
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boron-imidazolate coordination networks with 3d transition metals for enhanced CO2 adsorption capability†
Takeshi
Kato
,
Ikuho
Akiyama
,
Fumika
Mori
,
Ayumu
Shinohara
,
Yudai
Ogura
,
Akitaka
Ito
 and
Masataka
Ohtani
*
and
Masataka
Ohtani
*
School of Engineering Science, Kochi University of Technology, 185 Miyanokuchi, Tosayamada, Kami, Kochi 782-8502, Japan. E-mail: ohtani.masataka@kochi-tech.ac.jp
First published on 23rd January 2024
Abstract
The synthesis conditions of a sodalite (SOD)-topology coordination network with a boron-imidazolate framework (BIF), namely BIF-3-Cu, were systematically investigated to control the crystal growth process. By optimising the reaction time, temperature, solvent, reaction reagent, and additives, highly crystalline BIF-3-Cu crystals were obtained, showing high specific surface areas comparable to those of well-known SOD-topology network materials based on zeolitic imidazolate frameworks. Furthermore, we found that the valence of the metal ion plays a vital role in the molecular inclusion into the pores during the crystal nucleation and growth process. Taking advantage of this, new crystals with SOD topology were obtained by a similar reaction using divalent zinc (Zn2+), cobalt (Co2+), and manganese (Mn2+) ions. The resultant crystals contained several monovalent counterions in their pores to compensate for the charge of the cationic framework formed by the divalent metal cations and monovalent anionic boron-bridged ligands. Moreover, the SOD-Zn-BIF exhibits relatively high CO2 adsorption in the narrow micropores, despite the presence of counter anions.
Introduction
Metal–organic frameworks (MOFs) are porous crystalline materials that can form a wide variety of spatial structures from a combination of metal ions and organic ligands.1–3 Zeolitic imidazolate frameworks (ZIFs), consisting of metal ions and imidazolate ligands, are typical MOFs classified based on their pore structure, which is similar to zeolites. For example, ZIF-8 is composed of Zn ions and 2-methylimidazolate and exhibits a sodalite (SOD) topology. Differences in pore topology affect the gas adsorption properties, molecular sieving effect, and molecular recognition in the pores.The topology of the ZIF series depends on the coordination angle of the imidazolate ligand. When metal ions coordinate with two nitrogen atoms in the imidazolate ligand to form a bridging structure in ZIFs, the coordination angle is approximately 145°, which is similar to the Si–O–Si bond in the zeolite structure. Inspired by this characteristic coordination structure, variants of the ZIF series consisting of boron-imidazolate ligands have also been reported.4–6 These MOFs with boron-bridged structures are referred to as boron-imidazolate frameworks (BIFs). In BIFs, boron is located in the centre of the four imidazolate ligands, resulting in a monovalent anionic ligand. Boron-bridged ligands can form three-dimensional structures with various metal ions. To date, several types of BIFs have been synthesised from boron-bridged ligands and various metal ions such as Li,4,7,8 Co,9–14 Cu,15–22 Zn,23–31 Ru,32 and Cd ions.33–39
In this study, we focus on BIF-3-Cu as a typical example, which consists of Cu+ ions and [B(2-methylimidazolate)4]− ([B(2-MIm)4]−).4 As a main building unit, [B(2-MIm)4]− ligands comprise four 2-methylimidazolates bound to the boron centre, resulting in a rigid ligand structure with a tetrahedron form. This unique boron-centred anionic ligand leads to a three-dimensional network with zeolite-like SOD topology. However, ZIF-8 with a similar SOD topology shows a high specific surface area (1800 m2 g−1),40,41 whereas BIF-3-Cu generally exhibits a relatively low specific surface area (182.3 m2 g−1).4 In contrast, Lennox et al. reported that a mechanochemical synthesis method can significantly improve the specific surface area of BIF-3-Cu crystals (1196 m2 g−1).42 Notably, it remains unclear how the crystal formation process is different in the solid-phase and solution reactions, and whether this process affects the crystallinity and specific surface area of the resulting crystals. In this study, we systematically investigated the synthesis method of BIF-3-Cu to clarify the relationship between the solution-based reaction conditions for crystal formation/growth and the gas adsorption capacity of the obtained BIF-3-Cu crystals. In addition, under the optimised synthesis conditions, we aimed to develop a novel SOD-type BIF with different metal ions to examine the influence of metal ions on the gas adsorption properties.
Results and discussion
Optimisation of crystal growth conditions for crystalline BIF-3-Cu exhibiting a high specific surface area
In the previously reported synthesis method for BIF-3-Cu, copper iodide (CuI) was used as the metal salt, and Na[B(2-MIm)4] was used as the boron-bridging ligand.4 Considering the experimental conditions, the low specific surface area was likely due to the residual sodium iodide (NaI) produced as a reaction byproduct in the pores. Herein, to avoid residual NaI formation, we conducted a similar BIF-3-Cu synthesis with H[B(2-MIm)4] instead of Na[B(2-MIm)4].35 The mixture solution containing CuI and H[B(2-MIm)4] was reacted in DL-2-amino-1-butanol/acetonitrile (MeCN) at 120 °C for 5 days.4 As a result, single crystals with a size of approximately 5 μm were obtained, and the powder X-ray diffraction (PXRD) pattern was consistent with a simulated pattern of BIF-3-Cu (Fig. S1(I), ESI†Fig. 1a(I)). After systematically varying the reaction conditions, fine single crystals were obtained from the reaction in DL-2-amino-1-butanol at 100 °C for 4 days. The single crystals were approximately 100 μm in size (Fig. S1(II), ESI†). As a result of single-crystal X-ray crystallographic analysis, the obtained single crystal was completely consistent with the previously reported crystal structure of BIF-3-Cu, and the corresponding crystallographic parameters, as summarized in Table S1 (ESI†), were comparable to those of the previously reported structure of BIF-3-Cu. Then, N2 adsorption measurements at 77 K were conducted to confirm the specific surface area. According to Brunauer–Emmett–Teller (BET) analysis of the N2 adsorption isotherm, the specific surface area of the crystals was 30 m2 g−1 (Fig. 1b(II)), which is lower than that of the previously reported BIF-3-Cu (180 m2 g−1).4 In addition, although PXRD measurements of the obtained BIF-3-Cu crystals showed that the diffraction pattern (Fig. 1a(II)) was consistent with the 1D simulated pattern of the BIF-3-Cu single-crystal structure (CCDC 697959), the peak intensity around 2θ = 8° was relatively lower than that of the simulated pattern. Thus, the pores of BIF-3-Cu were probably occupied by some residual material. | ||
| Fig. 1 (a) PXRD patterns and (b) N2 adsorption measurements (77 K) of the prepared BIF-3-Cu crystals with different synthesis methods (I: previously reported BIF-3-Cu synthesis method, II: using DL-2-amino-1-butanol as the reaction solvent at 100 °C for 4 days, III: using CuCl as the metal salt, IV: heating for 2 days, V: heating 24 h and VI: adding NaI.). | ||
To determine the presence of iodide salt, X-ray fluorescence analysis was performed. However, a negligible amount of I− was observed in the obtained BIF-3-Cu crystals (Cu: 99.98 at%, I: 0.02 at%). This suggests that pore filling with an inorganic residue, such as iodide salt, was not the main reason for the low specific surface area. Furthermore, BIF-3-Cu crystals prepared from copper chloride (CuCl) instead of CuI show a similar PXRD pattern and low specific surface area (10 m2 g−1) (Fig. 1a and b(III)). These results indicate that the residual metal salt source or their derived inorganic by-products in the pores is not the main reason for the reduced specific surface area in BIF-3-Cu.
Another possibility is that organic residues remain in the pores after synthesis. Thus, thermogravimetry (TG) measurements were performed to determine the organic residue. The results showed that the weight loss below 200 °C, corresponding to the residual solvent, was approximately 1% (Fig. S2(I), ESI†). Therefore, the organic solvent, such as DL-2-amino-1-butanol, did not remain in the pore. The TG curve from 200 to 400 °C revealed a large weight loss (78%), indicating the degradation of organic parts in BIF-3-Cu crystals (Fig. S2(I), ESI†). According to the elemental composition in BIF-3-Cu, the theoretical degradation degree is 71%. This indicates that the observed TG weight loss partly contains the degradation of the organic residue in the pore. Scanning electron microscopy and energy-dispersive X-ray spectroscopy (SEM-EDX) measurements showed that an excess amount of boron was detected in BIF-3-Cu crystals compared with the copper composition (Fig. S3, ESI†). From these results, the low specific surface area of BIF-3-Cu crystals may be due to the presence of residual [B(2-MIm)4]− ligands in the pores of the BIF-3-Cu crystals. According to the crystal structure of BIF-3-Cu, the inner pore space (17.9 Å, Cu–Cu distance) is sufficiently larger than the molecular size of [B(2-MIm)4]− (∼6.3 Å). However, the excess amount of encapsulated [B(2-MIm)4] ligands could not be removed by repeated washings or solvent immersion, indicating that the [B(2-MIm)4]− was strongly adsorbed in the pore.
We assumed that the strong adsorption of [B(2-MIm)4]− is due to the ionic interaction between the framework and [B(2-MIm)4]−. One plausible reason is the partial oxidation of Cu+ to Cu2+ during the heating reaction. If the divalent metal ion partially replaces the monovalent metal ion in the BIF-3-Cu framework, the resultant framework becomes partially cationic, resulting in the incorporation of the anionic [B(2-MIm)4]− in the pore for charge compensation. The powder of BIF-3-Cu crystals obtained by the heating reaction was a light blue-white colour, which is similar to the blue colour observed in the solution of Cu2+ ion and [B(2-MIm)4]− (Fig. S4, ESI†). Furthermore, in cases of a higher reaction temperature and longer heating time, low crystallinity products of BIF-3-Cu were obtained. This suggests that the valence of copper ions in the obtained BIF-3-Cu crystals was partly oxidised from Cu+ to Cu2+. To examine this, the obtained BIF-3-Cu crystals were exposed to a MeCN solution of tetracyanoethylene (TCNE) as a weak oxidising agent. As the results of XRD measurements revealed, BIF-3-Cu crystals were easily decomposed upon immersion in the high concentration of TCNE solution (Fig. S5, ESI†). Therefore, the valence state of Cu ions in the crystal is important to obtain a highly crystalline porous structure in BIF-3-Cu crystals.
According to this hypothesis, we attempted to shorten the reaction time to prevent oxidation of Cu+. The reaction time was changed from 4 days to 2 days and 24 h. Judging from the optical microscopy and PXRD observations, no significant differences were observed in the crystal appearance and structure of the products obtained by changing the reaction time (Fig. 1a and b(IV, V)). Nitrogen adsorption–desorption measurements (77 K) of the resulting BIF-3-Cu crystals also showed low specific surface areas: 30 m2 g−1 at 4 days, 140 m2 g−1 at 2 days, and 170 m2 g−1 at 24 h (Fig. 1c(IV, V)). Thus, oxidation of monovalent copper ions may occur at an earlier stage of crystal formation.
According to the industrial production process of CuI, an excess amount of I− is necessary to prevent the oxidation of Cu+ to Cu2+. Therefore, we investigated the effect of adding NaI to the precursor solution. As a result, the peak intensity of the obtained BIF-3-Cu crystals was significantly increased in the PXRD pattern compared with that of BIF-3-Cu crystals synthesised without adding NaI (Fig. 1a and b(VI)), indicating that their crystallinity was improved by the addition of NaI. In addition, the yield of BIF-3-Cu crystals based on the starting Cu+ concentration was improved from 18% to 90% by adding NaI. However, a significant improvement in the specific surface area of the resulting BIF-3-Cu crystals was not observed (80 m2 g−1, Fig. 3c(VI)). Thus, these results suggest that the oxidation of Cu+ and subsequent reduction in crystallinity is not the main reason for the low specific surface area, although the addition of NaI enhances the crystal formation rate and crystallinity in the formation process of BIF-3-Cu crystals.
Next, we attempted to synthesise BIF-3-Cu at lower temperatures to prevent the diffusion and incorporation of excess boron-bridged ligands into the pores. A mixture of methanol (MeOH) and acetonitrile (MeCN) was used as the reaction solvent, considering the solubility of the starting materials and the formation rate of BIF-3-Cu crystals. Judging from the solubility and reactivity, CuCl was suitable for room-temperature synthesis of BIF-3-Cu. The mixing ratio of the reaction solvents (MeOH/MeCN) was also crucial. The obtained crystal shapes were different depending on the MeOH/MeCN ratio, as revealed by SEM (Fig. 2a(I–III)). PXRD results showed that a diffraction pattern of the obtained crystals was consistent with BIF-3-Cu (Fig. 2b(I)). Notably, the diffraction patterns of the octahedral and rod-shaped crystals did not match that of the BIF-3-Cu crystal (Fig. 2b(II, III)). Thus, the solvation environment of Cu+ and [B(2-MIm)4]− may be crucial to the rapid formation of BIF-3-Cu crystals. To promote the deprotonation and solvation of H[B(2-MIm)4] ligand in the precursor solution, we added a base (triethylamine) as an accelerator of deprotonation. As a result, BIF-3-Cu was obtained at any mixing ratio of MeOH/MeCN (Fig. 2a and b(IV–VI)). The BET specific surface area of BIF-3-Cu crystals obtained under the optimised room-temperature conditions was 1000 m2 g−1 (Fig. 3(II)) which was significantly improved compared with that of the BIF-3-Cu obtained under previous solvothermal conditions,4 and comparable to that of the mechanochemical products42 (Table S2, ESI†). The BIF-3-Cu crystals obtained at room temperature were also analysed by TG measurements to evaluate the presence of organic residue in the pores. As a result, the TG weight loss in the room-temperature products was approximately 70%, which is consistent with the theoretical value (71%) based on the BIF-3-Cu composition (Fig. S2(II), ESI†). Thus, the fast crystal nucleation and growth process at room temperature was effective for kinetically preventing the inclusion of ligands in the pores of BIF-3-Cu.
 | ||
| Fig. 2 (a) SEM images and (b) PXRD patterns of the BIF-3-Cu crystals prepared by room-temperature solution synthesis at different MeOH/MeCN ratios with and without adding triethylamine. MeOH/MeCN ratio (v/v); (I), (IV): 90/10, (II), (V): 50/50 and (III), (VI): 10/90. (I)–(III): without adding triethylamine, (IV)–(VI): with adding triethylamine. | ||
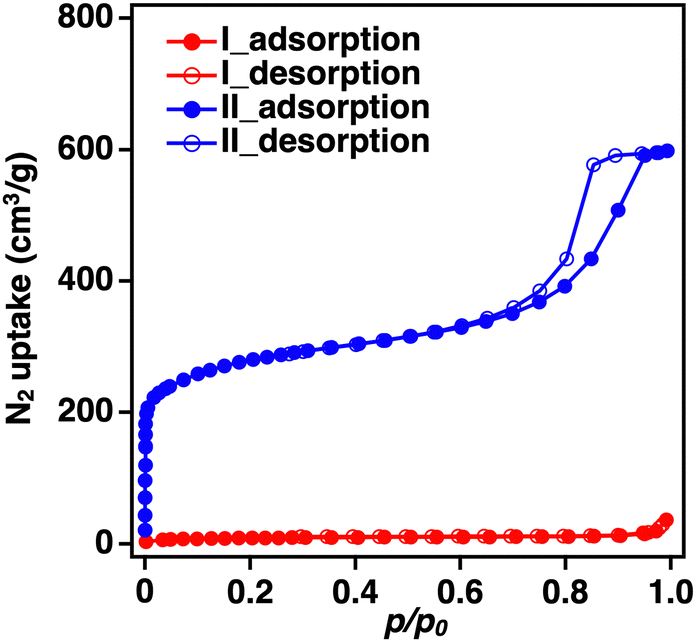 | ||
| Fig. 3 N2 adsorption–desorption measurements (77 K) of the prepared BIF-3-Cu crystals, I: using DL-2-amino-1-butanol as the reaction solvent at 100 °C for 4 days and II: with triethylamine addition and 50/50 (v/v, MeOH/MeCN) at room temperature for 24 h. | ||
The important aspects of BIF-3-Cu synthesis in solution can be summarised as follows. BIF-3-Cu exhibits a porous structure with SOD topology consisting of Cu+ ions and boron-bridged ligands. Although thermodynamically stable BIF-3-Cu can be obtained on a micron scale under heating conditions, slow crystal growth promotes the encapsulation of excess organic residue in the pores, resulting in a low specific surface area. In addition, solution heating causes partial Cu+ oxidation to Cu2+ ions, leading to crystal defects in the BIF-3-Cu framework and further anionic ligand encapsulation. In contrast, fast nucleation and crystal growth in the room-temperature solution reaction yields high-quality BIF-3-Cu crystals with larger specific surface areas, but with smaller crystal sizes.
Synthesis of SOD boron-bridged frameworks with divalent 3d-transition metals
As previously mentioned, ZIF-8 is a well-known MOF with SOD topology and is formed from Zn2+ and 2-MIm−.43 Herein, we considered the construction of a new BIF-3-Cu analogue that exhibits SOD topology, based on Zn2+ and B(2-MIm)4− (BIF-3-Zn). We attempted to synthesise BIF-3-Zn by following the optimised conditions used in the synthesis of BIF-3-Cu. After screening the reaction conditions using Zn nitrate [Zn(NO3)2] or Zn triflate [Zn(CF3SO3)2] as a Zn2+ source, the obtained crystals exhibited characteristic diffraction patterns similar to those of ZIF-8 (Fig. 4(I)). From Rietveld refinement of the observed PXRD pattern, the crystal structure of the obtained Zn-BIF was consistent with the BIF-3-Cu SOD topology (Fig. S6, ESI†). Additionally, the SEM-EDX results showed that B and Zn were detected in the obtained BIF-3-Zn crystals at approximately a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. S7, ESI†), demonstrating the formation of a 1:1 coordination network between Zn2+ and B(2-MIm)4−. Confirming the presence of B(2-MIm)4− in the resultant crystals, 1H-NMR measurement was also performed after dissolving the crystal by adding d-HCl aqueous solution (Fig. S8, ESI†). According to the 1H-NMR spectrum, the ligands assembled in the crystals were fully recovered and observed as B(2-MIm)4−, indicating that they were not decomposed during the formation process of the crystals. Considering the inclusion of excess ligands in the BIF-3-Cu pores, BIF-3-Zn may also contain counter anions in the pores owing to charge compensation. Thus, we also performed an X-ray fluorescence analysis to determine the inclusion of counter anions. In the case of the product obtained from Zn(CF3SO3)2, the presence of sulfur element (Zn: 80 at%, S: 20 at%) was confirmed, derived from the CF3SO3−, suggesting that counter anions were encapsulated in the BIF-3-Zn crystals. To further prove the inclusion of the counter anion in the inner space of the BIF-3-Zn crystal, we also examine an anion-exchange experiment. By immersing BIF-3-Zn crystals in a MeOH solution containing ZnCl2 for 17 hours at 75 °C, the encapsulated CF3SO3− can be partly replaced with Cl− (Zn: 76 at%, S: 10 at%, Cl: 14 at%), indicating that the counter anions were encapsulated in free space of the pore. Furthermore, the nitrogen gas adsorption isotherm of BIF-3-Zn at 77 K shows less gas adsorption capability compared with that of ZIF-8 because the BIF-3-Zn pores are occupied by counter ions (Fig. S9, ESI†). These results indicate that a stable SOD topology comparable to BIF-3-Cu was formed from divalent zinc ions and monovalent B(2-MIm)4−.
:1 ratio (Fig. S7, ESI†), demonstrating the formation of a 1:1 coordination network between Zn2+ and B(2-MIm)4−. Confirming the presence of B(2-MIm)4− in the resultant crystals, 1H-NMR measurement was also performed after dissolving the crystal by adding d-HCl aqueous solution (Fig. S8, ESI†). According to the 1H-NMR spectrum, the ligands assembled in the crystals were fully recovered and observed as B(2-MIm)4−, indicating that they were not decomposed during the formation process of the crystals. Considering the inclusion of excess ligands in the BIF-3-Cu pores, BIF-3-Zn may also contain counter anions in the pores owing to charge compensation. Thus, we also performed an X-ray fluorescence analysis to determine the inclusion of counter anions. In the case of the product obtained from Zn(CF3SO3)2, the presence of sulfur element (Zn: 80 at%, S: 20 at%) was confirmed, derived from the CF3SO3−, suggesting that counter anions were encapsulated in the BIF-3-Zn crystals. To further prove the inclusion of the counter anion in the inner space of the BIF-3-Zn crystal, we also examine an anion-exchange experiment. By immersing BIF-3-Zn crystals in a MeOH solution containing ZnCl2 for 17 hours at 75 °C, the encapsulated CF3SO3− can be partly replaced with Cl− (Zn: 76 at%, S: 10 at%, Cl: 14 at%), indicating that the counter anions were encapsulated in free space of the pore. Furthermore, the nitrogen gas adsorption isotherm of BIF-3-Zn at 77 K shows less gas adsorption capability compared with that of ZIF-8 because the BIF-3-Zn pores are occupied by counter ions (Fig. S9, ESI†). These results indicate that a stable SOD topology comparable to BIF-3-Cu was formed from divalent zinc ions and monovalent B(2-MIm)4−.
 | ||
| Fig. 4 PXRD patterns of (I) BIF-3-Zn, (II) BIF-3-Co, and (III) BIF-3-Mn. | ||
ZIF-67 is another SOD-type MOF, which is formed from Co2+ and 2-MIm−.44 Based on the above results using Zn2+ ions, we attempted to synthesise a Co2+-based SOD network (BIF-3-Co) and found that a reaction with a competitive ligand (1-methylimidazole) at ambient temperature was suitable for the crystal growth because of the highly reactive Co2+ ions. PXRD measurements and the Rietveld refinement results showed that the obtained crystals were consistent with the SOD-topology framework (Fig. 4(II), Fig. S10, ESI†). According to the SEM-EDX results, the boron and cobalt ratio in the crystal was approximately 1:1, as in the case of BIF-3-Zn (Fig. S11, ESI†). These results demonstrate that the formation of the counter-anion-encapsulated SOD topology consisting of a divalent metal ion and a boron-bridged ligand applies to a variety of metal ions. Microcrystals with manganese-based SOD topology (BIF-3-Mn) were also obtained in similar experiments, but the product yield was extremely low and not amenable to further measurements, except for PXRD (Fig. 4(III)). As a special example of a SOD-type MOF, the synthesis of Mn-ZIF-8 formed from Mn2+ and 2-MIm− was reported by Kadota et al.45 PXRD results of BIF-3-Mn showed a diffraction pattern similar to that of Mn-ZIF-8 (Fig. 4(III)). In this work, a series of experimental results showed that a stable SOD topological network can be formed from the combination of divalent metal ions and monovalent anionic B(2-MIm)4− ligands accompanied by the inclusion of counter ions in the pores.
To investigate the effects on the physical properties of the anionic boron-bridged structure with SOD topology, CO2 adsorption measurements at 300 K for the series of BIF-3 were performed (Fig. 5). Compared with that of ZIF-8 and ZIF-67, BIF-3-Cu and BIF-3-Co crystals show slightly higher CO2 uptake (ZIF-8: 0.64 mmol g−1, ZIF-67: 0.51 mmol, BIF-3-Cu: 0.72 mmol g−1, BIF-3-Co: 0.67 mmol g−1), but the CO2 uptake of BIF-3-Zn was significantly higher (1.2 mmol g−1).46,47 Furthermore, the rise of the CO2 adsorption isotherm for BIF-3-Zn occurs in the lower pressure region, indicating the characteristics of micropore adsorption. This high CO2 adsorption capacity of BIF-3-Zn was maintained at a comparable extent in three repeated tests (Fig. S12, ESI†), suggesting that the cationic framework of BIF-3-Zn with counter anions is sufficiently durable for the gas adsorption process. As mentioned above, the nitrogen gas adsorption isotherms at 77 K for BIF-3-Zn show negligible gas adsorption owing to the inclusion of the counter anion. At 77 K, the counter ions and framework are strongly interacting and there are few pores where N2 gas can be adsorbed. Conversely, during CO2 adsorption at 300 K, the counter ions and BIF-3-Zn framework flexibly changed positions and formed micropores, allowing CO2 to infiltrate. These results show that gas adsorption capacity can be controlled in the SOD topology depending on the combination of metal ions, counter anions, and anionic boron-bridging ligands.
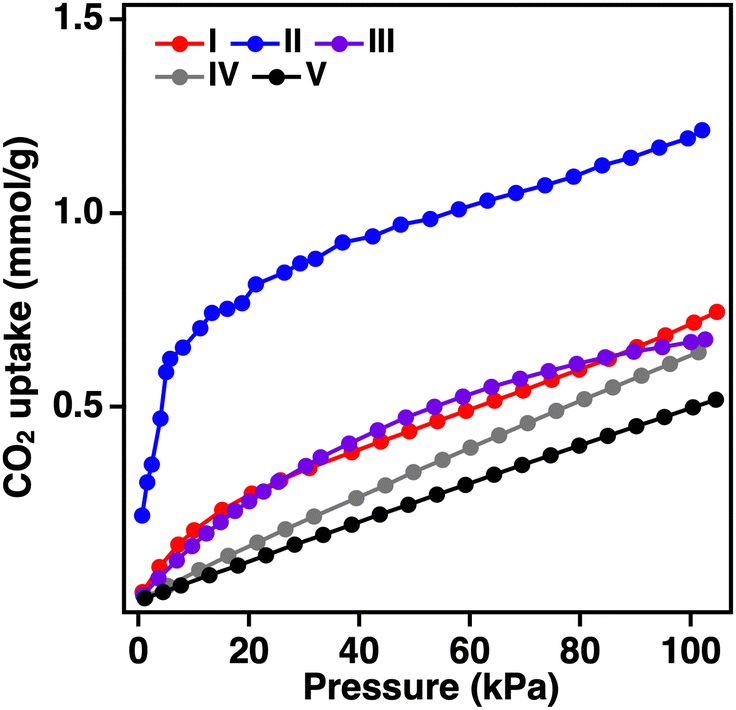 | ||
| Fig. 5 CO2 adsorption isotherms (300 K) of (I): BIF-3-Cu, (II): BIF-3-Zn, (III): BIF-3-Co, (IV): ZIF-8, and (V): ZIF-67 crystals. | ||
Conclusions
In conclusion, reaction conditions were examined to elucidate the crystal formation and growth process of BIF-3-Cu crystals in the liquid phase, including the influences of the boron-bridged ligand, counter ion, reaction solvent, metal salt, reaction time, additive, and reaction temperature. Maintaining the valence of the copper ions during the reaction was one of the important factors in improving the crystallinity of BIF-3-Cu. In addition, to increase the specific surface area of BIF-3-Cu crystals, it is necessary to consider the crystal nucleation and growth process to prevent the encapsulation of excess ligands within the pores of the crystals. As a result, BIF-3-Cu with a high specific surface area (1000 m2 g−1) was synthesised in solution at room temperature. Furthermore, we attempted to synthesise new BIF-3-Cu analogues with SOD topology by changing the metal ion from Cu+ to Zn2+ or Co2+ based on the optimised BIF-3-Cu synthesis conditions. By screening the reaction conditions, BIF-3-Zn, BIF-3-Co, and BIF-3-Mn were synthesised, and Rietveld analysis and elemental analysis revealed that they also exhibit a SOD-topology framework stabilised by counter anion encapsulation. Among the series of SOD-type BIFs, BIF-3-Zn exhibited a characteristic high CO2 adsorption ability. These findings may apply to the solution-based synthesis of numerous BIFs and facilitate the development of alternative porous materials with enhanced gas adsorption properties.Experimental
Materials
All the reagents and chemicals were obtained from commercial sources and used as received unless otherwise noted. CuCl, CuI, toluene, triethylamine, Tris(dimethylamino)borane, MeOH, MeCN, 1-methylimidazole (1-HMIm), 2-methylimidazole (2-HMIm), DL-2-amino-1-butanol, zinc(II) nitrate hexahydrate [Zn(NO3)2·6H2O], zinc(II) trifluoro-methanesulfonate [Zn(II) triflate], ZnCl2, dimethyl sulfoxide [DMSO], d-HCl, cobalt(II) nitrate hexahydrate [Co(NO3)2·6H2O] and tris(2,4-pentanedionato) manganese(III) [Mn(acac)3] were purchased from FUJIFILM Wako Pure Chemical Co., Tokyo Chemical Industry Co., Ltd, or Sigma Aldrich.Synthesis procedure of tetra(2-methylimidazol-1-yl)borate: H[B(2-MIm)4]
Toluene (50 mL) was added to a 100 mL round flask, and 2-HMIm (10 mol) was dispersed in the solvent. Tris(dimethylamino)borane (2.5 mol) was added to the dispersion, which was stirred with a magnetic stirrer (25 °C, 3 min). After heating at reflux (110 °C, 2 h), the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (white powder, yield: 91%). The product was dissolved in MeOH, and the MeOH was evaporated until it was a saturated solution. Then, the solution was quenched and recrystallised. 1H NMR (400 MHz, CD3OD): δ (ppm): 7.05 (d, J = 1.64 Hz, H), 6.74 (d, J = 1.60 Hz, H), and 1.79 (s, 3H). HRMS (ESI-MS) m/z: [M − H]−, for C16H21BN8, with an exact mass of 335.1904, and 335.1949 was observed.
Synthesis procedure of BIF-3-Cu using a previously reported method
CuI (4.1 mmol) was dissolved in MeCN (7.5 mL) and DL-2-amino-1-butanol (0.065 mol). H[B(2-MIm)4] (2.7 mmol) was dissolved in MeCN (7.5 mL) and DL-2-amino-1-butanol (0.065 mol). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After heating (120 °C, 5 days), the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN and MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 78%).
Synthesis procedure of BIF-3-Cu
CuI (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). H[B(2-MIm)4] (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After heating (100 °C, 4 days), the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN and MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 81%).
Synthesis procedure of BIF-3-Cu prepared by changing the metal salt
CuCl (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). H[B(2-MIm)4] (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After heating (100 °C, 4 days), the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN and MeOH, and dried under vacuum (30 °C, 24 hours) to obtain a powdery product.
Synthesis procedure of BIF-3-Cu prepared by changing the heating time
CuI (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). H[B(2-MIm)4] (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After heating (100 °C, 2 days or 24 h), the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN and MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 20% [2 days], 18% [24 h]).
Synthesis procedure of BIF-3-Cu with adding NaI
CuI (0.50 mmol) and NaI (0.25 mmol or 0.50 mmol) were dissolved in DL-2-amino-1-butanol (5 mL). H[B(2-MIm)4] (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After heating (100 °C, 24 h), the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN and MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 90% [0.25 mmol], 83% [0.50 mmol]).
Synthesis procedure of BIF-3-Cu at room temperature
CuI (0.30 mmol) was dissolved in MeCN (50 mL). H[B(2-MIm)4] (0.30 mmol) was dissolved in MeOH (50 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After 24 h, the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN and MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 80%).
Synthesis procedure of BIF-3-Cu prepared by changing the metal salt and solvent ratio at room temperature
CuCl (0.2985 mmol) was dissolved in MeCN (50 mL). H[B(2-MIm)4] (0.2985 mmol) was dissolved in MeOH (50 mL). Using MeOH/MeCN ratios (v/v) of 90/10, 50/50, and 10/90, the H[B(2-MIm)4] solution was poured into the CuCl solution while stirring with a magnetic stirrer (25 °C, 3 min). After 24 h, the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN and MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 20% [90/10], 54% [50/50], 80% [10/90]).
Synthesis procedure of BIF-3-Cu prepared by adding triethyl-amine at room temperature
CuCl (0.2985 mmol) was dissolved in MeCN. H[B(2-MIm)4] (0.2985 mmol) and triethylamine (0.2985 mmol) were dissolved in MeOH. Using MeOH/MeCN ratios (v/v) of 90/10, 50/50, and 10/90, the H[B(2-MIm)4] solution was poured into the CuCl solution while stirring with a magnetic stirrer (25 °C, 3 min). After 24 h, the product was centrifuged (10000 rpm, 10 min), washed several times with MeCN and MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 76% [90/10], 52% [50/50], 80% [10/90]).
Synthesis procedure of BIF-3-Zn
Zn(NO3)2·6H2O (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). H[B(2-MIm)4] (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After heating (100 °C, 1 day), the product was centrifuged (10000 rpm, 10 min), washed several times with MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 18%).
1H-NMR analysis of the boron-bridged ligand in the resultant BIF-3-Zn by d-HCl aqueous solution
BIF-3-Zn (0.1 mmol), D2O, d-HCl, and DMSO (0.1 mmol) were added to the vial and the resultant BIF-3-Zn dispersion was sonicated for few minutes. Then, the solution was analyzed by 1H-NMR spectroscopy.Synthesis procedure of BIF-3-Zn by using Zn(II) triflate
Zn(II) triflate (0.50 mmol) was dissolved in MeOH (5 mL). H[B(2-MIm)4] (1.0 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After heating (100 °C, 1 day), the product was centrifuged (10000 rpm, 10 min), washed several times with MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 46%).
Synthesis procedure of BIF-3-Co
Co(NO3)2·6H2O (0.2985 mmol) and 1-HMIm (4.776 mmol) were dissolved in MeOH (50 mL). H[B(2-MIm)4] (0.2985 mmol) was dissolved in MeOH (50 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After 24 h, the product was centrifuged (10000 rpm, 10 min), washed several times with MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 86%).
Synthesis procedure of BIF-3-Mn
Mn(acac)3 (0.50 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). H[B(2-MIm)4] (1.0 mmol) was dissolved in DL-2-amino-1-butanol (5 mL). The latter clear solution was poured into the former clear solution while stirring with a magnetic stirrer (25 °C, 3 min). After heating (80 °C, 2 days), the product was centrifuged (10000 rpm, 10 min), washed several times with MeOH, and dried under vacuum (30 °C, 24 h) to obtain a powdery product (yield: 1%).
Analytical methods
Author contributions
Takeshi Kato: data curation, formal analysis, investigation, methodology, visualisation, writing – original draft, writing – review & editing. Ikuho Akiyama, Fumika Mori, Yudai Ogura, Ayumu Shinohara: data curation, formal analysis, investigation, methodology. Akitaka Ito: data curation, formal analysis, investigation, methodology. Masataka Ohtani: conceptualisation, funding acquisition, formal analysis, investigation, methodology, project administration, resources, supervision, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of JSPS KAKENHI Grant Numbers 19K05186 and 22K04857. We thank Robert Ireland Robert Ireland, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.Notes and references
- S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. U. Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef PubMed.
- H. J. Peng, P. Q. Zheng, H. Y. Chao, L. Jiang and Z. P. Qiao, RSC Adv., 2020, 10, 551–555 RSC.
- J. Zhang, T. Wu, C. Zhou, S. Chen, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2009, 48, 2542–2545 CrossRef PubMed.
- H. X. Zhang, M. Liu, T. Wen and J. Zhang, Coord. Chem. Rev., 2016, 307, 255–266 CrossRef.
- V. I. Isaeva, K. E. Papathanasiou and L. M. Kustov, Crystals, 2020, 10, 617 CrossRef.
- I. A. Baburin, B. Assfour, G. Seifert and S. Leoni, Dalton Trans., 2011, 40, 3796–3798 RSC.
- J. Zhang, T. Wu, P. Feng and X. Bu, Dalton Trans., 2010, 39, 1702–1704 RSC.
- F. Wang, Yu. B. Shu, X. Bu and J. Zhang, Chem. – Eur. J., 2012, 18, 11876–11879 CrossRef.
- V. I. Isaeva, K. Papathanasiou, V. V. Chernyshev, L. Glukhov, G. Deyko, K. K. Bisht, O. P. Tkachenko, S. V. Savilov, N. A. Davshan and L. M. Kustov, ACS Appl. Mater. Interfaces, 2021, 13, 59803–59819 CrossRef.
- M. R. Liu, Q. L. Hong, Q. H. Li, Y. Du, H. X. Zhang, S. Chen, T. Zhou and J. Zhang, Adv. Funct. Mater., 2018, 28, 1801136 CrossRef.
- T. Wen, Y. Zheng, J. Zhang, K. Davey and S. Z. Qiao, Adv. Sci., 2019, 6, 1801920 CrossRef.
- Y. M. Wan, Q. L. Hong, H. X. Zhang and J. Zhang, Inorg. Chem., 2020, 59, 17851–17855 CrossRef PubMed.
- Z. Y. Chen, Q. L. Hong, H. X. Zhang and J. Zhang, ACS Appl. Energy Mater., 2022, 5, 1175–1182 CrossRef.
- Z. Li, Y. Han, A. Wang, D. Zhao, L. Fan, L. He, S. Zhang, P. Cheng, H. Liu, Z. Chaia and S. Wang, Dalton Trans., 2022, 51, 11625–11629 RSC.
- S. Banerjee, J. M. Gorham, P. B. Varela, H. G. Hackbarth, M. A. Siegler, N. Drichko, J. T. Wright, N. M. Bedford and V. S. Thoi, ACS Appl. Energy Mater., 2023, 18, 9044–9056 CrossRef.
- D. X. Zhang, H. X. Zhang, H. Y. Li, T. Wen and J. Zhang, Cryst. Growth Des., 2015, 15, 2433–2436 CrossRef.
- H. X. Zhang, Q. L. Hong, J. Li, F. Wang, X. Huang, S. Chen, W. Tu, D. Yu, R. Xu, T. Zhou and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 11752–11756 CrossRef PubMed.
- D. X. Zhang, H. X. Zhang, T. Wen, D. S. Li and J. Zhang, Inorg. Chem. Front., 2016, 3, 263–267 RSC.
- H. X. Zhang, M. Liu, G. Xu, L. Liu and J. Zhang, Chem. Commun., 2016, 52, 3552–3555 RSC.
- P. Shao, W. Zhou, Q. L. Hong, L. Yi, L. Zheng, W. Wang, H. X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687–16692 CrossRef.
- G. Wang, J. Ye, M. Wang, Y. Qi, S. Zhang, L. Shi, Y. Fang, Y. Tian and G. Ning, Carbohydr. Polym., 2022, 291, 119588 CrossRef PubMed.
- H. X. Zhang, F. Wang, H. Yang, Y. X. Tan, J. Zhang and X. Bu, J. Am. Chem. Soc., 2011, 133, 11884–11887 CrossRef.
- H. X. Zhang, H. R. Fu, H. Y. Li, J. Zhang and X. Bu, Chem. – Eur. J., 2013, 19, 11527–11530 CrossRef PubMed.
- T. Wen and J. Zhang, Inorg. Chim. Acta, 2017, 460, 89–92 CrossRef.
- G. Xu, Q. L. Hon, Y. Sun, M. Liu, H. X. Zhang and J. Zhang, Chin. Chem. Lett., 2015, 231, 185–189 Search PubMed.
- Q. L. Hong, H. X. Zhang, Y. H. Wen and J. Zhang, Inorg. Chem. Commun., 2018, 95, 130–133 CrossRef.
- Q. Hong, W. Wang, S. Chen, K. Chen, M. Liu, H. X. Zhang and J. Zhang, Chem. Mater., 2022, 34, 303–317 Search PubMed.
- M. Y. Bi, Q. L. Hong, M. Liu, F. Wang, H. X. Zhang and J. Zhang, Chem. Commun., 2021, 57, 5020–5023 RSC.
- Z. Y. Chen, Q. L. Hong, H. X. Zhang and J. Zhang, Inorg. Chem., 2022, 61, 6861–6868 CrossRef.
- Z. Y. Chen, Y. Chen, H. X. Zhang and J. Zhang, J. Solid State Chem., 2022, 310, 123001 CrossRef.
- P. J. Bailey, D. L. Gonzales, C. McCormack, F. Millican, S. Parsons, R. Pfeifer, P. P. Pinho, F. Rudolphi and A. S. Perucha, Chem. – Eur. J., 2006, 12, 5293–5300 CrossRef.
- E. Yang, L. Wang and J. Zhang, CrystEngComm, 2014, 16, 2889 RSC.
- D. X. Zhang, J. Liu, H. X. Zhang, T. Wen and J. Zhang, Inorg. Chem., 2015, 54, 6069–6071 CrossRef CAS.
- H. X. Zhang, M. Liu, G. Xu, L. Liu and J. Zhang, Chem. Commun., 2016, 52, 3552–3555 RSC.
- L. Liu, T. Wen, S. Chen and J. Zhang, J. Solid State Chem., 2015, 231, 185–189 CrossRef.
- T. Wen, M. Liu, S. Chen, Q. Li, Y. Du, T. Zhou, C. Ritchie and J. Zhang, Small, 2020, 16, 1907669 CrossRef PubMed.
- M. Liu, D. X. Zhang, S. Chen and T. Wen, J. Solid State Chem., 2016, 237, 32–35 CrossRef.
- M. Liu, S. Chen, T. Wen and J. Zhang, Chem. Commun., 2016, 52, 8577–8580 RSC.
- W. Morris, C. J. Stevens, R. E. Taylor, C. Dybowski, O. M. Yaghi and M. A. G. Garibay, J. Phys. Chem., 2012, 116, 13307–13312 Search PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O’Keeffe and O. M. Yaghi, Science, 2008, 319, 938 CrossRef PubMed.
- C. B. Lennox, J. L. Do, M. Arhangelskis, H. M. Titi, O. K. Farha and T. Friscic, Chem. Sci., 2021, 12, 14499–14506 RSC.
- J. Cravillon, R. Nayuk, S. Springer, A. Feldhoff, K. Huber and M. Wiebcke, Chem. Mater., 2011, 23, 2130–2141 CrossRef.
- H. T. Kwon, H. Jeong, A. S. Lee, H. S. An and J. S. Lee, J. Am. Chem. Soc., 2015, 137, 12304–12311 CrossRef PubMed.
- K. Kadota, E. Sivaniah, S. Bureekaew, S. Kitagawa and S. Horike, Inorg. Chem., 2017, 56, 8744–8747 CrossRef PubMed.
- A. Imtiaz, M. H. D. Othman, A. Jilani, I. U. Khan, R. Kamaludin and O. Samuel, J. Environ. Chem. Eng., 2022, 10, 108541 CrossRef.
- Y. Dai, Z. Niu, W. Luo, Y. Wang, P. Mu and J. Li, Sep. Purif. Technol., 2023, 15, 122752 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00996c |
| This journal is © The Royal Society of Chemistry 2024 |