DOI:
10.1039/D3MA00826F
(Paper)
Mater. Adv., 2024,
5, 4324-4344
A forsterite-reinforced polypropylene fumarate/methoxy polyethylene glycol-hydrogel enriched with flavonoid nanoparticles enhances osteoconductivity
Received
9th October 2023
, Accepted 26th March 2024
First published on 29th March 2024
Abstract
Tissue engineering offers potential solutions for degenerative bone diseases. In the current study hesperetin (a flavonoid) was loaded in gliadin (a natural protein) NPs and used to enrich an injectable hydrogel of PPF–MPEG reinforced with Fs (a bioceramic) to enhance osteoconductivity. The PPF–MPEG hydrogel (4–6% w/v) containing 3–5% w/w Fs NPs was prepared. Then, the optimum hydrogel concentration was selected based on its properties like degradation, swelling, rheologic behavior, injectability, porosity, and biomineralization. Finally, Hst–GNPs (10, 20, and 40% w/v) were added to the optimized hydrogel. The MG-63 cells were utilized to conduct cell proliferation and attachment analysis, alkaline phosphatase assay, alizarin red staining, and osteogenic gene expression analysis. Hst–GNPs in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (Hst 5 mg and GNPs 15 mg) showed an appropriate particle size (233.4 ± 3.6 nm), zeta potential (−13.5 ± 1.752 mV), EE% (97.98 ± 0.00%), and RE8h% (86.39 ± 0.10%) among other formulations. The 6% w/v PPF–MPEG hydrogel containing 3% w/w Fs showed appropriate injectability (17.41 ± 0.88 N), swellability, degradation, and mechanical properties. Moreover, the Fs NPs affected the degradation rate and swelling degree of the hydrogel; nevertheless, their overloading resulted in decreased Young's modulus and compressive strength of the hydrogel. The hydrogel containing 10% w/v Hst–GNPs showed the most significant rapid release among those containing other ratios. Also, significant enhancement was seen in mineralization, differentiation, and cell proliferation. In addition, higher expression of bone-specific genes, including collagen 1, osteocalcin, and osteopontin, was revealed in the cells treated with the PPF–MPEG/Fs/Hst–GNPs compared to the blank hydrogel.
:3 (Hst 5 mg and GNPs 15 mg) showed an appropriate particle size (233.4 ± 3.6 nm), zeta potential (−13.5 ± 1.752 mV), EE% (97.98 ± 0.00%), and RE8h% (86.39 ± 0.10%) among other formulations. The 6% w/v PPF–MPEG hydrogel containing 3% w/w Fs showed appropriate injectability (17.41 ± 0.88 N), swellability, degradation, and mechanical properties. Moreover, the Fs NPs affected the degradation rate and swelling degree of the hydrogel; nevertheless, their overloading resulted in decreased Young's modulus and compressive strength of the hydrogel. The hydrogel containing 10% w/v Hst–GNPs showed the most significant rapid release among those containing other ratios. Also, significant enhancement was seen in mineralization, differentiation, and cell proliferation. In addition, higher expression of bone-specific genes, including collagen 1, osteocalcin, and osteopontin, was revealed in the cells treated with the PPF–MPEG/Fs/Hst–GNPs compared to the blank hydrogel.
Introduction
Cancer, trauma, osteoporosis, bone fractures, and rheumatoid arthritis are the conditions that could lead to bone tissue destruction. The increasing incidence of bone defects is a major concern in middle-aged women and a major cause of disability in obese and elderly patients. Traditional treatments such as surgery, bone grafting, and implantation of permanent prostheses are invasive and risky and have limited sources.1–3 Bone tissue engineering involves three vital components, including mesenchymal stem cells (MSCs) or osteoblast-like cells (MG-63), scaffolding matrix, and growth factors or bioactive molecules like chemicals, peptides, proteins, nanoparticles (NPs) or phytochemicals.4,5
Nowadays, using biodegradable injectable composites and hydrogels is one of the emerging methods for bone tissue engineering. For example, graphene and its derivatives, bioceramics, carbon nanotubes, natural materials, and synthetic polymers are extensively applied in tissue engineering. Biodegradable synthetic polymers with acceptable mechanical, physicochemical, and biological properties are regarded as potential bone graft substitutes.6 Polyesters such as polycaprolactone (PCL), poly L-lactic acid (PLLA), and polyglycolic acid (PGA) are the most used polymers in tissue engineering. However, fumaric acid-based polyesters such as polypropylene fumarate (PPF) are more attractive in biomedical research due to their better biocompatibility and biodegradability. PPF is a biodegradable linear polyester characterized by one carbon–carbon unsaturated double bond and two ester bonds. The ester linkages facilitate both crosslinking across the double bond and hydrolysis through these bonds. The properties of PPF can be altered by adjusting its molecular weight and incorporating suitable crosslinkers. Also, PPF could be combined with other polymers such as polyethylene glycol (PEG) and copolymers could be formed to enhance its properties like injectability, hydrophilicity, and mechanical strength.7 Recent studies have shown that 3D-printed PPF scaffolds enhance cell proliferation and migration in bone marrow-derived human mesenchymal stem cells.8 In another study, PPF/PEG-modified graphene oxide nanocomposites not only showed antibacterial activity against Gram-negative and Gram-positive microorganisms without any toxicity to the human dermal cells, but also presented better stiffness, strength, and toughness.9,10 Another study on a PPF/PEG copolymer demonstrated that because of the presence of hydrophilic groups, this structure had great potential for water absorption and showed sol–gel behavior. The PPF/PEG copolymer could be in liquid (sol) form at room temperature (25 °C) and could form a gel at body temperature (35–37 °C), which could improve the drug–polymer scaffold's injectability.6 Bioceramics, mostly fabricated from porous ceramics, also have a vital role in bone tissue engineering and have been investigated as potential candidates for hard tissue regeneration due to their suitable strength and resistance against destruction.7,8 Calcium, magnesium, and silicon-based bioceramics are widely used in the field of bone tissue engineering. Bioactive silicate ceramics, including diopside (CaMgSi2O6), bredigite (Ca7MgSi4O16), wollastonite (CaSiO3), akermanite (Ca2MgSi2O7), and larnite (Ca2SiO4), have shown beneficial bioactivity in bone tissue engineering. Calcium phosphate (CaP) ceramics, including hydroxyapatite (HA) (Ca10(PO4)6(OH)2), dicalcium phosphate (DCP), tricalcium phosphate (TCP), and bioglasses, are commonly used owing to their high compressive strength, biocompatibility, and osteoconductivity.9–11 Forsterite (Mg2SiO4) (Fs) is dimagnesium silicate belonging to the olivine group with good bioactivity and better mechanical properties than HA and bioglasses.12,13 In addition, recent studies have shown that Fs has antibacterial activity, better degradability, and mechanical strength. Fs may also enhance adhesion to osteoblast cells, stimulate proliferation, and delay hydroxyapatite deposition.9,12
As previously mentioned, bioactive compounds are essential for bone tissue engineering, which can be NPs or phytochemicals. Flavonoids, the largest group of plant phenolic compounds, are broadly found in many plants, vegetables, and fruits and possess different biological and pharmacological properties such as antitumor, antiviral, antibacterial, antioxidative, and anti-inflammatory and thus have attracted the attention of many researchers.4,14 Hesperetin (Hst), 3′,5,7-trihydroxy-4-methoxy flavanone, the aglycone of hesperidin (β-7-rutinoside of hesperetin), is a flavonoid belonging to the flavanone subgroup and is commonly identified in citrus fruits (Rutaceae). It has various biological activities, such as antioxidant, anti-inflammatory, lipid profile improvement, analgesic, and neurological function protection.14,15 Liu et al.14 revealed that Hst alleviated LPS-induced bone loss, diminished osteoclast numbers, and lowered the receptor activator's ratio of nuclear factor-kB ligand (RANKL)/osteoprotegerin (OPG) in vivo. Additionally, the study proposed the use of Hst as a promising therapeutic approach for treating destructive bone diseases. It was demonstrated that Hst can improve glucocorticoid-induced inhibition of osteogenic differentiation in mesenchymal stem cells through activation of the ERK signal pathway.16 Moreover, research indicated that low concentrations (1–10 μM) of Hst could increase the mineralization, proliferation, and migration of the human MSCs. Also, hesperetin/gelatin scaffolds with the Hst concentration of 5 μM promoted fracture healing in vivo.17,18 Although Hst exhibits various beneficial properties, its clinical applications are limited due to poor water solubility, low in vivo bioavailability, short half-life, and rapid clearance from the body. However, the use of new drug delivery systems, such as NPs, holds promise in addressing these limitations.19
Nanotechnology plays a critical role in the development of drug formulations. The NPs are small-sized materials made of atomic or molecular materials and can easily enter the human body and present specific structural, biological, chemical, and mechanical features. The selection of NPs for drug delivery depends on the physicochemical properties of the target drugs being chosen for the treatment.20 Proteins possess many advantages to be used in NP preparation. They are biocompatible, biodegradable, cost-effective, able to modify surfaces, and exist in natural resources. Moreover, protein-based NPs do not have potential limitations of other NPs, including toxicity, fast body clearance, and accumulation. These nanostructures are synthesized using proteins like gelatin, casein, albumin, gliadin, elastin, and zein by different methods, including coacervation, emulsification, electrospray, and desolvation.21
Gliadin (G) and glutenin are the main components of the wheat gluten protein. Both proteins are insoluble in aqueous media at normal pH; however, monomeric G is soluble in 70% ethanol and is composed of single-chain polypeptides with intra-molecular disulfide bonds and 25–100 kDa molecular weight; nonetheless, the polymeric glutenin is insoluble in ethanol and is made of macropolymers with intermolecular disulfide bonds.22 The GNPs could be obtained using the desolvation method or the anti-solvent precipitation method. Studies have revealed that GNPs improve the bioavailability of encapsulated lipophilic molecules due to the bioadhesive ability of G particles towards the gastric mucosa.23 Previous studies have investigated the use of GNPs as a drug delivery system for encapsulated retinoic acid and vitamin E and obtained promising results related to controlled drug release.24,25
This research was designed to develop an optimized thermosensitive injectable hydrogel reinforced with Fs NPs containing Hst–GNPs as the active ingredient for implementation in bone tissue engineering. In this study, Hst was employed as the bioactive molecule due to its cost-effectiveness and role in bone regeneration. Also, GNPs were used to facilitate the smooth release of Hst in the cell environment and to improve cell adhesion and migration.26,27 Furthermore, these data encouraged current research on synthesis of a polypropylene fumarate/methoxy polyethylene glycol (PPF–MPEG) hydrogel reinforced with bioceramic Fs NPs, as the carrier for Hst loaded GNPs (Scheme 1) to enhance the physicochemical and biological properties and to develop a controlled-release injectable hydrogel for bone tissue engineering.
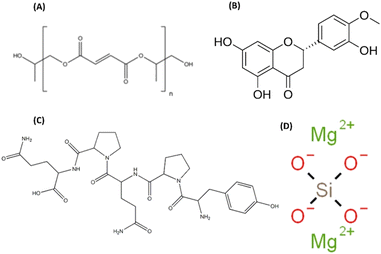 |
| | Scheme 1 Chemical structures of (A) poly(propylene fumarate) (PPF), (B) hesperetin (Hst), (C) gliadin (G), and (D) forsterite (Fs). | |
Experimental
Materials
Hesperetin (C16H14O6, purity ≥ 95%, Mw = 302.27 g mol−1), diphenyl tetrazolium bromide (MTT), alizarin red, ascorbic acid-2-phosphate, β-glycerophosphate, dexamethasone, zinc chloride, methoxy polyethylene glycol-2000, diethyl fumarate, hydroquinone, formaldehyde, hydrochloric acid, tetraethyl orthosilicate ((C2H5O)4Si, TEOS), cetyltrimethylammonium bromide (CTAB), ammonium hydroxide, calcium chloride, magnesium nitrate hexahydrate (Mg(NO3)2·6H2O), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (USA). Propanol-(1), acetone, sodium iodide, ethanol, sodium hydroxide, ammonium acetate, methanol, propylene glycol, acetic acid, citric acid, Triton X-100, and chloroform were provided by the Merck Chemical Company (Germany). The dialysis bag (mol. wt. cut-off ∼ 12–14 kDa) was purchased from the Betagen Company (Mashhad, Iran). A human osteoblast-like cell line (MG-63) was obtained from the National Cell Bank (Rooyan Institute, Iran). Dulbecco's Modified Eagle Medium-Low Glucose (DMEM-LG), fetal bovine serum (FBS), streptomycin/penicillin, trypsin-EDTA 0.25%, and phosphate buffered saline (PBS) were acquired from Bioidea (Iran). The alkaline phosphatase kit was provided by the Pars Azmoon Company (Iran). Moreover, the highly pure RNA isolation kit was supplied by the Roche Company (Germany). The SYBR Green PCR Master Mix and AddScript cDNA Synthesis Kit were obtained from Addbio (Korea). Primer genes included: osteocalcin (BGLAP), collagen 1 (COL1A1), osteopontin (SPP1), and beta-actin (ACTB) (as control), which were bought from Cinaclon (Iran).
Gliadin extraction
According to the prior study accomplished by Dupont et al.,28 wheat flour was used to extract G and then SDS-PAGE assay was performed to identify the extracted protein. For each extraction, 100 mg of wheat flour was mixed with 1 mL of a solution of 0.3 M sodium iodide and 7.5% 1-propanol and then centrifuged. The extraction process was repeated twice. Afterwards, the resulting supernatant was precipitated by adding a cold solution of methanol and ammonium acetate. The precipitated mixture was stored at −20 °C for 48 hours and then was subjected to centrifugation at 5000 rcf for 15 minutes at room temperature. The resulting pellets were carefully re-suspended using the minimum amount of acetic acid and subsequently freeze-dried to be utilized in the future procedures.
Synthesis of forsterite NPs
The Fs NPs were synthesized employing the surfactant-assisted sol–gel process, as previously reported by Hassanzadeh-Tabrizi et al.,29,30 albeit with minor modifications. Initially, 0.05 g mL−1 CTAB (as a surfactant) was dissolved in deionized water (DW) under stirring conditions to yield a clear and homogeneous solution. Subsequently, the pH was rapidly adjusted to approximately 12 through the addition of ammonium hydroxide, followed by an additional five minutes of stirring. Thereafter, a solution of TEOS–magnesium nitrate hexahydrate–water (1/2/7) was incrementally added over a period of 20 minutes. After 12 hours of continuous stirring, the resultant solution was washed with DW and subsequently placed in a dry oven at 65 °C for 12 hours. The synthesized viscous gel was then calcined at 700 °C at a heating rate of 10 °C min−1 for three hours in an air environment to eliminate the template (CTAB). The resultant Fs NPs were characterized by scanning electron microscopy (SEM, Zeiss Leo 1430, Germany), energy dispersive X-ray spectroscopy (EDX, Oxford instruments), and X-ray diffractometry (XRD, Philips X'Pert MPD).
Synthesis of a PPF–MPEG copolymer
PPF synthesis.
In a reaction setup, diethyl fumarate, zinc chloride, propylene glycol, and hydroquinone were combined in a three-necked round bottom flask in a molar ratio of 1:3:0.01:0.003. Zinc chloride was used as a catalyst for the reaction. The resulting mixture was heated up to 150 °C under nitrogen gas conditions. This heating process led to the formation of a diester called bis(2-hydroxypropyl)fumarate and ethanol as a by-product. To remove ethanol, a distillation process was carried out. Subsequently, the intermediate diester underwent transesterification for three hours under the vacuum conditions with the pressure less than 1 mmHg at 150 °C. This transformation resulted in the production of linearly unsaturated poly(propylene fumarate) (PPF) and propylene glycol as the final products. The final products were washed with ethanol to remove excess propylene glycol and other impurities and the pure product was stored in a refrigerator for use in the next reaction step.31
PPF–MPEG synthesis.
Methoxy polyethylene glycol (molecular weight = 500) was mixed with PPF in a molar ratio of 1:2 and the transesterification reaction was performed under vacuum at a reaction temperature of 160 °C. After the end of the reaction, the copolymer was rinsed with dichloromethane and ethyl ether. Hydroquinone and zinc chloride were dissolved in ethyl ether and the copolymer became waxy or solid in the solvent. The solvent was stirred when the copolymer continued to be waxy. Then, the polymer was dried under vacuum. To purify the copolymer, the resulting product was dissolved in dichloromethane. The solution was then filtered and precipitated into ethyl ether while vigorously stirring. This process helped to remove impurities and obtain a purified form of the copolymer. Zinc chloride and hydroquinone were removed during the washing process through their solubility in ethyl ether. The final product was tested using proton nuclear magnetic resonance spectroscopy (H-NMR 400 MHz, Bruker, Germany) and Fourier transform infrared spectroscopy (FT-IR) (JASCO-6300, Japan) to confirm its successful synthesis.321H-NMR was used to analyze PPF and PPF–MPEG. For analysis, the samples were dissolved in an appropriate solvent (DMSO-d6 for PPF and PPF–MPEG). The H-NMR spectra provided information about the chemical shifts and confirmed the presence of specific chemical groups within the polymers. FT-IR was also performed on all three components (PPF, PPF–MPEG, and PEG) using the KBr disc method (400–4000 cm−1) at a resolution of 4 cm−1. FT-IR analysis enabled the identification of characteristic absorption peaks corresponding to specific functional groups, providing further insights into the chemical structures and compositions of the materials. Scheme 2 shows the synthesis procedures of PPF and PPF–MPEG.
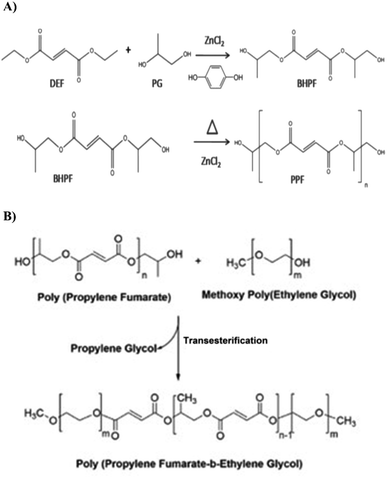 |
| | Scheme 2 The synthesis procedures of (A) PPF and (B) PPF–MPEG. | |
Preparation of the Hst–GNPs.
According to previous studies with small modifications, the GNPs were prepared by a desolvation method.24,33 First, G solutions were prepared at the concentrations of 2.5, 5, and 7.5 mg mL−1 with 96% ethanol (organic phase) and stirred for at least 30 minutes on a magnetic stirrer. Afterward, 2 mL of each solution was filtered with a 0.45 μm syringe filter and Hst powder was added to each solution in different ratios of Hst to G of 1:1, 1:2, and 1:3 and stirred for a further 15 minutes. On the other hand, a 1% Pluronic F127 solution was prepared by dissolving Pluronic F127 in 0.9% NaCl solution (aqueous phase). Then, 4 mL of this solution was filtered through a 0.45 μm syringe filter. The organic solution (2 mL) was added to the water-based phase (4 mL) with a syringe and the solution was subjected to sonication with a probe sonicator (UW 3200, Bandelin, Germany) at 30 W for one minute with on and off pulses for 1 second. Eventually, the resulting solutions were dialyzed in DW for 30 minutes, freeze-dried and kept at −20 °C for further experiments.
Physicochemical characterization of NPs
Particle size, zeta potential, and polydispersity index (PDI).
A dynamic light scattering method was employed to measure the particle size, PDI, and zeta potential of NPs at room temperature and pH 6.8 using a particle size analyzer (Zetasizer 3600, Malvern, UK). The optimized formulation was freeze-dried, then the NPs were redispersed in DW, and measurements were repeated. All assessments were carried out in triplicate, and the mean ± SD of the outcomes was reported.
Entrapment efficiency measurement.
The absorbance of the supernatant, obtained after centrifugation, was measured using a UV spectrophotometer (UV-mini-1240, Shimadzu, Japan) at a wavelength of 377.5 nm. This measurement was conducted to determine the concentration of free drug and the entrapment efficiency (EE%). EE% was determined using eqn (1), where M0 represents the initial amount of Hst and M1 represents the amount of free Hst in the medium after loading.34| | | EE% = [(M0 − M1)/M0] × 100 | (1) |
In vitro Hst release from NPs.
To calculate the release percentage of Hst from Hst–GNPs, 1 mL of NP dispersion was transferred into the dialysis bags and immersed in 50 mL of the release medium, which contained a 1:1 mixture of PBS (pH 7.4) and ethanol with 2% Pluronic F127, while it was kept under constant shaking at 37 °C. During the sampling process, 1 mL of the medium was collected at various time intervals (0.5–12 hours). The concentration of Hst in the samples was determined using a UV spectrophotometer (UV-mini-1240, Shimadzu, Japan) and a calibration curve based on the Hst standard was constructed, with measurements taken at a wavelength of 383 nm. To maintain sink conditions, after each sampling, fresh medium was exchanged. The cumulative release of Hst was calculated using eqn (2), where Mn represents the amount of Hst released at the time n and M0 represents the total amount of the Hst in the sample.30 Each formulation was analyzed three times.| | | Hst released (%) = (Mn/M0) × 100 | (2) |
NP morphology determined by FE-SEM.
Images obtained using a field emission scanning electron microscope (MIRA3, TESCAN, Czechia) were analyzed to determine the morphological properties of GNPs.
Fourier transform infra-red (FTIR) analysis.
The infrared spectra of freeze-dried Hst–GNPs, GNPs, and Hst were obtained using the KBr disc method (400–4000 cm−1) at a resolution of 4 cm−1 using an FTIR spectrophotometer (JASCO-6300, Japan).
Preparation of hydrogels.
Hydrogels were prepared and crosslinked by dispersing the synthesized polymer of PPF–MPEG (4 and 6% w/v) in a solution of citric acid and DW and were stirred at room temperature to obtain a homogenous suspension. Therefore, the copolymer was insoluble but dispersible in water and it could make the hydrogel when dispersed in water. Then, Fs NPs (3 and 5% w/w) were dispersed in the hydrogels under stirring and the final solution was kept at 4 °C for further studies. The best hydrogel concentration was selected according to the rheological and mechanical properties, injectability, porosity, and biomineralization tests. Then, the Hst–GNPs (10, 20, and 40% w/v) were added to the optimized hydrogel, and the optimum concentration of NPs was selected based on their release studies for further in vitro biological experiments.
Characterization of hydrogels
Injectability.
The universal testing machine (Santam, STM-20, Iran) was utilized to determine the injectability of the hydrogels. For each formulation, 1 mL of hydrogel was discharged from the 2 mL syringe connected to the 18-gauge needle and was compressed at 20 mm min−1 at room temperature. The maximum force at plateau (FMAX) was determined as the maximum injectability force and then the force curves were constructed. The test was performed in triplicates.35
Mechanical properties.
Mechanical properties were evaluated by conducting compressive tests on hydrogels using a standard testing machine (SANTAM, STM-20, Iran). The hydrogel samples were frozen in a 24-well silicon mould overnight and freeze-dried for 48 h. The freeze-dried cubic samples (12 mm × 12 mm × 4 mm) were compressed between two jaws of the machine at the rate of 10 mm min−1 with a 100 N load cell. The compressive test was conducted in triplicate for each sample, and the compressive strength and compressive modulus were determined based on the stress–strain curve.35,36
Rheology.
The rheometric mechanical spectrometer (RMS, Anton Paar, MCR 301, Austria) was used to examine the rheology of the hydrogels through oscillatory temperature, time, frequency, and viscosity sweep tests. The rheometer was loaded with the samples and the loss modulus (G′′), storage modulus (G′), and complex viscosity (η) were measured as the functions of time (2% amplitude gamma, 1 Hz frequency, 4–50 °C temperature, 5 °C min−1 heating rate) and temperature (2% amplitude gamma, 1 Hz frequency, 25 °C temperature). The gelation point was determined when the loss modulus and storage modulus became equal. G′ and G′′ were also measured against shear stress and strain at an angular frequency of 10 rad s−1 from 0.1 to 100% strain to determine the linear viscoelastic (LVE) region. Moreover, frequency sweeps were conducted at 1% strain throughout an angular frequency range of 600 to 0.1 Hz.37
In vitro swelling index and degradation assay.
The gravimetric method was performed to assess the swelling and degradation of hydrogels. Pre-weighed (W0) freeze-dried hydrogel samples were incubated in 2 mL of PBS (pH 7.4) at 37 °C under constant stirring. At different time intervals, PBS was removed, extra solution on the surface of the samples was absorbed with filter paper, the weight of samples (Wt) was recorded, and the medium was replaced with the fresh one. For the degradation test, after 1, 3, 7, 14, 21, and 28 days, the samples were freeze-dried and weighed (Wd). The weight loss percentage and the equilibrium degree of the swelling were computed through eqn (3) and (4):36| | | Weight loss (%) = [(W0 − Wd)/W0] × 100 | (3) |
| | | Equilibrium degree of swelling (%) = [(Wt − W0)/W0] × 100 | (4) |
Porosity ratio.
The porosity measurement was carried out following Archimedes’ principle. The pre-weighed dry (Wd) samples were soaked in normal hexane at 25 °C until saturation was achieved. The saturated weight (Wsat) and submerged weight (Wsub) of the samples in normal hexane were measured and porosity was determined according to eqn (5):38| | | Porosity (%) = [(Wsat − Wd)/(Wsat − Wsub)] × 100 | (5) |
In vitro mineralization study.
To evaluate the mineralization activity of the hydrogels, from each formulation, three freeze-dried samples (sample dry weight was 8.37 ± 0.07 g) were placed in individual wells of a 24-well culture plate. Afterwards, each well was filled with 2 mL of simulated body fluid (SBF-1×), and the plate was placed in an incubator set at 37 °C for a duration of 28 days. After 7 and 28 days of incubation, the samples were washed with distilled water (DW) and then dehydrated using a series of ethanol solutions. Finally, dried samples were assessed using SEM (Zeiss Leo 1430, Germany) followed by EDX (Oxford instruments) to investigate the apatite (Ca2+ and P) formation on the samples.35,39
Morphology.
The morphological assessments were carried out using a scanning electron microscope (SEM, Zeiss Leo 1430, Germany) on hydrogels. To prepare samples before imaging, the hydrogels were frozen in a 24-well silicon mould, freeze-dried, and then coated with gold under vacuum.
Hst release from hydrogels.
The Hst release profiles from the prepared hydrogels were determined using the Franz diffusion cell method. A cellulose filter membrane was affixed to the donor compartment of the apparatus, and 5 mL of the hydrogel samples were applied onto the membrane. The acceptor part was filled with a 25 mL release medium (a 1:1 mixture of PBS (pH 7.4) and ethanol, with 2% Pluronic F127) at 37 °C with continuous stirring. At specified time intervals, 1 mL of the medium was collected to measure the released Hst absorbance using a spectrophotometer at 383 nm. The medium was promptly replaced with fresh medium. The Hst concentration and cumulative drug release percentage were determined using the standard curve equation.40,41
Cell culture studies.
The MG-63 cells were used due to their close resemblance to human osteoblasts. The cells were cultured using Dulbecco's modified Eagle's medium/nutrient mixture F12 (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin antibiotics. The cell cultures were maintained in an incubator set at the temperature of 37 °C and a 5% CO2 atmosphere for optimal growth. To ensure healthy cellular growth, the culture medium was changed every three days. Once the cells reached approximately 70–80% confluency within the culture flask, they were detached through trypsinization before being counted for further experimental procedures.
Cell viability and proliferation (MTT assay).
To determine the sub-toxic concentration of the Hst–GNPs, an MTT assay was performed for 24 h, 72 h, and 7 days for the concentrations below IC50. After that, two sub-toxic concentrations of the Hst–GNPs with the optimum proliferation rate were selected and the Hst–GNPs were combined with hydrogel samples to study the proliferation of MG-63 cells in hydrogels. After treating hydrogel samples with UV light for 30 minutes, 50 μL of hydrogel samples were placed in a 12-well plate and incubated for 30 minutes at 37 °C for gel formation. Subsequently, 500 μL of cell suspension containing 1.5 × 104 cells per well was added to each well, followed by 30-minute incubation to facilitate cell penetration. Afterward, 1.5 mL of the culture medium was added to each well, and the plate was further incubated for the designated period. At specific time points (24 h, 72 h, and 7 days), 1.5 mL of the culture medium was evacuated, and 50 μl of MTT solution (5 mg mL−1) was introduced into each well. The formazan crystals were formed in the plate during a four-hour incubation. Subsequently, the culture medium was gently removed, and 500 μL of DMSO was added to each well. The plate was kept at room temperature in a dark location for two hours to facilitate DMSO penetration into the hydrogels and dissolution of formazan crystals. Finally, from each well, 150 μL was transferred to a 96-well plate, and the absorbance was assessed at 570 nm using a microplate reader (Biotek, 800-TS). The percentage of cell viability was calculated using eqn (6), where Xs, Xb, and Xc represent the sample mean absorbances, blank (tissue culture plate), and the control (cell suspension), respectively.37| | | Cell viability (%) = [(Xs − Xb)/(Xc − Xb)] × 100 | (6) |
Alizarin red staining (ARS).
The ARS method assessed the ability of MG-63 cells to form mineralized nodules in the presence of hydrogels. Each sample was seeded in a 12-well plate with 1 × 104 cells per well in an osteogenic medium (DMEM/F12 medium, 10% FBS, 1% penicillin/streptomycin solution, 100 nM dexamethasone, 10 mM β-glycerophosphate, and 0.2 mM vitamin C). After 7 and 14 days, the medium was removed, and the cell layer was washed with PBS three times. Then, the cells and hydrogels were fixed with formaldehyde (3.6%, 15 min, 37 °C) and the cells were stained with Alizarin Red dye (2%, pH 4.1–4.3, 30 min, 37 °C). Calcium deposition was detected and photographed using an Olympus microscope (Tokyo, Japan) after rinsing the cell layer with DW.42
Alkaline phosphatase (ALP) assay.
The ALP activity of MG-63 cells was measured using an ALP assay kit (Pars Azmoon, Iran) to assess the conversion of para-nitrophenyl phosphate to para-nitrophenol. First, 50 μL of each formulation containing 6.25 μg mL−1 Hst was added into each well of a 12-well plate and incubated at 37 °C for 30 min. Afterward, 500 μL of 1.5 × 104 cells were added to each well and after 30 minutes of incubation at 37 °C, 1.5 mL of osteogenic culture medium was added to each well. At days 7 and 14, the cells were rinsed twice with PBS after discarding the medium. Then, 1 mL of 1% Triton X-100 was added to each well to lyse the cells. Considering the ALP kit protocol, after centrifuging the contents of each well at 12000 rpm for three minutes, 8 μL of the sample's supernatant was added to 200 μL of the working solution in a 96-well plate. After one, two, three, and four minutes, the optical density was recorded at 405 nm through a microplate reader (Biotek, 800-TS), and the ALP activity was determined according to the kit protocol.43
Gene expression study (RT-PCR).
Cells were harvested and lysed after 14 days of cell culture to determine the protein expression levels. In accordance with the manufacturer's instructions, total RNA was extracted with a High Pure RNA Isolation Kit (Roche, Germany). Using a nano-spectrophotometer (WPA, England), the quality of the total RNA extracted from each sample was evaluated. Thereafter, each sample's complementary DNA (cDNA) was created using oligo dT primers and the AddScript cDNA Synthesis Kit (Addbio, Korea) according to the manufacturer's protocol. The cDNA was then amplified using specific primers and SYBR Green PCR Master Mix (Addbio, Korea) in the StepOneTM Real-time PCR Detection System (Applied Biosystems, USA) to perform a quantitative RT-PCR. Table 1 displays the primer sequences used in this study, including osteocalcin (BGLAP), collagen 1 (COL1A1), osteopontin (SPP1), and beta-actin (ACTB) as the internal control. The comparative cycle threshold (ΔΔCt) was employed to estimate the relative gene expression.
Table 1 Sequences of primers employed for RT-PCR analysis of different gene expressions in MG-63 osteoblast-like cells
| Gene |
Forward |
Reverse |
| Collagen 1 (COL1A1) |
5′-GAGGGCCAAGACGAAGACATC-3′ |
5′-CAGATCACGTCATCGCACAAC-3′ |
| Osteocalcin (BGLAP) |
5′-CACTCCTCGCCCTATTGGC-3′ |
5′-CCCTCCTGCTTGGACACAAAG-3′ |
| Osteopontin (SPP1) |
5′-GCCGAGGTGATAGTGTGGTT-3′ |
5′-TGAGGTGATGTCCTCGTCTG-3′ |
| Beta-actin (ACTB) |
5′-GTTGTCGACGACGAGCG-3′ |
5′-GCACAGAGCCTCGCCTT-3′ |
Statistical analysis.
All experiments were carried out three times, and the results were presented as mean ± SD. Also, the GraphPad Prism software (V.9) was used to perform one- and two-way analyses of variance (ANOVA), followed by Tukey's post hoc test for statistical analysis of the data. The p-values less than 0.05 (p ≤ 0.05) were considered statistically significant.
Results and discussion
Gliadin NP characterization
FE-SEM and TEM images of the GNPs are displayed in Fig. 1. Both FESEM and TEM images exhibit clusters of spherical GNPs with diameter below 400 nm. This is in accordance with the study performed by Frazan et al.,44 in which GNPs were used for targeted delivery of usnic acid in breast cancer. Also, in another study conducted by Voci et al.,45 TEM analysis evidenced a round-shaped morphology of GNPs, which were employed to encapsulate and transport doxorubicin. According to Duoduo et al.,46 TEM images of hollow GNPs displayed distinct core–shell structures. Also, the sizes of particles observed through a TEM were typically smaller than those measured by DLS, attributed to the nanoparticles shrinking during the drying process before TEM examination.
 |
| | Fig. 1 GNP characterization. (A) FE-SEM and (B) TEM images of the GNPs. | |
Forsterite characterization
Fig. 2 illustrates the SEM image and XRD pattern of the synthesized Fs NPs. The XRD pattern indicates the presence of distinct peaks that are the characteristic of highly crystalline Fs. The pattern was compared to the standard JCPDS data card and successfully matched and indexed. Also, the EDX pattern of the hydrogels combined with Fs shows Mg and Si existence compared to the hydrogel samples without Fs. The results of XRD and EDX analyses confirm that Fs is synthesized successfully, which is in line with previous studies.9,30 The SEM image of the Fs NPs indicates the spherical morphology and clusters of agglomerated Fs NPs attributed to the intrinsic nature of nanoparticles. The nano-sized particles can also be seen in some parts of the image.47 As presented in the study done by Hassanzadeh-Tabrizi et al.,29 using CTAB in the synthesis of forsterite nanoparticles resulted in the creation of pores inside the nanoparticles and it was found that samples with higher CTAB content showed slower drug release rates, likely due to smaller pore sizes. In the present study, our results indicated that the presence of these porous Fs NPs facilitates the production of highly porous hydrogels with favorable physicochemical and cellular properties such as sustained drug release from the gel matrix, good interconnectivity and the transport of vital nutrients to cells and elimination of metabolic by-products.
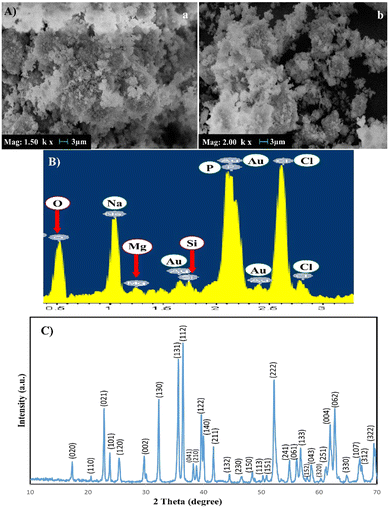 |
| | Fig. 2 (A) SEM images of Fs NPs under two magnifications: (a) ×1500, (b) ×2000, (B) EDX and (C) XRD of the forsterite NPs. | |
Characterization of the PPF–MPEG copolymer
The NMR spectrum of PPF, which can be seen in Fig. 3B-a, showed characteristic peaks at 1.4 ppm (methyl protons), 5.3 ppm (methylene protons adjacent to ester), and 4.1 ppm (methylene protons adjacent to hydroxyl). These peaks are consistent with the expected structure of PPF, confirming its successful synthesis. The NMR spectrum of PPF–MPEG (Fig. 3B-b) revealed all the characteristic peaks of PPF, along with additional peaks at 3.2 ppm corresponding to the methylene protons of MPEG. The appearance of these peaks indicates the successful conjugation of MPEG to PPF. The NMR results confirm the successful synthesis of PPF and its modification with MPEG. The presence of characteristic peaks in the expected regions of the NMR spectrum provides strong evidence for the proposed structures of these compounds. The FTIR spectra of the synthesized polymers are shown in Fig. 3A. The FTIR pattern of PPF showed the existence of unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C fumarate groups, which are necessary for core-crosslinking of the copolymer, with a strong peak at 1640 cm−1. The ester linkages in PPF were observed with strong characteristic stretching frequencies of carbonyl groups CO at 1720 cm−1 and C–O at 1100 cm−1. The successful esterification of MPEG with PPF's terminal carboxyl group was confirmed by the absence of the O–H signal in the FTIR spectrum of PPF–MPEG.32,48
C fumarate groups, which are necessary for core-crosslinking of the copolymer, with a strong peak at 1640 cm−1. The ester linkages in PPF were observed with strong characteristic stretching frequencies of carbonyl groups CO at 1720 cm−1 and C–O at 1100 cm−1. The successful esterification of MPEG with PPF's terminal carboxyl group was confirmed by the absence of the O–H signal in the FTIR spectrum of PPF–MPEG.32,48
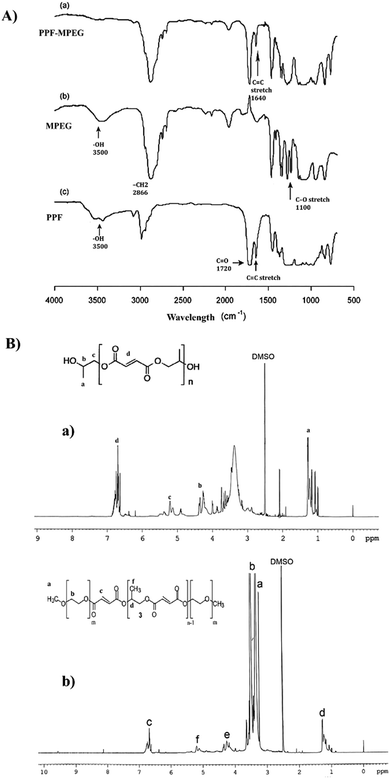 |
| | Fig. 3 (A) FTIR spectra of (a) PPF–MPEG, (b) MPEG, and (c) PPF, and (B) NMR spectra of (a) PPF and (b) PPF–MPEG. | |
Physicochemical characterization of NPs
Particle size, zeta potential, PDI, EE%, and drug release profile.
Different formulations of NPs and their characteristics, including particle size, zeta potential, PDI, and EE%, are depicted in Table 2. Fig. 4A shows release profiles and Fig. 4B the particle size and PDI. Hst2.5G5 and Hst10G10 were not selected because of their inappropriate particle size, which was significantly greater than those of other formulations (p ≤ 0.0001). Three formulations were specified, and their release profiles were studied. The Hst5G15 formulation was opted as the optimal formulation for subsequent experiments according to its desirable particle size and PDI, appropriate Hst entrapment efficiency, and better release profile than the other two formulations. Also, assessing the zeta potentials of Hst5G15 (−13.5 ± 1.752 mV) and Hst0G15 (−6.716 ± 0.779 mV) NPs after freeze-drying, dispersing them in DW and adjusting the pH at 7.4 showed a more negative zeta potential for both formulations, especially Hst5G15, which demonstrates that NPs are more stable after the freeze-drying process and the zeta potential is changed from neutral to negative along with increasing pH from 6.8 to 7.4.49
Table 2 Physicochemical characteristics of various formulations of GNPs loaded with Hst (n = 3, mean ± SD)
| Formulation |
Hesperetin (Hst) (mg) |
Gliadin (G) (mg) |
EE% ± SD |
RE8% ± SD |
Particle size (nm) ± SD |
Zeta potential (mV) ± SD |
PDI ± SD |
| Hst5G5 |
5 |
5 |
73.01 ± 0.11 |
61.01 ± 0.46 |
215.2 ± 4.2 |
0.57 ± 0.08 |
0.48 ± 0.04 |
| Hst5G10 |
5 |
10 |
96.65 ± 0.12 |
73.86 ± 2.14 |
225.5 ± 8.8 |
0.41 ± 0.04 |
0.39 ± 0.07 |
| Hst5G15 |
5 |
15 |
97.98 ± 0.00 |
86.39 ± 0.10 |
233.4 ± 3.6 |
−0.27 ± 0.06 |
0.38 ± 0.04 |
| Hst2.5G5 |
2.5 |
5 |
66.77 ± 2.47 |
— |
425.1 ± 21.1 |
0.83 ± 0.28 |
0.64 ± 0.32 |
| Hst10G10 |
10 |
10 |
96.43 ± 0.18 |
— |
516.5 ± 7.8 |
0.68 ± 0.19 |
0.21 ± 0.05 |
| Hst0G5 |
0 |
5 |
— |
— |
315.2 ± 6.0 |
−0.07 ± 0.02 |
0.40 ± 0.01 |
| Hst0G10 |
0 |
10 |
— |
— |
346.9 ± 5.5 |
0.05 ± 0.01 |
0.40 ± 0.07 |
| Hst0G15 |
0 |
15 |
— |
— |
357.2 ± 4.2 |
0.14 ± 0.03 |
0.38 ± 0.02 |
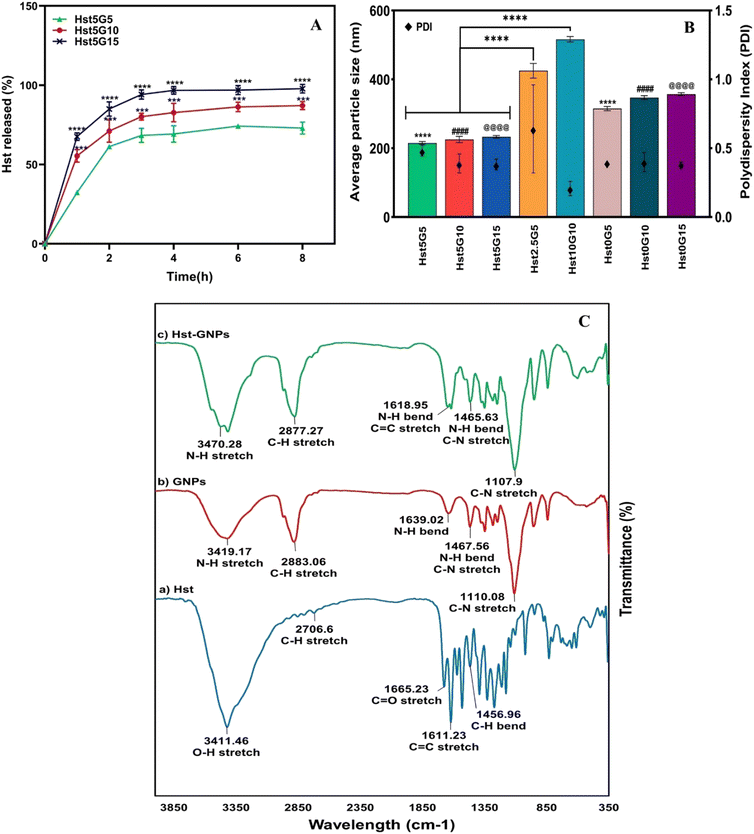 |
| | Fig. 4 (A) Hst release profiles of Hst–GNPs at different ratios (**** p ≤ 0.0001 between Hst5G15 and Hst5G5 and *** p ≤ 0.001 between Hst5G15 and Hst5G10) and (B) average particle size and PDI of NPs at different ratios (****, ####, @@@@ p ≤ 0.0001), and (C) FTIR spectra of Hst, blank GNPs, and Hst–GNPs. | |
The release profile of the Hst was assessed for different formulations in GNPs through a dialysis membrane in the release medium of a 1:1 mixture of PBS (pH 7.4) and ethanol, containing 2% Pluronic F127 at 37 °C. As seen in Fig. 4A, the samples showed an initial burst release during the first hour, which is related to the more superficially loaded Hst molecules. After that, the release slightly increased up to four hours and then reached a plateau and remained relatively constant. It can be observed from the figure that the formulation with more G (Hst5G15 i.e., the formulation with 1:3 Hst to G) illustrates a much more drug release (approximately 100%) than other formulations over time (p ≤ 0.0001). The cause may lie in the increased surface area and the porosity provided by GNPs, which allow more efficient diffusion and drug delivery. Additionally, higher G concentrations may facilitate greater interactions among Hst molecules within the release medium, leading to enhanced solubility and overall release of Hst.50
Fourier transform infra-red (FTIR) analysis of NPs.
FTIR spectroscopy was used to investigate the molecular compositions of the Hst, blank GNPs, and Hst–GNPs. As shown in Fig. 4C, the Hst spectrum includes a peak at 3411 cm−1 due to the stretching vibration of OH, a peak at 1665 cm−1 due to CO stretching in flavonoids, a peak at 1611 cm−1 due to CC stretching in the aromatic ring, and a peak at 1357 cm−1 due to C–H bending. In addition, two peaks were observed between O–H bending and C–O stretching in the phenolic compound (1520 cm−1 and 1562 cm−1).51–53 The GNP spectrum showed a peak at 3419 cm−1, which is associated with N–H stretching vibrations. It also depicted signature G bands such as the amide I band (1639 cm−1), which corresponds to the N–H bending, the amide II band (1467 cm−1), and the amide III band (1110 cm−1), which are associated with the combination of N–H bending and C–N stretching vibrations.54,55 The FTIR spectrum of Hst–GNPs indicates distinctive G peaks, demonstrating nearly complete incorporation of Hst into the GNPs. Small peaks of O–H vibration around 3400 cm−1 and CC around 1610 cm−1 in the spectrum of Hst–GNPs likely stem from surface adsorption of some Hst onto the NP surface due to surface interactions. These peaks underscore the fact that there was no chemical interaction between Hst and GNPs, and no significant shift was detected in the peak positions.
Characterization of hydrogels
Gel injectability.
Injectable hydrogels possess unique characteristics that make them highly suitable for bone tissue engineering applications. One of their key features is their injectability, which enabled minimally invasive delivery directly to the site of bone defects or injuries.56 These hydrogels are typically in the form of a liquid or gel that can be easily injected through a syringe, enabling precise placement and adaptation to irregular bone structures. The injectability of hydrogels was measured through the utilization of a compression test, which determines the maximum force (FMAX) required to expel the sample to the designated injection site. The injectability results of the designed hydrogels are illustrated in Table 3. The hydrogel PPF–MPEG and hydrogel with forsterite PPF–MPEG/Fs exhibited FMAX values below the threshold of 30 N. This value represents the maximum permissible injection force limit, as established in the prior study.37 Adding 3% w/w Fs to the hydrogel enhanced the FMAX of PPF–MPEG-6 from 10.25 N to 17.41 N in the case of PPF–MPEG-6/Fs-3 formulation (Table 3).
Table 3 Injectability of hydrogel formulations
| Formulation |
PPF–MPEG (w/v%) |
Forsterite (w/w%) |
Maximum force (N) |
| PPF–MPEG-4 |
4 |
0 |
9.86 ± 0.77 |
| PPF–MPEG-4/Fs-3 |
4 |
3 |
20.99 ± 1.88 |
| PPF–MPEG-4/Fs-5 |
4 |
5 |
17.07 ± 0.44 |
| PPF–MPEG-6 |
6 |
0 |
10.25 ± 0.43 |
| PPF–MPEG-6/Fs-3 |
6 |
3 |
17.41 ± 0.88 |
| PPF–MPEG-6/Fs-5 |
6 |
5 |
16.19 ± 1.26 |
This can be attributed to the higher particle concentration resulting from the addition of Fs enhancing particle–particle interactions, leading to increased resistance to flow and higher viscosity. Additionally, the tendency of Fs particles to agglomerate or form clusters further hinders the hydrogel flow and contributes to the increased viscosity. This was also observed in the hydrogels containing 4% w/v PPF–MPEG. Although FMAX was enhanced for formulations containing 5% w/w Fs compared to the hydrogels without Fs, FMAX was lower for hydrogels containing 5% Fs compared to 3% w/w Fs (Table 3). This may be attributed to the influence of particle–particle interactions and the overall particle concentration within the hydrogel. At higher concentrations, the particles may be more closely packed together, resulting in a more compact network structure that facilitates better flow compared to the lower concentration.57 Furthermore, other factors such as particle size distribution and formation of particle aggregates may also contribute to the observed changes in viscosity.58 These results indicate that adding the Fs has a significant impact on the flowability and viscosity of the hydrogel (p ≤ 0.05), and the optimal concentration may vary depending on the desired viscosity properties. The injectability properties of the PPF–MPEG-6/Fs-3 formulation are depicted in Fig. 5A.
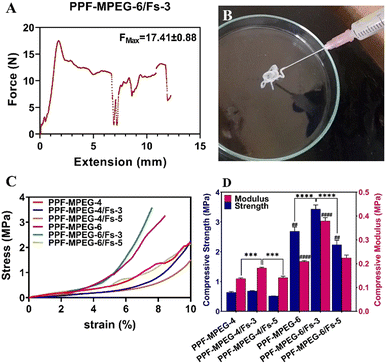 |
| | Fig. 5 (A) The Fmax of injection, (B) the injectability properties of the optimum hydrogel of PPF–MPEG-6/Fs-3, (C) stress–strain curves, and (D) compressive strength (CS) and modulus (CM) of the formulations (## indicates p ≤ 0.01 between CS of PPF–MPEG-6 and PPF–MPEG-6/Fs-5, *** indicates p ≤ 0.001 between CM of PPF–MPEG-4 and PPF–MPEG-4/Fs-3 and between CM of PPF–MPEG-4/Fs-3 and PPF–MPEG-4/Fs-5, **** indicates p ≤ 0.0001 between CS of PPF–MPEG-6 and PPF–MPEG-6/Fs-3 and between CS of PPF–MPEG-6/Fs-3 and PPF–MPEG-6/Fs-5, and #### indicates p ≤ 0.0001 between CM of PPF–MPEG-6 and PPF–MPEG-6/Fs-3). | |
Mechanical properties.
It is crucial for a hydrogel matrix to possess mechanical stability that allows it to endure physiological stress in vivo. The mechanical features of the hydrogel samples were assessed by conducting the compressive test and were analyzed through the stress–strain curve through calculating the compressive modulus and strength. The resulting graphs can be seen in Fig. 5C and D. The stress–strain curve displayed an exponential correlation between compression stress and strain in all samples. As this figure uncovers, the compressive modulus is the highest for the PPF–MPEG-6/Fs-3 composite, indicating that this mixture has the highest deformation resistance under compressive stress. Similarly, the PPF–MPEG-6/Fs-3 composition demonstrated the highest compressive strength, signifying its capacity to withstand against greater compressive stress without failure. Moreover, the compressive strength of the hydrogel formulation containing PPF–MPEG 6% w/v was in the range of the human trabecular bone compressive strength (between 2 and 12 MPa).59 The differences among the samples were significant (p ≤ 0.01) as illustrated in the graph. This reveals that the PPF–MPEG-6/Fs-3 mixture was the stiffest formulation among the tested compositions. Incorporation of the Fs into the PPF–MPEG hydrogel significantly (p ≤ 0.001) improved its mechanical properties, notably in the formulations with 3% w/w Fs. This is evidenced by the higher compressive modulus and compressive strength (Fig. 5D). Nevertheless, similar to the injectability results (Table 3), the samples with 5% w/w Fs showed lower stiffness and stability compared to the samples containing lower Fs concentration (p ≤ 0.001). This underlines the fact that Fs could be a promising additive to enhance the mechanical properties of the PPF–MPEG hydrogel to be used as an injectable bone tissue scaffold; however, the optimal concentration may be varied according to the favorable mechanical properties.60 According to previous studies, the presence of Fs as a rigid filler can lead to phase segregation and an increased risk of stress fractures within the polymer matrix at higher filling ratios.58,61
Rheology.
The rheology test could provide good insight into the properties of the hydrogel such as viscosity, gelation kinetics, shear thinning behavior and stability. Fig. 6A and B show the storage modulus (G′) and loss modulus (G′′) of the samples against temperature and time. The G (shear modulus) (Pa) is the response of a material to the applied sinusoidal stress or strain. G′ (storage modulus) and G′′ (loss modulus) show the elastic state and the viscous state of the gels, respectively. The results disclosed that G′ was initially lower than G′′ in all samples at low temperature; however, G′ surpassed G′′ with increasing temperature, indicating the thermal sensitivity of the PPF–MPEG-based gel. The point where G′ equaled G′′ was considered the onset of gelation temperature and time for each sample.62 The gelation temperature increased from 22 °C for PPF–MPEG-4/Fs-3 to 35 °C for PPF–MPEG-4/Fs-5. Also, the gelation time decreased from 150 s to 45 s. This suggested that incorporation of Fs into the hydrogel at a lower concentration could increase the stiffness of the gel matrix and help the hydrogel to convert to the gel in a shorter time and at lower temperature; however, the higher concentration of Fs can decrease the stability of the sample and lead to an increase in gelation temperature and time. Nonetheless, this is not a consistent trend in all samples, since the interaction and association of PPF and MPEG with each other and Fs in the hydrogel matrix could affect the time and temperature of gelation. In this study, the PPF–MPEG-6/Fs-3 formulation showed appropriate rheological behavior. It had a gelation time of 140 s and started to convert from a clear solution to a turbid gel at 25 °C, meaning that it can be injected easily at room temperature within two minutes.
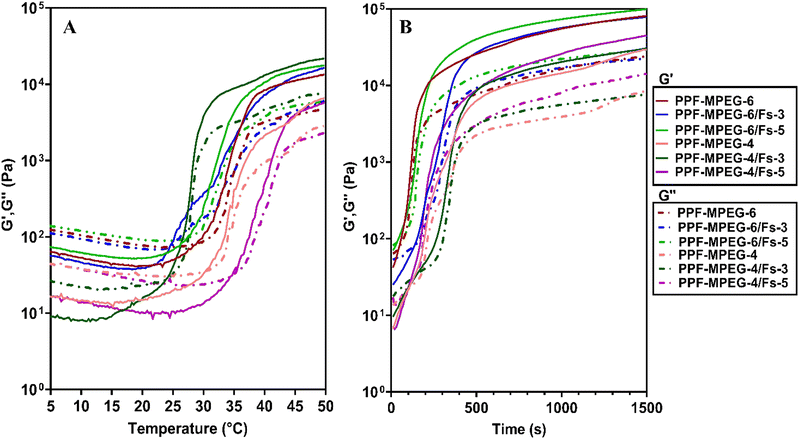 |
| | Fig. 6 Rheology test of the PPF–MPEG hydrogel at concentrations of 4 and 6% w/v with and without different concentrations of Fs (3 and 5% w/w), showing the storage moduli (G′) and the loss moduli (G′′) as a function of (A) temperature and (B) time. | |
Fig. 7A–C shows the changes in viscosity as a function of frequency, temperature, and time. The viscosity of all samples initially decreased with increasing temperature as the polymer chains unraveled and the hydrogen-bonded network around the polymers broke. As shown in Fig. 7B and C, a rapid increase in viscosity was observed as the gelation temperature and time were reached. This increment in viscosity can be ascribed to the aggregation of hydrophobic domains within the copolymer.37 A dynamic frequency sweep test was conducted to determine the linear viscoelastic range of the hydrogel, as shown in Fig. 7A. In this test a continuous excitation was applied to the hydrogel; while its structure was not destroyed, the viscosity properties changed via the alteration of frequency with a negative slope. In other words, the viscosity of all samples reduced with the variation of frequency from 0.1 to 100 Hz.
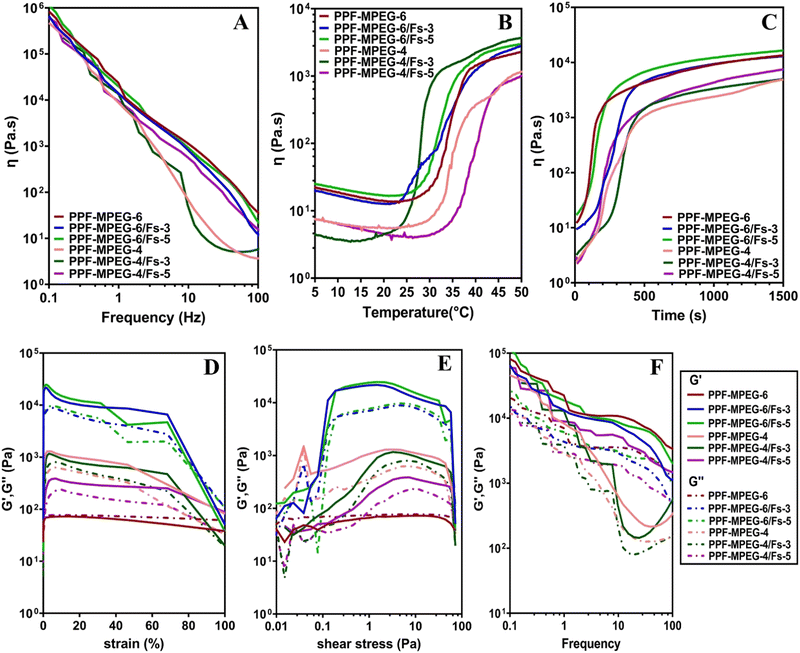 |
| | Fig. 7 Viscosity behaviour of the PPF–MPEG hydrogel at concentrations of 4 and 6% w/v with and without different concentrations of Fs (3 and 5% w/w) against (A) frequency, (B) temperature, and (C) time, and rheological analysis of different formulations of the PPF–MPEG hydrogel having different amounts of Fs: storage moduli (G′) and loss moduli (G′′) versus (D) strain, (E) shear stress, and (F) frequency. | |
The results of strain and shear stress sweeps are presented in Fig. 7D and E. These figures show the relationship among the strain, shear stress, frequency, and storage moduli (G′) of the PPF–MPEG based hydrogels at 37 °C. The G′ value, which represents the strength of the hydrogel, is higher for hydrogels with greater strength. The hydrogels of PPF–MPEG-6/Fs-3 and PPF–MPEG-6/Fs-5 showed obviously greater G′ and G′′ at similar strain percentages (Fig. 7D), thanks to the higher percentages of PP-MPEG. Fig. 7E illustrates that at the beginning of the non-linear response, a clear rise of G′ took place at the same time as G′′ raised. The peak seen for G′ at the onset of the nonlinear range possibly indicates the formation of a secondary structure in the nonlinear part of the strain and shear stress curves before collapsing (Fig. 7D and E). Based on the results of strain and shear stress sweeps (Fig. 7D and E), 1% strain chosen for the frequency sweep measurements is high enough to get valid data from stable hydrogel structures. Mechanical properties (G′ and G′′) of hydrogels determined from frequency sweep measurements at room temperature are shown in Fig. 7F. As this figure shows, all of the dynamic storage moduli G′ and the loss moduli G′′ show a dependency on frequency with an upward trend by increasing frequency. At the range of frequencies from 0.1 to 100 Hz, storage moduli G′ were always superior to G′′. Also, elastic response to frequency sweep was continuously higher than viscosity response.
In vitro degradation assay and swelling index.
The in vitro degradation behavior of the produced hydrogels was evaluated by measuring their weight loss over time in PBS. As presented in Fig. 8A, all samples exhibited increasing weight loss with increasing incubation time, indicating their ability to undergo hydrolytic degradation.63 Notably, PPF–MPEG-6 demonstrated a slower breakdown rate, roughly 32% after 28 days of incubation compared to the lower polymer concentration (PPF–MPEG-4), which exhibited a weight loss of 37% at the same period of time (p ≤ 0.01). This finding aligns with previous studies on PPF–PEG copolymer hydrogels, which showed gradual disintegration during incubation.64 PPF and MPEG are synthetic polyesters that degrade through the process of ester linkage hydrolysis and the rate of degradation is primarily influenced by two key processes, including water penetration into the polymer matrix and the random cleavage of the polymer chains. According to the previous studies, the incorporation of PEG into PPF enhances the degradation rate of the polymer by inducing the formation of small pores throughout the polymer network.65,66
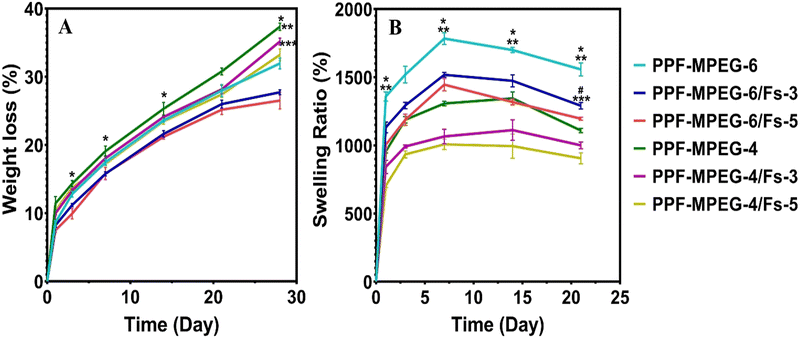 |
| | Fig. 8 The effects of incubation in PBS on the (A) weight loss and (B) swelling degree of different hydrogel formulations. The results revealed significant differences (* p ≤ 0.05) among PPF–MPEG-6 and PPF–MPEG-6/Fs-3, (** p ≤ 0.01) PPF–MPEG-6 and PPF–MPEG-4, (*** p ≤ 0.001) PPF–MPEG-6/Fs-3 and PPF–MPEG-4/Fs-3, (# p ≤ 0.05) PPF–MPEG-6/Fs-3 and PPF–MPEG-6/Fs-5. | |
Interestingly, in this study, the addition of 3 and 5% w/w Fs appears to have slowed down the deterioration rate and reduced the weight loss rate significantly compared to the plain hydrogels that lack Fs (p ≤ 0.05). This effect may be attributed to cross-metalation caused by cations present in the environment and creating a more stable network structure. This is in agreement with the degradation results of the study of Choudhary et al.,67 in which the composites containing more Fs showed a lower degradation rate. Although Fs is a hydrophilic material, in the case of good compatibility of the composite mixture and Fs, and appropriate designing of the composite to mitigate moisture-related degradation, the incorporation of Fs may not necessarily speed up the deterioration rate. There was not a significant difference between 3 and 5% w/w Fs (p ≥ 0.05) possibly as the percentages of Fs used in the hydrogels are quite close to each other. Focussing on the study of Tavangarians et al.68, the degradation of Fs ceramic can create pores and meshes that facilitate hydroxyapatite formation and accelerate overall tissue regeneration. Previous research on Fs composites has revealed that different degradation profiles depend on the factors such as particle size, shape, surface area, and the medium. According to Wei et al.,69 nanomaterials could degrade more quickly than micron-sized particles due to their smaller size and larger specific surface area. This increased surface area can lead to greater reactivity with the surrounding environmental factors, causing faster degradation of the material over time. Teimouri et al.70 indicated that the amount of Fs powder in the composites had a significant impact on their degradation rates. This could be due to increased breakdown or dissolution of Fs particles and the release of ions into solution. The swelling ratio of the hydrogel samples was evaluated to investigate their ability to absorb and retain water.71Fig. 8B illustrates the results of swelling and indicates that all hydrogel formulations exhibited an increasing trend in the swelling ratio in seven days. Notably, a rapid increase in water uptake was observed during the first 24 hours, which can be attributed to the hydrophilic nature of the polymer matrix and formation of hydrogen bonds.72 All samples exhibited great ability to absorb water; however, the hydrogels containing Fs significantly demonstrated lower swelling ratios compared to the pure PPF–MPEG hydrogels (p ≤ 0.05). This can be associated with a tighter network structure formed by Fs NPs and the hydrogen bond formation between the Fs NPs and the hydrogel network.30,35 As can be observed from the figure, the PPF–MPEG hydrogels demonstrated lower swelling ratios at a lower concentration (4% w/v) than PPF–MPEG-6. As the polymer concentration decreases, fewer groups are available to form hydrogen bonds, which can explain the lower swelling ratio observed. The water absorption of the samples reached a maximum value within 7 to 14 days followed by a decreasing trend in the swelling rate towards the end of the incubation period. This profile varied for the samples based on the content of Fs NPs and copolymer concentration. The observed differences in swelling behavior highlight the importance of controlling the cross-linking density to achieve desired swelling properties for specific applications.
Porosity ratio.
As depicted in Fig. 10B, all samples exhibited high porosity ratios, indicating a greater extent of pore formation within the hydrogel structure, which aligns with the SEM images of the hydrogel. Although adding Fs to the PPF–MPEG hydrogel decreased the porosity ratio, no significant reduction was seen in Fs concentrations of 3 and 5% w/w (p ≥ 0.05) hydrogels. In addition, incorporation of Hst–GNPs into the hydrogel matrix showed a significant increase in the porosity ratio compared to the hydrogel with or without Fs (p ≤ 0.01). In addition, the porosity ratio of the hydrogel with 4% w/v concentration of the copolymer was higher than 6%, due to a less compact structure. Overall, the results reveal that the hydrogels possess a favorable interconnectivity, which allows efficient flow to occur, promoting the diffusion of essential nutrients into the cells and facilitating the removal of metabolic waste.73 In this study, the hydrogel containing 6% w/v PPF–MPEG and 3% w/w Fs (PPF–MPEG-6/Fs-3) showed the best performance among the tested compositions and was selected as the optimum formulation.
In vitro mineralization study.
Biomineralization testing was performed by immersing the freeze-dried samples in simulated body fluid (SBF-1x) to evaluate the in vivo bone binding capacity of the hydrogel matrix for bone tissue engineering applications. The SEM images of scaffolds showed that both PPF–MPEG and PPF–MPEG/Fs samples displayed some degree of mineralization, with higher levels observed in the hydrogel containing Fs after 28 days of immersion, which is demonstrated in Fig. 9A-g and A-h. At day 7, the large cauliflower-like apatites can be seen on the surface of the PPF–MPEG/Fs scaffolds (Fig. 9A-c and A-d); however, the PPF–MPEG scaffolds showed only very small apatite crystals (Fig. 9A-a and A-b). On the other hand, on day 28, the SEM images of scaffolds showed the accumulation of apatite crystals over the entire surface of the scaffolds, and again, the crystals were more frequently located on the PPF–MPEG/Fs than on the PPF–MPEG scaffold. In addition, SEM analysis revealed that the mineralized material was deposited on the surface of hydrogel scaffolds, indicating favorable interactions between cells and gel matrices during the incubation periods. Also, Fig. 9B presents the EDX analysis of PPF–MPEG/Fs scaffolds after the immersion in SBF solution for 28 days. The EDX results confirm the formation of apatites through indicating the presence of Ca and P peaks. Also, the existence of Fs could be verified through Si and Mg peaks. The results are consistent with the recent research implemented in relation to bioceramics and their impact on the biomineralization activity of composites.58,74,75 Altogether, these results underscore the promising potential of hydrogels as biomimetic matrices that facilitate cell growth and stimulate new bone formation through biomineralization processes.
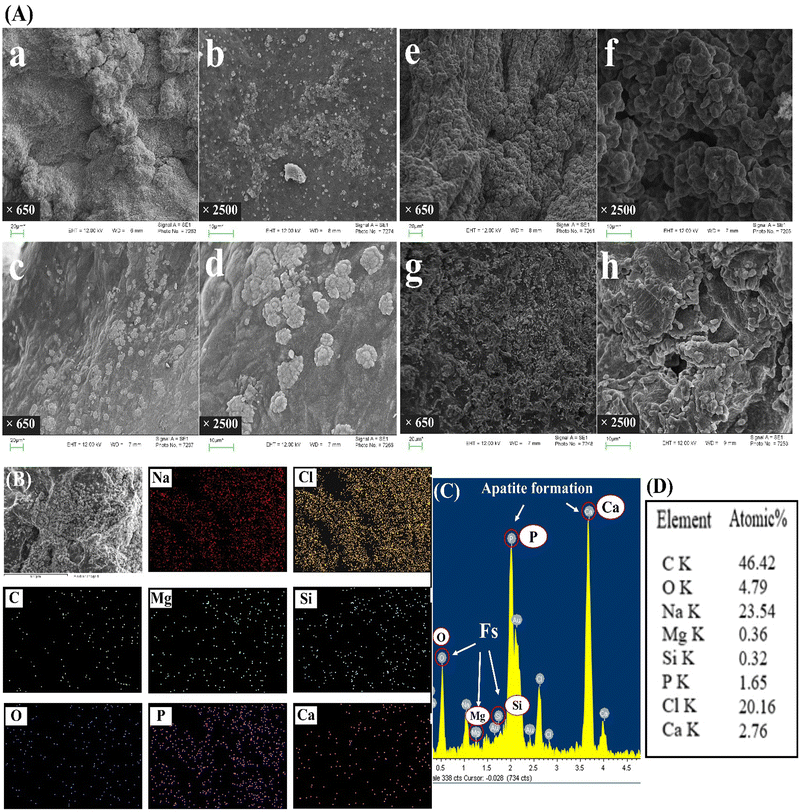 |
| | Fig. 9 Biomineralization study: (A) SEM images of the apatite formation on (a)–(f) PPF–MPEG and (c)–(h) PPF–MPEG/Fs hydrogels after (a)–(d) 7 days and (e)–(h) 28 days of immersion in SBF-1× at 650× and 2500×. (B) SEM-EDX mapping, (C) EDX spectra, and (D) Elements' atomic percentage of the PPF–MPEG/Fs hydrogel after 28 days of immersion in SBF-1×. | |
SEM morphology of hydrogels.
The scanning electron microscopy images of the freeze-dried PPF–MPEG-6 and PPF–MPEG-6/Fs-3/Hst–GNP hydrogels are illustrated in Fig. 10A. The images revealed the presence of a three-dimensional, highly porous, and layer network structure with interconnected pores. Similar structures have been reported for PPF and PPF–PEG based hydrogels, indicating their suitability for supporting cellular activities.76,77 Nevertheless, it was observed that incorporation of Fs into the PPF–MPEG hydrogel leads to changes in its overall morphology resulting in a rougher surface in texture, which is important for cell adhesion and proliferation.78 Moreover, it seems that adding Fs has provided more crosslinked sites and has decreased the pore sizes but has not reduced the number of pores. This highly porous structure is advantageous not only for the drug delivery applications as it expedites controlled drug release from the gel matrix, but also for various cellular processes, including cell growth, migration, proliferation, and attachment as well as feeding during the incubation.
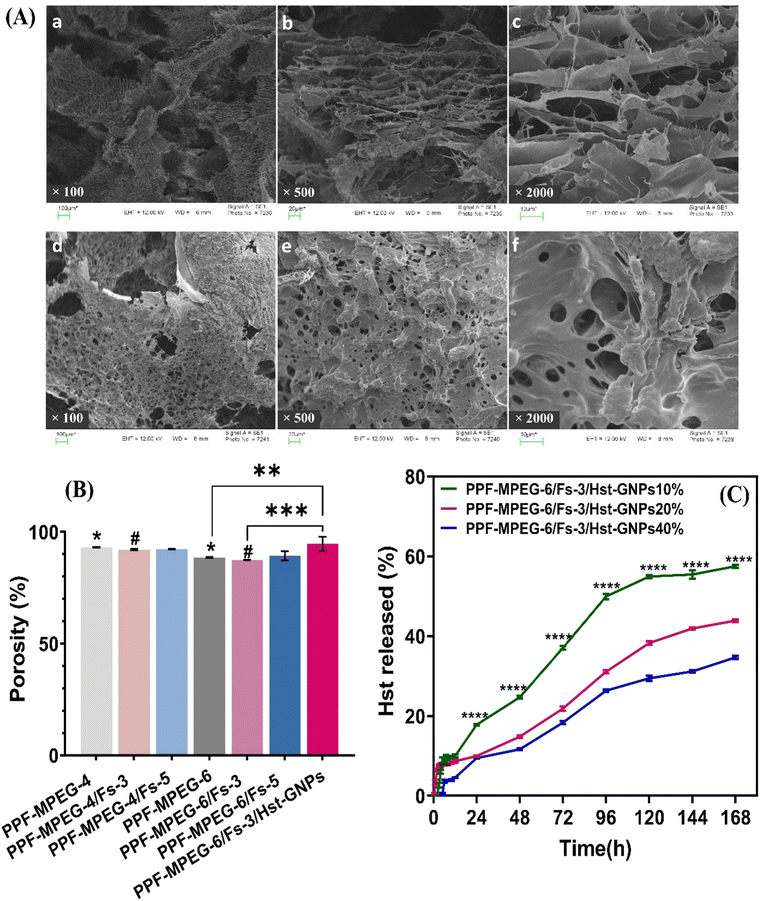 |
| | Fig. 10 (A) SEM images of the freeze-dried PPF–MPEG (a)–(c) and PPF–MPEG/Fs (d)–(f) hydrogels under three magnifications: (a) and (d) 100×, (b) and (e) 500×, (c) and (f) 2000×. (B) Porosity measurement of freeze-dried hydrogel samples, * indicates p ≤ 0.05 between PPF–MPEG-4 and PPF–MPEG-6, # indicates p ≤ 0.05 between PPF–MPEG-4/Fs-3 and PPF–MPEG-6/Fs-3, ** indicates p ≤ 0.01 between PPF–MPEG-6 and PPF–MPEG-6/Fs-3/Hst–GNPs, and *** indicates p ≤ 0.001 between PPF–MPEG-6/Fs-3 and PPF–MPEG-6/Fs-3/Hst–GNPs. (C) Cumulative release profile of Hst from PPF–MPEG-6/Fs-3 hydrogels containing different ratios of Hst–GNPs after seven days: **** indicates significant differences (p ≤ 0.0001) among PPF–MPEG-6/Fs-3/Hst–GNPs10% and two other formulations. | |
Hst release from the hydrogel.
The release behavior of Hst from PPF–MPEG-6/Fs-3/Hst–GNPs hydrogels containing 10, 20, and 40 v/v% Hst–GNPs was investigated over a 7-day period through a Franz-cell diffusion device. The findings of this study are presented in Fig. 10C. The samples did not show a noticeable burst release; nevertheless, during the first ten hours, the samples released about 5% to 10% of Hst, which can be attributed to the superficially loaded Hst in the GNPs and hydrogel matrix. The release has been sustained in all hydrogel formulations compared to the release profile of Hst from Hst–GNPs (Fig. 4A), which completely released during eight hours. The sustained drug release ability of an injectable medicine is a suitable property for bone repair and helps to improve patient adherence besides treating the damaged tissue with drugs for a longer period.30 Particularly, the hydrogel containing lower concentration of Hst–GNPs (10 v/v%) showed the most rapid release among other hydrogels significantly (p ≤ 0.0001). This may be attributed to the (i) saturation effect: at higher drug concentrations, the drug could approach or reach its saturation solubility limit within the hydrogel, which may limit the availability of drug molecules for the release, resulting in a slower release rate, (ii) diffusion barrier: a higher drug concentration can create a dense drug–polymer matrix within the hydrogel. This dense matrix can act as a diffusion barrier, making it more difficult for the drug molecules to diffuse out of the hydrogel and slowing down the release rate, and (iii) increased drug–polymer interactions: higher drug concentrations can lead to stronger interactions between the drug and the polymer matrix of the hydrogel. Stronger interactions can hinder the drug's mobility and release, resulting in a slower release rate. Also, the Hst–GNP incorporation ratio into the hydrogel can significantly impact the drug release kinetics within the composite system and this feature enables precise control over the drug delivery profiles in tissue engineering and drug delivery.
Cell culture studies
Cell viability and proliferation (MTT assay).
Based on the MTT assay of free Hst at different concentrations (1.56–300 μM), the IC50 of Hst was found to be 75 μM. According to Fig. 11A, MTT analysis of free Hst, GNPs, and Hst–GNPs at the IC50 concentrations of Hst showed that the cell proliferation rate increased at 0.78, 1.56, 3.125, 6.25, and 12.5 μM during seven days, and cell survival was higher than 90%, which underlines the cytocompatibility of the samples with MG-63 cells. According to the results, the optimum concentrations (6.25 and 12.5 μM) with significant proliferation rates (p ≤ 0.0001) were added to 50 μL of hydrogels, and an MTT assay was conducted to assess the cell proliferation rate in hydrogel samples. Results indicated that both concentrations were safe and cytocompatible for seven days when combined with hydrogels. Also, the hydrogels containing NPs indicated slower proliferation than the samples with the free drug, which is due to sustained drug release from NPs in the cell culture medium. This is a good feature for an injectable medicine and increases patient adherence for use in diseases like osteoporosis. As can be seen in Fig. 11A-a, the formulation that had 6.25 μM Hst in NPs showed a more remarkable proliferation than the formulation with 12.5 μM Hst in seven days. Therefore, the hydrogel sample with 6.25 μM Hst in GNPs was chosen for further biological tests.
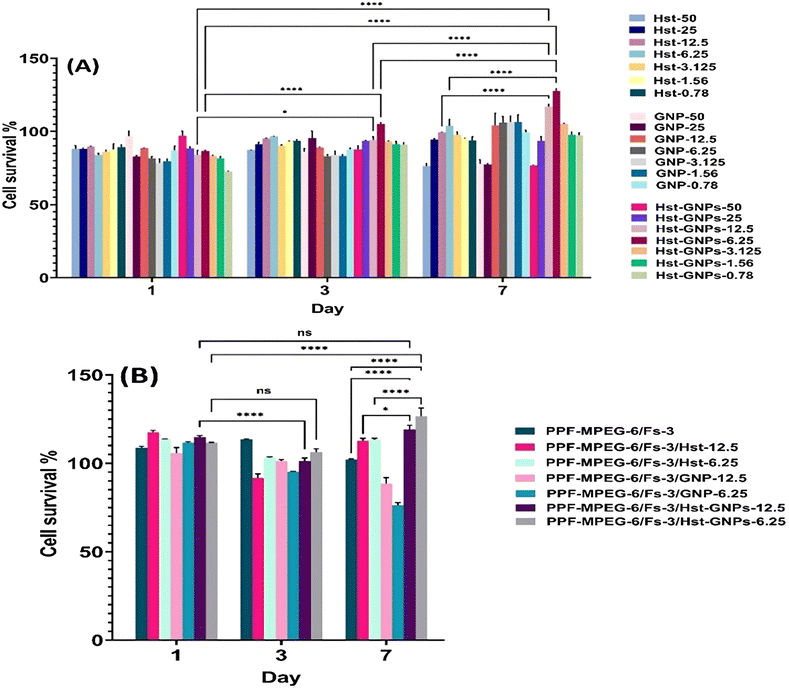 |
| | Fig. 11 MG-63 cell proliferation examined by MTT assay: (A) Hst, blank GNPs, and Hst–GNPs with different concentrations of Hst (50, 25, 12.5, 6.25, 3.125, 1.56, and 0.78 μM) and (B) hydrogel-Fs, hydrogel-Hst, and hydrogel-Fs-Hst–GNPs at Hst concentrations of 12.5 and 6.25 μM [ns: non-significant, * p ≤ 0.05, **** p ≤ 0.0001]. | |
As previously mentioned, the results of MTT assays showed that at utilized concentrations, Hst did not exhibit cell toxicity and did not reach IC50 at days 1, 3, and 7. However, from the perspective of cell growth proliferation, a concentration-dependent trend was observed on day 7, with the maximum cell viability at an Hst concentration of 6.25 μM, beyond which the proliferation rate diminished, indicating that the Hst–GNPs-6.25 formulation was optimal.17,79 The outcomes of drug-free NPs (GNPs) also revealed that no toxicity was observed at different concentrations. In GNPs with concentrations of 1.56–12.5 μM, NPs could stimulate cell growth on day 7 onward. Moreover, the mixture of GNPs and Hst (Hst–GNPs) did not exhibit any toxicity at all concentrations and confirmed the results of the two previous experiments (Hst and GNPs separately). The most effective mixtures used were Hst–GNP-6.25 and Hst–GNP-12.5, which significantly stimulated cell proliferation on day 7, showing the highest cell survival percentage.44 From the MTT results, three critical conclusions can be drawn:
(i) The effect of Hst on MG-63 cells was concentration-dependent, and beyond concentration 6.25 μM, the drug's effect decreased, possibly due to the saturation of the cell pathways influenced by Hst at higher concentrations. Furthermore, at higher concentrations, the cell toxicity of Hst metabolites may increase, resulting in non-linear effectiveness.80
(ii) Cell viability was observed both on day 3 and 7 possibly due to anti-oxidative, anti-stress, and anti-apoptotic effects of Hst. This highlights that Hst effectively reduces cell death over time, and with the reduction in cell death, cell proliferation may increase even after seven days.53,81
(iii) The slow release of Hst dramatically enhanced its effect on cell growth, indicating that the Hst effectiveness pathways have the capability to get saturated. Slow drug release can have a more prolonged and effective impact on cell growth. This conclusion is in agreement with the results of the free drug on day 1, where the lower concentrations showed a little higher effectiveness, but as the time passed, the effectiveness of the lower concentrations diminished due to drug deterioration. The slow release of the drug from GNPs reduced the drug degradation and increased cell access to the drug over time.82
According to Fig. 11A-b, the hydrogel and Fs mixture without drug and NPs neither exhibited cell toxicity nor significantly influenced cell proliferation over time. When Hst was added to the PPF–MPEG/Fs hydrogel, decreased cell viability from day 1 to 3 and increased cell proliferation from day 3 to 7 were observed. This pattern was repeated at both concentrations. This can be attributed to the Hst release from the hydrogel matrix, where initially a quick release of some drugs occurred and exerted their effects. The remaining drug was then gradually released into the surrounding environment as the gel structure degraded. As water and the culture medium infiltrate the gel matrix, the released drug became accessible to cells over time.83 Results of the Hst–GNPs combined with the hydrogel and Fs followed a similar pattern to the free drug. However, on day 7, there was significantly a greater increase in cell growth observed in the presence of Hst–GNPs compared to the free drug (p ≤ 0.05). This can be attributed to the reduced degradation of the drug resulting from its nanoencapsulation, as well as the accumulation of higher drug concentrations by the end of day 7.
Alizarin red staining.
The osteogenic activity of the hydrogel samples was assessed using ARS, a well-known marker used to measure calcium deposition in osteoblasts. This evaluation was conducted over a period of 7 and 14 days, comparing the samples with and without the presence of Hst, GNPs, and Hst–GNPs. As depicted in Fig. 12A, a variation in the intensity and amount of the reddish color can be observed in the calcium granules generated during ARS after 7 and 14 days of cell culture on different hydrogel samples. The surfaces of PPF–MPEG-6/Fs-3 and PPF–MPEG-6/Fs-3/Hst, as well as PPF–MPEG-6/Fs-3/GNP, displayed a red color with lower intensity compared to PPF–MPEG-6/Fs-3/Hst–GNPs at an Hst concentration of 6.25 μM. This indicates the fact that the hydrogel based on PPF–MPEG-6/Fs-3 provides a favorable bioactive surface for promoting bone mineralization. Moreover, the hydrogel-releasing Hst–GNPs demonstrated the formation of a higher amount of the Alizarin Red S–calcium complex compared to free Hst, GNPs, and Hst–GNPs alone. This finding confirms the synergistic effect of Hst–GNPs and PPF–MPEG-6/Fs-3 in promoting osteogenesis. Moreover, the presence of GNPs could have further facilitated the interaction between the hydrogel surface and cells, leading to increased calcium deposition and mineralization.
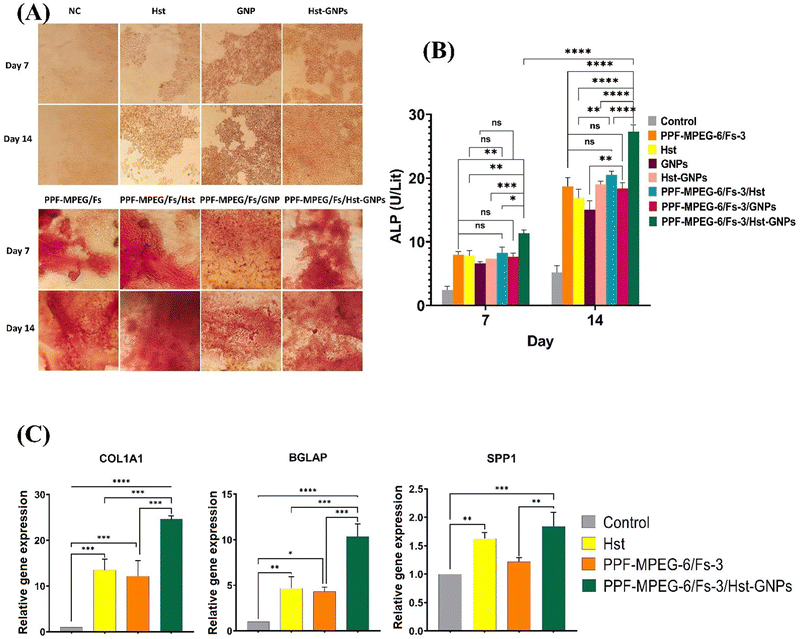 |
| | Fig. 12 Determination of the in vitro osteogenic activity of different studied hydrogels by (A) the alizarin red staining test and (B) alkaline phosphatase assay of different samples after 7 and 14 days of incubation with MG-63 cells [ns: non-significant, (* p ≤ 0.05, ** p ≤ 0.01, **** p ≤ 0.001, **** p ≤ 0.0001)]. (C) Expression of osteoblast-specific gene markers (COL1A1, BGLAP, and SPP1) of MG-63 cells after 14 days of cell culture on samples: [* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001]. | |
Alkaline phosphatase assay.
The activity of ALP, which serves as an early marker for osteoblast differentiation, is considered to be a key factor promoting osteointegration at the bone–implant interface.84 The results illustrated in Fig. 12B underline that the cells cultured on the PPF–MPEG-6/Fs-3/Hst–GNP hydrogel exhibited significantly higher ALP activity after 7 and 14 days of cell seeding (p ≤ 0.05), compared to the cells cultured on pure Hst, GNPs, Hst–GNPs, and pure hydrogel samples with and without free Hst and GNPs. The observed increase in ALP activity and subsequent mineralization can be attributed to the synergistic effect of the PPF–MPEG/Fs hydrogel and Hst–GNPs in simulating osteogenesis, indicating their beneficial role in promoting cell function and mineralization. Wu et al.85 reported that composite materials containing silk fibroin and MgO (magnesium oxide) NPs resulted in increased ALP activity levels compared to the control samples without these components. Also, Cai et al.86 reported a PPF–mPEGA injectable hydrogel, which amplified both the ALP activity and calcium content at the optimal mPEGA concentrations of 5% and 7%. In a separate study, it was found that Hst significantly improved ALP activity, indicating enhanced differentiation; however, it did not lead to the noticeable formation of calcium nodules.18
Gene expression study (RT-PCR).
The RT-PCR analysis was conducted to assess the expression levels of various genes associated with bone repair markers. These genes included beta-actin (ACTB) as an internal control, in addition to osteocalcin (BGLAP), collagen 1 (COL1A1), and osteopontin (SPP1). The application of Hst, PPF–MPEG-6/Fs-3, and PPF–MPEG-6/Fs-3/Hst–GNPs led to the upregulation of the target genes associated with bone repair markers. The results indicated that after 14 days, PPF–MPEG-6/Fs-3/Hst–GNPs caused the most significant increase in the expression of the studied genes compared to the other samples (p ≤ 0.05). Outstandingly, the treatment demonstrated a profound effect on the expression of the collagen 1 gene, indicating a remarkable upregulation (more than 20-fold) in relative gene expression compared to the control cells that were not subjected to any treatment (p ≤ 0.0001). These findings, as illustrated in Fig. 12C, highlight the significant impact of PPF–MPEG-6/Fs-3/Hst–GNPs on the expression of the bone repair-related genes. The observed positive correlation among gene upregulation, enhanced cell proliferation, and improved osteogenic outcomes suggests the potential utility of these interventions in promoting bone repair and regeneration.
Conclusion
In this study, a novel injectable hydrogel containing PPF–MPEG/Fs effectively encapsulated Hst-loaded GNPs, allowing controlled release of Hst for bone repair. This formulation significantly enhanced MG-63 cell functions and showed therapeutic benefits within a biocompatible hydrogel. Incorporating Fs into PPF–MPEG hydrogels notably improved mechanical properties and biomineralization but influenced hydrogel stability, porosity, and degradation based on Fs concentration. Similarly, GNP concentration impacted Hst release, requiring optimal levels determined by characterization and in vitro tests. The PPF–MPEG hydrogel shows great promise across various biomedical applications, boasting excellent biocompatibility, controlled drug release, adaptable mechanical properties for integration and stability, injectability for minimally invasive procedures, and versatility in modifying with bioactive molecules to enhance therapeutic effects and promote specific cellular responses. Despite its strengths, the PPF–MPEG hydrogel does have limitations. Its restricted load-bearing capacity could limit its utility in applications needing high mechanical strength. Also, the tendency of these hydrogels, including PPF–MPEG, to absorb water and swell might impact implant stability and long-term performance. Additionally, diffusion constraints could lead to slower release rates for larger molecules or drugs with low diffusivity, affecting the controlled release mechanism through the hydrogel matrix. Additionally, the biodegradation rate of the PPF–MPEG hydrogel might not match the intended therapeutic or regenerative timeline, potentially causing complications or requiring further interventions. Overall, the developed injectable hydrogel (PPF–MPEG-6/Fs-3/Hst–GNPs-6.25μM) is a promising candidate for bone tissue engineering. This innovative hydrogel has the potential to improve bone healing and regeneration in individuals affected by degenerative bone disease, particularly middle-aged and elderly women. Further investigations, including in vivo and clinical studies, are required to confirm and better realize the full clinical potential of this hydrogel system.
Author contributions
Conceptualization, design and coordination of the study, data curation and validation: JV; visualization: JV, MR, MM; investigation and data interpretation, methodology, project administration, collection and assembly of data, writing (original draft): MZ; supervision: JV, MR, MM; and writing (reviewing and editing): JV, MZ. All authors have read and approved the final version of the manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest in this research.
Acknowledgements
The authors appreciate the financial support of the project offered by the Isfahan University of Medical Sciences (grant number 399184).
References
- K. Zhang, S. Wang, C. Zhou, L. Cheng, X. Gao, X. Xie, J. Sun, H. Wang, M. D. Weir and M. A. Reynolds, Bone Res., 2018, 6, 1–15 CrossRef CAS PubMed.
- N. Salari, N. Darvishi, Y. Bartina, M. Larti, A. Kiaei, M. Hemmati, S. Shohaimi and M. Mohammadi, J. Orthop. Sur. Res., 2021, 16, 669 CrossRef PubMed.
- K. Dzobo, N. E. Thomford, D. A. Senthebane, H. Shipanga, A. Rowe, C. Dandara, M. Pillay and K. S. C. M. Motaung, Stem Cells Int, 2018, 2018, 2495848 CrossRef PubMed.
- S. Preethi Soundarya, V. Sanjay, A. Haritha Menon, S. Dhivya and N. Selvamurugan, Int. J. Biol. Macromol., 2018, 110, 74–87 CrossRef CAS PubMed.
- H. Kandil, B. Ekram and M. A. M. Abo-Zeid, Biomed. Mater., 2023, 18, 15002 CrossRef CAS PubMed.
- J. P. Fisher, S. Jo, A. G. Mikos and A. H. Reddi, J. Biomed. Mater. Res., Part A, 2004, 71, 268–274 CrossRef PubMed.
- M.-P. Ginebra, M. Espanol, Y. Maazouz, V. Bergez and D. Pastorino, EFORT Open Rev., 2018, 3, 173–183 CrossRef PubMed.
- S. Alidadi, Indian J. Vet. Sci. Biotechnol., 2020, 16, 7–11 CrossRef CAS.
- R. Choudhary, A. Chatterjee, S. K. Venkatraman, S. Koppala, J. Abraham and S. Swamiappan, Bioact. Mater., 2018, 3, 218–224 Search PubMed.
- O. Sunho, N. Oh, M. Appleford and J. Ong, Am. J. Biochem. Biotechnol., 2006, 2, 49–56 CrossRef.
- S. K. Venkatraman and S. Swamiappan, J. Biomed. Mater. Res., Part A, 2020, 108, 1546–1562 CrossRef CAS PubMed.
- M. Diba, M. Kharaziha, M. H. Fathi, M. Gholipourmalekabadi and A. Samadikuchaksaraei, Compos. Sci. Technol., 2012, 72, 716–723 CrossRef CAS.
- R. Choudhary, P. Manohar, J. Vecstaudza, M. J. Yáñez-Gascón, H. P. Sánchez, R. Nachimuthu, J. Locs and S. Swamiappan, Mater. Sci. Eng., C, 2017, 77, 811–822 CrossRef CAS PubMed.
- H. Liu, Y. Gao, Y. Dong, P. Cheng, A. Chen and H. Huang, Curr. Pharm. Des., 2017, 23, 1993–2001 CrossRef CAS.
- X. Bai, P. Yang, Q. Zhou, B. Cai, M. Buist-Homan, H. Cheng, J. Jiang, D. Shen, L. Li and X. Luo, Br. J. Pharmacol., 2017, 174, 41–56 CrossRef CAS.
- L. Liu, J. Zheng, Y. Yang, L. Ni, H. Chen and D. Yu, Med. Mol. Morphol., 2021, 54, 1–7 CrossRef CAS PubMed.
- D. Xue, E. Chen, W. Zhang, X. Gao, S. Wang, Q. Zheng, Z. Pan, H. Li and L. Liu, Oncotarget, 2017, 8, 21031 CrossRef PubMed.
- A. Trzeciakiewicz, V. Habauzit, S. Mercier, P. Lebecque, M.-J. Davicco, V. Coxam, C. Demigne and M.-N. Horcajada, J. Nutr. Biochem., 2010, 21, 424–431 CrossRef CAS PubMed.
- P. D. P. Menezes, L. A. Frank, B. D. S. Lima, Y. M. B. G. de Carvalho, M. R. Serafini, L. J. Quintans-Júnior, A. R. Pohlmann, S. S. Guterres and A. A. S. Araújo, Int. J. Nanomed., 2017, 12, 2069–2079 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- A. Jain, S. K. Singh, S. K. Arya, S. C. Kundu and S. Kapoor, ACS Biomater. Sci. Eng., 2018, 4, 3939–3961 CrossRef CAS PubMed.
- R. Urade, N. Sato and M. Sugiyama, Biophys. Rev., 2018, 10, 435–443 CrossRef CAS PubMed.
- I. J. Joye, V. A. Nelis and D. J. McClements, Food Hydrocolloids, 2015, 44, 86–93 CrossRef CAS.
- I. Ezpeleta, J. M. Irache, S. Stainmesse, C. Chabenat, J. Gueguen, Y. Popineau and A.-M. Orecchioni, Int. J. Pharm., 1996, 131, 191–200 CrossRef CAS.
- C. Duclairoir, A.-M. Orecchioni, P. Depraetere, F. Osterstock and E. Nakache, Int. J. Pharm., 2003, 253, 133–144 CrossRef CAS PubMed.
-
N. Reddy and Y. Yang, in Potential and Properties of Plant Proteins for Tissue Engineering Applications, ed. C. T. Lim and J. C. H. Goh, Springer, Berlin, Heidelberg, 2009, pp. 1282–1284 Search PubMed.
- N. Reddy and Y. Yang, J. Mater. Sci.: Mater. Med., 2008, 19, 2055–2061 CrossRef CAS PubMed.
- F. M. DuPont, R. Chan, R. Lopez and W. H. Vensel, J. Agric. Food Chem., 2005, 53, 1575–1584 CrossRef CAS PubMed.
- S. A. Hassanzadeh-Tabrizi, A. Bigham and M. Rafienia, Mater. Sci. Eng., C, 2016, 58, 737–741 CrossRef CAS PubMed.
- M. Kouhi, J. Varshosaz, B. Hashemibeni and A. Sarmadi, Mater. Sci. Eng., C, 2020, 115, 111114 CrossRef CAS PubMed.
- F. K. Kasper, K. Tanahashi, J. P. Fisher and A. G. Mikos, Nat. Protoc., 2009, 4, 518–525 CrossRef CAS PubMed.
- R. A. Dilla, C. M. M. Motta, S. R. Snyder, J. A. Wilson, C. Wesdemiotis and M. L. Becker, ACS Macro Lett., 2018, 7, 1254–1260 CrossRef CAS PubMed.
- C. Duclairoir, E. Nakache, H. Marchais and A.-M. Orecchioni, Colloid Polym. Sci., 1998, 276, 321–327 CrossRef CAS.
- W. Wu, X. Kong, C. Zhang, Y. Hua, Y. Chen and X. Li, LWT, 2020, 129, 109532 CrossRef CAS.
- J. Varshosaz, Z. S. Sajadi-Javan, M. Kouhi and M. Mirian, Int. J. Biol. Macromol., 2021, 192, 869–882 CrossRef CAS PubMed.
- T. Yu, Y. Hu, W. He, Y. Xu, A. Zhan, K. Chen, M. Liu, X. Xiao, X. Xu, Q. Feng and L. Jiang, Mater. Today Bio, 2023, 100558 CrossRef CAS PubMed.
- Z. S. Sajadi-Javan, J. Varshosaz, M. Mirian, M. Manshaei and A. Aminzadeh, Cellulose, 2022, 29, 2413–2433 CrossRef CAS.
- S. Mozammil, R. Verma, J. Karloopia and P. K. Jha, J. Mater. Res. Technol., 2020, 9, 8041–8057 CrossRef CAS.
- H. S. Sofi, T. Akram, N. Shabir, R. Vasita, A. H. Jadhav and F. A. Sheikh, Mater. Sci. Eng., C, 2021, 118, 111547 CrossRef CAS PubMed.
- P. Priyadarshini, P. Karwa, A. Syed and A. N. Asha, J. Drug Delivery Ther., 2023, 13, 33–43 CrossRef CAS.
- M. Špaglová, M. Papadakos, M. Čuchorová and D. Matušová, Polymers, 2023, 15, 329 CrossRef PubMed.
- R. Sedghi, A. Shaabani and N. Sayyari, Carbohydr. Polym., 2020, 230, 115707 CrossRef CAS PubMed.
- M. Farokhi, S. Sharifi, Y. Shafieyan, Z. Bagher, F. Mottaghitalab, A. Hatampoor, M. Imani and M. A. Shokrgozar, J. Biomed. Mater. Res., Part A, 2012, 100, 1051–1060 CrossRef CAS PubMed.
- M. Farzan, J. Varshosaz, M. Mirian, M. Minaiyan and A. Pezeshki, J. Drug Delivery Sci. Technol., 2023, 84, 104459 CrossRef CAS.
- S. Voci, A. Gagliardi, N. Ambrosio, M. C. Salvatici, M. Fresta and D. Cosco, Pharmaceutics, 2023, 15 Search PubMed.
- D. Li, Z. Wei and X. Li, Foods, 2023, 12 CAS.
- S. Manafi, F. Mirjalili and S. Joughehdoust, Adv. Ceram. Prog., 2020, 6, 35–42 Search PubMed.
- E. Behravesh, A. K. Shung, S. Jo and A. G. Mikos, Biomacromolecules, 2002, 3, 153–158 CrossRef CAS PubMed.
- I. J. Joye, V. A. Nelis and D. J. McClements, Food Hydrocolloids, 2015, 43, 236–242 CrossRef CAS.
- M. A. Arangoa, M. A. Campanero, M. J. Renedo, G. Ponchel and J. M. Irache, Pharm. Res., 2001, 18, 1521–1527 Search PubMed.
- M. Fathi, J. Varshosaz, M. Mohebbi and F. Shahidi, Food Bioprocess Technol., 2013, 6, 1464–1475 CrossRef CAS.
- L. M. Lazer, B. Sadhasivam, K. Palaniyandi, T. Muthuswamy, I. Ramachandran, A. Balakrishnan, S. Pathak, S. Narayan and S. Ramalingam, Int. J. Biol. Macromol., 2018, 107, 1988–1998 CrossRef PubMed.
- M. Alipour, S. Sharifi, M. Samiei, S. Shahi, M. Aghazadeh and S. M. Dizaj, Sci. Rep., 2023, 13, 2076 CrossRef CAS PubMed.
- Z. Bahrami, A. Pedram-Nia, M. Saeidi-Asl, M. Armin and M. Heydari-Majd, Food Sci. Nutr., 2023, 11, 307–319 CrossRef CAS PubMed.
- N. Sharif, M.-T. Golmakani, M. Niakousari, B. Ghorani and A. Lopez-Rubio, Food Res. Int., 2019, 116, 1366–1373 CrossRef CAS PubMed.
- M. Liu, X. Zeng, C. Ma, H. Yi, Z. Ali, X. Mou, S. Li, Y. Deng and N. He, Bone Res., 2017, 5, 17014 CrossRef CAS PubMed.
- K. Apmann, R. Fulmer, A. Soto and S. Vafaei, Materials, 2021, 14, 1291 CrossRef CAS PubMed.
- G. Furtos, M. Naghiu, H. Declercq, M. Gorea, C. Prejmerean, O. Pana and M. Tomoaia-Cotisel, J. Biomed. Mater. Res., Part B, 2016, 104, 1290–1301 CrossRef CAS PubMed.
- L.-C. Gerhardt and A. Boccaccini, Materials, 2010, 3, 3867–3910 CrossRef CAS PubMed.
- H. Ghomi, M. Jaberzadeh and M. H. Fathi, J. Alloys Compd., 2011, 509, L63–L68 CrossRef CAS.
- C. I. Vallo, P. E. Montemartini, M. A. Fanovich, J. M. P. Lopez and T. R. Cuadrado, J. Biomed. Mater. Res., 1999, 48, 150–158 CrossRef CAS PubMed.
-
C. Misra, S. K. Behera and R. Bandyopadhyay, arXiv, 2024, preprint, arXiv:2210.09694v2 DOI:10.48550/arXiv.2210.09694.
- L. J. Suggs, R. S. Krishnan, C. A. Garcia, S. J. Peter, J. M. Anderson and A. G. Mikos, J. Biomed. Mater. Res., 1998, 42, 312–320 CrossRef CAS PubMed.
- B. C. Kallukalam, M. Jayabalan and V. Sankar, Biomed. Mater., 2008, 4, 15002 CrossRef.
- F. von Burkersroda, L. Schedl and A. Göpferich, Biomaterials, 2002, 23, 4221–4231 CrossRef CAS PubMed.
- E. L. Hedberg, C. K. Shih, J. J. Lemoine, M. D. Timmer, M. A. K. Liebschner, J. A. Jansen and A. G. Mikos, Biomaterials, 2005, 26, 3215–3225 CrossRef CAS.
- R. Choudhary, S. K. Venkatraman, I. Bulygina, A. Chatterjee, J. Abraham, F. Senatov, S. Kaloshkin, A. Ilyasov, M. Abakumov, M. Knyazeva, D. Kukui, F. Walther and S. Swamiappan, J. Asian Ceram. Soc., 2020, 8, 1051–1065 CrossRef.
- F. Tavangarian and R. Emadi, Ceram. Int., 2011, 37, 2275–2280 CrossRef CAS.
- J. Wei, S. J. Heo, D. H. Kim, S. E. Kim, Y. T. Hyun and J.-W. Shin, J. R. Soc., Interface, 2008, 5, 617–630 CrossRef CAS PubMed.
- A. Teimouri, L. Ghorbanian, A. Chermahini and R. Emadi, Ceram. Int., 2014, 40, 6405–6411 CrossRef CAS.
- N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 26, 85–123 CrossRef CAS PubMed.
- L. J. Suggs, E. Y. Kao, L. L. Palombo, R. S. Krishnan, M. S. Widmer and A. G. Mikos, J. Biomater. Sci., Polym. Ed., 1998, 9, 653–666 CrossRef CAS PubMed.
- A. Leonés, M. Lieblich, R. Benavente, J. L. Gonzalez and L. Peponi, Nanomaterials, 2020, 10 Search PubMed.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12, 3323 CrossRef CAS PubMed.
- S. Ramesh, A. Yaghoubi, K. Y. Sara Lee, K. M. Christopher Chin, J. Purbolaksono, M. Hamdi and M. A. Hassan, J. Mech. Behav. Biomed. Mater., 2013, 25, 63–69 CrossRef CAS PubMed.
- H. Ueda, M. C. Hacker, A. Haesslein, S. Jo, D. M. Ammon, R. N. Borazjani, J. F. Kunzler, J. C. Salamone and A. G. Mikos, J. Biomed. Mater. Res., Part A, 2007, 83, 656–666 CrossRef CAS PubMed.
- A. M. Diez-Pascual and A. L. Diez-Vicente, ACS Appl. Mater. Interfaces, 2016, 8, 17902–17914 CrossRef CAS PubMed.
- Z. Wu, K. Zheng, J. Zhang, T. Tang, H. Guo, A. R. Boccaccini and J. Wei, J. Mater. Chem. B, 2016, 4, 7974–7988 RSC.
- H. Liu, Y. Dong, Y. Gao, L. Zhao, C. Cai, D. Qi, M. Zhu, L. Zhao, C. Liu and F. Guo, J. Cell. Physiol., 2019, 234, 11009–11022 CrossRef CAS PubMed.
- K. Gurushankar, M. Gohulkumar, N. R. Prasad and N. Krishnakumar, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2013, 5, 15006 Search PubMed.
- B. Li, A.-L. Huang, Y.-L. Zhang, Z. Li, H.-W. Ding, C. Huang, X.-M. Meng and J. Li, Molecules, 2017, 22, 1067 CrossRef PubMed.
- Q. Mu, Y. Zhang, Q. Cheng, H. Huang, C. Huang and L. Tang, Ann. Transl. Med, 2022, 10, 806 CrossRef CAS PubMed.
- A. M. Diez-Pascual, Polymers, 2017, 9, 260 CrossRef PubMed.
- Z. Wu, K. Zheng, J. Zhang, T. Tang, H. Guo, A. Boccaccini and J. Wei, J. Mater. Chem. B, 2016, 4, 7974–7988 RSC.
- Z. Wu, Z. Meng, Q. Wu, D. Zeng, Z. Guo, J. Yao, Y. Bian, Y. Gu, S. Cheng, L. Peng and Y. Zhao, J. Tissue Eng., 2020, 11, 2041731420967791 Search PubMed.
- L. Cai, K. Wang and S. Wang, Biomaterials, 2010, 31, 4457–4466 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mahboubeh
Rostami
*a,
Mahboubeh
Rostami













