
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous enzyme grafting on bio-inspired scaffolds for antibacterial protection
Baptiste
Arbez
a,
Chloé
Retourney
a,
Fabienne
Quilès
 a,
Gregory
Francius
a,
Henri-Pierre
Fierobe
b and
Sofiane
El-Kirat-Chatel
*ac
a,
Gregory
Francius
a,
Henri-Pierre
Fierobe
b and
Sofiane
El-Kirat-Chatel
*ac
aUniversité de Lorraine, CNRS, LCPME, 405 Rue de Vandoeuvre, 54600 Villers-lès-Nancy, France. E-mail: sofiane.el-kirat-chatel@u-bordeaux.fr; Fax: +33-383275444; Tel: +33-372747399
bLaboratoire de Chimie Bactérienne (LCB), CNRS, Université d'Aix-Marseille, UMR7283 31 Chemin Joseph Aiguier, 13009 Marseille, France
cCBMN, CNRS UMR 5248, IPB, Université de Bordeaux, Pessac, France
First published on 20th December 2023
Abstract
Surface bacterial contamination represents a crucial health and industrial concern which requires new strategies to be continuously developed. Successful antibacterial surfaces are characterized by a combination of durable and broad-spectrum antimicrobial actions. Herein, we present a bio-inspired strategy mimicking natural cellulosome to simultaneously immobilize multiple enzymes with antibacterial activity onto surfaces. The grafting strategy leverages the strong biomolecular interaction between receptors on a scaffold protein anchored on the substrate and ligands added to the enzymes. As a proof of concept, lysozyme and lysostaphin were chosen to target the bacterial cell wall, and DNase I to degrade DNA released during cell lysis, known to promote bacterial adhesion which can later lead to biofilm formation. The specificity of the ligand/receptor interaction was confirmed by biochemical and AFM-based single-molecule force spectroscopy assays, thus demonstrating successful co-immobilization of the three enzymes on the protein scaffold. Then, the antibacterial protection was evaluated against Staphylococcus aureus, Escherichia coli and Micrococcus luteus by viability tests which revealed long-term antimicrobial protection of the multi-enzymatic scaffold on both Gram-positive and Gram-negative bacteria. After 24 hours of contact, the system induced lysis of 71 to 85% of bacteria, and its antimicrobial properties remained effective after 5 days even with several cumulative waves of bacterial contamination. This work demonstrates the relevance of bio-inspired multi-enzymatic scaffolds for antibacterial protection, providing long-term and broad-spectrum action.
Statement of significance
With the increase of resistance among pathogenic bacteria, it became crucial to find alternatives to conventional antibiotics. Antimicrobial enzymes present many advantages as they are “re-usable”, less associated with resistance issues, non-toxic and can withstand a wide range of contamination. In this article, we focus on grafting antibacterial enzymes on materials surfaces. We used a bio-inspired approach to simultaneously graft different yet complementary enzymes on a protein scaffold and to achieve broad-spectrum antibacterial effect. The resulting multi-enzymatic scaffold enhanced antibacterial effects compared to a strategy where the enzymes were directly grafted on the material's surface. This was a proof of concept of antibacterial strategies bio-inspired by cellulosomes as our system provided long-lasting protection concurrently against Gram + and Gram − bacteria.1. Introduction
Bacterial infections pose a significant threat in both medical and industrial contexts. They represent an ever-growing global scourge which caused the death of 7.7 million patients in 2019, about one eighth of the total number of deaths worldwide.1 Infections often take the form of surface contamination forming on medical devices such as wound dressings, urethral and intravascular catheters, prosthetic grafts, prosthetic joints, and shunts.2,3 This leads to increased mortality and morbidity which causes an important economic burden on health care systems.2,4–6Surface contamination starts with the initial adhesion of pioneer bacteria to the substrate which then multiply to form microcolonies and later biofilm, the most predominant and successful lifestyle of bacteria and highly resistant to external stress, including antibiotics.7–9 Antibacterial effect (or contact-killing) is often used on surfaces to inactivate pioneer cells and therefore prevent infections by inhibiting bacterial growth and biofilm formation.10–12 Initially, antibiotics and synthetic/natural antimicrobial peptides were considered promising candidates but the emergence of multi-resistant strains has rapidly put a curb on this prospect.13,14 In this context, antimicrobial enzymes have been considered as an interesting alternative as they are less associated with resistance issues. Their activity is “re-usable” i.e. they can act several times provided their catalytic site is active which can ensure long-lasting antimicrobial effect. Moreover, some enzymes can withstand broad-spectrum contamination and are non-toxic.12,13,15–20
In this work, we present an antimicrobial strategy based on a multi-enzymatic system bio-inspired by natural cellulosomes, which are cell-associated supramolecular structures or “nanomachines” consisting in several cellulolytic enzymes organized on a protein scaffold.21,22 Enzymes displayed on cellulosomes have complementary roles that enable efficient degradation and absorption by cellulolytic bacteria of nutrients from cellulose, a highly recalcitrant polysaccharide. Our proposed bio-inspired strategy was to mimic the supramolecular organization of natural cellulosomes by simultaneously assembling three enzymes with complementary antimicrobial roles on a protein scaffold grafted on material surfaces. Enzymes were chosen to favor broad-spectrum antimicrobial protection while concurrently preventing the accumulation of lysed cell debris on the surface. The first enzyme is lysozyme which cleaves the β-(1,4) bond between the N-acetyl-muramic acid and the N-acetyl-D-glucosamine residue of the peptidoglycan (PG) chains present in bacterial cell walls.23,24 This enzyme was also reported to act against Gram-negative bacteria through membrane disruption. For instance, it has the capability to permeabilize both the outer and inner membranes of E. coli, achieving this effect with and without the formation of pores, respectively.24–27 The second enzyme is lysostaphin, an endopeptidase that hydrolyses the pentaglycine chains linking the PG fibrils in the cell wall of staphylococci, a major pathogenic genus.28 The third enzyme is deoxyribonuclease I (DNase I), which aims to degrade the extracellular DNA, an important polymer released during cell lysis by cell wall degrading enzymes, and known to promote microbial adhesion and bacterial cohesion.29
Immobilizing antimicrobial enzymes is a crucial and challenging step for antibacterial strategies. Ideally, the grafting technique should preserve the tridimensional conformation of the enzymes to ensure their antimicrobial effects. It should also provide an optimized accessibility of the catalytic site once loaded onto the surface. A wide range of immobilization methods rely on physical adsorption or direct covalent attachment on the substrate which often lead to steric hindrance of randomly orientated enzymes, loss of activity of antimicrobial compounds due to conformational changes and possibly leaching of adsorbed molecules.13,19,30 Previously, we demonstrated that strategies based on strong-affinity biomolecular ligand/receptor interactions present many advantages for antibacterial and antibiofilm properties as they ensure a controlled orientation of the grafted enzymes.30 This controlled immobilization preserves the orientation and the conformation of the enzymes and thus contributes to optimal and long-lasting antimicrobial properties. Lysozyme, for instance, was successfully grafted on a substrate and showed enhanced antimicrobial effects over several consecutive cycles of contamination when grafted in an ordered manner, via ligand/receptor interactions, rather than randomly orientated.30 Yet, this approach cannot be applied to the simultaneous and controlled grafting of several enzymes with complementary activities in order to broaden the antimicrobial protection towards various contaminants. In the strategy proposed herein, chimeric ligand-tagged enzymes were grafted on receptors located on a recombinant protein scaffold, a supramolecular organization similar to those of natural cellulosomes. Cellulosomal enzymes anchored on supramolecular structures found in natural cellulosomes show optimal synergistic activity which is significantly enhanced compared to the corresponding free enzymes.31 These supramolecular assembly relies on a highly specific and strong ligand/receptor interactions between domains named dockerin and cohesin, respectively.32 The calcium-dependent ligand/receptor pairs in cellulosomes were found to be one of the strongest non-covalent biomolecular bonds known in nature where a duplicated calcium-binding loop-helix motif of the ligand interacts with the receptor site.32–36
Here we demonstrated that bio-inspired multi-enzymatic scaffold is an attractive strategy to prevent surfaces from being colonized by biofilm-forming bacteria. Far western blot based on scaffold detection confirmed that all three enzymes displayed functional dockerin modules and were successfully immobilized on the scaffold via ligand/receptor (dockerin/cohesin) binding. Atomic force microscopy (AFM)-based single-molecule force spectroscopy (SMFS) revealed the calcium-dependent nature of the ligand/receptor interactions used to dock the enzymes on the protein scaffold, similarly to natural cellulosome. It also demonstrated a strong binding and the specificity of these interactions. Lastly, viability assays on planktonic bacteria indicated the antimicrobial effect of the multi-enzymatic scaffold over five consecutive cycles of contamination by biofilm-forming bacteria, namely Staphylococcus aureus, Escherichia coli and Micrococcus luteus.
2. Materials and methods
2.1. Reagents and proteins
Lysozyme from chicken egg white (>90%, >40 units mg−1), recombinant lysostaphin and DNase I (≥2000 Kunitz units mg−1 protein) from bovine pancreas were purchased from Sigma and BioVendor (France). Recombinant scaffold protein as well as lysozyme, lysostaphin and DNase I enzymes tagged with ligands were produced and purified by the protein production and purification plateforme of the Institut de Biologie Moléculaire et Cellulaire (IBMC)-CNRS of the Université de Strasbourg (details below).2.2. Scaffold expression and purification
BL21 (DE3) E. coli strain transformed with pET9d-Scaf6-6xHis plasmid was grown at 37 °C and in 2L lysogeny broth medium supplemented with 1.2% glycerol and Carbenincilin until reaching an optical density of 1.5. The temperature was lowered to 20 °C and the expression induced by 0.2 mM IPTG during 20 hours.Bacteria were collected by centrifugation at 5000g, resuspended in 50 mL of 50 mM NaPO4 (pH = 7) with 20 μg mL−1 DNase I and protease inhibitors mix (Roche), and lysed with a Microfluidizer LM-20 (Microfluidics).
The lysate was clarified by centrifugation at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C, the supernatant collected and incubated with 15 g of micro-crystalline AvicelPH101 cellulose (SIGMA) for 90 min at 4 °C under constant agitation. The cellulose was recovered by filtration on 2 μm glass fiber filter and washed three times with 50 mL of 50 mM NaPO4 (pH = 7) and twice with 50 mL of 12.5 mM NaPO4 (pH = 7).
000g for 20 min at 4 °C, the supernatant collected and incubated with 15 g of micro-crystalline AvicelPH101 cellulose (SIGMA) for 90 min at 4 °C under constant agitation. The cellulose was recovered by filtration on 2 μm glass fiber filter and washed three times with 50 mL of 50 mM NaPO4 (pH = 7) and twice with 50 mL of 12.5 mM NaPO4 (pH = 7).
Protein was sequentially eluted by three 50 mL volumes of cold water, followed by two sequential 50 mL volumes of 1% triethanolamine, immediately buffered by the addition of 0.1 volume of 500 mM Tris–HCl (pH = 8). Elution fractions were assessed by SDS-PAGE and those containing the protein were pooled and filtered at 0.2 μm. The protein was further purified on an Akta Pure FPLC system with a MonoQ 5/150 GL anion exchange column (Cytiva) equilibrated in 25 mM Tris–HCl (pH = 8), 10% glycerol and eluted with a gradient 0–1 M NaCl, 25 mM Tris–HCl (pH = 8) and 10% glycerol. Fractions containing Scaf6 were pooled and loaded onto a HiLoad 16/60 Superdex S200 size exclusion column (Cytiva) equilibrated in PBS (pH = 7.4). Fraction corresponding to the monomeric protein were pooled and concentrated on a Vivaspin 20 concentrator (MWCO 30 kDa; Sartorius) to a final concentration of 3.5 mg mL−1, aliquoted, snap frozen in liquid nitrogen and stored at −80 °C.
2.3. Tagged-enzymes expression and purification
BL21 (DE3) E. coli strains transformed with pT7 expression vectors bearing 6His-enzymes (lysozymes, lysostaphin, and DNase I) tagged with the ligand motifs were grown at 37 °C and in 1L Terrific Broth medium supplemented with kanamycin until reaching an optical density of 2.5. The temperature was lowered to 20 °C and the expression induced by 0.5 mM IPTG during 18 hours.Bacteria were collected by centrifugation at 5000g, resuspended in 100 mL 25 mM Tris–HCl (pH = 8), 150 mM NaCl, 0.1% CHAPS, 3 mM β-mercapto-ethanol, and lysed with a Microfluidizer LM-20 (Microfluidics).
The lysate was clarified by centrifugation at 18000g for 20 min at 4 °C, and the supernatant purified on an Akta Pure FPLC system by binding on a HisTrap HP Ni-NTA affinity column (Cytiva) with 25 mM imidazole. The protein was eluted by a 25 to 500 mM imidazole gradient in 25 mM Tris–HCl (pH = 8), 150 mM NaCl, 0.1% CHAPS, 3 mM β-mercapto-ethanol. Fractions enriched in tagged enzymes were further purified on a Hi-Load 16/60 Superdex 75 column (Cytiva) equilibrated in PBS (pH = 7.4). Fractions containing monomeric tagged enzymes were pooled and concentrated on an AMICON Ultra 4 (MWCO 3 kDa, Millipore) to a final concentration of 1.5 mg mL−1, aliquoted, snap frozen in liquid nitrogen and stored at −80 °C.
2.4. Far western blots
Ligand-tagged and non-tagged proteins (20 μL at 0.2 mg mL−1) were mixed with 6 μL of denaturing buffer, prior boiling for 5 min. 5-μL of boiled samples were subjected to denaturing polyacrylamide gel electrophoresis (SDS-PAGE) using Bio-rad precast gels (gradient 4–15%) and analyzed by far western blot, after transfer on nitrocellulose (GE healthcare), using biotinylated Scaf6 and streptavidin-POD (Roche) as formerly described in ref. 37.2.5. Surface and AFM tip functionalization
Silicon wafers and round glass coverslips (Ø12 mm) were coated by plasma sputtering with an under layer of chromium (∼10 nm) and an upper layer of gold (∼30 nm). They were then cleaned by UV-ozone treatment for 15 min, rinsed with ethanol and dried under N2 flow.Enzymes and scaffolds were covalently attached on self-assembled monolayers (SAMs) of carboxyl-terminated alkanethiols. Briefly, gold-coated substrates and gold-coated NPG-D (Bruker) AFM tips were immersed overnight in a solution of 0.1 mM 16-mercaptododecahexanoic acid and 0.9 mM 11-mercapto-1-undecanol to form SAMs. They were then rinsed with ethanol and dried under N2 flow. Next, carboxylic groups were activated by immersion 30 min in a solution of N-hydroxysuccinimide (NHS, 10 mg mL−1) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, 25 mg mL−1). AFM tips and substrates were then rinsed with ultrapure water and immersed in protein solutions (0.2 mg mL−1 in PBS) for 2 hours. Enzymes-decorated tips and scaffold-coated surfaces were stored in Tris-buffered saline (TBS) supplemented 1 mM CaCl2 and 1 mM ZnCl2 (TBS+) at 4 °C for 4 days maximum before they were used for SMFS. Calcium chloride and zinc chloride were used to ensure calcium-dependent ligand/receptor interactions and zinc-dependent lysostaphin activity.
For bacterial viability experiments, substrates coated with scaffolds were immersed in a solution containing 0.2 mg mL−1 of tagged lysozyme, tagged lysostaphin and tagged DNase I. Control surfaces were prepared by grafting directly non-tagged enzymes on SAMs activated substrates (without scaffold layer) or by grafting only the scaffold (no enzyme addition). All grafting lasted for 2 hours at 0.2 mg mL−1 in PBS. Prepared surfaces were rinsed in TBS+ and directly used for bacterial viability tests.
2.6. AFM imaging and force spectroscopy
All AFM experiments were achieved at 21 ± 1 °C in an air-conditioned room, in TBS+ buffer using a Bioscope Resolve AFM (Bruker corporation, Santa Barbara, CA). Cantilevers spring constants were determined by the thermal noise method.38 Measurement of scaffold coating thickness was performed by scanning a 1 × 1 μm2 area at high forces and high scan rate and then imaging a larger area (of 5 × 5 μm2) under small forces and reduced frequencies. To perform SMFS, adhesion maps of 32 × 32 force–distance (FD) curves on 5 × 5 μm2 areas were recorded on the scaffold-coated substrates with the enzyme-decorated AFM tips with an applied force of 500 pN, 1 μm s−1 constant approach and retraction speeds and a contact time of 500 ms. For control experiments, after acquiring adhesion in presence of calcium (TBS+), 20 mM of ethylenediaminetetraacetic acid (EDTA, Carlo Erba Reagents) was added in the buffer and SMFS was performed again to acquire the adhesion in the presence of the chelating agent. The media was then replaced by fresh TBS+ and SMFS was performed one last time. Positive control was achieved by performing the AFM measurements with the cellulase Cel9T from R. cellulolyticum tagged with its native dockerin.The adhesion force and rupture length of the last adhesive peaks were extracted from each force curve with Nanoscope Analysis 1.8 (Bruker). Adhesion forces and rupture lengths of the last adhesive peaks for each curve were ordered in bins to form the frequencies histograms displaying adhesion frequencies and rupture frequencies. These histograms were processed with MATLAB R2016b (MathWorks) and designed in figures with Origin 2023 (OriginLab). Results presented for SMFS correspond to the average over three independent experiments of 1024 force–distance curves each.
2.7. Bacterial growth and viability tests
All bacterial cells were grown routinely at 37 °C. Staphylococcus aureus strain 187 (HER 1239) were grown on tryptic soy broth (TSB, Sigma) plates and Micrococcus luteus (ATCC4698) and Escherichia coli (ATCC25922), on lysogeny broth Lennox (LB, Sigma) plates. For viability experiments, a single colony of S. aureus, M. luteus and E. coli was grown in 5 mL of respective broth media under gentle agitation (150 rpm on an agitation plate). Overnight cultures were washed twice in TBS+ by centrifugation at 5000g for 5 min before being retrieved. For the consecutive challenges, 1.2 mL of bacterial suspension containing ∼5 × 107 bacteria were deposited on functionalized surfaces in 12-well plates and gently agitated at 37 °C. Multiple waves of contamination (challenges) were performed by replacing the bacterial suspension in the plates with fresh suspension containing ∼5 × 107 bacteria every 24 h for a total of 5 challenges. Samples were pipetted (100 μL) at 0 h, 2 h, 4 h, 6 h and 24 h of every 24 h-challenge and serially diluted then plated on agar plates before culture at 37 °C to determine bacterial viability by colony forming units counting. Results presented for viability tests correspond to the average on four serial dilutions for each time and over four independent experiments. Results are presented with exponential decay fittings on Origin 2023 (OriginLab).2.8. Statistical analysis
All statistical analysis was performed using GraphPad Prism 7.0. Standard deviations of each data group of the adhesion frequency for each enzyme (n = 3) were not found significantly different with a Brown–Forsythe test. A one-way ANOVA was therefore used followed by a Tukey's multicomparison test for pair-wise comparison between conditions.3. Results
3.1. Ligand-tagged enzymes bound to the receptors located on the scaffolds
To concurrently graft three different enzymes on surfaces, we decided to construct and purify after heterologous expression an interspecific hybrid scaffold made of receptors (cohesin modules) from three different species, namely Acetivibrio thermocellus (formerly known as Clostridium thermocellum), Ruminiclostridium cellulolyticum and Ruminococcus flavefaciens, and separated by spacers of 30 amino acids. Enzymes of interests were tagged with the ligands corresponding to their scaffold receptors, i.e. lysostaphin, lysozyme and DNase I were tagged with ligands (dockerin modules) from A. thermocellus, R. cellulolyticum and R. flavefaciens, respectively. More precisely, Cel48S dockerin from A. thermocellus was anchored on the C-terminus of lysostaphin to form the tagged lysostaphin; lysozyme tagged enzymes carried Man5K dockerin from R. cellulolyticum on the N-terminus; and tagged DNase I was tagged with dockerin from R. flavefaciens on its C-terminus. The cellulase Cel9T from R. cellulolyticum with its native dockerin was used as positive control to confirm binding of native enzyme-ligand to scaffold receptor.39 Enzymes were tagged with ligand of different species to ensure that each tagged enzyme had a specific receptor on the scaffold and thus that each enzyme could bind to the scaffold. Far western blots were the first step of our work to evaluate the ability of the enzymes to bind to the scaffold. Electrophoresis gel with the tagged and non-tagged lysostaphin, lysozyme and DNase I were transferred onto a membrane and biotinylated scaffold was used to probe the membrane. Bands at 36 kDa, 23 kDa and 41 kDa were revealed and attributed to tagged lysostaphin, tagged lysozyme and tagged DNase I, respectively (Fig. 1). No bands were observed when the biotinylated scaffold was used to probe non-tagged enzymes, namely lysostaphin (27 kDa), lysozyme (16 kDa) and DNase I (31 kDa). This confirmed that tagged enzymes specifically bound to the scaffold whereas non-tagged ones were unable to bind to it. Based on these results, it was assumed that, all three tagged enzymes bound to the scaffold and that the receptor located on the scaffolds targeted specifically tagged enzymes.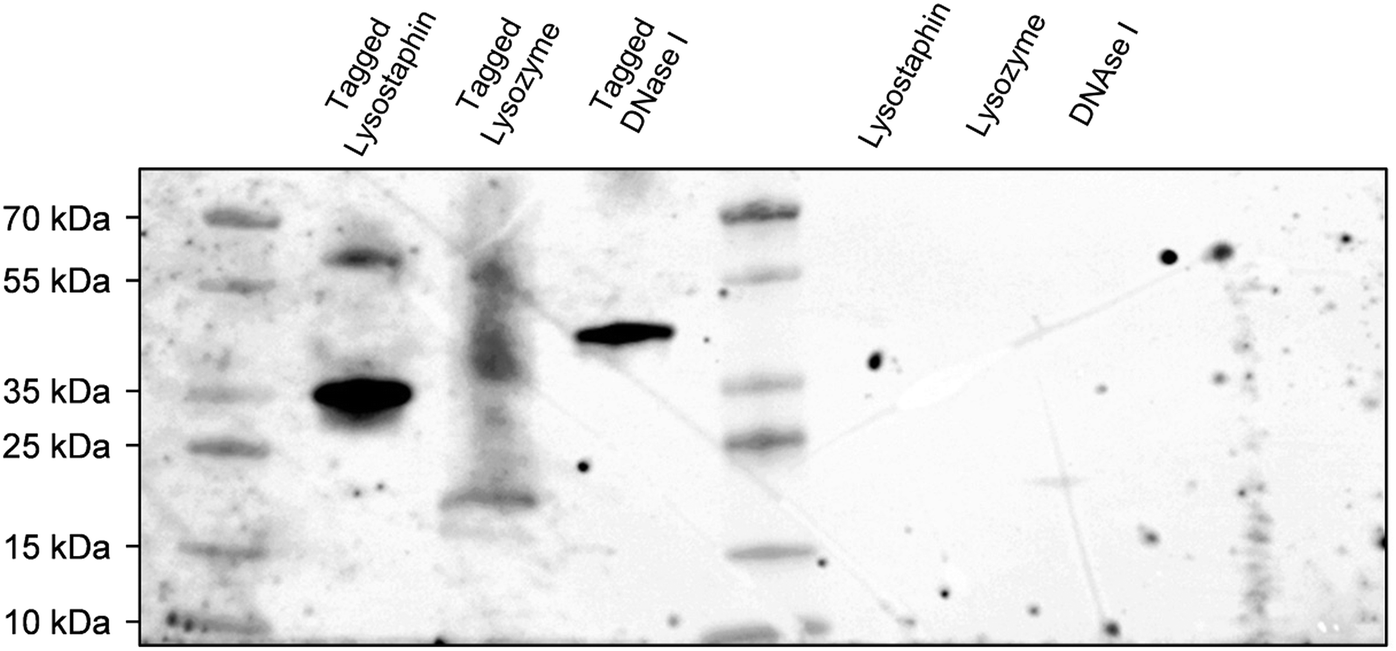 | ||
| Fig. 1 Far western blot using biotinylated scaffold (and Streptavidin-POD) to probe the membrane. Tagged lysostaphin, tagged lysozyme, tagged DNase I (left channels) were identified at 36 kDa, 23 kDa and 41 kDa, respectively. Non-tagged lysostaphin, lysozyme and DNase I (right channels) did not lead to any interaction with biotinylated scaffold as no bands were identified. | ||
3.2. Enzymes tagged with ligand interacted specifically with scaffold-coated surfaces
Probing the interaction between the enzymes and scaffold-coated surfaces was a crucial step in order to demonstrate that the surface functionalization strategy led to the immobilization of tagged enzymes through specific interactions between the ligands of the enzymes and the receptors located on the scaffolds.First, scaffolds were covalently grafted on material surfaces and topography AFM images were performed to confirm the grafting. In order to evaluate the thickness of the coatings, a part of each coating was removed by “scratching” a squared area of 1 × 1 μm2 at high forces (>10 nN) and high frequencies with AFM tips. A coating was considered present on the surface of the material when a scratched square was visible on the image. The depth of the scratched area was considered an estimation of the coating thickness (Fig. 2).
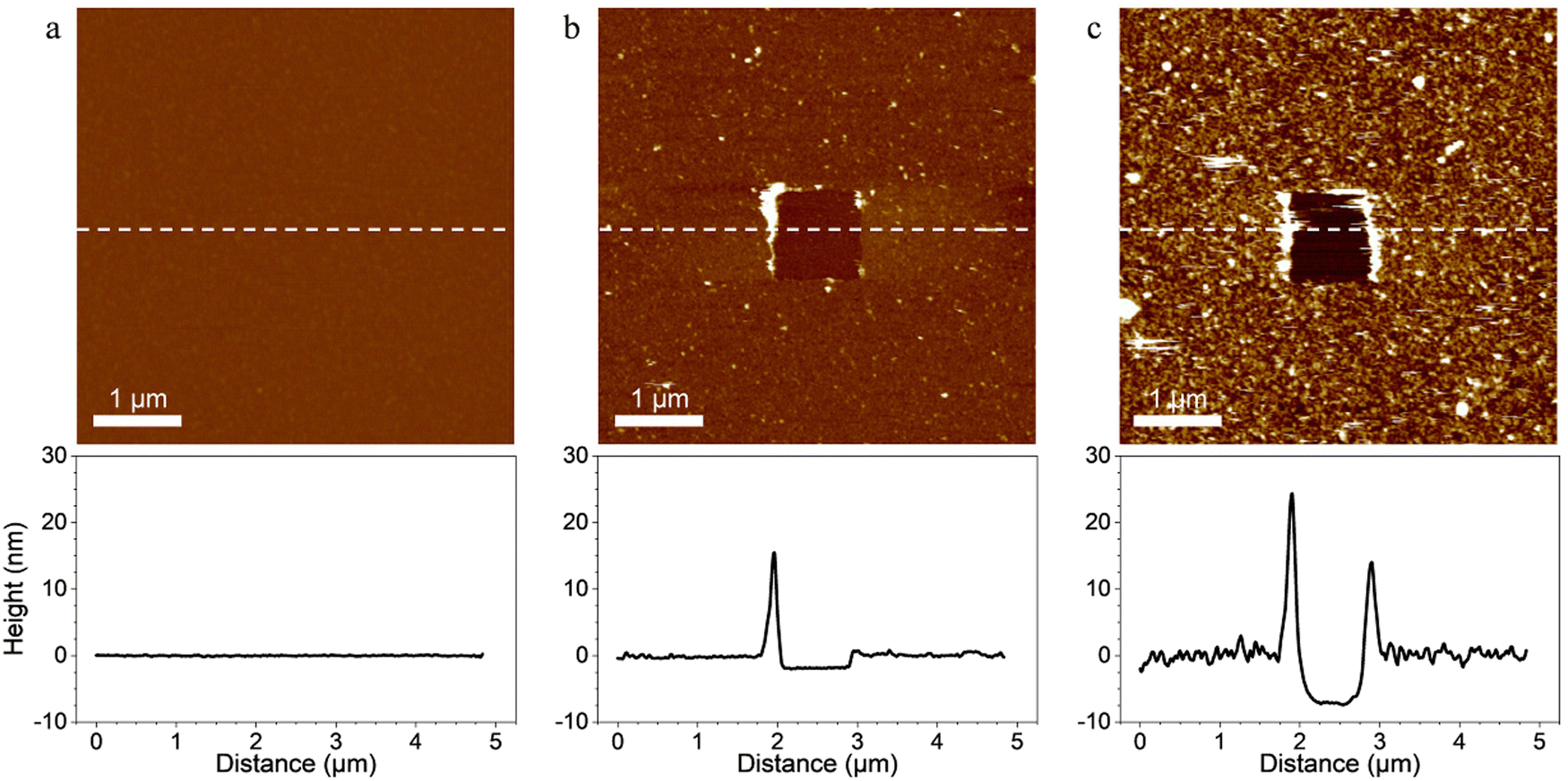 | ||
| Fig. 2 Topography of surfaces coated with (a) self-assembled monolayers (SAMs) only (b) SAMs and scaffold and (c) SAMs, scaffold and the three tagged enzymes. AFM height images (5 × 5 μm2; z values represented by the LUT with a difference of 40 nm between the maximum and the minimum values). A square of 1 × 1 μm2 was first scanned at high forces (>10 nN), then, the same area was imaged (5 × 5 μm2) with smaller forces. No scratched area was observed with the SAMs-coated surface. The AFM image revealed smooth and homogenous coatings of the surface with the scaffold and the scaffold coupled with the tagged enzymes. Scratched areas were observed and the thickness of the scaffold coating and scaffold + enzymes coating were evaluated at 1.95 ± 0.24 nm and 7.80 ± 0.67 nm, respectively by taking a vertical section (showed below the AFM images) along the dashed line drawn on the AFM image. | ||
Images of surfaces coated only with SAMs revealed a smooth and flat surface on which the coating was not visible even after using the “scratching” AFM technique (Fig. 2a). The relatively small size of the molecules composing the SAMs, the typical thickness of carboxylic acid terminated SAMs (few ångströms40) and the covalent nature of the coating made it difficult to evidence the SAMs with the “scratching” AFM technique. However, when the scaffold was added on the SAMs-coated surfaces, the resulting coating was observable on the AFM images and was homogeneous throughout the surface (Fig. 2b). Its thickness was evaluated at 1.95 ± 0.24 nm which confirmed that the scaffold bounded with the carboxyl groups of the SAMs. When the tagged-enzymes were added on the surface coated with the SAMs and the scaffold, the thickness of the coating increased and reached 7.80 ± 0.67 nm (Fig. 2c). This evidenced the binding of the enzymes’ ligands onto the scaffold's receptors as the increased thickness of the coating was attributed to the addition of the tagged enzymes. When scanned in contact mode at small forces, the coatings remained stable and no visible topography alterations were observed indicating a strong anchorage of the coatings on the substrate.
AFM imaging and far western blots showed that the enzymes had the ability to bind to the scaffold but to investigate these bonds further, the specificity of the receptor–ligand interactions was assessed by AFM-based SMFS. For that, each enzyme was grafted one at a time on AFM cantilevers which were then used to probe scaffold-coated surfaces (Fig. 3).
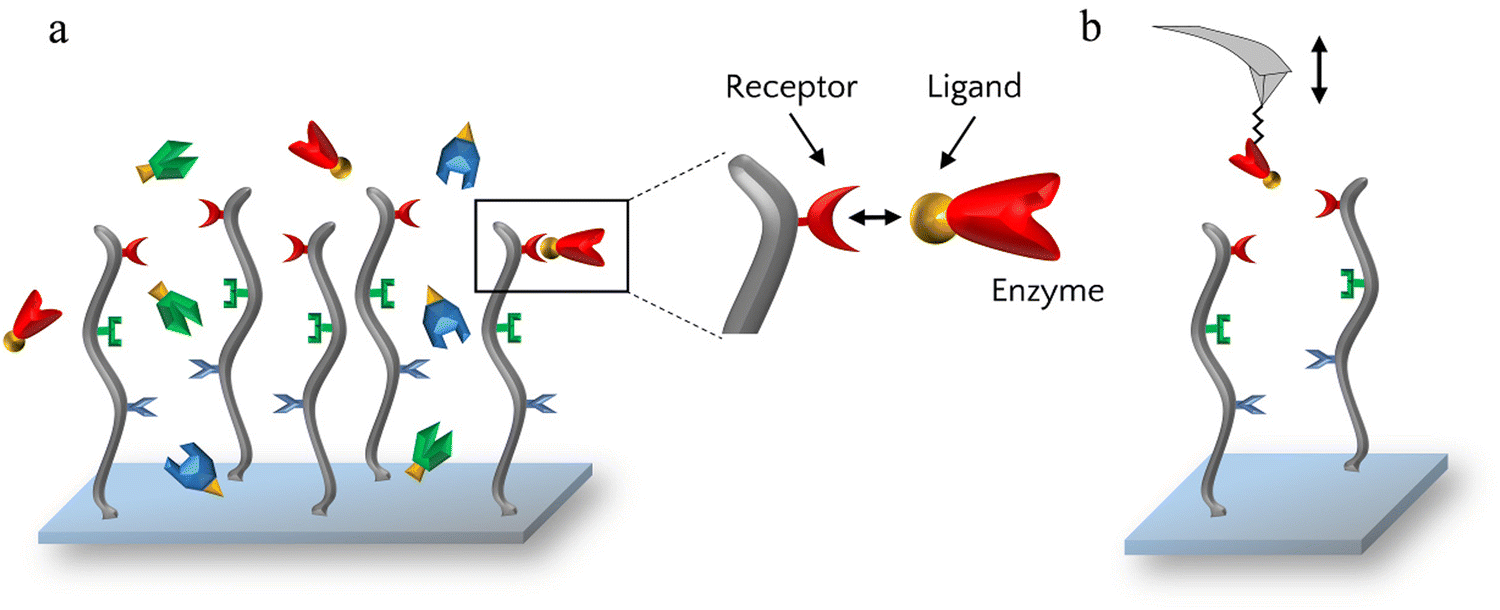 | ||
| Fig. 3 (a) Principle of enzyme grafting on scaffold-coated surfaces via the ligand–receptor interaction. (b) Principle of single-molecule force spectroscopy to probe the interaction and specificity between ligand-enzymes and receptors. | ||
For each enzyme-decorated AFM tip, adhesion in presence of calcium was first probed on the scaffold coating by recording adhesion maps. To test the calcium-dependency of the receptor/ligand pairs, a calcium chelating agent (EDTA) was added to the buffer to trap free calcium ions and SMFS measurements were subsequently performed a second time. Lastly, EDTA was removed, the buffer containing calcium was added and adhesion was recorded for the last time.
Fig. 4–6 show the adhesion frequencies and the rupture lengths histograms as well as representative force–distance (FD) curves for ligand-tagged lysostaphin, lysozyme and DNase I and their corresponding negative controls, i.e. non-tagged enzymes. For all enzymes, adhesive pixels on adhesion maps were randomly spread and adhesion maps did not show cluster patterns (data not shown). This suggests that the scaffolds were homogeneously grafted on the surfaces which matched the results observed above with the AFM topography images (Fig. 2).
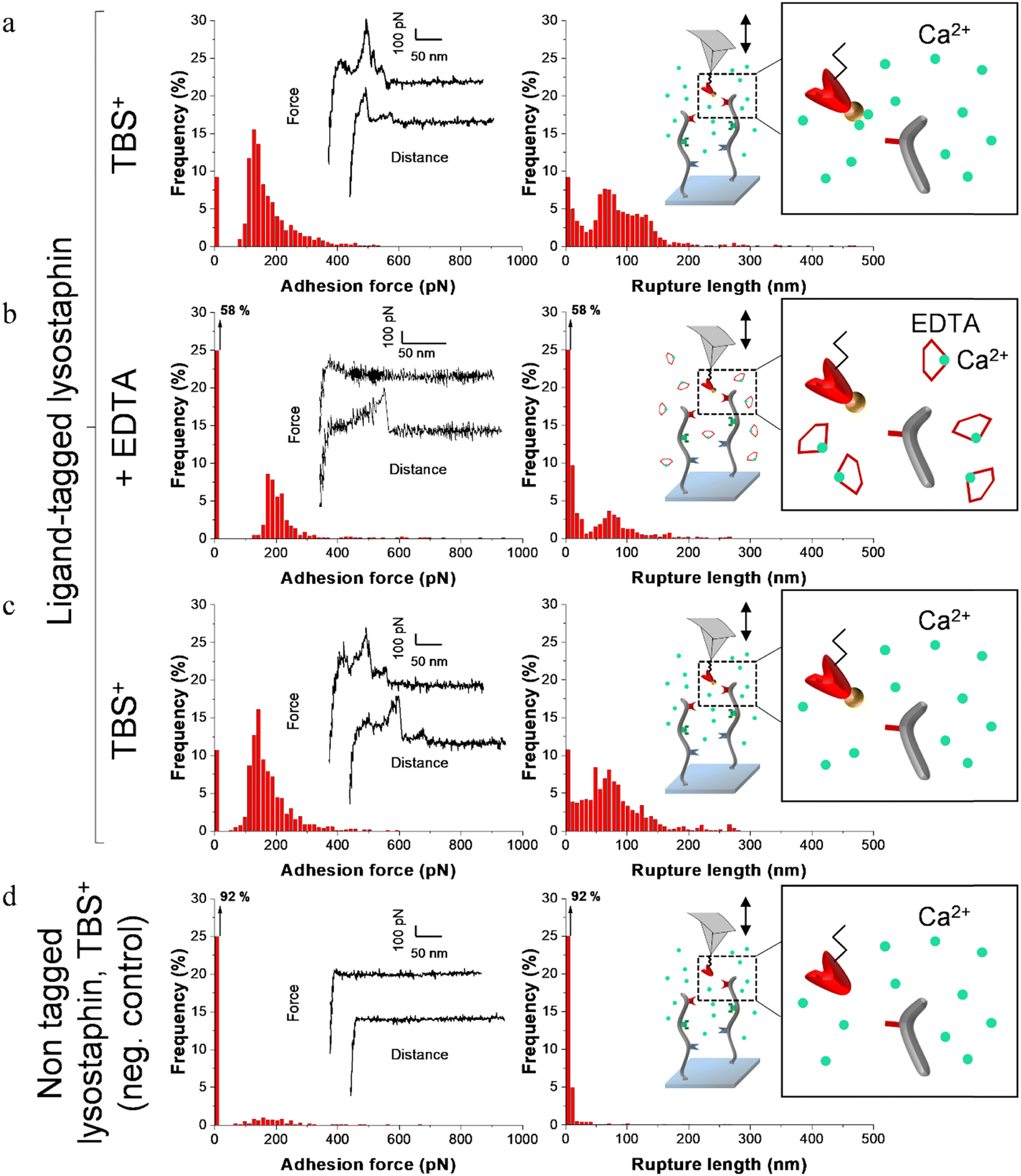 | ||
| Fig. 4 Adhesion frequency histograms with representative force curves (left) and corresponding rupture length histograms (right) extracted from adhesion maps (5 × 5 μm2) recorded on scaffold-coated surfaces with AFM cantilever decorated with tagged lysostaphin. Schematics on the right represent the different conditions: adhesion was consecutively recorded on one sample (a) in a calcium rich buffer before adding EDTA, (b) in presence of EDTA and (c) after removal of EDTA and addition of TBS+ containing calcium. (d) Negative control adhesion was recorded in TBS+ with tips decorated with lysostaphin lacking dockerin ligand. | ||
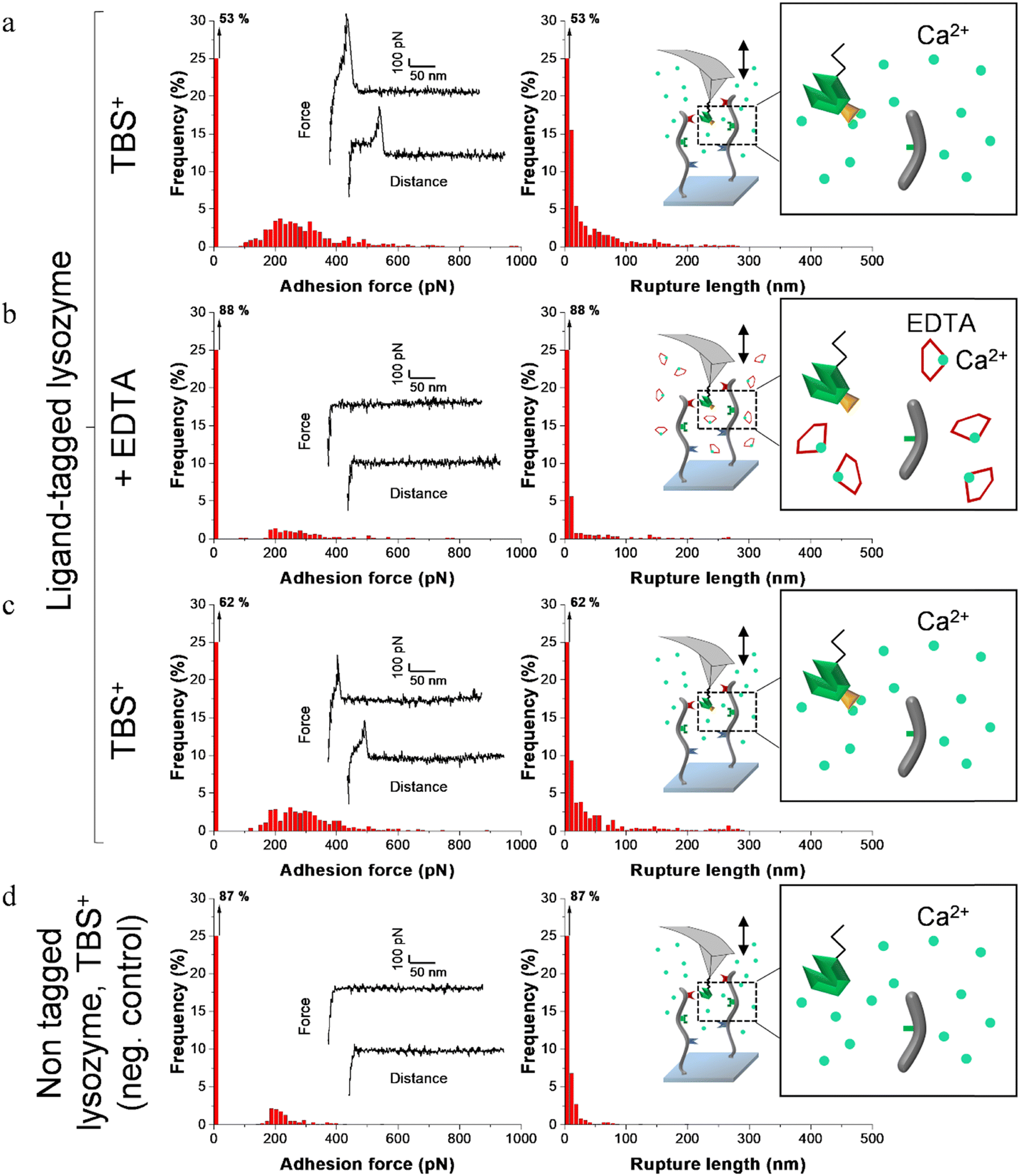 | ||
| Fig. 5 Adhesion frequency histograms with representative force curves (left) and corresponding rupture length histograms (right) extracted from adhesion maps (5 × 5 μm2) recorded on scaffold-coated surfaces with AFM cantilever decorated with tagged lysozyme. Schematics on the right represent the different conditions: adhesion was consecutively recorded on one sample (a) in a calcium rich buffer before adding EDTA, (b) in presence of EDTA and (c) after removal of EDTA and addition of TBS+ containing calcium. (d) Negative control adhesion was recorded in TBS+ with tips decorated with lysozyme lacking dockerin ligand. | ||
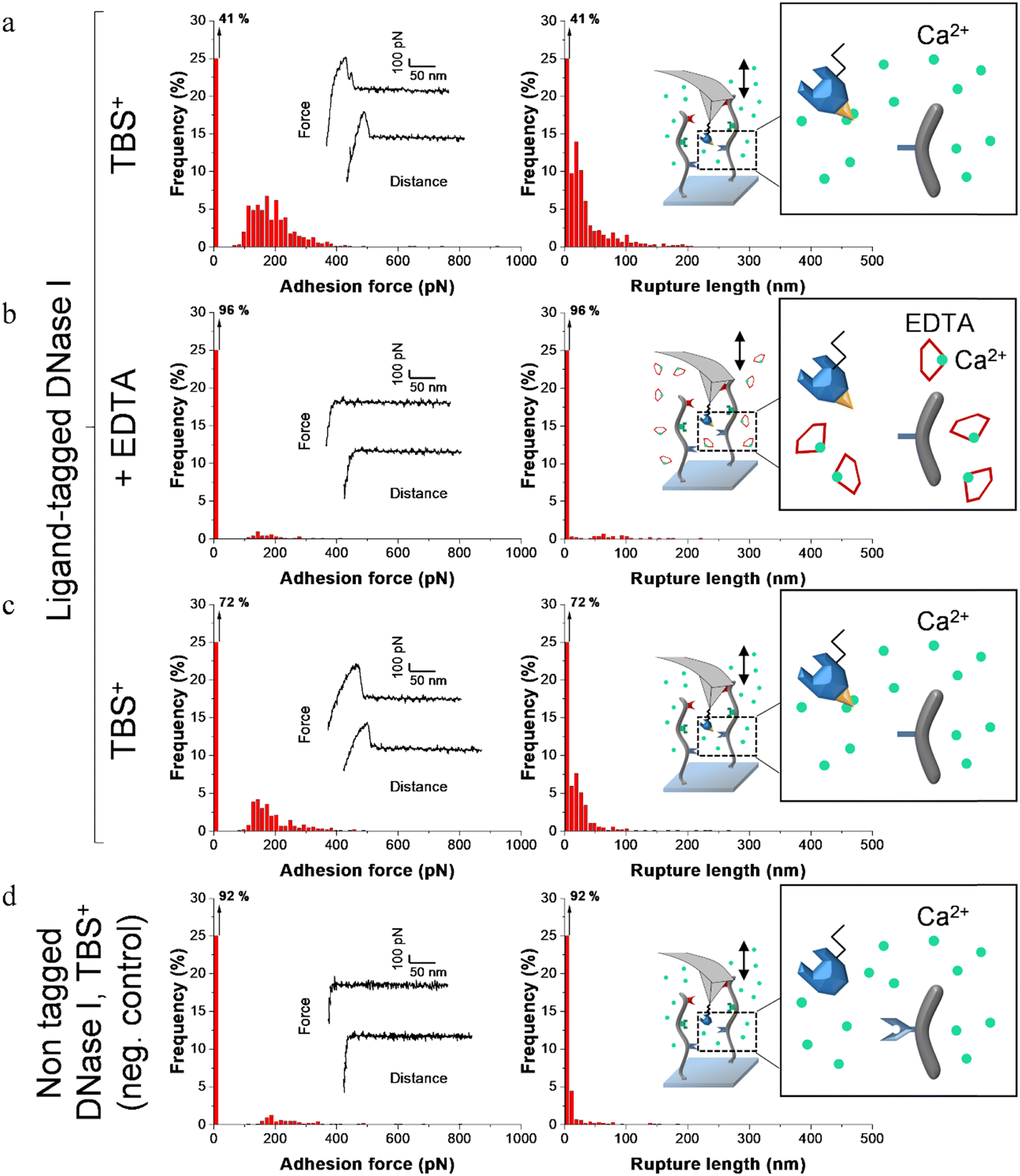 | ||
| Fig. 6 Adhesion frequency histograms with representative force curves (left) and corresponding rupture length histograms (right) extracted from adhesion maps (5 × 5 μm2) recorded on scaffold-coated surfaces with AFM cantilever decorated with tagged DNase I. Schematics on the right represent the different conditions: adhesion was consecutively recorded on one sample (a) in a calcium rich buffer before adding EDTA, (b) in presence of EDTA and (c) after removal of EDTA and addition of TBS+ containing calcium. (d) Negative control adhesion was recorded in TBS+ with tips decorated with DNase I lacking dockerin ligand. | ||
Before adding EDTA, representative FD curves showed adhesion peaks for all ligand-tagged enzymes (Fig. 4a, 5a and 6a). In contrast, less adhesion signatures were found on FD curves when EDTA was added with the ligand-tagged enzymes (Fig. 4b, 5b and 6b). However, once EDTA was removed, adhesion signatures were restored (Fig. 4c, 5c and 6c). Little to no adhesion signature were observed on FD curves of homologous enzymes lacking ligands (Fig. 4d, 5d and 6d). Adhesion frequency in presence of calcium for tagged lysostaphin was high since 86% of the FD curves showed an adhesion signature (Fig. 4a). Adhesion frequency then dropped to around 42% when EDTA was added (Fig. 4b) and increased again to 87% after EDTA was rinsed and calcium chloride was added (Fig. 4c). In contrast, only 8% of the FD curves showed adhesion signature for non-tagged lysostaphin (negative control, Fig. 4d) which we attributed to non-specific adhesion events. In presence of calcium, the average adhesion frequency of tagged lysozyme was 42% (Fig. 5a); it decreased to 12% with EDTA (Fig. 5b) and increased to 33% when EDTA was removed (Fig. 5c). Non-tagged lysozyme (negative control) adhesion frequency was 13% which shows that EDTA was able to completely inhibit ligand/receptor recognition in the case of this tagged lysozyme sample (Fig. 5d). Adhesion frequencies of tagged DNase I were 61% in presence of calcium (Fig. 6a), 4% when EDTA was added (Fig. 6b) and 25% when EDTA was removed (Fig. 6c). EDTA was also able to totally inhibit interactions for this sample since only 8% FD curves recorded with non-tagged DNase I presented adhesion peaks (Fig. 6d). Adhesion forces and rupture lengths of scaffold-enzymes interactions ranged from 100 to 400 pN and 5 to 150 nm, respectively. Tagged lysostaphin, tagged lysozyme and tagged DNase I mean rupture forces, in absence of EDTA and over three independent experiments of 1024 FD curves each, were on average 211 ± 2 pN, 297 ± 4 pN and 246 ± 3 pN, respectively (results are reported as mean ± standard error of the mean).
Fig. 7a represents adhesion frequencies for each condition and each enzyme on three independent experiments of 1024 FD curves each. Adhesion frequencies decreased significantly for all enzymes in presence of EDTA which confirmed the calcium-dependent nature of the ligand/receptor interactions. Before EDTA addition, differences were observed in the adhesion frequencies between the tagged-enzymes. Tagged lysostaphin reached an average of 87 ± 4% adhesion frequency whilst tagged lysozyme and DNase I were around 42% ± 4% and 61% ± 17%, respectively (Fig. 7a). Tagged lysostaphin led to the most adhesion compared to the other tagged enzymes before and after addition of EDTA.
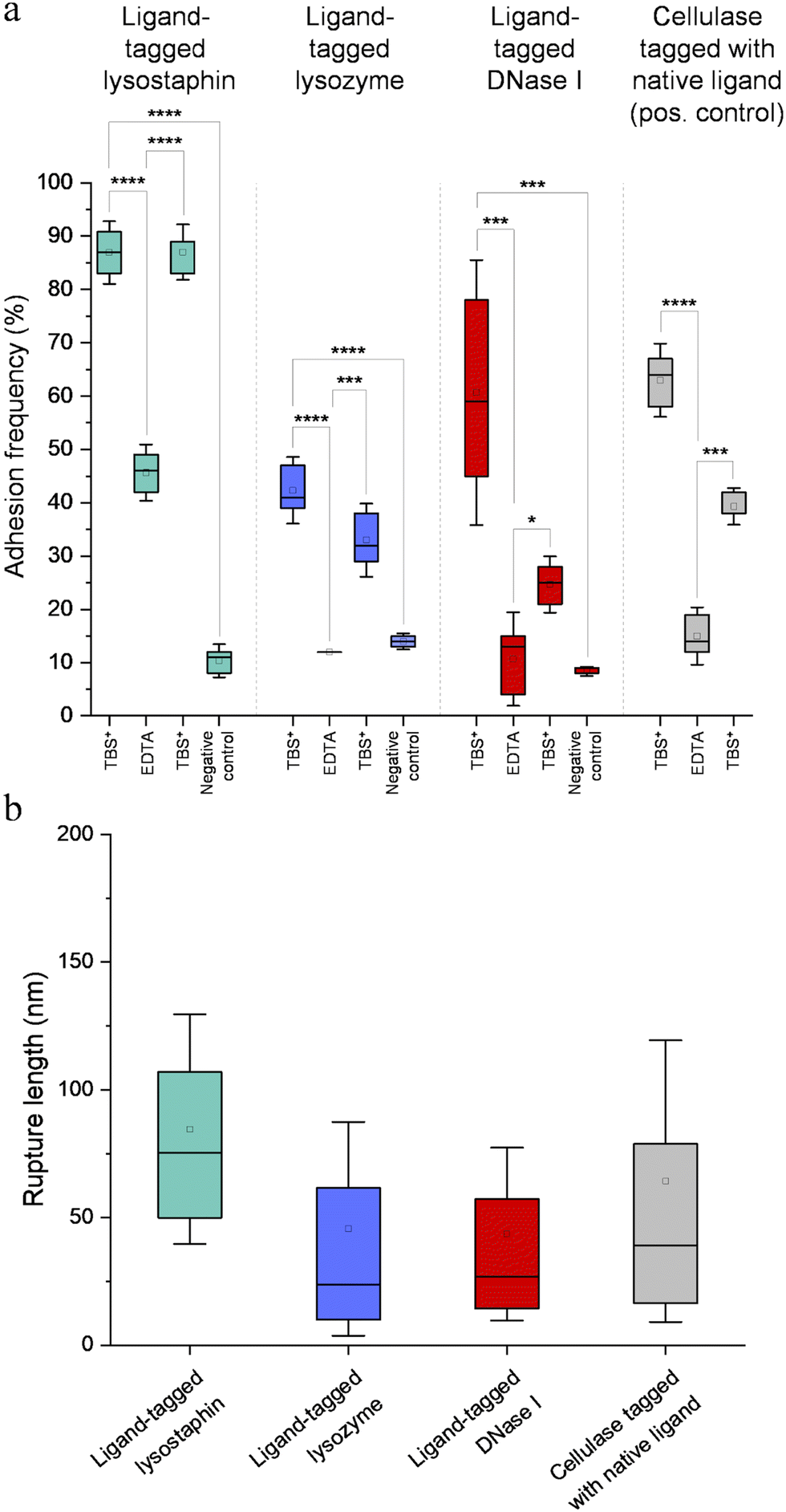 | ||
| Fig. 7 (a) Adhesion frequency for each enzyme obtained by scanning the scaffold by SMFS with enzyme-decorated AFM tips. Boxes represent the 25%–75% percentiles, error bars represent the mean ± 1.5 SD and horizontal bars represent the median of each group. Each box represents 3072 force–distance curves (n = 3). Statistical analysis was performed with a one-way ANOVA followed by a Tukey's multicomparison test for pair-wise comparison. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001. (b) Rupture lengths for each enzyme obtained by scanning the scaffold by SMFS with enzyme-decorated AFM tips. Boxes represent the 25%–75% percentiles, error bars represent mean ± 1.5 SD and horizontal bars represent the median of each group. Each box represents 3072 force–distance curves (n = 3). Statistical analysis performed with a one-way ANOVA followed by a Tukey's multicomparaison test for pair-wise comparaison (p < 0.01) revealed significant differences except between ligand-tagged lysozyme and ligand-tagged DNase I. | ||
No significant differences were found between the EDTA condition and the negative control (non-tagged enzymes) for tagged lysozyme and tagged DNase I. EDTA was therefore able to entirely inhibit ligand/receptor interactions for these tagged enzymes. As for the interactions between tagged lysostaphin and the scaffold, EDTA was able to inhibit by around half of them. Since tagged lysostaphin resulted in the most adhesion compared to other tagged enzymes, EDTA not being able to inhibit all interactions for tagged lysostaphin was attributed to the high affinity of the ligand/receptor pair and/or to a limited EDTA accessibility to the calcium-binding loop-helix motif of this ligand/receptor.
After removal of chelating agent, tagged lysostaphin adhesion frequency was entirely restored as no significant differences were found before adding EDTA and after removal of EDTA (Fig. 7a). Adhesion frequencies of tagged lysozyme and tagged DNase I were significantly higher before adding EDTA than after which showed that the adhesion was partially restored after the chelating agent was removed. This partial restoration of adhesion was also observed with the positive control achieved with the natural cellulosome cellulase Cel9T from R. cellulolyticum. Adhesion of the positive control was inhibited by EDTA and almost half of the adhesion was restored after removal of EDTA. However, for all enzymes, after EDTA was removed, a significant increase in adhesion frequencies was observed. These results confirmed that in the absence of free calcium in the buffer (i.e., when EDTA was added), the ligand/receptors interactions between the tagged-enzyme and the scaffolds were inhibited and therefore that these interactions were calcium dependent.
The comparison of rupture lengths obtained for the different tagged enzymes revealed interesting discrepancies that can be attributed to: (i) the position of the probed receptor on the molecular scaffold leading to different extension lengths of this molecule, i.e., the receptor of A. thermocellus for tagged lysostaphin positioned at the terminus of the scaffold led to longer rupture lengths and the receptor of R. cellulolyticum led to comparable extensions with both Cel9 cellulase (positive control) and tagged lysozyme that bear the same ligand for this site; and (ii) the position of the ligand on the enzyme and the molecular flexibility of each enzyme that can result in long or short extension (Fig. 7b).
Non-tagged enzymes displayed very low adhesion, compared to tagged enzymes (Fig. 7a). This showed the high specificity of the scaffold-enzymes interactions and this ensured that only the tagged enzymes were anchored on the scaffold and that substitution with other proteins were highly unlikely. As a result, very low adhesion observed on non-tagged enzymes were attributed to non-specific interactions.
3.3. Multi-enzymatic scaffold coating improved antimicrobial protection of surfaces
To assess surface protection achieved by our system, antibacterial effect was evaluated over multiple waves of contamination (challenges) with fresh bacteria by standard plating assays. Bacteria were exposed to surfaces coated with the multi-enzymatic scaffolds. To evaluate the relevance of the scaffold in the system, results were compared to samples onto which enzymes were directly grafted without any protein scaffolding. Surfaces only coated with the scaffolds were used as negative controls. Bacterial viability was evaluated after 0 h, 2 h, 4 h, 6 h and 24 h of exposition. Each 24 h, the media was replaced by a fresh suspension of bacteria. The same procedure was repeated over 5 consecutive days (Fig. 8).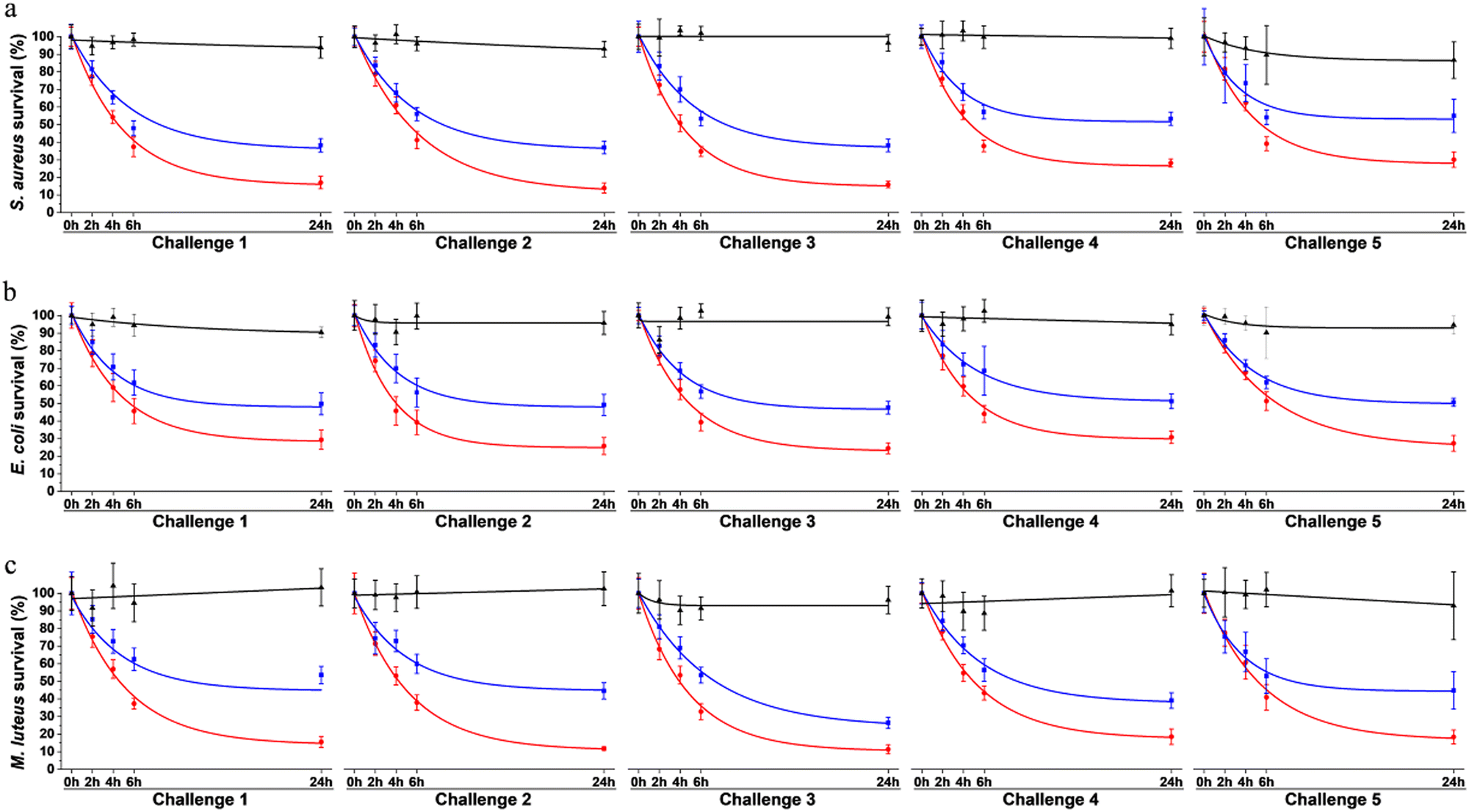 | ||
| Fig. 8 Viability of (a) S. aureus, (b) E. coli and (c) M. luteus exposed to surfaces coated with the scaffold alone (in black), with the three enzymes directly on the surface without any scaffolds (in blue) and with the multi-enzymatic scaffolds i.e. the three enzymes docked on the scaffolds (in red). Challenge represent consecutive waves of bacterial contamination. Each challenge started with the replacement of the media with fresh bacterial suspensions. Data was fitted with exponential decays. | ||
On the control surface, bacterial viability was around 90 to 100% after 24 h for most challenges (Fig. 8). Slight decreases (<10%) were observed over time for some samples and were attributed to: (i) bacteria adhering to the plastic well plate where the surfaces were immersed and thus not being collected when performing viability assays, and/or (ii) possible bacterial aggregation that reduce the number of CFU, i.e. one CFU could have been corresponding to aggregated bacteria instead of individual cells growing as a colony, thus artificially reducing the survival rate.41
The multi-enzymatic coating showed broad spectrum antimicrobial effects as both Gram-negative, namely E. coli, and Gram-positive bacteria, S. aureus and M. luteus were killed. Around 83% of S. aureus, 71% E. coli and 84% of M. luteus cells were lysed after the first 24 h when in contact with the multi-enzymatic scaffold (Fig. 8a–c). In contrast, enzymes directly grafted on surfaces without any scaffold support had a milder effect on bacterial lysis. Around 62% of S. aureus were lysed in contact with enzymes grafted directly on the surface after the first 24 h (Fig. 8a) and around 52% and 46% for E. coli and M. luteus, respectively (Fig. 8b and c). Multi-enzymatic scaffolds exhibited more antimicrobial effects throughout the five consecutive challenges than enzymes grafted directly on the material surface.
The antibacterial effects of multi-enzymatic scaffolds remained unchanged after five consecutive challenges for E. coli and M. luteus. Around 71% of E. coli bacteria were lysed at the end of the first challenge and 73% at the end of the fifth challenge for the multi-enzymatic scaffold. M. luteus lysing rate varied from 84% at 24 h to 82% at the end of the fifth challenge. A slight decrease in bacterial lysing rate was observed with S. aureus with the multi-enzymatic scaffold but the antimicrobial action remained high nonetheless. Around 70% of S. aureus were lysed by the multi-enzymatic scaffold after five consecutive challenges whereas the lysed rate was roughly 83% for the first challenge. A similar decrease in lysing rate was also observed for the unsupported enzymes for S. aureus dropping from 62% after 24 h to 45% after the fifth challenge.
4. Discussion
Key features of a successful protective surface reside in a long-lasting and broad-spectrum antibacterial effect. Aside from many crucial advantages in the fight against infections, antibacterial enzymes are known to be long-lasting as long as their catalytic site is active in comparisons of other strategies that often relies on a “one-shot” effect.12,13,15–20 However, antibacterial enzymes are specific and target a narrow range of pathogens. This presents the advantage of limiting adverse effects by avoiding the lyse of benign commensal microbiome threatened by broad spectrum antibiotics.42 But, at the same time, antibacterial surface treatment composed of a single antibacterial enzyme have intrinsically a limited spectrum of action. In order to increase the range of action of the antibacterial surfaces, our bio-inspired strategy in based on the simultaneous grafting of complementary active enzymes. Previous studies showed the relevance of bi- and trifunctional chimeric scaffolds combining different cellulosomal enzymes for the construction of designer cellulosomes.31,43 The proposed bio-inspired strategy was based on a similar approach to simultaneously assemble different yet complementary antibacterial enzymes on a recombinant protein scaffold. This allowed a broad-spectrum effect and a controlled positioning of each tagged enzyme on the scaffold.31 Ligand/receptor pairs, dockerin and cohesin respectively, from different species producing cellulosomes were used to anchor concurrently different ligand-tagged enzymes.Dockerin/cohesin pairs are considered to be one of the most stable ligand/receptor interactions known with rupture forces that occur via a tensile-based mechanism and above 120 pN,32,44 which is in concordance with our SMFS results. We concluded that unfolding of dockerin and cohesin which typically occurs at forces higher than 300 pN and up to 750 pN depending on the cohesin module,44–46 was unlikely since such high forces were not representative of our results. The SMFS conducted in the present study showed that ligand/receptor pairs used to dock the enzymes to the scaffold were calcium-dependent similarly to ligand/receptor pairs in natural cellulosome.32,47,48 This excluded the possibility of the tagged enzymes to be docked to the scaffold though unspecific interactions or any other interactions than through the binding of cohesin and dockerin. It also showed the highly specific interaction of our ligand/receptor strategy. The chimeric protein scaffold of our system was therefore not restricted to the anchorage of the lysozyme, lysostaphin and DNase I. As long as other enzymes of interest can be tagged with the ligand, the multienzymatic scaffold could be adapted to different applications to face other contaminants.
Discrepancies in adhesion frequencies of ligand and receptor interactions were observed. Tagged lysostaphin led to the highest adhesion frequencies in SMFS when in contact to its corresponding receptor on the scaffold compared to tagged lysozyme and tagged DNase I. These enhanced interaction frequencies might be attributed to species-dependent variations as it was shown that the affinity for the ligand and the receptor of A. thermocellum (used to bind lysostaphin to the scaffold in the present study) was higher than for ligand/receptor pairs of R. cellulolyticum and R. flavefaciens (used to dock lysozyme and DNase I, respectively, to the scaffold in our work).31 These variations in adhesion frequencies between tagged enzymes might also be explained by differences in accessibility between the ligand-tag and the receptor site and/or by a variation in steric hindrances due to the different positions of the receptors in the amino acid chain of the scaffold. The discrepancies observed in rupture lengths might also have been originated from species-dependent variations, lengths of the enzymes, steric hindrances and the positioning of the receptors in the amino acid chain of the scaffold.
The prerequisite for long-lasting antibacterial coatings is the constant degradation of bacteria and the elimination of contaminants that can accumulate and promote biofilm formation over time. One of the major advantages of using enzymes as antimicrobial agents is that they are “re-usable” and they continuously degrade bacteria as long as their catalytic site remains active. The proposed strategy combined cell wall degrading enzymes as well as DNase I which degraded DNA, an important cohesive component of the adhesion of microbial colonies and biofilm and the major polymer that would be released during cell lysis after cell wall degradation. S. aureus (Gram-positive), E. coli (Gram-negative) and M. luteus (Gram-positive) were chosen to assess the antimicrobial properties of the surfaces coated with the multi-enzymatic scaffolds. S. aureus and E. coli are some of the most etiologic agents in nosocomial infections.2,49,50 Despite its typical low virulence, M. luteus can increase S. aureus pathogenesis by acting as a “proinfectious agent” and present risks of contaminations.51–55 Previous studies evidenced that grafting enzymes directly on surfaces resulted in a non-oriented distribution of the enzymes.29,56–60 Supported enzymes were shown to have optimized antimicrobial activity since the orientation of their catalytic sites is improved as well as their 3D conformations.30 As expected in our system, when enzymes were not supported by a scaffold, antimicrobial properties were evidenced to some extent but they were enhanced when enzymes were anchored on the supporting scaffold. The reduced efficacy of the unsupported enzymes was attributed to the randomness of their orientation on the substrate and the possible modification of their conformation leading to some of them being denatured and inactivated. The fact that consecutive challenges did not appear to alter significantly the enzymes' ability to lyse bacteria whether supported or unsupported confirmed (i) the long-term “re-usable” antibacterial effect of the enzymes and (ii) the long-term properties of DNase I preventing the accumulation of lysed bacteria that are known to promote bacterial adhesion which can later lead to biofilm formation. The relevance of the use of a multi-enzymatic scaffold was confirmed throughout all the consecutive challenges since enzymes organized on the protein scaffold exhibited a higher antimicrobial response compared to the enzymes grafted directly on the surfaces. The enzymes' organization and immobilization on a molecular scaffold allowed a controlled orientation and better accessibility to their target and resulted in an improved protection.
5. Conclusions
Cellulosomes rely on a supramolecular organization to enhance enzymatic efficiency and degrade recalcitrant biopolymers. Our bio-inspired multi-enzymatic scaffold relied on a similar organization to lyse bacteria in order to protect surfaces from bacterial colony formation. Single-molecule force spectroscopy evidenced that enzymes were anchored on the scaffold through highly specific ligand/receptor interactions to form a supramolecular structure analogous to natural cellulosomes. The simultaneous use of complementary enzymes on the scaffold provided broad-spectrum protection concurrently against Gram + and Gram − bacteria. Our system exhibited substantial antimicrobial activity against planktonic biofilm forming bacteria, namely S. aureus, E. coli and M. luteus, by lysing 71% to 84% of bacteria within the first 24 hours. The “re-usable” nature of the lysozyme and lysostaphin as well as the properties of DNase I ensured long term antimicrobial effects after 5 consecutive 24 h challenges. The scaffold improved the protective antimicrobial activity compared to unsupported enzymes. This was attributed to an optimized orientation and spatial conformation of enzymes attained with the multi-enzymatic scaffold. Enzymes were anchored on the scaffold through highly specific ligand/receptor interactions that enabled a controlled organization and positioning of the enzymes. Our system had great adaptability potential as it offered the possibility to tailor enzymes cocktails to the contamination risk of specific applications. Furthermore, considering the rapidly expanding repertoire of specific cohesin/dockerin devices described in the literature, gathering a larger number of different antimicrobial enzymes/proteins should be straightforward, whereas elongation or reduction of the inter-cohesins linkers in the scaffold could help to gain efficiency.Author contributions
BA, FQ, GF, HPF and SEKC designed the research and wrote the manuscript; BA, CR, FQ, HPF and SEKC contributed to the acquisition, analysis, and interpretation of the data. BA, FQ, HPF and SEKC revised the paper and all authors approved the final version.Conflicts of interest
None.Acknowledgements
The authors would like to thank Nicolas Baumberger and Laurence Herrgott from the Protein Production and Purification Plateforme of the Institut de Biologie Moléculaire et Cellulaire (IBMC)-CNRS of the Université de Strasbourg for the protein expression and purification. We would like to acknowledge the spectroscopy and microscopy Service Facility SMI of LCPME (Université de Lorraine-CNRS – https://www.lcpme.ul.cnrs.fr). This work was supported by the French National Agency for Research ANR JCJC, grant ANR-20-CE06-0001.References
- K. S. Ikuta, L. R. Swetschinski, G. R. Aguilar, F. Sharara, T. Mestrovic, A. P. Gray, N. D. Weaver, E. E. Wool, C. Han and A. G. Hayoon, Lancet, 2022, 400, 2221–2248 CrossRef.
- M. Shirtliff, J. G. Leid and M. Shirtliff, The role of biofilms in device-related infections, Springer, 2009 Search PubMed.
- S. Percival, Br. J. Surg., 2017, 104, e85–e94 CrossRef CAS.
- E. M. Padegimas, M. Maltenfort, M. L. Ramsey, G. R. Williams, J. Parvizi and S. Namdari, J. Shoulder Elbow Surg., 2015, 24, 741–746 CrossRef PubMed.
- A. Premkumar, D. A. Kolin, K. X. Farley, J. M. Wilson, A. S. McLawhorn, M. B. Cross and P. K. Sculco, J. Arthroplasty, 2021, 36, 1484–1489 CrossRef PubMed.
- M. Assefa and A. Amare, Infect. Drug Resist., 2022, 15, 5061–5068 CrossRef CAS.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS.
- M. Otto, Bact. Biofilms, 2008, 207–228, DOI:10.1007/978-3-540-75418-3.
- K. Sauer, P. Stoodley, D. M. Goeres, L. Hall-Stoodley, M. Burmølle, P. S. Stewart and T. Bjarnsholt, Nat. Rev. Microbiol., 2022, 20, 608–620 CrossRef CAS PubMed.
- H.-C. Flemming, Water Res., 2020, 173, 115576 CrossRef CAS.
- D. Campoccia, L. Montanaro and C. R. Arciola, Biomaterials, 2013, 34, 8533–8554 CrossRef CAS PubMed.
- I. Francolini and G. Donelli, FEMS Immunol. Med. Microbiol., 2010, 59, 227–238 CrossRef CAS.
- K. Glinel, P. Thebault, V. Humblot, C.-M. Pradier and T. Jouenne, Acta Biomater., 2012, 8, 1670–1684 CrossRef CAS PubMed.
- S. Tambe, L. Sampath and S. Modak, J. Antimicrob. Chemother., 2001, 47, 589–598 CrossRef CAS.
- D. Alves and M. Olivia Pereira, Biofouling, 2014, 30, 483–499 CrossRef CAS.
- A. K. Muszanska, H. J. Busscher, A. Herrmann, H. C. van der Mei and W. Norde, Biomaterials, 2011, 32, 6333–6341 CrossRef CAS PubMed.
- A. Shah, J. Mond and S. Walsh, Antimicrob. Agents Chemother., 2004, 48, 2704–2707 CrossRef CAS PubMed.
- J. J. Swartjes, P. K. Sharma, T. G. van Kooten, H. C. van der Mei, M. Mahmoudi, H. J. Busscher and E. T. Rochford, Curr. Med. Chem., 2015, 22, 2116–2129 CrossRef CAS.
- C. Von Eiff, B. Jansen, W. Kohnen and K. Becker, Drugs, 2005, 65, 179–214 CrossRef CAS.
- X. Wu, K. Fraser, J. Zha and J. S. Dordick, ACS Appl. Mater. Interfaces, 2018, 10, 36746–36756 CrossRef CAS.
- R. H. Doi and A. Kosugi, Nat. Rev. Microbiol., 2004, 2, 541–551 CrossRef CAS PubMed.
- E. A. Bayer, L. J. Shimon, Y. Shoham and R. Lamed, J. Struct. Biol., 1998, 124, 221–234 CrossRef CAS PubMed.
- V. M. Dillon, Natural Anti-Microbial Systems, Preservative Effects During Storage, in Encyclopedia of Food Microbiology, ed. C. A. Batt and M. L. Tortorello, Academic Press, second edn, 2014, pp. 941–947 DOI:10.1016/B978-0-12-384730-0.00238-X.
- M. Derde, V. Vié, A. Walrant, S. Sagan, V. Lechevalier, C. Guérin-Dubiard, S. Pezennec, M. F. Cochet, G. Paboeuf and M. Pasco, Biopolymers, 2017, 107, e23040 CrossRef.
- M. Derde, V. R. Lechevalier, C. Guerin-Dubiard, M.-F. O. Cochet, S. Jan, F. Baron, M. Gautier, V. R. Vie and F. O. Nau, J. Agric. Food Chem., 2013, 61, 9922–9929 CrossRef CAS PubMed.
- A. Pellegrini, U. Thomas, R. von Fellenberg and P. Wild, J. Appl. Bacteriol., 1992, 72, 180–187 CrossRef CAS PubMed.
- A. Pellegrini, U. Thomas, P. Wild, E. Schraner and R. von Fellenberg, Microbiol. Res., 2000, 155, 69–77 CrossRef CAS PubMed.
- J. K. Kumar, Appl. Microbiol. Biotechnol., 2008, 80, 555–561 CrossRef CAS.
- J. J. Swartjes, T. Das, S. Sharifi, G. Subbiahdoss, P. K. Sharma, B. P. Krom, H. J. Busscher and H. C. van der Mei, Adv. Funct. Mater., 2013, 23, 2843–2849 CrossRef CAS.
- A. Beaussart, C. Retourney, F. Quiles, R. D. S. Morais, C. Gaiani, H.-P. Fiérobe and S. El-Kirat-Chatel, J. Colloid Interface Sci., 2021, 582, 764–772 CrossRef CAS.
- H.-P. Fierobe, F. Mingardon, A. Mechaly, A. Belaich, M. T. Rincon, S. Pages, R. Lamed, C. Tardif, J.-P. Belaïch and E. A. Bayer, J. Biol. Chem., 2005, 280, 16325–16334 CrossRef CAS.
- S. W. Stahl, M. A. Nash, D. B. Fried, M. Slutzki, Y. Barak, E. A. Bayer and H. E. Gaub, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20431–20436 CrossRef CAS.
- S. D. Jeon, J. E. Lee, S. J. Kim, S. W. Kim and S. O. Han, Biosens. Bioelectron., 2012, 35, 382–389 CrossRef CAS.
- K. Sakka, Y. Sugihara, S. Jindou, M. Sakka, M. Inagaki, K. Sakka and T. Kimura, FEMS Microbiol. Lett., 2011, 314, 75–80 CrossRef CAS.
- M. A. Jobst, L. F. Milles, C. Schoeler, W. Ott, D. B. Fried, E. A. Bayer, H. E. Gaub and M. A. Nash, eLife, 2015, 4, e10319 CrossRef PubMed.
- A. L. Carvalho, F. M. Dias, T. Nagy, J. A. Prates, M. R. Proctor, N. Smith, E. A. Bayer, G. J. Davies, L. M. Ferreira and M. J. Romão, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3089–3094 CrossRef CAS.
- J. Ravachol, R. Borne, I. Meynial-Salles, P. Soucaille, S. Pages, C. Tardif and H. P. Fierobe, Biotechnol. Biofuels, 2015, 8, 114 CrossRef PubMed.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef CAS.
- J. Ravachol, R. Borne, C. Tardif, P. de Philip and H.-P. Fierobe, J. Biol. Chem., 2014, 289, 7335–7348 CrossRef CAS.
- M. Carot, V. Macagno, P. Paredes-Olivera and E. Patrito, J. Phys. Chem. C, 2007, 111, 4294–4304 CrossRef CAS.
- P. Herman-Bausier, S. El-Kirat-Chatel, T. J. Foster, J. A. Geoghegan and Y. F. Dufrêne, mBio, 2015, 6, e00413–00415 CrossRef CAS PubMed.
- A. Langdon, N. Crook and G. Dantas, Genome Med., 2016, 8, 1–16 CrossRef.
- H. P. Fierobe, E. A. Bayer, C. Tardif, M. Czjzek, A. Mechaly, A. Belaich, R. Lamed, Y. Shoham and J. P. Belaich, J. Biol. Chem., 2002, 277, 49621–49630 CrossRef CAS.
- M. Gunnoo, P. A. Cazade, A. Galera-Prat, M. A. Nash, M. Czjzek, M. Cieplak, B. Alvarez, M. Aguilar, A. Karpol and H. Gaub, Adv. Mater., 2016, 28, 5619–5647 CrossRef CAS PubMed.
- A. Galera-Prat, S. Moraïs, Y. Vazana, E. A. Bayer and M. Carrión-Vázquez, J. Biol. Chem., 2018, 293, 7139–7147 CrossRef CAS.
- A. Valbuena, J. Oroz, R. Hervás, A. M. Vera, D. Rodríguez, M. Menéndez, J. I. Sulkowska, M. Cieplak and M. Carrión-Vázquez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13791–13796 CrossRef CAS PubMed.
- S. Pagès, A. Bélaïch, J. P. Bélaïch, E. Morag, R. Lamed, Y. Shoham and E. A. Bayer, Proteins: Struct., Funct., Bioinf., 1997, 29, 517–527 CrossRef.
- M. T. Rincón, J. C. Martin, V. Aurilia, S. I. McCrae, G. J. Rucklidge, M. D. Reid, E. A. Bayer, R. Lamed and H. J. Flint, J. Bacteriol., 2004, 186, 2576–2585 CrossRef PubMed.
- G. Sharma, S. Sharma, P. Sharma, D. Chandola, S. Dang, S. Gupta and R. Gabrani, J. Appl. Microbiol., 2016, 121, 309–319 CrossRef CAS PubMed.
- A. Reisner, M. Maierl, M. Jörger, R. Krause, D. Berger, A. Haid, D. Tesic and E. L. Zechner, J. Bacteriol., 2014, 196, 931–939 CrossRef PubMed.
- E. Boldock, B. G. Surewaard, D. Shamarina, M. Na, Y. Fei, A. Ali, A. Williams, E. J. Pollitt, P. Szkuta and P. Morris, Nat. Microbiol., 2018, 3, 881–890 CrossRef CAS.
- R. Peces, E. Gago, F. Tejada, A. Laures and J. Alvarez-Grande, Nephrol., Dial., Transplant., 1997, 12, 2428–2429 CrossRef CAS.
- U. Dürst, E. Bruder, L. Egloff, J. Wüst, J. Schneider and H. Hirzel, Z. Kardiol., 1991, 80, 294–298 Search PubMed.
- G. Rodriguez-Nava, A. Mohamed, M. A. Yanez-Bello and D. P. Trelles-Garcia, IDCases, 2020, 20, e00743 CrossRef.
- H. Seifert, M. Kaltheuner and F. Perdreau-Remington, Zentralbl. Bakteriol., 1995, 282, 431–435 CrossRef CAS PubMed.
- D. Alves, A. Magalhães, D. Grzywacz, D. Neubauer, W. Kamysz and M. O. Pereira, Acta Biomater., 2016, 44, 313–322 CrossRef CAS PubMed.
- G. Yeroslavsky, O. Girshevitz, J. Foster-Frey, D. M. Donovan and S. Rahimipour, Langmuir, 2015, 31, 1064–1073 CrossRef CAS PubMed.
- A. Caro, V. Humblot, C. Méthivier, M. Minier, L. Barbes, J. Li, M. Salmain and C.-M. Pradier, J. Colloid Interface Sci., 2010, 349, 13–18 CrossRef CAS PubMed.
- A. Caro, V. Humblot, C. Méthivier, M. Minier, M. Salmain and C.-M. Pradier, J. Phys. Chem. B, 2009, 113, 2101–2109 CrossRef CAS PubMed.
- M. Minier, M. Salmain, N. Yacoubi, L. Barbes, C. Méthivier, S. Zanna and C.-M. Pradier, Langmuir, 2005, 21, 5957–5965 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |