
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom rust to riches: phytochemically assisted synthesis of superparamagnetic Fe3O4 nanoparticles with Au for SERS sensing†
Dariush
Aligholizadeh‡
 a,
Landon
Bechdel
a,
Mansoor
Johnson
a,
Vera
Smolyaninova
b and
Mary Sajini
Devadas
*a
a,
Landon
Bechdel
a,
Mansoor
Johnson
a,
Vera
Smolyaninova
b and
Mary Sajini
Devadas
*a
aDepartment of Chemistry, Towson University, Towson, Maryland 21252, USA. E-mail: mdevadas@towson.edu
bDepartment of Physics, Astronomy, and Geosciences, Towson University, Towson, Maryland 21252, USA
First published on 26th June 2024
Abstract
Iron oxide nanoparticles are a prominent choice among applied nanotechnologies, with multiple iterations being FDA-approved therapeutics available to general consumers. While their applications are numerous, their syntheses occasionally involve reagents toxic to the ecosystem and potential human end-users. However, application of natural products to nanoparticle syntheses allows one to traverse these issues. Additionally, recent advances in hybrid nanoparticles have opened the door for possibilities to combine a noble-metal nanoparticle's plasmonic nature with those containing magnetic properties to create magneto-plasmonic materials. The pursuit of these plasmonic and magnetic nanoparticles arises from the emergent properties from the hybrid particle. For example, is the ability to magnetically concentrate the plasmonic nanoparticles to increase SERS intensity, or the ability to apply them for biological imaging and magnetic hyperthermia treatment without needing to use two separate nanoparticles. Here, we describe the synthesis and characterization of hybrid Au/Fe3O4 superparamagnetic nanoparticles combining the plasmonic properties of Au with the magnetic properties of Fe3O4 to enable surface-enhanced Raman scattering (SERS). We further demonstrate the ability to functionalize these particles with four bioactive compounds: histidine, glutathione, cysteamine, and tiopronin. Lastly, we show the ability to synthesize these magneto-plasmonic nanoparticles (68 emu g−1 magnetic saturation) using locally sourced plant metabolites from the Cercis canadensis, or Eastern redbud, tree. Our SERS measurements indicate the detection of nanogram quantities of an organothiol in solution, and SERS enhancement factors on the order of 102 and 103 while maintaining magnetic saturations of approximately 30–45 emu g−1 in the presence of Au.
Introduction
The ability to synthesize and apply nanoparticles of differing shapes, sizes, and properties was one of the hallmarks of chemistry in the twentieth century; Feynman's prediction1 that we would be able to build microscopic computers and store and transmit information on the nanoscale all came true with the advent of magnetic nanoparticles.2–7 Since the initial introduction of liquid suspensions of magnetic nanoparticles–ferrofluids in 1965, many groups have created magnetic nanoparticles with both size-tunability and high magnetization. Unfortunately, many of these syntheses are not feasible in aerobic conditions and readily combust if not done under anaerobic conditions. For exactly these reasons, the only magnetic nanoparticles (MNP) approved by the USA's Food & Drug Administration (FDA) are iron oxide nanoparticles8 which are not only air-stable but synthesized under atmospheric conditions from relatively simple starting materials. Among the MNPs available, iron oxide does not demonstrate the highest magnetic saturation, but it is simple and safe enough to administer to patients that it is used as both an MRI contrast agent and hyperthermia-inducing agent for cancer imaging and treatment. Therefore, when considering further applications of MNPs to other fields, one would ideally utilize iron oxide nanoparticles due to their success in FDA-approved applications and their facile synthesis.The main types of iron oxide nanoparticles available are magnetite (Fe3O4), maghemite (γ-Fe2O3), and hematite (α-Fe2O3). However, of the three types, α-Fe2O3 displays anti-ferromagnetism and γ-Fe2O3 displays 80% of the maximum magnetic saturation of Fe3O4,9 therefore Fe3O4 remains the best choice. As much work has been completed in the preparation of tunable and facile syntheses of Fe3O4, the next generation of MNPs involves the preparation of heterometal Fe3O4 complexes with properties beyond ferromagnetism. In the present work, we establish a protocol for the synthesis of gold magnetic nanoparticles (AuMNP) whereby we modify the Fe3O4 structure with Au in the presence of four biocompatible capping agents: cysteamine, L-histidine, L-glutathione, and tiopronin. The robustness of the synthesis across these four capping agents, or ligands, demonstrates the versatility of the available functionalization of these nanoparticles. Additionally, the synthesis utilized aqueous extracts obtained from locally-sourced plant matter. In recent years efforts have been taken to reduce the number of synthetic reagents in chemical synthesis, both for cost-benefits and for increased environmental compatibility. These “green” syntheses commonly swap either polymeric ligands or reducing agents for plant matter as the metal salt is in many cases irreplicable, especially by a naturally-occurring source.10–12 We further demonstrate the ability of these functionalized nanoparticles to generate a localized surface plasmon resonance (LSPR) in order to detect a model analyte with surface-enhanced Raman scattering.
Concurrently with the advancements in magnetic nanoparticles came advancements in noble metal nanoparticles. While unable to demonstrate ferromagnetism, the use of these nanoparticles comes from another important property: the ability to generate surface plasmons. At certain optical frequencies, noble metals can conduct electrons on their surface, eliminating the incident wave and forming a temporary plasma on their surface. This plasmonic surface can be coupled to vibronic or electronic transitions of an analyte adsorbed to the surface of the metal, leading to surface-enhanced spectroscopic detection. This plasmonic nature is exploited in multiple optical sensing techniques, namely surface-enhanced Raman scattering (SERS), metal-enhanced fluorescence (MEF), and surface-enhanced IR absorption spectroscopy (SEIRAS). These techniques focus on the confinement of the plasmonic surface to the nanoscale, as plasmonic oscillations when confined to the nanoscale can typically become localized and resonate at higher intensities generating an LSPR. The ability to enhance the spectroscopic signatures of many analytes allows for micro-, nano-, femto-, and attomole detection of organic and inorganic target compounds. The ability to couple plasmonic and magnetic properties in nanoparticles has been shown previously by our group13 and others. Some effort has been undertaken to successfully apply these hybrid nanoparticles to SERS sensing.14–16 The difficulties in synthesizing these hybrid nanoparticles come from the challenge of developing a protocol that does not sacrifice either the plasmonic or magnetic nature of the nanoparticle. Additionally, the surface ligands of the nanoparticle greatly dictate the ease or difficulty in later surface-based detection methods.17 For this reason, we demonstrate the synthesis of AuMNP with four different biocompatible ligands and their differing ability to allow SERS sensing while simultaneously stabilizing the molecule. The literature surrounding Au and Fe3O4 structures typically lacks any detailed magnetization data or SERS enhancement factors, warranting the narrowing of this knowledge gap. In this work, we not only demonstrate the simple synthesis and characterization of these novel functionalized AuMNP but also report magnetization data for all synthesized particles, as well as Au loading percentages, and SERS enhancement factors. We additionally synthesize these nanoparticles with an aqueous extract from locally sourced plants in a novel phytochemical route to create these Au/Fe3O4 heterometal nanoparticles. The plant extract is from the Cercis canadensis tree, common in many regions of eastern North America. This tree is chosen due to its magenta coloration which indicates a high number of anthocyanins which have been shown previously to assist in the chemical reduction and phytochemical synthesis of nanoparticles.18 The phytochemically-assisted synthesis of AuMNP will not only increase the biocompatibility of the final nanoparticle but will also allow decentralization of the synthesis to empower the creation of these hybrid functional materials in laboratories across the globe while enabling a green alternative to ecotoxic iron oxide dispersants and reducing agents.
Materials and methods
Materials
Tetrachloroauric acid (HAuCl4·3H2O; ACS Grade), L-glutathione reduced (C10H17N3O6S; 98%), and iron(II) chloride tetrahydrate (FeCl2·4H2O; 99+%) were purchased from Acros Organics. L-Histidine (C6H9N3O2; >98%), para-nitrothiophenol (p-NTP, C6H5NO2S; 96%), and iron(III) chloride anhydrous (FeCl3; Laboratory Grade) were purchased from Fisher Scientific. Trisodium citrate dihydrate (Na3C6H5O7; 99%) and ammonium hydroxide 30% (w/v) solution (NH4OH; 28–30%) were purchased from BDH Chemicals. Tiopronin (C5H9NO3S; >96.0%) was purchased from TCI Chemicals. Cysteamine (C2H7NS; ∼95%) was purchased from Sigma-Aldrich. 190 proof ethanol was purchased from Pharmco.Instruments
Raman and SERS measurements were acquired using a Rigaku Progeny X2 Raman spectrophotometer with a 785 nm excitation wavelength. UV-vis absorption spectra were obtained on a Cary 60 UV-vis spectrophotometer. For all UV-vis measurements, 250 μL of sample was diluted to 3.25 mL total volume with Milli-Q water. All solutions were centrifuged with an Eppendorf 5810 R centrifuge at 4000 RPM for 30 minutes. All IR spectra were taken with a Nicolet iS50 ATR-IR using a ∼100 μL droplet of solution or 10–100 mg of dry powder. Scanning electron microscopy (SEM) was performed on an FEI Apreo 2 SEM/STEM with a field-emission gun, 20 keV accelerating voltage, and a current of 13 pA to 0.1 nA. Scanning-transmission electron microscopy (STEM) was performed on the same SEM, with a STEM3+ detector and bright-field (BF) imaging mode unless otherwise specified. Magnetization measurements were conducted on a Quantum Design Versa Lab vibrating sample magnetometer (VSM) with a 50–300 K temperature range and 1–3 T or 30 kOe magnetic field strength. Milli-Q (18.2 MΩ cm) was prepared using a Thermo Scientific GenPure UV-TOC xCAD filtration system. All glassware and quartz cuvettes were cleaned in aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 12 M HCl:15.8 M HNO3) for 15 minutes before use.
:1 12 M HCl:15.8 M HNO3) for 15 minutes before use.
Preparation of Cercis canadensis extract
Collection and purification of the Cercis canadensis extract was conducted by acquiring locally sourced flower buds of the Cercis canadensis tree. Special attention was paid to reducing the number of branches and other non-flower components collected. The C. canadensis tree was chosen as it is extremely common in North America and could be locally sourced from our academic institution. To create the extract, 5.000 g of the flower was added to 50 mL of Milli-Q water in a 125 mL Erlenmeyer flask and then boiled for 20 minutes. After 20 minutes, the solution was brought to room temperature and the mixture was filtered through a 0.45-micron syringe filter and split into two 20 mL scintillation vials to be stored in the refrigerator until further use.Cercis canadensis extract characterization
The plant extract's antioxidant capacity was determined spectrophotometrically using a previously reported method by Murray et al.19 and Hughes et al.20 In brief, 1.5 g of C. canadensis and 9 mL of acidified methanol were combined and diluted to 25 mL. The blank was 1 mL of acidified methanol in lieu of plant matter. Then, 1.5 mL of this extract was vacuum-dried overnight to allow the sample to age and redissolved in 1.5 mL of acidified methanol after fully drying. Finally, 1 mL of this extract was diluted once again to 25 mL with acidified methanol, and absorbance at 530 nm (A530) and absorbance at 653 nm (A653) were recorded. Anthocyanin content was found using the following equation:where V1 = 0.5 mL, V2 = 12.5 mL, l = 1 cm, ε = 30
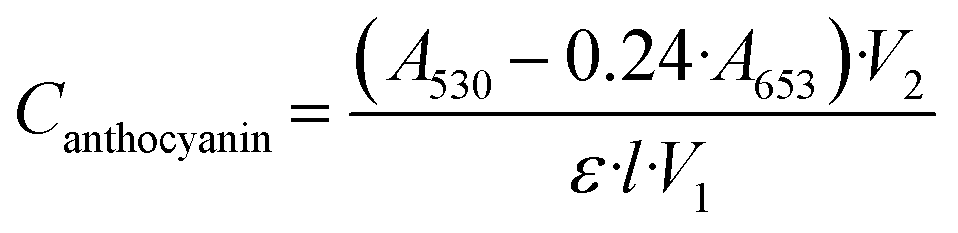 000 L mol−1 cm−1. A653 is considered due to chlorophyll absorbance at that wavelength.
000 L mol−1 cm−1. A653 is considered due to chlorophyll absorbance at that wavelength.
Synthesis of Fe3O4
The synthesis of Fe3O4 proceeded through a co-reduction method utilizing two chloride salts. Initially, 0.973 g of anhydrous FeCl3 and 0.994 g of FeCl2·4H2O were quickly dissolved in 40 mL of Milli-Q water under constant stirring at 900 RPM in a 250 mL round-bottom flask. Special attention was paid to the immediate dissolution of the chloride salts as they have the potential to oxidize the FexCly into Fe oxide. Next, 5 mL of 28% (w/v) NH3OH solution was quickly injected into the reaction mixture and then left to stir for 10 minutes. After 10 minutes, 4.4 g of trisodium citrate was added to the solution and the temperature of the flask was raised to 90 °C and then left to stir for an additional 30 minutes. The resulting black mixture was separated with an N40-grade magnet for <5 minutes to only collect the most magnetic product and the supernatant was disposed of. The mixture was then resuspended in an equal volume of fresh Milli-Q water, and this cleaning was repeated three times and stored as a dry powder in a vacuum desiccator.Synthesis of AuFe3O4 and AuNP
To create hybrid AuFe3O4 magnetic nanoparticles (AuMNP), 0.020 g of the previously synthesized Fe3O4 was added to a clean 20 mL scintillation vial and resuspended in 18 mL Milli-Q water and 2 mL aqueous C. canadensis extract. The mixture was treated with ultrasonication for 5 minutes or until the solution was visibly homogenous. This solution was then added to 20 mL of 3.25 mM HAuCl4·3H2O in a 125 mL round-bottom flask. The flask was then heated to a gentle boil under vigorous stirring. After boiling began, the stirring was continued for 10 minutes until a visible color change was noticed. During the final growth stage, the AuMNP were functionalized upon 30 min of stirring with 2 mL of one of the following ligands: (1) cysteamine 1.67 mg mL−1, (2) L-glutathione 6.63 mg mL−1, (3) L-histidine 3.35 mg mL−1, (4) tiopronin 3.52 mg mL−1. The solution was then centrifuged and resuspended in 10 mL of Milli-Q water via ultrasonication. The functionalized AuMNP were then labeled as cysAuMNP (cysteamine-functionalized AuMNP), gluAuMNP (glutathione-functionalized), hisAuMNP (histidine-functionalized), and tioAuMNP (tiopronin-functionalized).For comparative purposes, Au nanoparticles (AuNP) were also synthesized using a modified Turkevich method.21–25 Initially, 4 mL of a 1 mM solution of HAuCl4 in Milli-Q was added to a 20 mL scintillation vial. The flask was then brought to 100 °C while stirring at 900 RPM. Once the temperature of the vial reached 100 °C, 1 mL of C. canadensis extract was rapidly injected into the solution. This solution was stirred for 15 minutes, during which a noticeable color change from yellowish-red to deep purple was evident. The solution was used as synthesized without the need for further purification.
Magnetization measurements
The magnetization properties of the magnetic nanoparticles were measured using a Quantum Design Versa Lab vibrating sample magnetometer (VSM) to measure both saturation and potential hysteresis in the sample. A known sample mass was placed into the VSM in a plastic container and all measurements are reported as mass magnetization using electromagnetic units per gram (emu g−1) to report magnetic saturation. Magnetic field strength and magnetic flux density are reported in Oe and T, respectively. VSM measurements were conducted in temperatures starting at 50 K increasing to 300 K, with a step size of 5 K.Raman and SERS measurements
Raman and SERS measurements were obtained using a 785 nm portable Raman spectrometer. All measurements were taken at 300 mW incident laser power with an acquisition time of 20 seconds per scan. For Raman measurements, 2 mL of the sample was pipetted into a 4 mL optically transparent borosilicate vial. For SERS measurements, 1 mL of the nanoparticle at a fixed concentration of 0.3 g L−1 and 1 mL of the analyte for a total volume of 2 mL was added to a 4 mL optically transparent vial and measured. SERS enhancement factor (EF) was calculated using the following formula: EF = (NRaman/NSERS)(ISERS/IRaman) where NRaman and NSERS are the number of molecules probed in spontaneous Raman and SERS, respectively, and IRaman and ISERS are the intensities of the scattering in spontaneous Raman and SERS, respectively.Vibrational mode simulations
Due to the presence of a large number of simple organic substrates, IR simulations were carried out using Avogadro26 to simulate the initial geometry of the nanoparticle and GAMESS27 to calculate the optimized geometry and (using density-functional theory) determine the active vibrational modes in the molecule. We have previously reported the use of this method for Raman vibrational simulations for SERS enhancement.Results and discussion
Plant extract antioxidant capacity
The C. canadensis extract was initially quantified using a phytochemical assay to determine the total anthocyanin content. Quantification of this extract's anthocyanin content was completed as these compounds are known to have anti-inflammatory, anti-microbial, anti-tumor, and anti-diabetic properties,28 therefore knowledge of the total content within the extract, and by association, the end nanoparticle would open doors to future biomedical studies utilizing AuMNP. Anthocyanin content was hypothesized to be high in C. canadensis as it is a visibly purplish-pink flower, a coloration typically caused by high concentration of anthocyanins in flowers.29 Unfortunately, only a few reports exist in the literature on the anthocyanin content of C. canadensis,30 and thus little was understood about the use of C. canadensis in nanoparticle syntheses hitherto. From measurements on the aqueous extract, the anthocyanin content in 1 g of C. canadensis is roughly 105 ± 16 μM. For all future measurements, dilutions were made to ensure the C. canadensis extract did not fall outside of the 105 ± 16 μM anthocyanin content range as a standard test of concentration. The data for the four trials done on the C. canadensis extract can be found in Table S1.†Nanoparticle spectroscopic characterization
Initially, the synthesized nanoparticles were characterized using UV-vis spectroscopy. It is well understood that Fe3O4 nanoparticles lack significant absorbance at wavelengths higher than 600–700 nm.31–33 However, when combined with Au or Ag, an increase in absorbance in the visible region occurs, hinting towards the acquisition of plasmonic properties of the nanoparticle.34,35 After synthesis of the four functionalized AuMNP, UV-vis spectra were obtained and are compiled in Fig. 1. In Fig. 1a, the spectrum of cleaned Fe3O4 nanoparticles stabilized with citrate are shown, where absorbance is high in the UV and near-UV regions, and levels off quickly to near zero absorbance as λ reaches 700 nm. Differences are apparent when these particles are combined with Au: for example, a large background increase is seen in gluAuMNP (Fig. 1b) and tioAuMNP (Fig. 1c) indicating potentially larger particle sizes indicative of larger quasi spherical plasmonic structures. Ideally, the AuMNP formed would have a single plasmonic peak in the 400–800 nm region for spherical Au nanoparticle formation. The UV-vis spectrum of AuNP synthesized with C. canadensis extract can be found in Fig. S1† for comparison. The non-descript spectra of the four AuMNP when compared to the original Fe3O4 nanoparticles warranted further spectroscopic investigation.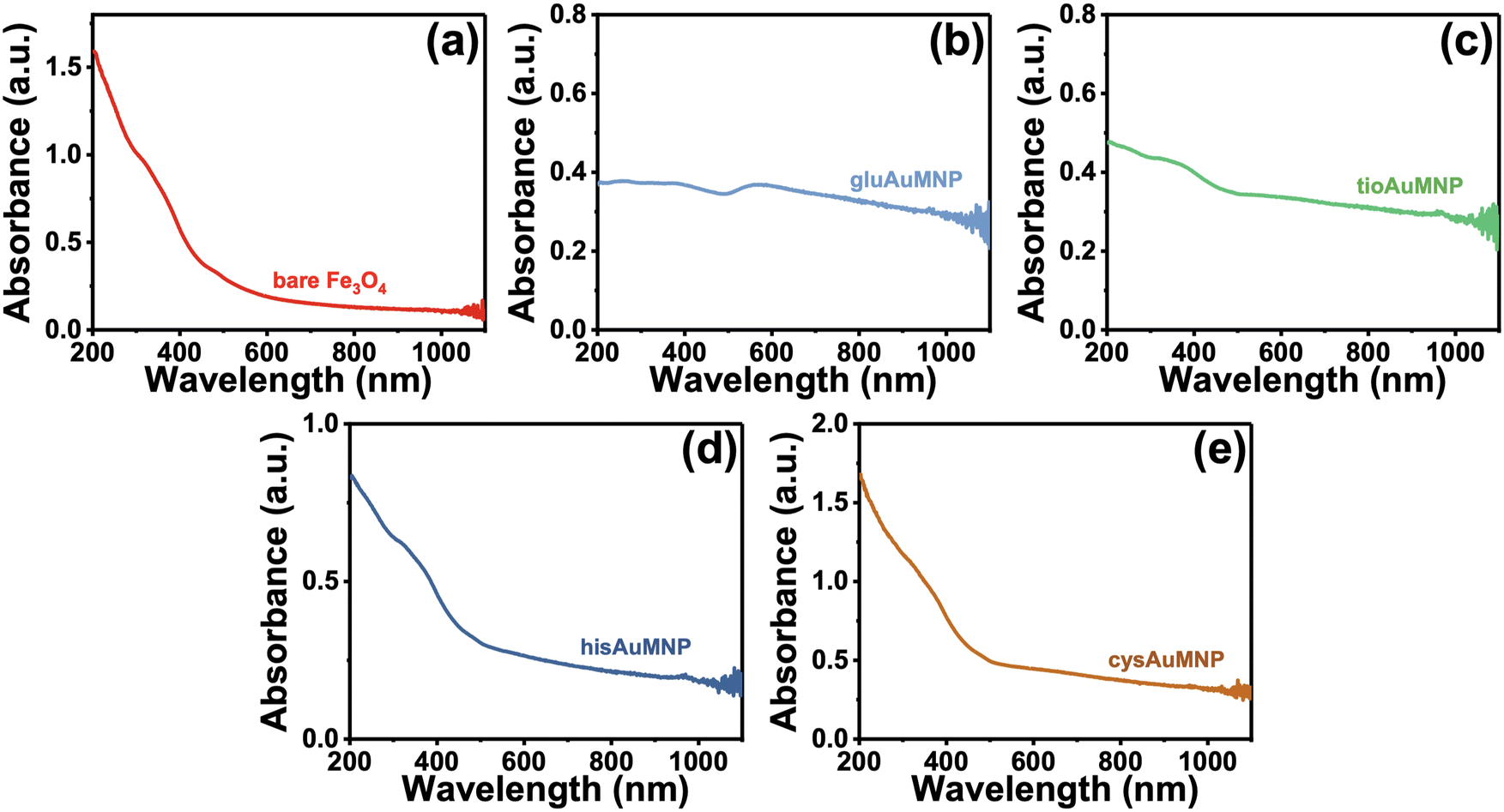 | ||
| Fig. 1 UV-vis spectra of all synthesized nanoparticles from 200–1100 nm. (a) Citrate-capped Fe3O4 nanoparticles synthesized demonstrating strong absorbance at 200–700 nm. (b) Glutathione-capped AuFe3O4 nanoparticles (gluAuMNP) demonstrating broad absorbance from 200–1000 nm with a general background increase and a short, broad peak appearing at ∼550 nm. (c) Tiopronin-capped AuFe3O4 nanoparticles (tioAuMNP) with broad absorbance from 200–1000 nm with a small shoulder at ∼960 nm and general background increase. (d) Histidine-capped AuFe3O4 nanoparticles (hisAuMNP) with an absorbance spectrum similar to that of (a), with a slight increase in background. (e) Cysteamine-capped AuFe3O4 nanoparticles (cysAuMNP) with an absorbance spectrum similar to that of (a) and large background increase. | ||
To determine information on the organic ligands attached to the nanoparticles synthesized, attenuated total reflectance IR (ATR-IR) spectroscopy was used. Vibrational spectroscopy techniques were chosen due to their ability to confer bond-specific information of the organic molecules that would be adsorbed to the nanoparticle surface. The ATR-IR spectrum of the Fe3O4 (Fig. 2a) demonstrates four sets of unique peaks visible in the functionalized AuMNP as well: (1) 535 cm−1 due to Fe–O stretching of the Fe3O4 nanoparticle, (2) 1048 cm−1 and 1071 cm−1 from C–O–C stretching of the citrate ligand, (3) 1389 cm−1 and 1589 cm−1 from Fe–COO− symmetric and asymmetric stretching, and (4) 3304 cm−1 due to O–H stretching from the citrate ligand. An annotated ATR-IR spectrum can be found in Fig. S2,† alongside the corresponding Table S2† for vibrational frequency assignments of the Fe3O4. Vibrational spectra were also collected for all analytes alongside vibrational frequency calculations for peak assignments and the spectrum of the analyte in bulk for comparison. The data for all the ligand-dependent AuMNP vibrational spectra can be found in Fig. 2b–d and annotated spectra of these nanoparticles can be found in Fig. S3–S6† alongside their corresponding tables for vibrational frequency assignments (Tables S3–S6†). Generally, across all analytes, the strong and narrow Fe–O stretch at 535 cm−1 is distinguishable in both AuFe3O4 and Fe3O4 products, allowing for the easy identification of Fe3O4 product. This Fe–O stretch is known to be specific to Fe3O4 magnetite samples, and not present in Fe2O3 maghemite. Additionally, vibrational stretches due to the organic ligand attached to the nanoparticle surfaces are present in every analyte allowing for easy characterization of the surface of the nanoparticle. For example, the gluAuMNP (Fig. 2b) has ten distinguishing peaks characteristic of the glutathione ligand attached to the AuMNP surface, with a majority being in the fingerprint region (500–1400 cm−1) of the IR spectrum allowing for unique characterization even when impurities arise in the solution. While the ATR-IR was effective in determining the characterization of organic ligands (i.e. glutathione, cysteamine, histidine, or tiopronin, on the surface of the AuMNP) it was unable to detect the presence of the C. canadensis extract due to various confounding factors. Firstly, the C. canadensis extract was a dilute solution in water and very little could be dried to obtain a spectrum. Additionally, the extract only demonstrated peaks that could be easily obscured with water, as the singular distinguishing peaks of a polyphenol solution would be aryl hydrogen stretching/bending and alkene stretching modes which are not strong and distinguishable peaks given the overwhelming presence of water. Therefore, a final modality of vibrational characterization was used: Raman spectroscopy.
 | ||
| Fig. 2 ATR-IR spectra of all synthesized nanoparticles alongside spectra of the bulk powdered form of their capping agent. (a) Bare Fe3O4 nanoparticles showing three primary distinguishing characteristics: a strong and narrow band at 535 cm−1, two medium and narrow bands at 1389 cm−1 and 1589 cm−1, and one medium and broad band at 3304 cm−1. (b) Glutathione-capped AuFe3O4 spectrum overlayed with the spectrum of powdered L-glutathione. The major 533 cm−1 band from Fe–O indicates Fe3O4 presence; nine bands from glutathione indicate glutathione presence on the AuMNP. A lack of the 2521 cm−1 medium and narrow band from the glutathione powder is absent in the gluAuMNP and indicates the presence of an Au–S bond, as 2521 cm−1 is due to ν(S–H). (c) Tiopronin-capped AuFe3O4 spectrum overlayed with the spectrum of powdered tiopronin. The 535 cm−1 is due to Fe–O, and four distinct tiopronin vibrational modes are present in the tioAuMNP spectrum. The lack of the 2528 cm−1ν(S–H) weak and narrow band indicates the presence of an Au–S bond. (d) Histidine-capped hisAuFe3O4 spectrum overlayed with the spectrum of powdered L-histidine. The presence of the 533 cm−1 band indicates Fe3O4 presence, and there exist three bands from L-histidine in the hisAuFe3O4 spectrum indicating its presence on the nanoparticle. (e) Cysteamine-capped cysAuFe3O4 spectrum overlayed with the spectrum of powdered cysteamine. The 536 cm−1 band indicated Fe3O4 presence, and the absence of the 2075 cm−1 strong and narrow band indicates the presence of an Au–S bond as 2075 cm−1 is due to ν(S–H). | ||
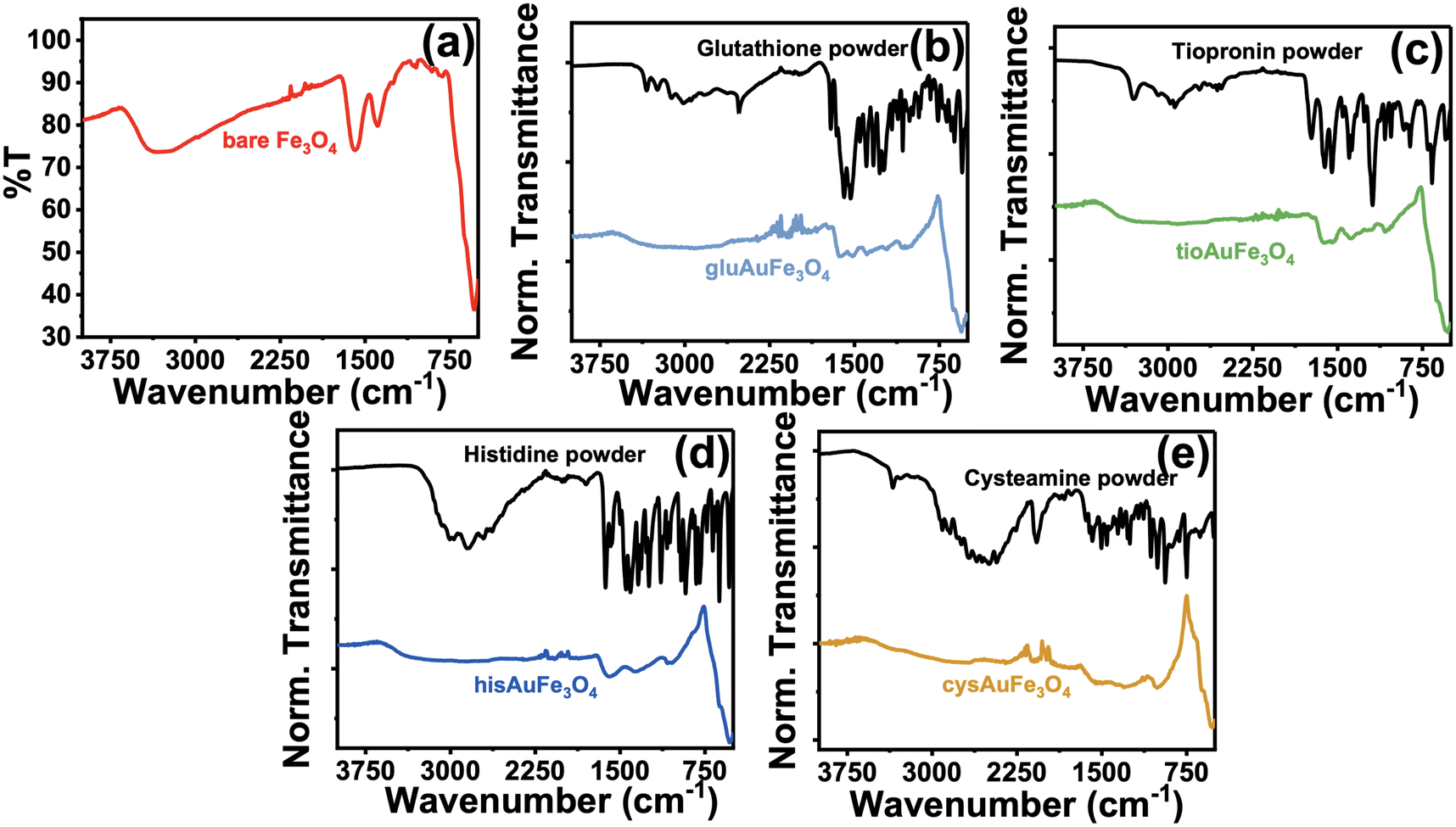
Raman spectroscopy allows for the unique and accurate identification of known compounds, with its primary disadvantage being that the peaks identified are very difficult to trace back to a single pure product unless that product's pure spectrum is known and annotated. However, the biggest advantage for the characterization of C. canadensis extract is that water does not have any significant Raman peaks in the liquid phase, allowing for the characterization of extract peaks in the absence of other obfuscating vibrational modes. In Fig. 3, a compiled Raman spectrum of all synthesized nanoparticle systems as well as the C. canadensis extract is shown. Due to the Raman instrument being portable, it is unable to filter contributions from the incident 785 nm excitation laser and therefore some peaks have major contributions from incident excitation. For clarity, the laser background is also provided; the spectrum was obtained without any sample loaded to only measure the contributions from the laser itself. As is evident in Fig. 3a, the C. canadensis extract has six characteristic bands at 419 cm−1, 562 cm−1, 667 cm−1, 768 cm−1, 1082 cm−1, and 1298 cm−1. Additional peaks from the organic ligands are also annotated on the spectrum. A region of interest spectrum over the 419 cm−1 and 667 cm−1 peaks is shown in Fig. 3b, demonstrating that in gluAuMNP, hisAuMNP, and cysAuMNP there exists a prominent band produced by the plant extract centered around 667 cm−1. The tioAuMNP demonstrates a similar band to a lesser extent and the laser background and Fe3O4 nanoparticles minimally scatter at this wavenumber. The 419, 667, and 1300–1350 cm−1 bands in the gluAuMNP, hisAuMNP, and cysAuMNP spectra as well as in the plant extract, hints to the presence of plant extract in the nanoparticle samples. The band around 610–630 cm−1 and 1324–1327 cm−1 are typically attributed to the presence of anthocyanins in violet-colored plants.36 A full set of Raman spectra with and without background subtraction can be found in Fig. S7.† From these results, it's clear that the plant extract not only has reducing capacity, as indicated by the anthocyanin content assay, but also associated strongly enough with the AuFe3O4 nanoparticles to not be washed away during centrifugation and purification steps. These results are strengthened by the experimental observation that upon addition of plant matter, the resuspension of Fe3O4 becomes far easier than in lieu of the additive. Due to the difficulty of resuspending Fe3O4 in water, many groups typically utilize some polymeric ligand, commonly cationic ones such as cetyltrimethylammonium bromide or sodium dodecyl sulfate, in order to aid in the resuspension and make the process more reproducible and user-friendly.37 Polymeric dispersants such as those previously mentioned increase stability of the final product and can open doors to later functionalization, such as with the four bioactive compounds currently described. Unfortunately, these cationic dispersants, and in fact many other dispersants as well, demonstrate considerable ecotoxicity.38–40 While non-cationic dispersants exist as well, the two available classes for water-soluble Fe3O4 dispersants are low molecular weight and charged molecules, or high molecular weight and uncharged molecules, both of which are incompatible with biological cells.41 Therefore, substitution of this dispersant with plant matter, and then functionalization of the nanoparticles with biocompatible ligands, garners potential biocompatibility for future biological applications. While the presence of ligand and plant matter on the nanoparticles was confirmed spectroscopically, the inability of the vibrational analyses to quantify the size and magnetic strength of the nanoparticles warranted detailed magnetization measurements to verify the successful synthesis of superparamagnetic Fe3O4 nanoparticles.
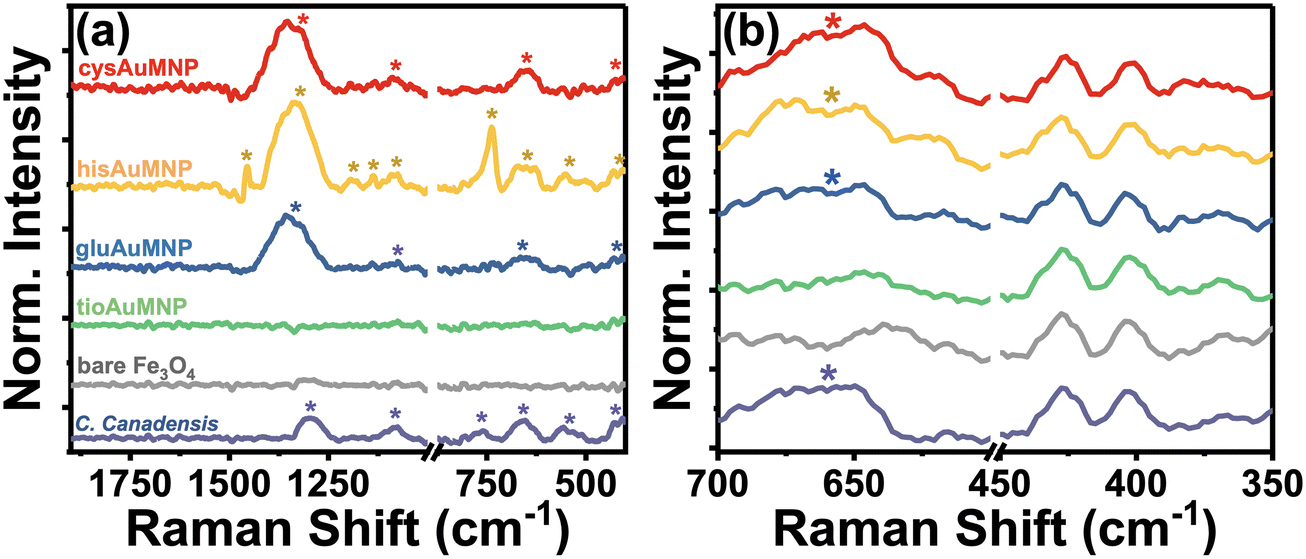 | ||
| Fig. 3 Raman spectra of all synthesized nanoparticles alongside the C. canadensis plant extract and the instrument's laser background. (a) Overlayed Raman spectra of the full window (1900–400 cm−1) for each analyte demonstrate three common bands: one centered at 419 cm−1, one centered at 667 cm−1, and one centered around 1300–1350 cm−1. A region of interest over both bands is shown (b) to demonstrate the absence or presence of those peaks in each sample. The 419, 667, and 1300–1350 cm−1 bands are all clearly present in the gluAuMNP, hisAuMNP, and cysAuMNP samples that are originating from C. canadensis. | ||
Magnetization data
After synthesizing the Fe3O4 and various AuMNP, magnetization data was collected for the bare Fe3O4 and the AuMNP. Also, hysteresis measurements were conducted on the sample to determine any potential superparamagnetic behavior of the nanoparticle. In Fig. 4a, a comparison of all the magnetic saturation measurements from the bare Fe3O4 and AuMNP is shown with their maximal magnetic moments reported at both 50 K and 300 K. Ubiquitously, the magnetic saturation of the products decreases by, on average, 13.2 ± 1.4% when the sample is heated from 50 to 300 K. For detailed graphical data of the magnetic saturation as a function of temperature, refer to Fig. S8a–e.† Hysteresis measurements were also conducted, and no hysteresis was seen in any of the samples across both Fe3O4 and AuFe3O4 products (Fig. 4b). Fe3O4 nanoparticles typically show hysteresis at diameters >20 nm.42 As nanoparticle diameter decreases below 10 nm, the Fe3O4 nanoparticle exhibits one magnetic domain for the entire nanoparticle and thus becomes superparamagnetic.43 From the magnetic hysteresis curves, it can be deduced that the synthesized nanoparticles exhibit potential superparamagnetic effects. To determine superparamagnetism, the Langevin function was used to demonstrate the agreement between the hysteresis curve and the ideal superparamagnetic behavior (Fig. S9a–e†). Langevin dynamics are typically used to determine superparamagnetic behavior in a sample,44 where the closer the experimental data agrees with the ideal Langevin the more likely the nanoparticle is to be superparamagnetic. For all nanoparticles synthesized, superparamagnetism is demonstrated by the close agreement between the experimental data and the Langevin fits. It is notable that even if size differences exist between the AuMNP and the original Fe3O4 template, the difference will only be due to added Au, and therefore if the Fe3O4 template is superparamagnetic it will make the AuMNP superparamagnetic as well. Additionally, as expected, magnetic saturation only decreases when Au is added to the structure. For example, the hisAuMNP (38 emu g−1 at 50 K) has the highest magnetic saturation of all AuFe3O4 products and only retains a little more than half of the magnetic saturation of the bare Fe3O4 (68 emu g−1 at 50 K). The magnetic saturation maintains high values all throughout the syntheses with the lowest value obtained being that of cysAuMNP at 27 emu g−1 when cooled to 50 K and 24 emu g−1 at 300 K. Overall, the Fe3O4 synthesized reaches roughly 68% of the theoretical maximum magnetic saturation for bulk Fe3O4 (100 emu g−1).45 Previous groups have shown that the magnetic saturation of the Fe3O4 nanoparticles decreases proportionally with size and nanoparticles of Fe3O4 typically have 45–80 emu g−1 magnetic saturation.46–48 This variance in magnetic saturation post-synthesis warranted further investigation into the amount of Au present per particle. To determine Au loading on the AuFe3O4 structure, imaging and elemental data were necessary.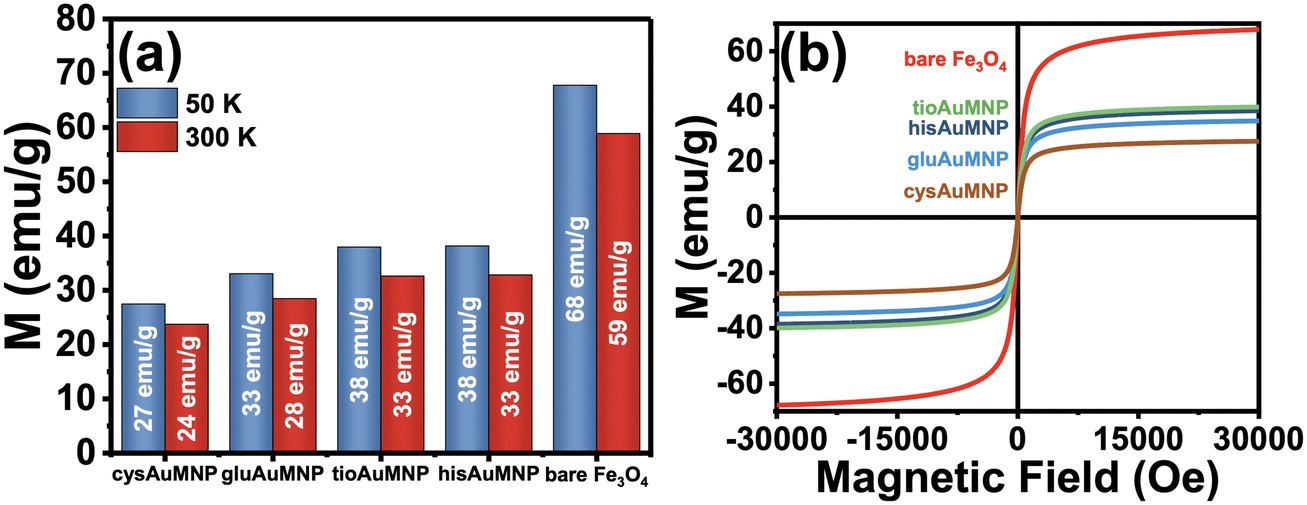 | ||
| Fig. 4 Magnetic saturation measurements of the Fe3O4 and various AuMNPs. A bar chart (a) showing the magnetic saturations at 50 K and 300 K for each nanoparticle synthesized is shown alongside the magnetic hysteresis curves (b) for all the nanoparticles at 50 K. No magnetic hysteresis can be seen, and the nanoparticles generally lose a small fraction of their magnetic saturation as the temperature increases due to expected temperature dependence of the saturation magnetization of a ferromagnetic material. | ||
Nanoparticle imaging
To determine the accurate size dimensions of the nanoparticles synthesized, a scanning-transmission electron microscope (STEM) was used. Representative STEM images are compiled in Fig. 5, with additional STEM images for each sample in the ESI† (Fig. S10–S14). From the STEM images, one can see roughly spherical dimensions for most nanoparticles synthesized as well as sub-10 nm dimensions for the Fe3O4 indicating the possibility for superparamagnetic nanoparticles to have been formed.49 Interestingly, for the cysAuMNP and hisAuMNP, anisotropic structures seem to form in high-yield which typically only occur when particle sizes are larger than 100 nm. This data is further corroborated by histograms collected from the STEM images (Fig. 6), where one can see that despite the Fe3O4 being 8.3 ± 2.5 nm in diameter (Fig. 6a), the cysAuMNP (Fig. 6b) and hisAuMNP (Fig. 6c) are much larger with an average diameter of 29.5 ± 20.0 nm and 130.2 ± 33.2 nm, respectively. The other two nanoparticles, gluAuMNP (Fig. 6d) and tioAuMNP (Fig. 6e) have size dimensions of 8.6 ± 5.6 nm and 14.3 ± 4.6 nm, respectively, showing sizes similar to the original Fe3O4 core template. From this data, one can see that cysAuMNP and hisAuMNP would have anisotropic sizes, as anisotropic shapes are formed more easily from larger nanoparticles.50 Additionally, from this data, one would expect larger nanoparticles to have more Au loading on the Fe3O4, with gluAuMNP having the smallest percentage of Au due to its small diameter difference from the Fe3O4, and hisAuMNP having the largest percentage of Au loading as it has the largest diameter. To verify the Au loading and the composition of the particles in general, energy-dispersive X-ray (EDX) spectroscopy was used. | ||
| Fig. 5 STEM images of all nanoparticles synthesized. (a) Bare Fe3O4, (b) cysAuMNP, (c) gluAuMNP, (d) hisAuMNP, and (e) tioAuMNP. All images were obtained in bright-field mode on a STEM3+ detector at 30 kV accelerating voltage and 13–50 pA of current. | ||
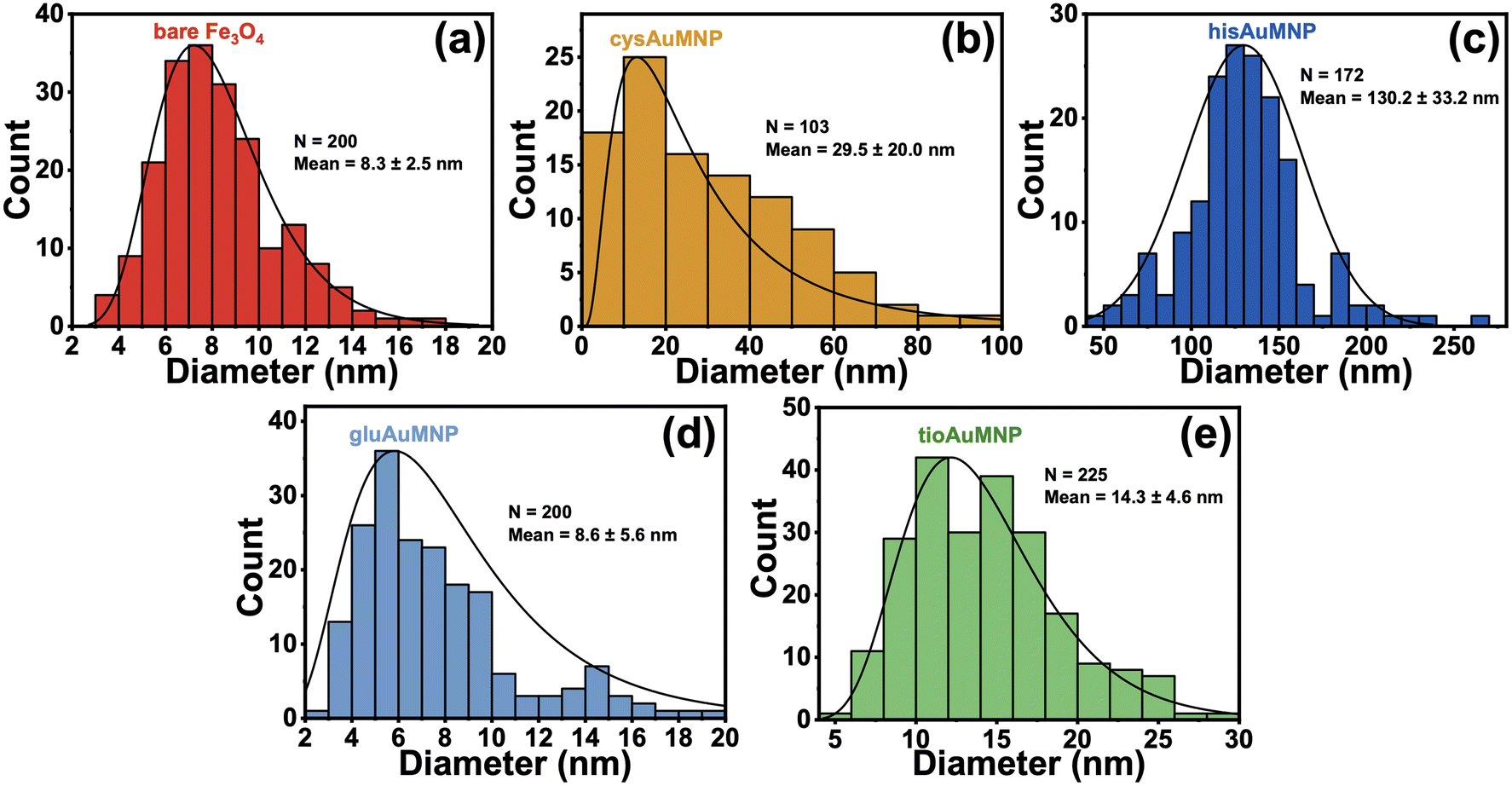 | ||
| Fig. 6 Histograms obtained from STEM micrographs of the various synthesized nanoparticles. The histograms a–e correspond to: (a) bare Fe3O4, (b) cysAuMNP, (c) hisAuMNP, (d) gluAuMNP, and (e) tioAuMNP. All size distributions were completed using ImageJ with a minimum of 100 particles and a maximum of 225 particles per histogram. | ||
EDX data was obtained using a scanning electron microscope (SEM) to achieve high-resolution elemental data that cannot be achieved using the STEM setting in the SEM. Per the histograms, the EDX data (Table 1) shows that hisAuMNP has the highest Au loading per AuMNP particle, and gluAuMNP has the lowest Au loading. Interestingly, cysAuMNP has a lower Au loading than tioAuMNP despite having a larger size. However, this can be reasoned through attention to the standard deviation in sizes for both particles. The cysAuMNP has a far larger spread than the tioAuMNP, with a relative standard deviation (RSD) of 67.8%, while the tioAuMNP has an RSD of 32.2%, indicating a much smaller spread of sizes. Accordingly, although the mean of the cysAuMNP nanoparticle dimensions is larger, the variability is also larger, and the histogram is skewed left towards the 10–20 nm size regime owing some explanation as to the smaller Au loading value. It should additionally be noted that EDX is a semi-quantitative measurement of elemental percent presence,51 and therefore the results confidently demonstrate the presence of Au, but less confidently predict the true percentage of Au in the nanoparticles. Given these variable Au loading percentages, the particles were then tested for their abilities to generate surface-enhanced Raman scattering (SERS) on a common Raman reporter molecule, para-nitrothiophenol (p-NTP).
| Element | Bare Fe3O4 | cysAuFe3O4 | tioAuFe3O4 | hisAuFe3O4 | gluAuFe3O4 |
|---|---|---|---|---|---|
| Average %Au | — | 0.50% | 4.07% | 2.95% | 1.09% |
| Average %Fe | 18.25% | 6.20% | 12.29% | 8.16% | 14.55% |
| Average %O | 49.41% | 31.23% | 50.00% | 39.88% | 44.66% |
| Au:Fe ratio |
— | 0.081:1 |
0.330:1 |
0.360:1 |
0.075:1 |
SERS detection of p-NTP
The ability of plasmonic nanoparticles to enhance Raman spectral signatures becomes vital in chemical detection at low milli- and micromolar concentrations. Due to the loading of these nanoparticles with Au, the AuMNP should in theory be plasmonic and thus be well-suited for a SERS detection methodology. To test this hypothesis, we decided to implement the widely used SERS reporter p-NTP.52–54 SERS detection typically involves p-NTP detection as a positive control due to the fact that as an organothiol p-NTP, can bind directly to the nanoparticle allowing the user simplicity in testing the potential enhancement factor of their nanoparticle. For this reason, we decided to test all four AuMNP with p-NTP, as well as the bare Fe3O4 as a negative control. We additionally tested Au nanoparticles (AuNP) synthesized through a modified Turkevich protocol. The data compiled in Fig. 7 shows the results of this experiment, whereby hisAuMNP and cysAuMNP demonstrated SERS enhancement of the p-NTP signal. For the complete Raman spectral data, see Fig. S15.† One can see that for the fingerprint peak of the analyte at 1567 cm−1, Fe3O4 does not enhance the signal as was expected, and gluAuMNP as well as tioAuMNP additionally demonstrate no enhancement. However, cysAuMNP and hisAuMNP are able to enhance the signal of the 50 μM p-NTP by 19.5 ± 1.6x and 15.0 ± 2.4x, respectively, demonstrating an ability of cysAuMNP and hisAuMNP to exhibit SERS enhancements. As the trend follows the size of the AuMNP, but does not follow the Au loading percentages, we posit that the difference is due to the ligands bound to the nanoparticle.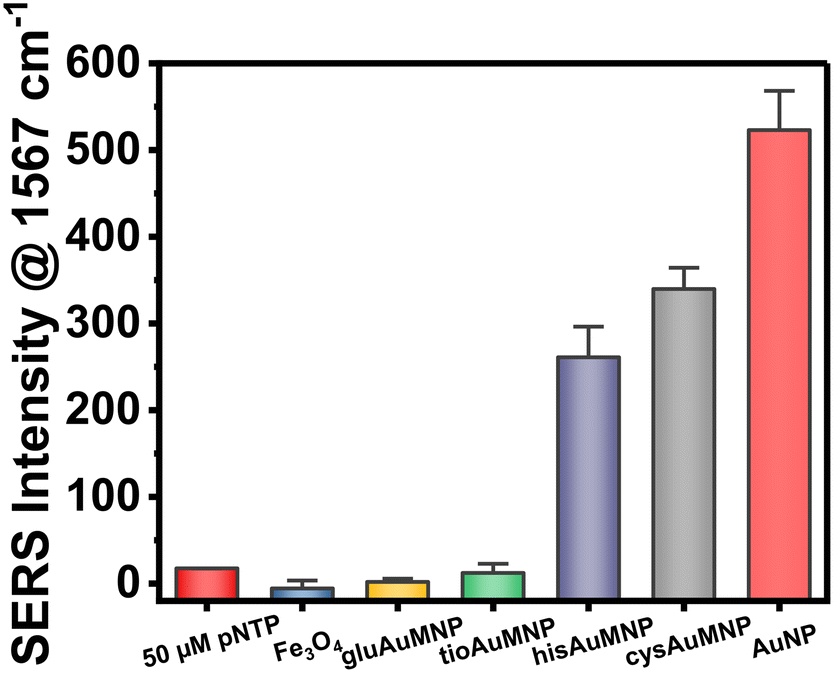 | ||
| Fig. 7 A bar chart depicting the Raman/SERS intensity from the 50 μM p-NTP on its own or when added to a solution of nanoparticle. The adsorption of p-NTP to the cysAuMNP, hisAuMNP, and AuNP were the only cases in which positive SERS enhancement was demonstrated, with AuNP having the highest increase overall. All measurements were completed in triplicate. | ||
It is well understood that for chemical-based SERS enhancement to occur, the ligand on the surface of the nanoparticle must first be displaced.55,56 A typical method for this is adding NaBH4 to remove all organothiols on the surface of the nanoparticle. However, in adding NaBH4, off-site reactions are common, and the reproducibility and simplicity of the protocol are both diminished. Therefore, in our technique, we chose to attempt SERS of the nanoparticle without the addition of any potentially harmful and non-specific molecular adsorbate removal techniques, as well as without the addition of any aggregating agents which would also hurt the reproducibility of the methodology. Thus, we hypothesize that the ability for cysAuMNP and hisAuMNP to provide SERS enhancement over the tioAuMNP and gluAuMNP is simply due to the ability for cysteamine and histidine to be removed from the surface of the nanoparticle after p-NTP is introduced. The ability for histidine to be a better leaving group than tiopronin and glutathione can very easily be rationalized, as it is the only ligand from the four used that does not have a thiol group on it, and thus, would have a less stable interaction with the Au than an organothiol.57 This means that when the p-NTP is introduced, the histidine is most likely very easily displaced by the thiolate group on the analyte, and thus we see SERS in this case. In the case of cysteamine, more rationalization is needed due to it containing a thiol group which would seemingly bind very stably with Au. However, when assessing the difference in pKa between cysteamine and the aromatic organothiol p-NTP, one can see that there exists a large difference between cysteamine's thiol group with a pKa of 8.2 (ref. 58) and p-NTP's thiol group with a pKa of 4.4 (ref. 59) where the latter has a far more acidic thiol. While contested by some groups,60 it has been shown previously that thiolated ligands bind to Au in the negatively charged thiolate form61,62 supporting the hypothesis that p-NTP would have a better interaction than cysteamine due to its readily acidic thiol group stabilized by the adjacent phenyl ring and aromaticity of the molecule. While the thiol groups present on glutathione and tiopronin would have similar pKa's to that of cysteamine, both are very bulky ligands, especially when compared to cysteamine and would thus have a much lower rate of substitution with p-NTP than cysteamine. Therefore, cysteamine and histidine seem to be the two rational candidates for chemical SERS enhancement. Furthering this rationality, we ascribe the differing surface ligands to why the AuNP would have the highest SERS enhancement compared to the AuMNPs. As the only ligand attached to the surface of the AuNP is the C. canadensis extract, it would be easy to displace these non-thiolated (anthocyanins and polyphenols) ligands with a thiolated p-NTP. While the Fe3O4 on its own is not necessarily plasmonic, under various conditions it has been shown that Fe3O4 can generate a plasmon resonance and is not a strong dampener, therefore no decrease in SERS enhancement should be seen between AuMNP and AuNP for any reason other than ligand adsorption strength. This would mean that AuMNP would theoretically be as good a SERS substrate as AuNP with far more applications possible with the added magnetic functionality. This difference in binding strength between phenol groups and thiol groups has been shown previously,63 verifying that phenol displacement would more energetically undemanding than thiol displacement. To verify that p-NTP was bound by the thiol group to the AuMNP, the 1080–1120 cm−1 can be studied. When bound to nanoparticles, the 1090 cm−1 band due to C–S stretching splits into two peaks: one at 1109 cm−1 and one at 1087 cm−1 wavenumbers.64 This phenomenon is visible in the AuNP, hisAuMNP, and cysAuMNP (Fig. S16†), as well as to a smaller extent in the tioAuMNP. It is interesting that tiopronin, which seems to be less charged and less bulky in aqueous solutions, also has some number of interactions with the p-NTP. From this data we can verifiably assert that cysAuMNP and hisAuMNP are able to interact with and enhance the signal of organothiols, thereby opening doors to future SERS detection with these magnetic nanoparticles. To fully quantify the potential for these nanoparticles to enhance the SERS spectrum, enhancement factor calculations were undertaken.
As SERS signal increase alone is not a reliable way to quantify the potential for a nanoparticle to enhance a Raman signal, quantifiable methods such as “enhancement factor” are typically used instead.65 While large variability exists in the available enhancement factors for a chemical enhancement methodology, the numbers typically fall in the 10–107 fold enhancement range.62 The most common way to report enhancement factor is by using the equation EF = (NRaman/NSERS)(ISERS/IRaman). Calculating the EF for both cysAuMNP and hisAuMNP allowed us to determine the values to be 7.73 × 103 and 5.14 × 102, respectively. For detailed descriptions on the calculation and methodology used, refer to Section S1 of the ESI.† The compiled data for Au loading, particle diameter, magnetization, and EF can be found in Table 2. Further efforts were taken to conceptualize this EF in terms of the instrument's limit of detection (LOD). For p-NTP alone, the portable Raman was found to have an LOD of roughly 566.5 μM (Fig. S16a†). For the cysAuMNP, the LOD of p-NTP (Fig. S17b and c†) was found to be 5.42 μM, a 102-fold decrease. With the theoretical substrate-based EF of 7.73 × 103, the LOD should be 73.3 nM, therefore the experimental EF for the nanoparticle seems to be in the range of 102–103 and represents a high enhancement for a nanoparticle with low Au loading. It should be noted that the EF utilized is not an analytically-based calculation, rather it is the conventional EF calculation that is based only on the analytes bound to the surface of the nanoparticle, assuming a core–shell structure of the AuFe3O4. Previous AuFe3O4 nanoparticles of similar sizes have demonstrated EF values of 104–106. While the nanoparticles synthesized have lower EF and magnetic saturation than those in literature, our methodology uses farless ecologically toxic chemicals (Table 3). Due to the high magnetic saturation of the AuFe3O4, and the ability for aggregating nanoparticles to increase SERS through the generation of interparticle hotspots,66 a magnet could be used to aggregate a large mass of AuFe3O4 together and generate a higher SERS signal.
| Bare Fe3O4 | cysAuMNP | tioAuMNP | hisAuMNP | gluAuMNP | |
|---|---|---|---|---|---|
| %Au in Fe3O4 | — | 3.69% | 14.65% | 15.95% | 3.21% |
| Particle diameter (nm) | 8.3 ± 2.5 | 29.5 ± 20.0 | 14.3 ± 4.6 | 130.2 ± 33.2 | 8.6 ± 5.6 |
| Magnetization@50 K (emu g−1) | 68 | 27 | 38 | 38 | 33 |
| Magnetization@300 K (emu g−1) | 59 | 24 | 33 | 33 | 28 |
| Au adjusted magnetization@50 K (emu g−1) | 68 | 28 | 45 | 45 | 36 |
| Enhancement factor | N/A | (7.73 ± 0.6) × 103 | N/A | (5.14 ± 0.8) × 102 | N/A |
| Substrate | Enhancement factor | Magnetic saturation | Reagents | Related references |
|---|---|---|---|---|
| Na3Cit = trisodium citrate, PVP = polyvinylpyrrolidone, PEI = polyethylenimine, NaAc = sodium acetate, PAA = polyacrylic acid, CTAB = cetyltrimethylammonium bromide, * = reagent of ecological concern. | ||||
| AuFe3O4 nanorods | 6 × 104 | — |
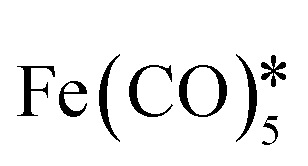 , hexadecylamine*, 1-octanol, oleic acid, , hexadecylamine*, 1-octanol, oleic acid, 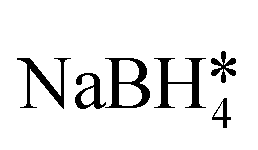 , HAuCl4, PVP, K2CO3 , HAuCl4, PVP, K2CO3 |
67 |
| Core–satellite AuFe3O4 | ∼3 × 105 | — | FeC6H7O7·NH4OH*, PAA, N2H4·H2O*, 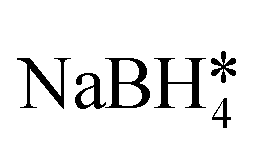 , CTAB* , CTAB* |
68 |
| Core–shell AuFe3O4 | 102–103 | 27–38 emu g−1, superparamagnetic | FeCl2/FeCl3, Na3Cit, NH3OH*, HAuCl4, organic plant matter, cysteamine, L-glutathione, L-histidine, tiopronin | This work |
| Au@Ag–Fe3O4@Au | 2 × 106 | ∼40 emu g−1, superparamagnetic | HAuCl4, PVP, NH2OH*, PEI*, Na3Cit, AgNO3 | 15, 69 |
| Fe3O4/Au nanocomposites | — | ∼50 emu g−1, superparamagnetic | FeCl3, ethylene glycol*, Na3Cit, NaAc, HAuCl4, 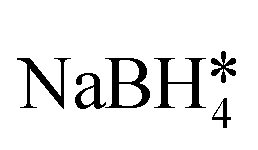 |
70 |
| AuFe3O4 dumbbells | — | 80 emu g−1, superparamagnetic |
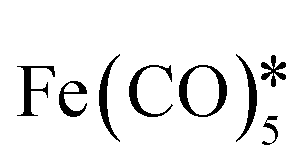 , octadecene, oleic acid, HAuCl4, oleylamine* , octadecene, oleic acid, HAuCl4, oleylamine* |
71 |
While aggregation-based SERS does increase signal response drastically (Fig. 8, full spectra in Fig. S18†), the disadvantage of this technique when compared to conventional liquid-phase or solid-substrate based SERS is the semi-quantitative nature of the technique. Upon aggregation, the lack of control of the distance between nanoparticles and the amount of particle gathered in the aggregate leads to irreproducibility in the technique, making it semi-quantitative at best unless efforts are taken to increase reproducibility.72 However, by simply aggregating the nanoparticles with a magnet one is able to detect the analyte far more sensitively. For cysAuMNP, the signal increases by 22.1 ± 9.6x; for hisAuMNP, the signal increases by 37.6 ± 3.7x. Therefore, by simple magnetic concentration of the nanoparticles to one area, one can quickly aggregate the nanoparticles for qualitative detection of an analyte, and then quantify its presence either using a solid-substrate or liquid-phase sample.
 | ||
| Fig. 8 Aggregation-based SERS of the AuMNP nanoparticles. The labels with asterisks indicate the SERS signal received after aggregation of the AuMNP sample. From left to right, the samples are: 50 μM p-NTP Raman, Fe3O4 liquid-phase SERS (Fe3O4SERS) and Fe3O4 aggregated SERS (Fe3O4SERS*), gluAMNP liquid-phase SERS (gluSERS) and then aggregated SERS (gluSERS*), tioAuMNP liquid-phase SERS (tioSERS) and aggregated SERS (tioSERS*), cysAuMNP liquid-phase SERS (cysSERS) and aggregated SERS (cysSERS*), and lastly hisAuMNP liquid-phase SERS (hisSERS) and aggregated SERS (hisSERS*). | ||
Conclusions
The creation of active plasmonic substrates for chemical sensing and the creation of magnetic nanoparticles for a variety of applications have historically occupied two distinct fields. In this work, we demonstrate the synthesis of both plasmonic and magnetic AuFe3O4 nanoparticles functionalized with four bioactive ligands for applications in chemical sensing using surface-enhanced Raman scattering (SERS). The novelty of this work lies in the simplicity of the protocol, both in its ability to reduce the necessity for harmful surfactants by instead using metabolites from the Cercis canadensis flower, as well as being able to create hybrid nanoparticles that retain magnetism while enabling SERS enhancement in the liquid phase. The metabolites of the C. canadensis flower are shown to interact with the nanoparticles and experimentally demonstrate an ability to substitute polymeric dispersants typically used during the resuspension of Fe3O4. With a method utilizing this locally sourced plant extract, we were able to create superparamagnetic nanoparticles with magnetic saturation values of >30 emu g−1 while maintaining SERS enhancement factors on the order of 102–103 for enhanced organothiol detection. From the four bioactive ligands tested, tiopronin- and histidine-stabilized AuFe3O4 allowed for the highest magnetic saturation; cysteamine- and histidine-stabilized AuFe3O4 allowed for the highest SERS enhancement factors. Typically, for chemically enhanced SERS, enhancement factors fall within the range of 10–107, therefore in the scope of available materials for interfacial chemical analysis our hybrid heterometallic nanoparticle is comparable with monometallic Au or Ag nanoparticles. Further improvements must be made in characterizing the biological compatibility of these biomaterial-capped AuFe3O4 to determine the role of both the small-molecule bioactive ligands and the adsorbed plant matter. This facile synthesis can aid in the investigation of compounds at the metal–air or metal–liquid interface allowing for a broad array of potential applications such as magnetic separation and analysis of target compounds.Data availability
All data can be found within the manuscript and ESI† file.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Rigaku Corporation for donating the Raman spectrometer to our lab. This work was supported by the TU Foundation Grants, the FCSM Undergraduate Research Grants, NSF MRI 1626326, DARPA AMEBA HR0011-17-C-0115. The authors thank Dr. Shiva Pokhrel for helping with magnetization measurements.References
-
R. Feynman, in Feynman and computation, CRC Press, 2018, pp. 63–76 Search PubMed
.
- S. Manipatruni, D. E. Nikonov, C.-C. Lin, T. A. Gosavi, H. Liu, B. Prasad, Y.-L. Huang, E. Bonturim, R. Ramesh and I. A. Young, Nature, 2019, 565, 35–42 CrossRef CAS PubMed
- S. Manipatruni, D. E. Nikonov, C.-C. Lin, B. Prasad, Y.-L. Huang, A. R. Damodaran, Z. Chen, R. Ramesh and I. A. Young, Sci. Adv., 2018, 4, eaat4229 CrossRef CAS PubMed
- S. Manipatruni, D. E. Nikonov and I. A. Young, Nat. Phys., 2018, 14, 338–343 Search PubMed
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS PubMed
- S. S. P. Parkin, N. More and K. P. Roche, Phys. Rev. Lett., 1990, 64, 2304–2307 CrossRef CAS PubMed
- E. E. Fullerton, D. T. Margulies, M. E. Schabes, M. Carey, B. Gurney, A. Moser, M. Best, G. Zeltzer, K. Rubin, H. Rosen and M. Doerner, Appl. Phys. Lett., 2000, 77, 3806–3808 CrossRef CAS
- L. Zhu, Z. Zhou, H. Mao and L. Yang, Nanomedicine, 2016, 12, 73–87 CrossRef PubMed
- G. S. Parkinson, Surf. Sci. Rep., 2016, 71, 272–365 CrossRef CAS
- M. Khan, F. Ahmad, J. T. Koivisto and M. Kellomäki, Colloid Interface Sci. Commun., 2020, 39, 100322 CrossRef CAS
- P. Raveendran, J. Fu and S. L. Wallen, J. Am. Chem. Soc., 2003, 125, 13940–13941 CrossRef CAS PubMed
- S. Ying, Z. Guan, P. C. Ofoegbu, P. Clubb, C. Rico, F. He and J. Hong, Environ. Technol. Innovation, 2022, 26, 102336 CrossRef CAS
- M. S. Devadas, V. Smolyaninova, L. Krushinski, D. Aligholizadeh, K. Langford, W. Korzi, C. Miller, N. R. Kadasala, M. Zhukovskyi and E. Hondrogiannis, Langmuir, 2023, 39, 1947–1956 CrossRef CAS PubMed
- S. V. Salihov, Y. A. Ivanenkov, S. P. Krechetov, M. S. Veselov, N. V. Sviridenkova, A. G. Savchenko, N. L. Klyachko, Y. I. Golovin, N. V. Chufarova, E. K. Beloglazkina and A. G. Majouga, J. Magn. Magn. Mater., 2015, 394, 173–178 CrossRef CAS
- C. Wang, J. Wang, M. Li, X. Qu, K. Zhang, Z. Rong, R. Xiao and S. Wang, Analyst, 2016, 141, 6226–6238 RSC
- M. Rincón-Iglesias, I. Rodrigo, L. B. Berganza, E. S. A. Serea, F. Plazaola, S. Lanceros-Méndez, E. Lizundia and J. Reguera, ACS Appl. Mater. Interfaces, 2022, 14, 7130–7140 CrossRef PubMed
- Y. Zhang, Z. Ye, C. Li, Q. Chen, W. Aljuhani, Y. Huang, X. Xu, C. Wu, S. E. J. Bell and Y. Xu, Nat. Commun., 2023, 14, 1392 CrossRef CAS PubMed
- I. S. Unal, A. Demirbas, I. Onal, N. Ildiz and I. Ocsoy, J. Photochem. Photobiol., B, 2020, 204, 111800 CrossRef CAS PubMed
- J. R. Murray and W. P. Hackett, Plant Physiol., 1991, 97, 343–351 CrossRef CAS PubMed
- N. M. Hughes, C. B. Morley and W. K. Smith, New Phytol., 2007, 175, 675–685 CrossRef CAS PubMed
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed
- J. Dong, P. L. Carpinone, G. Pyrgiotakis, P. Demokritou and B. M. Moudgil, Kona Powder Part. J., 2020, 37, 224–232 CrossRef CAS PubMed
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC
- J. Turkevich, Gold Bull., 1985, 18, 86–91 CrossRef CAS
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS
- G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina, E. Guidez, T. Harville, S. Irle, J. Ivanic, K. Kowalski, S. S. Leang, H. Li, W. Li, J. J. Lutz, I. Magoulas, J. Mato, V. Mironov, H. Nakata, B. Q. Pham, P. Piecuch, D. Poole, S. R. Pruitt, A. P. Rendell, L. B. Roskop, K. Ruedenberg, T. Sattasathuchana, M. W. Schmidt, J. Shen, L. Slipchenko, M. Sosonkina, V. Sundriyal, A. Tiwari, J. L. Galvez Vallejo, B. Westheimer, M. Włoch, P. Xu, F. Zahariev and M. S. Gordon, J. Chem. Phys., 2020, 152, 154102 CrossRef CAS PubMed
- K. He, X. Li, X. Chen, X. Ye, J. Huang, Y. Jin, P. Li, Y. Deng, Q. Jin, Q. Shi and H. Shu, J. Ethnopharmacol., 2011, 137, 1135–1142 CrossRef PubMed
- H. E. Khoo, A. Azlan, S. T. Tang and S. M. Lim, Food Nutr. Res., 2017, 61, 1361779 CrossRef PubMed
- P.-V. Penelope, M. Guoying and W. Dennis, Journal of Horticultural Science and Research, 2017, 1, 13–18 CrossRef
- X. Wang, R. Niessner and D. Knopp, Sensors, 2014, 14, 21535–21548 CrossRef PubMed
- Madhavi, M. Kumar, J. R. Ansari, V. Kumar, S. Nagar and A. Sharma, Metals, 2022, 12, 2145 CrossRef CAS
- A. Azizi, J. Inorg. Organomet. Polym. Mater., 2020, 30, 3552–3561 CrossRef CAS
- Y. Sheng and J. Xue, J. Colloid Interface Sci., 2012, 374, 96–101 CrossRef CAS PubMed
- R. Ramesh, M. Geerthana, S. Prabhu and S. Sohila, J. Cluster Sci., 2017, 28, 963–969 CrossRef CAS
- S. Bruni, M. Longoni, C. Minzoni, M. Basili, I. Zocca, S. Pieraccini and M. Sironi, Molecules, 2023, 28, 1709 CrossRef CAS PubMed
- E. Amstad, M. Textor and E. Reimhult, Nanoscale, 2011, 3, 2819–2843 RSC
- A. M. B. Hansen, J. L. Brill, K. A. Connors, S. E. Belanger, A. Baun and H. Sanderson, Environ. Res., 2023, 231, 116282 CrossRef CAS PubMed
- O. Kaczerewska, R. Martins, J. Figueiredo, S. Loureiro and J. Tedim, J. Hazard. Mater., 2020, 392, 122299 CrossRef CAS PubMed
- S. O. Badmus, H. K. Amusa, T. A. Oyehan and T. A. Saleh, Environ. Sci. Pollut. Res., 2021, 28, 62085–62104 CrossRef CAS PubMed
- A. M. Weiss, M. A. Lopez, II, B. W. Rawe, S. Manna, Q. Chen, E. J. Mulder, S. J. Rowan and A. P. Esser-Kahn, Macromolecules, 2023, 56, 7286–7299 CrossRef CAS PubMed
- C. Martinez-Boubeta, K. Simeonidis, A. Makridis, M. Angelakeris, O. Iglesias, P. Guardia, A. Cabot, L. Yedra, S. Estradé, F. Peiró, Z. Saghi, P. A. Midgley, I. Conde-Leborán, D. Serantes and D. Baldomir, Sci. Rep., 2013, 3, 1652 CrossRef PubMed
- T. Vangijzegem, V. Lecomte, I. Ternad, L. Van Leuven, R. N. Muller, D. Stanicki and S. Laurent, Pharmaceutics, 2023, 15, 236 CrossRef CAS PubMed
- G. Barrera, P. Allia and P. Tiberto, Phys. Rev. Appl., 2022, 18, 024077 CrossRef CAS
-
R. M. Cornell and U. Schwertmann, The iron oxides: structure, properties, reactions, occurrences, and uses, Wiley-vch Weinheim, 2003 Search PubMed
- H. Iida, K. Takayanagi, T. Nakanishi and T. Osaka, J. Colloid Interface Sci., 2007, 314, 274–280 CrossRef CAS PubMed
- D. L. Huber, Small, 2005, 1, 482–501 CrossRef CAS PubMed
- Q. Li, C. W. Kartikowati, S. Horie, T. Ogi, T. Iwaki and K. Okuyama, Sci. Rep., 2017, 7, 9894 CrossRef PubMed
- J. Dulińska-Litewka, A. Łazarczyk, P. Hałubiec, O. Szafrański, K. Karnas and A. Karewicz, Materials, 2019, 12, 617 CrossRef PubMed
- E. Ringe, R. P. Van Duyne and L. D. Marks, J. Phys. Chem. C, 2013, 117, 15859–15870 CrossRef CAS
- M. Scimeca, S. Bischetti, H. K. Lamsira, R. Bonfiglio and E. Bonanno, Eur. J. Histochem., 2018, 62, 2841 Search PubMed
- B. O. Skadtchenko and R. Aroca, Spectrochim. Acta, Part A, 2001, 57, 1009–1016 CrossRef PubMed
- S. Juergensen, P. Kusch and S. Reich, Phys. Status Solidi B, 2020, 257, 2000295 CrossRef CAS
- P. Miao, Y. Ma, M. Sun, J. Li and P. Xu, Faraday Discuss., 2019, 214, 297–307 RSC
- S. M. Ansar, F. S. Ameer, W. Hu, S. Zou, C. U. Pittman, Jr. and D. Zhang, Nano Lett., 2013, 13, 1226–1229 CrossRef CAS PubMed
- M. A. Mahmoud, B. Garlyyev and M. A. El-Sayed, J. Phys. Chem. C, 2013, 117, 21886–21893 CrossRef CAS
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed
- E. P. Serjeant and B. Dempsey, IUPAC Chem. Data Ser., 1979, 23, 160–190 Search PubMed
- K. N. Dalby and W. P. Jencks, J. Chem. Soc., Perkin Trans. 2, 1997, 1555–1564, 10.1039/A700205J
- C. S. Weisbecker, M. V. Merritt and G. M. Whitesides, Langmuir, 1996, 12, 3763–3772 CrossRef CAS
- S. M. Ansar, R. Haputhanthri, B. Edmonds, D. Liu, L. Yu, A. Sygula and D. Zhang, J. Phys. Chem. C, 2011, 115, 653–660 CrossRef CAS
- S. M. Ansar, X. Li, S. Zou and D. Zhang, J. Phys. Chem. Lett., 2012, 3, 560–565 CrossRef CAS PubMed
- S. M. Rehn, T. M. Gerrard-Anderson, Y. Chen, P. Wang, T. Robertson, T. P. Senftle and M. R. Jones, ACS Nano, 2023, 17, 6698–6707 CrossRef CAS PubMed
- Y. Ling, W. C. Xie, G. K. Liu, R. W. Yan, D. Y. Wu and J. Tang, Sci. Rep., 2016, 6, 31981 CrossRef PubMed
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794–13803 CrossRef CAS
- M. Das, D. Gangopadhyay, R. Pelc, R. Hadravová, J. Šebestík and P. Bouř, Talanta, 2023, 253, 123940 CrossRef CAS PubMed
- L. B. Berganza, L. Litti, M. Meneghetti, S. Lanceros-Méndez and J. Reguera, ACS Omega, 2022, 7, 45493–45503 CrossRef CAS PubMed
- F. Hu, H. Lin, Z. Zhang, F. Liao, M. Shao, Y. Lifshitz and S.-T. Lee, Sci. Rep., 2014, 4, 7204 CrossRef CAS PubMed
- D. Song, R. Yang, S. Fang, Y. Liu, F. Long and A. Zhu, Microchim. Acta, 2018, 185, 491 CrossRef PubMed
- F. Yan and R. Sun, Mater. Res. Bull., 2014, 57, 293–299 CrossRef CAS
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS PubMed
- D.-B. Grys, R. Chikkaraddy, M. Kamp, O. A. Scherman, J. J. Baumberg and B. de Nijs, J. Raman Spectrosc., 2021, 52, 412–419 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00121d |
| ‡ Current address: University of Maryland Baltimore-County, Baltimore, Maryland 21250, USA. |
| This journal is © The Royal Society of Chemistry 2024 |