
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn iron(III) oxide-anchored conductive polymer–graphene ternary nanocomposite decorated disposable paper electrode for non-enzymatic detection of serotonin†
Sharmila
Prashanth
a,
Raifa Abdul
Aziz
b,
Shamprasad Varija
Raghu
 bc,
Yoon-Bo
Shim
d,
K.
Sudhakara Prasad
*a and
Airody Vasudeva
Adhikari
*ae
bc,
Yoon-Bo
Shim
d,
K.
Sudhakara Prasad
*a and
Airody Vasudeva
Adhikari
*ae
aNanomaterial Research Laboratory (NMRL), Smart Materials And Devices, Yenepoya Research Centre and Centre for Nutrition Studies, Yenepoya (Deemed to be University), Deralakatte, Mangalore 575018, India. E-mail: avachem@gmail.com; ksprasadnair@yenepoya.edu.in
bNeurogenetics Lab, Department of Applied Zoology, Mangalore University, Mangalagangothri, 574199, Karnataka, India
cDivision of Neuroscience, Yenepoya Research Centre, Yenepoya (Deemed to be University), Deralakatte, Mangalore 575018, India
dDepartment of Chemistry and Institute of Biophysio Sensor Technology, Pusan National University, Busan 46241, Republic of Korea
eDepartment of Chemistry, National Institute of Technology Karnataka, Surathkal, Mangalore 575025, India
First published on 11th December 2023
Abstract
Serotonin, also known as 5-hydroxytryptamine (5-HT), is an important neurotransmitter that regulates many physiological processes. Both low and high concentrations of 5-HT in the body are associated with several neurological disorders. Hence, there is an urgent need to develop fast, accurate, reliable, and cost-effective disposable sensors for 5-HT detection. Herein, we report the sensing of 5-HT using a disposable paper-based electrode (PPE) modified with a ternary nanocomposite comprising poly(pyrrole) (P(py)), reduced graphene oxide (rGO), and iron oxide (Fe2O3). The sensor material was well characterized in terms of its structural, morphological, and chemical attributes using electron microscopy, spectral techniques, and electrochemical studies to prove the robust formation of the electroactive ternary nanocomposite and its suitability for 5-HT detection. The developed sensor exhibited an impressive limit of detection (LOD) of 22 nM with a wide linear range of 0.01 to 500 μM, which falls in the recommended clinically relevant range. The analytical recovery, spike sample analysis, and interference studies with ascorbic acid (AA), uric acid (UA), and epinephrine (E) showed satisfactory results, wherein the sensor could detect simultaneously both 5-HT and dopamine (DA). The potential practical utility of the developed sensor was further assessed by quantifying the concentration of 5-HT in the brain samples of Drosophila melanogaster, a versatile genetic model organism employed for modeling different neural disorders in humans, and validated by gold-standard HPLC-UV experiments. The as-fabricated single-run disposable sensor with a ternary nanocomposite exhibits excellent stability with good reproducibility and is a promising platform for identifying clinically relevant concentrations of 5-HT.
Introduction
Serotonin (5-HT) is a key excitatory neurotransmitter belonging to the catecholamine family, with a crucial role in physical and psychological health. Its deficiency can result in biological, psychotic, and neurodegenerative disorders.1 Conversely, its increased level may enhance positive social interactions and reduce aggression.2,3 Frequent over-activation of post-synaptic 5HT1A and 5HT2A receptors caused by certain serotonergic medications can lead to serotonin syndrome (SS), a potentially life-threatening condition characterized by neuromuscular, autonomic, and mental status changes. The diagnosis can be challenging due to its various clinical presentations, and so physicians must be highly vigilant in monitoring such patients to prevent harm and improve patient outcomes.4 To achieve this, monitoring the concentration of 5-HT in the human body is crucial, which requires sensitive and reliable biosensors with high temporal resolution at low levels. Tracking of 5-HT facilitates disease detection and improved treatment outcomes. Several techniques have been employed for the detection of 5-HT, which include high-performance liquid chromatography with ultraviolet spectroscopy (HPLC-UV),5 the colorimetric method,6 EG-Au-gate-FET biosensing,7 LSPR-based sensing using Au NPs8 and near-infrared fluorescence nanosensing.9 However, they are unsuitable due to their time-consuming and complicated procedures, high cost, and requirement for trained personnels to operate. Consequently, electrochemical methods are desirable because of the rapid response, relatively high sensitivity, affordability, possibility for in situ analysis, and ease of operation by non-specialists, thus making them valuable methods for monitoring 5-HT levels in biological systems.10,11Electrochemical methods comprise a collection of extremely useful measurement tools, and the right choice of electrode materials plays a pivotal role, i.e., achieving electrodes with the best possible properties can lead to products with a wide range of potential applications.12,13 Paper-based electrodes (PPEs) have recently gained much attention as prospective alternatives to traditional three-electrode systems because of their low cost, easy disposability, portability, ion accessibility, flexibility, and facile manufacturing methods.11,14 Normally, the PPE design consists of hydrophobic and hydrophilic regions, along with 3-in-1 electrode patterns incorporating working, counter, and reference electrodes.15 To the best of our knowledge, there have been limited studies on disposable sensors for the detection of 5-HT, which can detect 5-HT in brain fluids and can be validated using gold standard HPLC-UV methods. In view of this, in the present study, we have chosen an inexpensive PPE for developing an electrochemically active ternary nanocomposite for detecting 5-HT levels.
Polymer-modified electrodes (PMEs) have gained significant attention for their ability to enhance sensitivity and selectivity. Conducting polymers are potential candidates for such electrodes, as they possess features responsible for their anomalous electronic properties like high electrical conductivity, low electronegativity, and high electron affinity due to their π-electron conjugated backbones, making them excellent materials for the immobilization of biomolecules.16,17 P(py), a biocompatible polymer, synthesized via electro-polymerization, has emerged as a promising electronic sensing material, garnering attention in the sensor field. Owing to its inherent electrical conductivity, favourable stability, redox characteristics, and extensive surface area, P(py) is a promising electronic sensing material, particularly for use in neutral pH environments.17–19 However, P(py) exhibits certain limitations, including poor mechanical properties, relatively poor thermal stability in air, non-biodegradability, and lack of mechanical stability, which have hindered a more comprehensive range of applications.20 Subsequently, previous research endeavours have proposed various strategies to enhance the properties of P(py). One such strategy involves incorporating appropriate materials like reduced graphene oxide (rGO) and metal nanoparticles to achieve the desired characteristics of the electrode.21
Electrochemical detection of 5-HT is essentially based on the oxidation on a suitable electrocatalyst surface. Recently, it has been demonstrated that P(py)–rGO, a binary composite electrode, performs better when compared to the P(py) electrode alone, exhibiting higher capacitance and lower charge transfer resistance with excellent cyclability and renewability.22 rGO is a highly versatile material, effective for various applications, including electrocatalysis and electrochemical sensing. Its outstanding properties, such as high conductivity, large surface area, and tunable electronic structure, make it an attractive material for these applications. However, the primary drawback associated with binary nanocomposites is their inadequate stability during catalysis. Therefore, developing a straightforward method for creating nanocomposites that demonstrate superior catalytic performance is crucial.23
Transition metal nanoparticles are widely recognized for their exceptional conductivity and catalytic properties, which increase the electron transfer between the redox center in the target analyte and the electrode surface.24 It is well-established that nanocomposites comprising conducting polymers and transition metal nanoparticles are advantageous for achieving enhanced sensitivity and stability. Also, the incorporated metal nanoparticles serve as redox mediators, facilitating efficient electron transfer. Simultaneously, the polymer component acts as an adsorbent for biomolecules, further improving the performance of the nanocomposite.23,24 Over the last decade, considerable attention has been given to transition metal oxide nanoparticles due to their notable advantages, which include their ability to be functionalized, exceptional thermal and chemical stability, tunable oxidation states, unique optical properties, and large surface area. These characteristics make them highly appealing for various scientific and technological applications.25,26 Several research groups have investigated the applications of ternary nanocomposites, such as Ag–P(py)–Cu2O,25 rGO–Ag2Se,27 SPCE–ZnONR–PMB(DES),28 poly(bromocresol l green)–Fe3O4,29 P(py)–Fe3O4,30 and FeC–AuNPs–MWCNTs31 for 5-HT sensing. Even though ternary nanocomposites have been explored for 5-HT sensing, most of the reported articles failed to validate the sensors using gold standard methods, and also the real sample analysis was conducted with spiked blood or urine samples in the absence of 5-HT. Hence, it is imperative to develop a sensor that has the potential to detect 5-HT directly in clinical samples or in brain fluids.
Drosophila melanogaster, commonly referred to as the fruit fly, is a significant and simplified model organism employed for modelling different neural disorders in humans.32 The utilization of Drosophila in neuroscience research is favoured due to its genetic tractability, complex behavioural patterns, well-documented and straightforward neuroanatomy, and possession of many orthologous genes to humans.33 So, in our experimental design, we used the brain samples of Drosophila melanogaster for the quantitative and sensitive detection of 5-HT employing the newly fabricated electrode.
In the present work, a simple and cost-effective method for determining 5-HT was proposed using a P(py)–rGO–Fe2O3 ternary nanocomposite decorated PPE. The fabrication was achieved using a step-by-step procedure, where layer-by-layer assembly of the ternary nanocomposite is established. At first by depositing P(py) as a base coat using amperometry, a conductive platform is established followed by the electro-reduction of drop-coated graphene oxide (GO), and finally, the electro-deposition of Fe2O3 was carried out through cyclic voltammetry. The fabricated PPEs were characterized using electron microscopy (FE-SEM) and spectral techniques (FT-IR and XPS) and subjected to electrochemical studies such as impedance spectroscopy (EIS) and cyclic voltammetry (CV) to understand the robust formation of the sensor. The sensor was further tested with different concentrations of 5-HT and investigated to satisfy the requirements of a real-world sensor for clinical applications. The newly developed sensor is further validated using the gold standard HPLC-UV method and explored to detect the limited quantity of 5-HT in the Drosophila melanogaster brain samples.
Experimental section
Chemicals and apparatus
The required chemicals like 5-HT, DA, AA, UA, E, graphite, sodium nitrate, potassium permanganate, ferrous chloride, sodium acetate, pyrrole, graphite, potassium ferricyanide (K3Fe(CN)6), potassium ferrocyanide (K4Fe(CN)6), and hexamine ruthenium(III) chloride (Ru(NH3)6Cl3) were purchased from Merck-Sigma-Aldrich. The SU-8 photoresist material, acquired from Gersteltec in Pully, Switzerland, Whatman CHR paper, provided by GE Healthcare in the UK, and the conductive carbon ink and Ag/AgCl ink, supplied by Kayaku Advanced Materials, Inc. in Massachusetts, USA, were employed in the process of fabricating the paper electrodes. A Shimadzu LC-2050C i-Series (Shimadzu Corporation, Kyoto, Japan) liquid chromatograph system configured with quaternary low-pressure pumps, a degasser unit, a dual wavelength UV detector, a column oven, and an auto sampler was used for the validation study. All experimental protocols were carried out utilizing a 0.1 M phosphate buffer solution (PB) with a pH of 7.4. The buffer solution was prepared by accurately combining the required sodium phosphate monobasic (NaH2PO4) and sodium phosphate dibasic (Na2HPO4) concentrations. Prior to use, the buffer solution was purged with nitrogen to ensure the removal of any dissolved gases or impurities.Electrochemical experiments, including electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV), and amperometry, were conducted using a CHI 708E electrochemical workstation from CH Instruments and a Zensor simulator potentiostat (Zensor®, Ecas 100 from Taiwan). Surface morphological analyses of the fabricated sensor were performed using Fourier transform infrared-attenuated total reflectance (ATR-FTIR) spectroscopy with a Shimadzu instrument from Japan and a field emission scanning electron microscope (FE-SEM) equipped with an Oxford energy-dispersive X-ray spectrometer (EDS). The FE-SEM utilized was a model EVO MA18 with a magnification range of 1× to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. X-ray photoelectron spectroscopy (XPS) experiments were carried out using a K-alpha X-ray photoelectron spectrometer manufactured by Thermo Scientific in the United Kingdom. Curve fitting of XPS data was performed using the XPS PEAK 41 system software.
000×. X-ray photoelectron spectroscopy (XPS) experiments were carried out using a K-alpha X-ray photoelectron spectrometer manufactured by Thermo Scientific in the United Kingdom. Curve fitting of XPS data was performed using the XPS PEAK 41 system software.
Paper electrode fabrication
Fabrication of disposable paper-based electrodes (PPEs) involved the implementation of in-house photolithography and punching techniques, as previously described by our research group.15,34 In brief, the photolithography method was used to create electrode patterns on the paper substrate by selectively forming hydrophobic and hydrophilic regions using an SU-8 photoresist and a suitable photomask. The SU-8-treated paper, in conjunction with the photomask, was subjected to UV light exposure for 12 minutes to induce the desired hydrophilic region, while creating hydrophobic barriers. Subsequently, the treated paper underwent a thorough cleaning process employing isopropyl alcohol and acetone. After drying at ambient temperature, the electrode patterns were etched onto the hydrophobic region using a simple crown punching technique. The working and counter electrodes were the conductive carbon ink, while the reference electrode was the Ag/AgCl ink. The resulting 3-in-1 PPE was dried in an oven at 90 °C for an hour. Geometric calculations determined the working electrode area to be 0.07 cm2.Electrochemical deposition of PPE–P(py)–rGO–Fe2O3
The PPE–P(py)–rGO–Fe2O3 nanocomposite fabrication on a paper electrode involved a stepwise procedure (Scheme 1). Firstly, P(py) was electrodeposited onto the PPE using an amperometric technique at a potential of 0.8 V for 600 s relative to the pseudo-Ag/AgCl reference electrode in 0.1 M phosphate buffer (PB) solution containing 0.01 M pyrrole,35 resulting in a significant increase in current, indicating the successful polymerization of P(py) on the negatively charged paper electrode15 (Fig. 1(A)). Subsequently, 2 μL of graphene oxide (GO), prepared via a modified Hummers’ method,36 was drop-coated onto the P(py)-coated PPE and electrochemically reduced to reduced graphene oxide (rGO) by performing cyclic-voltammetry (CV) with a scan rate of 0.05 V s−1 for 20 cycles, at a potential ranging from 0 to −1.4 V (Fig. 1(B)).37 The anodic and cathodic peaks associated with the oxidation–reduction pair of electroactive oxygen-containing groups on graphene planes are visible, and the pattern closely resembles the previously reported results.37 Briefly, the first CV curve shows a cathodic reduction wave at around −0.9 V, which is attributed to the reduction of surface oxygen groups. Furthermore, the CV curves exhibited increased capacitance behaviour with each new cycle, confirming that a conductive film has been electrodeposited on the working electrode. All CV curves clearly show a cathodic wave starting at −1.0 V, which is generally attributed to the irreversible electrochemical reduction of GO. Other, less apparent reduction and oxidation peaks at approximately −0.42 and −0.28 V are due to the activity of oxygen-based functional surface groups.38 Following the electro-reduction step, the PPE–P(py)–rGO electrode was rinsed with deionized water and dried at 30 °C for 10 minutes. Finally, the electrodeposition of Fe2O3 onto PPE–P(py)–rGO was carried out following the previously reported method.38Fig. 1(C) depicts the electrodeposition of Fe2O3 nanoparticles on PPE–P(py)–rGO. All electrode deposition experiments were performed under constant nitrogen purging unless otherwise specified. During the electrodeposition of Fe2O3, the anodic and cathodic scans displayed two prominent reduction and oxidation peaks. The peak located at −0.73 V was attributed to the oxidation of Fe2+ to Fe3+39 as shown in Fig. 1(C). The CV curves depicted an increased capacitance behaviour of the electrode due to the presence of the conducting rGO–P(py) layer, and interestingly there was no peak for Fe2+ to Fe0 reduction. However, as the electrochemical cycles continued, the delayed deposition of Fe3+ occurred at −0.73 V during the reverse scan, suggesting that the deposition of iron (in the form of Fe(III)-oxide) sequentially happened on PPE–P(py)–rGO.
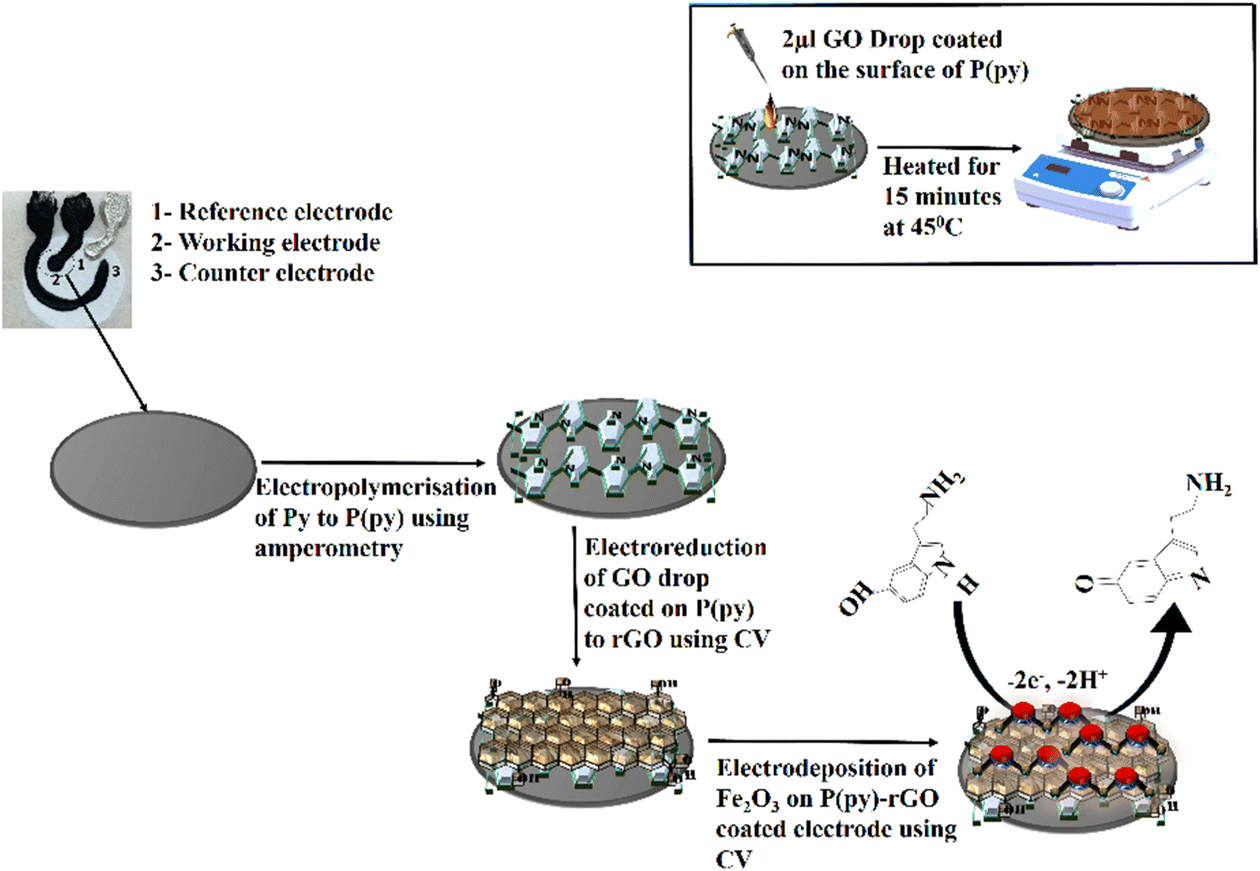 | ||
| Scheme 1 Schematic illustration of the synthesis of the P(py)–rGO–Fe2O3 ternary nanocomposite on the surface of the PPE, and the inset shows the drop-coating method. | ||
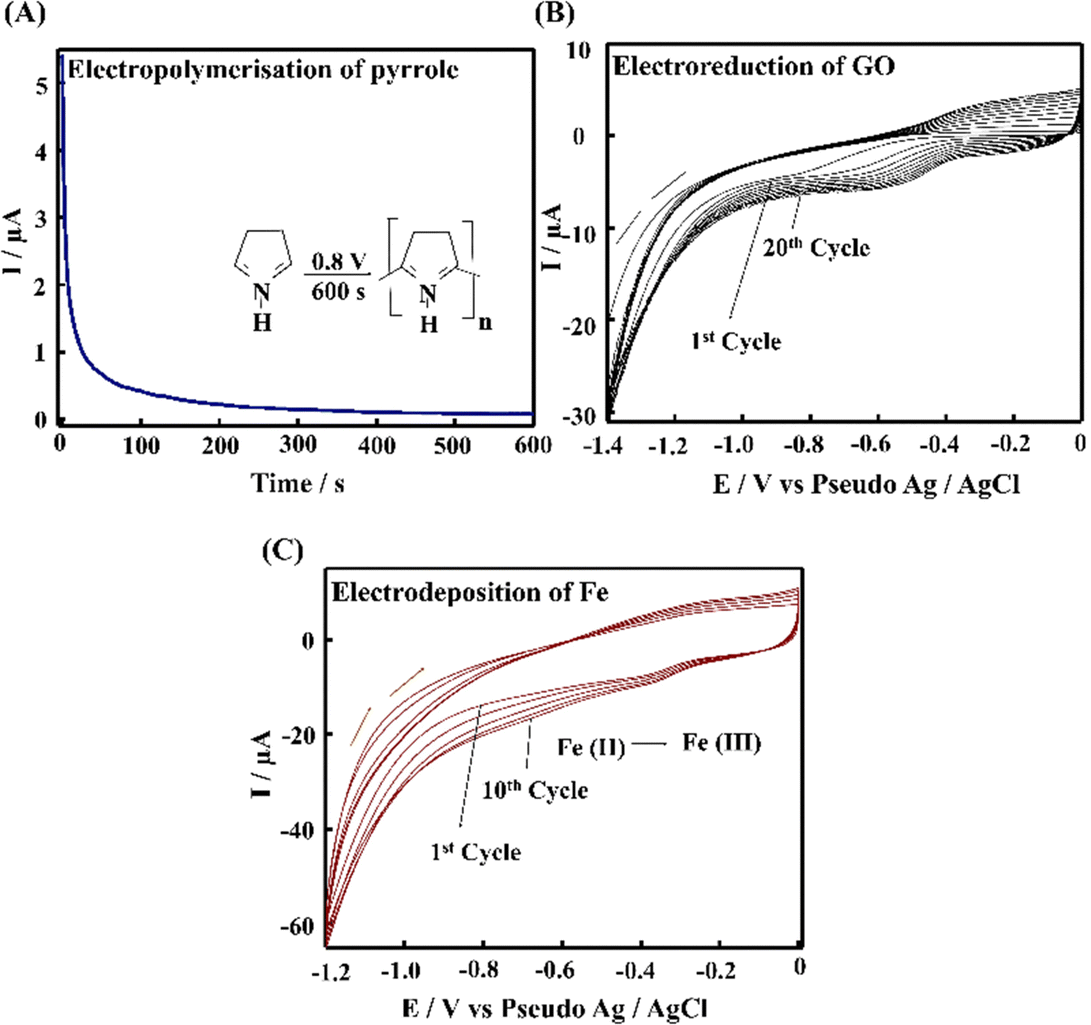 | ||
| Fig. 1 (A) Amperometry profile of electrodeposition of P(py) in solution containing 0.01 M pyrrole in 0.1 M PB solution (pH 7.4). (B) Cyclic voltammograms of the electro-reduction of 2 μL GO drop coated on PPE–P(py). (C) Cyclic voltammograms of the electrodeposition of Fe2O3 on PPE–P(py)–rGO. | ||
Extraction of Drosophila melanogaster brain samples
Scheme 2 illustrates the step-by-step process for extracting brain samples from Drosophila melanogaster. Initially, the flies were cultured under controlled environmental conditions. After three days of age, the flies were collected and subjected to anesthesia to immobilize them. Subsequently, decapitation carefully removed the head tissue of interest. The collected heads were homogenized using pellet pestles in 0.1 M phosphate buffered saline to facilitate further analysis. This homogenization process ensured the breakdown of the head tissue and the release of its contents into the buffer solution. The resulting homogenate was centrifuged at 5000 rpm for 10 minutes to separate the solid debris from the supernatant. The supernatant containing the extracted 5-HT was meticulously collected for subsequent analysis, allowing for a comprehensive quantification of the 5-HT levels in the Drosophila melanogaster brain samples.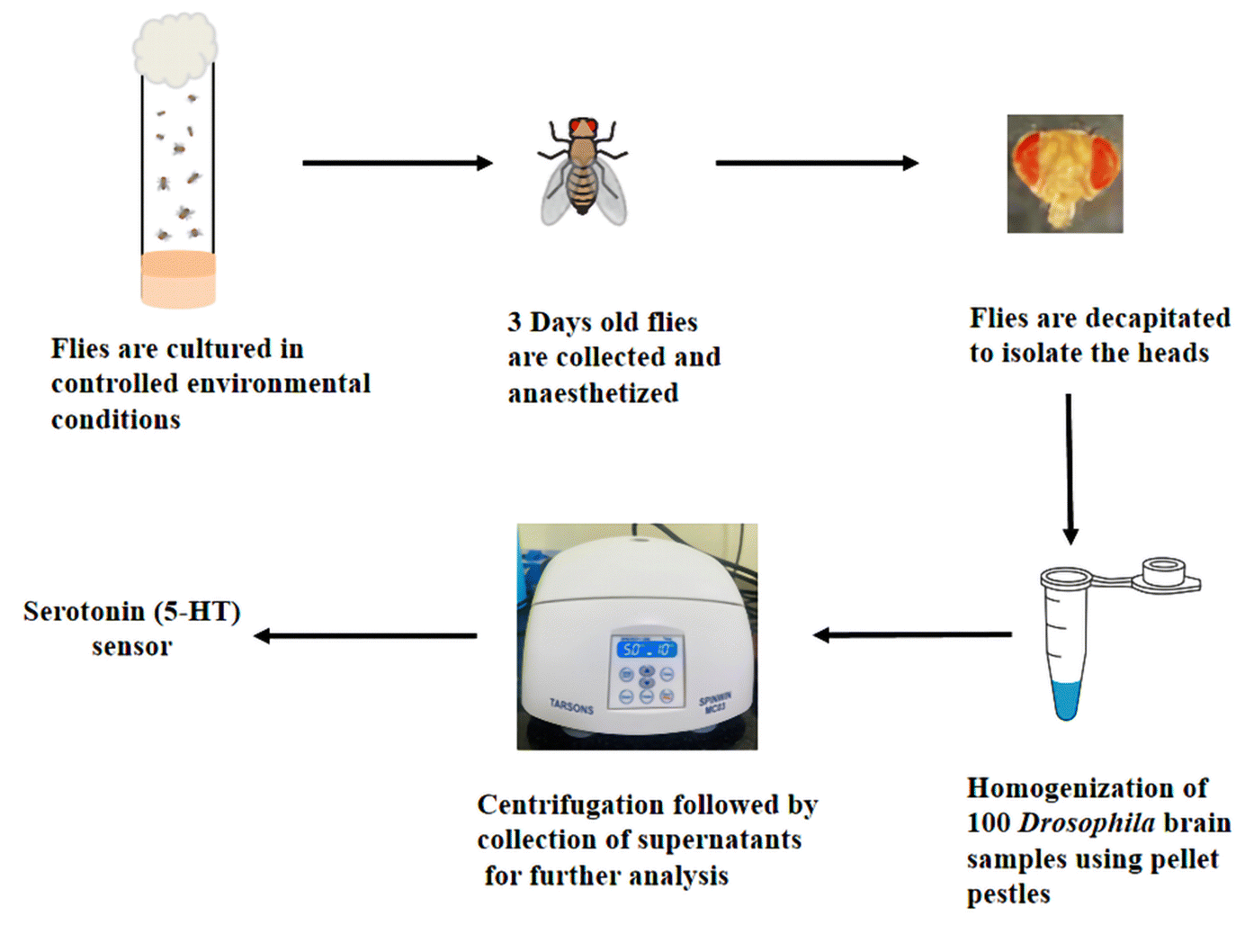 | ||
| Scheme 2 Schematic illustration of the extraction of Drosophila brain samples. | ||
Results and discussion
Characterization of PPE–P(py)–rGO–Fe2O3
To confirm the successful assembly of the electrochemical sensor, surface morphological characterization methods, such as field emission-scanning electron microscopy (FE-SEM), X-ray photoelectron spectroscopy (XPS) analysis, and Fourier transform infrared (FTIR) spectroscopy, were effectively carried out. Fig. 2 displays the images obtained using a SEM to visualize the difference in the PPE working electrode after each modification. Here, the PPE demonstrated the presence of micropores in typical cellulose fibers rather than a stacked arrangement. However, it is essential to note that the FE-SEM image of the bare PPE presented in Fig. 2(A) showed graphite flakes,15 while the interconnected network of coral cauliflower-like P(py) nanoparticles over the surface of the working electrode is identified in Fig. 2(B). Cauliflower-shaped P(py) has garnered much attention in various applications, including sensors and energy storage devices, due to its high surface area and porosity, which can enhance its sensitivity and electrochemical performance.40Fig. 2(C) displays a SEM image of electro-reduced graphene oxide (rGO), which showcases a wrinkled, sheet-like structure. When iron oxide particles are deposited onto a sheet-like reduced graphene oxide (rGO) surface, they tend to form agglomerated structures, as clearly illustrated in Fig. 2(D). These agglomerates result in the formation of clusters, where multiple iron oxide particles aggregate together on the rGO sheet.41 In addition, elemental mapping, as shown in Fig. 2(E)–(H), revealed the presence of C, N, O, and Fe at various modified electrodes, such as PPE, PPE–P(py), PPE–P(py)–rGO, and PPE–P(py)–rGO–Fe2O3, which invariably shows the elemental distribution in the nanocomposite at each step of modification involved. | ||
| Fig. 2 SEM images of different modified electrodes: (A) PPE, (B) PPE–P(py), (C) PPE–P(py)–rGO, and (D) PPE–P(py)–rGO–Fe2O3 and EDS elemental mapping of (E) PPE, (F) PPE–P(py), (G) PPE–P(py)–rGO, and (H) PPE–P(py)–Fe2O3. | ||
Furthermore, to elucidate the alterations in surface functionality during the fabrication, we conducted a thorough analysis employing Fourier-Transform Infrared Spectroscopy (ATR-FTIR), and X-ray Photoelectron Spectroscopy (XPS). Fig. S2 (ESI†) presents the outcomes derived from the AT-FTIR spectrum of the modified electrodes. The PPE modified with P(py) exhibits additional peaks at 1577 cm−1 and 1438 cm−1, which indicate the characteristic vibrations associated with C–C and C–N in the P(py) ring.42 The peaks observed at 2967 cm−1 and 3347 cm−1 correspond to stretching vibrations of C–H and O–H bonds, respectively, suggesting the incorporation of P(py) on the electrode surface. PPE–P(py)–rGO displays a slight shift in the peak, indicating a change in the chemical environment. In summary, the FTIR analysis provided compelling evidence supporting the existence of nanocomponents in the modified paper electrodes. In addition, XPS analysis was carried out to understand each modified electrode's elemental electronic states and surface composition. The XPS spectra of the newly formed nanocomposites are depicted in Fig. 3. The investigation involved wide scan and deconvoluted XPS spectra of C 1s, O 1s, N 1s, and Fe 2p for PPE–P(py) (Fig. S1A, ESI†), PPE–P(py)–rGO (Fig. S1B, ESI†), and PPE–P(py)–rGO–Fe2O3 (Fig. 3). The atomic percentages of carbon (C), oxygen (O), nitrogen (N), and iron (Fe) were determined from the XPS spectra to gain insights into the compositional changes induced by the surface modifications.
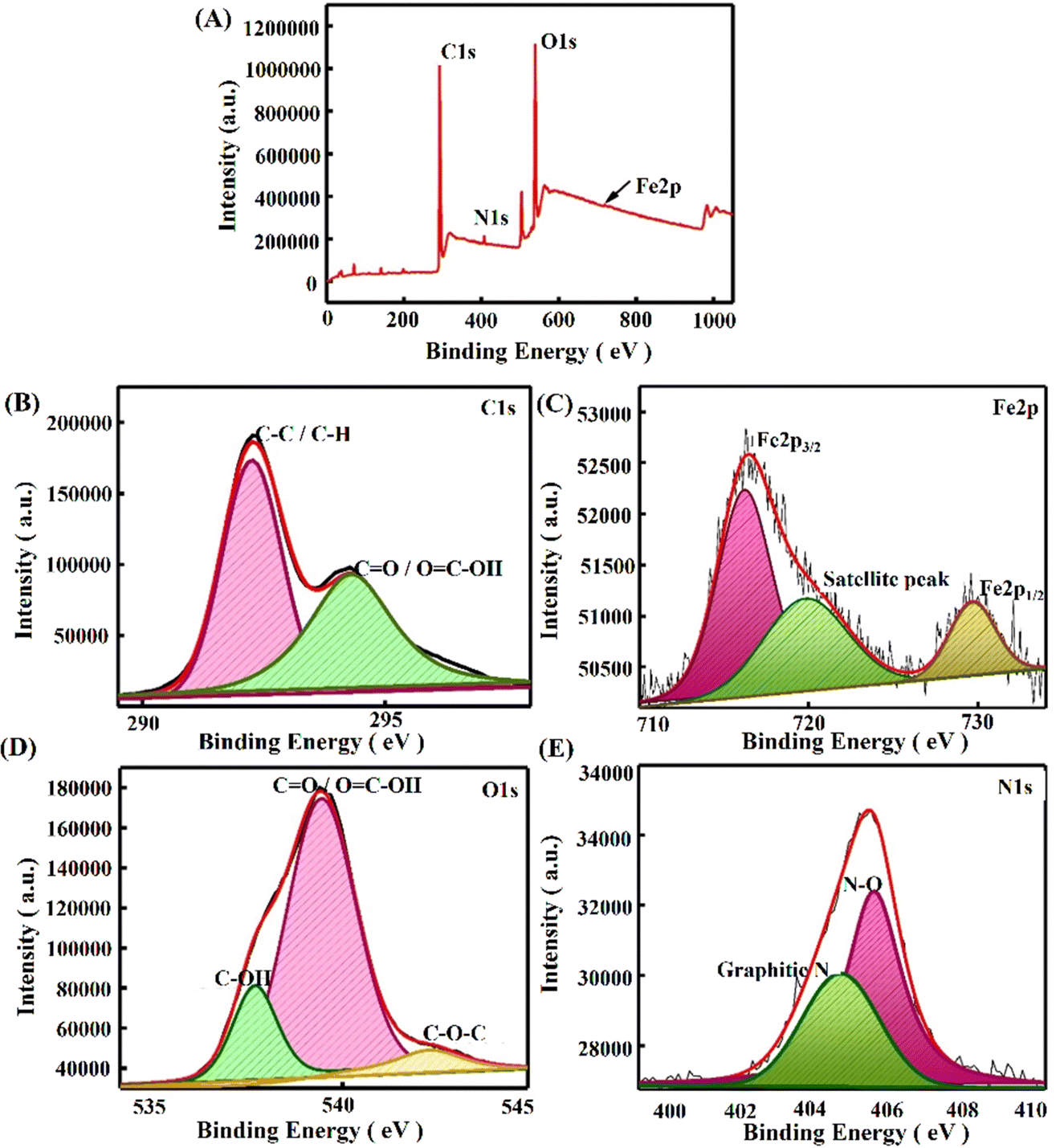 | ||
| Fig. 3 XPS wide scan deconvoluted spectrum of PPE–P(py)–rGO–Fe2O3. (A) XPS survey scan. (B) Deconvoluted C1s spectrum. (C) Deconvoluted Fe2p spectrum. (D) Deconvoluted O1s spectrum. (E) Deconvoluted N1s spectrum. | ||
The PPE exhibited a predominant composition of carbon (90.29%) and oxygen (9.71%), with no detectable nitrogen. This composition suggests that the PPE mainly consists of a carbonaceous material, likely having oxygen-containing functional groups. Upon the electrodeposition of P(py) onto the PPE, the atomic composition revealed a decrease in carbon content (87.7%), a slight increase in oxygen content (10.01%), and the appearance of nitrogen (2.29%). The presence of nitrogen indicates the successful incorporation of P(py) molecules on the PPE surface, forming nitrogen-containing functional groups. In the PPE–P(py)–rGO–Fe2O3 sample, the carbon content further decreased (81.29%) compared to the previous sample, while the oxygen content increased (16.7%). The nitrogen content also decreased to 0.81%. These observations suggest that the interaction between P(py) molecules and the PPE extends to include rGO in the system, resulting in surface chemistry modifications. Furthermore, the PPE–P(py)–rGO–Fe2O3 ternary nanocomposite exhibited a notable decrease in carbon content (75.31%), accompanied by a significant increase in oxygen content (21.38%). Minor amounts of iron (0.32%) and nitrogen (0.72%) were also detected. These findings strongly indicate the successful integration of iron oxide nanoparticles (Fe2O3) into the system, likely interacting with both rGO and P(py) on the PPE surface. The observed changes in carbon, oxygen, nitrogen, and iron contents suggest the formation of new chemical bonds and functional groups, resulting in altered surface properties. The XPS results thus provide valuable insights into the layer-by-layer modification of the PPE.
Notably, an increase in the O1/C1 atomic ratio was consistently observed throughout the layer modifications, signifying a higher proportion of oxygen atoms relative to carbon atoms. The increasing oxygen content and the elevated O1/C1 ratios observed for PPE–P(py), PPE–P(py)–rGO, and PPE–P(py)–rGO–Fe2O3 (measured as 0.114, 0.205, and 0.285, respectively) demonstrated the progressive formation of oxygen–carbon bonds or oxygen-containing functional groups, such as C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, and the incorporation of oxygen into the carbon-based materials through interactions with the oxygen atoms present in Fe2O3 in each modification step. These findings suggest that oxygen-rich groups are generated on the PPE surface during layering. Conversely, the N1/C1 ratios showed a decreasing trend with each modification, with values of 0.026, 0.01, and 0.0095 for PPE–P(py), PPE–P(py)–rGO, and PPE–P(py)–rGO–Fe2O3, respectively. This decrease in the N1/C1 ratio indicated a reduction in the nitrogen content on the PPE surface during each successive modification step.15,43,44
O bonds, and the incorporation of oxygen into the carbon-based materials through interactions with the oxygen atoms present in Fe2O3 in each modification step. These findings suggest that oxygen-rich groups are generated on the PPE surface during layering. Conversely, the N1/C1 ratios showed a decreasing trend with each modification, with values of 0.026, 0.01, and 0.0095 for PPE–P(py), PPE–P(py)–rGO, and PPE–P(py)–rGO–Fe2O3, respectively. This decrease in the N1/C1 ratio indicated a reduction in the nitrogen content on the PPE surface during each successive modification step.15,43,44
The deconvoluted C1s spectrum of the PPE–P(py) sample exhibited binding energy peaks at 289 and 291 eV, indicating the presence of C–C/C–H bonds and π–π transitions.45 However, in the case of PPE–P(py)–rGO and PPE–P(py)–rGO–Fe2O3, the deconvoluted C1s peaks shifted to 292 eV and 294 eV, respectively. The observed higher binding energy shifts are associated with functional groups on rGO and the charge distribution on P(py), which influence the electron density of the carbon atoms.46 The deconvoluted O1s spectra for all three modifications (PPE–P(py), PPE–P(py)–rGO, and PPE–P(py)–rGO–Fe2O3) showed peaks at 537 and 538 eV, indicating the presence of –COOH and –C–O–C groups. However, in the O1s spectrum of PPE–P(py)–rGO, additional binding energy peaks at 535 eV corresponded to –C–OH groups.15 In the deconvoluted N1s spectra, binding energy peaks at 404 eV and 405 eV were observed, which correspond to nitrogen defects/graphitic nitrogen and oxidized nitrogen species.45,47,48 Additionally, the N1s spectrum of PPE–P(py)–rGO exhibited an additional binding energy peak at 402 eV, indicating the presence of imine groups. Regarding the Fe (iron) spectrum, the XPS binding energy peaks observed at 716 eV, 719 eV, and 729 eV indicated the presence of iron in different oxidation states, including Fe2+ and Fe3+.47,49 The peak at 716 eV is characteristic of the Fe3+ form of Fe2O3. These binding energy peaks arise from interactions between iron and other elements or functional groups, which influence the electron density and shift in the binding energy. The presence of both Fe2+ and Fe3+ states, as indicated by the Fe 2p XPS spectrum and the peak at 729 eV (corresponding to the Fe 2p1/2 core level binding energy), supports the identification of iron(III) oxide (Fe2O3) in the analysed sample.50 The increase in the O1/C1 ratio and the decrease in the N1/C1 ratio confirmed the successful layer-by-layer modification of the PPE. These changes in elemental composition and surface chemistry indicate the successful incorporation of P(py), rGO, and Fe2O3 nanoparticles on the PPE, leading to potentially enhanced properties and new applications for the resulting nanocomposite materials. These combined EDS and XPS results have provided strong evidence for the successful PPE–P(py)–rGO–Fe2O3 sensor fabrication.
Electrochemical behaviour of 5-HT at PPE–P(py)–rGO–Fe2O3
The electrochemical performance of the modified electrodes was assessed via voltammetric experiments using a commonly employed cationic redox probe Ru (NH3)6Cl3, which exhibited a redox peak with a high current as depicted in Fig. 4(A). The perceived voltammograms revealed an increase in the oxidation peak potential after each modification with a slight anodic shift. The conductivity of the formed films of P(py) is initially due to the transfer of electrons along the conjugated π-molecular orbital backbone and the movement of charge carriers within the material. Upon oxidation, electrons are removed from the π-system of the backbone, creating cations and causing local distortions in every four pyrrole units. These distortions result in entities known as polarons. Polarons combine as the oxidation progresses to higher levels, forming energetically favourable structures called bipolarons. Bipolarons play a crucial role in the conducting polymer framework as primary charge transport carriers, facilitating electrical conductivity.38,51 Introducing rGO on PPE–P(py) enhances the specific capacitance due to improved conductivity and improves ion diffusion into the electroactive materials on the electrode surface.52 Including Fe2O3 in the nanocomposite enables efficient interfacial electron transport, promoting faster movement of electrons across the material. Additionally, the nanocomposite offers a significant amount of active sites, which enhance its capacity to participate in electrochemical reactions, thereby contributing to increased specific capacitance. This shift in peak potential indicated that the modified surface acted as a catalyst, enhancing the electrochemical reaction rate and resulting in a higher peak current (Fig. 4(A)). The modified electrode displayed a ΔEp greater than 60 mV and Ipa/Ipc exceeding unity, demonstrating a quasi-reversible process at the electrode surface. The observed asymmetry in anodic and cathodic currents suggested that the electrochemical reaction is not entirely symmetric, with a higher ratio magnitude indicating the greater dominance of the anodic reaction.53 The enhanced electron transfer properties observed for the nanocomposite are primarily attributed to P(py) and rGO, leading to the observed rise in the active area. This increase in the active area is attributed to the surface irregularities and defects on the modified electrode surface, providing more reaction sites for the analyte to interact with, causing an amplified signal output.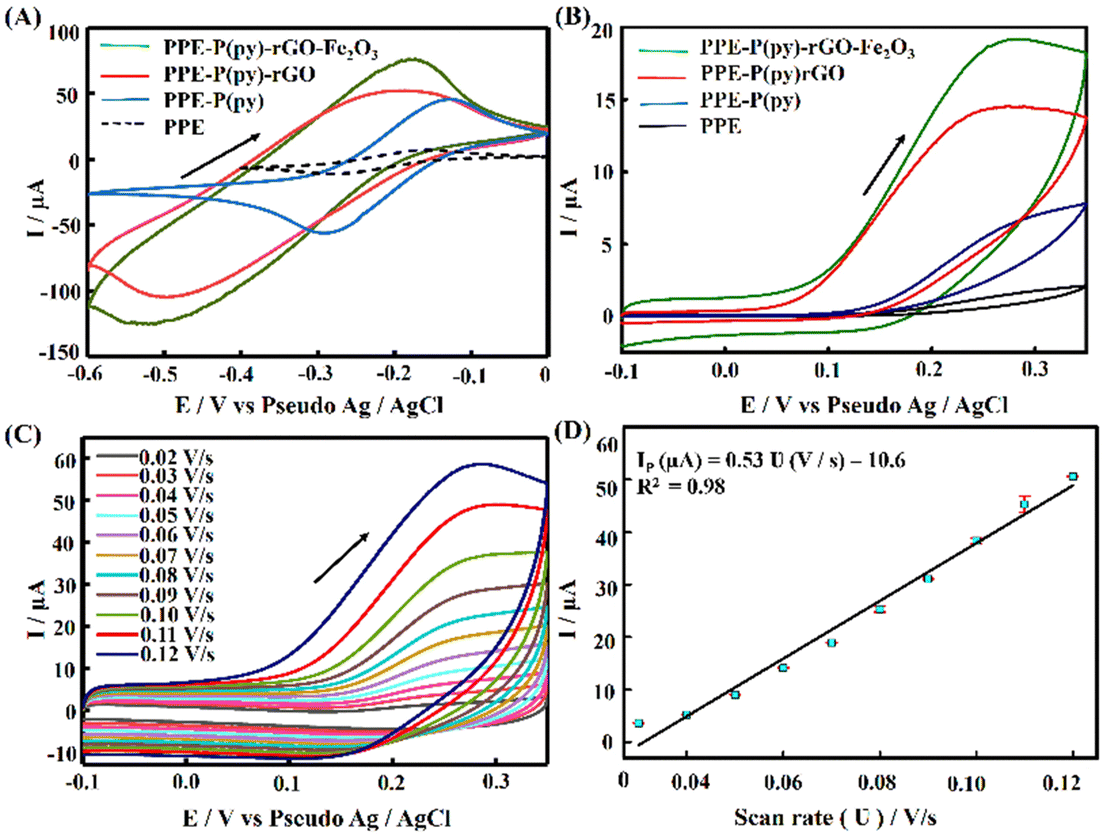 | ||
| Fig. 4 Cyclic voltammetry (CV) analysis of various modified electrodes in different electrochemical environments. (A) CVs of PPE, PPE–P(py), PPE–P(py)–rGO, and PPE–P(py)–rGO–Fe2O3 with 1 mM Ru(NH3)6Cl3 in 0.1 M KCl at a scan rate of 0.05 V s−1. (B) CVs of PPE, PPE–P(py), PPE–P(py)–rGO, and PPE–P(py)–rGO–Fe2O3 with 1 mM 5-HT in 0.1 PB solution (pH 7.4) at 0.05 V s−1. (C) CV analysis of PPE–P(py)–rGO–Fe2O3 at different scan rates from 0.02 to 0.12 V s−1 in 0.1 M PB solution (pH 7.4) containing 1 mM 5-HT and the corresponding (D) linear relationship between the peak current (Ip) and scan rate (ν). | ||
To determine the electroactive area of the electrode, we employed the Randles–Sevcik equation for quasi-reversible electrochemical systems,15,54
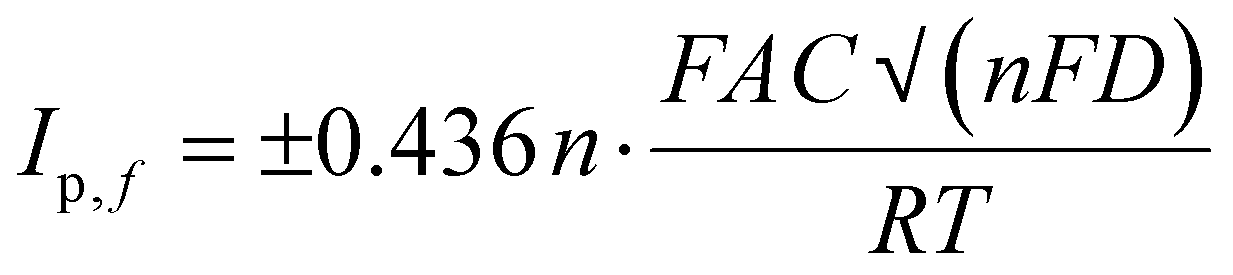
Furthermore, PPE–P(py)–rGO–Fe2O3 was evaluated for its electrochemical behaviour using cyclic voltammetry with 1 mM 5-HT in 0.1 M PB solution at pH 7.4 and a scan rate of 0.05 V s−1. The cyclic voltammograms obtained for PPE, PPE–P(py), PPE–P(py)–rGO, and PPE–P(py)–rGO–Fe2O3 are summarized in Fig. 4(B). The unmodified PPE exhibited an ill-defined peak meant for 5-HT oxidation, and on the other hand, PPE–P(py) displayed a remarkable oxidation peak with increased peak current. The oxidation peak current intensity was further enhanced for PPE–P(py)–rGO, which was further increased for PPE–P(py)–rGO–Fe2O3, indicating the importance of rGO and Fe2O3 in the electrochemical detection of 5-HT.55 Combining P(py) with rGO and Fe2O3 nanoparticles resulted in a 2.4-fold increase in oxidation peak current intensity compared to PPE–P(py) and a 10.8-fold increase compared to the PPE, attributed to the good electron transport behaviour of P(py) and rGO. Moreover, the 5-HT response of PPE–P(py)–rGO–Fe2O3 was 1.3 times higher than that of PPE–P(py)–rGO, demonstrating the strong electrocatalytic behaviour of Fe2O3. The anodic peak potential (Epa) of 5-HT at PPE–P(py)–rGO–Fe2O3 was observed to be 0.28 V. The peak current is due to the oxidation reaction of 5-HT to 5-HT quinone imine. Here, 5-HT, an electro-active neurotransmitter, can undergo electrochemical oxidation in a solvent with physiological pH.
In general, the oxidation process involves a two-step mechanism, characterized by two-electron, two-proton transfer,56 wherein 5-HT is initially oxidized to form a carbocation, followed by a subsequent oxidation step leading to the formation of quinone imine, as depicted in Scheme 3.56
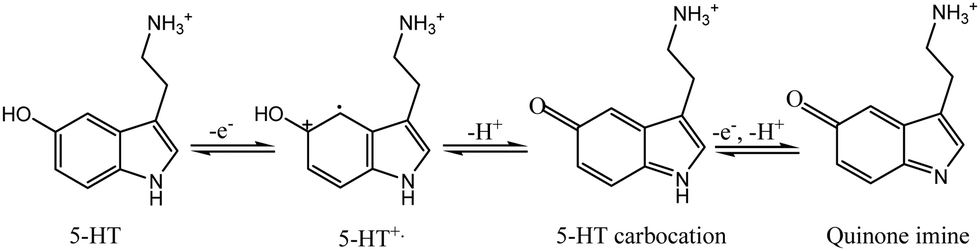 | ||
| Scheme 3 Illustration of the experimentally validated two-step oxidation mechanism of 5-HT. | ||
The remarkable enhancement of the electrocatalytic current observed at the PPE–P(py)–rGO–Fe2O3 electrode can be attributed to the catalytic properties exhibited by Fe2O3. Fe2O3 facilitates electron transfer between the electrode and 5-HT molecules, facilitating their oxidation. By accepting electrons from the electrode, Fe2O3 promotes the transfer of electrons to the 5-HT molecules, thereby promoting their oxidation process. Moreover, the paper electrode modified with P(py)–rGO indeed plays an important role in the electrode performance towards 5-HT. The P(py)–rGO composite provides a conductive pathway for electron transfer from the electrode to the Fe2O3 nanoparticles and a high surface area for the adsorption of serotonin molecules and their subsequent oxidation. The electrochemical oxidation of 5-HT at the electroactive sites of Fe3+ results in the formation of quinone imine, generating Fe2+ ions and resulting in an increased anodic peak current.57 The increase in oxidation current for 5-HT observed at the Fe2O3-modified electrode (rGO and P(py)), serving as a catalyst, promotes the electrochemical oxidation reaction and facilitates electron transfer. Iron(III) oxide (Fe2O3) is a good catalyst for the oxidation of 5-hydroxytryptamine (5-HT) to quinone imine. This is because the Fe(III)/Fe(II) ion center in Fe2O3 can catalyse the oxidation of 5-HT.58 The observed oxidation peak for 5-HT is due to the two-electron oxidation of 5-HT. The electrochemical response of the 5-HT electrode involves a two-step process: an initial electrochemical oxidation followed by a subsequent chemical reaction. In the first step, ferrous ions (Fe(II)) undergo electrochemical oxidation to form ferric ions (Fe(III)) (eqn (1)). This electrochemical process is responsible for the generation of the electrical signal. In the second step, the chemically reactive Fe(III) ions act as an oxidant, causing the oxidation of 5-hydroxytryptamine (5-HT) to form the corresponding quinone derivative (eqn (2)).
| Fe(II) + 2e− → Fe(III) | (1) |
| 5-HT + Fe(III) → 5-HT quinone imine + Fe(II) + H2O | (2) |
O bond in the quinone imine is observed.59 The appearance of this peak in the spectrum after 5-HT electrochemical oxidation indicates that the quinone is formed as a product of the oxidation reaction. The product analysis was carried out by conducting electrochemical bulk oxidation of 5-HT with an applied potential of 0.28 V for 900 s.
A scan rate study was conducted to gain a deeper understanding of the electrochemical behaviour of 5-HT on the surface of PPE–P(py)–rGO–Fe2O3. The study aimed to investigate the influence of the scan rate on the oxidation peak current of 5-HT in a 0.1 M PB solution. The results, presented in Fig. 4(C), revealed a gradual increase in the anodic oxidation peak current (Ipa) as the scan rate ranged from 0.02 V s−1 to 0.12 V s−1. The plot of the scan rate against anodic peak current intensity (Fig. 4(D)) yielded a linear relationship. This observation suggests a surface-controlled electrochemical reaction60 on the PPE–P(py)–rGO–Fe2O3 sensor. From the above information, the concentration of the adsorbed analytes (C) on PPE–P(py)–rGO–Fe2O3 was calculated using the Brown–Anson model equation Ipa = (n2F2CAϑ)/4RT and was found to be 0.137 × 10−9 mole cm−2.61 Furthermore, the electronic transport properties of individual layers were validated by electrochemical impedance spectroscopy (EIS) in a 0.1 M KCl solution containing 5 mM [Fe(CN)6]3−/4− at open circuit potential (OCP) over a frequency range of 100 mHz to 1 MHz (Fig. 5(A)). The Randles-equivalent circuit is a model that consists of four components: solution resistance (Rs), double-layer capacitance (Cdl), charge transfer resistance (Rct), and Warburg impedance (Zw). The initial point of the semicircle in the Nyquist plot corresponds to solution resistance (Rs), while the diameter of the semicircle represents the charge transfer resistance (Rct) (Fig. 5(A)). The bare electrode displayed a higher charge transfer resistance (1317.48 Ω), while PPE–P(py)–rGO–Fe2O3 showed a lower Rct (34.61 Ω), indicating its excellent conductivity due to high electron transfer efficiency. Rct is mathematically related to the thermodynamic gas constant (R), the absolute temperature (T), the standard heterogeneous electron transfer rate constant (K0), the number of transferred electrons (N), Faraday's constant (F), the electrode surface area (A), and the concentration of the redox species in the solution (C), as expressed by the equation, Rct = RT/K0N2F2AC.61 The calculated K0 values of PPE and PPE–P(py)–rGO–Fe2O3 were found to be 0.005 × 10−4 and 0.6 × 10−4, respectively. The observed combination of a low charge-transfer resistance (Rct) value and a high standard heterogeneous electron transfer rate constant (K0) value can be attributed to a synergistic effect that enhances the sensor's performance, making it highly suitable for 5-HT detection.38,62
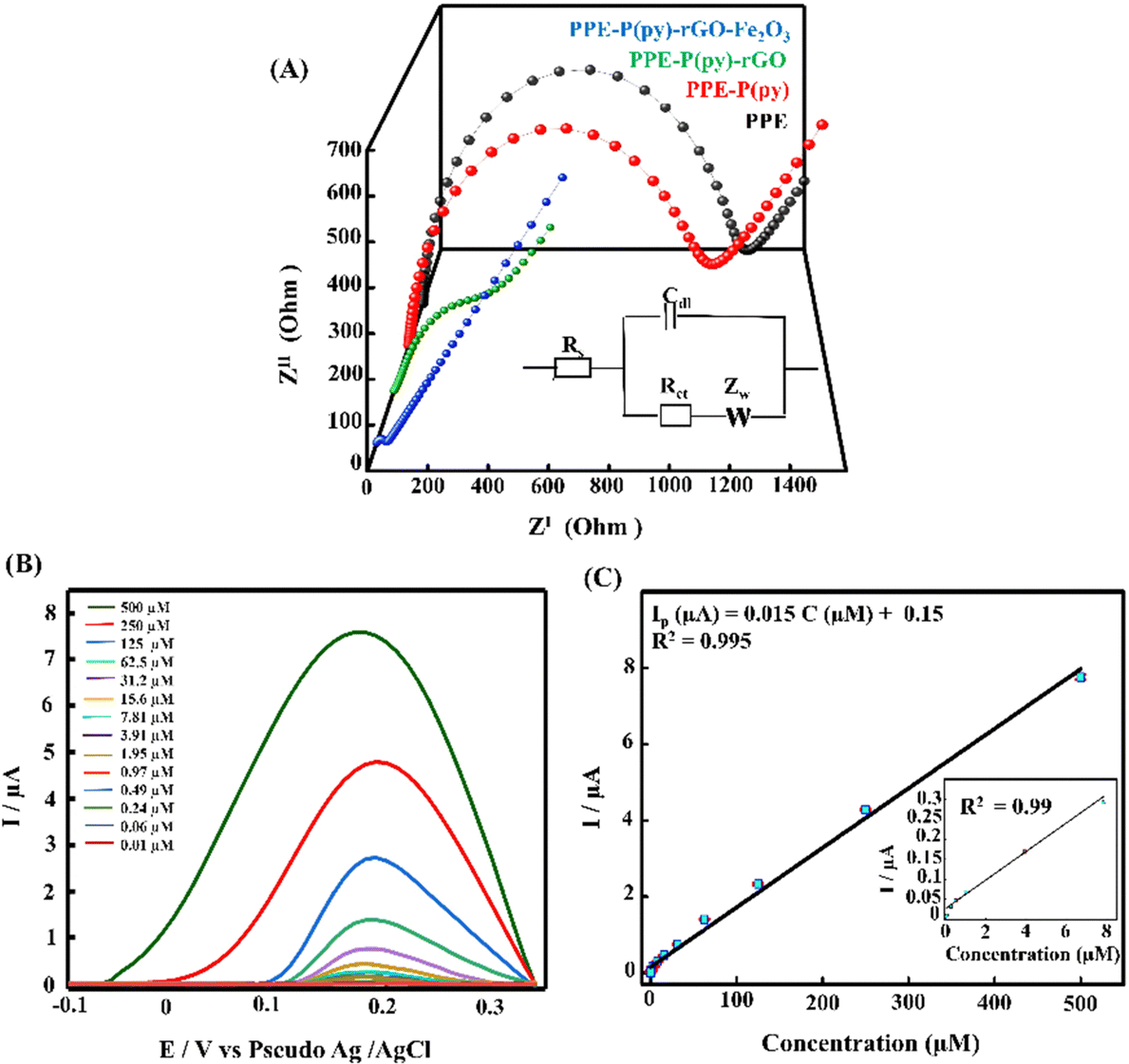 | ||
| Fig. 5 (A) Electrochemical impedance spectroscopy of the bare PPE (black) and PPE–P(py) (red), PPE–P(py)–rGO (green), and PPE–P(py)–rGO–Fe2O3 (blue) modified electrodes in 0.1 M KCl solution containing 5 mM [Fe(CN)6]3−/4−. (B) DPV for the different concentrations of 5-HT from 500 μM to 0.01 μM. (C) The corresponding calibration plot (inset shows the linear calibration plot from 7.81 μM to 0.01 μM for 5-HT). | ||
Sensitivity and selectivity study of the developed 5-HT sensor
Differential pulse voltammetry (DPV) analysis was employed to evaluate the analytical performance of PPE–P(py)–rGO–Fe2O3 for detecting 5-HT in 0.1 M PB solution at pH 7.4. Fig. 5(B) displays the DPV voltammogram response for different concentrations of 5-HT, indicating an amplified DPV current response signal. The shift of the anodic peak potential towards a less positive value with increasing 5-HT concentration can be attributed to enhanced mass transport arising from the elevated concentration of 5-HT molecules in the solution, leading to an increase in current and a corresponding shift in the peak potential.63 The average of peak current responses was recorded and plotted against concentration and the obtained linearity between the 5-HT concentration and peak current (Ip) is demonstrated in Fig. 5(C). The linear regression equation for the analyte can be expressed as Ip (μA) = 0.015C (μM) + 0.15 (R2 = 0.995). The direct correlation between the oxidation peak current and the concentration of 5-HT indicates the excellent detection capability of the proposed electrode, yielding a limit of detection (LOD) of 22 nM (3SD blank/slope of the calibration plot). Notably, the wide linear range of detection starting from 0.01 μM to 500 μM invariably shows the potential of the as-developed sensor for clinical use. It should be noted that the clinically found levels of 5-HT in blood and urine fall well below the detection range obtained in the present work.64 The results mentioned above proved that PPE–P(py)–rGO–Fe2O3 showed excellent sensitivity towards 5-HT, and the results were comparable to the 5-HT sensors reported so far, as indicated in Table S2 (ESI†).Evaluating the anti-interference capability of PPE–P(py)–rGO–Fe2O3 is essential for its successful application in the electrochemical analysis of real samples. Optimization experiments were conducted to investigate the effects of potential interferences on the measurement of 5-HT at a concentration of 50 μM, including 50 μM concentration of ascorbic acid (AA), uric acid (UA), dopamine (DA), and epinephrine (E). The performance of the modified electrode was assessed, and the resulting data were carefully analysed and summarized in Fig. 6(B) and Fig. S3A (ESI†). Interestingly, adding interfering chemicals had a minimal impact on the anodic peak current response of the modified electrode, indicating its excellent anti-interference properties. However, the presence of interfering E showed a slight variation in peak current responses. Interestingly, the sensor exhibited dual detection behaviour when tested with 5-HT and DA, where simultaneous detection of both biomolecules was possible without any substantial change in the oxidative peak current for 5-HT (Fig. S3A, ESI†).65,66,67 Despite the slight effect, the modified electrode still demonstrated remarkable anti-interference properties, indicating its potential for accurate and reliable electrochemical measurements of 5-HT in the presence of interfering species. It should be noted that Fe2O3 nanoparticles are known to have a high affinity for serotonin.57 Fe2O3 has a strong electrostatic interaction with the positively charged amino group on the serotonin molecule. The modified electrode surface exhibited electrostatic repulsive behaviour towards other negative analytes such as AA and UA. Unlike DA, epinephrine showed slight interference with 5-HT detection (Fig. S3A, ESI†). Interestingly the DA oxidation occurred at a much lower potential than the common oxidation potential known for DA. This could be attributed to the chemical modification of the electrode surface with P(py)–rGO and Fe2O3. The selective electrochemical oxidation process of 5-HT at the modified electrode is similar to the oxidation of folic acid reported at the Fe2O3 modified electrode.58,67
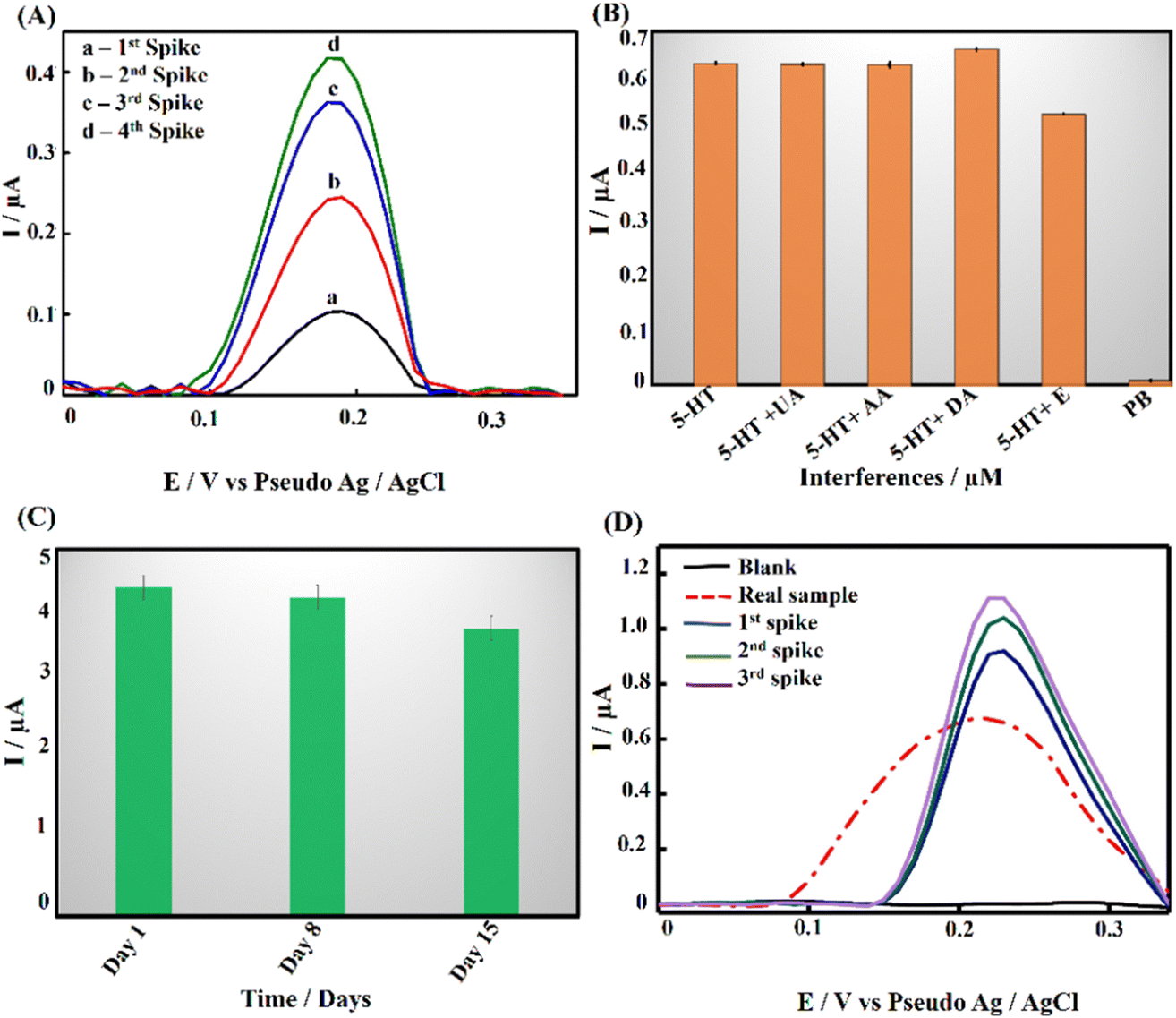 | ||
| Fig. 6 (A) DPV voltammogram for the recovery study. (B) Histogram showing the selectivity of the developed PPE–P(py)–rGO–Fe2O3 sensor towards interfering species (AA, UA, DA, E). (C) Histogram depicting the results of the stability study conducted on the PPE–P(py)–rGO–Fe2O3 sensor stored at room temperature for a period of 15 days. (D) DPV response for Drosophila melanogaster brain samples. | ||
Recovery, repeatability/reproducibility, and stability of the developed sensor
A recovery analysis of standard 5-HT was conducted to assess the accuracy and reliability of the developed method. Fig. 6(A) displays DPV voltammograms for the recovery study. Known amounts of the target analyte were measured, and the observed percentage recovery ranged from 96.7% to 104% (Table S1, ESI†). These results indicate that the developed method showed satisfactory results suitable for real-world applications. To further support the above-said features, repeatability tests were performed with 500 μM 5-HT using 3 different electrodes and the results are summarized in Fig. S3B (ESI†). The results revealed insignificant discrepancies that may be ascribed to non-uniform electrodeposition, potentially caused by irregularities and pores. Nonetheless, an outstanding response was observed in three of the four electrodes. Additionally, the stability of PPE–P(py)–rGO–Fe2O3 (Fig. 6(C)) was assessed by preparing 12 electrodes under identical conditions and storing them at room temperature. The electrodes were tested for stability at seven-day intervals with 250 μM 5-HT. The obtained graph showed that the oxidation peak current remained stable for up to two weeks, with a slight reduction observed on the 15th day. Therefore, the sensor can remain stable for two weeks and could be successfully used for the detection of 5-HT through a single-run approach. Overall, the findings demonstrated the feasibility of using the as-developed ternary nanocomposite as a promising electrode material for 5-HT sensing.Detection of 5-HT in Drosophila brain samples and validation
The real-time application of PPE–P(py)–rGO–Fe2O3 was investigated using differential pulse voltammetry (DPV) measurements with brain samples of Drosophila melanogaster. 100 brain samples were homogenized in a 0.1 M PB solution and centrifuged to obtain the supernatant for analysis. The modified electrode responded excellently when exposed to the real sample (Fig. 6(D)). Remarkably, adding a known quantity of 5-HT to the sample increased the current signal, as shown in Fig. 6(D). Based on this experiment, the concentration of 5-HT in the real sample was determined to be 33.3 μM.Furthermore, the findings of the modified electrode were validated by comparing them with the results obtained from the high-performance liquid chromatography (HPLC) method (Fig. S6, ESI†). The concentration of the analyte, in this case, was determined to be 32 μM. The close agreement between the results obtained for the modified electrode and the HPLC method showcased the accuracy and reliability of the modified electrode in quantifying 5-HT in real samples (Table 1). In order to check the analytical feasibility, the spike sample analysis in Table 2 depicts the mean obtained recoveries of 101.05%, 98.22%, and 100.78% for 5-HT analysis, accompanied by the respective relative standard deviation (RSD) of 0.054%, 0.99%, and 1.32%. These findings confirmed the exceptional precision of the newly developed electrode in quantifying 5-HT. As a result, the developed sensor exhibits significant potential for precisely identifying 5-HT in human samples, rendering it an appealing candidate for potential applications in analytical chemistry and biosensing.
| Real sample | Determined concentration by the EC method | Determined concentration by the HPLC-UV method | Recovery |
|---|---|---|---|
| Brain sample of Drosophila melanogaster | 33.3 μM | 31.84 μM | 95.6% |
| Sample | Added amount (μM) | Found amount (μM) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Drosophila brain sample | — | 33.3 | 95.6 | 0.13 |
| 1st spike | 10 | 42.1 | 97.2 | 0.054 |
| 2nd spike | 20 | 53.2 | 99.8 | 0.99 |
| 3rd spike | 30 | 64.7 | 102.2 | 1.32 |
Conclusions
In this study, we successfully fabricated a ternary nanocomposite P(py)–rGO–Fe2O3 decorated paper electrode as a disposable electrochemical sensor for 5-HT and tested its efficiency in monitoring 5-HT in the brain samples of Drosophila melanogaster. The sensor was well characterized using electron microscopy and spectral techniques and subjected to in-depth CV, DPV, and EIS studies. The results revealed the effective formation of the ternary nanocomposite on the PPE surface and its improved electron transfer properties, because of the synergistic effect due to P(py), rGO and Fe2O3 composites. The DPV analysis further confirmed the remarkable analytical characteristics with an impressive LOD of 22 nM and a wider linear detection range spanning from 0.01 μM to 500 μM, which was found to be clinically relevant. The sensor exhibited excellent sensitivity and selectivity even in the presence of interferents such as AA, UA and DA. However, a slight decrease in peak current was observed in the presence of a neurotransmitter, E. The validation of the fabricated sensor was carried out by comparing the levels of 5-HT in the brain samples of Drosophila melanogaster with the gold standard HPLC-UV technique, ensuring the reliability and accuracy of our developed sensor. The sensor showed good stability and reproducibility, making it a promising tool for the rapid detection of 5-HT levels in different neurological conditions or disorders in humans.Abbreviations
| PPE | Paper electrode |
| P(py) | Poly(pyrrole) |
| rGO | Reduced graphene oxide |
| GO | Graphene oxide |
| 5-HT | 5-Hydroxytryptamine |
| DPV | Differential pulse voltammetry |
| HPLC-UV | High-performance liquid chromatography with ultra-violet spectroscopy |
| SS | Serotonin syndrome |
| 5-HT-1A | Serotonin 1A receptor |
| 5-HT-2A | Serotonin 2A receptor |
| EG-Au-gate-FET | An extended-gold-gate-field effect transistor |
| PME | Polymer-modified electrode |
| ZnONR | Zinc oxide nanorods |
| PMB (DES) | Polymethylene blue (deep eutectic solvent) |
| FeC | Ferrocene |
| MWCNTs | Multiwalled carbon nanotubes |
| AuNPs | Gold nanoparticles |
| Ag2Se | Silver selenide |
| Fe2O3 | Ferric oxide |
| EIS | Electrochemical impedance spectroscopy |
| CV | Cyclic voltammetry |
| ATR-FTIR | Attenuated total reflectance-Fourier transform infrared spectroscopy |
| FE-SEM | Field emission scanning electron microscopy |
| EDS | Energy dispersive X-ray spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| PB | Phosphate buffer |
| ECE | Electron transfer-chemical reaction-electron transfer mechanism. |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. is grateful to DST for the Women Scientist-A Fellowship (ref. no. DST/WOS-A/CS-53/2021). KSP is thankful for the BIRAC-ETA, India grant (YTI/ETA/SL/PR009) and the Yenepoya Technology Incubator MedTech rapid prototyping facility supported by National Biopharma Mission (NBM), DBT-BIRAC.References
- N. L. Baganz and R. D. Blakely, ACS Chem. Neurosci., 2013, 4(1), 48–63 CrossRef CAS PubMed.
- D. H. Edwards and E. A. Kravitz, Curr. Opin. Neurobiol., 1997, 7(6), 812–819 CrossRef CAS PubMed.
- M. Berger, J. A. Gray and B. L. Roth, Annu. Rev. Med., 2009, 60(1), 355–366 CrossRef CAS PubMed.
- N. Mikkelsen, P. Damkier and S. A. Pedersen, Basic Clin. Pharmacol. Toxicol., 2023, 133(2), 124–129 CrossRef CAS PubMed.
- J. Thomas, R. Khanam and D. Vohora, Pharm. Biol., 2015, 53(10), 1539–1544 CrossRef CAS.
- S. G. Chavan, A. K. Yagati, H. T. Kim, E. Jin, S. R. Park and D. V. Patil, et al. , Biosens. Bioelectron., 2021, 191, 113447 CrossRef CAS.
- S. Nishitani and T. Sakata, ACS Appl. Mater. Interfaces, 2020, 12(13), 14761–14769 CrossRef CAS.
- P. Q. Tran Do, V. Thi Huong, N. T. Truc Phuong, T. H. Nguyen, H. K. Thi Ta and H. Ju, et al. , RSC Adv., 2020, 10(51), 30858–30869 RSC.
- M. Dinarvand, E. Neubert, D. Meyer, G. Selvaggio, F. A. Mann and L. Erpenbeck, et al. , Nano Lett., 2019, 19(9), 6604–6611 CrossRef CAS PubMed.
- K. Khoshnevisan, E. Honarvarfard, F. Torabi, H. Maleki, H. Baharifar and F. Faridbod, et al. , Clin. Chim. Acta, 2020, 501, 112–119 CrossRef CAS.
- S. Sharma, N. Singh, V. Tomar and R. Chandra, Biosens. Bioelectron., 2018, 107, 76–93 CrossRef CAS.
- X. Wan, H. Du, D. Tuo, X. Qi, T. Wang and J. Wu, et al. , ACS Appl. Nano Mater., 2023, 6(20), 19403–19413 CrossRef CAS.
- Y. Xia, G. Li, Y. Zhu, Q. He and C. Hu, Microchem. J., 2023, 190, 108726 CrossRef CAS.
- C. J. Valentine, K. Takagishi, S. Umezu, R. Daly and M. De Volder, ACS Appl. Mater. Interfaces, 2020, 12(27), 30680–30685 CrossRef CAS.
- J. Sonia, G. M. Zanhal and K. S. Prasad, Microchem. J., 2020, 158, 105164 CrossRef CAS.
- M. H. Naveen, N. G. Gurudatt and Y. B. Shim, Appl. Mater. Today, 2017, 9, 419–433 CrossRef.
- G. Li, J. Wu, X. Qi, X. Wan, Y. Liu and Y. Chen, et al. , Mater. Today Chem., 2022, 26, 101043 CrossRef CAS.
- S. Geetha, C. R. K. Rao, M. Vijayan and D. C. Trivedi, Anal. Chim. Acta, 2006, 568(1), 119–125 CrossRef CAS PubMed.
- R. Jain, N. Jadon and A. Pawaiya, TrAC, Trends Anal. Chem., 2017, 97, 363–373 CrossRef CAS.
- T. Qian, C. Yu, X. Zhou, P. Ma, S. Wu and L. Xu, et al. , Biosens. Bioelectron., 2014, 58, 237–241 CrossRef CAS PubMed.
- C. Tlili, H. Korri-Youssoufi, L. Ponsonnet, C. Martelet and N. J. Jaffrezic-Renault, Talanta, 2005, 68(1), 131–137 CrossRef CAS.
- S. Dhibar and C. K. Das, J. Appl. Polym. Sci., 2017, 134(16), 44724 CrossRef.
- J. Xue, Q. Sun, Y. Zhang, W. Mao, F. Li and C. Yin, ACS Omega, 2020, 5(19), 10995–11004 CrossRef CAS PubMed.
- V. Balakumar and P. Prakash, J. Catal., 2016, 344, 795–805 CrossRef CAS.
- K. Ghanbari and S. Bonyadi, New J. Chem., 2018, 42(11), 8512–8523 RSC.
- R. R. Poolakkandy and M. M. Menamparambath, Electrochem. Sci. Adv., 2021, 1(2), e2000024 CrossRef CAS.
- S. Selvarajan, A. Suganthi and M. Rajarajan, Ultrason. Sonochem., 2018, 44, 319–330 CrossRef CAS PubMed.
- S. Panneer Selvam and K. Yun, Sens. Actuators, B, 2020, 302, 127161 CrossRef CAS.
- T. Atici, M. Bilgi Kamaç, M. Yilmaz and A. Yilmaz Kabaca, Electrochim. Acta, 2023, 458, 142484 CrossRef CAS.
- G. Ran, X. Chen and Y. Xia, RSC Adv., 2017, 7(4), 1847–1851 RSC.
- G. E. Uwaya and O. E. Fayemi, Sens. Bio-Sens. Res., 2020, 28, 100338 CrossRef.
- B. Wu, S. Yeasmin, Y. Liu and L. J. Cheng, Sens. Actuators, B, 2022, 354, 131216 CrossRef CAS.
- A. Jeibmann and W. Paulus, Int. J. Mol. Sci., 2009, 10(2), 407–440 CrossRef CAS.
- M. Shin, J. M. Copeland and B. J. Venton, ACS Chem. Neurosci., 2018, 9(8), 1872–1883 CrossRef CAS.
- H. Manisha, J. Sonia, S. Shashikiran, S. Yuvarajan, P. D. Rekha and K. S. Prasad, Electrochem. Commun., 2022, 137, 107259 CrossRef CAS.
- P. M. Nia, W. P. Meng, F. Lorestani, M. R. Mahmoudian and Y. Alias, Sens. Actuators, B, 2015, 209, 100 CrossRef.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3(2), 101–105 CrossRef CAS PubMed.
- R. M. Alonso, M. I. San-Martín, A. Sotres and A. Escapa, Sci. Rep., 2017, 7(1), 13726 CrossRef CAS PubMed.
- P. Singh, S. Mandal, D. Roy and N. Chanda, ACS Biomater. Sci. Eng., 2021, 7(7), 3446–3458 CrossRef CAS.
- G. Kulkarni, P. Kandesar, N. Velhal, H. Kim and V. Puri, J. Appl. Polym. Sci., 2021, 138 Search PubMed.
- A. Kobyliukh, K. Olszowska, U. Szeluga and S. Pusz, Adv. Colloid Interface Sci., 2020, 285, 102285 CrossRef CAS PubMed.
- M. Xiang, Z. Yang, J. Chen, S. Zhou, W. Wei and S. Dong, ACS Omega, 2020, 5(46), 29955–29962 CrossRef CAS PubMed.
- R. Yadav, P. Joshi, M. Hara, T. Yana, S. Hashimoto and M. Yoshimura, SN Appl. Sci., 2020, 2(10), 1630 CrossRef CAS.
- K. Sapna, J. Sonia, Y. B. Shim, A. B. Arun and K. S. Prasad, ACS Appl. Nano Mater., 2022, 5(9), 12454–12463 CrossRef CAS.
- G. Greczynski and L. Hultman, Prog. Mater. Sci., 2020, 107, 100591 CrossRef CAS.
- I. Matanovic, K. Artyushkova, M. B. Strand, M. J. Dzara, S. Pylypenko and P. Atanassov, J. Phys. Chem. C, 2016, 120(51), 29225–29232 CrossRef CAS.
- A. Moyseowicz, K. Pająk, K. Gajewska and G. Gryglewicz, Materials, 2020, 13(10), 2273 CrossRef CAS.
- K. S. Prasad, M. C. Chuang and J. A. A. Ho, Talanta, 2012, 88, 445–449 CrossRef CAS PubMed.
- B. Wang, J. J. Yin, X. Zhou, I. Kurash, Z. Chai and Y. Zhao, et al. , J. Phys. Chem. C, 2013, 117(1), 383–392 CrossRef CAS.
- A. Moyseowicz, A. Śliwak, E. Miniach and G. Gryglewicz, Composites, Part B, 2017, 109, 23–29 CrossRef CAS.
- T. Wang, X. Xu, Z. Ren, B. Gao and H. Wang, RSC Adv., 2016, 6(6), 5089–5099 RSC.
- J. Heinze, Electronically conducting polymers, in Electrochemistry IV, Topics in Current Chemistry, ed. E. Steckhan, Springer, Berlin, Heidelberg, 1990, pp. 1–47 Search PubMed.
- K. S. Prasad, X. Cao, N. Gao, Q. Jin, S. T. Sanjay and G. Henao-Pabon, et al. , Sens. Actuators, B, 2020, 305, 127516 CrossRef CAS.
- A. García-Miranda Ferrari, C. W. Foster, P. J. Kelly, D. A. C. Brownson and C. E. Banks, Biosensors, 2018, 8(2), 53 CrossRef PubMed.
- R. Santhosh Kumar, K. Govindan, S. Ramakrishnan, A. R. Kim, J. S. Kim and D. J. Yoo, Appl. Surf. Sci., 2021, 556, 149765 CrossRef CAS.
- M. Z. Wrona and G. Dryhurst, Bioorg. Chem., 1990, 18(3), 291–317 CrossRef CAS.
- T. Maiyalagan, J. Sundaramurthy, P. S. Kumar, P. Kannan, M. Opallo and S. Ramakrishna, Analyst, 2013, 138(6), 1779 RSC.
- A. Taeuk, L. Namhun, C. Hong-Jun, K. Seongsoo, S. Dong-Sik and L. Sang-Myung, RSC Adv., 2017, 7(49), 30582–30587 RSC.
- Y. H. Kim, T. Kim, J. H. Ryu and Y. J. Yoo, Biosens. Bioelectron., 2010, 25(5), 1160–1165 CrossRef CAS PubMed.
- C. Fan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(16), 9134–9137 CrossRef CAS.
- A. Ch Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3(3), 162–193 CrossRef CAS.
- A. J. Steckl and P. Ray, ACS Sens., 2018, 3(10), 2025–2044 CrossRef CAS.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2022, p. 1112 Search PubMed.
- H. Kaur, S. S. Siwal, R. V. Saini, N. Singh and V. K. Thakur, ACS Nanosci. Au, 2023, 3(1), 1–27 CrossRef CAS.
- W. F. Sokol and D. H. Evans, Anal. Chem., 1981, 53(4), 578–580 CrossRef CAS.
- T. Pradhan, H. S. Jung, J. H. Jang, T. W. Kim, C. Kang and J. S. Kim, Chem. Soc. Rev., 2014, 43(13), 4684–4713 RSC.
- J. Oni and T. Nyokong, Anal. Chim. Acta, 2001, 434(1), 9–21 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00777d |
| This journal is © The Royal Society of Chemistry 2024 |