
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast carbon dioxide–epoxide cycloaddition catalyzed by metal and metal-free ionic liquids for designing non-isocyanate polyurethanes†
Marwa
Rebei
ab,
Ctirad
Červinka
c,
Andrii
Mahun
ab,
Petra
Ecorchard
d,
Jan
Honzíček
 e,
Sébastien
Livi
f,
Ricardo K.
Donato
g and
Hynek
Beneš
*a
e,
Sébastien
Livi
f,
Ricardo K.
Donato
g and
Hynek
Beneš
*a
aInstitute of Macromolecular Chemistry of the Czech Academy of Sciences, Heyrovského nám.2, Prague 6, 162 00, Czech Republic. E-mail: benesh@imc.cas.cz
bDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Hlavova 8, 12843 Prague, Czech Republic
cDepartment of Physical Chemistry, University of Chemistry and Technology Prague Technická 5, CZ-166 28, Prague 6, Czech Republic
dInstitute of Inorganic Chemistry of the Czech Academy of Sciences, Husinec-Řež 1001, 250 68 Řež, Czech Republic
eInstitute of Chemistry and Technology of Macromolecular Materials, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10, Pardubice, Czech Republic
fUniversité de Lyon, CNRS, Université Claude Bernard Lyon 1, INSA Lyon, Université Jean Monnet, UMR 5223, Ingénierie des Matériaux Polymères, F-69621 Cédex, France
gNational University of Singapore, Center for Advanced 2D Materials, Singapore 117546, Singapore
First published on 28th March 2024
Abstract
The recycling of industrially produced greenhouse gases, such as CO2, into high-value-added chemicals is one of the most relevant strategies for reaching climate targets. Herein, we present a two-step strategy for how to efficiently convert renewable carbon dioxide (CO2) into β-hydroxyurethanes using imidazolium ionic liquids (ILs) bearing metal ([ZnCl4]2− and [CoCl4]2−) or Cl− IL-anions as all-in-one catalysts. The first step involves the rapid (1 h) conversion of phenyl glycidyl ether using ILs and supercritical carbon dioxide (7.7 MPa, 80 °C) to cyclic carbonates in high yield (98%). The DFT calculations suggested a comprehensive mechanistic pathway for the IL-catalyzed CO2-epoxy reaction showing a rate-determining step of the initial epoxide ring opening and the direct participation of IL-anions. Moreover, the applied ILs are also able to catalyze the subsequent reaction of the formed cyclic carbonates with butylamine resulting in fast formation of β-hydroxyurethanes. Thus, the present concept seems to be a promising strategy for designing non-isocyanate polyurethanes (NIPUs).
Introduction
Nowadays, advanced polymeric materials are at the forefront of tackling global challenges, such as environmental issues, while pursuing research on high-performance materials. Thus, the development of new synthetic methods is critical for designing innovative polymer materials and proposing sustainable solutions to meet the requirements of the circular economy, which are durability, reusability, and recyclability. Therefore, scientists must propose functional materials using a design approach to develop molecular brick platforms that allow integration of the required functions at the molecular scale in the initial synthesis steps, for example, the use of industrially produced greenhouse gases, such as CO2. Their conversion into high-value-added chemicals has become one of the most relevant strategies to reduce global warming and thus limit climate change (the CO2 concentration in the atmosphere reached levels above 412 ppm in 2020).1–4CO2 is an inexpensive, non-flammable, and abundant carbon resource, rendering its use in polymer synthesis economically viable on an industrial scale.1,5–7 In this context, the chemical fixation of CO2 by cycloaddition of the epoxide ring represents an environmentally benign route to produce polyfunctional cyclic carbonate-based monomers.8–11 Naturally, the synthesis of cyclic carbonates via CO2 cycloaddition requires catalysts, e.g. aluminium porphyrin derivatives12 or metal-containing catalysts such as zinc and cobalt complexes, usually active at high reaction temperatures and requiring the use of a solvent.13–15 One of the promising applications of cyclic carbonates is the preparation of so-called non-isocyanate polyurethanes (NIPUs), which are synthesized by ring-opening polymerization of polyfunctional cyclic carbonate monomers with polyamines. NIPUs represent a promising alternative to replace conventional polyurethanes (PUs).12,16 Nevertheless, the fabrication of NIPUs from cyclic carbonates17–19 usually requires the addition of suitable catalysts and solvents,20,21 long reaction time22 and high temperature,23 which decreases the reaction sustainability features and environmental aspects. Therefore, the development of new materials enabling efficient CO2 absorption, capture and NIPU synthesis presents a challenging task to overcome the above-mentioned drawbacks.
Recently, imidazolium, phosphonium, and ammonium-based ionic liquids (ILs) were found to be suitable catalysts for the CO2 addition to epoxides due to their high thermal stability, tunable structure and simple synthetic routes.18–20,24–26 Furthermore, ILs offer value by stabilizing metal atoms within their structure, without deactivating the metal's catalytic functionality and synergizing it with its own properties. The result is a functionalized-metal-containing IL (MIL) with high catalytic activity, where the metal atom can be incorporated by the anion while the cation is, e.g., a catalytically active imidazolium ring.27 These MILs have demonstrated their efficacy as catalysts/initiators for ring-opening polymerization (ROP) reactions of lactones28–30 and epoxides.31 Furthermore, the utilization of metal–organic frameworks (MOFs) incorporating ILs has emerged as a promising strategy for the preparation of recyclable porous catalysts employed in the CO2 cycloaddition to epoxide.32 However, this approach targets the synthesis of catalysts with desirable properties such as high surface area and porosity, which facilitates the CO2 adsorption into epoxide compounds efficiently while allowing the fast and facile regeneration of the catalyst; however, such catalysts require high temperatures, long reaction times (12–24 h), and the use of an organic solvent, resulting in less environmentally friendly processes.33–35
Based on previous findings showing the remarkable catalytic activity of imidazolium ILs for epoxy ring opening,36 herein we further investigate the potential of imidazolium ILs in CO2 cycloaddition. Moreover, we explore the catalytic potential of discrete MILs for the copolymerization of CO2 and epoxides.37–39 Herein, we hypothesize that the MIL introduced in the initial step for the synthesis of cyclic carbonates can also catalyse the subsequent reaction between the cyclic carbonates and amines leading to hydroxyurethanes.
In this study, three distinct ILs, a metal-free IL (1-butyl-2-methylimidazolium chloride, BMIMCl) and two imidazolium MILs bearing [ZnCl4]2− and [CoCl4]2− anions, were tested as all-in-one catalysts for both the formation of cyclic carbonates and their subsequent aminolysis (ring opening) producing β-hydroxyurethanes. First, the conversion of epoxy groups of monofunctional phenyl glycidyl ether (PGE) to cyclic carbonates catalyzed by ILs was studied using FTIR and NMR spectroscopy. Based on the obtained experimental data and DFT calculations, a comprehensive mechanism of the IL-catalyzed CO2-epoxy reaction was suggested. The subsequent reaction of the synthesized cyclic carbonates with butylamine without additional catalysis was then studied to verify the future potential of this approach for the rapid solvent-free preparation of NIPU materials.
Experimental
Materials and synthesis
PGE was purchased from Sigma-Aldrich (99%, 150 g mol−1), 1-butyl-3-methylimidazolium chloride (BMIMCl, 99%, 174.67 g mol−1) was purchased from Iolitech and purified before use as described in our previous paper.36n-Butylamine was purchased from Fluka Chemicals (73.14 g mol−1). The structures of epoxy monomers, amines, and all ILs used are shown in Fig. 1.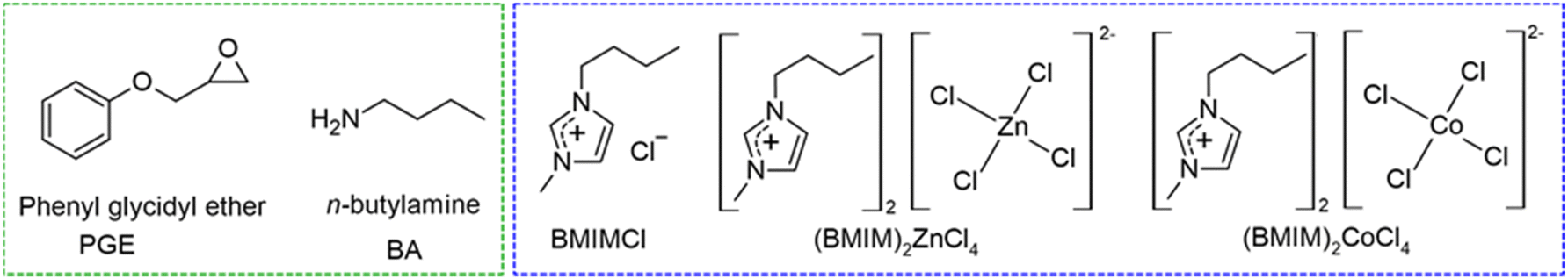 | ||
| Fig. 1 Structures and acronyms of epoxy monomers, amines and ionic liquids used. | ||
Synthesis of bis(1-butyl-3-methylimidazolium) tetrachlorozincate(II), (BMIM)2ZnCl4
(BMIM)2ZnCl4 was synthesized by treating BMIMCl (10 g, 57.3 mmol) with ZnCl2 (3.9 g, 28.6 mmol). The reaction mixture was heated to 80 °C for 6 h. The obtained (BMIM)2ZnCl4 appears as a yellow viscous liquid. Yield: 13.75 g (28.3 mmol, 99%). The 13C and 1H NMR spectra are given in the ESI† (Fig. S1 and S2).Synthesis of bis(1-butyl-3-methylimidazolium) tetrachlorocobaltate(II), (BMIM)2CoCl4
(BMIM)2CoCl4 was synthesized by treating BMIMCl (10 g, 57.3 mmol) with CoCl2 (3.8 g, 29.3 mmol). The reaction mixture was heated to 150 °C for 2 h. The unreacted CoCl2 was removed by filtration over a short Celite pad heated to ∼100 °C. The obtained (BMIM)2CoCl4 appears as a blue viscous liquid. Yield: 13.3 g (27.8 mmol, 97%). The synthesized (BMIM)2CoCl4 was not characterized using NMR spectroscopy due to its magnetic properties.Synthesis of cyclic carbonates
The CO2 addition reactions were performed under supercritical CO2 (sCO2) using a stainless-steel high-pressure reactor equipped with a pressure control regulator under solvent-free conditions. PGE (1a) was reacted with ILs, i.e, BMIMCl, (BMIM)2ZnCl4, or (BMIM)2CoCl4, added at various concentrations (1, 5, or 10 mol%) to an open aluminium pan, as shown in Scheme 1(a). The mixture was manually mixed to obtain a transparent homogeneous liquid, which was introduced inside the CO2 reactor. Then, air and moisture were eliminated by purging the reactor with low-CO2 pressure at RT for 5 min. The reactor containing the reaction mixture was connected to the sCO2 gas cylinder for the entire reaction time (1 h) at a pressure of 7.7 MPa and a temperature of 80 °C. Afterward, sCO2 was slowly released, and the reactor was cooled down at room temperature.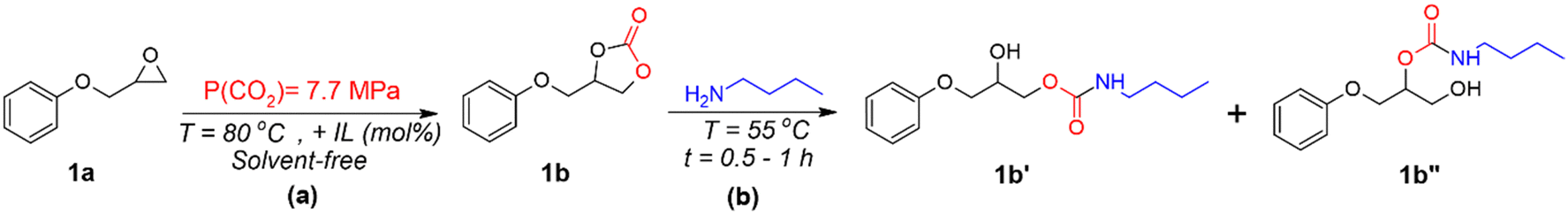 | ||
| Scheme 1 Simplified schemes of (a) cycloaddition reaction for PGE using ILs, and (b) synthesis of β-hydroxyurethanes from cyclic carbonate. | ||
The reactions were performed at different times, temperatures, and pressures to reach the final optimal reaction conditions (catalyst loading = 10 mol%, T = 80 °C, and P(sCO2) = 7.7 MPa) and were subject to FTIR and NMR characterizations.
Synthesis of β-hydroxyurethanes
The cyclic carbonates (1b) obtained from the PGE/CO2/IL reaction were mixed in a stoichiometric amount of n-butylamine (BA) without the addition of any solvent or co-catalyst, as shown in Scheme 1(b). The reaction mixture was stirred under mild conditions (T = 55 °C) for 0.5 to 1 h. The reaction progress was followed by FTIR spectroscopy, by following the disappearance of the peak at approximately 1780 cm−1 related to the carbonate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, of the cyclic carbonate.31 The disappearance of such a peak proves the ring-opening of the cyclic carbonate and the proceeding of the reaction with functional groups (primary amines). The obtained products were measured in 1H NMR to confirm the structure and provide the β-hydroxyurethane yields.
O, of the cyclic carbonate.31 The disappearance of such a peak proves the ring-opening of the cyclic carbonate and the proceeding of the reaction with functional groups (primary amines). The obtained products were measured in 1H NMR to confirm the structure and provide the β-hydroxyurethane yields.
Characterization
Metal-based ionic liquids characterization techniques
FTIR measurements were performed using a Nicolet iS50 or Nicolet Nexus 670 FTIR spectrometer (Thermo Fisher Scientific). Mid-FTIR was acquired using a built-in diamond ATR (attenuated total reflection) crystal in the region of 4000–400 cm−1 (data spacing = 0.5 cm−1). FTIR spectroscopy is considered an easy tool that provides fast proof of the formed characteristic functions, such as the imidazolium ring and alkyl chains. 1H and 13C NMR spectra were recorded using a JEOL 600 MHz (14.1 T) spectrometer, to confirm the structure and characteristic bands of the formed MIL. TGA was performed using a thermogravimetric analyzer Pyris 1 TGA (PerkinElmer) under nitrogen flow (25 cm3 min−1). A sample of ca. 5 mg was heated from 30 to 800 °C at a heating rate of 10 °C min−1. TGA was carried out to study the thermal stability of the MIL. Elemental analysis (AAS) was used to determine the metal content (Co and Zn) in each MIL. The amount of the sample adjusted according to the expected content of the measured element (2–10 mg) was weighed into pre-weighed Eppendorf tubes under dry air in a glove box. The weighed samples were then quantitatively transferred into the water of MilliQ purity in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50. The content of Co and Zn was measured by atomic absorption spectrometry (AAS) using an atomic absorption spectrometer (PerkinElmer, model 3110) with a hollow cathode lamp emitting the spectrum specific for Co and Zn as the light source. An external calibration was used.
:50. The content of Co and Zn was measured by atomic absorption spectrometry (AAS) using an atomic absorption spectrometer (PerkinElmer, model 3110) with a hollow cathode lamp emitting the spectrum specific for Co and Zn as the light source. An external calibration was used.
Cyclic carbonates’ characterization techniques
FTIR measurements of the samples were performed using a Spectrum 100 spectrometer (PerkinElmer) equipped with a mercury–cadmium–telluride (MCT) detector and a universal ATR (attenuated total reflectance) accessory with a diamond prism. The spectra were averaged over 4 scans at a resolution of 4 cm−1. The disappearance of the peak at ∼915 cm−1 is followed during the reaction as it is attributed to the C–O stretching of the oxirane group.36,40 Also, the formation of the cyclic carbonates is followed by the intensity increase of the band at ∼1790 cm−1.17 The conversions of epoxy functions were calculated from the integral intensity of the absorption band at 915 cm−1 (A) and the reference band at 1600 cm−1 (Aref) according to eqn (1):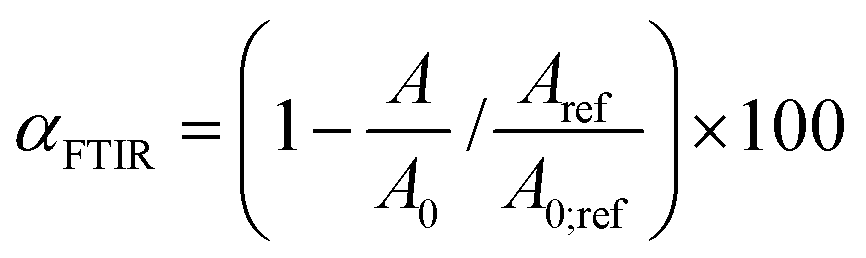 | (1) |
Parameters A0 and A0;ref are the intensities of the absorption bands for neat epoxy resin at given wavenumbers.
1H NMR spectra were recorded using a Bruker NEO 400 spectrometer operating at 400.1 MHz. NMR spectroscopy was utilized to investigate the structure of organic compounds, determine the components and products of reactions, calculate the reaction yields and provide the molar ratio of isomeric products. All the investigated samples were dissolved in DMSO-d6 at 295 K. 1H NMR spectra were acquired by applying a 90° pulse (width = 18 μs) with a 10 s recycle delay and 32 scans. The chemical shifts were relative to TMS using hexamethyldisiloxane (HMDSO, 0.05 ppm from TMS in 1H spectra) as an internal standard. In general, the conversion of the initial reagent (i.e. epoxides or cyclic carbonates) can be calculated using the following equation (eqn (2)):
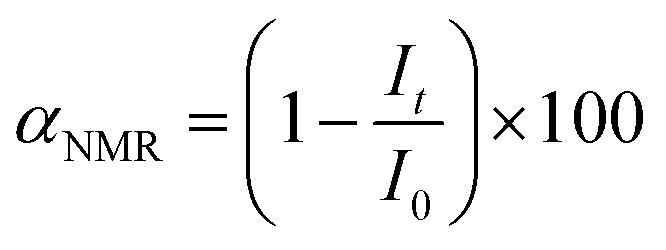 | (2) |
Computational models
The initial modelling setup assumed a three-molecular gas-phase (isolated) cluster composed of a PGE molecule, a carbon dioxide molecule, and a chloride anion. To investigate the impact of the solvent on the reaction barriers, up to two BMIM cations and an additional chloride anion were optionally included in the simulated clusters for comparison.Generation of initial structures
First, to obtain a reasonable initial estimate of the geometry of molecular clusters of reactants, intermediate molecules, and products, all participating in individual reaction steps, a simulation annealing on the level of classical molecular dynamics (MD) was performed. This consists of simulating the motion of clusters of several target molecules upon a gradual slow cooling. It starts at an elevated temperature of 227 °C allowing the simulated clusters to escape any spurious potential energy wells. The temperature of the simulated clusters near absolute zero, −273.15 °C, was then reached after simulating its trajectory covering a 2 μs period. Such a procedure typically yields cluster conformations that correspond to the global minimum of their potential energy,41 which is important to preselect the most beneficial intermolecular contacts and intramolecular conformations of the relevant flexible molecules out of the large space of all theoretical conformations and mutual arrangements of molecules in such clusters. Performing this initial step using the cheap classical MD simulations saves computational resources in the latter stage.All simulated annealing runs were performed in Lammps software,42 using an all-atom non-polarizable OPLS force field and a 1 fs time integration step. For BMIMCl, the established CL&P model was used,43 whereas a flexible force field model was used for carbon dioxide.44 For PGE, being the primary reactant, and for 1-chloro-3-phenoxypropan-2-olate and 1-chloro-3-phenoxypropan-2-yl carbonate, both expected to occur in the reaction mechanism, the LigParGen utility45 was used to generate all the required harmonic bonding parameters, OPLS torsion parameters and Lennard-Jones parameters for dispersion interactions according to the OPLS force field library.46 Atomic charges in the given molecular entities were adopted from the CM1A model, scaling the atomic charges by the default 1.0 factor.47
DFT calculations
Geometries of the simulation-annealed clusters were further optimized using density functional theory in Gaussian 1648 at the B3LYP-D3(BJ)/6-311+G(d,p) level of theory49,50 to verify the dynamical stability of all optimized configurations of reactants, intermediates and products. Tentative relaxed scans of the potential energy upon variation of the reaction coordinates were attempted to glimpse the possible geometries of the relevant transition states. Finally, geometries of the transition states, corresponding to saddle points on the potential energy surface, and thus determining the reaction energy barriers, were optimized using the Synchronous Transit-Guided Quasi-Newton51 method as implemented in Gaussian 16. Frequencies of the fundamental vibrational modes were calculated for the clusters of relevant species within the harmonic approximation to assess the thermal and entropic effects on the activation energies. These were plugged together with molecular masses and moments of inertia (based on the optimized geometries) to the rigid-rotor harmonic-oscillator model,52 yielding enthalpies and Gibbs free energies of all target molecules at ambient conditions. This DFT-based procedure consistently yields relatively accurate energies, allowing us to qualitatively rank the relative stability and thermochemical relationships among the present molecules.Solvation models
Since the proposed reactions take place in a highly polar medium, a combination of implicit and explicit solvation models was considered to address the solvation effects. The polarizable continuum model using the integral equation formalism53 was used for modelling the solvent implicitly. Solvent parameters had to be newly defined according to an analysis of the experimental bulk properties of BMIMCl. Namely, its relative permittivity was estimated to be 8.500, resulting from an interpolation of literature data within a homologous series of similar ionic liquids;54,55 its refractivity was estimated at 1.532, resulting from extrapolations of data measured for binary mixtures, and an effective radius 4.001 Å of the ion pair was estimated from its liquid phase density.56 As a step towards the explicit solvation, all calculations were performed for comparison also in the explicit presence of either only a single chloride anion, or a single BMIMCl ion pair, or two BMIMCl ion pairs for comparison.Results and discussion
Synthesis of cyclic carbonates
PGE was reacted with supercritical CO2 and two types of ILs: (i) the metal-free ILs (BMIMCl) or (ii) the MILs denoted (BMIM)2ZnCl4 and (BMIM)2CoCl4 under solvent-free and co-catalyst-free conditions to receive cyclic carbonates. The synthesized MILs were characterized by various techniques, such as FTIR, TGA, NMR, and a detailed discussion of their confirmed structures and thermal stabilities is given in the ESI† (Fig. S1–S3). First, PGE/CO2/BMIMCl mixtures with IL concentrations of 1, 5 and 10 mol% were reacted in supercritical CO2 conditions (1 h at 80 °C, 7.7 MPa). The reaction yielded solid materials with melting temperatures (Tm) between 75 and 88 °C, confirmed by DSC (Table 1).| Entry | IL | IL loading [mol%] | α FTIR (%) | α NMR (%) | TOFd | T m (°C)e |
|---|---|---|---|---|---|---|
| a Reaction conditions: catalyst loading [mol%] with respect to PGE,7.7 MPa of CO2, 80 °C, 1 h. b The cyclic carbonates yield calculated via FTIR spectra (eqn (1)). c The cyclic carbonates yield was calculated from the 1H NMR spectra (eqn (2)). d TOF related to IL: yield/[(IL loading in mol%) × time]. e Melting temperature of cyclic carbonates determined as the onset of melting endotherm from the DSC runs. | ||||||
| 1 | BMIMCl | 5 | — | 97 | 9.7 | 87 |
| 2 | BMIMCl | 10 | 94 | 98 | 9.8 | 75 |
| 3 | (BMIM)2CoCl4 | 10 | 99 | 99 | 9.9 | 88 |
| 4 | (BMIM)2ZnCl4 | 10 | 93 | 98 | 9.8 | 76 |
The FTIR spectroscopy allowed fast monitoring of the reaction progress as it constitutes an easy tool to follow the formation of specific functional groups, such as the carbonate bond CO of cyclic carbonates (structure 1b, in Scheme 1). Thus, the FTIR spectra of the product with 10 mol% of BMIMCl showed the disappearance of the characteristic peak at 915 cm−1 confirming the opening of the oxirane ring.56–58 This proves at first glance the catalytic ability of imidazolium-based ILs to initiate the ring-opening of epoxy monomers, which is in line with our previous findings using BMIMCl.36 The use of a lower concentration of ILs (1 and 5 mol%) revealed only a partial opening of epoxy groups (Fig. S4, ESI†). Nevertheless, the formation of cyclic carbonates was observed in all the formed products with all IL concentrations. This is evidenced by the appearance of a new distinct peak at 1787 cm−1, attributed to the carbonate group CO of cyclic carbonate.59–62 In summary, it seems that an IL composition of 10 mol% is ideal for obtaining a full epoxy consumption within 1 h in favor of cyclic carbonate formation. Therefore, this concentration was adopted to investigate the proposed systems further. Accordingly, the yield of cyclic carbonates calculated from 1H NMR (eqn (2)) was determined to be 98% and 97% for 10 and 5 mol% of BMIMCl, respectively, while the use of only 1 mol% BMIMCl was not sufficient to achieve the desired cyclic carbonate function. Other studies also reported similar high cyclic carbonate yields using binary DBU catalytic systems or imidazolium-based ILs.59,62–65 However, a high temperature (T ≥ 100 °C)66–68 and a long reaction time are generally needed.26,67,69 Meanwhile, our approach does not depend on the conditions mentioned above, yet, we speculate that the high efficiency of the IL obtained could be due to the use of CO2 pressure. Moreover, in this study, DFT calculations are performed, allowing us to suggest the CO2 cycloaddition mechanism catalysed by ILs.
To compare the catalytic activity of synthesized MILs with the metal-free BMIMCl, the reactions of PGE/CO2 mixtures with 10 mol% of (BMIM)2ZnCl4 and (BMIM)2CoCl4 were performed by applying the previous conditions (80 °C, 1 h, 7.7 MPa). FTIR and 1H NMR spectroscopy were used to investigate the interactions between the MILs and PGE/CO2 mixtures (Fig. 2).
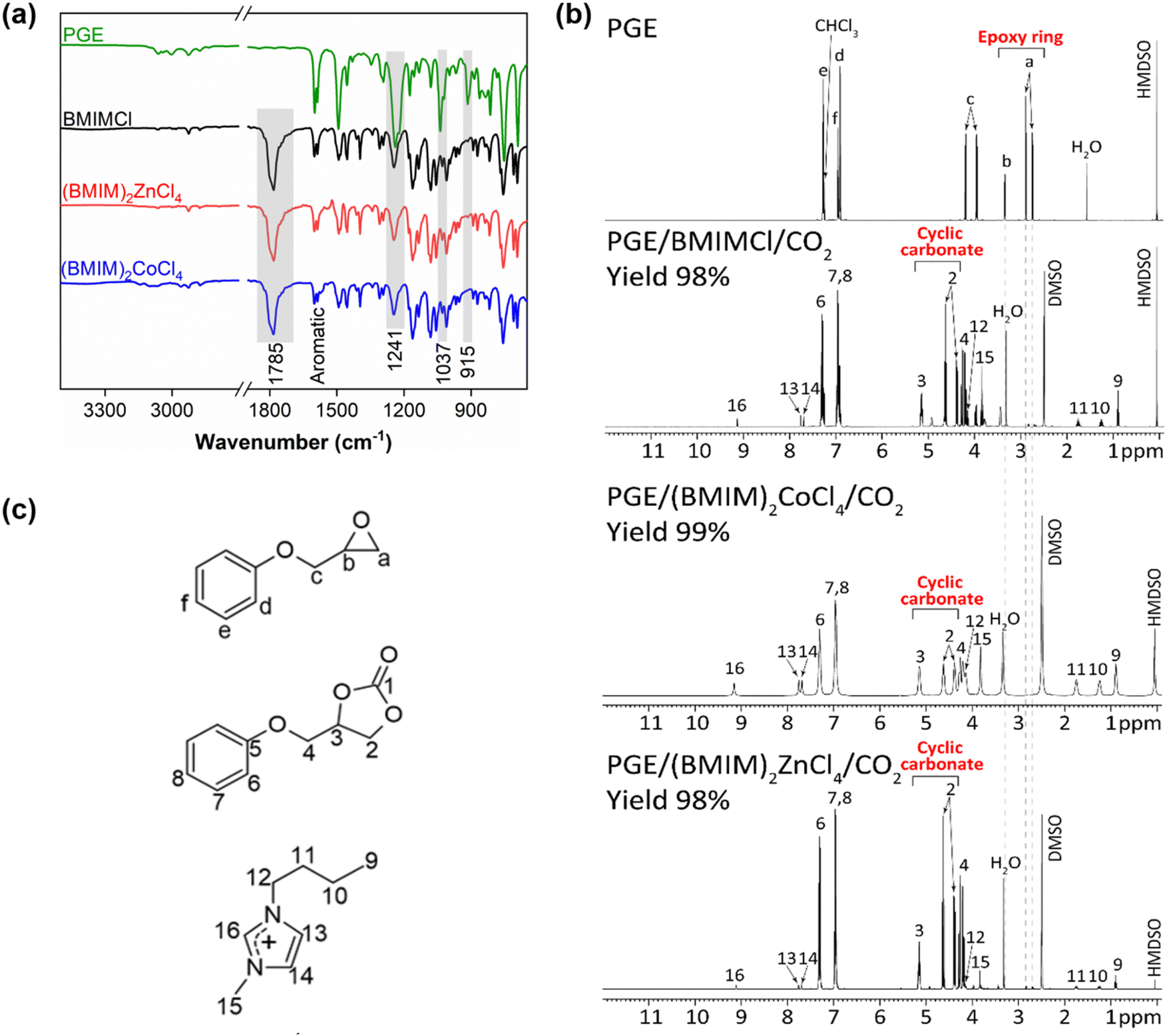 | ||
| Fig. 2 (a) FTIR and (b) 1H NMR spectra of PGE/CO2/IL reaction products after 1 h and at 10 mol% of BMIMCl, (BMIM)2CoCl4, and (BMIM)2ZnCl4 (c) structures of reactants and products detected in 1H NMR spectra. | ||
The FTIR spectra of reaction products showed a full disappearance of the C–O epoxy band at 915 cm−1 suggesting the epoxy ring-opening.56,57 Additionally, the formation of the CO band at 1785 cm−1 corresponding to the carbonyl of cyclic carbonate was observed (Fig. 2a). Other FTIR bands confirmed the presence of the aromatic backbone of PGE (the bands in the region of 1600 and 1400 cm−1)70 and the C–O ether group of the cyclic carbonate structure (the stretching vibration bands at 1274 and 1037 cm−1).26
The full consumption of epoxy rings was also confirmed using the 1H NMR spectroscopy by the disappearance of the respective NMR signals, marked as “a” and “b” (Fig. 2b and c) at 2.74 ppm, 2.89 ppm and 3.34 ppm. The appearance of new signals “2” and “3” at 4.38 ppm, 4.62 ppm and 5.15 ppm revealed the formation of cyclic carbonates (Fig. 2b and c).71 Remarkably, the yields of cyclic carbonates obtained from PGE/CO2 with all ILs were between 98–99% as calculated by 1H NMR, which was further confirmed by FTIR spectroscopy showing similar high yields of the respective cyclic carbonates (93–99%) (Table 1, entries 2–4). A similar result was reported previously by Chen et al., where (BMIM)2ZnBr4 was used as a catalyst for CO2 cycloaddition to propylene oxide by obtaining a cyclic carbonate yield of 92.6% after 5 h at room temperature; however, only a yield of 50.5% was obtained when using (HMIM)2ZnCl4.72 These results proved the catalytic ability of MILs for the CO2 cycloaddition. The high catalytic activity of MILs can be explained by their enhanced Lewis acid character due to the presence of an anionic metallic moiety.73
Considering the use of PGE as a monomer, the type of the IL's anion (Cl−, CoCl42−, ZnCl42−) exerted little effect on the CO2 conversion to cyclic carbonates. Conversely, when comparing our results with other studies where metal complexes or organometallic catalysts are used for the CO2 cycloaddition, such as Zn- and Co-based, we found that our MILs deliver similar if not higher yields of cyclic carbonates, but in a short time frame and under milder reaction conditions.39,59,74 Moreover, in most of these studies, a combination of a metallic compound, such as dichlorometals or MOFs, and an organic salt in a solvent medium was applied to give cyclic carbonates.75,76 For example, A. Sibaouih et al. used CoCl2 in combination with onium salts and dichloromethane to produce cyclic carbonates from CO2/propylene oxide reaction at 120 °C.75 In another study, Yuan et al. synthesized and used a Co-based MOF complex for CO2/epoxy reaction yielding 12.6% of cyclic carbonates when used alone, but 99% after 4 h of reaction, when used with tetrabutylammonium bromide (TBABr).76
Computational investigation of the mechanism
To reach a better insight into the mechanism of CO2 cycloaddition to PGE via ILs, a density functional theory (DFT) study was performed on the model reaction PGE/BMIMCl/CO2. Important points along the proposed reaction path within the potential energy surface, such as any stable intermediate species and transition states, imposing energy barriers for individual reaction steps, were investigated using DFT, microsolvation, and polarizable continuum models (PCM). The analysis of the vibration frequencies calculated for the reacting molecular clusters serves to distinguish unstable entities, exhibiting imaginary vibrational frequencies, corresponding thus to transition states, or stable intermediates, exhibiting only real frequencies.The computational analysis revealed two transition states that occurred along the fixation of a CO2 molecule from the initial epoxide to the final cyclic carbonate, along with a stable chlorinated carbonate intermediate (Fig. 3 and Fig. S5, ESI†). The first transition state corresponds to a concurrent opening of the epoxide ring, a nucleophilic attack of the chloride anion on the terminal carbocation, and another attack of the epoxide oxygen on carbon dioxide (TS1 in Fig. 3a). Notably, the chlorinated alcoholate, initially expected to represent another stable intermediate structure along the reaction path, could not be optimized to a minimum on the potential energy surface in the presence of CO2. All optimization attempts and energy scans of the chlorinated alcoholate immediately converged to the chlorinated carbonate, representing a local minimum on the potential energy surface. Vibrational analysis of the optimized geometry of the alcoholate moiety interacting with CO2 revealed a single imaginary normal mode, the eigenvectors of which correspond to a scissoring mode of the C–C–O epoxide moiety, approached by the chloride in the nearby presence of CO2. This confirms the transition state character of the alcoholate moiety in the presence of CO2. The chlorinated carbonate exhibited only real positive vibrational frequencies, indicating its dynamic stability. Following the torsional mode of its carbonate moiety, an oxygen atom can approach the terminal carbon atom bearing a chlorine substituent. This process can lead to another transition state with a planar arrangement of the terminal CH2 moiety that is surrounded from up and down, by the chloride and carbonate moieties, respectively. As depicted in Fig. S5 (ESI†), this structure exhibits a single imaginary mode that corresponds to the oscillations of the CH2 moiety between the respective chlorine and oxygen atoms. This transition state then decays to a cyclic carbonate upon the release of a chloride anion.
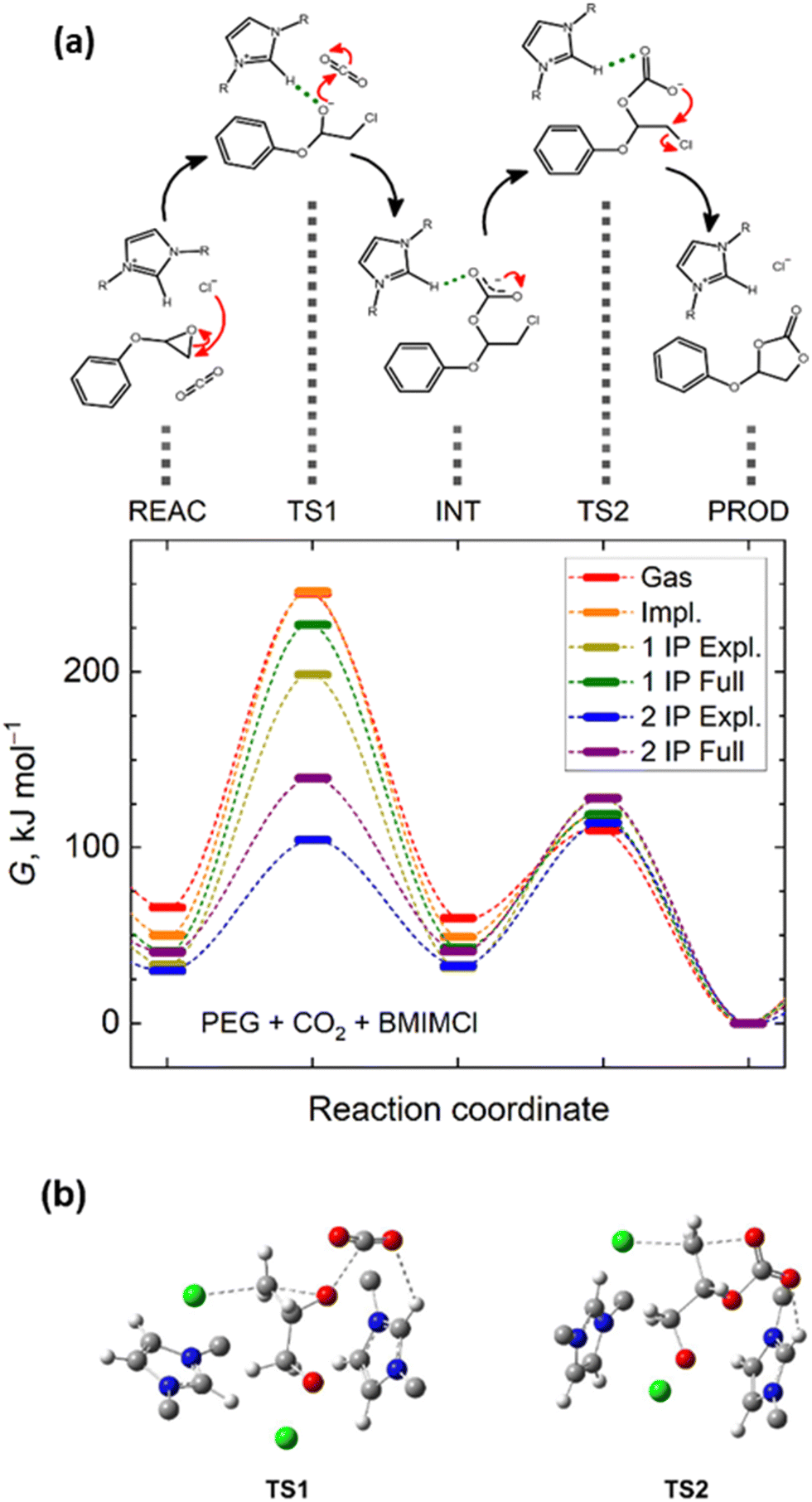 | ||
| Fig. 3 (a) Important points of the Gibbs energy profiles along the suggested reaction path modelled at the B3LYP-D3/6-311+G(d,p) level of theory. Energies of the reactants (REAC), intermediates (INT), products (PROD), and both transition states (TS) are compared for multiple solvation models: no solvation (Gas); implicit solvation in BMIMCl (Impl.); explicit solvation (Expl.) departing from reactants solvated by either a single ion pair (1 IP) or two ion pairs (2IP); and full solvation as a combination of the implicit and explicit models. Lines tentatively interconnecting Gibbs energies of individual states are only a guide-for-the-eye to illustrate what the whole Gibbs energy profile along the reaction coordinate could look like (the reaction coordinate scale is only schematic). Grey dashed lines relate the Gibbs energy profiles with particular chemical entities while the reaction mechanism among those is marked by black arrows, (b) 3D images of the key molecular fragments representing transition states TS1 and TS2 visualizing the spatial arrangement of the reacting species. | ||
Owing to the polar or ionic nature of the reacting species, solvation effects can severely affect the optimum structures along the reaction path or its energetics. Fig. S6 (ESI†) depicts analogous structures corresponding to the localized minima and maxima along the studied reaction pathway, as shown in Fig. S5 (ESI†), in the explicit presence of a [BMIM]+ cation. Notably, the key molecular contacts of the reactants were not altered upon the inclusion of an explicit cation solvating the reacting species; however, new interactions of the reacting species with the explicit cations stabilize the transition states, leading to a lowering of the energy barriers. Both transition states again exhibited a single imaginary mode, with eigenvectors always corresponding to the desired conversions, as depicted in Fig. S6 (ESI†). Turning on both implicit and explicit solvation models, the first transition state is predicted to exhibit distances of 2.10 Å and 2.60 Å from the terminal carbon atom to the epoxide oxygen and chloride anion, respectively, while the carbon atom of CO2 is separated from the epoxide oxygen atom by 2.60 Å. For the latter transition state, the key atomic distances amount to 2.39 Å and 2.34 Å for the separation of the terminal carbon atom from the carbonate oxygen atom and chloride, respectively.
Importantly, DFT calculations predicted the existence of a strong hydrogen bond between the –CH– imidazolium bridge and any of the relevant oxygen-anion species. The corresponding O⋯H distances are of 1.95 Å for the first alcoholate transition state and 2.18 Å for the latter carboxylate transition state. Since halide anions are not particularly active in forming hydrogen bonds with imidazolium cations,55,77 this observation is crucial for interpreting the mechanism by which the given IL can facilitate the course of this reaction pathway. Hydrogen bonding of the otherwise labile and high-energy transition state geometry to the solvent cation lowers its energy as well as the activation energy barrier for the entire CO2 fixation pathway.
A comparison of the Gibbs energies of the key states along the reaction path, as well as the results of the individual solvation models, is presented (Fig. 3a). It is obvious that in the gas phase, the primary fixation of CO2 into a carbonate intermediate is hindered by a substantially larger energy barrier than the mere cyclization of the carbonate intermediate into the cyclic carbonate product. The explicit presence of a single BMIMCl ionic pair stabilizes the first transition state. Nevertheless, the explicit presence of two BMIMCl ionic pairs in the computational model brings a game-changing stabilization of the first transition state. Both the chlorine and epoxide oxygen atoms possess a negative charge but are located on the opposite sides of the epoxide moiety. As such, they can beneficially interact with an individual cation at the respective sides of the reacting PGE molecule. Explicitly two BMIMCl entities lower the first reaction-free energy barrier below 75 kJ mol−1, representing less than a third of the energy barrier for the same process to occur in a vacuum (Table S1, ESI†). Thus, these DFT calculations contribute to proving that the initial epoxide ring opening is the rate-determining step of the overall process in the absence of a catalyst.
Concerning the latter energy barrier, the variation in height observed among the individual models is not significant. In this case, the presence of explicit ions near the carbonate intermediate renders its cyclization more energy-demanding, as the hydrogen bond of the carbonate to the cation has to be disrupted upon the carbonate torsion that is required for the cyclization. However, since the former barrier is the rate-determining bottleneck, an unfavorable alteration of the latter barrier by ILs is not important.
Interestingly, applying the implicit solvation model mimicking BMIMCl as a polarizable continuum around the reactant molecules and two explicit BMIMCl species led to a 25 kJ mol−1 increase in the first energy barrier. This can be understood as an adverse effect of the polar environment, which screens out the otherwise beneficial electrostatic stabilization of the alcoholate transition state. The overall positive impact of BMIMCl on the reaction course can be understood as an interplay between the beneficial interference of the reactants with explicit ions and the somewhat adverse effect of the moderately polar solvent. Current DFT calculations thus suggest that the given CO2 fixation pathway should be performed in a less polar solvent that is, however, capable of formation of strong hydrogen bonds, stabilizing the key transition states.
While the first barrier is more than two times higher than the second one in the gas phase, concurrent application of the implicit solvation and the explicit presence of BMIMCl brings both energy barriers to a comparable magnitude of approximately 87–99 kJ mol−1, greatly facilitating the reaction to proceed. All the Gibbs energy barriers modelled for the reaction of PGE with CO2 in BMIMCl are summarized in Table S1 (ESI†).
Concurrent interpretation of our DFT and NMR results, and corroborating with previous reports,31,36,56,57,78,79 the proposed reaction mechanism steps for the BMIMCl-catalyzed cyclic carbonate formation is herein presented (Fig. 3a). The strong hydrogen bonding between the –CH– imidazolium bridge and the oxygen-anion species (–O⋯H–, see DFT discussion) enables the nucleophilic attack of the chloride anion on the terminal least hindered carbon of the epoxy ring of the PGE monomer (TS1, in Fig. 3a). Thus, this forms the first chlorinated alcoholate transition state and enables a quick CO2 fixation resulting in the formation of a stable chloro-carbonate intermediate (INT, in Fig. 3a). Then, the oxygen atom approaches the terminal carbon atom bearing the chlorine substituent, forming the second transition state consisting of the terminal methylene moiety in a planar arrangement surrounded by the chloride and carbonate moieties (TS2, in Fig. 3a). Finally, this transition state decays to a cyclic carbonate upon releasing a chloride anion, as confirmed by 1H NMR analysis (Fig. 2b).
Analogous DFT calculations of the reaction mechanism in MILs could not be performed to the same extent using the PCM model due to the unavailability of properties (mainly the dielectric constant) required by the PCM model for these novel MILs. To compare the potential energy profile of the reaction occurring in various solvents, we created a computational model of the reaction in (BMIM)2ZnCl4, assuming the explicit presence of a single (BMIM)2ZnCl4 entity interacting with the reactants, without any implicit solvation model. This analysis revealed that the reaction of PGE with CO2 in (BMIM)2ZnCl4 proceeds via a mechanism similar to that of BMIMCl. Fig. S7 (ESI†) depicts the geometries of the two transition states and intermediate entities. The first transition state corresponds to the ring-opening of the epoxide and a nucleophilic attack on it by the [ZnCl4]2− anion, and the other corresponds to a nucleophilic attack by the carbonate oxygen on the terminal PGE carbon atom bearing a chlorine substituent at that time. Interestingly, DFT calculations suggest that the stable intermediate species is a chlorinated carbonate coordinated via a chlorine substituent to a nearly planar [ZnCl3]− residue.
The comparison of the Gibbs energy profiles for the reaction proceeding in either BMIMCl or (BMIM)2ZnCl4 suggests that the explicit presence of the latter IL has a beneficial effect on lowering the principal reaction energy barrier (Fig. S8, ESI†). This may be due to the higher flexibility of the first solvation shell around the reacting PGE and CO2 molecules, enabling additional structural relaxation of the reaction center and thus an improved energetic stabilization of the transition state species. The microscopic explanation for this larger flexibility can be twofold: (i) the larger size of the [ZnCl4]2− anion compared to Cl− which contributes to a larger inter-cationic spatial separation, imparting less electrostatic repulsion; and (ii) unlike when using Cl−, an additional [ZnCl3]− anionic entity remains in the solution after the nucleophilic attack by the [ZnCl4]2− anion, which enables more beneficial configurations in terms of electrostatic interactions. Most likely, these phenomena contributed to the higher catalytic efficiency of MILs for CO2 fixation. Furthermore, the reaction intermediate predicted to occur in the (BMIM)2ZnCl4 catalyzed system appears to be significantly destabilized with respect to the mere chloro-carbonate occurring in the case of BMIMCl catalysis, which decreases the activation energy for the second reaction step in turn.
The DFT results and the proposed mechanism correspond well to the other studies showing the key role of the hydrogen bonding of the epoxide oxygen atom when the IL-functionalized MOFs were used for catalysis of the CO2-epoxide cycloaddition.80,81 Our calculations show that the IL anions directly participate in the CO2 fixation process and the cations play a highly important role in terms of supporting the reaction. These computed results agree well with the experimentally observed trend where too low a concentration of the IL hinders the reaction and leads to an incomplete conversion whereas a high enough concentration accelerates the process.
It is worth mentioning that there are two limitations to our computational methodology that are related to our efforts to rationalize the cost and complexity of the current computational model. First, it includes only two explicit ion pairs around the active reaction site to mimic the solvent effects, while the solvated cluster of the reaction center is probably larger in reality. Nevertheless, our models were sufficient to demonstrate the beneficial presence of the hydrogen bonding from the cation near the active reaction site. Sterically, there is only little space for additional IL cations to closely interact with the active reaction site directly. We thus expect the impact of the explicit presence of additional cations on the reaction Gibbs energy profiles to be rather minor. Also, our model relies on a single, presumably the most stable conformation of each significant state along the reaction pathway. These findings are based on the initial simulated annealing procedure and are verified further by additional scans of the potential energy related to conformational variations. Both aspects could be captured in significantly more costly ab initio molecular dynamics simulations. Since our aim was a qualitative interpretation of the reaction mechanism and identification of the key states herein, our model proved to be sufficient and other costly simulations remain out of our current scope.
From cyclic carbonates to non-isocyanate urethanes
Conventional PUs represent a market value of 51.2 Billion €, and they are usually processed from a polyol, an isocyanate and a chain extender. The synthesis and use of isocyanates such as methylene diphenyl 4,4′-diisocyanate (MDI) and toluene diisocyanate (TDI) are well-known to be harmful to human health. Therefore, cyclic carbonates are more environmentally friendly alternatives to standard monomers for NIPU production.82 Among the reported NIPUs, polyhydroxyurethanes (PHUs) synthesized by ROP of polyfunctional cyclic carbonate monomers with di- or polyamines represent a promising alternative to replace conventional PUs.83,84 For this reason, as a proof-of-concept for the preparation of potential NIPUs, or a pre-polymer for a NIPU network, the corresponding cyclic carbonates (1b) allowed easy mixing with a monofunctional amine at a low reaction temperature (55 °C). Butylamine (BA) was used, as it is a commercially available amine, and in the liquid state at room temperature to study the formation of urethane linkages (Fig. 1). Cyclic carbonates generally present low reactivity to aliphatic amines, and the reaction requires the addition of organic solvents and catalysts.84–86 However, as observed by FTIR spectra after 0.5 h, the reactions of BA towards the monocyclic carbonate 1b containing BMIMCl, (BMIM)2ZnCl4, and (BMIM)2CoCl4, display a wide band between 3100–3500 cm−1 attributed to O–H and N–H stretching vibrations, as result of cyclic carbonates ring opening (Fig. 4a–c).82,86 The reactions show a rapid opening of cyclic carbonates by aliphatic amine to form β-hydroxyurethanes without the addition of a (co)catalyst or (co)solvent, indicating the universal catalytic abilities of the selected and designed ILs. This is proved by the disappearance of the carbonyl group band from cyclic carbonate at 1780 cm−1 with time, proving the reaction between cyclic carbonates and amine groups (Fig. 4a–c). This was further confirmed by the appearance of the new strong band at approximately 1700 cm−1 corresponding to the carbonyl of the urethane group.86,87 The presence of other bands is also indicative of the formation of urethane bonds, such as the bands at 1532, 1245, and 1040 cm−1 corresponding to N–H (in the plane bend), C–N (stretch), and C–O (stretch) bonds, respectively.88,89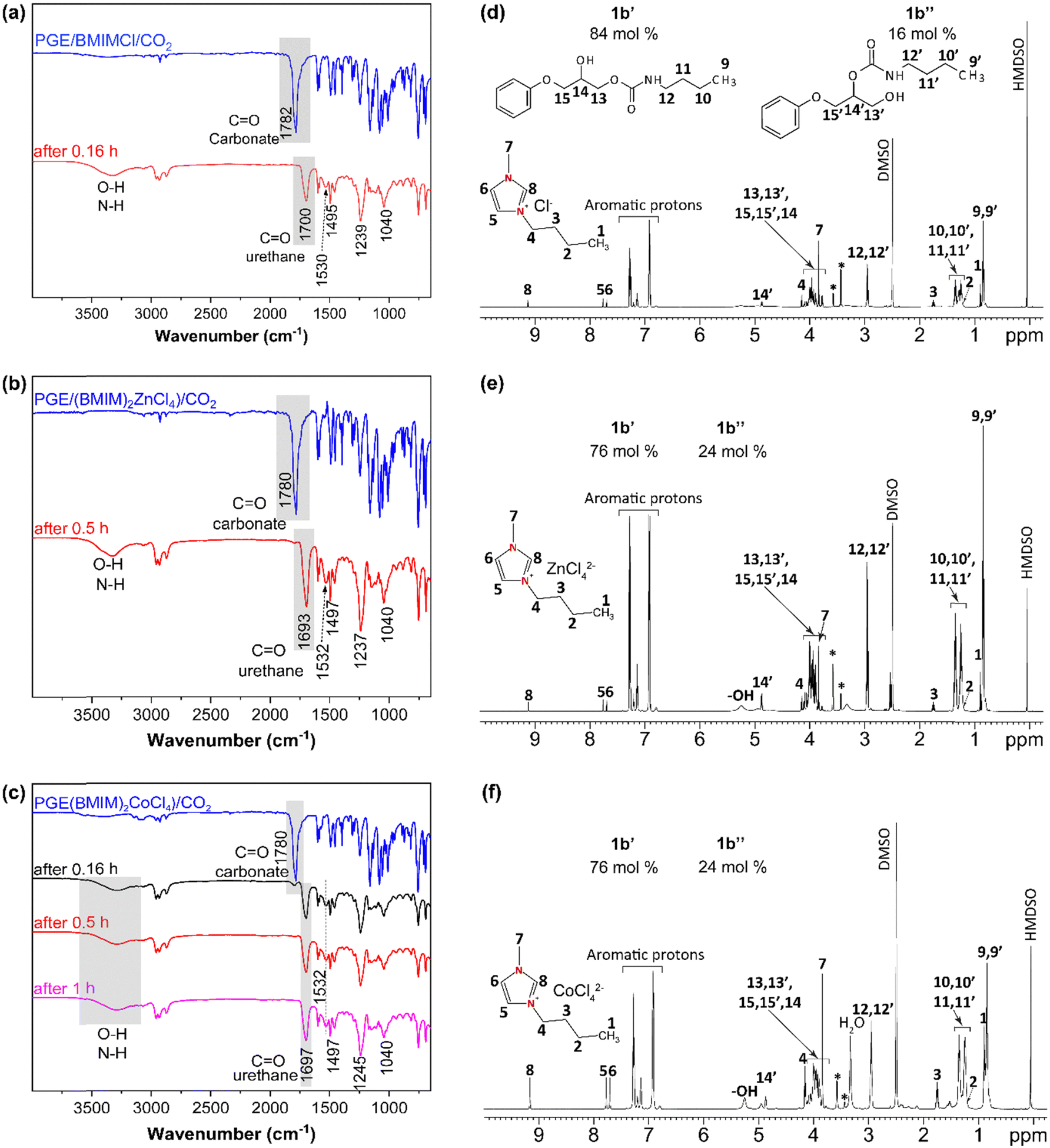 | ||
| Fig. 4 FTIR spectra of obtained β-hydroxyurethanes via reaction of cyclic carbonates from PGE and (a) BMIMCl, (b) (BMIM)2ZnCl4, (c) (BMIM)2CoCl4, and NMR spectra of β-hydroxyurethanes isomers (1b’ and 1b”) from PGE cyclic carbonates using (d) BMIMCl, (e) (BMIM)2ZnCl4, and (f) (BMIM)2CoCl4. Impurities are denoted by an asterisk (*). | ||
The 1H NMR analysis has supported these results by showing the urethane groups’ formation and complete conversion of cyclic carbonate groups confirmed by the presence of the respective signals corresponding to –CH2– protons adjacent to –NH–(CO)– (marked as 12 and 12′, Fig. 4d–f) at around 2.95 ppm and the absence of the NMR signals corresponding to cyclic carbonate at 4.38 ppm, 4.62 ppm and 5.15 ppm.21,90,91
Importantly, 1H NMR spectroscopy allowed the distinction of two formed β-hydroxyurethane isomers: one with a secondary hydroxyl group (denoted as 1b′, in Scheme 1) and one with a primary hydroxyl group (denoted as 1b′′, in Scheme 1) and calculating their corresponding yields. According to the NMR analysis, the isomer with a secondary hydroxyl constitutes the majority of the product, which is in accordance with previous studies suggesting that the reaction between 5-membered ring cyclic carbonates and amines preferentially gives secondary hydroxyl groups.92 In the presence of BMIMCl, the obtained isomers 1b′ and 1b′′ exhibited yields of 84 and 16%, respectively (Fig. 4d). On the other hand, (BMIM)2ZnCl4 or (BMIM)2CoCl4, yielded 76% for the isomer 1b′ and 24% of the isomer 1b′′ (Fig. 4e and f). Thus, it seems that the type of ILs initially present in the cyclic carbonate influences the ratio of the formed hydroxyurethane isomers. This impact is probably caused by the high Lewis acidity character73,93 of MILs and by the coordination effect of MIL anions which further promotes the formation of primary hydroxyl groups (the isomer 1b′′). Previous studies have also reported the influence of catalyst type on the ratio of β-hydroxyurethane isomers.94–96 In contrast to other studies requiring the addition of specific catalyst promoting cyclic carbonate ring-opening, herein we have shown that MILs may act as dual catalysts for the CO2 cycloaddition reaction as well as for the aminolysis of cyclic carbonates in bulk (Scheme 1(b)). Due to the catalytic effect of the IL present in cyclic carbonate, the reactivity of BA is increased, and a Lewis acid-activated cyclic carbonate intermediate is formed. The presence of the inductive effect of the substituent (phenyl group) increased the acidity of the negatively charged oxygen and stabilized the transition state.92 Then, the reaction proceeds by cleavage of the cyclic carbonate ring leading to a deprotonated amine and formation of two types of β-hydroxyurethane isomers, 1b′ (favorable, with secondary hydroxyl group), and 1b′′ (less probable, with primary hydroxyl group) (Scheme 1(b)).92
Conclusions
In this study, we successfully used a metal-free imidazolium IL (1-butyl-2-methylimidazolium chloride, BMIMCl) and two imidazolium MILs bearing [ZnCl4]2− and [CoCl4]2− anions as all-in-one catalysts for two-step conversion of carbon dioxide into β-hydroxyurethanes. For the first step of the supercritical CO2-epoxy cycloaddition of monofunctional phenyl glycidyl ether (PGE) to cyclic carbonates, the concentration of ILs was optimized (10 mol%) producing the cyclic carbonates with a high yield of ca 98% within 1 h at 80 °C. A feasible mechanism for the CO2-epoxy cycloaddition catalyzed by ILs was proposed using DFT model calculations. The DFT calculations clearly identified the epoxide ring opening as the rate-determining step and formation of the chlorinated carbonate species as the stable intermediate, which proves the direct participation of the chloride-based anion in the cycloaddition.In the second step, the formed cyclic carbonates, containing the used ILs as catalysts, exhibited high reactivity towards a monofunctional butylamine. Under solvent-free and co-catalyst-free mild conditions (35 min at 55 °C), the reaction yielded β-hydroxyurethane isomers, with the dominant one bearing the secondary OH groups (76–84%). Moreover, the results further showed that the different structure of the IL-anion affects the formation of individual isomers. Due to the high variability and facile tunability of the IL structure, this effect may be further elaborated for the tailor-made preparation of NIPU networks with variable architecture and properties, which will be the subject of further research studies in the future.
Altogether, the tested ILs can be beneficially considered as promising all-in-one catalysts for CO2 conversion into NIPUs through cyclic carbonate intermediates. On the other hand, the challenge seems to be the scale-up of the process involving the use of supercritical CO2, which would allow the transfer of the technology to an industrial scale.
Authors’ contributions
Marwa Rebei contributed to the project conceptualization, performed all reaction experiments and characterizations, and wrote the original manuscript. Ctirad Červinka performed the DFT calculations and interpreted the results. Andrii Mahun performed and interpreted the NMR data. Petra Ecorchard and Jan Honzíček synthesized the metal-based ionic liquids and characterized them via FTIR and NMR spectroscopy and TGA. Sébastien Livi supervised supercritical CO2 experiments. Ricardo K. Donato contributed to the conceptualization of the project. Hynek Beneš conceptualized and supervised the project and performed editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Czech Science Foundation (GACR No. 22-05244S). M.R. acknowledges the Grant Agency of Charles University (GAUK No. 413022). Computational resources were provided by the e-INFRA CZ project (ID:90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic. C. Č. acknowledges the support of the project “The Energy Conversion and Storage”, funded as project No. CZ.02.01.01/00/22_008/0004617 by Programme Johannes Amos Commenius, call Excellent Research.References
- M. Aresta, Michele Aresta-Carbon Dioxide as Chemical Feedstock, 2010 Search PubMed.
- A. Ballamine, Sci. Technol. Energy Transition, 2022, 1, 2–6 Search PubMed.
- L. Plasseraud, ChemSusChem, 2010, 3, 631–632 CrossRef CAS.
- R. Ghanbaralizadeh, H. Bouhendi, K. Kabiri and M. Vafayan, J. CO2 Util., 2016, 16, 225–235 CrossRef CAS.
- Y. Qin, X. Sheng, S. Liu, G. Ren, X. Wang and F. Wang, J. CO2 Util., 2015, 11, 3–9 CrossRef CAS.
- Y. Li, Y. Y. Zhang, L. F. Hu, X. H. Zhang, B. Y. Du and J. T. Xu, Prog. Polym. Sci., 2018, 82, 120–157 CrossRef CAS.
- A. J. Hunt, E. H. K. Sin, R. Marriott and J. H. Clark, ChemSusChem, 2010, 3, 306–322 CrossRef CAS PubMed.
- K. Kiatkittipong, M. A. A. M. Shukri, W. Kiatkittipong, J. W. Lim, P. L. Show, M. K. Lam and S. Assabumrungrat, Processes, 2020, 8, 548 CrossRef CAS.
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS PubMed.
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- A. Steblyanko, W. Choi, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2375–2380 CrossRef CAS.
- T. S. Anderson and C. M. Kozak, Eur. Polym. J., 2019, 120, 109237 CrossRef CAS.
- C. Yang, Y. Chen, P. Xu, L. Yang, J. Zhang and J. Sun, Mol. Catal., 2020, 480, 110637 CrossRef CAS.
- L. Qin, Y. Li, F. Liang, L. Li, Y. Lan, Z. Li, X. Lu, M. Yang and D. Ma, Microporous Mesoporous Mater., 2022, 341, 112098 CrossRef CAS.
- X. Liu, X. Yang, S. Wang, S. Wang, Z. Wang, S. Liu, X. Xu, H. Liu and Z. Song, ACS Sustainable Chem. Eng., 2021, 9, 4175–4184 CrossRef CAS.
- G. Beniah, D. J. Fortman, W. H. Heath, W. R. Dichtel and J. M. Torkelson, Macromolecules, 2017, 50, 4425–4434 CrossRef CAS.
- Z. Karami, K. Kabiri and M. J. Zohuriaan-Mehr, J. CO2 Util., 2019, 34, 558–567 CrossRef CAS.
- R. H. Lambeth and T. J. Henderson, Polymer, 2013, 54, 5568–5573 CrossRef CAS.
- J. Ke, X. Li, S. Jiang, C. Liang, J. Wang, M. Kang, Q. Li and Y. Zhao, Polym. Int., 2019, 68, 651–660 CrossRef CAS.
- O. Lamarzelle, P. L. Durand, A. L. Wirotius, G. Chollet, E. Grau and H. Cramail, Polym. Chem., 2016, 7, 1439–1451 RSC.
- M. Malik and R. Kaur, Polym. Adv. Technol., 2018, 29, 1078–1085 CrossRef CAS.
- J. J. Santos, J. H. Lopes, K. M. F. R. De Aguiar, M. B. Simões, J. C. Mpeko, R. G. Jasinevicius, E. T. Cavalheiro, H. Imasato and U. P. Rodrigues-Filho, J. CO2 Util., 2023, 27, 102303 CrossRef.
- S. Yue, P. Wang and X. Hao, Fuel, 2019, 251, 233–241 CrossRef CAS.
- J. Peng, S. Wang, H. J. Yang, B. Ban, Z. Wei, L. Wang and B. Lei, Fuel, 2018, 224, 481–488 CrossRef CAS.
- X. Yang, Q. Zou, T. Zhao, P. Chen, Z. Liu, F. Liu and Q. Lin, ACS Sustainable Chem. Eng., 2021, 9, 10437–10443 CrossRef CAS.
- A. G. Zazybin, K. Rafikova, V. Yu, D. Zolotareva, V. M. Dembitsky and T. Sasaki, Russ. Chem. Rev., 2017, 86, 1254–1270 CrossRef CAS.
- T. Wang, C. Shen, G. Yu and X. Chen, Polym. Degrad. Stab., 2021, 194, 109751 CrossRef CAS.
- I. A. Berezianko, I. V. Vasilenko and S. V. Kostjuk, Eur. Polym. J., 2019, 121, 109307 CrossRef CAS.
- A. Bouyahya, S. Balieu, R. Beniazza, M. Raihane, A. El Kadib, D. Le Cerf, P. Thébault, G. Gouhier and M. Lahcini, New J. Chem., 2019, 43, 5872–5878 RSC.
- R. R. Henriques, C. P. De Oliveira, R. Stein, K. Pontes, A. A. Silva and B. G. Soares, ACS Appl. Polym. Mater., 2022, 4, 1207–1217 CrossRef CAS.
- D. Liu, G. Li and H. Liu, Appl. Surf. Sci., 2018, 428, 218–225 CrossRef CAS.
- J. Liang, Y. Q. Xie, Q. Wu, X. Y. Wang, T. T. Liu, H. F. Li, Y. B. Huang and R. Cao, Inorg. Chem., 2018, 57, 2584–2593 CrossRef CAS PubMed.
- M. Ding and H. L. Jiang, ACS Catal., 2018, 8, 3194–3201 CrossRef CAS.
- J. Liang, Y. Q. Xie, X. S. Wang, Q. Wang, T. T. Liu, Y. B. Huang and R. Cao, Chem. Commun., 2018, 54, 342–345 RSC.
- M. Rebei, A. Mahun, Z. Walterová, O. Trhlíková, R. K. Donato and H. Beneš, Polym. Chem., 2022, 13, 5380–5388 RSC.
- G. P. Wu, D. J. Darensbourg and X. B. Lu, J. Am. Chem. Soc., 2012, 134, 17739–17745 CrossRef CAS PubMed.
- D. J. Darensbourg, J. Chem. Educ., 2017, 94, 1691–1695 CrossRef CAS.
- X. Wu, C. Chen, Z. Guo, M. North and A. C. Whitwood, ACS Catal., 2019, 9, 1895–1906 CrossRef CAS.
- N. A. Noorhisham, D. Amri, A. H. Mohamed, N. Yahaya, N. M. Ahmad, S. Mohamad, S. Kamaruzaman and H. Osman, J. Mol. Liq., 2021, 326, 115340 CrossRef CAS.
- C. Červinka, M. Klajmon and V. Štejfa, J. Chem. Theory Comput., 2019, 15, 5563–5578 CrossRef PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- J. N. Canongia Lopes and A. A. H. Pádua, Theor. Chem. Acc., 2012, 131, 1–11 Search PubMed.
- R. T. Cygan, V. N. Romanov and E. M. Myshakin, J. Phys. Chem. C, 2012, 116, 13079–13091 CrossRef CAS.
- L. S. Dodda, I. C. De Vaca, J. Tirado-Rives and W. L. Jorgensen, Nucleic Acids Res., 2017, 45, W331–W336 CrossRef CAS PubMed.
- W. L. Jorgensen and J. Tirado-Rives, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6665–6670 CrossRef CAS PubMed.
- L. S. Dodda, J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2017, 121, 3864–3870 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegelet al., Gaussian 16, Revision C. 01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- L. G. Stefan Grimme and S. Ehrlich, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- C. Červinka, M. Fulem and K. Růžčka, J. Chem. Eng. Data, 2012, 57, 227–232 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- E. L. Bennett, C. Song, Y. Huang and J. Xiao, J. Mol. Liq., 2019, 294, 111571 CrossRef CAS.
- M. M. Huang, Y. Jiang, P. Sasisanker, G. W. Driver and H. Weingärtner, J. Chem. Eng. Data, 2011, 56, 1494–1499 CrossRef CAS.
- M. Součková, J. Klomfar and J. Pátek, Fluid Phase Equilib., 2017, 454, 43–56 CrossRef.
- T. K. L. Nguyen, S. Livi, B. G. Soares, S. Pruvost, J. Duchet-Rumeau and J. F. Gérard, ACS Sustainable Chem. Eng., 2016, 4, 481–490 CrossRef CAS.
- T. K. L. Nguyen, S. Livi, S. Pruvost, B. G. Soares and J. Duchet-Rumeau, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3463–3471 CAS.
- J. Byun and K. A. I. Zhang, ChemCatChem, 2018, 10, 4610–4616 CrossRef CAS.
- K. Błażek, H. Beneš, Z. Walterová, S. Abbrent, A. Eceiza, T. Calvo-Correas and J. Datta, Polym. Chem., 2021, 12, 1643–1652 RSC.
- A. Ghosh, G. N. Reddy, M. Siddhique, P. K. S. Chatterjee, S. Bhattacharjee, R. Maitra, S. E. Lyubimov, A. V. Arzumanyan, A. Naumkin, A. Bhaumik and B. Chowdhury, Green Chem., 2022, 24, 1673–1692 RSC.
- C. Pronoitis, M. Hakkarainen and K. Odelius, ACS Sustainable Chem. Eng., 2022, 10, 2522–2531 CrossRef CAS.
- E. I. Privalova, E. Karjalainen, M. Nurmi, P. Mäki-Arvela, K. Eränen, H. Tenhu, D. Y. Murzin and J. P. Mikkola, ChemSusChem, 2013, 6, 1500–1509 CrossRef CAS PubMed.
- J. Avila, L. F. Lepre, C. C. Santini, M. Tiano, S. Denis-Quanquin, K. Chung Szeto, A. A. H. Padua and M. Costa Gomes, Angew. Chem., Int. Ed., 2021, 60, 12876–12882 CrossRef CAS PubMed.
- Y. Li, B. Dominelli, R. M. Reich, B. Liu and F. E. Kühn, Catal. Commun., 2019, 124, 118–122 CrossRef CAS.
- Z. Zhang, F. Fan, H. Xing, Q. Yang, Z. Bao and Q. Ren, Innovations of Green Process Engineering for Sustainable Energy Environment, 2017 - Top. Conf. 2017 AIChE Annu. Meet., 2017, 24–29.
- A. Comès, R. Poncelet, P. P. Pescarmona and C. Aprile, J. CO2 Util., 2021, 48, 101529 CrossRef.
- W. de Almeida, J. L. S. Milani, C. H. de J. Franco, F. T. Martins, A. de Fatima, A. F. A. da Mata and R. P. das Chagas, Mol. Catal., 2022, 530, 112632 CrossRef.
- M. H. Anthofer, M. E. Wilhelm, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2015, 7, 94–98 CrossRef CAS.
- B. C. Smith, Spectroscopy, 2016, 31, 34–37 CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- G. Chen, J. Zhang, X. Cheng, X. Tan, J. Shi, D. Tan, B. Zhang, Q. Wan, F. Zhang, L. Liu, B. Han and G. Yang, ChemCatChem, 2020, 12, 1963–1967 CrossRef CAS.
- J. Estager, J. D. Holbrey and M. Swadźba-Kwaśny, Chem. Soc. Rev., 2014, 43, 847–886 RSC.
- A. L. Girard, N. Simon, M. Zanatta, S. Marmitt, P. Gonçalves and J. Dupont, Green Chem., 2014, 16, 2815–2825 RSC.
- A. Sibaouih, P. Ryan, M. Leskelä, B. Rieger and T. Repo, Appl. Catal. A Gen., 2009, 365, 194–198 CrossRef CAS.
- R. Yuan and H. He, Inorg. Chem. Commun., 2020, 121, 108235 CrossRef CAS.
- C. Červinka and V. Štejfa, Phys. Chem. Chem. Phys., 2021, 23, 4951–4962 RSC.
- F. D. Bobbink, W. Gruszka, M. Hulla, S. Das and P. J. Dyson, Chem. Commun., 2016, 52, 10787–10790 RSC.
- F. Siragusa, E. Van Den Broeck, C. Ocando, A. J. Muller, G. De Smet, B. U. W. Maes, J. De Winter, V. Van Speybroeck, B. Grignard and C. Detrembleur, ACS Sustainable Chem. Eng., 2021, 9, 1714–1728 CrossRef CAS.
- L. J. Zhou, W. Sun, N. N. Yang, P. Li, T. Gong, W. J. Sun, Q. Sui and E. Q. Gao, ChemSusChem, 2019, 12, 2202–2210 CrossRef CAS PubMed.
- S. M. Masoom Nataj, S. Kaliaguine and F. G. Fontaine, ChemCatChem, 2023, 15, e202300079 CrossRef CAS.
- J. Ke, X. Li, F. Wang, M. Kang, Y. Feng, Y. Zhao and J. Wang, J. CO2 Util., 2016, 16, 474–485 CrossRef CAS.
- F. Camara, S. Benyahya, V. Besse, G. Boutevin, R. Auvergne, B. Boutevin and S. Caillol, Eur. Polym. J., 2014, 55, 17–26 CrossRef CAS.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- X. Sheng, Y. Wang, Y. Qin, X. Wang and F. Wang, RSC Adv., 2014, 4, 54043–54050 RSC.
- C. Carré, H. Zoccheddu, S. Delalande, P. Pichon and L. Avérous, Eur. Polym. J., 2016, 84, 759–769 CrossRef.
- A. Gomez-Lopez, B. Grignard, I. Calvo, C. Detrembleur and H. Sardon, ACS Appl. Polym. Mater., 2020, 2, 1839–1847 CrossRef CAS.
- Brian C. Smith, Spectroscopy, 2023, 38, 14–16 Search PubMed.
- Brian C. Smith, Spectroscopy, 2023, 38, 14–18 Search PubMed.
- A. Cornille, M. Blain, R. Auvergne, B. Andrioletti, B. Boutevin and S. Caillol, Polym. Chem., 2017, 8, 592–604 RSC.
- T. Quérette, E. Fleury and N. Sintes-Zydowicz, Eur. Polym. J., 2019, 114, 434–445 CrossRef.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- W. Wu, Y. Lu, H. Ding, C. Peng and H. Liu, Phys. Chem. Chem. Phys., 2015, 17, 1339–1346 RSC.
- C. D. Diakoumakos and D. L. Kotzev, Macromol. Symp., 2004, 216, 37–46 CrossRef CAS.
- S. Benyahya, M. Desroches, R. Auvergne, S. Carlotti, S. Caillol and B. Boutevin, Polym. Chem., 2011, 2, 2661–2667 RSC.
- B. Ochiai, S. Inoue and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6613–6618 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00852e |
| This journal is © The Royal Society of Chemistry 2024 |