
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMelanoma immunotherapy enabled by M2 macrophage targeted immunomodulatory cowpea mosaic virus†
Zhongchao
Zhao‡
 abc,
Young Hun
Chung‡
cd and
Nicole F.
Steinmetz
*abcdefgh
abc,
Young Hun
Chung‡
cd and
Nicole F.
Steinmetz
*abcdefgh
aDepartment of NanoEngineering, University of California, 9500 Gilman Dr, La Jolla, San Diego, CA 92093, USA. E-mail: nsteinmetz@ucsd.edu
bCenter for Nano-ImmunoEngineering, University of California, 9500 Gilman Dr, La Jolla, San Diego, CA 92093, USA
cMoores Cancer Center, University of California, 9500 Gilman Dr, La Jolla, San Diego, CA 92093, USA
dDepartment of Bioengineering, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA 92093, USA
eDepartment of Radiology, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA 92093, USA
fInstitute for Materials Discovery and Design, University of California, 9500 Gilman Dr, La Jolla, San Diego, CA 92093, USA
gCenter for Engineering in Cancer, University of California, 9500 Gilman Dr, La Jolla, San Diego, CA 92093, USA
hShu and K.C. Chien and Peter Farrell Collaboratory, University of California, 9500 Gilman Dr, La Jolla, San Diego, CA 92093, USA
First published on 26th January 2024
Abstract
We have developed nanoparticle formulations targeting M2 macrophages for cancer immunotherapy by conjugating high-affinity binding peptides to cowpea mosaic virus as an immunostimulatory adjuvant. We confirmed the targeting of and uptake by M2 macrophages in vitro and the therapeutic efficacy of the nanoparticles against murine melanoma in vivo.
Main
Cancer immunotherapy is now the fourth pillar of cancer treatment following the approval of immune checkpoint blockade drugs targeting PD-1, PD-L1, and CTLA-4.1–3 However, its efficacy is hindered by primary resistance, acquired resistance, poor CD8+ T-cell infiltration, and irreversible T-cell exhaustion,4,5 which can be attributed to the immunosuppressive tumor microenvironment (TME).6 The TME is composed of tumor cells, tumor associated macrophages (TAMs), and various secreted immunosuppressive factors.7 Notably, M2-type pro-tumor TAMs (hereafter, referred to as M2 macrophages) play a pivotal role in the immunosuppressive TME by hindering antigen presentation, promoting angiogenesis, tissue remodeling, and repair, suppressing the anti-tumor immune response, and promoting tumor progression and metastasis.7–9 M2 macrophages have therefore become a major target for the development of cancer immunotherapy.10,11Next-generation nanomedicines that target M2 macrophages can be based on synthetic or inorganic nanoparticles such as PLGA, chitosan, dextran, silica, gold, lipids and carbon.12 One key mechanism is to reprogram pro-tumor M2 macrophages into anti-tumor M1 macrophages by incorporating Toll-like receptor (TLR) agonists, cytokines, antibodies, or RNAs.13 Many such formulations achieve the effective polarization of M2 macrophages, including β-cyclodextrin nanoparticles loaded with TLR7/8 agonists such as R848,14 poly(β-amino ester) nanoparticles loaded with IL-1215 or mRNA,10 and hyaluronic acid nanoparticles loaded with microRNA.16 We have developed nanoparticles based on the plant virus cowpea mosaic virus (CPMV), leveraging its natural immunostimulatory potential.17,18 While non-infectious toward mammals, the nucleoprotein assemblies are immunogenic and stimulate innate immune cells through multiple pathways: the highly organized and repetitive nature of the viral capsid assembly itself can be viewed as a pathogen-associated geometric pattern similar to pathogen-associated molecular patterns (PAMPs); further immunostimulation is provided through engagement with complement, presentation of Thelper epitopes, and engagement with TLRs.19–21
Here we describe the development of an intratumorally injected immunotherapy based on CPMV, which we modified to target M2 macrophages. CPMV has shown promising efficacy as an intra-tumoral agent for the treatment of murine melanoma, breast cancer, colorectal cancer, glioma, and ovarian cancer, as well as canine patients with oral melanoma and breast cancer.22–27 Mechanistic studies have shown that CPMV acts as a triple TLR agonist (TLRs 2, 4, and 7) to stimulate the innate immune system, thereby inducing an anti-tumor immune response.28,29 Intratumorally injected CPMV can reprogram the immunosuppressive TME into immunostimulatory by recruiting and activating N1-type neutrophils, natural killer (NK), dendritic cells, and converting M2 macrophages to M1 macrophages.23 These activated and mature innate immune cells process tumor-associated antigens and neoantigens in the tumor to generate tumor-specific CD4+ and CD8+ effector and memory T cells.29–33 Therefore, to further improve efficacy by targeting and repolarization of M2 macrophages, we selected high-affinity peptides that target these cells34–38 and conjugated them to the surface of CPMV. We focused on two such linear peptides that bind strongly to the M2 macrophage mannose receptor, namely CSPGAK (CD206s, 561.7 Da) and CSPGAKVRC (CD206, 920.1 Da).39 We evaluated CMPV particles conjugated with CD206s and CD206 for their efficacy in the treatment of murine melanoma through intra-tumoral administration.
CPMV was propagated in black eyed pea plants and purified as previously described.40,41 CPMV is 30 nm in diameter and consists of 60 copies of the small (S, 24 kDa) and large (L, 42 kDa) coat proteins encapsulating genomic single-stranded RNAs. Each particle also has 300 surface-exposed lysine residues suitable for bioconjugation via N-hydroxysuccinimide (NHS) chemistry.42 A two-step conjugation approach was used to attach the CD206s and CD206 peptides (Scheme 1). CD206s has a C-terminal cysteine residue allowing us to use a thiol-maleimide reaction in the second step for CD206s conjugation. First, CPMV was conjugated to the bispecific linker SM(PEG)8 to introduce maleimide groups, and the subsequent cysteine and maleimide reaction allowed the formation of CPMV-CD206s particles. CD206 has cysteine residues at both the C-terminus and N-terminus, so we introduced a C-terminal azide group to ensure conjugation of CPMV to the CD206 C-terminus. CPMV was conjugated to the bispecific linker DBCO-PEG4-NHS ester to introduce DBCO groups, and DBCO-azide click chemistry43 then allowed the formation of CPMV-CD206 particles. To enable us to track particles in cell interaction studies, we also conjugated Cy5 to the surface using the same NHS chemistry with the SM(PEG)8 and DBCO-PEG4-NHS ester linkers. Accordingly, we produced five modified CPMV particles: CPMV-CD206, CPMV-CD206s, CPMV-Cy5, CPMV-CD206-Cy5, and CPMV-CD206s-Cy5. Detailed conjugation protocols are provided in the ESI.†
 | ||
| Scheme 1 Overview of the two-step conjugation methods to produce CMPV particles displaying the peptides CD206s and CD206. | ||
Successful conjugation was confirmed by conducting a range of analytical tests, described in more detail in the ESI.† Native agarose gel electrophoresis confirmed the particles were intact because RNA bands detected under UV light co-migrated with protein bands detected by staining with Coomassie Brilliant Blue (Fig. 1a and b). Denaturing NuPAGE revealed two native bands (representing the S and L coat proteins at ∼24 kDa and 42 kDa, respectively) – additional higher molecular weight bands were observed that corresponded to CD206(s)-modified CPMV. CD206s has a molecular weight of 561.1 Da and CD206 has a molecular weight of 920.1 Da. CPMV-CD206 resulted in two bands are around 25 kDa and 26 kDa which corresponds to S with one or two peptides conjugated. The CD206s formulation resulted in an additional band at ∼24.5 kDa indicating that one peptide per S was conjugated (Fig. 1a and b). No additional bands were observed for the L protein, which is a phenomenon we often observe for modified CPMV and may indicate that the total number of peptides per CPMV is underestimated, i.e. the resolution of the NuPAGE is not sufficient to quantify the peptides on the L protein – or peptides were preferably conjugated to the S protein. Densitometry revealed that ∼39 CD206 peptides and ∼21 CD206s peptides were conjugated per CPMV particle (Fig. S1, ESI†). The elution profiles of the modified particles in size exclusion chromatography (SEC) experiments were similar to native CPMV, with a single absorbance peak synchronizing the 260 and 280 nm signals (representing RNA and proteins, respectively) thus indicating no broken particles in the column volume (Fig. 1c). However, CPMV-CD206 and CPMV-CD206s eluted at ∼9 mL compared to 11 mL for native CPMV, reflecting the size increase due to peptide conjugation. The absorbance ratio at 260/280 nm was ∼1.7 in all cases, indicating intact particles with packaged RNAs.44 The size increase was confirmed by dynamic light scattering (Fig. 1d). Although the size distribution of CPMV-CD206 and CPMV-CD206s was similar to native CPMV, the diameter increased from 38.2 nm to 62.1 nm for CPMV-CD206 and to 46.6 nm for CPMV-CD206s. This was concordant with the SEC data, again confirming that peptide conjugation increased the particle size. Specifically for the size increase for CPMV-CD206, it was possible that there is some degree of interparticle crosslinking – which may be possible through disulfide formation between the N-terminal cystines of conjugated CD206 peptides. As demonstrated by transmission electron microscopy (Fig. 1e), all formulations showed intact particles and most CPMV and CPMV-CD206s remained monodisperse, however, more CPMV-CD206s were associated together possibly due to the interparticle crosslinking. The Cy5-labeled particles were characterized in the same manner. The Cy5 signal co-migrated with the CPMV proteins in agarose gel electrophoresis and NuPAGE experiments (Fig. S2a and b, ESI†) and the absorbance peak at 647 nm corresponded to the 260 and 280 nm peaks in SEC experiments (Fig. S2c, ESI†). UV-Vis spectrophotometry confirmed that ∼32 Cy5 molecules were conjugated per CPMV particle, ∼17 per CPMV-CD206-Cy5 particle, and ∼15 per CPMV-CD206s-Cy5 particle (Fig. S2d, ESI†).
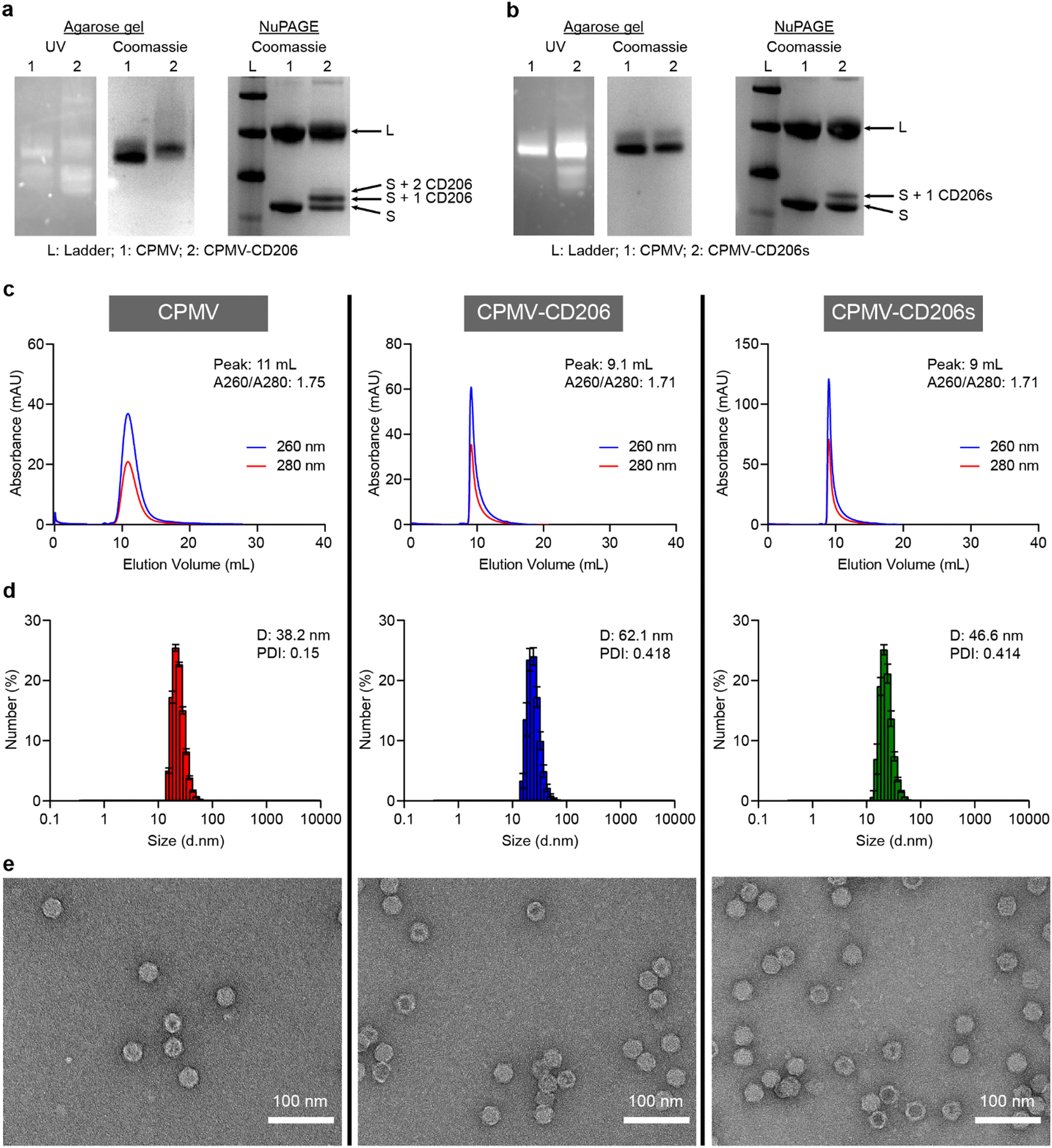 | ||
| Fig. 1 Characterization of modified CPMV particles. (a) and (b) Comparison of CPMV to (a) CPMV-CD206 and (b) CPMV-CD206s by agarose gel electrophoresis and NuPAGE. (c) Analysis of CPMV, CPMV-CD206, and CPMV-CD206s by size exclusion chromatography. (d) Analysis of CPMV, CPMV-CD206, and CPMV-CD206s by dynamic light scattering. (e) Transmission electron microscopy images of CPMV, CPMV-CD206, and CPMV-CD206s. | ||
Having confirmed the integrity of the conjugated particles, we next evaluated their ability to target M2 macrophages in vitro. We prepared M1 and M2 macrophages by polarizing murine RAW264.7 cells (M0) in vitro using lipopolysaccharide (LPS) or interleukins IL-4 and IL-13, respectively.45 First, we confirmed the identity of the polarized M1 and M2 macrophages by assessment of nitric oxide synthase (iNOS) expression which is characteristic for the M1 phenotype and Arginase 1 which is characteristic for the M2 phenotype.46 The M1/M2 polarization was confirmed (Fig. 2a and b); we also observed that only M2 macrophages expressed CD206 on the cell surface post IL-4 and IL-13 simulation (Fig. 2c). M0, M1 and M2 macrophages were then incubated with the same quantity of CPMV-Cy5, CPMV-CD206-Cy5 or CPMV-CD206s-Cy5 particles in 12-well plates. After 24 h, the cells were harvested and analyzed by flow cytometry. CPMV-CD206-Cy5 and CPMV-CD206s-Cy5 were able to target M2 macrophages more efficiently than M0 and M1 macrophages as confirmed by the right-shift in Cy5 fluorescence (Fig. 2d and e). The mean fluorescence intensities (MFIs) confirmed that both peptides targeted M2 macrophages preferentially, increasing the Cy5 signal (Fig. 2g and h). CPMV-CD206s-Cy5 was taken up more efficiently than CPMV-CD206-Cy5 (Fig. 2f), as confirmed by comparing the MFIs (Fig. 2i), even though more CD206 peptides were conjugated per CPMV particle. Overall, in vitro analysis confirmed that the conjugation of CD206s and CD206 to CPMV promoted the targeting of M2 macrophages.
 | ||
| Fig. 2 Flow cytometry. (a) iNOS, (b) arginase 1, and (c) CD206 expression within or on the surface of M0, M1, and M2 macrophages. (d) and (e) Analysis of M0, M1 and M2 macrophages incubated with (d) CPMV-CD206-Cy5 and (e) CPMV-CD206s-Cy5 particles. (f) Analysis of M2 macrophages incubated with CPMV-Cy5, CPMV-CD206-Cy5 and CPMV-CD206s-Cy5 particles. (g)–(i) Mean fluorescence intensities (MFIs) of each sample in (d) and (e) plotted as bar graphs. Statistical significance was determined by ordinary one-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
Next, we evaluated the efficacy of the formulations in the treatment of dermal B16F10-OVA melanoma in C57BL/6J mice. On day 0, 2 × 105 B16F10-OVA cells were intra-dermally (i.d.) injected into the right flank of each mouse to inoculate tumors. Starting on day 9, when all tumors had grown to ∼30 mm3, mice received three weekly intra-tumoral (i.t.) doses of the formulations in six treatment groups (n = 7 mice per group). The treatments were (1) CPMV, (2) CPMV-CD206, and (3) CPMV-CD206s (in each case, 100 μg CPMV in 30 μL PBS); (4) CD206 and (5) CD206s as free peptides matched to the peptide dose in groups (2) and (3); and (6) PBS as a control (Fig. 3a). Tumor sizes were measured every 2 days and mice were euthanized when the tumor exceeded 1000 mm3 (Fig. S3, ESI†). The CPMV, CPMV-CD206, and CPMV-CD206s treatments slowed the tumor growth and improved the survival rate (Fig. 3b and c). There was no significant difference in tumor growth between the CPMV, CPMV-CD206, and CPMV-CD206s groups, but 3/7 mice (43%) in the CPMV-CD206 group remained alive at the end of the study on day 60, compared to 1/7 (14%) in the CPMV group and 0/7 (0%) in the CPMV-CD206s group. More importantly, tumors were completely eradicated in two mice from the CPMV-CD206 group, with no sign of tumors at the end of the study. The CD206 and CD206s peptides did not inhibit tumor growth, with similar results to the PBS group. Detailed analysis of the survival data revealed that CPMV-CD206 treatment prolonged the median survival to 43 days compared to 37 days for CPMV treatment, 30 days for CPMV-CD206s treatment, and 20 days for PBS and the free peptides. Overall, CPMV conjugated to the CD206 peptide was shown to improve treatment efficacy in vivo by slowing down the growth of murine melanoma and prolonging median survival. More importantly, by targeting M2 macrophages using CPMV-CD206 particles, it was possible to completely eradicate the inoculated tumors.
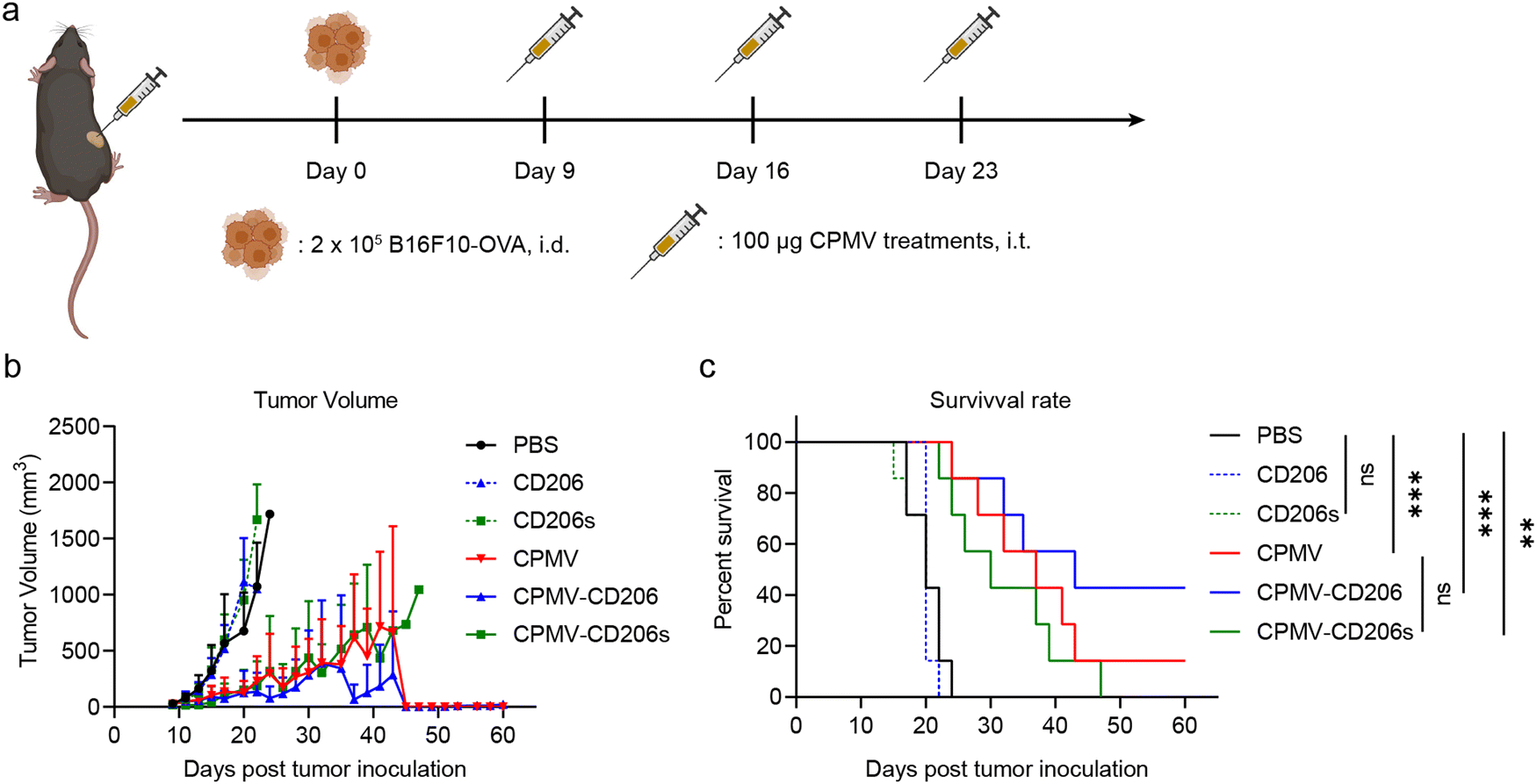 | ||
| Fig. 3 Analysis of treatment efficacy in vivo. (a) Treatment schedule for intradermal B16F10-OVA melanoma. (b) Tumor volume of all treatment groups. (c) Survival curves of all treatment groups. Statistical significance in (c) was calculated using the log-rank (Mantel-Cox) test (**p < 0.01, ***p < 0.001). | ||
In conclusion, we found that CPMV-CD206 and CPMV-CD206s significantly improved the efficiency of M2 macrophage targeting and uptake in vitro, and that CPMV formulations inhibited murine melanoma. CPMV-CD206 appeared somewhat more efficacious in vivo compared to native CPMV and CPMV-CD206s. In contrast, CPMV-CD206s was less effective at targeting M2 macrophages in vitro; therefore this data indicates that treatment efficacy is not solely dependent on targeting or uptake. Differences in in vitro and in vivo targeting also must be considered. Further work is therefore required to determine the factors that contributed to the observed treatment efficacy of CPMV-CD206. We have demonstrated that the conjugation of peptides (CD206 and CD206s) to a plant virus (CPMV) provides the basis of a promising cancer immunotherapy that targets M2 macrophages.
Author contributions
Z. Z., Y. C., and N. F. S. designed the experiments and analyzed the data. Z. Z. and Y. C. performed the experiments. Z. Z. and N. F. S. wrote the manuscript.Conflicts of interest
The authors declare the following competing financial interests: Dr Steinmetz is a co-founder of, has equity in, and has a financial interest in Mosaic ImmunoEnginering Inc. Dr Steinmetz is a co-founder of, and serves as manager of, Pokometz Scientific LLC, under which she is a paid consultant to Mosaic ImmunoEngineering Inc., Flagship Labs 95 Inc., and Arana Biosciences Inc. The other authors declare no potential conflicts of interest.Acknowledgements
This work was supported in part by the NIH (R01-CA253615 and R01-CA224605) to N. F. S. and the Shaughnessy Family Fund for Nano-ImmunoEngineering (nanoIE) at UCSD. We thank the Nano3 core facility at UCSD for transmission electron microscopy imaging. Nano3 is the San Diego Nanotechnology Infrastructure (SDNI) of the University of California, San Diego, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542148).References
- A. J. Korman, S. C. Garrett-Thomson and N. Lonberg, The foundations of immune checkpoint blockade and the ipilimumab approval decennial, Nat. Rev. Drug Discovery, 2021, 1–20 Search PubMed.
- N. Lonberg and A. Korman, Masterful antibodies: Checkpoint blockade, Cancer Immunol. Res., 2017, 5(4), 275–281, DOI:10.1158/2326-6066 CIR-17-0057.
- D. R. Littman, Releasing the brakes on cancer immunotherapy, Cell, 2015, 162(6), 1186–1190 CrossRef CAS PubMed.
- P. Sharma, S. Hu-Lieskovan, J. A. Wargo and A. Ribas, Primary, adaptive, and acquired resistance to cancer immunotherapy, Cell, 2017, 168(4), 707–723 CrossRef CAS PubMed.
- A. J. Schoenfeld and M. D. Hellmann, Acquired resistance to immune checkpoint inhibitors, Cancer Cell, 2020, 37(4), 443–455 CrossRef CAS PubMed.
- K. E. Pauken, M. Dougan, N. R. Rose, A. H. Lichtman and A. H. Sharpe, Adverse events following cancer immunotherapy: obstacles and opportunities, Trends Immunol., 2019, 40(6), 511–523 CrossRef CAS PubMed.
- N. M. Anderson and M. C. Simon, The tumor microenvironment, Curr. Biol., 2020, 30(16), R921–R925 CrossRef CAS PubMed.
- C. Caux, R. N. Ramos, G. C. Prendergast, N. Bendriss-Vermare and C. Ménétrier-Caux, A milestone review on how macrophages affect tumor growth, Cancer Res., 2016, 76(22), 6439–6442 CrossRef CAS PubMed.
- A. Mantovani and A. Sica, Macrophages, innate immunity and cancer: balance, tolerance, and diversity, Curr. Opin. Immunol., 2010, 22(2), 231–237 CrossRef CAS PubMed.
- F. Zhang, N. Parayath, C. Ene, S. Stephan, A. Koehne, M. Coon, E. Holland and M. Stephan, Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers, Nat. Commun., 2019, 10(1), 1–16 CrossRef PubMed.
- Y. Qian, S. Qiao, Y. Dai, G. Xu, B. Dai, L. Lu, X. Yu, Q. Luo and Z. Zhang, Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages, ACS Nano, 2017, 11(9), 9536–9549 CrossRef CAS PubMed.
- G. Hu, M. Guo, J. Xu, F. Wu, J. Fan, Q. Huang, G. Yang, Z. Lv, X. Wang and Y. Jin, Nanoparticles targeting macrophages as potential clinical therapeutic agents against cancer and inflammation, Front. Immunol., 2019, 10, 1998 CrossRef CAS PubMed.
- F. J. Van Dalen, M. H. Van Stevendaal, F. L. Fennemann, M. Verdoes and O. Ilina, Molecular repolarisation of tumour-associated macrophages, Molecules, 2018, 24(1), 9 CrossRef PubMed.
- C. B. Rodell, S. P. Arlauckas, M. F. Cuccarese, C. S. Garris, R. Li, M. S. Ahmed, R. H. Kohler, M. J. Pittet and R. Weissleder, TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy, Nat. Biomed. Eng., 2018, 2(8), 578–588 CrossRef CAS PubMed.
- Y. Wang, Y.-X. Lin, S.-L. Qiao, H.-W. An, Y. Ma, Z.-Y. Qiao, R. Y. J. Rajapaksha and H. Wang, Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment, Biomaterials, 2017, 112, 153–163 CrossRef CAS PubMed.
- N. N. Parayath, A. Parikh and M. M. Amiji, Repolarization of tumor-associated macrophages in a genetically engineered nonsmall cell lung cancer model by intraperitoneal administration of hyaluronic acid-based nanoparticles encapsulating microRNA-125b, Nano Lett., 2018, 18(6), 3571–3579 CrossRef CAS PubMed.
- A. E. Czapar and N. F. Steinmetz, Plant viruses and bacteriophages for drug delivery in medicine and biotechnology, Curr. Opin. Chem. Biol., 2017, 38, 108–116 CrossRef CAS PubMed.
- Y. H. Chung, H. Cai and N. F. Steinmetz, Viral nanoparticles for drug delivery, imaging, immunotherapy, and theranostic applications, Adv. Drug Delivery Rev., 2020, 156, 214–235 CrossRef CAS PubMed.
- M. O. Mohsen and M. F. Bachmann, Virus-like particle vaccinology, from bench to bedside, Cell. Mol. Immunol., 2022, 1–19 Search PubMed.
- M. F. Bachmann and G. T. Jennings, Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns, Nat. Rev. Immunol., 2010, 10(11), 787–796 CrossRef CAS PubMed.
- G. T. Jennings and M. F. Bachmann, The coming of age of virus-like particle vaccines, 2008.
- P. H. Lizotte, A. M. Wen, M. R. Sheen, J. Fields, P. Rojanasopondist, N. F. Steinmetz and S. Fiering, In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer, Nat. Nanotechnol., 2016, 11(3), 295–303 CrossRef CAS PubMed.
- C. Wang, S. N. Fiering and N. F. Steinmetz, Cowpea Mosaic Virus Promotes Anti-Tumor Activity and Immune Memory in a Mouse Ovarian Tumor Model, Adv. Ther., 2019, 2(5), 1900003 CrossRef PubMed.
- S. Shukla, C. Wang, V. Beiss, H. Cai, T. Washington 2nd, A. A. Murray, X. Gong, Z. Zhao, H. Masarapu, A. Zlotnick, S. Fiering and N. F. Steinmetz, The unique potency of Cowpea mosaic virus (CPMV) in situ cancer vaccine, Biomater. Sci., 2020, 8(19), 5489–5503 RSC.
- P. J. Hoopes, R. J. Wagner, K. Duval, K. Kang, D. J. Gladstone, K. L. Moodie, M. Crary-Burney, H. Ariaspulido, F. A. Veliz, N. F. Steinmetz and S. N. Fiering, Treatment of Canine Oral Melanoma with Nanotechnology-Based Immunotherapy and Radiation, Mol. Pharm., 2018, 15(9), 3717–3722 CrossRef CAS PubMed.
- D. Alonso-Miguel, G. Valdivia, D. Guerrera, M. D. Perez-Alenza, S. Pantelyushin, A. Alonso-Diez, V. Beiss, S. Fiering, N. F. Steinmetz and M. Suarez-Redondo, Neoadjuvant in situ vaccination with cowpea mosaic virus as a novel therapy against canine inflammatory mammary cancer, J. Immunotherapy Cancer, 2022, 10(3), e004044 CrossRef PubMed.
- Y. H. Chung, J. Park, H. Cai and N. F. Steinmetz, S100A9-Targeted Cowpea Mosaic Virus as a Prophylactic and Therapeutic Immunotherapy against Metastatic Breast Cancer and Melanoma, Adv. Sci., 2021, 8(21), 2101796 CrossRef CAS PubMed.
- C. Mao, V. Beiss, J. Fields, N. F. Steinmetz and S. Fiering, Cowpea mosaic virus stimulates antitumor immunity through recognition by multiple MYD88-dependent toll-like receptors, Biomaterials, 2021, 275, 120914 CrossRef CAS PubMed.
- C. Wang, V. Beiss and N. F. Steinmetz, Cowpea Mosaic Virus Nanoparticles and Empty Virus-Like Particles Show Distinct but Overlapping Immunostimulatory Properties, J. Virol., 2019, 93(21), e00129-19 CrossRef PubMed.
- C. Wang and N. F. Steinmetz, A Combination of Cowpea mosaic virus and Immune Checkpoint Therapy Synergistically Improves Therapeutic Efficacy in Three Tumor Models, Adv. Funct. Mater., 2020, 30(27), 2002299 CrossRef CAS PubMed.
- C. Wang and N. F. Steinmetz, CD47 Blockade and Cowpea Mosaic Virus Nanoparticle In Situ Vaccination Triggers Phagocytosis and Tumor Killing, Adv. Healthcare Mater., 2019, 8(8), e1801288 CrossRef PubMed.
- C. Wang, S. Fiering and N. F. Steinmetz, Cowpea Mosaic Virus Promotes Anti-Tumor Activity and Immune Memory in a Mouse Ovarian Tumor Model, Adv. Ther., 2019, 1900003 CrossRef PubMed.
- A. A. Murray, C. Wang, S. Fiering and N. F. Steinmetz, In Situ Vaccination with Cowpea vs Tobacco Mosaic Virus against Melanoma, Mol. Pharm., 2018, 15(9), 3700–3716 CrossRef CAS PubMed.
- M. Cieslewicz, J. Tang, J. L. Yu, H. Cao, M. Zavaljevski, K. Motoyama, A. Lieber, E. W. Raines and S. H. Pun, Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(40), 15919–15924 CrossRef CAS PubMed.
- E. K. Asciutto, S. Kopanchuk, A. Lepland, L. Simón-Gracia, C. Aleman, T. Teesalu and P. Scodeller, Phage-display-derived peptide binds to human CD206 and modeling reveals a new binding site on the receptor, J. Phys. Chem. B, 2019, 123(9), 1973–1982 CrossRef CAS PubMed.
- Y. Zhao, B. Han, J. Hao, Y. Zheng, J. Chai, Z. Zhang, Y. Liu and L. Shi, Bi-specific macrophage nano-engager for cancer immunotherapy, Nano Today, 2021, 41, 101313 CrossRef CAS.
- C. Lee, H. Jeong, Y. Bae, K. Shin, S. Kang, H. Kim, J. Oh and H. Bae, Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide, J. Immunotherapy Cancer, 2019, 7(1), 1–14 CrossRef CAS PubMed.
- Z. Duan and Y. Luo, Targeting macrophages in cancer immunotherapy, Signal Transduction Targeted Ther., 2021, 6(1), 127 CrossRef CAS PubMed.
- P. Scodeller, L. Simón-Gracia, S. Kopanchuk, A. Tobi, K. Kilk, P. Säälik, K. Kurm, M. L. Squadrito, V. R. Kotamraju and A. Rinken, Precision targeting of tumor macrophages with a CD206 binding peptide, Sci. Rep., 2017, 7(1), 14655 CrossRef PubMed.
- A. M. Wen, K. L. Lee, I. Yildiz, M. A. Bruckman, S. Shukla and N. F. Steinmetz, Viral nanoparticles for in vivo tumor imaging, J. Visualized Exp., 2012,(69), e4352 Search PubMed.
- Z. Zhao, O. A. Ortega-Rivera, Y. H. Chung, A. Simms and N. F. Steinmetz, A co-formulated vaccine of irradiated cancer cells and cowpea mosaic virus improves ovarian cancer rejection, J. Mater. Chem. B, 2023, 11(24), 5429–5441 RSC.
- A. Chatterji, W. F. Ochoa, M. Paine, B. R. Ratna, J. E. Johnson and T. Lin, New addresses on an addressable virus nanoblock; uniquely reactive Lys residues on cowpea mosaic virus, Chem. Biol., 2004, 11(6), 855–863 CrossRef CAS PubMed.
- J. Park, P. L. Chariou and N. F. Steinmetz, Site-specific antibody conjugation strategy to functionalize virus-based nanoparticles, Bioconjugate Chem., 2020, 31(5), 1408–1416 CrossRef CAS PubMed.
- I. Yildiz, K. L. Lee, K. Chen, S. Shukla and N. F. Steinmetz, Infusion of imaging and therapeutic molecules into the plant virus-based carrier cowpea mosaic virus: cargo-loading and delivery, J. Controlled Release, 2013, 172(2), 568–578 CrossRef CAS PubMed.
- C. Lee, H. Jeong, Y. Bae, K. Shin, S. Kang, H. Kim, J. Oh and H. Bae, Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide, J. Immunotherapy Cancer, 2019, 7, 1–14 CrossRef CAS PubMed.
- H. Cai, S. Shukla and N. F. Steinmetz, The antitumor efficacy of CpG oligonucleotides is improved by encapsulation in plant virus-like particles, Adv. Funct. Mater., 2020, 30(15), 1908743 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00820g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |