
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithium-ion battery functionality over broad operating conditions via local high concentration fluorinated ester electrolytes†
Calvin D.
Quilty
ab,
Edelmy J.
Marin Bernardez
ab,
Andrew
Nicoll
ab,
MD Jamil
Hossain
 c,
Arun
Kingan
ab,
David J.
Arnot
bd,
Hafsa A.
Mohamed
ef,
Ciara L.
O'Connor
eg,
Xiao
Tong
h,
Cherno
Jaye
i,
Daniel A.
Fischer
i,
Lei
Wang
bj,
Yue
Qi
c,
Esther S.
Takeuchi
abdj,
Amy C.
Marschilok
abdj,
Shan
Yan
bj,
David C.
Bock
*bj and
Kenneth J.
Takeuchi
*abdj
c,
Arun
Kingan
ab,
David J.
Arnot
bd,
Hafsa A.
Mohamed
ef,
Ciara L.
O'Connor
eg,
Xiao
Tong
h,
Cherno
Jaye
i,
Daniel A.
Fischer
i,
Lei
Wang
bj,
Yue
Qi
c,
Esther S.
Takeuchi
abdj,
Amy C.
Marschilok
abdj,
Shan
Yan
bj,
David C.
Bock
*bj and
Kenneth J.
Takeuchi
*abdj
aDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, USA. E-mail: kenneth.takeuchi.1@stonybrook.edu
bInstitute of Energy: Sustainability, Environment, and Equity (I:SEE), Stony Brook University, Stony Brook, NY 11794, USA
cSchool of Engineering, Brown University, Providence, RI 02912, USA
dDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, NY 11794, USA
eScience Undergraduate Laboratory Internship (SULI) Program, Brookhaven National Laboratory, Upton, NY 11973, USA
fDepartment of Chemistry, University of Rochester, Rochester, NY 14627, USA
gDepartment of Chemistry, The College of Wooster, Wooster, OH 44691, USA
hCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA
iMaterial Measurement Laboratory, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA
jInterdisciplinary Science Department, Brookhaven National Laboratory, Upton, NY 11973, USA. E-mail: dbock@bnl.gov
First published on 21st May 2024
Abstract
Facilitating widespread adoption of electric vehicles will require next-generation battery systems that can operate reliably over a large temperature range, at high operating voltage, and under fast charging rates. Herein, a novel class of nonflammable fluorinated ester-based local high concentration electrolytes (LHCEs) are described. When cycled in commercially relevant graphite/LiNi0.8Mn0.1Co0.1O2 (NMC811) pouch cells, these electrolytes demonstrate improved capacity retention compared to carbonate-based electrolytes under multiple usage conditions including high voltage (4.5 V), fast charge (4C, 15 minutes), and low temperature (−20 °C) without sacrificing capacity retention at elevated temperature (40 °C). Low carbon, high fluoride cathode electrolyte interphases formed by the LHCE system yield a significant reduction in charge transfer impedance during cycling and contribute to capacity retention. These results demonstrate that using fluorinated esters in a LHCE modality enables creation of a new class of nonflammable electrolytes that can successfully operate over broad operating conditions.
Introduction
There is increasingly a need for lithium-ion batteries (LIBs), especially for electric vehicles, that can operate under a wide range of conditions such as wide operating potential and high rate charging as well as low and elevated temperatures; the demand for lithium-ion batteries (LIBs) continues to increase, especially in the electric vehicle industry. Specifically, LIBs with wide operating potentials, high rate charging, and outstanding performance in low and high temperature environments are sought after.1–4 This is because use under such operating conditions can lead to compromised cycling stability when using industry standard carbonate-based electrolytes. Therefore, new electrolytes are required to meet the needs and operating conditions of next-generation LIBs.A desirable electrolyte for utilization in a battery that can operate under an array of extreme conditions should possess moderate (de)solvation energy to allow for Li+ dissolution and transport to the electrode interface, chemical compatibility with the materials inside the battery, low viscosity, and wide ranges of electrochemical stability and liquid phase behavior.2 Prior strategies to develop electrolytes with these characteristics include: (1) low viscosity, low melting point esters5–10 for improving low temperature and fast charge performance; (2) carbonate based electrolytes with functional salts or solvents as additives (e.g., vinylene carbonate (VC), lithium bis(oxalate)borate (LiBOB), and fluoroethylene carbonate (FEC)) to form chemically robust and thermally stable interfaces for high temperature2,11,12 and high voltage13 operation; (3) electrolytes incorporating flame-resistant additives such as trimethyl phosphate (TMP), triphenyl phosphate (TPP), and triethyl phosphate (TEP)14 or liquid polymer electrolytes15 for reducing flammability; and (4) non-flammable, non-volatile ionic liquids with high voltage stability.16,17
The major drawback with the prior approaches is that each often improves functionality for a specific usage regime at the expense of other desirable characteristics. For instance, the use of flame retardant additives can reduce electrolyte conductivity,18 and often ionic liquids have unacceptably high viscosity and low conductivity.19 Similarly, esters including methyl acetate,5,7 ethyl acetate,10 methyl butyrate,20 and methyl propionate20 have demonstrated improved deliverable capacity under fast charge at room temperature6,7 and low temperature operation at practical rates,9 but are severely limited by poor oxidative stability at high voltages during charge, resulting in decreased cell functional lifetime, particularly at higher temperatures.7,20,21
Intriguingly, recent reports investigating fluorinated ester solvents have demonstrated that fluorination improves oxidative stability compared to non-fluorinated ester analogs while imparting non-flammability and conserving functionality at low temperature.22–25 The electron withdrawing nature of fluorine is responsible for the improved oxidative stability, while the low polarizability of the molecules maintains the liquid phase at lower temperature.22,26 Fluorinated esters reported in the literature as solvents include methyl trifluoropropionate,23,25 ethyl trifluoroacetate,22,24 trifluoroethyl acetate24 and trifluoroethyl trifluoroacetate.24 Electrolytes based on these solvents can provide significant enhancement in low temperature delivered capacity and retention compared to state of the art carbonate electrolytes.22–25 However, demonstration of the fluorinated ester based electrolytes in Li-ion cells with graphite electrodes has been limited with observations including high first cycle irreversible capacity25 and low (<140 mA h g−1) capacity under high rate (>4C) in graphite/NMC622 cells.24
Incorporation of fluorinated ester solvents into a local high concentration electrolyte (LHCEs)27–29 provides a new opportunity to tune the functional properties of the electrolyte. In LHCEs, use of miscible but non Li-ion coordinating diluents enable formation of local domains of high salt concentration dominated by contact ion pairs and cation-ion aggregates. The unique solvation structures facilitate the formation of robust anion derived interphase layers while mitigating the low wettability and high viscosity disadvantages inherent to high concentration electrolytes (HCE) without diluent.30–33 LHCEs based on fluorinated carbonate solvents with fluoroether diluent have demonstrated low and high temperature function with good stability against high voltage cathodes, although cycling was only demonstrated at a low C/3 rate.34 For fast charge applications, an LHCE with ether solvent (dimethoxyethane, DME) and fluorinated ether diluent demonstrated improved delivered capacity under 4C current, but voltage stability was poor.28 Thus, investigation of new solvents for use in LHCEs is warranted to achieve operation under expanded usage conditions.
Experimental
Electrolyte formulation
Electrolytes were prepared by mixing anhydrous salts, solvents, and diluents in an argon-filled glovebox. Several electrolytes were tested. 1 mol L−1 lithium hexafluorophosphate in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 v:v ethylene carbonate:dimethyl carbonate (LiPF6 EC:DMC) was used as a control electrolyte, with solvents and salts procured from Gotion. An alternative lithium bis(fluorosulfonyl)imide (LiFSI, Gotion) containing control electrolyte was formulated using 1 mol L−1 LiFSI in 3:7 v:v EC:DMC (LiFSI EC:DMC). Local high concentration electrolytes (LHCEs) were prepared by initially preparing a 2.25 M HCE by dissolving LiFSI in methyl 3,3,3-trifluoropropionate (MTFP, Synquest) to yield a 2.25 mol L−1 solution. Diluent, either 2,2,2-trifluoroethyl 1,1,2,2-tetrafluoroethyl ether (TFETFE, Synquest), 2,2,3,3-tetrafluoropropyl trifluoroacetate (TFPTFA, Synquest), or 1H,1H,5H-octafluoropentyl 1,1,2,2-tetrafluoroethyl ether (OFPTFE, Synquest) was then added in a 1:2 molar ratio of diluent:solvent, to form the final LHCE. Finally, 10% by weight of fluoroethylene carbonate (FEC) or vinylene carbonate (VC) was added to the LHCEs. This yielded: [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:FEC 9:1 (MOF), [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:FEC 9:1 (MTF), [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:VC 9:1 (MOV), and [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:VC 9:1 (MTV). All solvents, diluents, and additives used in the electrolytes had water content <20 ppm.
:7 v:v ethylene carbonate:dimethyl carbonate (LiPF6 EC:DMC) was used as a control electrolyte, with solvents and salts procured from Gotion. An alternative lithium bis(fluorosulfonyl)imide (LiFSI, Gotion) containing control electrolyte was formulated using 1 mol L−1 LiFSI in 3:7 v:v EC:DMC (LiFSI EC:DMC). Local high concentration electrolytes (LHCEs) were prepared by initially preparing a 2.25 M HCE by dissolving LiFSI in methyl 3,3,3-trifluoropropionate (MTFP, Synquest) to yield a 2.25 mol L−1 solution. Diluent, either 2,2,2-trifluoroethyl 1,1,2,2-tetrafluoroethyl ether (TFETFE, Synquest), 2,2,3,3-tetrafluoropropyl trifluoroacetate (TFPTFA, Synquest), or 1H,1H,5H-octafluoropentyl 1,1,2,2-tetrafluoroethyl ether (OFPTFE, Synquest) was then added in a 1:2 molar ratio of diluent:solvent, to form the final LHCE. Finally, 10% by weight of fluoroethylene carbonate (FEC) or vinylene carbonate (VC) was added to the LHCEs. This yielded: [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:FEC 9:1 (MOF), [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:FEC 9:1 (MTF), [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:VC 9:1 (MOV), and [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:VC 9:1 (MTV). All solvents, diluents, and additives used in the electrolytes had water content <20 ppm.
Electrolyte characterization
Viscosity measurements were performed using a RheoSense microVisc viscometer and associated temperature controller. Ionic conductivity was measured by electrochemical impedance spectroscopy (EIS) as described in previous reports.35–37 Symmetric conductivity cells for EIS were used. EIS measurements were performed using a BioLogic VSP potentiostat with a frequency range of 500 kHz to 100 mHz, amplitude = 5 mV. Eqn (1) was used to calculate the conductivity. The cell constant (l/S) used for the measurements is 0.21 cm−1.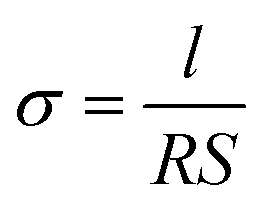 | (1) |
The voltage stability window of the solvents and LHCEs was assessed using cyclic voltammetry in a 3-electrode cell configuration with Pt working electrode versus Li/Li+. Solutions of 100 mM LiFSI in the appropriate solvent were used for the solvent measurements. Cyclic voltammograms were collected at a scan rate of 20 mV s−1, with the voltage incrementally increased or decreased for upper or lower voltage limit testing, respectively, until the current density reached 1 × 10−4 A cm−2. Differential scanning calorimetry (DSC) data of the electrolytes were determined using a DSC250 differential scanning calorimeter (TA Inc.). Approximately 10 mg of the sample was enclosed in an aluminum hermetic pan. The samples were prepared in an argon filled glove box. The electrolytes were cooled from 30 °C to −80 °C at a rate of 5 °C min−1.
Materials characterization
Polycrystalline NMC811 (LiNi0.8Mn0.1Co0.1O2) with Li2CO3 and LiOH content of 0.64% and 0.43%, respectively, was procured from Targray while natural graphite was procured from Superior Graphite. The active materials were analyzed by X-ray diffraction with data collected using beamline 28-ID-2 (XPD) at the National Synchrotron Light Source II (NSLS II) at Brookhaven National Laboratory (BNL). Data calibration, integration, and Rietveld refinements were performed using GSAS-II.38 The wavelengths were determined to be 0.1847 Å and 0.1885 Å for the XPD experiments in which the NMC and the graphite data were collected, respectively. The wavelength was determined by calibration of the data from a LaB6 standard, and all data was collected using a 16 in amorphous silicon detector with CsI-scintillator. A JEOL JSM-6010PLUS/LA scanning electron microscope (SEM) with an accelerating voltage of 10 kV was used to collect SEM images. SEM and energy dispersive X-ray spectroscopy (EDX) analysis of the cycled NMC811 electrodes were acquired using a JEOL 7600F instrument equipped with an EDX detector. SEM images were acquired using an accelerating voltage of 5 kV with an energy r-filter to allow acquisition in both secondary electron (SE) and backscattered electron (BE) imaging modes. EDX maps were acquired at an accelerating voltage of 20 kV. All the electrodes were rinsed with dimethyl carbonate (DMC) and dried in a glovebox prior to data collection.Cell fabrication and electrochemical testing
NMC811 composite cathodes were fabricated by tape-casting a N-methyl pyrrolidone (NMP) slurry of 90% NMC811, 5% carbon black, and 5% polyvinylidene difluoride (PVDF) on carbon-coated aluminum foil. Graphite composite anodes were fabricated by tape-casting a slurry of 90% graphite, 3% carbon black, and 7% PVDF in NMP on carbon-coated copper foil. A mass loading of 8.5 mg cm−2 (1.7 mA h cm−2) of NMC811 was targeted and graphite electrodes were matched to NMC811 such that the N/P ratio was 1.21 ± 0.04 (uncertainties are described throughout as the mean ± the standard deviation); using reversible capacities of 360 mA h g−1 and 200 mA h g−1 for graphite and NMC811, respectively. Single layer replicate pouch-type cells were fabricated with 20 cm2 cathode area, 23.76 cm2 anode area. Polyethylene separator was used (25 μm thick, 36% porosity). The dimensions of the pouch cells are 9.25 cm × 7.90 cm and they have a capacity of 34 mA h. 1.6 mL of electrolyte was added to each cell (9.4 mL gNMC−1).All electrochemical testing was performed at 30 °C unless otherwise specified. All cells underwent four formation cycles at a C/10 rate (assuming a theoretical capacity of 200 mA h g−1 for NMC811) from 3.0 V to 4.3 V at a C/10 rate for four cycles to ensure the formation of an effective SEI.39 Cells were then cycled according to one of five conditions (1C/1C CCCV, 40 °C, −20 °C, 4.5 V charge, 15 min charge). 1C/1C CCCV cycling was performed under a constant current constant voltage (CCCV) protocol with 1C charge rate until an upper voltage limit of 4.3 V was reached, with the voltage held at 4.3 V until the current dropped below the equivalent C/10 rate. The discharge rate was 1C with a lower voltage limit of 3.0 V. 40 °C cycling and −20 °C cycling was performed under the same protocol with variation of the temperature. For the 4.5 V charge cycling protocol the upper voltage limit was increased to 4.5 V. For the 15 min charge cycling, a constant current CCCV protocol with 4C charge rate until an upper voltage limit of 4.3 V was reached, with the voltage held at 4.3 V until the total charge time was fifteen minutes. Every 100 cycles, cells received two recovery cycles at 3–4.3 V, C/10, 30 °C. EIS was collected on cells using a BioLogic VSP multichannel potentiostat (10 mHz to 1 MHz, Va = 5 mV) after formation and after the completion of cycling, with the data collected in the charged state (4.3 V).
Soft X-ray Absorption Spectroscopy (sXAS) characterization
Cathodes were recovered from graphite/NMC811 pouch cells after 200 fast charge cycles in the discharged state, and were rinsed, dried, and kept under inert atmosphere prior to analysis. sXAS measurements were collected at NSLS-II (BNL) using beamline 7-ID-1. Measurements were carried out under vacuum at the Ni L-edge (840 to 880) eV using both partial electron yield (PEY) and fluorescence yield (FY) detectors. The Athena software package was used to merge and calibrate data.40X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) measurements were collected on recovered NMC811 cathodes and graphite anodes after 200× cycles under the baseline condition. Measurements were collected at the Center for Functional Nanomaterials (CFN) at BNL. Measurements were performed under a base pressure of 5 × 10−6 Pa in an ultra-high vacuum chamber. The X-ray source and detector were a non-monochromatized Al Kα (hν = 1486.6 eV, V = 15 kV, I = 20 mA) source and a SPECS Phoibos 100 MCD analyzer, respectively. An inert transfer suitcase was used to transfer all samples from an argon-filled glovebox to the XPS instrument. A step size of 0.05 eV and an Epass of 20 eV were used for the measurement of the C 1s, O 1s, F 1s, P 1s, Li 1s, S 2p, and N 1s spectral regions.All peak fitting of the XPS spectra was performed with CasaXPS software.41 A Shirley background function was used and the line shape was Gaussian–Lorentzian. Eqn (2) was used to determine the relative concentrations (xi) of all the surface components.
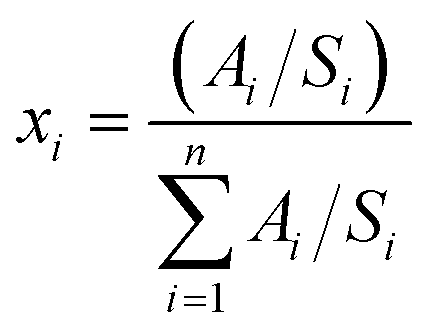 | (2) |
DFT calculations of solvent reduction potentials
The reduction potentials vs. Li+/Li0 for different electrolyte constituents were calculated using DFT. Gaussian 09 code was used to minimize the energies of salt–solvent–diluent complexes for all calculations.42 The calculations utilized the double hybrid functional M06-2X,43 the basis set 6-31+G**, and the D3 dispersion correction.44 The implicit SMD model was employed to account for the solvation environment for all solvation energy calculations.45 A dielectric constant of 7.2 was used for all electrolytes.46 The reduction and oxidation potentials with respect to Li+/Li were calculated using the equations47,48 ER (vs. Li/Li+) = −ΔGR/F − 1.4 and EO (vs. Li/Li+) = ΔGO/F − 1.4, where ΔGR and ΔGO are the Gibbs free energy change for the one-electron reduction reaction and the one-electron oxidation reaction, respectively and F is the Faraday constant.Results and discussion
In this work, a new class of LHCEs using fluorinated esters as the primary solvating species in the system was investigated. The voltage stability of four fluorinated esters (Fig. 1a): ethyl trifluoroacetate (ETFA), methyl 3,3,3-trifluoropropionate (MTFP), 2,2,2-trifluoroethylacetate (TFEA), and 2,2,2-trifluoroethylbutyrate (TFEB), was assessed using cyclic voltammetry (CV) in a 3-electrode cell configuration with Pt working electrode versus Li/Li+. Solutions used 100 mM lithium bis(fluorosulfonyl)imide (LiFSI) where a limiting current of 1 × 10−4 A cm−2 was used to define the electrochemical stability.49,50 Voltage stability windows are shown in Fig. 1c and the corresponding cyclic voltammograms are in Fig. S1a and c.† All solvents were found to have an oxidative voltage stability of >4.5 V vs. Li/Li+. However, MTFP was the only fluorinated ester to exhibit reductive voltage stability at 0.05 V. Density Functional Theory (DFT) was used to augment the experimental findings where computations for the free solvent MTFP indicated reductive voltage stability −0.01 V versus Li/Li+ and computed oxidative stability of 6.17 V. Based on these findings, MTFP was selected as the solvent for further studies in LHCEs.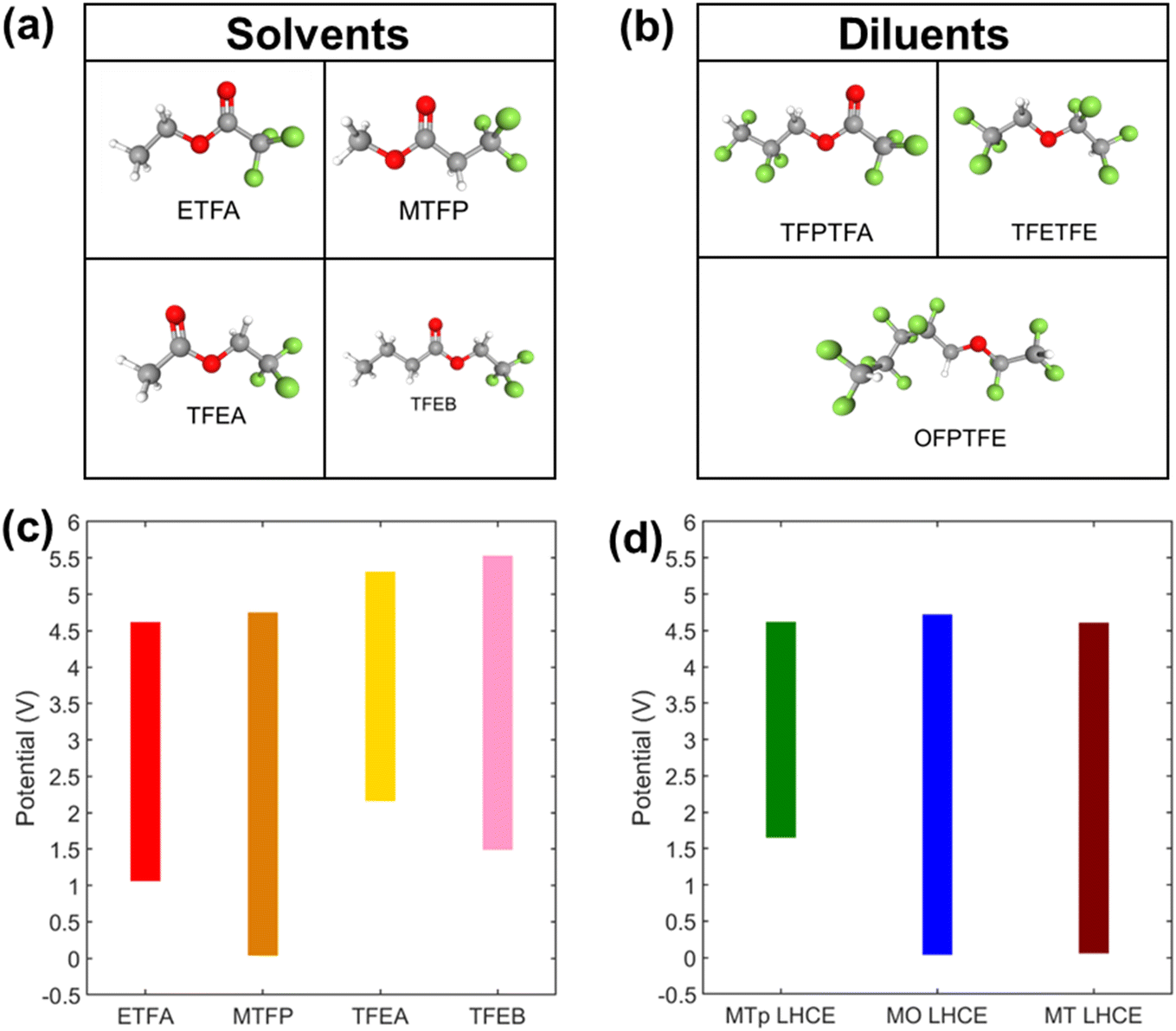 | ||
| Fig. 1 (a) Solvents investigated for LHCEs: ethyl trifluoroacetate (ETFA), methyl 3,3,3-trifluoropropionate (MTFP), 2,2,2-trifluoroethylacetate (TFEA), and 2,2,2-trifluoroethylbutyrate (TFEB). (b) Diluents for LHCEs: 2,2,3,3-tetrafluoropropyl trifluoroacetate (TFPTFA), 2,2,2-trifluoroethyl 1,1,2,2-tetrafluoroethylether (TFETFE), and 1H,1H,5H-octafluoropentyl 1,1,2,2-tetrafluoroethylether. (c) Experimental voltage stability window of fluorinated ester solvents in (a). (d) Experimental voltage stability of LHCEs: 2.25 M LiFSI in MTFP with 2,2,3,3-tetrafluoropropyl trifluoroacetate (MTp LHCE), 2,2,2-trifluoroethyl 1,1,2,2-tetrafluoroethylether (MT LHCE), or 1H,1H,5H-octafluoropentyl 1,1,2,2-tetrafluoroethylether (MO LHCE) in a 2:1 molar ratio. | ||
LHCEs were formulated using the MTFP solvent, with lithium bis-(fluorosulfonyl)imide (LiFSI) due to its chemical stability at high temperature compared to LiPF6 (ref. 51) and its ability to form ionically conductive interfaces.34 A HCE was prepared by dissolving LiFSI in MTFP at an experimentally determined maximum concentration of 2.25 M, corresponding to a salt:solvent molar ratio of 1:3.25. Our recently reported molecular dynamics (MD) simulations of the LiFSI-MTFP HCE system52 indicate that the solvation structure of the HCE is comprised mainly of contact ion pairs (FSI− anions coordinating to 1 Li+) and aggregates (FSI− coordinating to 2 and 3 Li+), rather than solvent separated ion pairs (FSI− coordinating to 0 Li+).
Suitable diluents for an LHCE should be miscible with the primary solvent but not dissolve the Li-based salt on their own to not disrupt the local salt/solvent coordination environment of the HCE.32 Three molecules were explored as diluents (Fig. 1b): fluorinated ester 2,2,3,3-tetrafluoropropyl trifluoroacetate (TFPTFA), and fluorinated ethers 2,2,2-trifluoroethyl 1,1,2,2-tetrafluoroethyl ether (TFETFE) and 1H,1H,5H-octafluoropentyl 1,1,2,2-tetrafluoroethyl ether (OFPTFE). TFPTFA was investigated as a diluent due to its increased number of electron withdrawing fluorine atoms relative to MTFP and thus weakened coordination strength of the solvating oxygen atoms, as confirmed by MD simulations.52 The fluorinated ethers TFETFE and OFPTFE were investigated as they have been utilized successfully in fluorinated carbonate-based LHCE formulations.34,53
The diluents were added to the ester based HCE using a 2:1 molar ratio of MTFP:diluent, and voltage stability of the resulting LHCEs was investigated using CV (Fig. 1d and S1b and d†). The oxidative voltage stability limits of all three LHCEs was between 4.6–4.7 V; however, the reductive voltage stability limits were favorable for diluents TFETFE and OFPTFE (<0.05 V), while the limit for the TFPTFA containing LHCE was >1.5 V. Thus, the 2.25 mol L−1 LiFSI in MTFP:OFPTFE (MO) and 2.25 mol L−1 LiFSI in MTFP:TFETFE (MT) electrolytes were selected for electrochemical evaluation in graphite/NMC pouch cells. In addition to the two LHCE formulations, conventional carbonate-based electrolytes 1 mol L−1 LiPF6 in 3:7 v:v ethylene carbonate:dimethyl carbonate (LiPF6 EC:DMC) and 1 mol L−1 LiFSI in 3:7 v:v EC:DMC were tested as control electrolytes. Formation cycling of the cells was performed at a C/10 rate (based on 200 mA h g−1 NMC811) for four cycles with voltage limits of 3.0–4.3 V. Voltage profiles for cycles 1 and cycle 4 are shown in Fig. 2a and b. Cells with both LHCE formulations exhibited a voltage plateau during the initial charge profile that was absent in the cells with the control electrolytes. This is suggestive of capacity associated with electrolyte reduction at the graphite anode above the lithium intercalation voltage (0.05–0.1 V vs. Li/Li+ (ref. 54)). The subsequent discharges of cells with the LHCE formulations showed suppressed capacity, indicating loss of Li inventory during the first charge. First cycle irreversible capacities (Table S1†) were 63 and 76 mA h g−1 for MO LHCE and MT LHCE cells, respectively, compared with only 37–38 mA g−1 for the control cells.
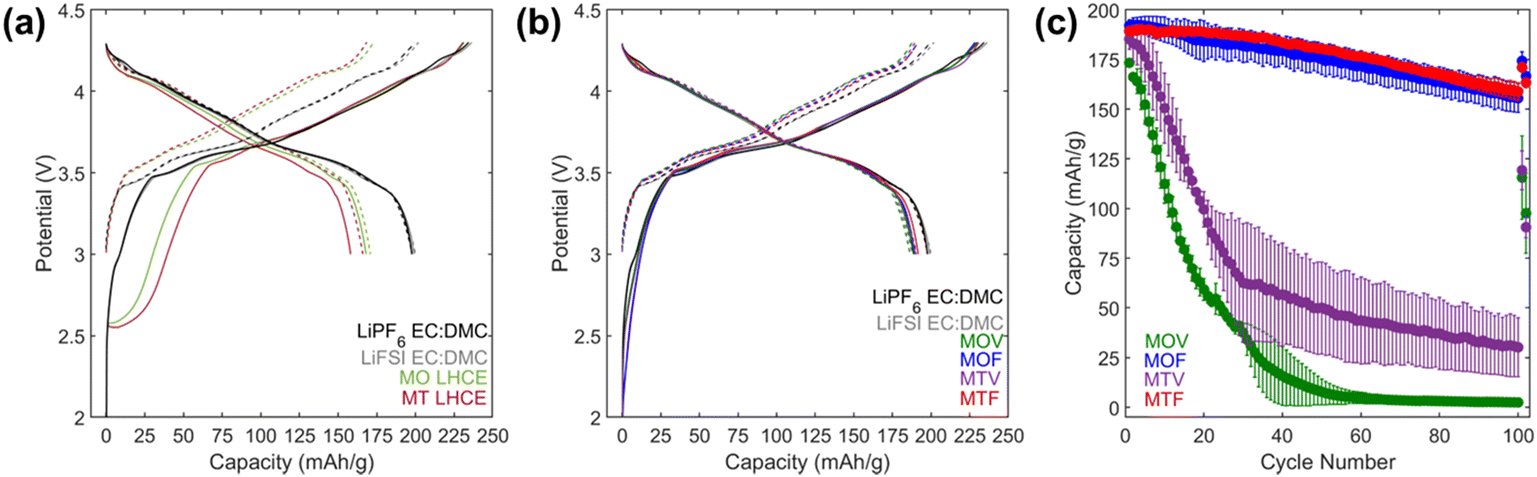 | ||
| Fig. 2 (a and b) 1st (solid) and 4th (dashed) (dis)charge galvanostatic cycling curves for graphite/NMC811 pouch cells during initial formation cycling between 3.0–4.3 V at C/10 rate at 30 °C. (a) Cells with 1 M LiPF6 EC:DMC, 1 M LiFSI EC:DMC, 2.25 mol L−1 LiFSI in MTFP):OFPTFE (MO), and 2.25 mol L−1 LiFSI in MTFP):TFETFE (MT) electrolytes. (b) Cells with [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:FEC 9:1 (MOF), [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:FEC 9:1 (MTF), [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:VC 9:1 (MOV), [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:VC 9:1 (MTV) electrolytes. (c) Discharge capacities vs. cycle number for graphite/NMC811 pouch cells during 1C/1C CCCV cycling at 3.0–4.5 V with the MOV, MOF, MTV, and MTF LHCEs. | ||
Modification of the negative solid electrode interphase (SEI) to suppress the irreversible capacity observed in the LHCE containing cells was done through the addition of fluoroethylene carbonate (FEC) and vinylene carbonate (VC), both previously reported as SEI stabilizers.7,9,20,55 The FEC and VC additives were added to the MO and MT LHCEs with a 9:1 ratio of LHCE:additive by mass, resulting in four formulations: [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:FEC 9:1 (MOF), [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:FEC 9:1 (MTF), [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:VC 9:1 (MOV), [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:VC 9:1 (MTV). Formation testing of graphite/NMC811 pouch cells using the LHCEs with VC or FEC additives, Fig. 2b, showed significant reduction of the first cycle voltage plateau at ∼2.5 V observed without FEC or VC and the first cycle irreversible capacities were reduced to ∼40 mA h g−1 (Table S1†), comparable to the control electrolytes.
Further testing of the graphite/NMC811 pouch cells with the MOV, MOF, MTV, and MTF electrolytes was performed between 3.0–4.5 V for 100 cycles at a 1C rate (Fig. 2c). The MOF and MTF electrolytes maintained >150 mA h g−1 of capacity for 100 cycles, however, capacity fade of the VC containing formulations MOV and MTV was observed within the first 20 cycles. The poor capacity retention of the VC containing formulations is consistent with previous reports that suggest the oxidative stability of VC is not sufficient for use at high voltage.56,57 A comparable MTV electrolyte with 2% VC was also tested (Fig. S2†); however, even charged to 4.3 V the cells with this electrolyte delivered limited capacity. Thus, the MOF and MTF electrolytes were selected as the two LHCE formulations for further electrochemical evaluation under other operating conditions. It is noted that the primary purpose of FEC in these formulations is as a film forming additive as it can form a stable interphase on graphite that is rich in LiF.7,9,20 However, it also likely enters the coordination sphere of Li+ due to its high dielectric constant.58,59 As the molar ratio of FEC and Li+ is 1:1; the coordination sphere of Li+ will be a combination of MTFP and FEC molecules coordinated by the carbonyl oxygen in a tetrahedral arrangement.60,61
The conductivity and viscosity of the relevant electrolytes are presented in Fig. 3a and b and S3–S5.† The conductivities of conventional carbonate-based electrolytes 1 mol L−1 LiPF6 in 3:7 v:v ethylene carbonate:dimethyl carbonate (LiPF6 EC:DMC) and 1 mol L−1 LiFSI in 3:7 v:v EC:DMC were 11 mS cm−1 and 16 mS cm−1 at 25 °C, respectively, while viscosities of these electrolytes were below 3 mPa s. In comparison to the EC:DMC electrolytes, the LHCEs have lower conductivities (4.3 mS cm−1 for MOF, 7.0 mS cm−1 for MTF) and higher viscosities (4.8 mPa s for MOF, 3.1 mPa s for MTF), where conductivity of the electrolytes are inversely proportional to the viscosity. The conductivities of these LHCEs are significantly higher than previously reported LHCEs with conductivities between 1 and 3 mS cm−1.27,32,62–71 All electrolytes exhibit an Arrhenius relationship of viscosity and conductivity with temperature. It is also noted that the measured viscosities and conductivities of the MOF and MTF LHCEs are related to the viscosity of their component diluents (OFPTFE = 2.6 mPa s, TFETFE = 0.70 mPa s at 25 °C).
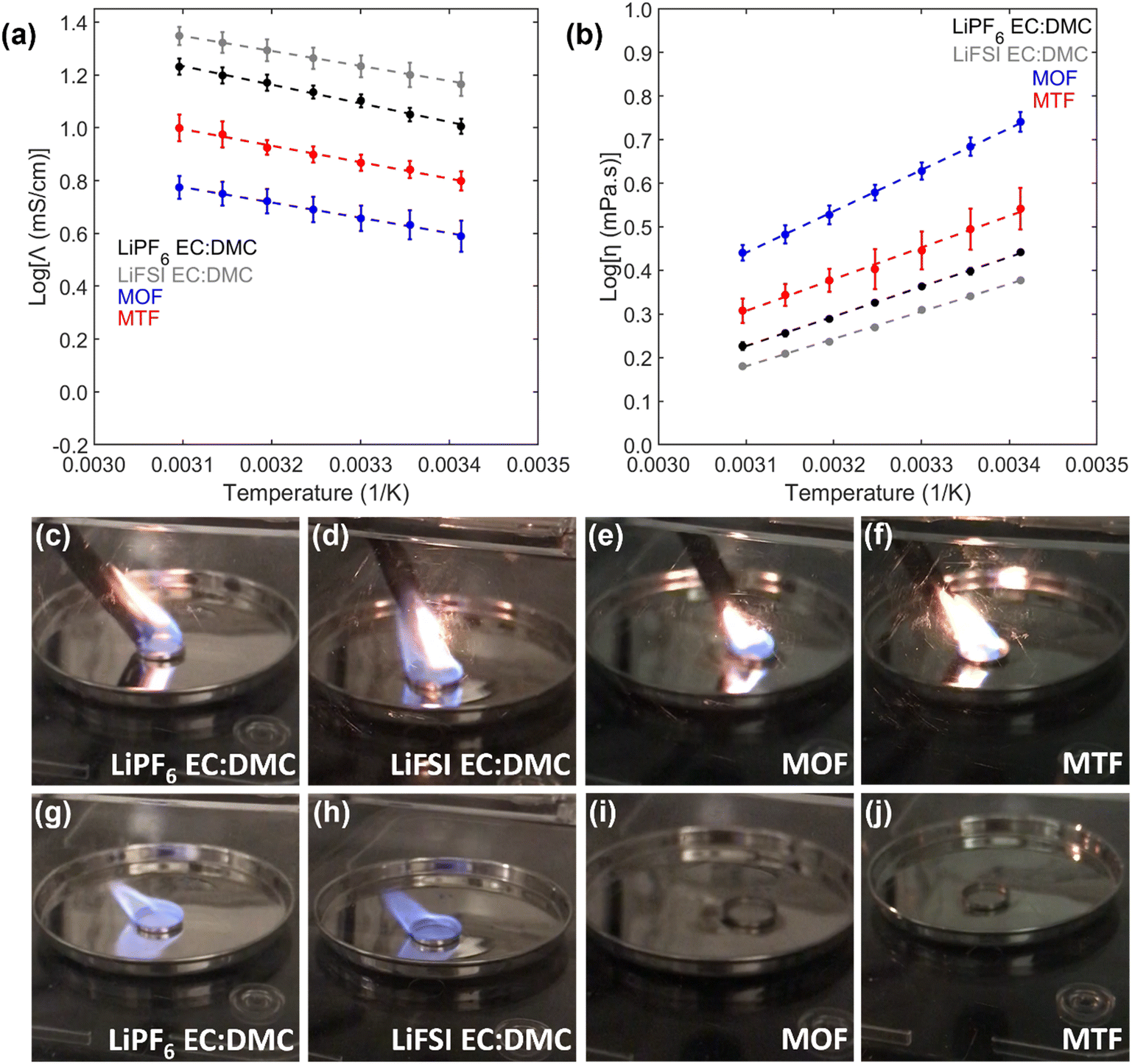 | ||
| Fig. 3 Properties of the LHCE electrolytes [(2.25 mol L−1 LiFSI in MTFP):OFPTFE 2:1]:FEC 9:1 (MOF) and [(2.25 mol L−1 LiFSI in MTFP):TFETFE 2:1]:FEC 9:1 (MTF) compared with control formulations (1 mol L−1 LiPF6 in 3:7 EC:DMC and 1 mol L−1 LiFSI in 3:7 EC:DMC). Arrhenius plots of (a) conductivity and (b) viscosity. (c–j) Flammability testing of the electrolytes. (c) LiPF6 EC:DMC, (d) LiFSI EC:DMC (e) MOF, and (f) MTF electrolytes with flame held to them. (g) LiPF6 EC:DMC, (h) LiFSI EC:DMC (i) MOF, and (j) MTF electrolytes after flame is removed. | ||
The flammability of the electrolytes is shown in Fig. 3c–j, and ESI† videos 1–4. Both EC:DMC electrolytes immediately ignite when exposed to a flame and burn until a highly viscous residue remains. On the other hand, both MOF and MTF, as well as the parent high concentration electrolyte, could not be ignited when a flame was applied; demonstrating reduced electrolyte flammability compared to the conventional carbonate-based electrolytes. This should have a beneficial effect on the safety of the overall battery;62,63,71–73 but would need to be verified at the battery level.68
Pouch cells with NMC811 cathodes and graphite anodes were fabricated with MTF, MOF and two EC:DMC electrolytes where the nominal areal capacity was 1.7 mA h cm−2; details of the cell construction are included in the experimental section of the ESI.† Characterization of the electrode active materials by X-ray diffraction (XRD) and scanning electron microscopy (SEM) is in Fig. S6.† XPS analysis of the pristine NMC811 cathode material (Fig. S7†) qualitatively reveals the presence of Li2CO3 on the particle surfaces. The cells underwent four formation cycles from 3.0 V to 4.3 V at a C/10 rate, followed by further cycling under one of five protocols. Testing was performed at 30 °C under a 1C constant current discharge and 1C constant current constant voltage (CCCV) charge protocol until an upper voltage limit of 4.3 V was reached with a voltage hold until the current dropped below a C/10 rate (termed 1C/1C CCCV). The same protocol was used at temperatures of 40 °C and −20 °C. Cycling was also conducted where the upper voltage limit was 4.5 V (4.5 V charge). Finally, a constant current CCCV protocol with a 4C charge rate and upper voltage limit of 4.3 V was used, with the voltage held at 4.3 V until the total charge time reached fifteen minutes (15 min charge).
The cycling results are presented in Fig. 4 and 5, and S8.† Cycling at C/10 at 30 °C from 3.0 V to 4.3 V was performed after every 100 cycles with corresponding capacity retention values in Table S2.† Under 1C/1C CCCV cycling (Fig. 4a) cells with the LHCEs exhibited significantly improved capacity retention over 200 cycles compared to the EC:DMC electrolytes (78% LiPF6 EC:DMC, 48% LiFSI EC:DMC, 87% MOF, and 93% MTF) with MTF exhibiting slightly less fade compared with MOF. Coulombic efficiencies were >99.6% for all electrolytes except for the LiFSI EC:DMC electrolyte which experienced a slow decline in efficiency which will be discussed below (Fig. S8†). Notably, addition of FEC to the EC:DMC electrolytes did not result in comparable capacity retention to the LHCEs, indicating that the presence of FEC additive alone does not induce the improved electrochemical behavior (Fig. S9†). Similarly, high concentration EC:DMC and conventional concentration MTF electrolytes do not yield results comparable to the LHCEs presented here (Fig. S2†). Although capacities were initially somewhat lower for the LHCEs at a higher areal capacity loading of 3.0 mA h cm−2 (Fig. S10†) likely due to the moderate conductivities and higher viscosities compared to the EC:DMC electrolytes, after 300 cycles the LHCE containing cells delivered higher capacities (127 mA h g−1 LiPF6 EC:DMC, 1 mA h g−1 LiFSI EC:DMC, 130 mA h g−1 MOF, and 148 mA h g−1 for MTF).
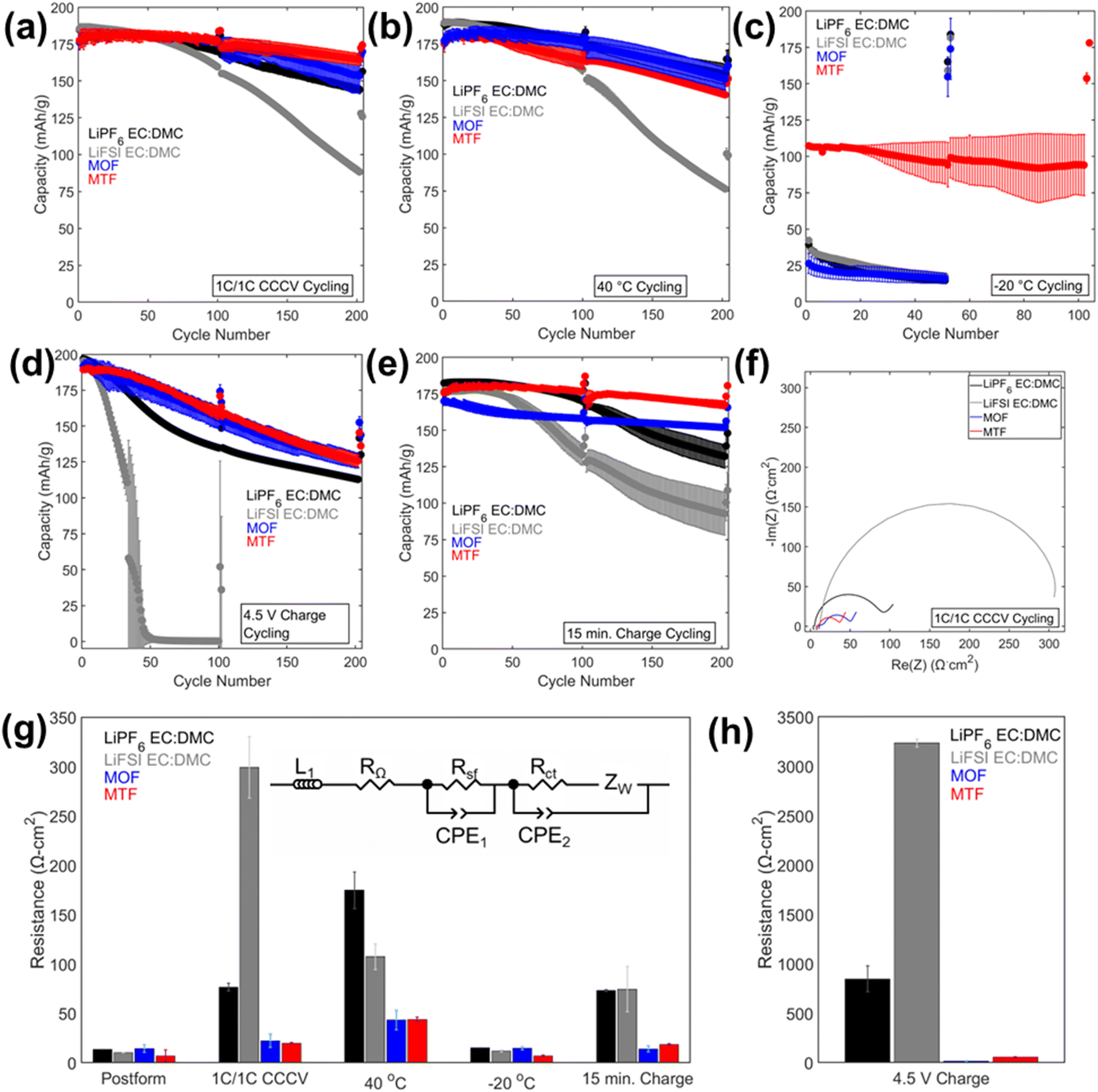 | ||
| Fig. 4 Discharge capacities vs. cycle number for graphite/NMC811 pouch cells cycled under the 1C/1C CCCV cycling condition at 30 °C (a), 40 °C (b), −20 °C (c), high voltage at 30 °C (d), and fast charge at 30 °C (e). (f) EIS spectra post 200 cycles under the condition shown in (a). (g and h) Charge transfer resistance (Rct) as determined from equivalent circuit fitting (inset) of the EIS spectra post formation and after cycling under (g) 1C/1C CCCV, 40 °C, −20 °C, 15 min charge, and (h) 4.5 V charge conditions. EIS measurements were collected at 30 °C after charging to 4.3 V at a C/10 rate. | ||
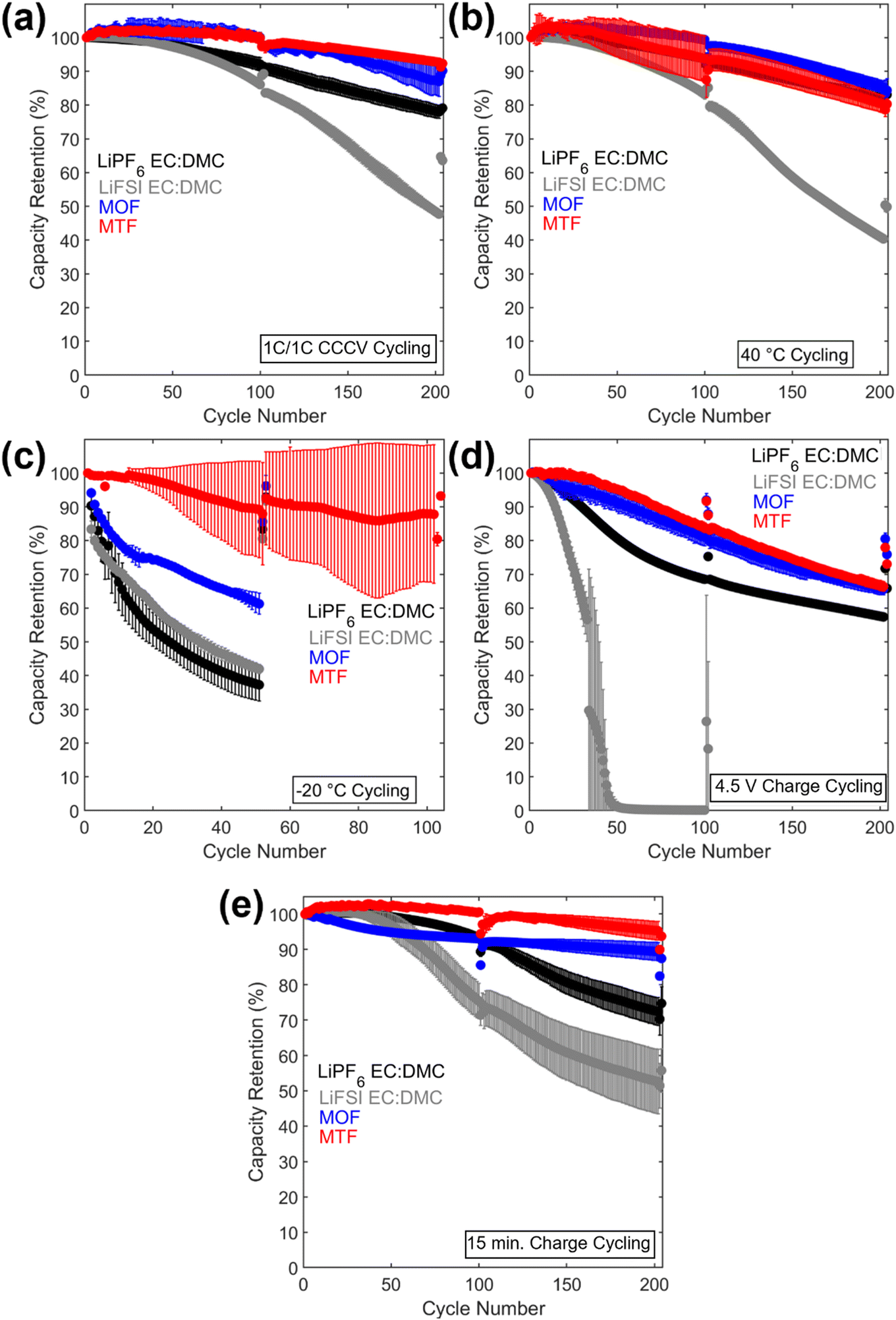 | ||
| Fig. 5 Discharge capacity retention vs. cycle number for the (a) 1C/1C CCCV, (b) 40 °C, (c) −20 °C, (d) 4.5 V charge, and (e) 15 min charge condition. | ||
Electrochemical impedance spectroscopy (EIS) spectra were collected on the cells after formation and cycling (Fig. 4f–h and 6) and fit using an equivalent circuit model (Fig. 4g) provided in Fig. S11.† After cycling under the 1C/1C CCCV condition, the charge transfer resistance (Rct) of the cells with LHCEs increased only slightly (1.6× for MOF and 3× for MTF) while the LiPF6 and LiFSI EC:DMC containing cells increased by 6× and 30×, respectively, Fig. 4g. These findings indicate more resistive interfaces formed by the EC:DMC electrolytes compared to the LHCEs, likely both at the cathode and the anode. There was some variation in OCV of the cells when EIS was collected (between 3.9–4.3 V for cells cycled under all conditions); this is due to the variation in state of charge as even though all cells were charged to 4.3 V prior to EIS collection. EIS was collected on a cell after formation at 3.9–4.3 V (Fig. S12†), there is a slight increase in impedance response as SOC increases but it is orders of magnitude less than the changes observed after cycling. This suggests that changes in state of charge cannot explain the considerable changes in impedance observed after cycling.
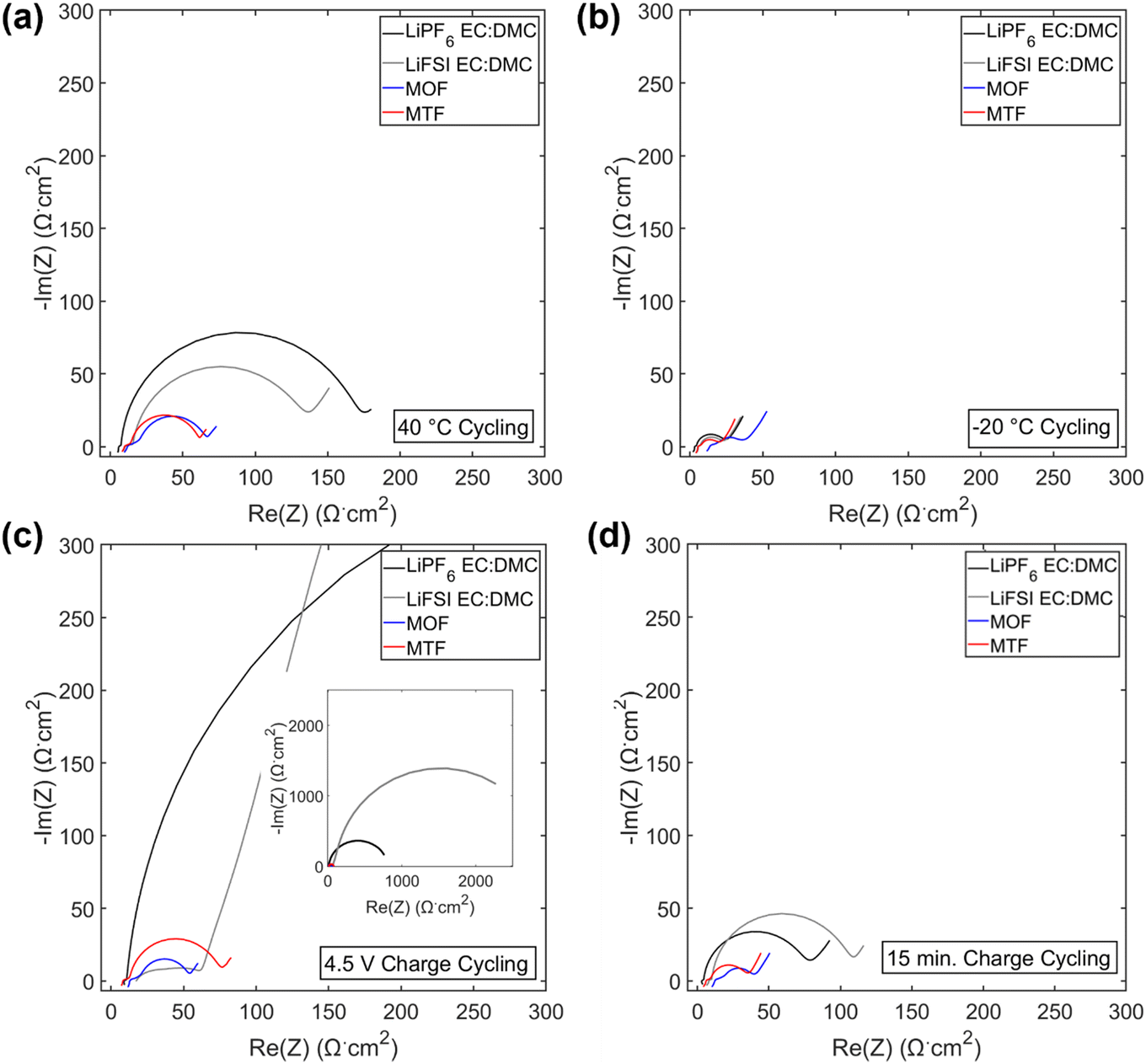 | ||
| Fig. 6 EIS spectra post 200 cycles under 40 °C cycling (a), −20 °C cycling (b), 4.5 V charge cycling (c) and 15 min charge cycling (d). Inset c: zoom out EIS spectra of 4.5 V charge cycling. | ||
Under testing at elevated temperature (40 °C, Fig. 4b), capacity retentions were 82%, 40%, 85%, and 80% for cells utilizing LiPF6 EC:DMC, LiFSI EC:DMC, MOF, and MTF, respectively. Cells utilizing the LHCEs and LiPF6 EC:DMC electrolytes exhibited higher charge transfer impedances as compared to both after formation cycling and after cycling under the 1C/1C CCCV condition. The increase in charge transfer impedance for the LHCEs, not observed after cycling under baseline conditions, is likely a factor in the increased capacity fade observed at high temperature. At an even higher temperature (60 °C), the LiPF6 EC:DMC and MTF electrolytes exhibit 89% and 72% retention over only 50 cycles, respectively, (Fig. S13†); while the MOF is more stable (95% retention over 50 cycles). The improved retention of the MOF electrolyte may be related to the lower vapor pressure of the diluent OFPTFE (boiling point, b.p. = 133 °C) compared to TFETFE (b.p. = 57 °C) or DMC (b.p. = 90 °C). The boiling point of the MTFP solvent used in both MOF and MTF is 96 °C. However, it is noted that the number of free MTFP molecules is minimized in the MTFP-based LHCE structure,52 and the coordinated MTFP molecules will have a lower vapor pressure.74
Cycling at low temperature (−20 °C, Fig. 4c) led to striking differences in electrochemical behavior. Cells utilizing MTF electrolyte had much higher initial capacities compared to the other electrolytes (39 mA h g−1, 42 mA h g−1, 26 mA h g−1, and 107 mA h g−1 for LiPF6 EC:DMC, LiFSI EC:DMC, MOF, and MTF, respectively), and faded less (88% capacity retention after 100 cycles for MTF, compared with <45% retention for LiPF6 and LiFSI EC:DMC electrolytes). For all the electrolytes tested, C/10 cycling at 30 °C post low temperature cycling resulted in recovery of delivered capacity, suggesting that the low capacities observed were due to kinetic limitations at low temperature. Optical images of the electrolytes after storage at −20 °C (Fig. S14†) reveal that the EC:DMC electrolytes freeze while the LHCEs remain liquid. Low temperature differential scanning calorimetry (DSC) measurements of the electrolytes (Fig. S15†) confirm these observations where the LHCEs show no thermal transitions through −90 °C. Furthermore, of the two LHCE formulations, MTF has >2× higher conductivity at −20 °C compared to MOF (Table S3†), explaining its superior low temperature electrochemical functionality. The differences in cycling behavior at low and high temperature in the LHCEs highlight the importance of tailoring the diluent for the specific operating condition where low temperature, lower viscosity/higher conductivity is important to alleviate mass transport limitations.75
When cycling under high voltage charge (3.0 V to 4.5 V) (Fig. 4d), capacity fade was observed over 200 cycles for all four electrolytes but was mitigated in the LHCEs. After 200 cycles, capacity retention values were 57%, 0.2%, 66%, and 66% for LiPF6 EC:DMC, LiFSI EC:DMC, MOF and MTF electrolytes, respectively. The capacity loss observed for the LiFSI EC:DMC is likely due to LiFSI corrosion of Al current collector at high voltage,76,77 observed by SEM images and EDS maps of the recovered Al current collectors (Fig. S16†). This Al corrosion would lead to side reactions during charge that would reduce coulombic efficiency as observed (Fig. S8†). Notably, MD simulations of the MTF electrolyte show that the LHCE reduces uncoordinated solvent molecules52 in the electrolyte preventing their oxidation.78,79 Post cycling EIS results (Fig. 4h) indicate that cells utilizing the LiPF6 EC:DMC had a >60× higher charge transfer resistance compared to after formation, versus a <3× increase for the MOF or MTF based cells. Notably, ethylene carbonate has been reported to decompose at high voltage on nickel-rich surfaces resulting in resistive interfaces.80
Fast charge (CCCV, 4C CC, 15 minute total charge time) cycling results are presented in Fig. 4e. After 200 cycles, cells utilizing the EC:DMC electrolytes faded significantly (capacity retention of 72% and 54% for LiPF6 EC:DMC and LiFSI EC:DMC, respectively), while the cell utilizing the MOF electrolyte exhibited 89% capacity retention and the MTF-based cell showed 95% capacity retention. Post fast charge cycling, there was a minimal increase in charge transfer impedance for cells utilizing the LHCE formulations but a significant increase for the cells utilizing the EC:DMC electrolytes. Lithium plating on the graphite anode is a primary cause of capacity fade under fast charging conditions as the anode is polarized below the thermodynamic potential for Li plating.81 Optical images of the anodes after fast charge cycling are shown in Fig. S17.† Anode polarization is influenced by three processes (i) migration of solvated Li+ in the bulk electrolyte through the pores of the electrode, (ii) desolvation at the interface, and, (iii) conductivity of Li+ ions across the solid electrolyte interface (SEI).82 Interestingly, the ionic conductivity of the bulk electrolyte may not always be the limiting factor,82 as electrolytes with lower bulk conductivity have demonstrated improved function at low temperatures and fast rates,83–86 suggesting that the de-solvation kinetics and SEI transport of Li+ are more critical. The results here support this premise, where the MTF cycled cells with lower charge transfer impedance exhibit reduced Li plating compared to the EC:DMC cells, despite having lower bulk conductivity and higher viscosity. It is also noted that the cell utilizing the MOF formulation, with the highest amount of Li plating, was significantly more polarized compared to MTF at the fast rate (Fig. S18†).
The cathodes were collected in the discharged state after fast charge cycling and were analyzed using soft X-ray absorption spectroscopy (sXAS) to assess changes in the surface oxidation state of NMC811. The measurements were made using both partial electron yield (PEY) and fluorescence yield (FY) detectors with depth sensitivities of ca. (1 to 2) nm and (50 to 100) nm, respectively.87,88 Measurements of the Ni L2,3-edges were carried out corresponding to 2p–3d excitations and are sensitive to the occupancy of these orbitals89 shown in Fig. 7a and b.† The Ni L3-edge is a doublet and the intensity ratio of the Ni L3 High to L3 Low peaks, denoted as γ, is an indicator for the Ni oxidation state, where γ decreases as Ni is reduced.87,90 The γ values for the samples, a pristine NMC811 electrode, and a Ni3+ standard are presented in Fig. 7c. All cycled samples had higher γ values and thus increased oxidation state in the discharged state compared to the pristine NMC811 electrode. The increased oxidation state may be related to degradation of the NMC811 with compromised ability to cycle effectively. However, contributing factors may also include loss of active lithium due to electrolyte degradation as well as irreversible Li plating at the negative electrode under the fast-charging condition. PEY γ values were 20 to 30% higher than FY γ values, indicating greater Li loss at the particle surface, in agreement with reported scanning tunneling electron microscope (STEM) measurements of cycled NMC811 cathodes.91 The relative order of gamma values was LiFSI control > MOF > LiPF6 control > MTF, signifying that the MTF cycled electrode had the least quantity of active lithium loss, consistent with the high capacity retention of MTF cycled cells.
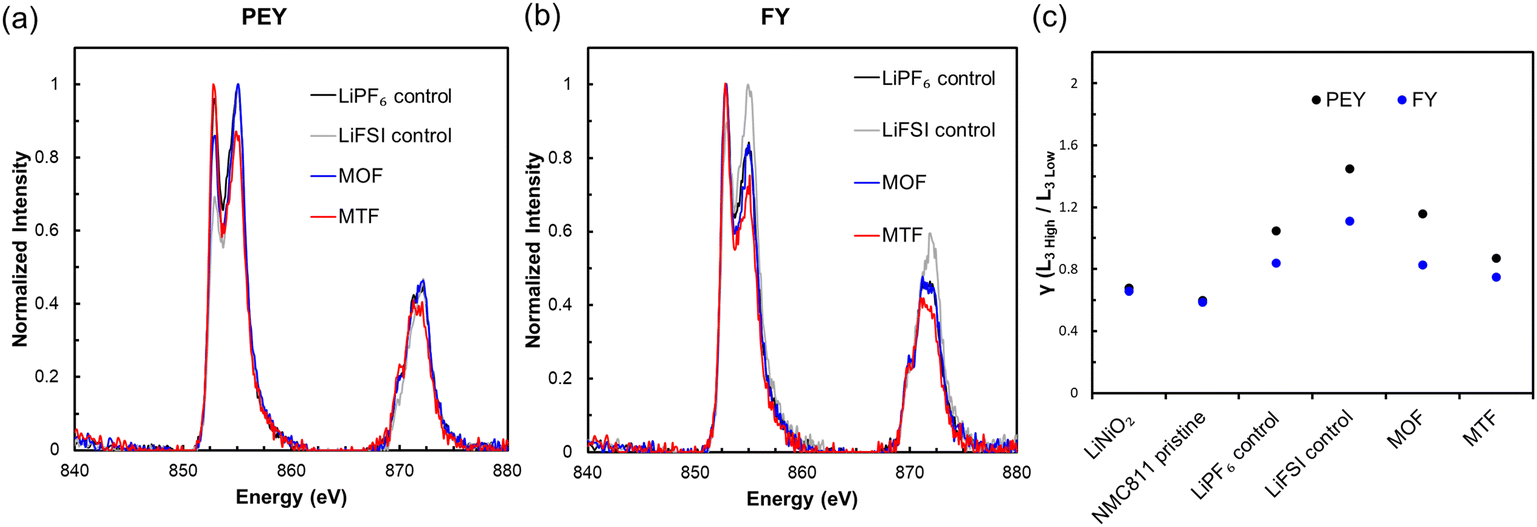 | ||
| Fig. 7 Soft XAS Ni L-edge spectra for NMC811 electrodes recovered from graphite/NMC811 cells after 200 fast charge cycles: (a) partial electron yield and (b) fluorescence yield. (c) The ratio between the peak intensities of the Ni L3 High and L3 Low peaks of the Ni L-edge, γ, for the four electrode samples as well as a LiNiO2 standard and a pristine NMC811 coating. | ||
X-ray photoelectron spectroscopy (XPS) was used to characterize the composition of the cathode electrolyte interphase (CEI) formed on the NMC811 cathodes and the solid electrolyte interphase (SEI) formed on the graphite anodes after 200 cycles under the baseline cycling condition. Comparison of the C 1s, F 1s and Li 1s regions for the cells with LiPF6 EC:DMC and MTF electrolytes are shown in Fig. 8a–c and e–g, along with elemental compositions for all electrolytes (Fig. 8d and h). Full fitting results for all cells and spectral regions are presented in the ESI† (Fig. S19–S25 and Tables S4 and S5).
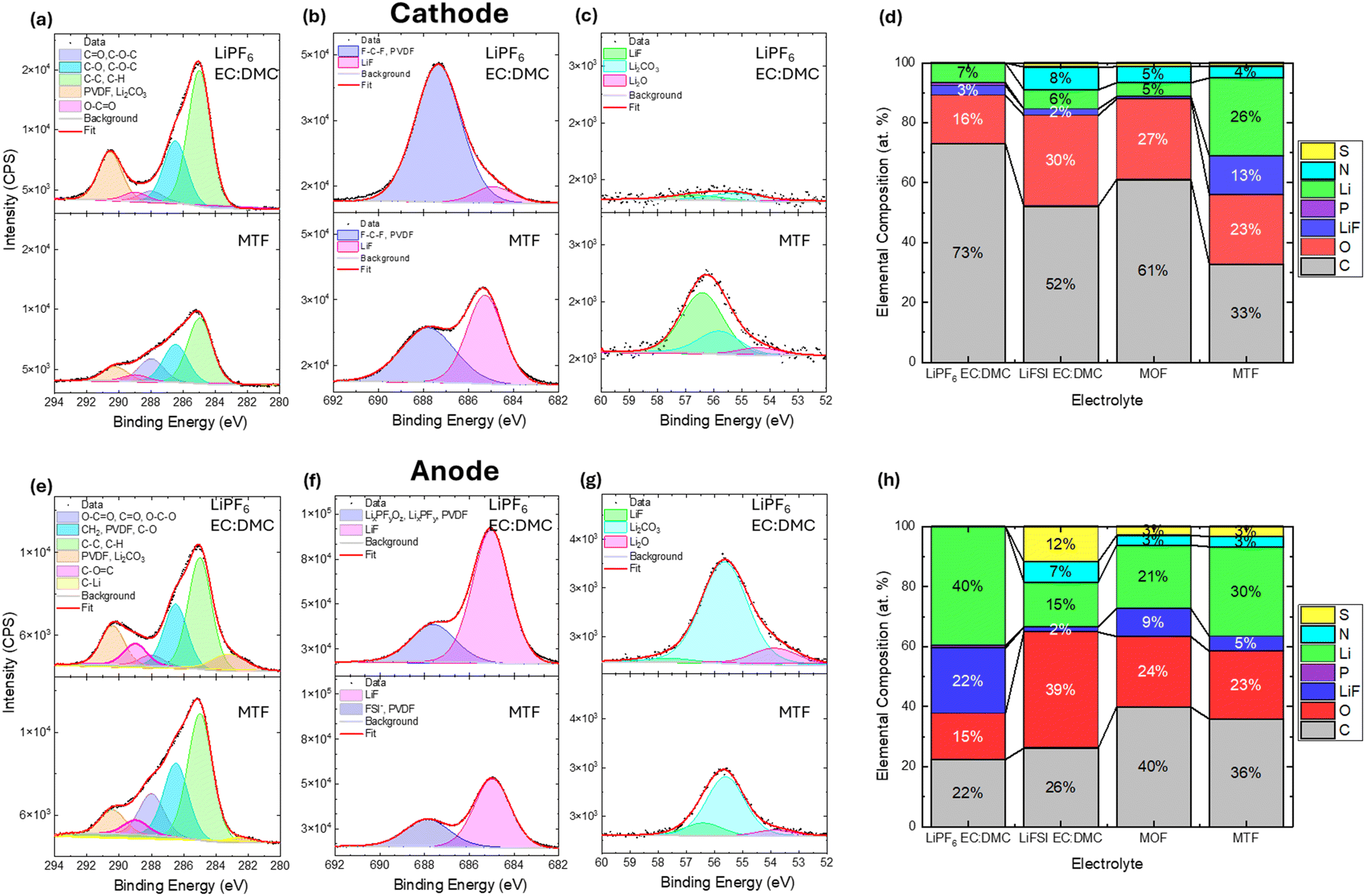 | ||
| Fig. 8 XPS (a and e) C 1s (b and f) F 1s and (c and g) Li 1s spectra of recovered (a–c) NMC811 cathodes and (e–g) graphite anodes after 200× cycles under 1C/1C CCCV cycling condition with either LiPF6 EC:DMC or MTF electrolytes. (d and h) Elemental composition of (d) cathodes and (h) anodes cycled with LiPF6 EC:DMC, LiFSI EC:DMC, MTF or MOF electrolytes. | ||
Notable differences in the CEI (Fig. 8a–d) were observed after cycling in the LiPF6 EC:DMC vs. MTF electrolytes with the EC:DMC electrolyte exhibiting almost double the carbon content (73 atomic (at) % for the LiPF6 EC:DMC vs. 33 at% for MTF), the carbon component assignments were based on a previous report,92 with the MTF electrolyte in particular showing a substantial reduction in carbonate type species at ≈291 eV.93–95 Comparison of the F1s spectra indicate ∼5× higher levels of LiF (≈685 eV) species for the MTF electrolyte (3 at% for LiPF6 EC:DMC vs. 13 at% for MTF). Meanwhile, the Li 1s spectra indicate a higher Li content for the MTF derived CEI (7 at% for LiPF6 EC:DMC vs. 26 at% for MTF). The increased LiF in the MTF derived CEI likely comes from a combination of decomposition of the FEC, FSI−, and fluorinated ester and ether.64,67,96,97 Analysis of the S2p, N1s regions (Fig. S23 and S24†) reveal that the MTF CEI also contains contributions from FSI− anions as well as FSI− decomposition species (SOx and Snx−)53,98–100 while the LiPF6 EC:DMC contains fluorophosphate (LixPOyFz and LixPFy) species (Fig. S25†).101–103 Investigation of the CEIs formed with MOF and LiFSI EC:DMC electrolytes reveal that they have similar Li, F and C contents as the LiPF6 EC:DMC (Fig. 8d), while also incorporating of S and N species associated with the FSI− anion. Overall, the analysis reveals that the LHCE derived CEI is rich in inorganic species including LiF and FSI− anion derived species, which have been shown to be associated with improved capacity retention for metal oxide cathodes.67,104,105 This LiF and FSI− derived CEI can improve capacity retention by producing a more compact and fully passivating CEI layer compared to a more organic-rich CEI as found for the LiPF6 EC:DMC electrolyte that may be less dense and provide incomplete coverage of the cathode surface.72,96,97,106 The MTF electrolyte likely contributes to the LiF-rich CEI as DFT calculations suggest that when fluorinated esters decompose they do so through C–F bond breakage yielding F− and F-rich fragments that can incorporate as LiF in the CEI.107 Further, DFT suggests that the Li+FSI−-solvent aggregates found in an LiFSI containing LHCE can preferentially oxidize at the cathode surface compared to free solvent molecules leading to a CEI that is rich in LiF and FSI− derived species rather than organic species.96
Analysis of the SEI chemistry (Fig. 8e–h) reveals differences in the Li and F 1s spectra among the samples. The LiPF6 EC:DMC derived SEI has somewhat higher LiF and Li2CO3 content than the LHCE and LiFSI EC:DMC, where LiF formation can occur by direct anion reduction and by electrocatalytic transformation of HF,55 with both mechanisms being significant.108,109 In contrast, the LiFSI salt used in the LHCE electrolyte has superior stability against hydrolysis,110 thus the LiF on the anode is attributed to reductive decomposition of the FEC additive111 and the FSI− anion.112 An additional M–F peak was observed at (≈686.3 eV) for the LiFSI EC:DMC anode, which could be assigned to AlF3 (≈686.3 eV)113–115 or MnF2 (≈686.1 eV),114,116–118 this observation is consistent with the corrosion of the Al current collector which was observed for the cycled cathode (Fig. S16†). Al and Mn were also observed as present for the cycled anode in the EDS spectrum (Fig. S26†) for the LiFSI EC:DMC cell.
Morphology of the electrode surfaces was characterized using scanning electron microscopy (SEM) of electrodes recovered after cycling 200× under the baseline condition. SEM images of the cathodes cycled in the LiPF6 EC:DMC and MTF electrolytes, Fig. 9, reveal CEI formation for both electrolytes. For cathode cycled in LiPF6 EC:DMC electrolyte, a dark, amorphous film can be seen on top of the particles (Fig. 9a–c). An energy-r filter was used which allows for acquisition of both backscattered and secondary electrons; the dark color suggests that the film is composed of lower z atoms (i.e. organic matter). The large quantity of dark, amorphous film could be some organic species from the SEI. On the other hand, the cathode particles cycled in MTF have no significant dark areas but are covered in particles (Fig. 9d–f) that are significantly smaller than the NMC811 primary crystallites, 210 ± 50 nm vs. 520 ± 80 nm, these may be carbon black. SEM-energy dispersive X-ray spectroscopy (EDS) was used to determine the relative atomic percentages of species formed on the two cathodes (Fig. S27†). The LiPF6 EC:DMC sample showed significantly more C and O while the MTF electrolyte showed higher fluorine content. The SEM and EDS mapping indicates the CEI film on the LiPF6 EC:DMC cathode is an organic rich film while the CEI on the MTF cathode is fluoride rich consistent with the XPS findings.
 | ||
| Fig. 9 SEM images of NMC811 cathodes cycled 200 times under the 1C/1C CCCV condition. A LiPF6 EC:DMC cathode at (a) 1000×, (b) 2000×, and (c) 5000× magnification. An MTF cathode at (d) 1000×, (e) 2000×, and (f) 5000× magnification. | ||
SEM images of the recovered anodes are presented in Fig. S28.† Similar morphology was observed for both the anodes cycled in the LiPF6 EC:DMC and the MTF electrolytes, with 9 ± 3 μm plate-like graphite particles surrounded by ∼100–200 nm spherical carbon black particles. EDS measurements of the anode (Fig. S29†) revealed similar surface composition, with the major difference being the detection of S and N species associated with the LiFSI anion and anion decomposition products on the anode cycled in MTF electrolyte. Overall, the XPS and SEM analysis indicate that differences in composition and morphology of the CEI are much greater than that of the SEI when comparing the control and LHCE cycled electrodes.
Conclusion
In summary, fluorinated ester solvents, ester and ether diluents, and film forming additives were investigated to develop LHCEs capable of operation under broad operating cycling conditions. The formulations are non-flammable and can demonstrate improved capacity retention compared to carbonate-based control electrolytes in a pouch cell form factor under a broad array of operating conditions including: cycling to high voltage (4.5 V), under fast charge (15 minute), and at low temperature (−20 °C), without sacrificing capacity retention at elevated temperature (40 °C). While the negative electrode (anode) interfaces and corresponding SEIs formed in the electrolyte systems studied are more similar, the composition of the CEI at the positive electrode (cathode) interface varies more significantly. The fluorinated electrolyte/electrode CEI formed by the LHCE systems with demonstrated reduced charge transfer impedance facilitate improved interfacial Li+ transport kinetics. The electrochemical behavior of the new electrolytes suggests it may be possible for an electrolyte system to meet the needs of next-generation Li-ion batteries requiring operation under a diverse set of operating conditions. These results represent an important advance as LHCE systems may operate well in one or two extreme conditions, but compromise others noted in a compilation of prior literature (Table S6†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U. S. Department of Energy Office of Energy Efficiency and Renewable Energy under the Vehicle Technologies program, award DE-EE0009643. Analysis of the XPS results to determine interfacial composition was supported by the Center of Mesoscale Transport Properties, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award No. DE-SC0012673. The research used resources of beamline 28-ID-2 and 7-ID-1 of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE), Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. In addition, this research used resources of the Center for Functional Nanomaterials, a U.S. DOE Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704. The pouch cell assembly equipment was supported under Award No. 75039 from the New York State Energy Research and Development Authority (NYSERDA) and Award No. 76890 from the New York State Department of Economic Development (DED), which were provided as matching funds to the Center of Mesoscale Transport Properties, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award No. DE-SC0012673. NYSERDA has not reviewed the information contained herein, and the opinions expressed do not necessarily reflect those of NYSERDA, or the State of New York. E. J. M. B. acknowledges the support of the National Science Foundation Graduate Research Fellowship under grant no. 2234683. E. S. T. acknowledges support as the William and Jane Knapp Chair in Energy and the Environment. Commercial equipment, instruments, or materials identified in this paper or ESI† are intended to specify the experimental procedure adequately and are not intended to imply recommendation or endorsement by the National Institute of Standards and Technology.References
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- X. Lin, G. Zhou, J. Liu, J. Yu, M. B. Effat, J. Wu and F. Ciucci, Adv. Energy Mater., 2020, 10, 2001235 CrossRef CAS.
- A. Tomaszewska, Z. Chu, X. Feng, S. O'Kane, X. Liu, J. Chen, C. Ji, E. Endler, R. Li, L. Liu, Y. Li, S. Zheng, S. Vetterlein, M. Gao, J. Du, M. Parkes, M. Ouyang, M. Marinescu, G. Offer and B. Wu, eTransportation, 2019, 1, 100011 CrossRef.
- C. E. Hendricks, A. N. Mansour, D. A. Fuentevilla, G. H. Waller, J. K. Ko and M. G. Pecht, J. Electrochem. Soc., 2020, 167(9), 090501 CrossRef CAS.
- E. R. Logan, E. M. Tonita, K. L. Gering, J. Li, X. Ma, L. Y. Beaulieu and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A21–A30 CrossRef CAS.
- E. R. Logan and J. R. Dahn, Trends Chem., 2020, 2, 354–366 CrossRef CAS.
- E. R. Logan, D. S. Hall, M. M. E. Cormier, T. Taskovic, M. Bauer, I. Hamam, H. Hebecker, L. Molino and J. R. Dahn, J. Phys. Chem. C, 2020, 124, 12269–12280 CrossRef CAS.
- L. Ma, S. L. Glazier, R. Petibon, J. Xia, J. M. Peters, Q. Liu, J. Allen, R. N. C. Doig and J. R. Dahn, J. Electrochem. Soc., 2016, 164, A5008–A5018 CrossRef.
- J. Holoubek, Y. Yin, M. Li, M. Yu, Y. S. Meng, P. Liu and Z. Chen, Angew. Chem., Int. Ed., 2019, 58, 18892–18897 CrossRef CAS PubMed.
- X. Dong, Z. Guo, Z. Guo, Y. Wang and Y. Xia, Joule, 2018, 2, 902–913 CrossRef CAS.
- X. Q. Zhang, X. Chen, X. B. Cheng, B. Q. Li, X. Shen, C. Yan, J. Q. Huang and Q. Zhang, Angew. Chem., 2018, 130, 5399–5403 CrossRef.
- J. Zheng, M. H. Engelhard, D. Mei, S. Jiao, B. J. Polzin, J.-G. Zhang and W. Xu, Nat. Energy, 2017, 2, 1–8 Search PubMed.
- L. Chen, X. Fan, E. Hu, X. Ji, J. Chen, S. Hou, T. Deng, J. Li, D. Su and X. Yang, Chem, 2019, 5, 896–912 CAS.
- Y. Zhu, X. Luo, H. Zhi, Y. Liao, L. Xing, M. Xu, X. Liu, K. Xu and W. Li, J. Mater. Chem. A, 2018, 6, 10990–11004 RSC.
- Q. Zhou, S. Dong, Z. Lv, G. Xu, L. Huang, Q. Wang, Z. Cui and G. Cui, Adv. Energy Mater., 2020, 10, 1903441 CrossRef CAS.
- G. A. Elia, U. Ulissi, S. Jeong, S. Passerini and J. Hassoun, Energy Environ. Sci., 2016, 9, 3210–3220 RSC.
- M. T. F. Rodrigues, K. Kalaga, H. Gullapalli, G. Babu, A. L. M. Reddy and P. M. Ajayan, Adv. Energy Mater., 2016, 6, 1600218 CrossRef.
- Y. Zhu, X. Luo, H. Zhi, Y. Liao, L. Xing, M. Xu, X. Liu, K. Xu and W. Li, J. Mater. Chem. A, 2018, 6, 10990–11004 RSC.
- W. Zhou, M. Zhang, X. Kong, W. Huang and Q. Zhang, Adv. Sci., 2021, 8, 2004490 CrossRef CAS PubMed.
- X. Ma, J. Li, S. L. Glazier, L. Ma, K. L. Gering and J. R. Dahn, Electrochim. Acta, 2018, 270, 215–223 CrossRef CAS.
- X. Ma, J. E. Harlow, J. Li, L. Ma, D. S. Hall, S. Buteau, M. Genovese, M. Cormier and J. R. Dahn, J. Electrochem. Soc., 2019, 166, A711–A724 CrossRef CAS.
- Y. Yang, P. Li, N. Wang, Z. Fang, C. Wang, X. Dong and Y. Xia, Chem. Commun., 2020, 56, 9640–9643 RSC.
- J. Holoubek, M. Yu, S. Yu, M. Li, Z. Wu, D. Xia, P. Bhaladhare, M. S. Gonzalez, T. A. Pascal, P. Liu and Z. Chen, ACS Energy Lett., 2020, 5, 1438–1447 CrossRef CAS.
- D.-J. Yoo, Q. Liu, O. Cohen, M. Kim, K. A. Persson and Z. Zhang, Adv. Energy Mater., 2023, 13, 2204182 CrossRef CAS.
- Y. Ugata, K. Yukishita, N. Kazahaya, S. Takahashi and N. Yabuuchi, Chem. Mater., 2023, 35, 3686–3693 CrossRef CAS.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132–A141 CrossRef CAS.
- X. Zhang, L. Zou, Y. Xu, X. Cao, M. H. Engelhard, B. E. Matthews, L. Zhong, H. Wu, H. Jia, X. Ren, P. Gao, Z. Chen, Y. Qin, C. Kompella, B. W. Arey, J. Li, D. Wang, C. Wang, J.-G. Zhang and W. Xu, Adv. Energy Mater., 2020, 10, 2000368 CrossRef CAS.
- L.-L. Jiang, C. Yan, Y.-X. Yao, W. Cai, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3402–3406 CrossRef CAS PubMed.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J. G. Zhang, Adv. Mater., 2018, 30, e1706102 CrossRef PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- D. Di Lecce and J. Hassoun, ACS Omega, 2018, 3, 8583–8588 CrossRef CAS PubMed.
- V. Nilsson, A. Kotronia, M. Lacey, K. Edström and P. Johansson, ACS Appl. Energy Mater., 2020, 3, 200–207 CrossRef CAS.
- Y. Yang, R. Xu, K. Zhang, S.-J. Lee, L. Mu, P. Liu, C. K. Waters, S. Spence, Z. Xu, C. Wei, D. J. Kautz, Q. Yuan, Y. Dong, Y.-S. Yu, X. Xiao, H.-K. Lee, P. Pianetta, P. Cloetens, J.-S. Lee, K. Zhao, F. Lin and Y. Liu, Adv. Energy Mater., 2019, 9, 1900674 CrossRef.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- S. J. An, J. Li, Z. Du, C. Daniel and D. L. Wood, J. Power Sources, 2017, 342, 846–852 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- Y. Li and Y. Qi, Energy Environ. Sci., 2019, 12, 1286–1295 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Wohlfarth, Selection and arrangement of static dielectric constants of pure liquid substances and their binary liquid mixtures, in Static Dielectric Constants of Pure Liquids and Binary Liquid Mixtures, Springer-Verlag Berlin Heidelberg, Berlin, 2015, Supplement to Volume IV/17, pp. 1–2 Search PubMed.
- L. E. Roy, E. Jakubikova, M. G. Guthrie and E. R. Batista, J. Phys. Chem. A, 2009, 113, 6745–6750 CrossRef CAS PubMed.
- O. Borodin, W. Behl and T. R. Jow, J. Phys. Chem. C, 2013, 117, 8661–8682 CrossRef CAS.
- T. Yim, C. Y. Choi, J. Mun, S. M. Oh and Y. G. Kim, Molecules, 2009, 14, 1840–1851 CrossRef CAS PubMed.
- R. A. Di Leo, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, Electrochim. Acta, 2013, 109, 27–32 CrossRef CAS.
- C. L. Campion, W. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152, A2327 CrossRef CAS.
- M. J. Hossain, Q. Wu, E. J. Marin Bernardez, C. D. Quilty, A. C. Marschilok, E. S. Takeuchi, D. C. Bock, K. J. Takeuchi and Y. Qi, J. Phys. Chem. Lett., 2023, 14, 7718–7731 CrossRef CAS PubMed.
- S. Chae, W.-J. Kwak, K. S. Han, S. Li, M. H. Engelhard, J. Hu, C. Wang, X. Li and J.-G. Zhang, ACS Energy Lett., 2021, 6, 387–394 CrossRef CAS.
- Q. Liu, C. Du, B. Shen, P. Zuo, X. Cheng, Y. Ma, G. Yin and Y. Gao, RSC Adv., 2016, 6, 88683–88700 RSC.
- Q. Q. Liu, D. J. Xiong, R. Petibon, C. Y. Du and J. R. Dahn, J. Electrochem. Soc., 2016, 163, A3010–A3015 CrossRef CAS.
- D. Pritzl, S. Solchenbach, M. Wetjen and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A2625 CrossRef CAS.
- R. Petibon, J. Xia, L. Ma, M. K. G. Bauer, K. J. Nelson and J. R. Dahn, J. Electrochem. Soc., 2016, 163, A2571 CrossRef CAS.
- J. Lee, Y.-J. Kim, H. S. Jin, H. Noh, H. Kwack, H. Chu, F. Ye, H. Lee and H.-T. Kim, ACS Omega, 2019, 4, 3220–3227 CrossRef CAS PubMed.
- P. Hilbig, L. Ibing, M. Winter and I. Cekic-Laskovic, Energies, 2019, 12(15), 2869 CrossRef CAS.
- T. Afroz, D. M. Seo, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Phys. Chem. C, 2015, 119, 7022–7027 CrossRef CAS.
- D. M. Seo, T. Afroz, J. L. Allen, P. D. Boyle, P. C. Trulove, H. C. De Long and W. A. Henderson, J. Phys. Chem. C, 2014, 118, 25884–25889 CrossRef CAS.
- X. Cao, Y. Xu, L. Zhang, M. H. Engelhard, L. Zhong, X. Ren, H. Jia, B. Liu, C. Niu, B. E. Matthews, H. Wu, B. W. Arey, C. Wang, J.-G. Zhang and W. Xu, ACS Energy Lett., 2019, 4, 2529–2534 CrossRef CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- H. Jia, P. Gao, L. Zou, K. S. Han, M. H. Engelhard, Y. He, X. Zhang, W. Zhao, R. Yi, H. Wang, C. Wang, X. Li and J.-G. Zhang, Chem. Mater., 2020, 32, 8956–8964 CrossRef CAS.
- H. Jia, L. Zou, P. Gao, X. Cao, W. Zhao, Y. He, M. H. Engelhard, S. D. Burton, H. Wang, X. Ren, Q. Li, R. Yi, X. Zhang, C. Wang, Z. Xu, X. Li, J.-G. Zhang and W. Xu, Adv. Energy Mater., 2019, 9, 1900784 CrossRef.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- H. Jia, Z. Yang, Y. Xu, P. Gao, L. Zhong, D. J. Kautz, D. Wu, B. Fliegler, M. H. Engelhard, B. E. Matthews, B. Broekhuis, X. Cao, J. Fan, C. Wang, F. Lin and W. Xu, Adv. Energy Mater., 2023, 13, 2203144 CrossRef CAS.
- C. Zhu, C. Sun, R. Li, S. Weng, L. Fan, X. Wang, L. Chen, M. Noked and X. Fan, ACS Energy Lett., 2022, 7, 1338–1347 CrossRef CAS.
- Y. Watanabe, Y. Ugata, K. Ueno, M. Watanabe and K. Dokko, Phys. Chem. Chem. Phys., 2023, 25, 3092–3099 RSC.
- Z. Xu, K. Deng, S. Zhou, Z. Liu, X. Guan and D. Mo, ACS Appl. Mater. Interfaces, 2022, 14, 48694–48704 CrossRef CAS PubMed.
- H. Jia, L. Zou, P. Gao, X. Cao, W. Zhao, Y. He, M. H. Engelhard, S. D. Burton, H. Wang, X. Ren, Q. Li, R. Yi, X. Zhang, C. Wang, Z. Xu, X. Li, J.-G. Zhang and W. Xu, Adv. Energy Mater., 2019, 9, 2000368 Search PubMed.
- J. Langdon, R. Sim and A. Manthiram, ACS Energy Lett., 2022, 7, 2634–2640 CrossRef CAS.
- Y.-X. Zuo and W. Fürst, Fluid Phase Equilib., 1997, 138, 87–104 CrossRef CAS.
- A. Belgibayeva, A. Rakhmetova, M. Rakhatkyzy, M. Kairova, I. Mukushev, N. Issatayev, G. Kalimuldina, A. Nurpeissova, Y.-K. Sun and Z. Bakenov, J. Power Sources, 2023, 557, 232550 CrossRef CAS.
- X. Wu and Z. Du, Electrochem. Commun., 2021, 129, 107088 CrossRef CAS.
- S.-M. Bak, E. Hu, Y. Zhou, X. Yu, S. D. Senanayake, S.-J. Cho, K.-B. Kim, K. Y. Chung, X.-Q. Yang and K.-W. Nam, ACS Appl. Mater. Interfaces, 2014, 6, 22594–22601 CrossRef CAS PubMed.
- D. M. Lutz, A. H. McCarthy, S. T. King, G. Singh, C. A. Stackhouse, L. Wang, C. D. Quilty, E. M. Bernardez, K. R. Tallman, X. Tong, J. Bai, H. Zhong, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and D. C. Bock, J. Electrochem. Soc., 2022, 169, 090501 CrossRef CAS.
- D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle and W. A. Henderson, Energy Environ. Sci., 2014, 7, 416–426 RSC.
- Y. Zhang, Y. Katayama, R. Tatara, L. Giordano, Y. Yu, D. Fraggedakis, J. G. Sun, F. Maglia, R. Jung, M. Z. Bazant and Y. Shao-Horn, Energy Environ. Sci., 2020, 13, 183–199 RSC.
- N. Legrand, B. Knosp, P. Desprez, F. Lapicque and S. Raël, J. Power Sources, 2014, 245, 208–216 CrossRef CAS.
- S. S. Zhang, InfoMat, 2021, 3, 125–130 CrossRef CAS.
- S. Zhang, K. Xu and T. Jow, Electrochem. Commun., 2002, 4, 928–932 CrossRef CAS.
- S. Zhang, K. Xu and T. Jow, J. Solid State Electrochem., 2003, 7, 147–151 CrossRef CAS.
- X. Zhang, L. Zou, Y. Xu, X. Cao, M. H. Engelhard, B. E. Matthews, L. Zhong, H. Wu, H. Jia and X. Ren, Adv. Energy Mater., 2020, 10, 2000368 CrossRef CAS.
- Y. Yamada, M. Yaegashi, T. Abe and A. Yamada, Chem. Commun., 2013, 49, 11194–11196 RSC.
- C. Tian, D. Nordlund, H. L. Xin, Y. Xu, Y. Liu, D. Sokaras, F. Lin and M. M. Doeff, J. Electrochem. Soc., 2018, 165, A696–A704 CrossRef CAS.
- J. W. Smith and R. J. Saykally, Chem. Rev., 2017, 117, 13909–13934 CrossRef CAS PubMed.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 5459–5468 CrossRef CAS PubMed.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef PubMed.
- B. G. Chae, S. Y. Park, J. H. Song, E. Lee and W. S. Jeon, Nat. Commun., 2021, 12, 3814 CrossRef CAS PubMed.
- M. C. Biesinger, Appl. Surf. Sci., 2022, 597, 153681 CrossRef CAS.
- D. Briggs and G. Beamson, Anal. Chem., 1992, 64, 1729–1736 CrossRef CAS.
- S. Leroy, F. Blanchard, R. Dedryvere, H. Martinez, B. Carre, D. Lemordant and D. Gonbeau, Surf. Interface Anal., 2005, 37, 773–781 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, ULVAC-PHI, 1995 Search PubMed.
- Z. Li, H. Rao, R. Atwi, B. M. Sivakumar, B. Gwalani, S. Gray, K. S. Han, T. A. Everett, T. A. Ajantiwalay, V. Murugesan, N. N. Rajput and V. G. Pol, Nat. Commun., 2023, 14, 868 CrossRef CAS PubMed.
- F. Guo, X. Chen, Y. Hou, W. Wei, Z. Wang, H. Yu and J. Xu, Small, 2023, 19, 2207290 CrossRef CAS PubMed.
- W. Xue, Z. Shi, M. Huang, S. Feng, C. Wang, F. Wang, J. Lopez, B. Qiao, G. Xu, W. Zhang, Y. Dong, R. Gao, Y. Shao-Horn, J. A. Johnson and J. Li, Energy Environ. Sci., 2020, 13, 212–220 RSC.
- J. He, A. Bhargav, W. Shin and A. Manthiram, J. Am. Chem. Soc., 2021, 143, 20241–20248 CrossRef CAS PubMed.
- C. D. Wagner, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble, NIST X-ray Photoelectron Spectroscopy Database, 2003 Search PubMed.
- X. Li, M. Xu, Y. Chen and B. L. Lucht, J. Power Sources, 2014, 248, 1077–1084 CrossRef CAS.
- A. Keefe, R. Weber, I. G. Hill and J. R. Dahn, J. Electrochem. Soc., 2020, 167, 120507 CrossRef CAS.
- L. Madec, J. Xia, R. Petibon, K. J. Nelson, J.-P. Sun, I. G. Hill and J. R. Dahn, J. Phys. Chem. C, 2014, 118, 29608–29622 CrossRef CAS.
- P. Bai, X. Ji, J. Zhang, W. Zhang, S. Hou, H. Su, M. Li, T. Deng, L. Cao, S. Liu, X. He, Y. Xu and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202202731 CrossRef CAS PubMed.
- Y.-L. Heng, Z.-Y. Gu, J.-Z. Guo, X.-T. Wang, X.-X. Zhao and X.-L. Wu, Energy Mater., 2022, 2, 2000017 Search PubMed.
- S. Kuang, H. Hua, P. Lai, J. Li, X. Deng, Y. Yang and J. Zhao, ACS Appl. Mater. Interfaces, 2022, 14, 19056–19066 CrossRef CAS PubMed.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X.-Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- C. Cao, T. P. Pollard, O. Borodin, J. E. Mars, Y. Tsao, M. R. Lukatskaya, R. M. Kasse, M. A. Schroeder, K. Xu, M. F. Toney and H.-G. Steinrück, Chem. Mater., 2021, 33, 7315–7336 CrossRef CAS.
- M.-T. F. Rodrigues, C. Liao, K. Kalaga, I. A. Shkrob and D. P. Abraham, ACS Appl. Energy Mater., 2019, 2, 5380–5385 CrossRef CAS.
- H.-B. Han, S.-S. Zhou, D.-J. Zhang, S.-W. Feng, L.-F. Li, K. Liu, W.-F. Feng, J. Nie, H. Li, X.-J. Huang, M. Armand and Z.-B. Zhou, J. Power Sources, 2011, 196, 3623–3632 CrossRef CAS.
- Y. Okuno, K. Ushirogata, K. Sodeyama and Y. Tateyama, Phys. Chem. Chem. Phys., 2016, 18, 8643–8653 RSC.
- E. P. Kamphaus, S. A. Gomez, X. Qin, M. Shao and P. B. Balbuena, ChemPhysChem, 2020, 21, 1310–1317 CrossRef CAS PubMed.
- L. Qiao, U. Oteo, M. Martinez-Ibañez, A. Santiago, R. Cid, E. Sanchez-Diez, E. Lobato, L. Meabe, M. Armand and H. Zhang, Nat. Mater., 2022, 21, 455–462 CrossRef CAS PubMed.
- H. Hemmelmann, J. K. Dinter and M. T. Elm, Adv. Mater. Interfaces, 2021, 8, 2002074 CrossRef CAS.
- V. I. Nefedov, Zh. Neorg. Khim., 1973, 18, 931–934 CAS.
- X. Wu, D. Ruan, S. Tan, M. Feng, B. Li and G. Hu, Ionics, 2020, 26, 5427–5434 CrossRef CAS.
- Y. Lu, Y. Zhou, Q. Yan and E. Fong, J. Mater. Chem. A, 2016, 4, 2691–2698 RSC.
- R. Hayes and N. Edelstein, Electron Spectroscopy, North-Holland Publishing Company, Amsterdam, 1972 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lf00259d |
| This journal is © The Royal Society of Chemistry 2024 |