
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of preceramic ferrocene-modified microparticles for the development of uniform porous iron oxide particles and their sustainable recycling†
Deborah
Schmitt
 a,
Oliver
Janka
b,
Regina
Leiner
a,
Guido
Kickelbick
bc and
Markus
Gallei
*ac
a,
Oliver
Janka
b,
Regina
Leiner
a,
Guido
Kickelbick
bc and
Markus
Gallei
*ac
aPolymer Chemistry, Saarland University, Campus C4 2, 66123 Saarbrücken, Germany. E-mail: markus.gallei@uni-saarland.de
bInorganic Solid State Chemistry, Saarland University, Campus C4 1, 66123 Saarbrücken, Germany
cSaarene, Saarland Center for Energy Materials and Sustainability, Campus C4 2, 66123 Saarbrücken, Germany
First published on 15th February 2024
Abstract
Structural design strategies at the nano- and micrometer length scale have attracted increasing interest in recent decades as they offer the possibility to fine-tune advanced properties for numerous applications. In this context, closing the cycle for the usability of (nano)materials, starting from their organic components leading to their applications, is of paramount importance today. Moreover, instead of recycling the materials for the same applications, other applications of the materials can also be followed. Iron oxides such as magnetite, maghemite, or hematite are materials that can be considered for many interesting applications in the field of batteries, drug delivery, or magnetic separation. The present study demonstrates the tailored design of microscaled iron oxide particles based on tailored molecular functionalization of organic microparticles. Ferrocene was used for the preceramic functionalization of iron-containing polymers to modify the microparticles and also as precursors with different degrees of functionalization and hydrophilicity. The precursor particles were calcined under different conditions such as nitrogen or oxygen gas flow or temperature. It was demonstrated that tuning the surface morphology, composition, and size of the ceramics could be realized very precisely by an appropriate choice of precursor functionalization, allowing universal tailored fabrication for various applications. As an example, the electrochemical addressability and pH-dependent behavior of the ceramics were demonstrated, enabling their application in batteries.
Introduction
The development of iron oxide-based ceramics has received considerable attention for decades owing to their wide range of technical applications, high biocompatibility, and harmless environmental properties. Due to these properties, the ceramics could be used in theranostic applications,1 as multidrug carriers,2 or for magnetic separation processes.3,4 In addition, the application of iron-based materials in batteries represents a growing field.5–7 In general, electrochemical applications for ferrous materials are of increasing interest both in the literature and society. For example, they have been used in ion sorption processes for the selective removal of Pb2+ by Lei et al.8 However, the long-term stability of iron-containing electrodes is restricted by the volume expansions of iron by switching process, leading to a fracturing of the electrodes.9 To overcome this issue, homogeneous distributions of the iron oxide crystallites, e.g. in a carbon matrix, will be required.10,11 At the same time, high porosity will be essential to ensure high accessibility of the electrolyte to the iron-based material.12 However, the targeted synthesis of such electrodes is still a challenge and part of ongoing research.The size, morphology, composition, and shape of the ceramics are key factors in determining the application range of the respective materials. The significant influence of structure and crystallite size of ceramics on the properties, e.g., in electrochemical or magnetic investigations, has already been described in the literature.13–15 Especially the synthesis and application of nanometer-sized ceramics have been studied in detail. For instance, nanospheres, tetrahedrons, cubes, octahedrons, wires, or tubes could be synthesized.1,16,17 Nanospheres represent the most examined structures used as superparamagnetic materials,18 in drug-conjugated processes19 or batteries.20 The monodisperse particle synthesis was realized even on a scale of 40 g by temperature annealing processes.21 However, examples of monodisperse spherical micrometer-sized iron or iron oxide-based ceramic particles are rare. Li et al. synthesized flower-like nanostructured particles by decomposition of iron oxide precursors.22 Moreover, the preparation of particles based on the aggregation of nanoplate-like building blocks into Fe3O4 hollow microspheres was accomplished by Wang et al. to achieve anode materials with highly reversible capacitance.23 Ma et al. confirmed that a porous and hierarchical hollow spherical structure provides the advantages of excellent electrochemical properties as an anode material.24 In addition to self-assembly processes, however, template routes for the preparation of microscale particles have been reported. Microscale organic particles could be modified with FeCl3·6H2O via precipitation reaction and subsequently converted into non-porous ceramics by calcination.25 Porous ceramics were synthesized by quasi-emulsion-templated formation for electrochemical investigation as an anode material.26 Also, the introduction of ferrocene into organic particles during synthesis could be carried out to produce microscale Fe3O4 ceramics.27
In general, ferrocenes have been described more frequently as feasible ceramic precursors. In particular, the ferrocene-based polymers have proven to be preceramic materials, for example, by the Manners group by using poly(ferrocenylsilanes) (PFS).28,29 In addition to bulk materials, PFS could also be immobilized on polystyrene nanoparticles to obtain ceramic nanoparticles.30 Mazurowski et al. used polystyrene nanoparticles covalently functionalized with poly(2-(methacryloyloxy)ethyl ferrocene carboxylate) (PFcMA) brushes as a precursor for iron oxide nanoparticles.31 The most common functionalization strategies for covalent anchoring of metallopolymers on nanoparticles include controlled radical polymerizations based on reversible radical deactivation reactions like atom transfer radical polymerization31 or reversible addition–fragmentation chain transfer.32,33 But other methods like emulsion polymerization were also described for the preparation of preceramic materials. In this context, Scheid et al. synthesized magnetically responsive nanocapsules by incorporating preceramic polymers such as PFcMA in organic films. This enabled the fabrication of magnetic opal films.34,35 In addition to metallocene-containing polymers, silicon-containing compounds were mainly used as preceramic polymers.36 Both block copolymers37 and nanoparticle-based37 systems have been described in the literature. Since the functionalization of microscale particles with preceramic polymers has rarely been described, no microscale ceramics have been prepared in this way.
The main challenge in the preparation of microscale ceramic particles is to develop a synthesis strategy that allows monodisperse particle synthesis and the adjustment of particle size and surface structure. The application of self-assembly processes usually resulted in low uniformities and few monodisperse particles, while well-structured particles separated from each other are almost impossible to produce by precipitation reactions. In contrast, covalent bonding of precursors would overcome this problem due to the homogeneous distribution of iron in the template. In this work, covalent immobilization of various ferrocene-based precursors to organic particles was used to produce uniform ceramics. The surface modifications of organic polystyrene-divinylbenzene microparticles (PSDVB) were investigated by attenuated total reflection infrared spectroscopy (ATT-IR), cyclic voltammetry (CV), and thermogravimetric analysis (TGA). The influence of the degree of functionalization as well as the chosen ferrocene precursor (polymer-based or surface monolayer-functionalization) on the morphology and size of the ceramics was investigated with scanning electron microscopy (SEM). The incorporation of oxygen-containing groups into the particles and their influence on the composition of the ceramics was examined by energy-dispersive X-ray (EDS), Raman spectroscopy, and powder X-ray diffraction (PXRD). In addition, cyclic voltammetric studies of the ceramic particles were carried out under different pH values to investigate the electrochemical behavior and addressability.
Experimental
Materials
All reagents and solvents were purchased from Sigma-Aldrich, Fisher Scientific, TCI Chemicals, or Alfa Aesar and used as received unless otherwise noted. Polystyrene-divinylbenzene (PSDVB) particles (55 wt% DVB in ethyl vinyl benzene) and strongly hydrophilized polystyrene (PSOH) particles38 were donated from Metrohm AG. High-purity multi-walled carbon nanotubes (MWCNT, NC7100) were purchased from Nanocyl. n-Hexane was dried over n-butyl lithium (n-BuLi, 1.6 M solution in n-hexane) using diphenylhexyl lithium as the indicator and distilled before use. Pyridine was dried over CaH2 and distilled. 3-Ferrocenyl propylamine was synthesized according to the literature, starting from ferrocenyl carbaldehyde, which was reacted first to 2-cyanovinyl ferrocene.39 The Preussmann test was carried out according to the literature in ethanol40 and the poly(ferrocenyl methyl methacrylate) functionalized polystyrene-divinylbenzene particles (PSDVB@PFMMA) were synthesized and characterized as described previously.41Instrumentation
Fourier transformation infrared spectra (FTIR) in an attenuated total reflection mode (ATR) were recorded using a Bruker Alpha II FT-IR spectrometer (Bruker Optics, Ettlingen, Germany) in transmittance. OPUS 8.5 software was used for evaluation and baseline correction. All spectra were normalized between 0 and 1.Raman spectroscopy was performed on a Renishaw InVia microscope (Wotton-under-Edge, Gloucestershire, United Kingdom) using an excitation laser with a 633 nm wavelength. The samples were deposited on a glass slide, and the measurements were carried out with an exposure time of 30 s and accumulated five times.
Powder X-ray diffraction (PXRD) patterns of the pulverized ceramic samples were recorded at room temperature on a D8-A25-Advance diffractometer from Bruker (Bruker AXS, Karlsruhe, Germany). Bragg–Brentano θ–θ-geometry (280 mm radius) with Cu Kα-radiation (λ = 154.0596 pm) was used, and a 12 μm Ni foil, working as Kβ filter, as well as a variable divergence slit, was mounted at the primary beam side. At the secondary beam side, a LYNXEYE detector with 192 channels was used. The experiments were carried out in a 2θ range of 7 to 120° with a step size of 0.013° and a total scan time of 2 h. The evaluation was carried out with the Rietveld method42 by using the Bruker TOPAS 5.0 software. Lattice parameters and the microstructure were refined, while instrument-based line broadening effects and peak shape descriptions were determined empirically according to the fundamental parameters approach43 after reference scans of LaB6, elemental Si NIST 640a, and Al2O3 supplied by Bruker AXS.
Thermogravimetric analyses were performed on a NETZSCH TGA 209 Libra (Selb, Germany) in an Al crucible for measurements up to 580 °C and Al2O3 for measurements up to 800 °C. During the measurements, the calculated differential thermal analysis (c-DTA) was used to estimate the difference between the calculated temperature signal and the measured signal of the samples.
Elemental analyses for quantification of the oxygen content (CHNO measurements) were carried out at a pyrolysis temperature of 1450 °C with a rapid OXY cube from Elementar (Langenselbold, Germany) after calibration with benzoic acid. The samples were placed in a combustion tube and compressed to exclude air.
Cyclic voltammetry was performed with a BioLogic SP-150 potentiostat (Göttingen, Germany) in a three-electrode configurated cell. A platinum wire was used as the counter electrode, and a glassy carbon electrode with an inner diameter of 5 mm was used as the working electrode. The measurements of ferrocene-based precursors and ceramics were carried out in a 0.1 M solution with NaClO4 as an electrolyte in water and an Ag/AgCl reference electrode (equilibrated in 3 M NaCl). In alkaline solutions, a HgO/Hg electrode (equilibrated in 1 M NaOH) was used as a reference and the measurements were performed in a 1 M solution with NaOH as a supporting electrolyte. The samples were mixed with MWCNTs (ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), dispersed in ethanol, and drop-cast on the working electrode. All measurements were carried out with a scan rate of 20 mV s−1, calibrated with ferrocene, and evaluated with EC-Lab V11.46.
:1), dispersed in ethanol, and drop-cast on the working electrode. All measurements were carried out with a scan rate of 20 mV s−1, calibrated with ferrocene, and evaluated with EC-Lab V11.46.
Scanning electron microscopy (SEM) was carried out on a Zeiss Sigma VP GeminiSEM 500 (Oberkochen, Germany) device using SmartSEM Version 6.07 as software. All samples were mounted on an Al stud using carbon tape and coated with 6 nm platinum using an Automatic Turbo Coater Plasmatool 125 SIN 2020_131 from Ingenieurbüro Peter Liebscher. Micrograph images were collected with an acceleration voltage of 1 kV in high current mode and a 20 μm aperture using an in-lens detector. Energy-dispersive X-ray spectroscopy (EDS) was performed using an SE2 detector and an acceleration voltage of 15 kV. The particle size determination was carried out with ImageJ software based on the analysis of 100 particles.
Exemplary PSDVB particle oxidation for conversion into PS-oxirane
1 g PSDVB particles were placed in a reaction flask equipped with a KPG stirrer and dispersed in 50 mL DCM (technical grade). After 1 h of swelling, 1.4 g (8.1 mmol) meta-chloroperoxy benzoic acid (mCPBA, ≤77%) was added at 0 °C and stirred for an additional hour. The suspension was stirred for 18 h at room temperature before the PS-oxirane particles were washed five times with DCM, water, acetone, and THF, respectively. The particles were dried in vacuo.Exemplary surface modification with 3-ferrocenyl propylamine
200 mg PS-oxirane particles were dispersed in acetonitrile (HPLC grade) and stirred for 1 h. The dispersion was cooled to 0 °C and 500 mg of 3-ferrocenyl propylamine (FcPA, 2.1 mmol) was added. After 1 h, the mixture was stirred for 24 h at 60 °C. The particles were washed five times with ethanol and THF, respectively. The PS-ox@Fc particles were dried in vacuo. The reaction was also carried out in ethanol as a solvent.Exemplary surface modification with ferrocenyl acid chloride
1 g strongly hydroxylated polystyrene (PSOH) particles were dispersed in 7.5 mL dry pyridine under an argon atmosphere and stirred for 1 h. 240 mg (1.0 mmol) ferrocenyl acid chloride in 2.5 mL dry n-hexane was added at 0 °C and stirred for 24 h at room temperature. The orange particles were washed repeatedly five times with n-hexane, acetone, ethanol, and THF. The PSOH@Fc particles were dried in vacuo.Calcination of the precursor PSOH@Fc, PS-ox@Fc and PSDVB@PFMMA
∼15 mg precursor particles were placed in an aluminum or Al2O3 crucible and heated to 580 °C or 800 °C, respectively. A heating rate of 10 °C min−1 in synthetic air or nitrogen atmosphere was used with a gas flow set to 20 or 40 mL min−1.Results and discussion
Surface modification and polymer precursor
Two different types of particle precursors were investigated for the preparation of iron oxide ceramics. First, we used polymer-functionalized polystyrene particles prepared as described in the literature.41 The particles in this study were polystyrene-divinylbenzene particles (PSDVB) functionalized by using surface-initiated living anionic polymerization (SI-LAP) of ferrocenyl methyl methacrylate (FMMA) entitled PSDVB@PFMMA particles (Fig. 1a). The PFMMA functionalization was shown at the particles’ surface and the particles’ interior inside the pores. The exact degrees of functionalization are given in Table S1 (ESI†). Second, surface-modified PSDVB particles with a single layer of covalently bonded ferrocene derivatives were used. For this purpose, two synthesis strategies were applied (Fig. 2). PSDVB particles with a high content of available double bonds were epoxidized by using meta-chloroperoxy benzoic acid (mCPBA) to achieve PS-oxirane particles (Fig. 2a). The epoxide groups obtained were demonstrated by the Preussmann test using 4-(4-nitrobenzyl) pyridine (NBP), which reacted in an efficient ring-opening reaction by nucleophilic attack (Fig. S1, ESI†) accompanied by a purple coloration of the particles (Fig. 1b).40,44 Quantification of the epoxide groups was carried out by CHNO element analyses where the oxygen content was found to be between 3 to 5 wt% (Table S2, ESI†). | ||
| Fig. 1 (a) Graphical representation of the structure of PSDVB@PFMMA functionalized particles and (b) images of Preussmann test results with PS-oxirane01 before and after epoxidation of PSDVB: (1) PSDVB particles in ethanol, (2) PSDVB particles and NBP in ethanol showing no reaction of PSDVB with the Preussmann reagent, (3) PS-oxirane particles in ethanol and (4) PS-oxirane particles after reaction with NBP in ethanol showing the positive Preussmann test. | ||
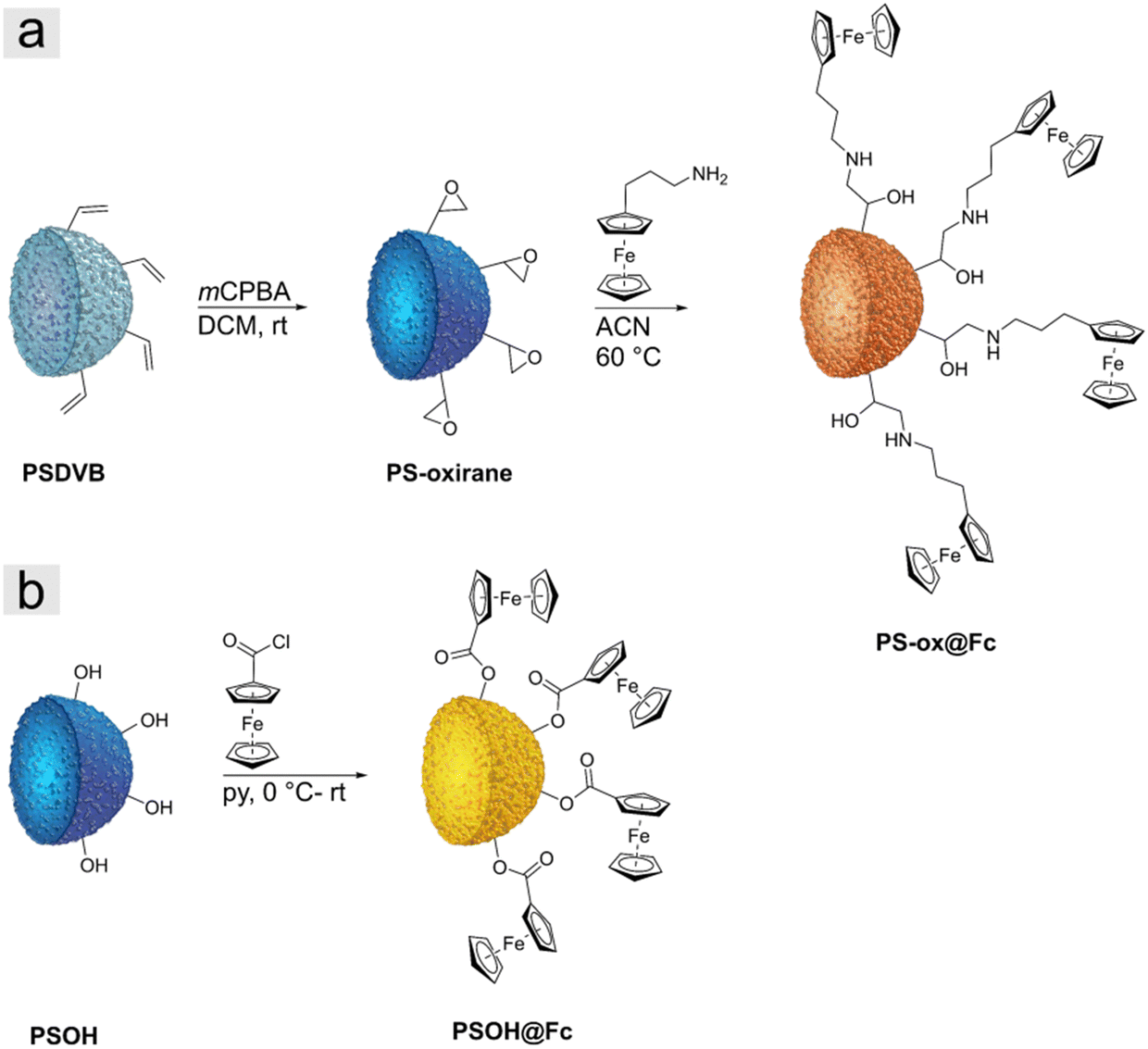 | ||
| Fig. 2 Graphical representation of the synthesis of (a) PS-ox@Fc particles starting from PSDVB particles by double bond epoxidation with mCPBA and ring-opening reaction with FcPA and (b) PSOH particles with ferrocene acid chloride leading to PSOH@Fc particles. | ||
The epoxidized particles were converted using a polymer-analogous reaction at the surface (Fig. 2a). As a ferrocene precursor, 3-ferrocenyl propylamine (FcPA) was synthesized according to the literature.39 For the investigation of different solvents, acetonitrile (ACN) or ethanol has been proven to be suitable for functionalization (Table 1). In THF, on the other hand, degradation of the particle matrix could be observed, leading to irregular and ill-defined particles (Fig. S2a–d, ESI†).
| Particle | Ferrocene precursor per particle (mg mg−1) | Solvent |
|---|---|---|
| PSOH@Fc-1 | 1.00 | Pyridine |
| PSOH@Fc-2 | 0.60 | Pyridine |
| PSOH@Fc-3 | 0.24 | Pyridine |
| PSOH@Fc-4 | 0.10 | Pyridine |
| PS-ox@Fc-1 | 1.20 | ACN |
| PS-ox@Fc-2 | 1.00 | EtOH |
| PS-ox@Fc-3 | 0.50 | EtOH |
As an additional ferrocene-modified precursor, highly hydroxylated polystyrene particles (PSOH) were functionalized (Fig. 2b).38 The PSOH particles were treated with ferrocene acid chloride synthesized according to the literature, starting from ferrocene carboxylic acid and oxalyl chloride (Fig. S3, ESI†).45 In a polymer-analogous nucleophilic substitution in the presence of pyridine, the precursor could be used for particle functionalization. In both synthesis strategies, the degree of functionalization was adjusted and varied by the ratio between ferrocene precursor and particles to investigate the influence on the iron oxide ceramics (Table 1). The PS-ox@Fc-1 and the PSOH@Fc-1, respectively, exhibited the highest ferrocene content, with a decreasing trend until the lowest content of functionalization was reached for PS-ox@Fc-3 and PSOH@Fc-4.
To prove the successful modification of polystyrene-based microparticles, ATR-FTIR spectroscopy measurements were performed. For comparison, measurements of the PS-oxirane particles, ferrocene precursor FcPA, and modified particles were recorded for the PS-ox@Fc particles. As shown in Fig. 3a, the PS-oxirane particles exhibited the characteristic bands of aliphatic and aromatic –C–H stretching vibration of PSDVB at 3000–2790 cm−1. The –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration at 1605 cm−1 and the –C–C stretching vibration at 1515–1415 cm−1 were detected. The C–H bending vibration at 900 cm−1 and the ring bending vibrations of substituted benzenes at 708 cm−1 can be observed in all particle samples. Moreover, the characteristic oxirane breathing at 1267 cm−1, the asymmetrical ring vibration at 890 cm−1, and the bands at 744 cm−1 can be observed after oxidation with mCPBA.46 The FcPA exhibited the –NH2 bending vibrations at 1647 cm−1 and the –C–N stretching vibrations at 1103 cm−1 as well as the C–H band of cyclopentadiene (cp) at 3093 cm−1 and the out-of-plane vibration of FeII-cp-H at 821 cm−1.47 However, the functionalized particles revealed both, the bands of FcPA besides the –NH2 band and PSDVB. The oxirane vibrations could not be observed any longer, indicating the successful ring-opening reaction. To verify the functionalization of PSOH particles, the measurements were compared to those of ferrocene carboxylic acid. In addition to the ferrocene bands, the characteristic –CO vibration of the carboxylic acid could be detected at 1653 cm−1. The expected shift to higher wavenumbers (1708 cm−1) could be observed according to the particles’ surface modification. The –C–O stretching vibrations could be detected at 1281 cm−1, and the –C–O vibration at 1156 cm−1 of ferrocene carboxylic acid revealed a shift to 1135 cm−1 by the modified particles. Moreover, the –C–O stretching vibration of the primary alcohol of the PSOH particles at 1031 cm−1 as well as the –O–H band at 3160–3640 cm−1 significantly decreases after functionalization. As expected, the intensity of the bands strongly correlated with the degree of functionalization. Further, ATR-FTIR spectra of PSDVB@PFMMA particles, different particle batches, and additional magnifications can be found in Fig. S4–S6 (ESI†).
C stretching vibration at 1605 cm−1 and the –C–C stretching vibration at 1515–1415 cm−1 were detected. The C–H bending vibration at 900 cm−1 and the ring bending vibrations of substituted benzenes at 708 cm−1 can be observed in all particle samples. Moreover, the characteristic oxirane breathing at 1267 cm−1, the asymmetrical ring vibration at 890 cm−1, and the bands at 744 cm−1 can be observed after oxidation with mCPBA.46 The FcPA exhibited the –NH2 bending vibrations at 1647 cm−1 and the –C–N stretching vibrations at 1103 cm−1 as well as the C–H band of cyclopentadiene (cp) at 3093 cm−1 and the out-of-plane vibration of FeII-cp-H at 821 cm−1.47 However, the functionalized particles revealed both, the bands of FcPA besides the –NH2 band and PSDVB. The oxirane vibrations could not be observed any longer, indicating the successful ring-opening reaction. To verify the functionalization of PSOH particles, the measurements were compared to those of ferrocene carboxylic acid. In addition to the ferrocene bands, the characteristic –CO vibration of the carboxylic acid could be detected at 1653 cm−1. The expected shift to higher wavenumbers (1708 cm−1) could be observed according to the particles’ surface modification. The –C–O stretching vibrations could be detected at 1281 cm−1, and the –C–O vibration at 1156 cm−1 of ferrocene carboxylic acid revealed a shift to 1135 cm−1 by the modified particles. Moreover, the –C–O stretching vibration of the primary alcohol of the PSOH particles at 1031 cm−1 as well as the –O–H band at 3160–3640 cm−1 significantly decreases after functionalization. As expected, the intensity of the bands strongly correlated with the degree of functionalization. Further, ATR-FTIR spectra of PSDVB@PFMMA particles, different particle batches, and additional magnifications can be found in Fig. S4–S6 (ESI†).
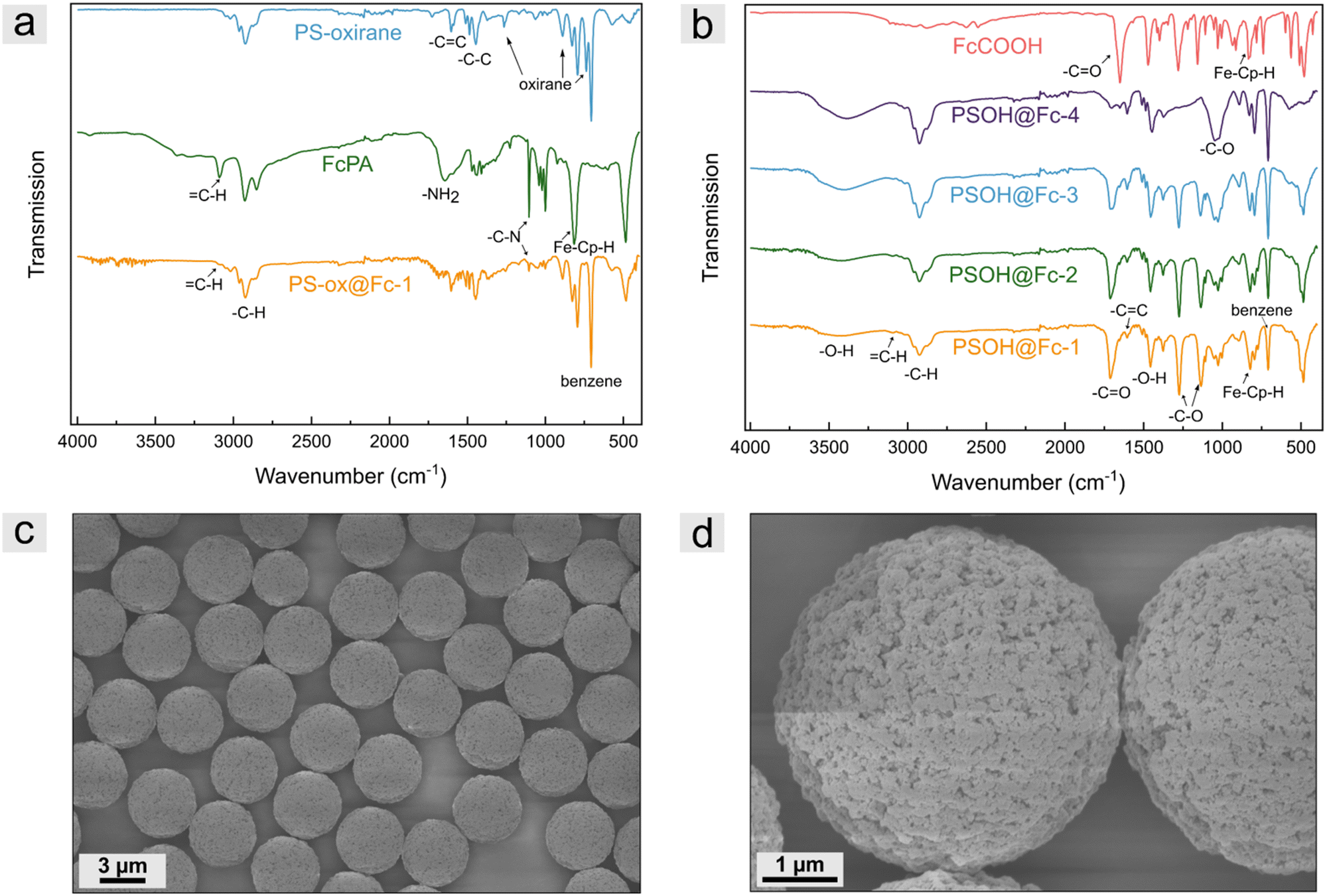 | ||
| Fig. 3 Infrared spectroscopic data of (a) PS-oxirane particles, FcPA, and representative PS-ox@Fc-1 particles and (b) PSOH@Fc particles with different degrees of functionalization and ferrocene carboxylic acid (FcCOOH) for comparison. (c) and (d) Representative scanning electron micrographs of highly functionalized PSOH@Fc-2 particles in different magnifications. | ||
The presence of the redox-responsive ferrocene moieties was also shown by the electrochemical addressability of the ferrocene units (Fig. S7, ESI†). For this purpose, cyclic voltammetry was performed in aqueous media using NaClO4 as the electrolyte. The particles were mixed with multi-walled carbon nanotubes (MWCNT) for a high contact area to a conductive material and drop-coated to a glassy carbon working electrode. Due to the low flexibility of the ferrocene molecules on the surface and thus the low contact ability with the conductive material, only low signal intensities were detected. Nevertheless, the particles of the two functionalization strategies could be electrochemically addressed. Scanning electron microscopy (SEM) measurements were performed to investigate the particle structure and morphology after functionalization. Fig. 3c and d, for example, depict micrographs of PSOH@Fc-2 particles. Compared to previous studies given in the introduction, by the presented functionalization strategy, no changes in terms of surface morphology or particle structure could be observed, which is a clear advantage compared to other template strategies so far.
Synthesis of iron oxide ceramics by calcination process of ferrocene-based precursors
The calcination of the different ferrocene-based particles PSOH@Fc, PS-ox@Fc, and PSDVB@PFMMA were investigated under different atmospheric and temperature conditions (Fig. 4a). All particle samples were investigated after thermal treatment in synthetic air with an oxygen-to-nitrogen ratio of 80 to 20% up to 580 °C. In addition, the influence of the oxygen flow and the temperature was demonstrated with polymer functionalized particles by investigation with higher synthetic air flow (40 compared to 20 mL min−1) and at different temperature maxima (580 °C and 800 °C). Furthermore, all three kinds of precursor materials were calcined under inert conditions in a nitrogen atmosphere for comparison and for obtaining other ceramics.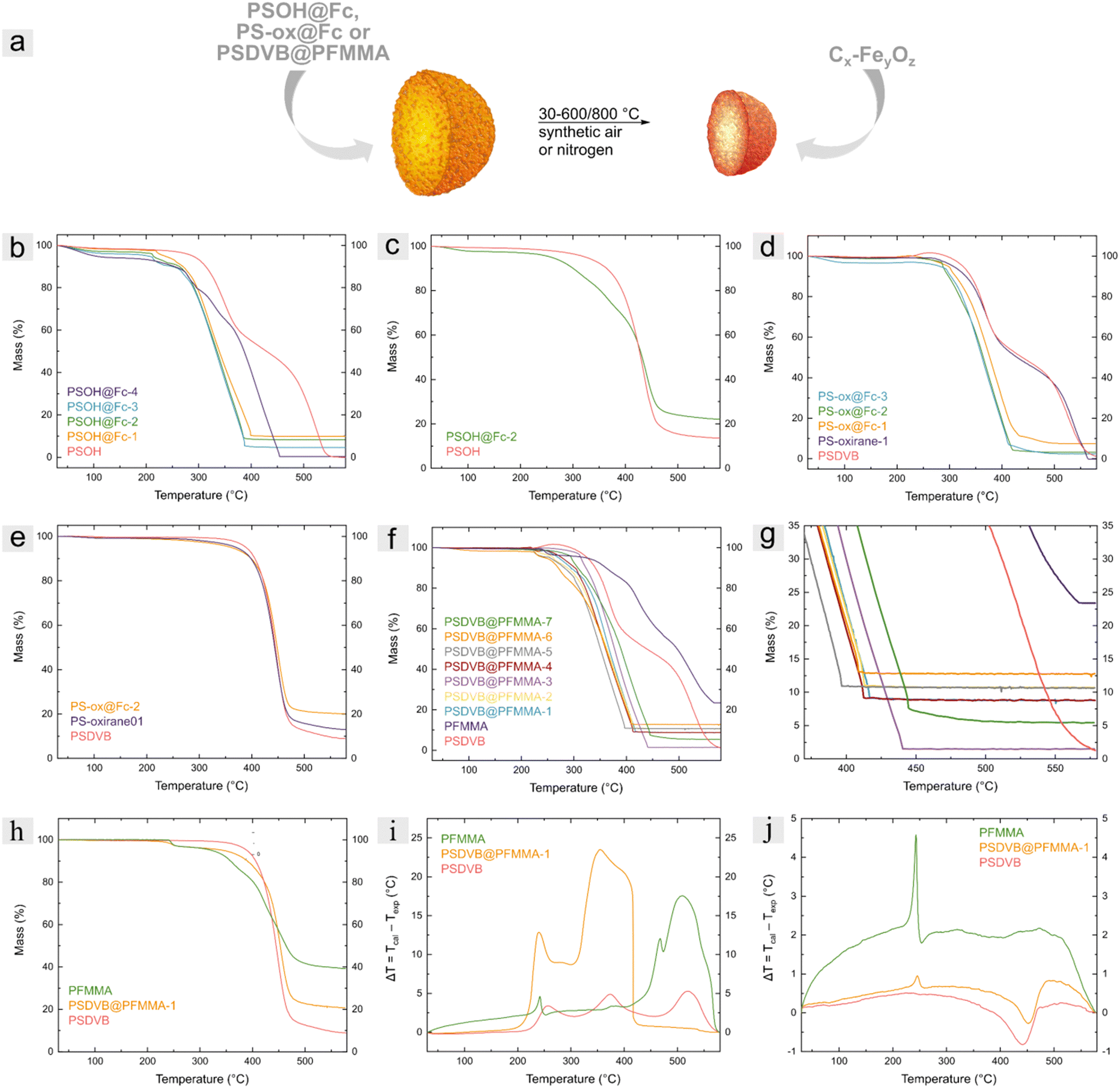 | ||
| Fig. 4 (a) Graphic representation of the synthesis of Cx–FeyOz ceramic particles beginning with different (poly)ferrocene-functionalized particles. Thermogravimetric analyses of (b) non-functionalized PSOH and PSOH@Fc particles in synthetic air and (c) nitrogen atmosphere, (d) PSDVB, PS-oxirane and PS-ox@Fc particles in synthetic air and (e) nitrogen atmosphere, (f) PSDVB particles, PSDVB@PFMMA particles and PFMMA in synthetic air atmosphere, (g) a partial spectrum of resulting mass (%) and (h) in nitrogen atmosphere conditions. (i) c-DTA data PSDVB particles, PSDVB@PFMMA particles, and PFMMA in synthetic and (j) nitrogen atmosphere. | ||
To investigate the influence of the thermal treatment, the mass loss during the reaction was detected using thermogravimetric analysis (TGA; Fig. 4b–h), and the difference between the calculated temperature and the sample temperature was measured using the calculated differential thermal analysis (c-DTA; Fig. 4i and j). The samples were heated with a heating rate of 10 K min−1. For comparison, the non-functionalized particles and the PFMMA homopolymer for PSDVB@PFMMA particles were considered. The ferrocene precursors could not be analyzed without immobilization on the surface due to the sublimation of the ferrocene compounds, leading to a significant error within TGA.
Thermogravimetric analysis of PSOH particles showed a two-step exothermic reaction starting at around 300 °C with a complete mass loss after 560 °C. After the modification process with the different ferrocene-based functionalizations, the onset temperature of PSOH@Fc particles shifted to lower temperatures between 210–225 °C for PSOH@Fc-1 to PSOH@Fc-3 and 258 °C for the less functionalized PSOH@Fc-4. This is attributed to the catalytic activity of iron, which accelerates the degradation of the carbon matrix, especially in the presence of oxygen.48–50 The catalytic influence of iron could be recognized by a lower offset temperature of the functionalized particles, too. The end of the mass loss was calculated for the PSOH@Fc-1 to PSOH@Fc-3 particles between 387–402 °C while PSOH@Fc-4 revealed an offset by 454 °C. The percentage of iron oxide residue was between 9.9 wt% for PSOH@Fc-1 and 0.4 wt% for PSOH@Fc-4. Because of small mass losses before 200 °C, a corrected value of the effective mass loss between 200 and 580 °C was considered, besides the residual mass (Table 2). The calcination under nitrogen atmosphere was obtained in a single, endothermic step for both functionalized and non-functionalized PSOH particles. Nevertheless, the catalytic activity of iron was observed and the onset temperature was shifted from 375 °C for PSOH particles to 269 °C for PSOH@Fc-2. On the other hand, the catalytic activity was lower due to the lower oxygen content induced only by the oxygen content of the particles. The observations were confirmed by calcining the PS-ox@Fc particles under the nitrogen and oxygen atmosphere. For example, the offset under synthetic air atmosphere was shifted from 564 °C by PS-oxirane to 413–431 °C for the PS-ox@Fc particles. A characteristic signal was observed for the PSDVB particles as an endothermic reaction starting at 220 °C due to oxidation of the double bonds.41 This double bond oxidation was not observed after epoxidation or functionalization of the particles, verifying again the covalent functionalization. The residue was determined to be between 7.5 wt% for PS-ox@Fc-1 and 2.5 wt% for PS-ox@Fc-3. Under nitrogen atmosphere, the PS-ox@Fc-2 particles exhibited a residue of 20.1 wt% and an onset temperature of 284 °C compared to 405 °C for PSDVB particles.
| Particle | Residual mass (%) | Effective mass loss (%) | Particle | Residual mass (%) | Effective mass loss (%) |
|---|---|---|---|---|---|
| PSOH@Fc-1a | 9.9 | 87.7 | PSDVB@PFMMA-3a | 1.5 | 98.4 |
| PSOH@Fc-2a | 8.3 | 88.1 | PSDVB@PFMMA-4a | 8.8 | 91.1 |
| PSOH@Fc-3a | 4.6 | 90.5 | PSDVB@PFMMA-5a | 10.6 | 88.5 |
| PSOH@Fc-4a | 0.4 | 92.9 | PSDVB@PFMMA-6a | 12.8 | 85.3 |
| PS-ox@Fc-1a | 7.5 | 92.1 | PSDVB@PFMMA-7a | 5.4 | 93.7 |
| PS-ox@Fc-2a | 3.3 | 95.7 | PSOH@Fc-2b | 22.1 | 75 |
| PS-ox@Fc-3a | 2.5 | 94.3 | PS-ox@Fc-2b | 20.1 | 78.9 |
| PFMMAa | 23.4 | 76.1 | PFMMAb | 39.2 | 60.8 |
| PSDVB@PFMMA-1a | 8.8 | 90.5 | PSDVB@ PFMMA-1b | 20.7 | 78.7 |
| PSDVB@PFMMA-2a | 10.7 | 89.1 |
For the consideration of the polymer functionalized particles, PFMMA was investigated first. Under a synthetic air atmosphere, PFMMA first revealed an exothermic signal at 240 °C, followed by a multistep degradation ending at 566 °C with a residual mass of 23.4 wt%. The onset temperature was found to be between 220 and 305 °C, with the particles with higher degrees of functionalization exhibiting a similar onset. However, the lower functionalized particles PSDVB@PFMMA-7 revealed an onset at 305 °C. The offset of the polymer functionalized particles could be observed between 395–445 °C compared to the 556 °C for PSDVB particles. In addition, the residues of iron oxides were detected between a minimum of 1.5 wt% for PSDVB@PFMMA-7 (containing 7% PFMMA) and a maximum of 12.8 wt% for PSDVB@PFMMA-6 (containing 51% PFMMA). Furthermore, the calcination process of PFMMA under a nitrogen atmosphere revealed an exothermic reaction at 240 °C followed by material degradation, leading to a residue of 39.2 wt%. However, the functionalized PSDVB@PFMMA-1 particles both revealed the exothermic reaction of PFMMA and the endothermic reaction of the PSDVB particles at about 440–450 °C in a single-step mass loss. The residue reached 20.7 wt% compared to 8.8 wt% under synthetic air due to the higher carbon content. Additionally, an increased oxygen flow from 20 to 40 mL min−1 as well as a higher end temperature at 800 °C did not lead to a significant change in the residual mass or the calcination process (Table S3, ESI†). A similar behavior was observed when the PFMMA-functionalized precursor ceramic was investigated using the PSDVB@PFMMA-1 precursor (Fig. 6 and Fig. S8, ESI†). The ceramics under nitrogen exhibited a size of 2.31 ± 0.06 μm and a rougher surface due to higher crystallite density compared to the surface-modified precursor ceramics. The size of the oxide ceramics exhibited 1.63 ± 0.05 μm with a similar surface morphology as the PS-ox@Fc-based materials. This could be attributed to the covalent bonding of the polymer in the precursor particles. In the case that a non-bound polymer was present within the samples, the porosity of the ceramic particles was reduced by a film-forming layer on the surfaces. Moreover, the presence of homopolymer resulted in non-particulate residues (Fig. S9–S11, ESI†). For this reason, the particulate and porous form of the ceramics could furthermore confirm the binding of the polymers to the surface.
Regarding the PFMMA content of the precursor, it could be observed that the TGA results led to an underestimation of the ferrocene content due to the sublimation of the ferrocene groups. While the exact determination of the ferrocene content of the PFMMA-based precursor was performed by UV Vis spectroscopy, the determination of the precursor containing OH-groups by this method turned out to be difficult. This was due to the changing refractive index of the particles depending on the OH groups, which complicated the adjustment of an isorefractive medium to prevent the scattering effects of the particles themselves. Nevertheless, using the PFMMA-containing precursors, it was demonstrated that despite underdetermination, the absolute content of ferrocenes (Table S1, ESI†) and the residual mass observed via TGA (Table 2) correlated.
Influence of atmospheres during calcination
It is well known that variations in conditions during the calcination process of iron-based precursors affected both the composition and the structure of the ceramics obtained.25,34,51,52 Scanning electron micrographs were used to provide information about the ceramic particles dependent on calcination conditions within this study. First, the materials based on surface-modified PS-ox@Fc-2 particles were examined (Fig. 5). For both calcination conditions, the high monodisperse properties of the precursors could be maintained. Moreover, strongly uniform ceramics without any defective particles could be observed. In the case of inert calcination conditions, the ceramics revealed 2.16 ± 0.05 μm size with an almost closed surface structure. Partly, small crystallites could be observed on the surface. In contrast, the surface morphology of the ceramics calcined under synthetic air was shown to be porous. These particles exhibited a significantly smaller size of 1.02 ± 0.03 μm. The CO2 formed during the decomposition of the organic particle matrix is expected to evaporate out of the particle's interior and thus shape the pore structure in particular. However, in comparison with the literature,1,2,25 it is evident that the covalent bonding of the ferrocene precursors throughout the polystyrene particle matrix significantly affected the uniformity and the surface structure. | ||
| Fig. 5 Scanning electron micrographs of ceramic particles resulting from PS-ox@Fc-2 precursors by synthesis in (a)–(c) nitrogen and (d)–(f) synthetic air. | ||
 | ||
| Fig. 6 Scanning electron micrographs of ceramic particles resulting from PSDVB@PFMMA-1 precursors by synthesis in (a)–(d) nitrogen and (e) and (f) synthetic air. | ||
To obtain information about the composition of the particles, energy dispersive X-ray spectroscopy was recorded for PSDVB@PFMMA-1 ceramics (Fig. 7a–c). The iron content of the particles calcined under inert conditions was 19 ± 2 w-% (mean value of 3 measurement results), and thus, significantly lower than under synthetic air with 36 ± 3 wt%. Nevertheless, the oxygen content was low (6 ± 1 wt%) and the carbon content was high (74 ± 3 wt%). This is due to the carbonization of the organic precursors, which resulted in carbon-embedded iron microparticles. The particles calcined under synthetic air revealed a low carbon content of 26 ± 3 wt% and a high oxygen content (36 ± 1 wt%).
 | ||
| Fig. 7 (a) Representative energy dispersive X-ray spectra of PSDVB@PFMMA-1 based ceramic particles and corresponding mapping images of the ceramic particles synthesized in (b) nitrogen and (c) synthetic air conditions. (d) X-ray diffractogram of calcined (1) PFMMA, (2) PSDVB@PFMMA-1, (3) PSOH@Fc-2, and (4) PS-ox@Fc-2 particles synthesized under synthetic air. (e) Raman spectra of calcined ceramics synthesized under synthetic air. | ||
Raman and X-ray diffraction (XRD) measurements were carried out to obtain more detailed information regarding the composition of the particles (Fig. 7d and e). The crystallographic data for the refined phases were taken from the Pearson database53 and the Raman active phonons were attributed according to Chamritski et al.54 The results of the investigated samples are summarized in Table S4 (ESI†).
X-ray diffractograms of ceramics synthesized under a nitrogen atmosphere revealed mainly elemental iron (besides amorphous carbon) and low quantities of oxide-containing iron species, according to the literature (Fig. S12, ESI†).25 An exception was the strongly hydrophilized PSOH@Fc particles, which, in addition to carbon, consisted of only 7 wt% elemental iron. Due to the high amount of hydroxy groups, 93 wt% of the crystalline iron-based species were Fe3O4. Raman spectroscopy (Fig. S13, ESI†) showed no active phonon signals of any iron oxides but a high amorphous carbon content. During the carbonization of the iron precursors, small amounts of iron oxides could be formed by the intrinsic oxygen of the particles or the PFMMA polymer. However, these were mainly reduced by the high carbon content:
| Fe3O4 + 2 C → 3 Fe + 2 CO2 |
| Fe3O4 + 2 C → 3 FeO + CO |
The composition of the ceramics calcined under synthetic air was strongly dependent on the precursor used (Fig. 7d and e). For example, ceramics based on both oxidized particles PS-ox@Fc and PSOH@Fc were composed of pure hematite. Due to the high oxygen content of the materials, which was homogeneously distributed within the precursor particle, the initially formed magnetite was oxidized. A difference between the more hydrophilic PSOH@Fc and the PS-ox@Fc precursors could not be observed. Note that maghemite (Fe2.67O4) and magnetite (Fe3O4) cannot be differentiated in XRD,25 but by using Raman spectroscopy. Based on Mössbauer spectra in the literature, which allowed the determination based on investigation of the hyperfine structure in similar calcination studies25 and the Raman spectra performed here, the mass content determination by XRD was related to Fe3O4. The ceramics based on polymer precursor PSDVB@PFMMA also consisted of some hematite (Fe2O3). The major part of the mixture consisted of maghemite and magnetite, as confirmed by XRD and Raman measurements. The high carbon content of the non-hydrophilized PSDVB particles, in combination with polymer functionalization, led to the formation of maghemite. Simultaneously, the high carbon content could have performed as a reducing agent to reduce significant amounts of Fe2O3, e.g., as follows:
| 6 Fe2O3 + C → 4 Fe3O4 + CO2 |
The distribution of oxygen within the precursor particles was found to have a significant role in the resulting ceramics. However, when the synthetic air flow was increased from 20 mL min−1 to 40 mL min−1 during calcination, no compositional difference to higher Fe2O3 content was observed in Raman spectroscopy (Fig. S14, ESI†). Additionally, larger crystallites were observed during calcination under oxygen-rich conditions (Table S4, ESI†).
Degree of precursor functionalization and morphology
The influence of the different degrees of functionalization of the precursors was investigated by scanning electron microscopy. First, the surface-modified particles were studied for PSOH@Fc1–4 (Fig. 8). It was found that the degree of functionalization of the precursors affects the size of the ceramic particles. A higher degree of functionalization resulted in larger ceramic particles. The correlation between size and degree of functionalization showed a high linearity within the individual functionalization procedures (Fig. 9j and Table S5, ESI†). The use of different precursors led to slight deviations regarding the linearity compared to other kinds of precursors, which was due to the different compositions of iron oxides. Overall, this method enables the control of the size of the ceramics, especially by using the same precursor. | ||
| Fig. 8 Scanning electron micrographs of ceramic particles based on PSOH@Fc precursor with different degrees of functionalization: (a) and (b) PSOH@Fc-1, (c) and (d) PSOH@Fc-2, (e) and (f) PSOH@Fc-3, (g) and (h) PSOH@Fc-4. | ||
 | ||
| Fig. 9 (a)–(i) Scanning electron micrographs of ceramic particles based on PSDVB@PFMMA precursor with different degrees of functionalization: (a)–(c) PSDVB@PFMMA-3 (residual mass of 1.5%), (d)–(f) PSDVB@PFMMA-4(residual mass of 8.8%), (g)–(i) PSDVB@PFMMA-2 (residual mass of 10.7%). (j) Overview of resulting particle sizes dependent on the residual mass and the different types of precursors. | ||
For the polymer functionalized precursors, the degree of functionalization also affected the size of the particles (Fig. 9a–i). In addition, a change in the surface morphology could be observed. In addition to the degree of functionalization, this could be attributed to the polymer structure on the surface. For example, longer polymer chains lead to a more closed surface (Fig. S15, ESI†). This enabled targeted adjustment of the size and porosity of the ceramics.
Previous studies have shown that ceramization leads to hollow materials, for example, in cubes or nanoparticles.1 Our investigations also revealed a hollow structure of the microscale particles. Some broken particles clearly showed a hollow structure (Fig. 10a). Moreover, the hollow particles could be shown particularly distinctly on already defective precursors. As described above, the structure of PS-ox particles was destroyed during the reaction with FcPA in THF (Fig. S2, ESI†). Consequently, the resulting ceramics also showed a high percentage of defective particles, where the hollow character of the ceramics can be seen (Fig. 10b and c). This is due to the diffusion of the molecules to the outside during calcination, which is induced by the formed gases during thermal treatment.1 It can be assumed based on previous studies that solid particles without hollow-structure were formed during calcination under inert conditions. This is due to the absence of diffusion of CO2 to the outside during thermal treatment. In particular, the absence of porous structure of the ceramics under inert conditions confirms this assumption further.55
 | ||
| Fig. 10 Scanning electron micrographs proving a hollow structure by (a) broken ceramic particle based on PS-ox@Fc-2 and (b) and (c) broken particles based on PS-ox@Fc-4. | ||
For the polymer-functionalized precursor particles, this could also be shown by changing the chain length, and thus, the degree of functionalization. While PSDVB@PFMMA-5 contained 39% PFMMA, further monomer addition via sequential anionic polymerization yielded longer chains, and thus, a higher degree of functionalization (51% PFMMA).41 The ceramics exhibited a highly ordered structure at the lower functionalization degree, while the breakdown of the structure occurred at higher PFMMA contents. The surface morphology was significantly denser. The higher degree of functionalization results in the failure to form a sufficient number of pore channels. As a result, the gases formed could not escape without fracturing the particles. This also shows that an excessive degree of functionalization of the precursor is not effective for the preparation of ceramic particles.
Electrochemical characterization
It is well known that the structural properties of ceramics affect the reaction rate of electrode materials. Cyclic voltammetry measurements were carried out to prove that the synthesized particles are in principle suitable for electrochemical applications. For this purpose, measurements in an alkaline solution (1 M NaOH) were performed in an aqueous solution representing standard conditions where iron-based ceramics are used in electrochemical devices.56Despite multiple electrochemical investigations of the system of iron and iron oxides, the discussed mechanism is often different, but most authors agree with the described evaluation.20,57–59 The ceramics based on the PSOH@Fc particles synthesized in synthetic air revealed in the first cycle a broad cathodic signal overlapping of several reduction signals between −0.84 and −1.04 V (vs. Fc/Fc+), which could be associated with the formation of Fe(OH)2 through different intermediate compounds (Fig. 11a).58 The following sharp signal at −1.48 V showed the reduction of Fe2+ to Fe0 and was shifted to higher potentials (−1.33 V) in the second cycle. This was due to the formation of a passivation layer during cycling in more positive potentials at high pH values,60,61 which is of significant importance for the stability of batteries.62 On the other hand, the formation of the passivation layer led to a decrease in capacitance in the following cycles. After reversing the potential, the anodic signal occurred at −0.84 V in the first cycle and shifted to −0.92 V in the following, associated with the oxidation of Fe0 to Fe3+.59 The oxidation of Fe0 to Fe2+ could not be observed in an alkaline solution but under slightly acidic conditions with NaClO4 as a supporting electrolyte (0.1 M in aqueous solution, Fig. 11b). Besides the oxidation to Fe2+ by −0.91 V, further oxidation of Fe2+ to Fe3+ could be detected at 0.21 V. While the oxidation increased with continued cycling, an increase in the reduction signal to Fe2+ was recorded that could not be observed in the first cycle. The electrochemical investigations of the ceramics obtained from PS-ox@Fc precursors exhibited a similar behavior (Fig. S16a and b, ESI†).
 | ||
| Fig. 11 Cyclic voltammetric investigation of (a) and (b) PSOH@Fc-2 and (c) and (d) PSDVB@PFMMA-1 based ceramics synthesized in synthetic air conditions. The measurements in (a) and (c) were carried out in an alkaline solution (1 M NaOH) and (b) and (d) were measured in 0.1 M NaClO4 with a scan rate of 20 mV s−1. The 1, 2, 3, 5, 10, and 20 cycles were shown, respectively. | ||
The ceramics based on PSDVB@PFMMA particles containing a lower amount of Fe3+ showed slight differences (Fig. 11c). The cathodic signals indicating the formation of Fe(OH)2 were found to be weak in the first cycle and significant only from the second cycle onwards. Reduction to Fe0 was the dominant reaction at −1.59 V in the first cycle and shifted to −1.33 V in the following. In the anodic region, a slight oxidation of Fe0 to Fe2+ was initially observed in the first cycle at −1.29 and −1.17 V, which could not be detected in the following cyclic investigations. The oxidation of Fe0 to Fe3+ at −0.78 V, which initially showed an increase and a shift to −0.87 V and then a decrease in intensity as in all ceramic kinds. Larger differences between the two types of ceramics were particularly evident in the measurements in NaClO4 (Fig. 11d). In the first cycle, only a reduction of Fe2+ to Fe0 could be observed at −1.28 V, indicating an absence of formation of the passivation layer. This was confirmed by the lack of reduction in capacitance in the following cycles, where an increase in signal intensity was observed with further cycling. Indeed, this concerns both the reduction to Fe0 and the formation of Fe(OH)2 at −0.67 V, which, however, was only slightly detected. The anodic region showed both the formation of Fe(OH)2 (−0.90 V) and FeOOH (−0.64 V) increasing initially. The oxidation of Fe2+ to Fe3+ was not detectable in the first cycle but also increased at 0.18 to 0.23 V (at the following cycles) with continued cycling, and in addition, led to the detection of the associated reduction of Fe3+ to Fe2+ at 0.13 to 0.10 V. Cyclic voltammetric measurements of the ceramics calcined under nitrogen atmosphere revealed a low addressability of the iron species (Fig. S16c–e, ESI†), which was due to high amorphous carbon content and the closed surface structure. Due to the lack of porous structure, there was no accessibility to the iron-containing species within the particles. Therefore, only the surface crystallites can be addressed to a limited extent. This showed the importance of the structure for electrochemical applicability. Further studies could investigate the material's capacitance in more detail and the influence of the different sizes and structures on electrochemical properties to demonstrate the performance of battery materials.
Conclusions
In this work, we have presented a universal method for the preparation of iron and iron oxide microparticles with tailored structural morphology, size, and composition. First, PSDVB particles were modified with ferrocene-containing precursors by covalent bonding. In addition, particles with PFMMA functionalization were used. Under different calcination conditions, such as gas flow, atmosphere conditions, and temperature, the particles were ceramicized. Starting from identical precursors, this affected the composition in terms of carbon-iron or carbon-iron oxide particles, as described in the literature. The different precursor methods resulted in different iron oxide compositions of hematite (Fe2O3) for surface functionalization with hydrophilic groups or maghemite (Fe2.67O4) and magnetite (Fe3O4) for polymer-functionalized precursors. Depending on the degree of functionalization of the precursors, the size of the particles could be controlled. The polymer functionalized precursors enabled influence and control over the surface morphology and porosity. High monodispersity and uniformity could be achieved, which can be attributed to the covalent bonding of the ferrocene molecules compared to the literature. Cyclic voltammetric measurements were able to show the addressability of the ceramics and the importance of the porous structure. The measurements especially of the particles calcined under oxygen atmosphere, demonstrated the suitability of the materials for energy applications.Author contributions
M. G. and D. S. conceived the idea, and D. S. performed the experiments and the majority of analyses. O. J. carried out XRD measurements and data acquisition, R. L. carried out TGA, M. G., and D. S. carried out the data interpretations and wrote the manuscript, and D. S., G. K. and M. G. did the manuscript revision. M. G. was in charge of the project administration and supervision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Claudia Fink Straube from the Leibniz Institute for New Materials (INM) for CHNO measurements and Prof. Volker Presser from the INM for the possibility of using SEM and Raman spectroscopy. The authors thank Metrohm AG (Switzerland) for donating the PSDVB and PSOH particles. Instrumentation and technical assistance for this work were provided by the Service Center X-ray Diffraction, with financial support from Saarland University and the German Science Foundation (project number INST 256/349-1)Notes and references
- W. Xie, Z. Guo, F. Gao, Q. Gao, D. Wang, B. S. Liaw, Q. Cai, X. Sun, X. Wang and L. Zhao, Theranostics, 2018, 8, 3284–3307 CrossRef CAS PubMed.
- K. El-Boubbou, R. Ali, H. Al-Zahrani, T. Trivilegio, A. H. Alanazi, A. L. Khan, M. Boudjelal and A. Alkushi, Sci. Rep., 2019, 9 Search PubMed.
- G. D. Moeser, K. A. Roach, W. H. Green, P. E. Laibinis and T. A. Hatton, Ind. Eng. Chem. Res., 2002, 41, 4739–4749 CrossRef CAS.
- P. Pageni, P. Yang, M. Bam, T. Zhu, Y. P. Chen, A. W. Decho, M. Nagarkatti and C. Tang, Biomaterials, 2018, 178, 363–372 CrossRef CAS PubMed.
- N. Kang, J. H. Park, J. Choi, J. Jin, J. Chun, I. G. Jung, J. Jeong, J.-G. Park, S. M. Lee, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2012, 51, 6626–6630 CrossRef CAS PubMed.
- B. Koo, H. Xiong, M. D. Slater, V. B. Prakapenka, M. Balasubramanian, P. Podsiadlo, C. S. Johnson, T. Rajh and E. V. Shevchenko, Nano Lett., 2012, 12, 2429–2435 CrossRef CAS PubMed.
- S.-H. Yu, D. E. Conte, S. Baek, D.-C. Lee, S.-K. Park, K. J. Lee, Y. Piao, Y.-E. Sung and N. Pinna, Adv. Funct. Mater., 2013, 23, 4293–4305 CrossRef CAS.
- L. Wang, L. Deligniere, S. Husmann, R. Leiner, C. Bahr, S. Zhang, C. Dun, M. M. Montemore, M. Gallei, J. J. Urban, C. Kim and V. Presser, Nano Res., 2023, 16, 9352–9363 CrossRef CAS.
- Y. Xia, T. Zhao, X. Zhu, Y. Zhao, H. He, C.-T. Hung, X. Zhang, Y. Chen, X. Tang, J. Wang, W. Li and D. Zhao, Nat. Commun., 2021, 12 CAS.
- S. Arnold, L. Wang and V. Presser, Small, 2022, 2107913 CrossRef CAS PubMed.
- Z. Qiang, Y.-M. Chen, B. Gurkan, Y. Guo, M. Cakmak, K. A. Cavicchi, Y. Zhu and B. D. Vogt, Carbon, 2017, 116, 286–293 CrossRef CAS.
- Y. S. Choi, W. Choi, W.-S. Yoon and J. M. Kim, ACS Nano, 2022, 16, 631–642 CrossRef CAS PubMed.
- Q. Li, C. W. Kartikowati, S. Horie, T. Ogi, T. Iwaki and K. Okuyama, Sci. Rep., 2017, 7, 9894 CrossRef PubMed.
- G. F. Dewald, Z. Liaqat, M. A. Lange, W. Tremel and W. G. Zeier, Angew. Chem., Int. Ed., 2021, 60, 17952–17956 CrossRef CAS PubMed.
- Y. Domi, H. Usui, K. Sugimoto and H. Sakaguchi, Energy Technol., 2019, 7, 1800946 CrossRef.
- L. Zhang, H. B. Wu and X. W. D. Lou, Adv. Energy Mater., 2014, 4, 1300958 CrossRef.
- G. Sharma and P. Jeevanandam, RSC Adv., 2013, 3, 189–200 RSC.
- W. Cheng, K. Tang, Y. Qi, J. Sheng and Z. Liu, J. Mater. Chem., 2010, 20, 1799 RSC.
- B. Banerji, S. K. Pramanik, S. Mandal, N. C. Maiti and K. Chaudhuri, RSC Adv., 2012, 2, 2493–2497 RSC.
- C. Zhang, Z. Chen, H. Wang, Y. Nie and J. Yan, ACS Appl. Nano Mater., 2021, 4, 8744–8752 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- X. Li, B. Zhang, C. Ju, X. Han, Y. Du and P. Xu, J. Phys. Chem. C, 2011, 115, 12350–12357 CrossRef CAS.
- B. Wang, H. B. Wu, L. Zhang and X. W. D. Lou, Angew. Chem., Int. Ed., 2013, 52, 4165–4168 CrossRef CAS PubMed.
- F.-X. Ma, H. Hu, H. B. Wu, C.-Y. Xu, Z. Xu, L. Zhen and X. W. David Lou, Adv. Mater., 2015, 27, 4097–4101 CrossRef CAS PubMed.
- D. Amara, J. Grinblat and S. Margel, J. Mater. Chem., 2010, 20, 1899–1906 RSC.
- B. Wang, J. S. Chen, H. B. Wu, Z. Wang and X. W. Lou, J. Appl. Chem. Sci., 2011, 133, 17146–17148 CAS.
- D. Amara and S. Margel, Nanotechnol. Rev., 2013, 2, 333–357 CAS.
- R. Petersen, D. A. Foucher, B.-Z. Tang, A. Lough, N. P. Raju, J. E. Greedan and I. Manners, Chem. Mater., 1995, 7, 2045–2053 CrossRef CAS.
- K. R. Thomas, A. Ionescu, J. Gwyther, I. Manners, C. H. W. Barnes, U. Steiner and E. Sivaniah, J. Appl. Phys., 2011, 109, 073904 CrossRef.
- J. Elbert, H. Didzoleit, C. Fasel, E. Ionescu, R. Riedel, B. Stühn and M. Gallei, Macromol. Rap. Commun., 2015, 36, 597–603 CrossRef CAS PubMed.
- M. Mazurowski, M. Gallei, J. Y. Li, H. Didzoleit, B. Stühn and M. Rehahn, Macromolecules, 2012, 45, 8970–8981 CrossRef CAS.
- P. Pageni, P. Yang, Y. P. Chen, Y. Huang, M. Bam, T. Zhu, M. Nagarkatti, B. C. Benicewicz, A. W. Decho and C. Tang, Biomacromolecules, 2018, 19, 417–425 CrossRef CAS PubMed.
- P. Yang, P. Pageni, M. A. Rahman, M. Bam, T. Zhu, Y. P. Chen, M. Nagarkatti, A. W. Decho and C. Tang, Adv. Healthcare Mater., 2019, 8, e1800854 CrossRef PubMed.
- D. Scheid, G. Cherkashinin, E. Ionescu and M. Gallei, Langmuir, 2014, 30, 1204–1209 CrossRef CAS PubMed.
- D. Scheid, D. Stock, T. Winter, T. Gutmann, C. Dietz and M. Gallei, J. Mater. Chem. C, 2016, 4, 2187–2196 RSC.
- J. J. Bowen, S. Mooraj, J. A. Goodman, S. Peng, D. P. Street, B. Roman-Manso, E. C. Davidson, K. L. Martin, L. M. Rueschhoff, S. N. Schiffres, W. Chen, J. A. Lewis and M. B. Dickerson, Mater. Today, 2022, 58, 71–79 CrossRef CAS.
- L. M. Rueschhoff, L. A. Baldwin, R. Wheeler, M. J. Dalton, H. Koerner, J. D. Berrigan, N. M. Bedford, S. Seifert, M. K. Cinibulk and M. B. Dickerson, ACS Appl. Nano Mater., 2018, 2, 250–257 CrossRef.
- A. Seubert, J. T. R. Aeschlimann and O. Michael, (EP3721998A1), 2020.
- H. Vapnik, J. Elbert and X. Su, J. Mater. Chem. A, 2021, 9, 20068–20077 RSC.
- T. Winter, M. Bitsch, F. Müller, S. Voskian, T. A. Hatton, K. Jacobs, V. Presser and M. Gallei, ACS Appl. Polym. Mater., 2021, 3, 4651–4660 CrossRef CAS.
- D. Schmitt, S. M. Abdel-Hafez, M. Tummeley, V. Schünemann, M. Schneider, V. Presser and M. Gallei, Macromolecules, 2023, 56, 7086–7101 CrossRef CAS.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS.
- R. W. Cheary, A. A. Coelho and J. P. Cline, J. Res. Natl. Inst. Stand. Technol., 2004, 109, 1–25 CrossRef CAS PubMed.
- S. Mehlhase, C. G. Schäfer, J. Morsbach, L. Schmidt, R. Klein, H. Frey and M. Gallei, RSC Adv., 2014, 4, 41348–41352 RSC.
- S. Schöttner, R. Hossain, C. Rüttiger and M. Gallei, Polymers, 2017, 9, 491 CrossRef PubMed.
- W. A. Patterson, Anal. Chem., 1954, 26, 823–835 CrossRef CAS.
- J. A. Kramer and D. N. Hendrickson, Inorg. Chem., 1980, 19, 3330–3337 CrossRef CAS.
- H. B. Dunford, Free Radic. Biol. Med., 1987, 3, 405–421 CrossRef CAS PubMed.
- J. A. Zazo, A. F. Fraile, A. Rey, A. Bahamonde, J. A. Casas and J. J. Rodriguez, Catal. Today, 2009, 143, 341–346 CrossRef CAS.
- M. Tepluchin, M. Casapu, A. Boubnov, H. Lichtenberg, D. Wang, S. Kureti and J. D. Grunwaldt, Chem. Cat. Chem., 2014, 6, 1763–1773 CAS.
- G. Mera, M. Gallei, S. Bernard and E. Ionescu, Nanomaterials, 2015, 5, 468–540 CrossRef CAS PubMed.
- C. Zhou, C. Fasel, R. Ishikawa, M. Gallei, Y. Ikuhara, S. Lauterbach, H.-J. Kleebe, R. Riedel and E. Ionescu, J. Eur. Ceram. Soc., 2017, 37, 5193–5203 CrossRef CAS.
- Pearson's Crystal Data: Crystal Structure Database for Inorganic Compounds, 2022/23.
- I. Chamritski and G. Burns, J. Phy. Chem. B, 2005, 109, 4965–4968 CrossRef CAS PubMed.
- D. Amara and S. Margel, J. Mater. Chem., 2011, 21, 15764–15772 RSC.
- A. Accogli, L. Bertoli, G. Panzeri, E. Gibertini, R. Pesce, G. Bussetti and L. Magagnin, ACS Omega, 2021, 6, 26880–26887 CrossRef CAS PubMed.
- R. S. Schrebler Guzmán, J. R. Vilche and A. J. Arvía, Electrochim. Acta, 1979, 24, 395–403 CrossRef.
- S. T. Amaral, E. M. A. Martini and I. L. Müller, Corros. Sci., 2001, 43, 853–879 CrossRef CAS.
- K. Duschek, M. Uhlemann, H. Schlörb, K. Nielsch and K. Leistner, Electrochem. Commun., 2016, 72, 153–156 CrossRef CAS.
- I. Song, D. Gervasio and J. H. Payer, J. Appl. Electrochem., 1996, 26, 1045–1052 CrossRef CAS.
- B. Philippe, M. Valvo, F. Lindgren, H. Rensmo and K. Edström, Chem. Mater., 2014, 26, 5028–5041 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, Comput. Mater., 2018, 4, 15 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01131c |
| This journal is © The Royal Society of Chemistry 2024 |