temperature (400 °C) at which, based on the XRD results in Fig. 1, the film is still amorphous, suggesting that crystallisation is not a pre-requisite for passivation, consistent with our prior work on ultra-thin hafnium oxide films.18 Above this temperature, the passivation quality of O3–HfOx gradually degrades – this decline is likely due to degradation of the surface passivation rather than the bulk, as there is no evidence of degradation in samples of the same substrate type with the other two HfOx films. We have previously verified the stability of the substrate bulk carrier lifetime with O2–HfOx passivation of nominally identical substrates using a superacid-based re-passivation method.6
Importantly, films grown with all three co-reactants require annealing at 350–425 °C to allow for millisecond-level effective lifetimes (SRVs at 1 × 1015 cm−3 < 10 cm s−1) to be achieved. ∼100 μs effective lifetimes (equivalent to an average SRV of 76.7 cm s−1, an order of magnitude lower than recent reports of H2O-based HfOx (ref. 36)) were recorded for as-deposited H2O–HfOx – which is orders of magnitude better than those determined for as-deposited O3–HfOx and O2–HfOx. Indeed, O3–HfOx and O2–HfOx require annealing at 400 °C to reach the same level of passivation. The increase in effective lifetime at 1 × 1015 cm−3 for O2–HfOx and O3–HfOx following annealing at this temperature coincides with a change in injection dependence, as evident from Fig. 3(b) and (d). The passivation quality of H2O–HfOx can be improved with a similar activation anneal of ≤350 °C, lower than reported previously.11
An important property of dielectrics is the character of the fixed charges, the undefined nature of which in HfOx has attracted much attention.9–11,25,27,37 Consequently, it is necessary to understand the effects of any charge present in the ultra-thin films with changing deposition co-reactant. Charge polarity can be assessed from the direction of shift under illumination when making surface photovoltage (SPV) measurements using a Kelvin probe setup, where negative SPV corresponds to negative charge and vice versa.38,39 KP analysis in Fig. 4(a) shows that all the HfOx films are negatively charged, irrespective of ALD co-reactant. Consistent charge polarity with different co-reactants concurs with the work of Park et al. on HfCl4-based HfOx, although they observe positive charges.24 Negative SPV is observed prior to any annealing steps, indicating the negative charge is present from deposition, rather than induced on annealing, as has previously been suggested.11
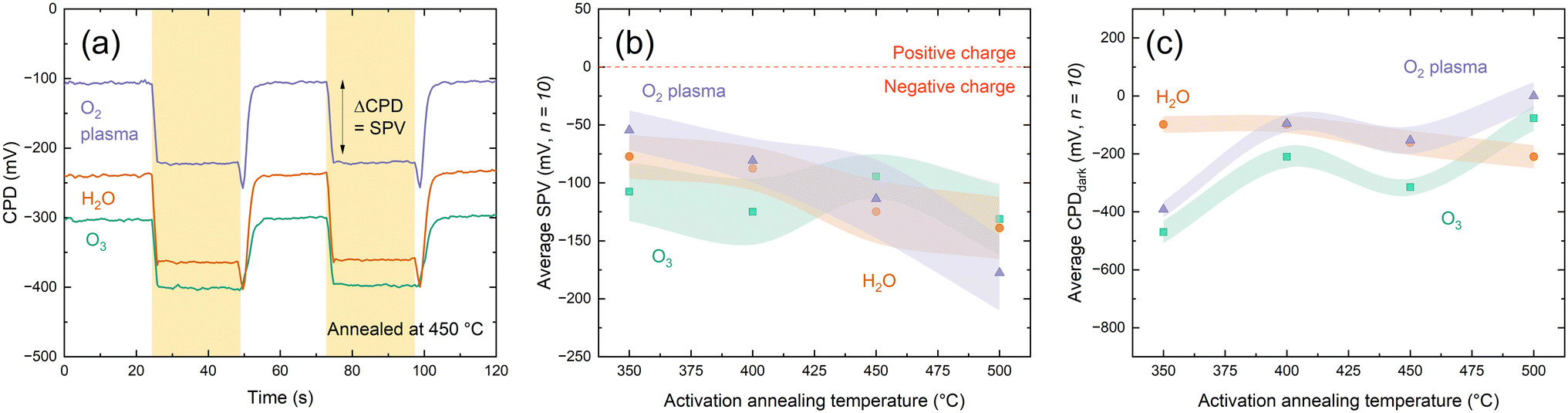 | ||
| Fig. 4 (a) Representative surface photovoltage measurement for O2–HfOx (purple), H2O–HfOx (orange) and O3–HfOx (green) after annealed in air at 450 °C. The yellow shaded regions correspond to periods of illumination. Surface photovoltage (b) and CPDdark (c) for O2–HfOx (purple triangles), O3–HfOx (green squares) and H2O–HfOx (orange circles), as deposited and annealed in air at temperatures 350–600 °C. In both cases, the plotted value is the average of ten measurements, and the shaded region corresponds to the standard deviation of these values. | ||
SPV values are similar for each film type (within one standard deviation) in all cases. Beyond SPV, contact potential difference (used in determination of SPV) can provide additional insight into material properties, with greater CPD corresponding to more highly charged films. Whereas the differences in SPV between the films plotted in Fig. 4(b) were within experimental variation, the same cannot be said for CPDdark plotted in Fig. 4(c). Following deposition, CPDdark differs for each co-reactant, with a separation of ∼500 mV in CPDdark between each HfOx film type (−800, −300, 200 mV for HfOx–H2O, HfOx–O3 and HfOx–O2, respectively). With annealing, this difference reduces, with all film types reaching a CPDdark of ∼−100–300 mV, irrespective of co-reactant or annealing temperature.
The magnitude of SPV can be indicative of the quantity of charge present but results can be highly variable and dependent on both material properties and surface defects.14,41 Thus, positive corona charging was used to characterize the negative fixed charge (Qfixed) present in HfOx grown by each method more reliably. This is based on adding extrinsic charge Qcorona to counteract the intrinsic Qfixed.42 Corona charging also allows an estimate of the level of chemical passivation at the interface from τmin, which is the lowest effective lifetime that is expected to indicate the point at which Qfixed is negated by Qcorona, leaving only chemical passivation and hence a proxy for interface trap density Dit.43 Following neutralisation of fixed charge, applying further extrinsic positive charge causes an increase in measured effective lifetime, as the applied charge provides field effect passivation.
The result of such investigation of the differently deposited layers annealed at varied temperatures is presented in Fig. 5. The effect of successive corona charging on measured effective lifetime is demonstrated in Fig. 6(a) for O2–HfOx, O3–HfOx and H2O–HfOx annealed at 450 °C. From this procedure, values for Qcorona (−Qfixed) and τmin can be extracted. These parameters are summarised in Fig. 5(b) and (c), respectively. The recovery of measured effective lifetime past the minimum point in Fig. 5(a) is more pronounced for O2 and H2O–HfOx than for O3–HfOx. Corona charging having minimal impact on effective lifetime, has been attributed to charge leakage.6,7,19,44 As such, we speculate that the lack of improvement in O3–HfOx lifetime could be attributed to this, noting we did not characterise charge leakage in this study.
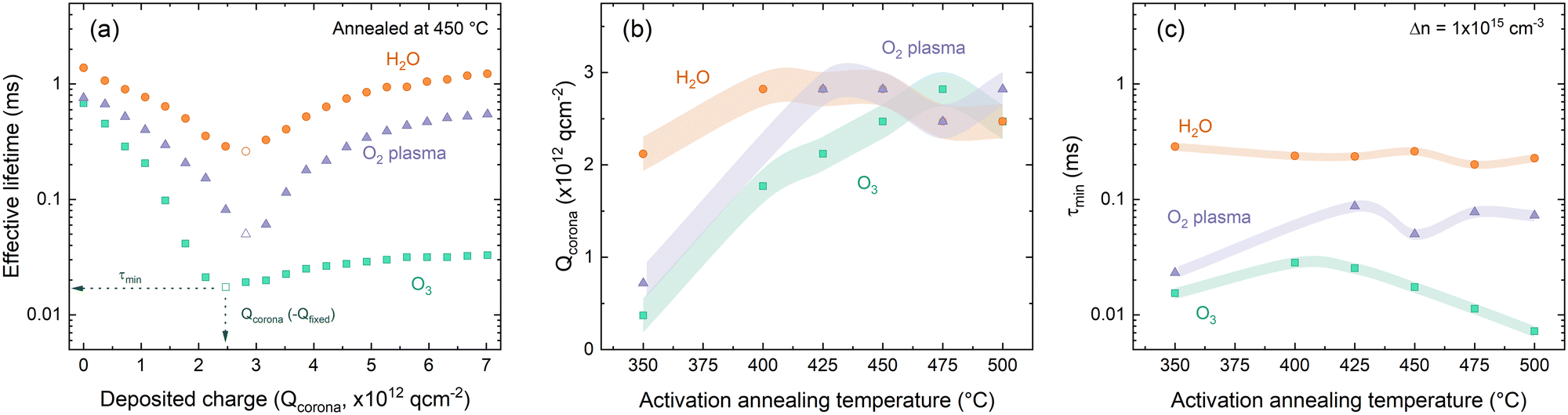 | ||
| Fig. 5 (a) Effective lifetime as a function of Qcorona for O2–HfOx (purple triangles), O3–HfOx (green squares) and H2O–HfOx (orange circles), annealed in air at 450 °C. The datapoint at which Qfixed and τmin are extracted is indicated in each case. Qcorona (b) and τmin (c) for O2–HfOx (purple triangles), O3–HfOx (green squares) and H2O–HfOx (orange circles), annealed in air at temperatures 350–600 °C. In both cases, the shaded region corresponds to the relative uncertainty of the measurement: (b) Qcorona of 1.85 × 1011 q cm−2, 50% of Qcorona deposited in each step and (c) 8% under short flash conditions, and 11% under long flash conditions.46 | ||
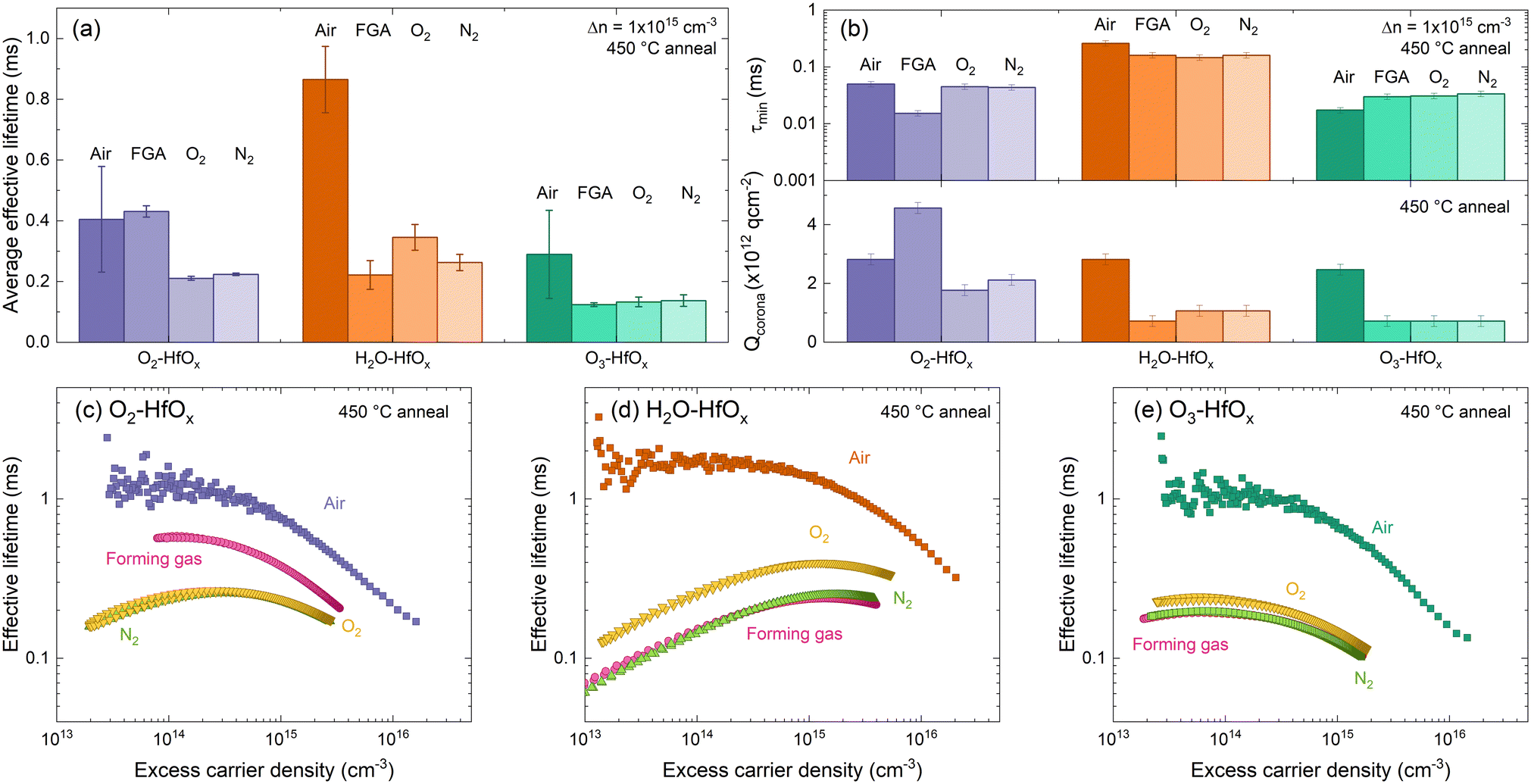 | ||
| Fig. 6 (a) Average effective lifetimes for O2–HfOx (purple), H2O–HfOx (orange) and O3–HfOx (green) after annealing in air, forming gas (FGA), O2 and N2 environments (dark to light shades) at 450 °C for 30 min. Error bars are the experienced variation between samples and plotted effective lifetimes were extracted at 1 × 1015 cm−3. Duplicate samples of O2–HfOx, H2O–HfOx and O3–HfOx were measured per annealing condition. (b) τmin (top) Qcorona (bottom) for O2–HfOx (purple), H2O–HfOx (orange) and O3–HfOx (green) after annealing in air, forming gas, O2 and N2 environments (dark to light shades) at 450 °C for 30 min In both cases, the shaded region corresponds to the relative uncertainty of the measurement (11% in the case of τmin,46 and Qcorona of 1.85 × 1011 q cm2, 50% of Qcorona deposited in each step in the bottom case). Optimum injection-dependent effective lifetimes of (c) O2–HfOx, (d) H2O–HfOx, and (e) O3–HfOx after annealing in air, forming gas (pink), O2 (yellow) and N2 (green) environments at 450 °C for 30 min. | ||
H2O–HfOx has similar values of both Qfixed and τmin at all studied annealing temperatures, demonstrating that in this temperature range both field- and chemical-effect passivation are relatively constant. There is a slight increase in Qfixed between 350–400 °C, but at all temperatures Qfixed is −2 × 1012 to −3 × 1012 q cm−2 (cf., ∼−1011 q cm−2 previously reported for H2O–HfOx grown with this precursor,11 and ∼−1012 to −1013 q cm−2 for the more conventional Al2O3).7,15,16 The trends observed with H2O–HfOx differ from HfOx grown with other co-reactants. As shown in Fig. 5(b), O3–HfOx annealed at 350 °C has Qfixed ≤−7 × 1011 q cm−2, a value which increases monotonically with annealing temperature up to 475 °C at which point there is a slight decline. Increasing Qfixed with annealing temperature has previously been observed for O2–HfOx,6 an observation which was correlated with film crystallisation at similar temperatures. As O3–HfOx does not appear to crystallise in this temperature range, it follows that the increase in charge magnitude is not connected with HfOx crystallisation.
In terms of τmin, plotted in Fig. 5(c) at all annealing temperatures, O3–HfOx has the lowest levels of chemical passivation of the three materials studied. The greatest τmin (i.e., best chemical passivation) is observed at 400–425 °C, but there is only a marginal difference between this and the other values. Above this temperature, there is a monotonic decrease in τmin. The ‘activation’ anneal appears to mainly impact fixed charge levels, with a minor increase in chemical passivation, although beyond ∼400 °C increasing fixed charge further is counterbalanced by reduced chemical passivation. O2–HfOx has intermediary behaviour. Like for O3–HfOx, Qfixed is initially low, but improves with annealing.
With annealing at 425–500 °C Qfixed is similar, akin to H2O–HfOx. O2–HfOx also exhibits τmin between that of H2O–HfOx and O3–HfOx at all temperatures studied. The difference in τmin between H2O–HfOx and O3–HfOx, with O2–HfOx falling in between, implies different degrees of chemical passivation of the Si/HfOx interface. The lower quality chemical passivation with O2–HfOx and O3–HfOx may arise from interfacial damage accrued during deposition from the reactive plasma and ozone radicals, as suggested by Dingemans et al.45
An alternative could be that the interfaces grow similarly but that H2O–HfOx is inherently improved by the presence of hydrogen in the H2O precursor,11 with the hydrogen content of ALD-grown films reported to be greater for those grown with a H2O co-reactant versus O2 plasma.16 Hydrogen is well-known to passivate interfacial defects and may be provided in situ from the co-reactant. It is important to note that the co-reactant is not the sole source of hydrogen in the deposition process. The hafnium precursor TDMAH is also a potential hydrogen source, cf., trimethylaluminium (TMA), a common Al2O3 ALD precursor, which contributes to the overall hydrogen content of the film.47 The good passivation quality observed for H2O–HfOx relative to O3–HfOx and O2–HfOx without any annealing step (shown in Fig. 3), demonstrates that the difference in passivation is linked to the deposition process, rather than any post-treatment. At low (∼350 °C) annealing temperatures, H2O–HfOx reaches Qfixed of order −1012 q cm−2, whilst the other HfOx films require higher annealing temperature to reach the same level. H2O–HfOx and O2–HfOx appear to have greater Qfixed than O3–HfOx at most annealing temperatures.
The origin of the fixed charges in HfOx are debated, although it is thought to be linked to oxygen sites/vacancies, with positive charges attributed to oxygen vacancies or under-coordinated oxygen sites,8,13 while negative charges are attributed to oxygen interstitials and hafnium vacancies.37 The XPS data presented in Fig. 1, collected from HfOx annealed at 450 °C demonstrates that at the film surface, O3–HfOx is more oxygen-rich than H2O–HfOx and O2–HfOx, but at the dielectric/Si interface this is reversed. H2O–HfOx and O2–HfOX films annealed at 450 °C have both higher interfacial oxygen concentrations and interfacial charge density than comparable O3–HfOx, suggesting the negative charge observed in this case may be related to oxygen interstitials/hafnium vacancies.
To assess whether the improved passivation with H2O–HfOx was related to additional hydrogenation (beyond that potentially provided by the metal precursor) provided from the H2O co-reactant, forming gas anneals were performed, as these have been reported to provide additional interface hydrogenation.48 Duplicate samples of O2–HfOx, O3–HfOx and H2O–HfOx were annealed in different environments, namely forming gas (H2/N2), N2 and O2, for 30 min in a tube furnace at 450 °C.
Comparing the average effective lifetimes extracted at 1 × 1015 cm−3 for each film type and annealing environment, shown in Fig. 6(a), yields some insight. For each film type, the highest passivation quality is generally achieved with annealing in an air environment (consistent with both our prior work6 and recent reports13,49), although for O2–HfOx similar passivation quality is achieved for air as for a forming gas environment. For all co-reactants, similar passivation is obtained when annealing in either an O2 or N2 environment (lower than that obtained with annealing in an air environment), consistent with our prior work on O2–HfOx.6 Of all films studied, the most amenable to forming gas annealing is O2–HfOx, with this approach offering no advantage for H2O–HfOx and O3–HfOx.
Focusing on extracted lifetimes at a single injected minority charge density – such as 1 × 1015 cm−3 – only tells part of the story. More information can be gleaned from the injection dependent lifetime curves.50 Curves measured on the samples annealed in different environments are shown in Fig. 6(b). The gradient of H2O–HfOx lifetime curves annealed in forming gas, O2 and N2 suggest a low field effect contribution relative to chemical effect. To extract more information around the relative effects of chemical and field effect passivation, corona charging was used. As all samples exhibited the behaviour shown in Fig. 5(a) on corona charging, it is clear that the annealing environment does not affect film charge polarity, with negative charges exhibited in each case.
In air, annealing at 450 °C resulted in similar levels of field effect passivation (determined from –Qcorona) but different levels of chemical effect (determined from τmin) passivation, with chemical passivation increasing in the order H2O–HfOx > O2–HfOx > O3–HfOx. This trend in chemical passivation is maintained with changing annealing environment, as shown in Fig. 6(c), but field effect passivation differs considerably.
Between annealing environments there is minimal variation in τmin for each film type. For O2–HfOx and H2O–HfOx, changing annealing environment away from air reduces τmin, whilst the converse is true for O3–HfOx, for which there is increased chemical passivation.
For all film types, the highest levels of field effect passivation (∼−3 × 1012 q cm−2) were achieved with annealing in air, excepting O2–HfOx, for which greater field effect passivation (∼−4.5 × 1012 q cm−2) was observed. In all other cases, Qcorona reduced considerably relative to that obtained after annealing in air.
The data presented in Fig. 5 demonstrated that ALD co-reactant does not influence charge polarity but does influence charge magnitude and chemical passivation. The data shown in Fig. 6 demonstrates that that the same can be said of annealing environment, which has a considerable impact on resulting passivation quality. It should be noted though, that different furnaces were used for annealing in air versus the defined environments, although the same temperature and process duration were targeted.
Experimental
Film preparation
Substrates for effective charge carrier lifetime and Kelvin probe measurements were high quality, ∼150 μm thick, (100) orientation, monocrystalline 5 Ω cm resistivity, phosphorus doped n-type Czochralski-grown silicon wafers with a chemically etched surface finish. Samples were prepared following a previously reported chemical cleaning and etching procedure,51 based on the ‘RCA’ standard clean. This procedure has previously been shown to enable good quality HfOx passivation.11,52 The final step in the cleaning process (immersion in 2% HF for 60 s) was modified to immersion in 1% HF/1% HCl for 5 min with no subsequent water rinse, as this has been found to improve final passivation quality.53 Substrates for film thickness measurements were ∼725 μm thick, mirror polished (100) wafers, and underwent an identical cleaning process, with the omission of etching.53Coatings were grown via ALD using a Veeco Fiji G2 system (with plasma-enhanced or thermal capability) with an external load lock. HfOx films were deposited on both sides of each wafer at 200 °C from a tetrakis(dimethylamido)hafnium (TDMAH) precursor (Pegasus Ltd.). 200 ALD cycles were used for each film with varying co-reactants – either O2 plasma, ozone (O3) or water (H2O). In all cases, the precursor was heated to 75 °C and Ar was used as an inert carrier gas. Relevant deposition parameters are summarised in Table 1. O3 is generated prior to deposition from flowing O2via a generator within the Fiji G2 system.
| Conditions | O2 plasma | H2O | O3 |
|---|---|---|---|
| Pulse duration (s) | 0.25 | 0.25 | 0.25 |
| Precursor purge duration (s) | 5 | 10 | 10 |
| Co-reactant pulse duration (s) | 6 | 0.06 | 0.15 |
| Co-reactant intensity (W) | 300 | — | — |
| Co-reactant purge duration (s) | 5 | 10 | 15 |
A post-deposition anneal in air was performed for 30 min in a quartz tube furnace at temperatures between 350–600 °C, unless otherwise specified. Samples annealed in different environments (forming gas, O2, N2) were annealed in a sintering furnace under a gas flow of 1 slm at 450 °C for 30 min.
Safety considerations
Dilute HF is used in this work as part of a standard industrial silicon cleaning process. It is important to note that HF is corrosive and toxic,55 and must be handled only by those trained in HF handling, hazards, and spill response. Exposure to HF and its fumes, even small quantities, can be fatal. HF work should be conducted in a well-ventilated fume hood with appropriate personal protective equipment: face shield, apron, and HF-resistant gloves. HF etches glass, so must be contained in HF-compatible beakers.The reagents used within the ALD deposition also introduce their own hazards – TDMAH (used as a hafnium precursor) is flammable, pyrophoric and corrosive,56 while ozone (used as a co-reagent) has to be kept at low concentrations (ideally controlled with an ozone generator and sensor), lest explosive decomposition reactions occur.57
Characterisation
Fitting procedures to extract peak positions and relative stoichiometries were performed using the Casa XPS software suite, linear backgrounds, and mixed Gaussian–Lorentzian (Voigt) line shapes. These were fitted and corrected using their corresponding sensitivity factors, considering the photoelectron mean free paths and photoionization cross sections of these core levels. The spectrometer work function and binding energy scale were calibrated using the Fermi edge and Ag 3d5/2 peak from a clean polycrystalline Ag sample measured prior to the experiments.
Depth profiles were collected by etching samples via monoatomic Ar+ sputtering for a total duration of 9200 s. To calibrate the milling rate, thickogram method was used, which determines film thickness based on the intensity and kinetic energy of overlayer and substrate peaks, sensitivity factors (7.12 for the HfOx overlayer and 0.772 for the Si substrate) and the attenuation length of photoelectrons in overlayer (2.17 nm).30–32 Thicknesses calculated using this method have an estimated uncertainty of ±30%, based on the uncertainty in the attenuation length,32 peak energies, and intensities.59 Relative atomic concentrations were determined from core level peaks (Hf 4f, O 1s, Si 2p) identified from survey scans at multiple milling depths. Subsequent high-resolution core level spectra were collected, on which fitting and chemical state analysis, supported by the NIST XPS database,35 were used to investigate the chemical composition of the film.
Effective lifetime measurements made using the quick decaying and slow decaying flash are assumed to be accurate to ±8% and ±11%, respectively.46 Effective lifetime measurements were made on samples following annealing in a forming gas, O2 and N2 environment using a spatially-resolved photoluminescence imaging modulum tool calibrated to carrier lifetimes, described in ref. 60. Measurements were made on 5 cm × 5 cm samples, considered sufficiently large to avoid strong impacts of edge recombination on the experiment.61 Passivation quality was quantified in terms of SRV as:
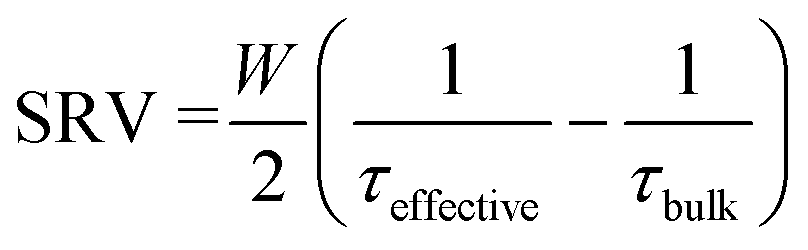 | (1) |
Conclusions
We have studied ALD-grown hafnium oxide with three different co-reactants: O2 plasma, O3 and H2O. Herein, HfOx is deposited on n-type silicon from a tetrakis(dimethylamido)hafnium (TDMAH) precursor at 200 °C, and characterised both in the as-deposited state and as a function of annealing temperature. The choice of co-reactant has a considerable impact on resultant film properties, with differences in crystallinity, composition, passivation quality, interfacial quality, and fixed charge magnitude. Depth profiling conducted with X-ray photoelectron spectroscopy reveals differences in composition between the HfOx surface and the HfOx/Si interface. We find that H2O-based HfOx gives rise to the best passivation quality, even at low annealing temperatures, as a result of high fixed negative charge levels (−1012 q cm−2) and good chemical passivation. Neither the co-reactant choice nor annealing environment change the resulting charge polarity.Data availability
Data underpinning figures in this paper can be freely downloaded from https://wrap.warwick.ac.uk/181742/. Requests for additional data should be made directly to the corresponding authors.Author contributions
The research concept was devised by S. L. P. Experimental work, sample processing, characterization, and data analysis were largely conducted by S. L. P. with assistance from A. Y. and B. F. M. H. A. L. performed annealing under forming gas, nitrogen and oxygen environments. XPS data were collected and analysed by E. K. and M. W. N. E. G. developed the corona charging methodology and D. W. provided support with XRD data collection. S. L. P., E. K., T. N., N. E. G., and J. D. M. contributed to discussions and data analysis. The manuscript was written by S. L. P., with input from T. N., E. K., M. W., N. E. G. and J. D. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work was supported by the EPSRC Charged Oxide Inversion Layer (COIL) solar cells project (EP/V037749/1), and the Leverhulme Trust (RPG-2020-377). M. W. and D. W. acknowledge financial support from the EPSRC-funded Warwick Analytical Science Centre (EP/V007688/1). A. Y. was in receipt of an ISIS Facility Development Studentship from the Science and Technology Facilities Council. XPS and XRD analysis were conducted via the Warwick Photoemission and X-ray Diffraction Research Technology Platforms, respectively.References
- R. S. Bonilla, B. Hoex, P. Hamer and P. R. Wilshaw, Phys. Status Solidi A, 2017, 214, 1700293 CrossRef.
- M. Leskelä and M. Ritala, Angew. Chem., Int. Ed., 2003, 42, 5548–5554 CrossRef PubMed.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS.
- H. B. Profijit, S. E. Potts, M. C. M. van de Sanden and W. M. M. Kessels, J. Vac. Sci. Technol., A, 2011, 29, 050801 CrossRef.
- J. Hendrickson, A. Homyk, A. Scherer, T. Alasaarela, A. Säynätjoki, S. Honkanen, B. C. Richards, J.-Y. Kim, Y.-H. Lee, R. Gibson, M. Gehl, J. D. Olitzky, S. Zandbergen, H. M. Gibbs and G. Khitrova, in Quantum Optics with Semiconductor Nanostructures, ed. F. Jahnke, Woodhead Publishing, 2012, pp. 433–434 Search PubMed.
- A. Wratten, S. L. Pain, D. Walker, A. B. Renz, E. Khorani, N. E. Grant and J. D. Murphy, IEEE J. Photovolt., 2023, 13, 40–47 Search PubMed.
- N. E. Grant, S. L. Pain, E. Khorani, R. Jefferies, A. Wratten, S. McNab, Y. Han, R. Beanland, R. S. Bonilla and J. D. Murphy, Appl. Surf. Sci., 2024, 645, 158786 CrossRef CAS.
- F. Lin, B. Hoex, Y. H. Koh, J. Lin and A. G. Aberle, Energ. Proc., 2012, 15, 84–90 CrossRef CAS.
- F. Lin, B. Hoex, Y. H. Koh, J. Lin and A. G. Aberle, ECS J. Solid State Sci. Technol., 2013, 2, N11–N14 CrossRef CAS.
- J. Gope, Vandana, N. Batra, J. Panigrahi, R. Singh, K. K. Maurya, R. Srivastava and P. K. Singh, Appl. Surf. Sci., 2015, 357, 635–642 CrossRef CAS.
- X. Cheng, P. Repo, H. Halvard, A. P. Perros, E. S. Marstein, M. Di Sabatino and H. Savin, IEEE J. Photovolt., 2017, 7, 479–486 Search PubMed.
- J. Cui, Y. Wan, Y. Cui, Y. Chen, P. Verlinden and A. Cuevas, Appl. Phys. Lett., 2017, 110, 021602 CrossRef.
- A. B. Gougam, B. Rajab and A. B. Afif, Mater. Sci. Semicond. Process., 2019, 95, 42–47 CrossRef CAS.
- V. Aubriet, K. Courouble, M. Gros-Jean and Ł. Borowik, Rev. Sci. Instrum., 2021, 92, 083905 CrossRef CAS PubMed.
- B. Hoex, S. B. B. Heil, E. Langereis, M. C. M. van de Sanden and W. M. M. Kessels, Appl. Phys. Lett., 2006, 89, 042112 CrossRef.
- G. Dingemans and W. M. M. Kessels, J. Vac. Sci. Technol., A, 2012, 30, 040802 CrossRef.
- E. Khorani, C. Messmer, S. L. Pain, T. Niewelt, B. Healy, A. Wratten, M. Walker, N. E. Grant and J. D. Murphy, IEEE J. Photovolt., 2023, 13, 682–690 Search PubMed.
- A. Wratten, S. L. Pain, A. Yadav, E. Khorani, T. Niewelt, L. Black, G. Bartholazzi, D. Walker, N. E. Grant and J. D. Murphy, Sol. Energy Mater. Sol. Cells, 2023, 259, 112457 CrossRef CAS.
- S. L. Pain, E. Khorani, T. Niewelt, A. Wratten, G. J. P. Fajardo, B. Winfield, R. S. Bonilla, M. Walker, L. F. J. Piper, N. E. Grant and J. D. Murphy, Adv. Mater. Interfaces, 2022, 9, 2201339 CrossRef CAS.
- H. Mulaosmanovic, E. T. Breyer, S. Dünkel, S. Beyer, T. Mikolajick and S. Slesazeck, Nanotechnology, 2021, 32, 502002 CrossRef CAS.
- M. Pešić, F. P. G. Fengler, L. Larcher, A. Padovani, T. Schenk, E. D. Grimley, X. Sang, J. M. LeBeau, S. Slesazeck, U. Schroeder and T. Mikolajick, Adv. Funct. Mater., 2016, 26, 4601–4612 CrossRef.
- A. Wratten, B. F. M. Healy, D. Walker, E. Khorani, N. E. Grant and J. D. Murphy, AIP Adv., 2023, 13, 065113 CrossRef CAS.
- C. Richter, T. Schenk, U. Schroeder and T. Mikolaijick, J. Vac. Sci. Technol., A, 2014, 32, 01A117 CrossRef.
- H. B. Park, M. Cho, J. Park, S. W. Lee, C. S. Hwang, J.-P. Jim, J.-H. Lee, N.-I. Lee, H.-K. Kang, J.-C. Lee and S.-J. Oh, J. Appl. Phys., 2003, 94, 3641–3647 CrossRef CAS.
- R. Sreenivasan, P. C. McIntyre, H. Kim and K. C. Saraswat, Appl. Phys. Lett., 2006, 89, 112903 CrossRef.
- M. Dalberth, M. Sowa, E. Deguns, R. Bhatia, A. Bertuch, G. Liu, G. Sundaram and J. Becker, ECS Meeting Abstracts, 2010, MA2010-02, 1451 CrossRef.
- S. Tomer, M. Devi, A. Kumar, S. Laxmi, C. M. S. Rauthan and Vandana, IEEE J. Photovolt., 2020, 10, 1614–1623 Search PubMed.
- J.-W. Park, D.-K. Lee, D. Lim, H. Lee and S.-H. Choi, J. Appl. Phys., 2008, 104, 033521 CrossRef.
- B.-O. Cho, J. P. Chang, J.-H. Min, S. H. Moon, Y. W. Kim and I. Levin, J. Appl. Phys., 2003, 93, 745–749 CrossRef CAS.
- S. Tougaard, QUASES-IMFP-TPP2MM (3.0), 2016 Search PubMed.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165–176 CrossRef CAS.
- P. J. Cumpson, Surf. Interface Anal., 2000, 29, 403–406 CrossRef CAS.
- C. E. Curtis, L. M. Doney and J. R. Johnson, J. Am. Ceram. Soc., 1954, 37, 458–465 CrossRef CAS.
- I. Zrinski, C. C. Mardare, L.-I. Jinga, J. P. Kollender, G. Socol, A. W. Hassel and A. I. Mardare, Appl. Surf. Sci., 2021, 548, 149093 CrossRef CAS.
- National Institute of Standards and Technology, NIST X-ray Photoelectron Spectroscopy Database 20, National Institute of Standards and Technology, Maryland, USA, 4.1 edn, 2000.
- S. Tomer, M. Devi, A. Kumar, S. Laxmi, S. Satapthy, K. K. Maurya, P. Singh, P. Pathi and Vandana, IEEE J. Photovolt., 2023, 13, 691–698 Search PubMed.
- R. Singh, Vandana, J. Panigrahi and P. K. Singh, RSC Adv., 2016, 100, 97720–97727 RSC.
- L. Kronik and Y. Shapira, Surf. Sci. Rep., 1999, 37, 1–206 CrossRef CAS.
- R. S. Bonilla, Mater. Res. Express, 2022, 9, 085901 CrossRef.
- T. Niewelt, B. Steinhauser, A. Richter, B. Veith-Wolf, A. Fell, B. Hammann, N. E. Grant, L. Black, J. Tan, A. Youssef, J. D. Murphy, J. Schmidt, M. C. Schubert and S. W. Glunz, Sol. Energy Mater. Sol. Cells, 2022, 235, 111467 CrossRef CAS.
- R. J. Hamers and K. Markert, Phys. Rev. Lett., 1990, 64, 1051–1054 CrossRef CAS.
- B. Hoex, J. Schmidt, M. C. M. van de Sanden and W. M. M. Kessels, presented in part at the 33rd IEEE Photovoltaic Specialist Conference (PVSEC 2008), California, USA, 2008 Search PubMed.
- Y. Zhao, C. Zhou, X. Zhang, P. Zhang, Y. Dou, W. Wang, X. Cao, B. Wang, Y. Tang and S. Zhou, Nanoscale Res. Lett., 2013, 8, 114 CrossRef PubMed.
- R. S. Bonilla, C. Reichel, M. Hermle and P. R. Wilshaw, Phys. Status Solidi RRL, 2017, 11, 1600307 CrossRef.
- G. Dingemans, N. M. Terlinden, D. Pierreux, H. B. Profijit, M. C. M. van De Sanden and W. M. M. Kessels, Electrochem. Solid-State Lett., 2010, 14, H1 CrossRef.
- A. L. Blum, J. S. Swirhun, R. A. Sinton, F. Yan, S. Herasimenka, T. Roth, K. Lauer, J. Haunschild, B. Lim, K. Bothe, Z. Hameiri, B. Seipel, R. Xiong, M. Dhamrin and J. D. Murphy, IEEE J. Photovolt., 2014, 4, 525–531 Search PubMed.
- C. Guerra-Nuñez, M. Döbeli, J. Michler and I. Utke, Chem. Mater., 2017, 29, 8690–8703 CrossRef.
- J.-I. Polzin, B. Hammann, T. Niewelt, W. Kwapil, M. Hermle and F. Feldmann, Sol. Energy Mater. Sol. Cells, 2021, 230, 111267 CrossRef CAS.
- X.-Y. Zhang, J. Han, Y.-T. Wang, Y.-J. Ruan, W.-Y. Wu, D.-S. Wuu, J. Zuo, F.-M. Lai, S.-Y. Lien and W.-Z. Zhu, Sol. Energy Mater. Sol. Cells, 2023, 257, 112384 CrossRef CAS.
- L. E. Black, in New perspectives on surface passivation: Understanding the Si-Al2O3 interface, Springer, 2016, ch. 2.6, p. 27 Search PubMed.
- N. E. Grant, P. P. Altermatt, T. Niewelt, R. Post, W. Kwapil, M. C. Schubert and J. D. Murphy, Sol. RRL, 2021, 5, 2000754 CrossRef CAS.
- A. Wratten, PhD thesis, University of Warwick, 2023 Search PubMed.
- N. E. Grant, A. I. Pointon, R. Jefferies, D. Hiller, Y. Han, R. Beanland, M. Walker and J. D. Murphy, Nanoscale, 2020, 12, 17332–17341 RSC.
- Ultratech/Cambridge Nanotech, Fiji HfO2 Thermal and Plasma, 2015.
- Sigma Aldrich, MSDS - 339261, https://www.sigmaaldrich.com/GB/en/sds/sigald/339261, (accessed 25th October, 2023).
- Sigma Aldrich, MSDS 455199 - Tetrakis(dimethylamido)hafnium(IV), https://www.sigmaaldrich.com/GB/en/sds/aldrich/455199, (accessed 16th November 2023).
- H. Park, J. Park, S. Shin, G. Ham, H. Choi, S. Lee, N. Lee, S. Kwon, M. Bang, J. Lee, B. Kim and H. Jeon, J. Vac. Sci. Technol., A, 2018, 36, 051509 CrossRef.
- Filmetrics Inc, Operations Manual for the FILMETRICS F20 Thin-Film Analyzer, Filmetrics Inc., 7.3.2 edn., 2013.
- R. Hesse, T. Chasse, P. Streubel and R. Szargan, Surf. Interface Anal., 2004, 36, 1373–1383 CrossRef CAS.
- H. Hoffler, F. Schindler, A. Brand, R. Eberle, R. Post, A. Kessel, J. Greulich and M. C. Schubert, presented in part at the 37th European PV Solar Energy Conference and Exhibition, 2020 Search PubMed.
- M. Kessler, T. Ohrdes, P. P. Altermatt and R. Brendel, J. Appl. Phys., 2012, 111, 054508 CrossRef.
- D. E. Kane and R. M. Swanson, presented in part at the 18th IEEE Photovoltaic Specialists Conference, Las Vegas, 1985 Search PubMed.
- B. Hammann, B. Steinhauser, A. Fell, R. Post, T. Niewelt, W. Kwapil, A. Wolf, A. Richter, H. Höffler and M. C. Schubert, IEEE J. Photovolt., 2023, 13, 535–546 Search PubMed.
- D. Kang, H. C. Sio, J. Stuckelberger, D. Yan, S. P. Phang, R. Liu, T. N. Truong, T. Le, H. T. Nguyen, X. Zhang and D. Macdonald, Progr. Photovolt.: Res. Appl., 2022, 30, 970–980 CrossRef CAS.
- I. D. Baikie, S. Mackenzie, P. J. Z. Estrup and J. A. Meyer, Rev. Sci. Instrum., 1991, 62, 1326–1332 CrossRef CAS.
- R. S. Bonilla, C. Reichel, M. Hermle, P. Hamer and P. R. Wilshaw, Appl. Surf. Sci., 2017, 412, 657–667 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Thickness measurements and optical characterisation of the deposited layers, XPS data corresponding to depth profiling of O2–, O3– and H2O–HfOx, and representative signal fitting. See DOI: https://doi.org/10.1039/d3lf00210a |
| This journal is © The Royal Society of Chemistry 2024 |