
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional polysaccharide-coated SPIONs for in vitro mRNA delivery in breast cancer cells†
Olga
Tsave
a,
Maria
Psarrou
bc,
Georgia
Kastrinaki
ad,
Eleni
Papachristou
e,
Rigini
Papi
e,
Vassilios
Zaspalis
ad,
Lori
Nalbandian‡
d,
Charalampos
Sarafidis
 f,
Theodora
Choli-Papadopoulou
e,
Maria
Vamvakaki
bc and
Christos
Chatzidoukas
*a
f,
Theodora
Choli-Papadopoulou
e,
Maria
Vamvakaki
bc and
Christos
Chatzidoukas
*a
aDepartment of Chemical Engineering, Aristotle University of Thessaloniki (AUTH), Thessaloniki, 54124, Greece. E-mail: chatzido@auth.gr; Tel: +30 2310 99 6167
bDepartment of Materials Science and Technology, University of Crete, Vasilika Vouton, 70013, Heraklion, Crete, Greece
cInstitute of Electronic Structure and Laser, Foundation of Research and Technology-Hellas, Vasilika Vouton, 70013, Heraklion, Crete, Greece
dChemical Process & Energy Resources Institute (CPERI), Centre for Research & Technology Hellas (CERTH), Thessaloniki, Thermi, Greece
eDepartment of Chemistry, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece
fDepartment of Physics, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece
First published on 11th May 2024
Abstract
RNA-based therapeutics have recently attracted great attention as a novel platform for the prevention and treatment of several pathological conditions, including COVID-19, cancer, diabetes, cardiovascular diseases, and others. However, despite their advantages compared to formerly established treatment modalities, which include an increased effectiveness and specificity and reduced toxicity, the efficient delivery of nucleic acids into the cells remains a major challenge. In the present study, novel mRNA carriers, based on superparamagnetic iron oxide nanoparticles (SPIONs) functionalized with modified natural polysaccharide, were designed and synthesized. More specifically, two polysaccharide derivatives, namely oxidized dextran and quaternized chitosan, bearing aldehyde and cationic quaternary ammonium salt groups, respectively, were deposited onto the SPION surface. The hybrid nanoparticles were characterized in terms of their size, surface charge, magnetization and polymer content, and were next used to bind green fluorescent protein–mRNA and produce mRNA delivery vehicles. Biological assays on the T47D breast cancer cell line were employed to assess the nucleic acid binding effectiveness and cellular uptake of the nanoparticles and the cell proliferation, viability, migration and protein expression of the SPION treated cells. The hybrid particles coated with oxidized dextran showed higher stability, mRNA binding capacity, via Schiff base formation, and gene transfection efficiency, due to their smaller size, high polymer content and the effective cleavage of the polymer–gene covalent bonds in the acidic cancer cell compartment. The superior performance of these gene vectors, compared to the well-established cationic nanoparticle-gene complexes, render them highly attractive as mRNA therapeutics for further testing in (pre)clinical in vivo studies.
1. Introduction
Cancer is a non-communicable disease and is the second leading cause of death worldwide.1,2 Breast cancer is the most common malignancy in women, with a total of 1.7 million new cases and ∼522![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths being reported worldwide since 2008, corresponding to an increase of the mortality rate of ∼18%. These epidemiological data clearly reflect the urgent need for efficient early diagnostic, preventive and treatment modalities.3 To date, the treatment of breast cancer has been mainly based on local surgery and radiation therapy in combination with drug administration.4–6 Although a patient's survival rate has steadily increased over the past few decades, systemic chemotherapy often results in numerous side effects, including chemoresistance and toxicity towards other healthy tissues, which are responsible for most cases of relapse.7 To improve the efficiency of breast cancer therapy and effectively minimize its harmful side effects on healthy tissues and organs, new therapeutic strategies are urgently required.
000 deaths being reported worldwide since 2008, corresponding to an increase of the mortality rate of ∼18%. These epidemiological data clearly reflect the urgent need for efficient early diagnostic, preventive and treatment modalities.3 To date, the treatment of breast cancer has been mainly based on local surgery and radiation therapy in combination with drug administration.4–6 Although a patient's survival rate has steadily increased over the past few decades, systemic chemotherapy often results in numerous side effects, including chemoresistance and toxicity towards other healthy tissues, which are responsible for most cases of relapse.7 To improve the efficiency of breast cancer therapy and effectively minimize its harmful side effects on healthy tissues and organs, new therapeutic strategies are urgently required.
The development of RNA-based pharmaceuticals has emerged as a novel platform for the prevention and treatment of numerous pathological conditions, including COVID-19, cancer, diabetes, heart disease, and others.8–10 To date, these approaches have been mainly based on RNAi gene regulation mechanisms, microRNA modulators, long-non coding RNAs and siRNA drugs.8 RNA-based therapies are expected to prevail over other already established antibody-based cancer treatments (e.g., herceptin/trastuzumab delivery in female breast cancer),11–13 which suffer from ineffective targeting to the cancer tumor sites, significant side effects, including undesired immune-responses and chemoresistance, and high production costs.14 The induction of antibody synthesis by the patient's cellular synthetic mechanism, by the administration of the appropriate mRNA molecules instead of the antibody medication, has been recently applied to alleviate the above weaknesses via a personalized healthcare approach.
Despite the great potential of nucleic acid-based therapies for the treatment of a broad array of important diseases, including breast cancer, the effective delivery of the gene to the target destination remains a fundamental challenge. Various non-viral gene vectors have been proposed for the efficient complexation and transfection of the nucleic acids within the cytosolic area; however certain issues, such as the side effects and the toxicity of the vector, the bioavailability and the stability of the gene-vector complex, the non-effective escape of the gene from the carrier, as well as the non-biodegradability of the vector, hinder their clinical application.15
The majority of gene carriers are based on positively charged synthetic or natural macromolecules, which can interact electrostatically with the negatively charged genes, to form polyplexes. Cationic synthetic polymers of various molecular weights and architectures, including poly(ethylene imine),16,17 poly(beta amino esters),18,19 polyamidoamine dendrimers20,21 and poly(2-(dimethylamino)ethyl methacrylate),22,23 have been extensively used in gene delivery applications; however their cytotoxicity, even at low polymer concentrations, high cost, harsh and complicated synthetic methodologies have hindered their clinical utility. On the other hand, natural polysaccharides offer important advantages, owing to their abundance, lack of inherent toxicity, being often FDA approved, and their facile chemical modification methods. Chitosan, collagen and gelatin have been explored as nonviral natural gene carriers, because of the cationic groups along the polymer backbone, which allow their effective interaction with RNA constructs.24–26 Despite the extensive use of electrostatic interactions between the oppositely charged macromolecules as a facile and efficient method for gene-carrier complexation, the stability of the polyplexes in the bloodstream and the mechanism of gene release from the polyplex within the cell remain unclear and are still under investigation.27
During the last few years, hybrid nanoparticle-based therapeutics have gained increased attention as promising candidates for diagnosis and targeted drug delivery.28–34 Among them, superparamagnetic iron oxide nanoparticles (SPIONs) are particularly advantageous for clinical use, due to their multifunctionality and biocompatibility.35 For instance, hyperthermia, using tumor-accumulated particles in the presence of an external alternating magnetic field, and magnetic drug targeting (MDT), using SPION–drug conjugates, have been reported.36 Moreover, SPIONs can serve as theranostic agents, combining imaging and therapy in precision medicine.37 Therefore, the design and development of magnetic nanocarriers conjugated with mRNA molecules that encode the production of antibodies by the patient (e.g., trastuzumab) is a highly promising approach for the treatment of breast cancer.
Despite the numerous reports in the literature addressing promising nanoparticle types for gene/drug delivery,38–40 only very few of them have been employed in clinical use to date.41–44 The application of SPIONs in biology and nanomedicine requires the nanoparticles to be nontoxic and biocompatible, while exhibiting good aqueous dispersion under physiological conditions and increased stability in vivo.45 They are also required to effectively bind biomolecules, such as genes and antibodies, or small organic molecules, such as drugs and active compounds. The colloidal stability of the construct depends strongly on the size of the particles, which must be sufficiently small to prevent their fast precipitation in the medium, whereas superparamagnetic behavior is observed for nanoparticles with an average size of less than 20 nm.46 The surface chemistry of the SPIONs plays a crucial role in their biocompatibility, stability as well as their binding properties. To control their surface properties (e.g., surface charge and functional groups) SPIONs are often coated with small molecules or biocompatible polymers,47 including polysaccharides (dextran, chitosan, and others). Interestingly, dextran coated SPIONs, such as FERIDEX, have been approved by the FDA for clinical use as MRI contrast agents. The hydroxyl-rich chemical structure of dextran allows its facile oxidation to bear aldehyde groups, which can bind the desired bioactive molecules through covalent bonds. In this context, Tseng et al. have used oxidized dextran (Ox-dextran) to chemically bind cetuximab and spermine onto the polysaccharide via Schiff base formation.33,48–51 However, to the best of our knowledge Ox-dextran has not been employed so far for the binding of mRNA. Another natural polymer that has been extensively used to coat the surface of iron oxide nanoparticles is chitosan.52–56 Chitosan has several advantages including low toxicity, low immunogenicity, and good biocompatibility; however its poor water solubility under neutral conditions is an important limitation for biomedical use.57 To overcome this issue chitosan is often derivatized by various modification routes.51 A water-soluble synthetic derivative of chitosan is quaternized chitosan (Q-chitosan), which bears permanent positively charged moieties along the polymer chain enabling the binding of negatively charged macromolecules, such as nucleic acids.57,58 Despite the superior physicochemical characteristics of Q-chitosan, its use in gene delivery is limited, and to the best of our knowledge, it has not been reported so far for the functionalization of SPIONs.59–61
Given the plethora of available materials and synthetic approaches as well as the complexity of the biological/molecular environment and their diverse uses, certain design criteria should be considered for the development of effective SPION-based mRNA delivery systems. In this work, we aim to elucidate the effect of the binding mode, covalent vs. ionic, of nucleic acids onto hybrid polysaccharide-coated SPIONs, on the efficiency of mRNA-based delivery systems. For this, two natural polysaccharide derivatives, namely Ox-dextran and Q-chitosan, bearing aldehyde and cationic quaternary ammonium salt groups, respectively, were prepared and coated onto the SPIONs. The hybrid particles were characterized in terms of their size, surface change, magnetization and polymer content, and were next used to bind green fluorescent protein–mRNA (GFP–mRNA) and prepare the mRNA delivery vehicles. GFP–mRNA was used as a model gene, due to the ease of protein detection, to assess the novel carriers. Challenges addressed were related to the mRNA instability and immunogenicity with associated toxicity, and the improvement of the therapeutic efficacy of the gene. The binding efficiency of the gene, the nanocarrier cellular uptake, the cell viability, migration and adhesion and the cell protein expression, were biologically assessed, aiming to develop a highly promising mRNA delivery system for (pre)clinical in vivo applications.
2. Materials and methods
Chitosan (Mn = 30000 g mol−1, 90% deacetylated) was purchased from Glentham Life Science. Dextran (Mn = 40000 g mol−1) and silver nitrate (AgNO3, 99.9% purity) were purchased from Alfa Aesar. Sodium periodate (99.8% purity) and glycidyl trimethyl ammonium chloride (GTMAC) (>90% purity) were obtained from Sigma-Aldrich. Acetic acid (Fluka), acetone (Scharlau), potassium chromate (K2CrO4, 99.5% purity, Merck), phenolphthalein (98% purity, Riedel-de Haen), sodium chloride (NaCl, 99.5% purity, Merck), ferric chloride hexahydrate (FeCl3·6H2O, 99% Merck), ferrous chloride tetrahydrate (FeCl2·4H2O, 99% AppliChem Panreac), ammonia (NH4OH, 25%, Merck), and citric acid ((CH2CO2H)2, 99%, Sigma Aldrich) were used as received. All solvents were used as received. Milli-Q water, with a resistivity of 18.2 MΩ cm at 298 K, was obtained from Millipore apparatus and was used for the preparation of all samples. RPMI-1640, Dulbecco's Modified Eagle's Medium (DMEM), Fetal Bovine Serum (FBS) and penicillin–streptomycin solution were purchased from Biochrom (Berlin, Germany). Optimem was obtained from Gibco. Prussian blue iron stain kit was purchased from Sigma Aldrich. Magnetofection Polymag Kit was purchased from Oz Biosciences. CellTiter-Glo® Luminescent Cell Viability kit was obtained from Promega Corporation, WI, USA.
2.1 Synthesis of iron oxide nanoparticles
The synthesis of bare iron oxide nanoparticles (NPs) and nanoparticles coated with citric acid (NPs-CA) was carried out as described in a previous study.62 In brief, ferric chloride hexahydrate (FeCl3·6H2O) and ferrous chloride tetrahydrate (FeCl2·4H2O) were dissolved at a Fe3+:Fe2+ molar ratio of 2:1 in 10 ml double deionized water. NH4OH, the precipitation agent, was added dropwise in 100 ml water, until pH 12 was obtained, under vigorous stirring and a nitrogen atmosphere (nitrogen gas flow) to prevent oxidation, followed by the dropwise addition of the aqueous solution of the iron precursors. The precipitate was magnetically separated and washed until the pH of the final aqueous solution was close to 7.5. To prepare the NP-CA, 20 ml citric acid aqueous solution (0.5 M) were added to a 100 ml dispersion of the NPs (3% w/v Fe3O4) under vigorous stirring for 30 min and the final product was magnetically separated and washed 3 times with deionized water.
2.2 Synthesis of Q-chitosan
5 g chitosan (Mn = 30000 g mol−1) were added into 200 ml Milli-Q water containing 2% v/v acetic acid and the solution was heated at 70 °C until complete polymer dissolution. Next, 25 ml (186 mmol) GTMAC were added into the polymer solution in portions (2.5 mL h−1) and the reaction was left under magnetic stirring at 70 °C for 12 h. Afterwards, the reaction was allowed to cool down to room temperature and the modified polymer was recovered by precipitation with the addition of cold acetone enabling the removal of the unreacted GTMAC. Finally, the product was freeze dried and stored under a nitrogen atmosphere at 4 °C.
The degree of quaternization of chitosan was determined by conductometric titration using a conductivity meter (WTW, cond 3310, TetraCon 325 and 325/C). Briefly, Q-chitosan (0.0373 g) was dissolved in water (200 ml) and 5 drops of a 5% w/v potassium chromate (K2CrO4) solution were added as a color indicator. Next, the solution was titrated with a 0.09 mol L−1 aqueous silver nitrate (AgNO3) solution until a color change from yellow to brown occurred indicating the endpoint of the titration. The degree of quaternization (DQ) was calculated using the following equations:
| nQchi = VAgNO3·CAgNO3 | (1) |
Considering the 90% degree of deacetylation of chitosan and the mass of Q-chitosan, mchi (g), used in the titration:
| mchi = MwQchi·nQchi + MwAcet·nAcet + MwDeacet·nDeacet | (2) |
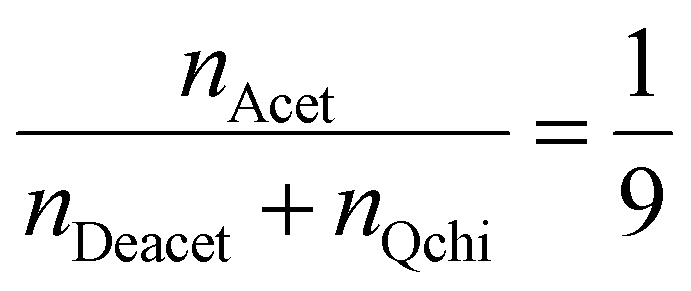 | (3) |
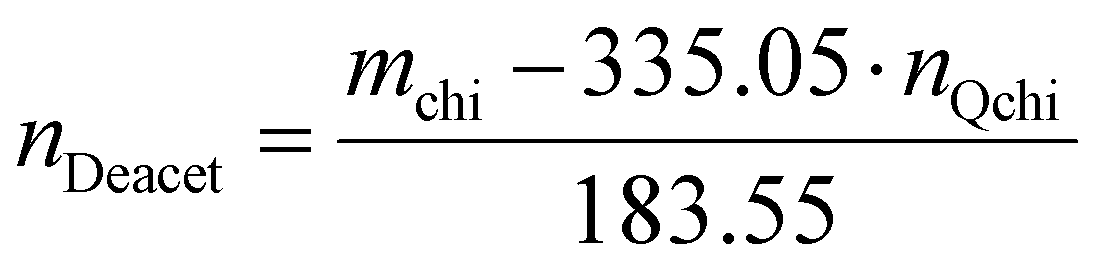 | (4) |
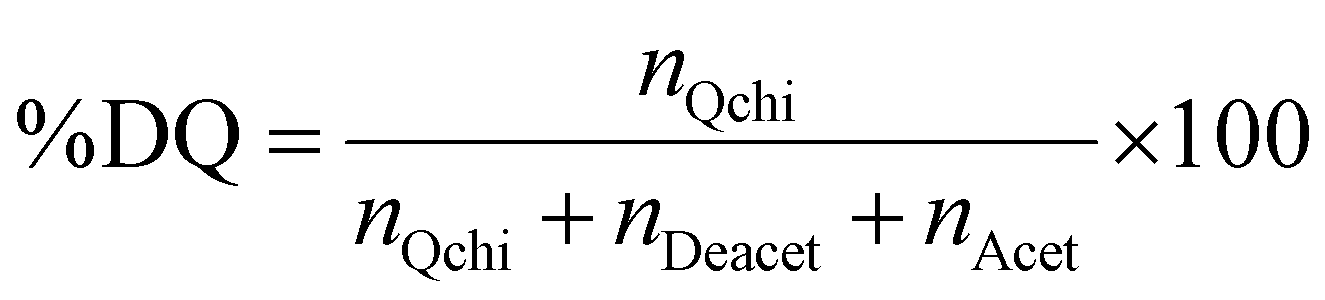 | (5) |
2.3 Synthesis of Ox-dextran
5 g dextran were dissolved in Milli-Q water at a 2% w/v concentration. The solution pH was adjusted at 5.5 using a 0.1 M HCl solution. The polymer solution was stirred at 30 °C for 2 h and then sodium periodate (5.9 g) was added. The reaction mixture was kept in the dark at 30 °C for 24 h under magnetic stirring, before being transferred into a dialysis bag (MW cut-off 14000 g mol−1) and dialyzed against water for 4 days, while the water was changed 4 times per day. Finally, the modified dextran was freeze dried and stored under a nitrogen atmosphere at 4 °C.
The degree of oxidation (%DO) was calculated by a potentiometric titration using phenolphthalein as the pH indicator.63 Briefly, Ox-dextran (0.125 g) was dissolved in 10 ml NaOH solution (0.25 M) and stirred for 15 min. Next, 15 ml HCl (0.25 M) were added to the solution, followed by the addition of 50 ml Milli-Q water and 1 ml of an aqueous phenolphthalein solution (0.2% w/v). The final solution was titrated with a 0.25 M NaOH solution until a color change from light yellow to purple occured. The initial dextran before oxidation was titrated following a similar process. The %DO was calculated according to the following equation:
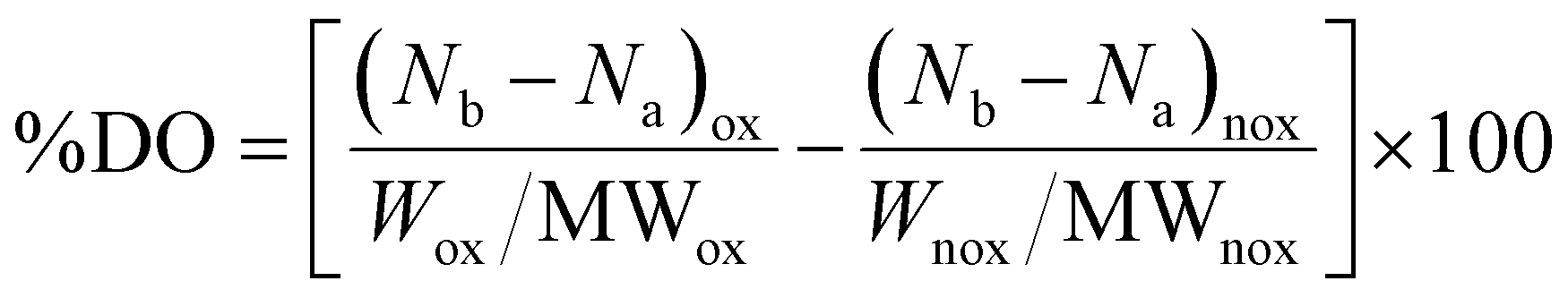 | (6) |
2.4 Synthesis of NPs and NPs-CA coated with Q-chitosan (NPs-Ch and NPs-CA-Ch)
Q-chitosan was dissolved in Milli-Q water to a final concentration of 2.5% w/v. A dispersion of the bare iron oxide nanoparticles in Milli-Q water (30 mg ml−1) was sonicated for 30 min in a sonication bath, before the addition of 1 g of the nanoparticles (33 ml dispersion) to the Q-chitosan solution (100 ml). The reaction was left under vigorous stirring at 60 °C for 24 h. Next, the mixture was cooled down to room temperature and finally, the polymer coated nanoparticles were recovered and purified from the non-bound polymer chains via several centrifugation/redispersion cycles in nanopure water. The polymer modified nanoparticles were redispersed in water (pH 7.4) and were stored at 4 °C until use. A similar procedure was followed for the coating of the NPs-CA with Q-chitosan.2.5 Synthesis of NPs and NPs-CA coated with dextran (NPs-Dx and NPs-CA-Dx)
An aqueous solution of Ox-dextran was prepared at a concentration of 10 mg ml−1. The polymer solution was stirred at 30 °C until complete dissolution. A dispersion of the bare iron oxide nanoparticles in Milli-Q water (30 mg ml−1) was sonicated for 30 min in a sonication bath, before the addition of 1 g of the nanoparticles (33 ml dispersion) to the Ox-dextran polymer solution. The reaction was left under vigorous stirring for 24 h at 30 °C. Finally, the Ox-dextran coated nanoparticles were purified to remove the non-bound dextran chains via several centrifugation/redispersion cycles in water. The polymer modified nanoparticles were redispersed in water (pH 7.4) and were stored at 4 °C until use. A similar procedure was followed for the coating of the NPs-CA with Ox-dextran.2.6 Physicochemical characterization techniques
The functionalized polysaccharides and the polysaccharide-coated NPs were characterized in terms of their physicochemical properties and morphological characteristics using a variety of characterization techniques, including proton nuclear magnetic resonance (1H NMR), attenuated total reflectance-Fourier transform infrared (ATR-FTIR) and Raman spectroscopies, thermogravimetric analysis (TGA), dynamic light scattering (DLS), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), vibrating sample magnetometry (VSM), X-ray diffraction (XRD) and BET surface area analysis.The modified polymers were characterized by 1H NMR spectroscopy using a Bruker AMX-500 NMR spectrometer. d6-DMSO and D2O were used as the deuterated solvents for the dextran and the chitosan samples, respectively. ATR-FTIR spectra were recorded on a Thermo Scientific Nicolet 6700 spectrometer and scans were collected for each measurement. Raman spectra (in Via, Renishaw plc, New Mills, UK) were obtained using a 532 nm solid-state diode laser at 0.5 mW power and 200 scans were collected for each measurement. The polymers, the iron oxide nanoparticles and the polymer-coated nanoparticles were characterized by thermogravimetric analysis (TGA) using a PerkinElmer Diamond TG/DTA instrument. Measurements were carried out in the temperature range between 30–550 °C, at a heating rate of 10 °C min−1, under a nitrogen atmosphere. Dynamic light scattering measurements (DLS, Cordouan Technologies SAS, Pessac, France) were carried out using a 657 nm laser and the number size distribution of the particles was determined using the SBL algorithm (Mie scattering model, real part: 2.3, imaginary part: 0.1). The Zetasizer NanoZS instrument (Malvern Instruments) was used for the determination of the zeta potential values of the nanoparticles. Aqueous dispersions of the nanoparticles (0.05 mg mL−1) in a 25 mM NaCl solution were prepared and sonicated for 15 min before each measurement. The average zeta potential value from three measurements was recorded for each sample. Finally, the morphology of the bare and the polymer-coated nanoparticles, either with or without citric acid, was determined using field emission scanning electron microscopy (FESEM), using a JEOL JSM-7000F microscope, and with transmission electron microscopy (TEM), using a JEOL JEM-2100 microscope. The samples for FESEM and TEM were prepared by the deposition of one drop of a dilute aqueous dispersion of the nanoparticles, on a glass substrate (for FESEM) and onto a carbon coated Cu grid (for TEM) and were allowed to dry overnight at room temperature.
Vibrating sample magnetometry (VSM) measurements were carried out at room temperature using a PAR Model 151 (Princeton Applied Research Corporation, Princeton, NJ). The VSM was calibrated against a NIST-certified Ni standard with a maximum applied field of about 2 T (μ0H). The samples were prepared by gently drying a magnetic nanoparticle suspension in a desiccator under ambient conditions. Mass magnetization was determined by extrapolation to an infinite external field. XRD patterns were obtained on a Siemens D 500 (Bruker, Massachusetts, US) X-ray diffractometer to analyse the crystal structure of the bare and polymer-coated nanoparticles, using a CuKα source. Finally, the BET surface area of the nanoparticles was measured by nitrogen adsorption and desorption measurements using a Tristar 3000 instrument (Micromeritics, Atlanta, US) at 150 °C degassing temperature.
2.7 GFP–mRNA synthesis
The green fluorescent protein (GFP) was cloned in a pT77 plasmid vector, which contains the T7 promoter, as described in ref. 64. The recombinant pT77-GFP vector was linearized using the restriction enzyme HindIII (Takara), downstream to the T7 promoter. The linearized plasmid was used as the template for the GFP–mRNA synthesis by in vitro transcription reactions, with the HiScribe T7 ARCA mRNA Kit following the manufacturer's protocol (New England, BioLabs). The synthesized mRNA was purified using a spin column-based method (Monarch RNA Cleanup Kits, BioLabs).2.8 Cell culture
T47D (epithelial human breast cancer, HER2+) cells were used in the present study to test the biological behavior of the newly synthesized nanoparticles. T47D cells serve as a well-established and known in vitro model to test the biological profile of the newly synthesized materials. Cells were seeded in 75 cm2 cell culture flasks and incubated under appropriate conditions (5% v/v CO2 at 37 °C and standard humidity) as 2D monolayers. Precisely, the T47D cells were cultured in RPMI-1640 supplemented with 10% v/v FBS and 1% penicillin–streptomycin. All used cells had a passage number of less than 20.:1) without removing the supernatant.65 The luminescence signal intensity, produced by the luciferase reaction, was determined using a Glomax 96 microplate luminometer (Promega Corporation, WI, USA).65 In all cases, cell exposure was performed with a colloidally stable, freshly prepared, homogenous dispersion of the nanoparticles.
min at room temperature. Then, the cells were washed with distilled water and incubated with a mixture of 10% w/v potassium ferrocyanide in water and 20% v/v hydrochloride solution (1:1 ratio) for 20min. After washing with distilled water, the cells were counterstained with nuclear fast red staining. Quantification was performed spectrophotometrically after washing with 1 mM EDTA solution in 1× PBS and extraction of the stain at 450 nm.
2.8.7.1 Preparation of the NPs-mRNA constructs. A nanoparticle suspension was diluted in cell culture medium and sonicated using a sonication bath for 10–15 min. The suspension was sterilized using UV light for approximately 10 min before being sonicated again for 5 min. Then, GFP–mRNA was added at the appropriate amount and gravimetric ratio (see Table 1) and the samples were vortexed for 10 s. Next, the samples were left under gentle stirring for 10–15 min to allow the formation of the complexes. Free mRNA was decanted under a magnetic field restraining the complexes. In the case of the commercially available magnetic beads, the binding process was performed following the manufacturer's instructions (Magnetofection, Polymag kit). Briefly, the appropriate amounts of mRNA and PolyMag magnetic beads were mixed in Optimem medium and left at room temperature for 20 min prior to their addition to the cells.
| Sample | w/w ratio | mRNA (μg) | Particle (μg) |
|---|---|---|---|
| 1 | 1:0.5 |
3 | 1.5 |
| 2 | 1:1.5 |
3 | 4.5 |
| 3 | 1:3 |
3 | 9 |
| 4 | 1:5 |
3 | 15 |
| 5 | 1:8 |
3 | 24 |
| 6 | 1:10 |
3 | 30 |
The binding efficiency was tested using agarose gel electrophoresis. The nanoparticles were isolated from the aqueous stock solution using a magnet and were washed with water. Samples were centrifuged for 1 min and the supernatant was discarded. The pellets were redissolved in 5 μL ddH2O water and 1× loading dye (1 μL). 1% w/v agarose gel in 1× TBE with 0.1% w/v ethidium bromide (EtBr) was used for the electrophoresis.
2.8.7.2 Magnetofection and translation tests. For the magnetofection experiments, cells were exposed to the magnetic field (Polymag kit) for 30 min in the presence of the NPs-mRNA constructs. The transfection mixture, prepared in the previously described step (Section 2.8.7.1), was added in a dropwise manner into the culture plates. 48–72 h post-transfection, the transfection efficiency was determined using an inverted fluorescent microscope at 10× magnification for GFP–mRNA. At least 500 cells were examined for each sample. Cells were rinsed once with PBS and incubated for an additional 15 min with DAPI stain (stock solution 1 mg mL−1) for the fluorescent detection of the nuclei. Cells were fixed with 4% paraformaldehyde (PFA) for 20 min and visualized using an Axio Observer Z1 microscope, with a 20× phase contrast (Carl Zeiss, GmbH Lena, Germany). Images were captured, using an AxioCam Hc camera. Fluorescence intensity was calculated as the fluorescence intensity of the NPs divided by the fluorescence intensity of the control group (mean fluorescence intensity) using ImageJ software. Statistical analysis was performed using one-way ANOVA followed by post hoc analyses (Tukey).
2.9 Statistical analysis
Data are presented as average values with standard error means (SEMs) of several independent measurements. Mean survival rates and SEMs were calculated for each individual group. Percent change of survival rates were calculated for each control group and one way analysis of variance (ANOVA) was performed for all pair comparisons, followed by post hoc analyses (Tukey). Two-way ANOVA was used in the case of cellular uptake studies using GraphPad Prism v.6. Degrees of significance were assessed by different rating values: *p < 0.05 (significant), **p < 0.01 (highly significant), ***p < 0.001 (extremely significant), ****p ≤ 0.0001 (extremely significant) and p > 0.05 (non-significant). The percentage contribution of the main factors effecting cell survival was calculate as the ratio of the adjusted sum of squares (Adj SS) calculated from the respected experimental values for each factor over the summation of Adj SS of all factors using a Minitab statistical software package.3. Results and discussion
The surface properties of the SPIONs play a crucial role on their dispersibility in aqueous media, on their size and on the binding efficiency of biomolecules of interest, including genes (RNAs, DNAs), antibodies and proteins, therefore significantly affecting their performance in biological applications. Herein, different types of polysaccharide coated SPIONs (see Table 2) were prepared and evaluated in terms of their colloidal stability, magnetic properties, binding of nucleic acids, cytotoxicity and transfection efficiency as gene carriers.| SPION-symbolic name | Description – type of coating |
|---|---|
| NPs | Ferrite nanoparticles – no coating |
| NPs-CA | Ferrite nanoparticles – citric acid coating |
| NPs-Ch | Ferrite nanoparticles – quaternized chitosan coating |
| NPs-Dx | Ferrite nanoparticles – oxidized dextran coating |
| NPs-CA-Ch | Ferrite nanoparticles – citric acid and quaternized chitosan coating |
| NPs-CA-Dx | Ferrite nanoparticles – citric acid and oxidized dextran coating |
| NPs-CA-ChGFPmRNA | mRNA bound on ferrite nanoparticles with citric acid and quaternized chitosan coating |
| NPs-CA-DxGFPmRNA | mRNA bound on ferrite nanoparticles with citric acid and oxidized dextran coating |
3.1 Synthesis, morphological and structural characterization of the SPIONs
First, the SPIONs were synthesized using the co-precipitation method as described in Section 2.1, and next the bare nanoparticles were coated with citric acid, which was found in previous studies to increase their colloidal stability and prevent nanoparticle aggregation due to electrostatic repulsions. The Raman spectrum of the bare NPs exhibited three dominant bands at 300, 510, and 690 cm−1, which correspond to the Eg, T2g(2), and A1g modes of magnetite (Fe3O4), respectively (Fig. 1(a)).69 The high intensity peak at 690 cm−1 was further deconvoluted to the maghemite peak at 700 cm−1 and a magnetite peak at 670 cm−1 that is shifted to higher values due to interactions of the Fe–O bonds with the surface bound organic molecules (Fig. S1, ESI†). The second order peaks between 1000 and 1700 cm−1 are related to the maghemite structure.70 The NPs-CA sample exhibited a similar spectrum to that of the bare NPs (Fig. 1(a)). The presence of the organic coating on the surface of the SPIONs was verified by the intense characteristic band at 540 cm−1 assigned to the surface Fe–O vibrations as well as the vibration bands attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of the free carboxyl groups of citric acid at 1584 cm−1 and the band at 1380 cm−1 corresponding to the CO bonds of the surface bound carboxyl groups (Fig. 2(a), red dashed line). The amount of citric acid deposited on the surface of the SPIONs was calculated at 3.4% w/w using TGA (Fig. 2(b), red dashed line).
O bonds of the free carboxyl groups of citric acid at 1584 cm−1 and the band at 1380 cm−1 corresponding to the CO bonds of the surface bound carboxyl groups (Fig. 2(a), red dashed line). The amount of citric acid deposited on the surface of the SPIONs was calculated at 3.4% w/w using TGA (Fig. 2(b), red dashed line).
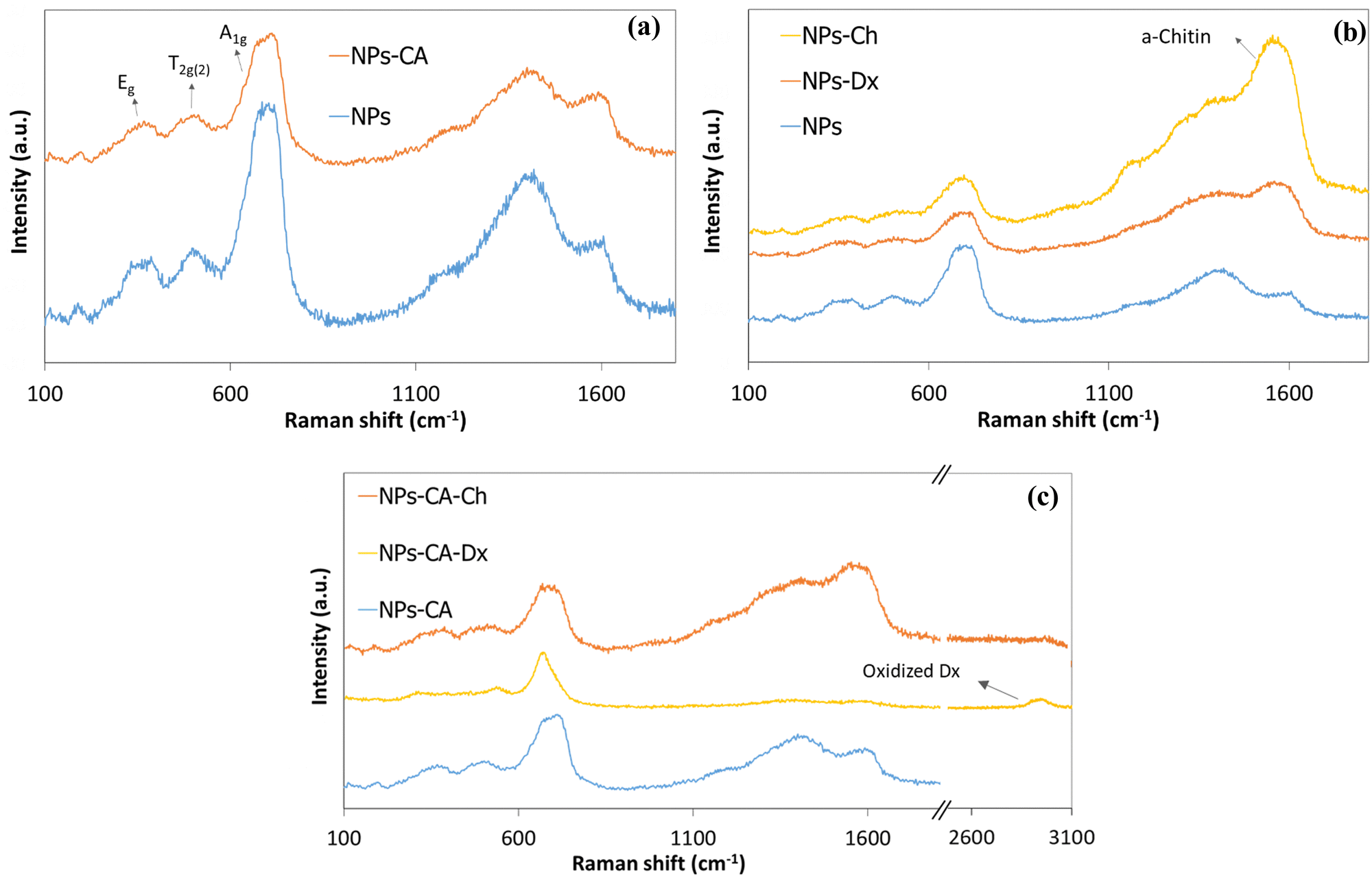 | ||
| Fig. 1 Comparative Raman spectra of the (a) NPs and NPs-CA, (b) NPs, NPs-Dx and NPs-Ch and (c) NPs-CA, NPs-CA-Dx and NPs-CA-Ch samples. | ||
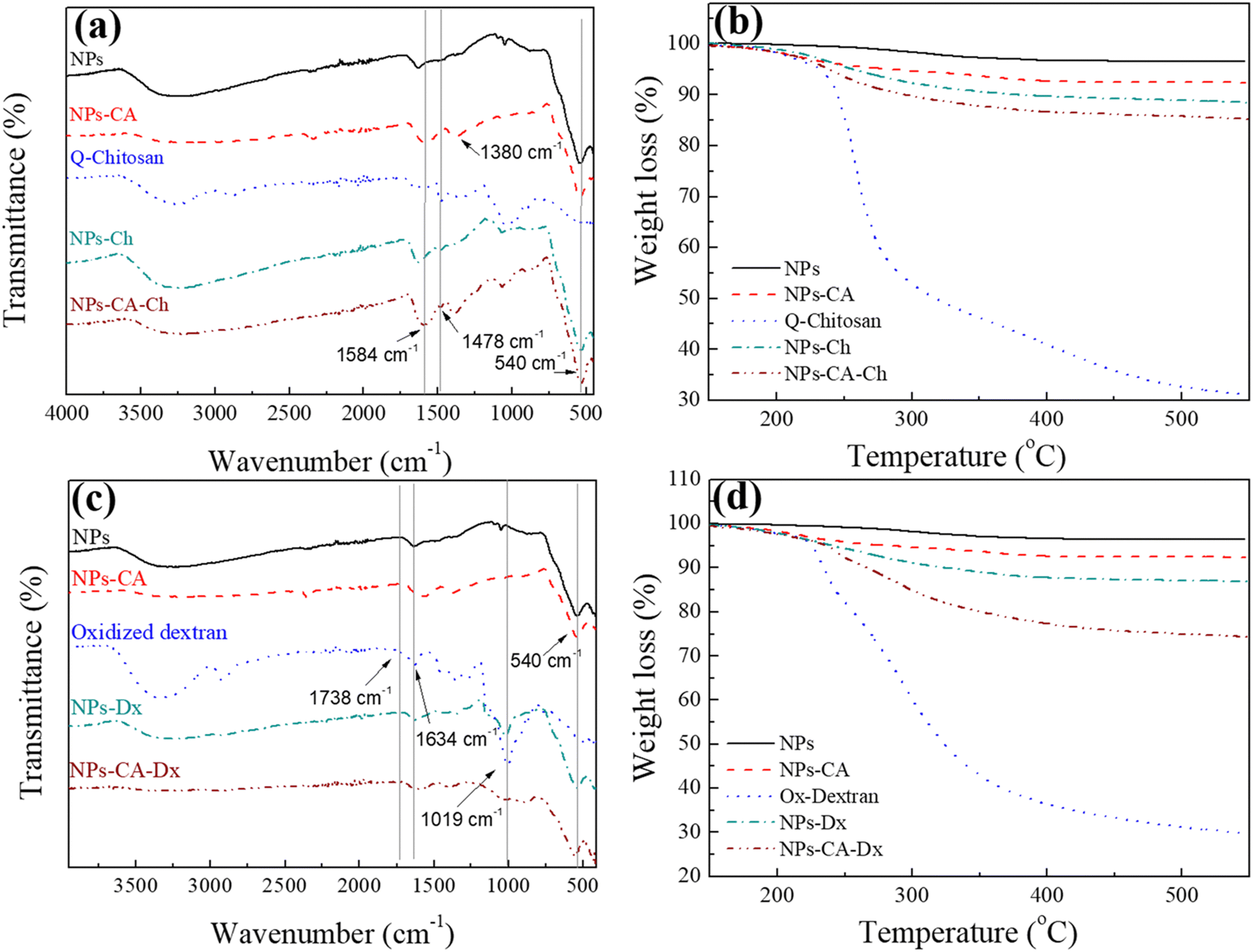 | ||
| Fig. 2 Comparative (a) and (c) ATR-FTIR spectra, and (b) and (d) TGA curves of the NPs. For (a) and (b) NPs (black solid line), NPs-CA (red dashed line), Q-chitosan (blue dotted line), NPs-Ch (green dash-dotted line) and NPs-CA-Ch (wine dash-dot-dotted line). For (c) and (d) NPs (black solid line), NPs-CA (red dashed line), Ox-dextran (blue dotted line), NPs-Dx (green dash-dotted line) and NPs-CA-Dx (wine dash-dot-dotted line). | ||
Next, the polysaccharides, chitosan and dextran, were chemically modified to form the appropriate functionalities for the conjugation of the GFP–mRNA gene. Chitosan underwent quaternization using GTMAC to bear cationic quaternary ammonium salt moieties, thereby conferring water solubility to the polymer under neutral conditions.71,72 Moreover, the cationic sites of chitosan enable its complexation with the anionic mRNA chains via electrostatic interactions. The successful modification of chitosan was confirmed by 1H NMR and ATR-FTIR spectroscopies (see Fig. S2 and S3 and the relevant text in the ESI†). The degree of quaternization of the polymer, determined by conductometric titration, was found to be 73% by mole. A high degree of quaternization (60–70%) was targeted to enhance the solubility of chitosan under neutral conditions and simultaneously provide an excess of positively charged amino groups for complexation with the mRNA chains. Q-chitosan was also characterized by TGA and showed a greater weight loss (68%) compared to the non-modified polymer (56%) (Fig. S3b, ESI†). This disparity arises from the introduction of the quaternary ammonium salt moieties, which render the polymer more susceptible to thermal degradation by also lowering its degradation temperature by approximately 50 °C.57,73
For the dextran oxidation, the well-known oxidizing agent, sodium periodate, was employed to introduce aldehyde moieties along the polymer chains. The 1H NMR and ATR-FTIR spectra proved the successful oxidation of dextran (see Fig. S4 and S5 and relevant text in the ESI†). The degree of oxidation, determined by potentiometric titration, was found to be 53%, which aligns well with the values reported in the literature.74,75 TGA showed a reduced weight loss of 70% for the modified polymer compared to 80% for the precursor material, which was attributed to the polymer mass loss during oxidation (Fig. S5b, ESI†).
The bare NPs and NPs-CA were next coated with the modified polymers by simply mixing the magnetic nanoparticles with a polymer solution. The appearance of a vibration peak at 1478 cm−1 in the ATR-FTIR spectra of NPs-Ch and NPs-CA-Ch verified the presence of the trimethyl ammonium groups of the polymer and confirmed the successful coating of the nanoparticles (Fig. 2(a)). Moreover, the Raman spectrum of NPs-CA-Ch, depicted in Fig. 1(c) showed a notable peak at 1600 cm−1 associated with a-chitin. Additionally, the 700 cm−1 peak linked to the vibration modes of magnetite and maghemite has been subject to deconvolution in Fig. S1 (ESI†). The resulting values as presented in Table 3, delineate the individual contribution of magnetite and maghemite within each construct. In particular, the NPs-CA-Ch construct exhibited a single peak at 683 cm−1 attributed to magnetite, contrasting with the two peaks at 683 cm−1 and 721 cm−1 observed in the NPs-CA sample, where these peaks correspond to magnetite and maghemite, respectively. Notably, the increase of the 1000–1700 cm−1 peaks was more pronounced in the case of NPs-Ch as illustrated in Fig. 1(b). The amount of polymer deposited on the surface of the NPs was quantified by TGA and was found to be 12% w/w and 11% w/w for NPs-Ch and NPs-CA-Ch, respectively (Fig. 2(b)). Interestingly, the TGA results suggested that the presence of the citric acid ligand on the surface of the nanoparticles did not affect the amount of polymer deposited on the NPs. Similarly, the successful coating of the bare and CA-functionalized NPs with Ox-dextran was verified by ATR-FTIR and Raman spectroscopies. The ATR-FTIR spectra of NPs-Dx and NPs-CA-Dx displayed the characteristic band of magnetite at 540 cm−1 along with the vibration bands of the polymer at 1019 cm−1 and 1634 cm−1, denoting the presence of the polymer coating on the surface of the NPs (Fig. 2(c)). In addition, the Raman spectrum of NPs-Dx, displayed an intense peak at 1550 cm−1 related to the vibration modes of both the maghemite and the organic coating (Fig. 1(b)), while the spectrum of NPs-CA-Dx exhibited a characteristic peak at 2900 cm−1 attributed to the oxidized polymer (Fig. 1(c)). TGA analysis indicated weight losses of 14% w/w and 26% w/w for NPs-Dx and NPs-CA-Dx, respectively, underscoring a significantly higher amount of polymer deposited on the surface of the NPs-CA-modified SPIONs (Fig. 2(d)), which was attributed to their improved dispersion in the aqueous medium, as discussed further in the TEM data below, as well as the effective binding of the Ox-dextran polymer chains via hydrogen bonding with the carboxylate groups of CA. This effect was less pronounced for the Q-chitosan sample, due to the electrostatic repulsions among the polymer chains, which impeded their multilayer coating of the NP surface.
| Sample | Fe3O4 A1g peak (cm−1) | γ-Fe2O3 A1g peak (cm−1) | M s (emu g−1) |
M
s(n) normalizeda (emu g−1) |
M
sFe3O4 − Ms(n) variation |
Organic content (wt%) | Surface area (m2 g−1) |
|---|---|---|---|---|---|---|---|
| a Normalized to the calculated iron oxide content found by TGA. | |||||||
| NPs | 673 | 723 | 68.7 | 68.7 | — | — | 112.59 |
| NPs-CA | 683 | 721 | 63 | 65.21 | 3.4 | 3.4 | 124.14 |
| NPs-Ch | 652 | 708 | 59.9 | 68.06 | 0.63 | 12 | 64.52 |
| NPs-Dx | 635 | 699 | 56.4 | 65.58 | 3.11 | 14 | 76.27 |
| NPs-CA-Ch | 683 | 54.8 | 61.57 | 7.12 | 11 | 37.8 | |
| NPs-CA-Dx | 666 | — | 40.8 | 55.13 | 13.56 | 26 | — |
| 675 | |||||||
The size and morphology of the bare, CA- and polymer-coated NPs were characterized by TEM microscopy. As observed in the TEM images, the bare NPs were spherical in shape with sizes in the 4–20 nm range (Fig. 3(a)). Moreover, the SPIONs appeared agglomerated, which was attributed to the attractive van der Waals forces between the nanoparticles as well as the drying of the sample for the measurement. Compared to the bare NPs, the CA-NPs appeared less aggregated, due to the presence of citric acid on their surface, which induced electrostatic repulsions among the colloids and facilitated their dispersion (Fig. 3(b)). Similarly, the TEM images of the NPs-CA-Ch and NPs-CA-Dx samples showed the aggregation of the NPs even in the presence of the polymer coating due to the attractive van der Waals forces between the nanoparticles and the dry state of the sample (Fig. S6a and b, ESI†). The nanoparticle structure was assessed by XRD measurements, exhibiting the characteristic peaks of magnetite (Fig. S7, ESI†). Furthermore, the crystallite size was determined between 5–10 nm using the Scherrer formula. This finding provides additional evidence, supported by TEM analysis, that the nanoparticles are monocrystalline and possess a crystal size below 20 nm.
 | ||
| Fig. 3 TEM images of the (a) NPs and (b) NPs-CA samples. | ||
The number size distribution of the NPs in aqueous dispersion was determined by DLS (Fig. 4). The bare NPs showed a peak at ∼75 nm and the NPs-CA sample had a peak at ∼60 nm, while both samples exhibited a second population of significantly lower intensity at 390 nm. The larger sizes measured by DLS, compared to the primary NP size of 5–10 nm determined by TEM (Fig. 3(a)), were attributed to smaller and larger agglomerates formed due to van der Waals forces between the nanoparticles in the aqueous suspension. Notably, the bare NPs formed agglomerates of the single crystallite Fe3O4 nanoparticles, while the NPs-CA sample exhibited single crystallites of Fe3O4 dispersed in the CA matrix, as shown in Fig. 3(b). It is interesting to note that the CA-coated NPs exhibited a smaller size, as well as a lower intensity for the larger agglomerates, signifying that the dispersion of the NPs is improved in the presence of the negatively charged citric acid ligand, due to the electrostatic repulsions among the nanoparticles. Fig. S14 (ESI†) exhibits the respective DLS denoting the volume fraction of the particle size distribution, showing the expected increase of volume at higher particle sizes, especially at 390 nm and a very limited particle aggregation at 650 and 860 nm.
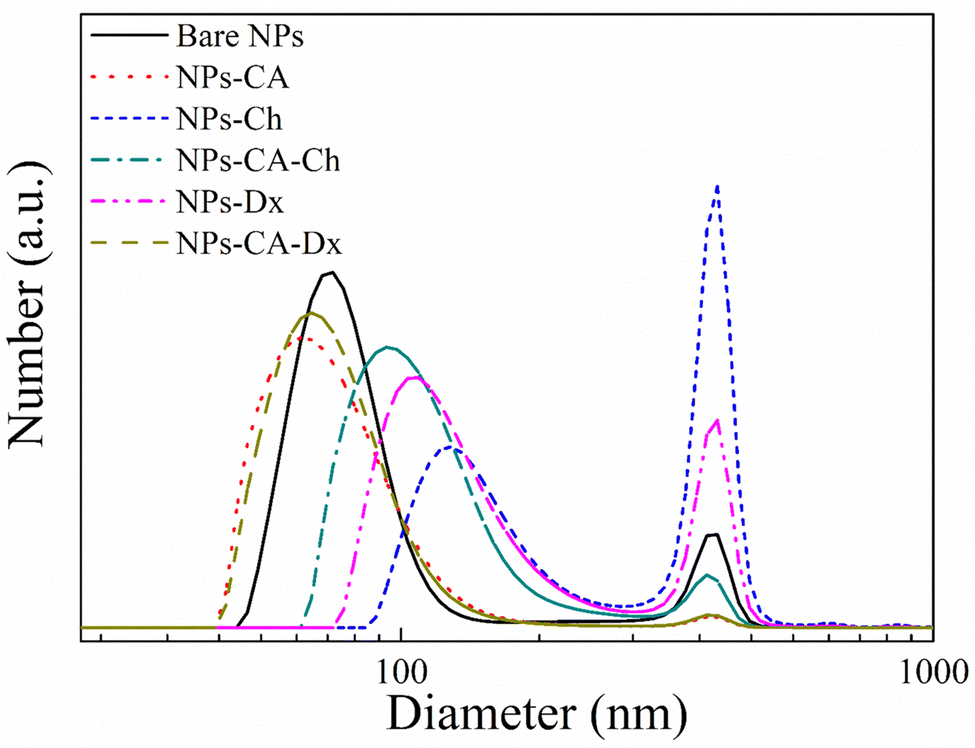 | ||
| Fig. 4 DLS measurements of the bare NPs (black solid line), NPs-CA (red dotted line), NPs-Ch (blue short-dashed line), NPs-CA-Ch (green dashed-dotted line), NPs-Dx (magenta dashed dotted-dotted line) and NPs-CA-Dx (dark yellow dashed line) samples. | ||
Fig. 4 also shows the size distribution of the polymer-coated NPs in the absence and presence of CA. The NPs-Ch and NPs-Dx samples, showed size distributions centered around 100 nm and 120 nm, respectively, which are larger compared to the bare NPs and were attributed to the presence of the polymer layer coating on the NP agglomerates. The size of the larger agglomerates remained constant at 390 nm, but increased in intensity for the polymer coated SPIONs verifying further NP aggregation. Similarly, the NPs-CA-Ch sample exhibited an increase in size from approximately 60 nm for the NPs-CA before polymer coating to approximately 95 nm for the polymer-coated NPs. However, the size distribution of NPs-CA-Dx did not increase compared to the NPs-CA sample, indicating that the higher polymer fraction found by TGA, resulted in an improved dispersion of the NPs in the aqueous medium. From the above results it is evident that the NPs-CA-Dx sample exhibited superior dispersion properties and the smaller particle size distribution, with the absence of large aggregates, compared to the other SPIONs. It is also important to note, that all the NPs developed in this study presented sizes well below 200 nm, which is known to be optimal for effective cell uptake and the absence of cell cytotoxic events.
Zeta potential is another important parameter that not only influences the stability of a NP-based delivery system under physiological conditions, but also determines the type of interactions of the nanoparticles with the mRNA chains and the cell membrane and therefore, the transfection efficiency. The zeta potential value of the bare NPs was found −8 mV; slightly negative under physiological conditions (pH 7.4) as expected (Fig. 5).76,77 However, the CA-functionalized nanoparticles exhibited a significantly lower zeta potential value of −37 mV, due to the presence of the anionic surfactant on the surface of the particles, which explained the improved colloidal stability of the NPs in the dispersion medium. The Ox-dextran coated NPs showed a zeta potential value of −5.5 mV and −31 mV for the NPs-Dx and the NPs-CA-Dx sample, respectively, signifying that the neutral polymer coating, did not alter the surface charge of the NPs, as expected (Fig. 5). On the other hand, coating the NPs with the positively charge Q-chitosan had a considerable effect on their surface charge and caused charge reversal, with a zeta potential value of +32 mV and +34 mV for the NPs-Ch and NPs-CA-Ch samples, respectively, verifying the presence of the cationic polymer coating on the surface of the NPs.
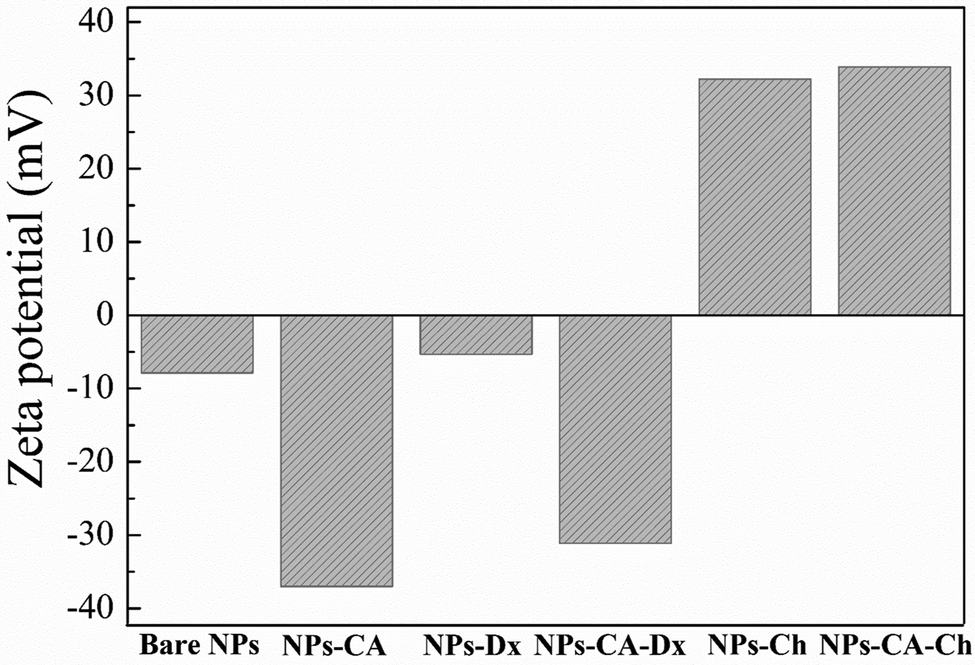 | ||
| Fig. 5 Zeta potential values of the NPs in aqueous dispersion at pH 7.4. | ||
The magnetization of the NPs was measured by vibrating sample magnetometry (VSM). Fig. 6. shows the hysteresis loops and Table 3 presents the respective saturation magnetization values, Ms, for the SPIONs. The lack of a hysteresis loop in all samples indicated their superparamagnetic nature, whereas, the lower Ms values found for the coated samples were attributed both to the lower effective weight fraction of the magnetic material (Fe3O4) and to the interactions of the surface atoms of Fe3O4 with the polymer coatings. These interactions, facilitated by hydrogen and/or covalent bonds, lead to the formation of a magnetically disordered layer that diminished the magnetic order and subsequently reduced the magnetization of the NPs. In order to evaluate these effects, the Ms values of the coated samples, measured by VSM, were normalized with respect to the magnetite mass fraction, calculated by TGA, and the respective values are reported in Table 3. The normalized Ms values (Ms(n)) should coincide with the Ms value of the bare Fe3O4 (68.7 emu g−1), while any variation of the Ms(n) was attributed to the magnetically disordered layer formed by the interactions of the iron oxide surface with the organic coating. The intrinsic Fe3O4 lattice disorders were analyzed by the deconvolution of the main magnetite peak at 690 cm−1 to the A1g bands of Fe3O4 and γ-Fe2O3 (see Fig. S8, ESI†). The shift of the A1g peak from 673 cm−1 for the bare Fe3O4 NPs to lower values (i.e., 635 cm−1 for the NPs-Dx SPIONs), was attributed to the distinct surface interactions of the iron oxide particles with the organic coatings.
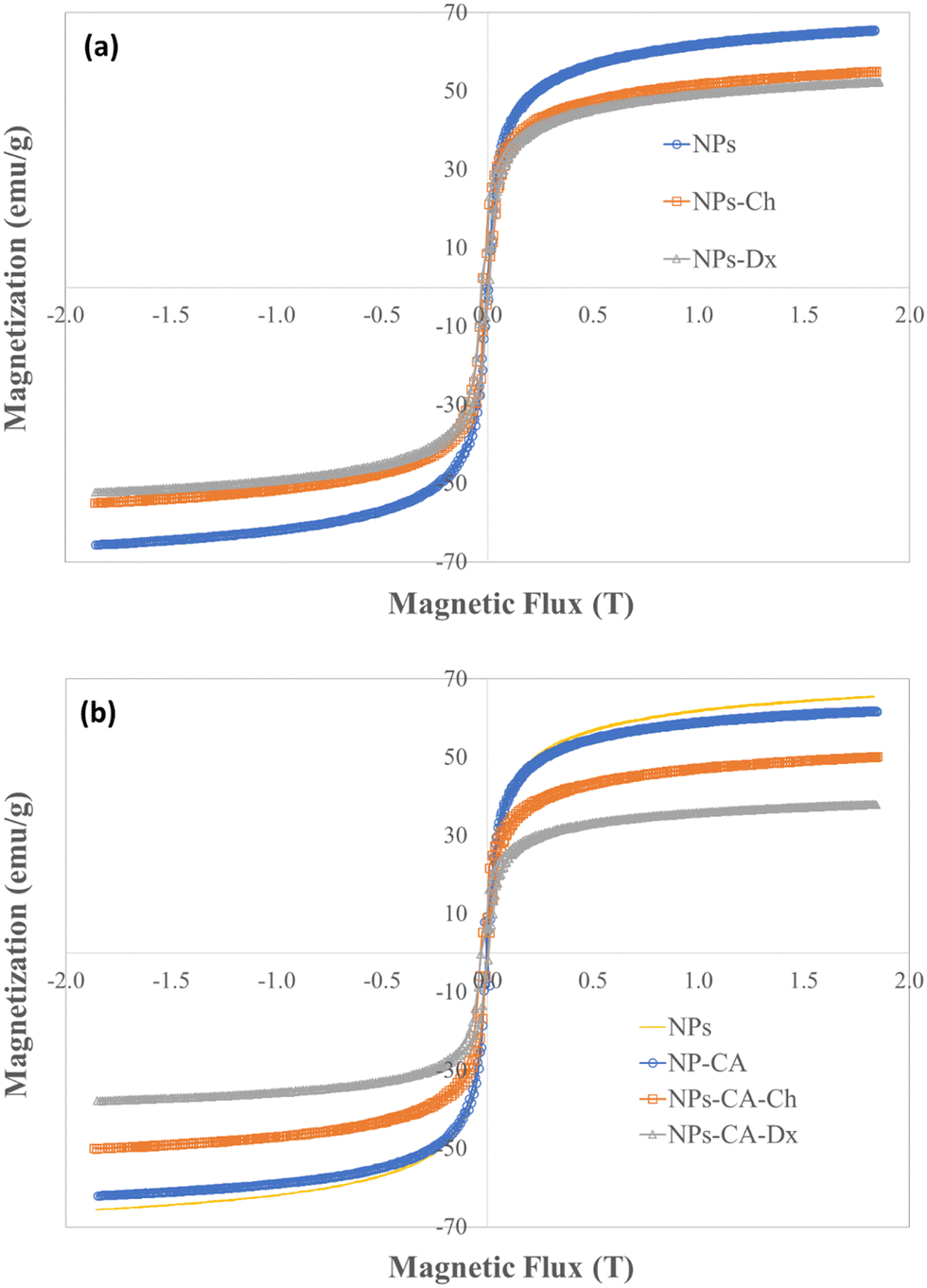 | ||
| Fig. 6 Hysteresis loops measured by VSM for the (a) NPs, NPs-Ch, NPs-Dx and (b) NPs, NPs-CA, NPs-CA-Ch and NPs-CA-Dx samples. | ||
The surface area of the SPIONs was measured by BET analysis on dehydrated samples. The bare NPs exhibited a surface area of 112.59 m2 g−1, which increased to 124 m2 g−1 for NPs-CA due to their improved dispersion and smaller particle size. In contrast, the polymer coated particles exhibited lower surface areas of 64 and 76 m2 g−1 for NPs-Ch and NPs-Dx, respectively. The surface area further decreased to 37 m2 g−1 for NPs-CA-Ch and was completely eliminated for NPs-CA-Dx. These results are explained since the polymer coated samples formed dense films when dehydrated, that inhibit nitrogen penetration and reduce the measurable surface area during BET analysis.
3.2 Biological profile and biocompatibility studies
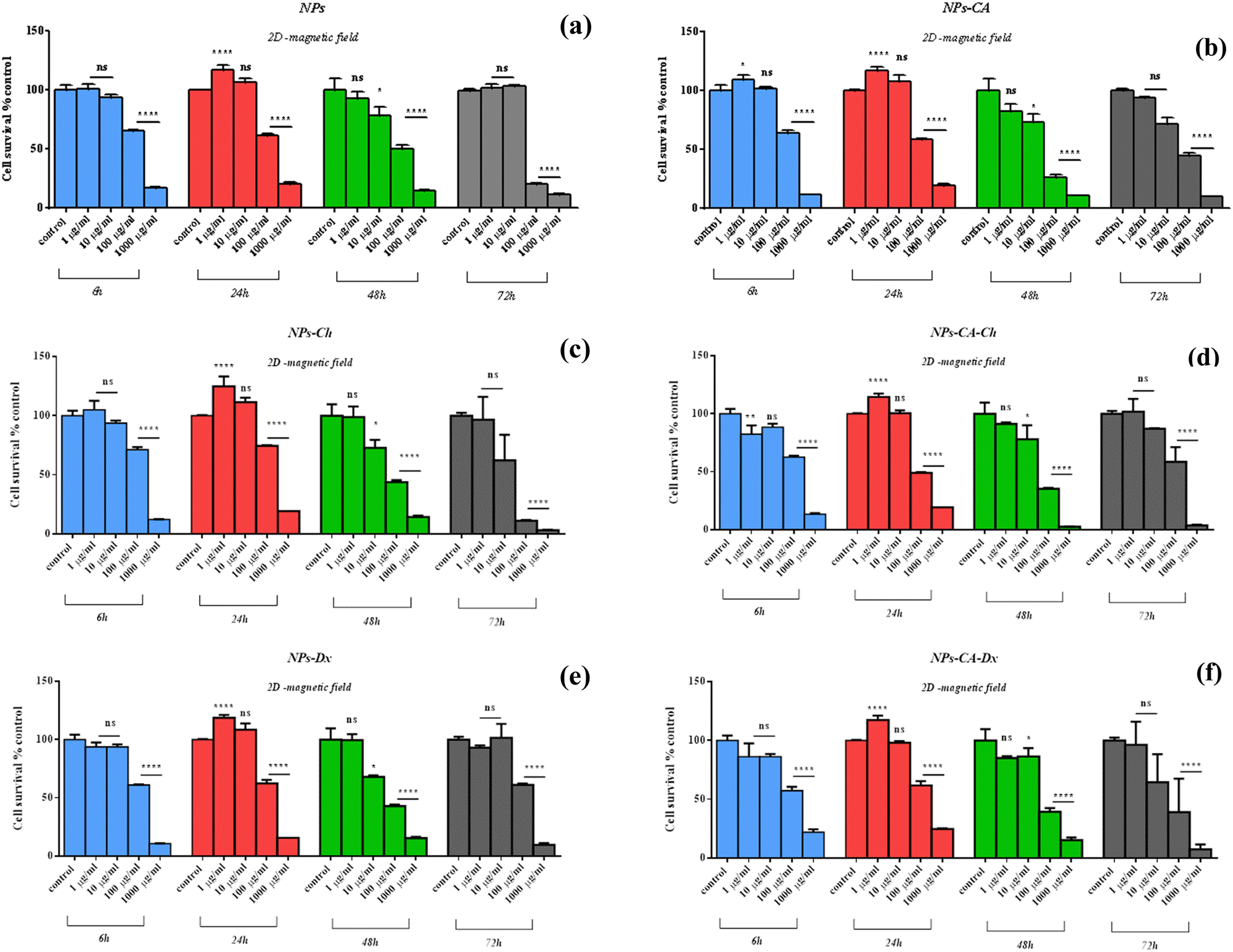 | ||
| Fig. 7 Percentage change of cell survival of the T47D cells (2D monolayers, without magnetic field) following treatment with various concentrations (1–1000 μM) of NPs (a), NPs-CA (b), NPs-Ch (c), NPs-Dx (d), NPs-CA-Ch (e), and NPs-CA-Dx (f) for 6, 24, 48, and 72 h. Values represent the mean value of n = 3 independent experiments. The vertical bars represent the standard error mean values, SEMs. *p < 0.05 (significant), **p < 0.01 (highly significant), ***p < 0.001 (extremely significant) and ****p ≤ 0.0001 (extremely significant) and p > 0.05 (non-significant). | ||
The NPs led to a cell survival rate variability, and quantitative assessment of the contribution of the main effectors to cell survival was carried out. Fig. 8 lists the contribution percentages of the different factors (surface modification, concentration and time) for each carrier separately. It is worth noting that the most important factor was the concentration for the NPs, whereas surface modification did not have an explicit effect on cell survival. However, in the case of the NPs-Ch and NPs-CA-Ch carriers, the presence of citric acid appeared to influence the intensity of the concentration. More specifically, the concentration in the first case contributed by ∼64%, while in the latter by ∼80%, indicating that for the SPIONs coated with chitosan, time emerges as an important factor compared to those also bearing citrate on their surface. This observation may be due to the fact that citric acid improves the dispersion of the NPs in the solution. In general, it has been shown that citric acid can enhance the solubility of the compounds it binds to and facilitate the delivery of active molecules to target points with strong biological activity.79 Similar enhancement of the role of the SPION concentration was not evident in the case of the NPs-Dx and NPs-CA-Dx samples, and this was attributed to the fact that the Ox-dextran-modified NPs exhibited anywise superior dispersion properties in solution compared to the Q-chitosan coated NPs.
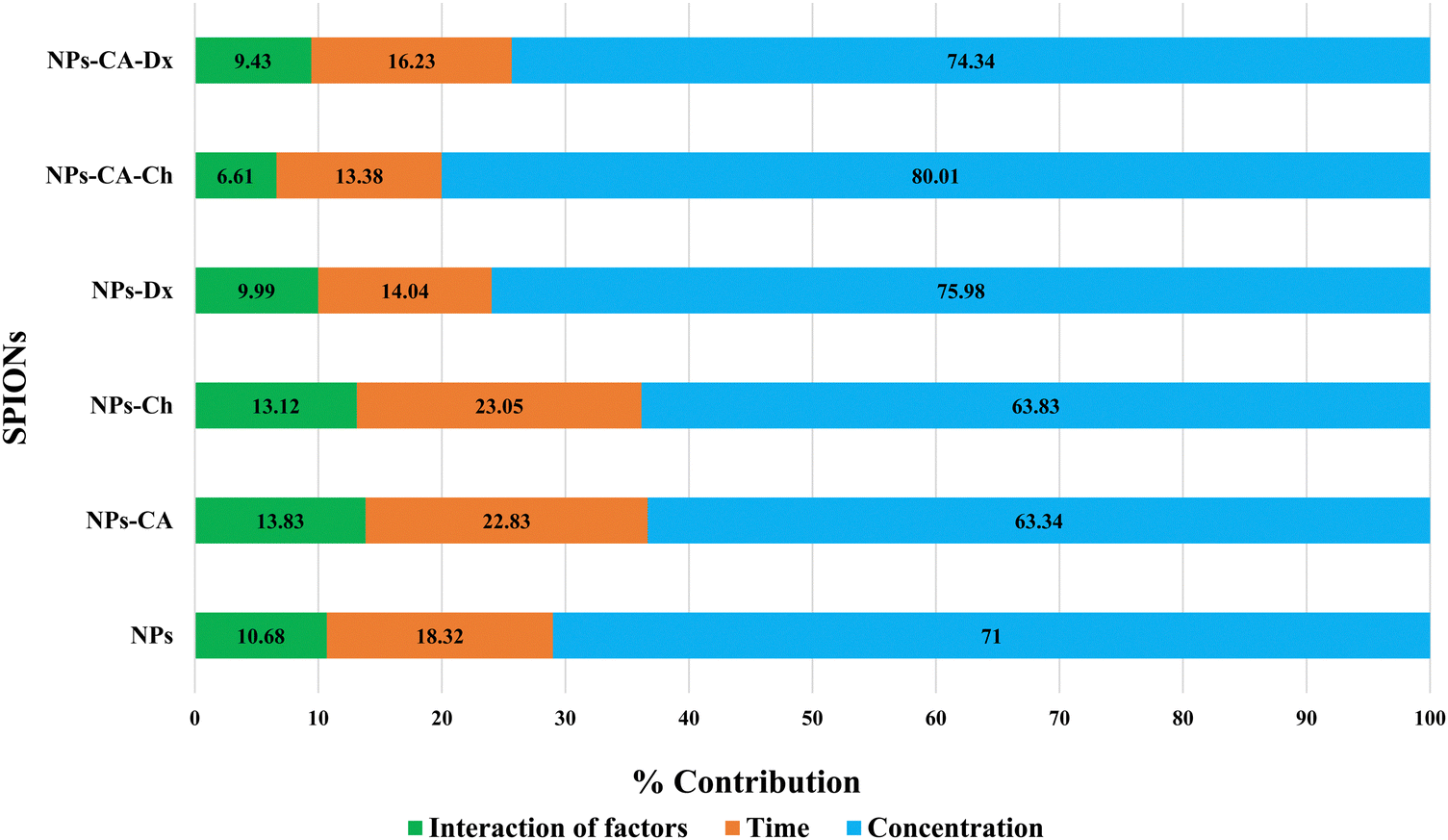 | ||
| Fig. 8 Percentage contribution of the main factors effecting cell survival. | ||
Following the above results on 2D cell cultures, the NPs were also examined for their potential cytotoxicity on 3D cell cultures to better simulate the in vivo scenario. To this end, 3D cell spheroids were formed and were allowed to grow using the hanging drop method. The formation process lasted for 12 days, and an initial cell loading of 700 cells per drop was selected as the optimum case. Evaluation of the spheroids’ viability, integrity, shape, and appearance was performed, with their diameters recorded in pixels and then converted to μm using ImageJ, based on an average of four measurements (Fig. S9, ESI†). The maximum cell viability in the spheroids was monitored on the 12th day of culture, and their diameters were found to be around 500 μm. Next, the spheroids were carefully transferred to a 96-well plate using a 100 μL pipet tip and were treated with the NPs for either 24 or 48 h at concentrations ranging from 1–100 μg mL−1.
Comparing the data presented in Fig. 7 and 9, it becomes evident that in the case of the 2D cultures, the concentrations of 1–10 μg mL−1 are not toxic toward the cells, whereas higher concentrations of 100 and 1000 μg mL−1 cause a decrease in cell viability already within the first 24 h of their contact with the cells (time-dependent and concentration-dependent). On the other hand, in the 3D cultures the observed cytotoxicity does not seem to depend on the concentration of the NPs during the first 24 h (time-dependent only), but contributes to the cytotoxicity after this time and up to 48 h (time-dependent and concentration-dependent). Furthermore, it appears that the cytotoxicity of the NPs in the 2D cultures is lower compared to the 3D counterparts. This observation suggests the possible sensitivity of the beads compared to the monolayer cultures. It is also noted that the viability measurement refers only to the metabolically active cells and therefore, does not account for the total number of cells in the 3D cultures, as this includes the necrotic nucleus. In contrast, in 2D cell cultures, the concentration required to induce a viability change depends on the density of cells on the surface of the culture (percentage of surface coverage), as reported in previous studies.80 The observation of increased particle toxicity in 3D cultures contrasts with the literature, where it has been observed that monolayer cultures show a sharper decrease in viability compared to 3D cultures, when exposed to magnetic nanoparticles.66,81 However, a direct comparison between these studies and the data obtained herein is not entirely feasible due to differences in the surface modification of the particles and the cell lines used.
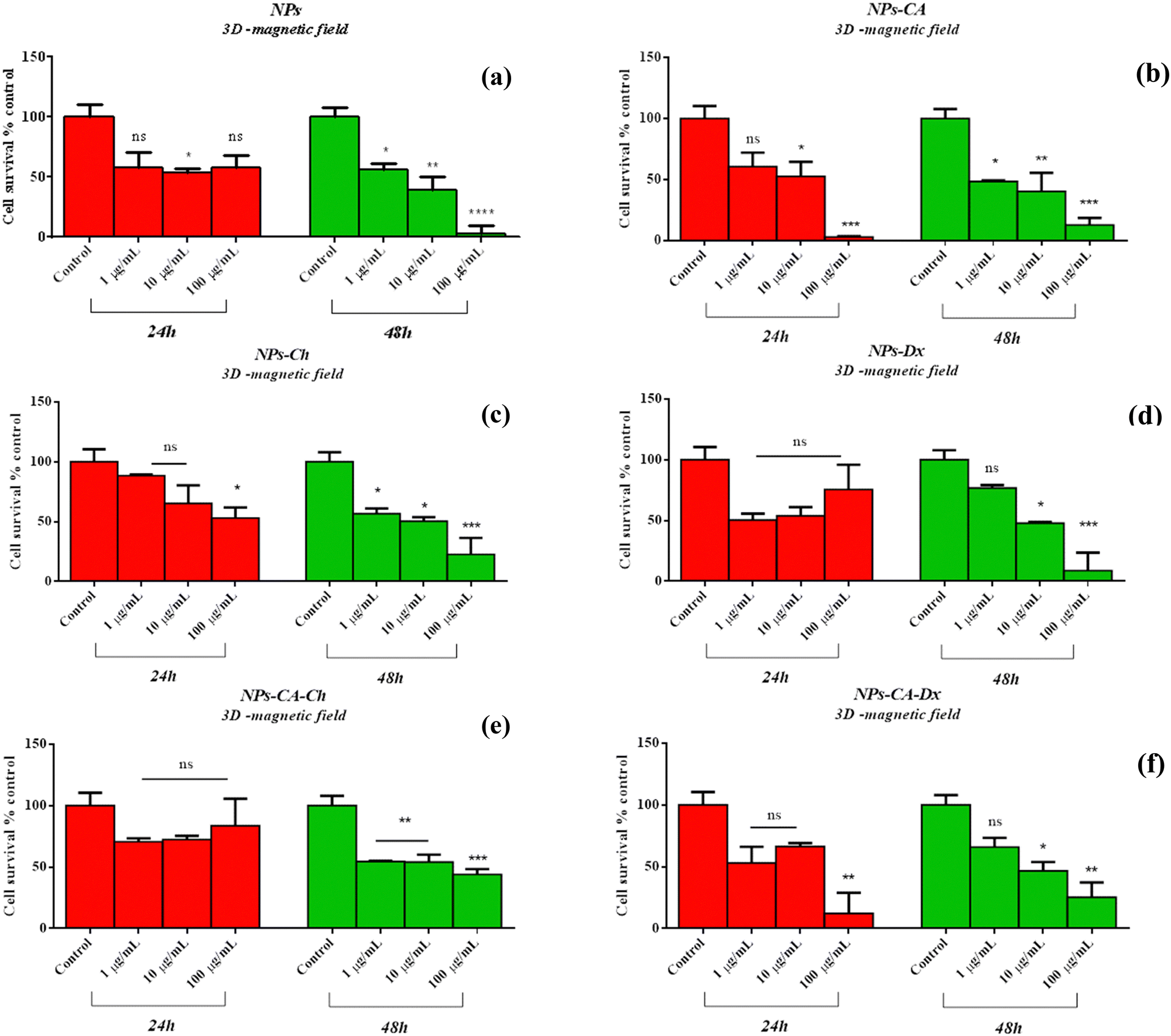 | ||
| Fig. 9 Percentage change of cell survival of the T47D cells (3D spheroids, without magnetic field) following treatment with various concentrations (1–100 μg mL−1) of NPs (a), NPs-CA (b), NPs-Ch (c), NPs-Dx (d), NPs-CA-Ch (e), and NPs-CA-Dx (f) for 24 and 48 h. Values represent the mean value of n = 3 independent experiments. The vertical bars represent the SEMs. *p < 0.05 (significant), **p < 0.01 (highly significant), ***p < 0.001 (extremely significant) and ****p ≤ 0.0001 (extremely significant) and p > 0.05 (non-significant). | ||
Based on the above cytotoxicity results, it can be concluded that the surface modification of the NPs does not alter their biological impact towards cell survival. Time and concentration of incubation emerge as the key parameters to consider. Taking into account that cell transfection and expression of the nucleic acid of interest might take as long as 72 h, the concentrations used in the transfection experiments below will range from 1–10 μg mL−1 for all carriers.
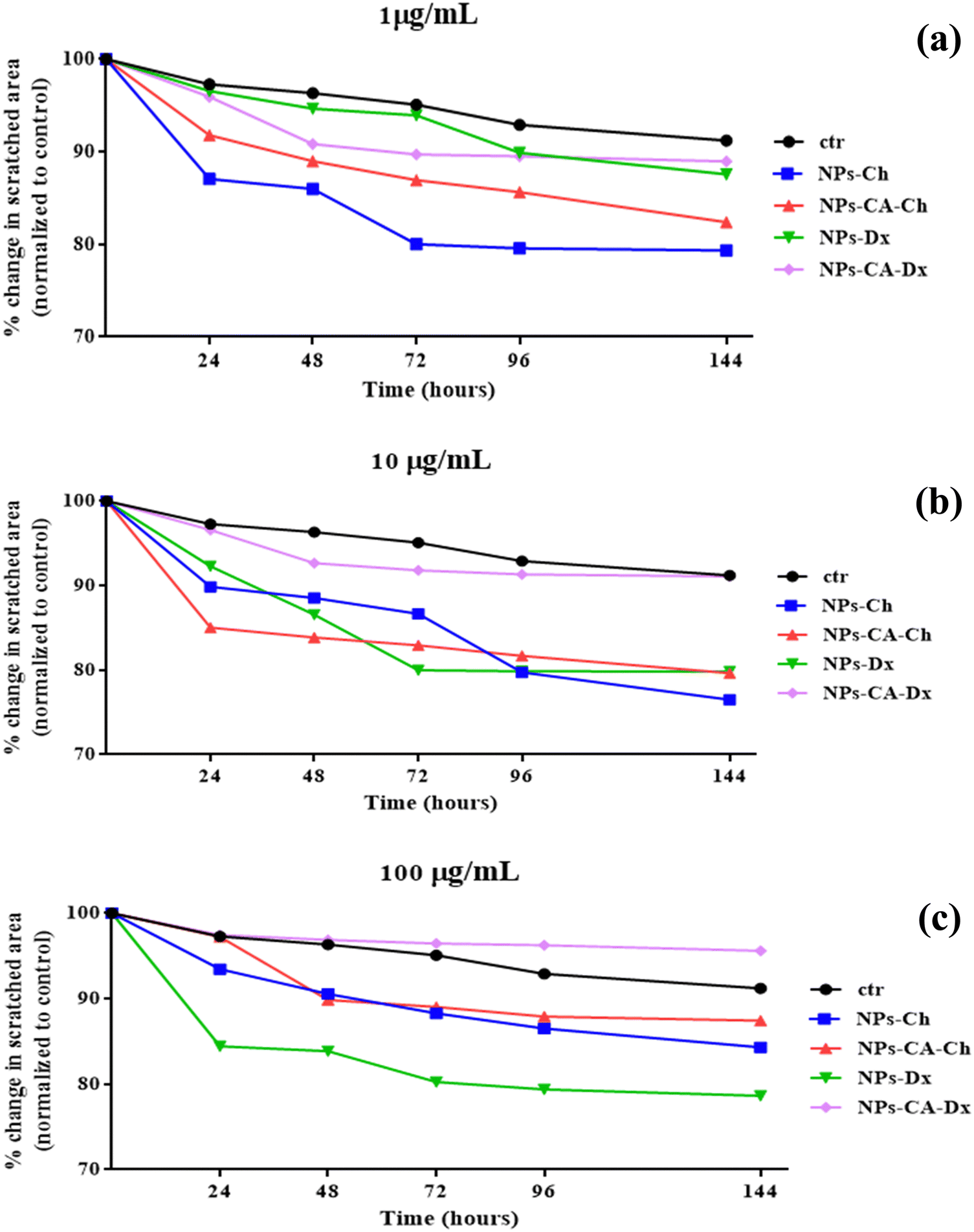 | ||
| Fig. 10 Percent change of the covered area (wound healing) after the in vitro scratch assay for (a) 1, (b) 10 and (c) 100 μg mL−1 of NPs, NPs-CA, NPs-Ch, NPs-Dx, NPs-CA-Ch, and NPs-CA-Dx for 24, 48, 72, 96 and 144 h. | ||
The results showed that the presence of the magnetic field did not induce cell death. Specifically, the EC50 value for NPs-CA-Ch was 123.4 μg mL−1 in the absence and 184.2 μg mL−1 in the presence of the magnetic field. Similarly, the EC50 value for the NPs-CA-Dx sample was found to be ∼220.0 μg mL−1 for both treatment types (presence and absence of the magnetic field) (Fig. 11). As a control sample, cell survival was also assessed solely in the presence of the magnetic field (without NPs), revealing that the cell number is not affected by the exposure to the static magnetic field (data not shown). This observation aligns with the literature where cell viability was generally maintained at high levels upon exposure to a static magnetic field.85 Our data further support the fact that although the application of a static magnetic field may induce faster internalization of the NPs, cell survival is directly proportional to the NP concentration and time of exposure, rather than the presence of the field. Similar results were obtained for all the nanocarriers tested in this study (see Fig. S12 in the ESI†). A slight increase in cell survival in the presence of a magnetic field was also observed, which is intriguing given the anticipated increase in uptake and associated cell toxicity under magnetic field exposure. However, the observed effect can be partially explained considering that the magnetic field could induce the formation of larger aggregates that remain deposited on the cell membrane without actively entering the cell, as also depicted in Fig. S10 and S11 (ESI†).86,87
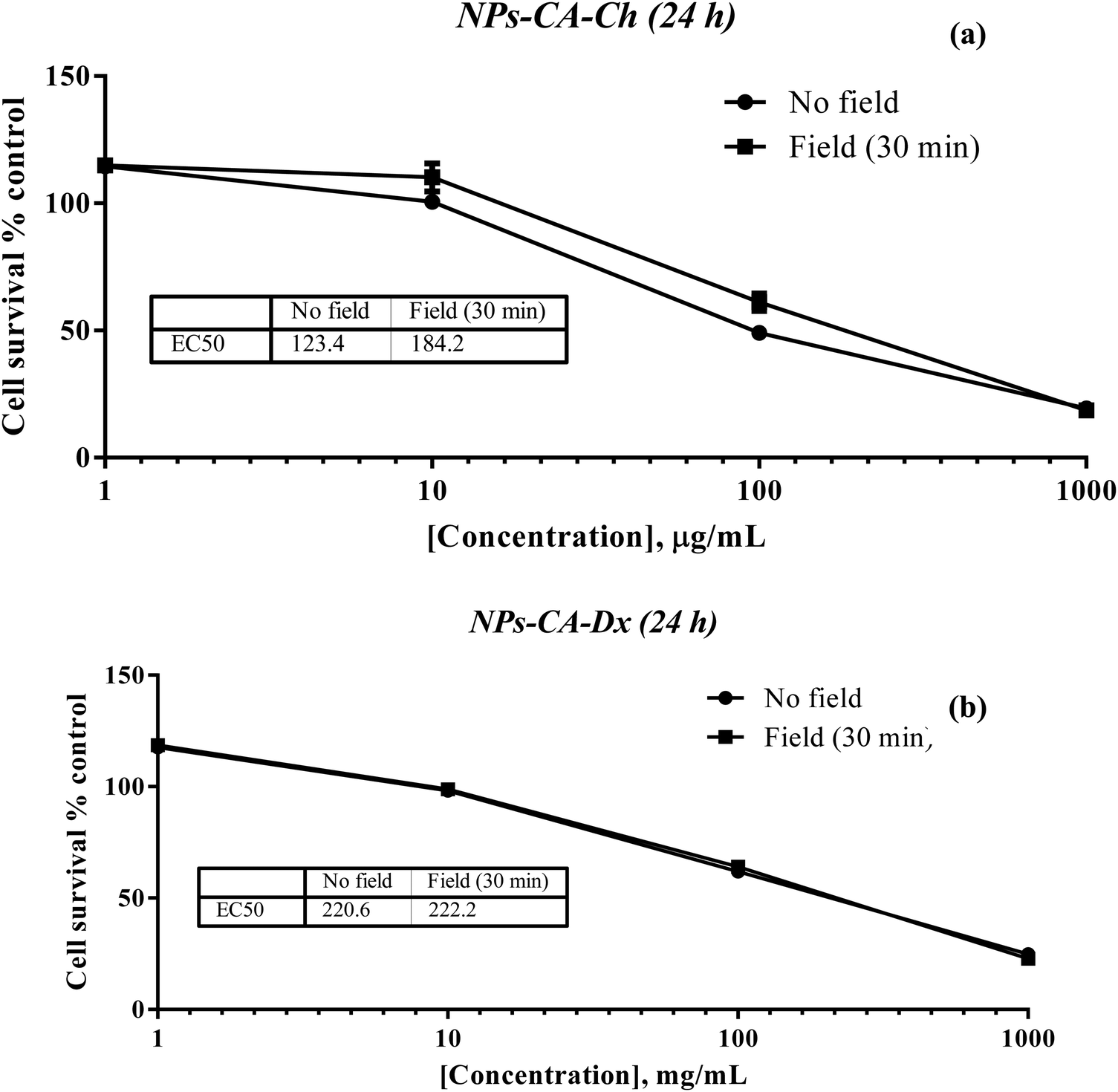 | ||
| Fig. 11 Percentage change of cell survival of the T47D cells (2D monolayers) following treatment with various concentrations (1–1000 μg mL−1) of (a) NPs-CA-Ch and (b) NPs-CA-Dx for 24 h in the presence and absence of a magnetic field. | ||
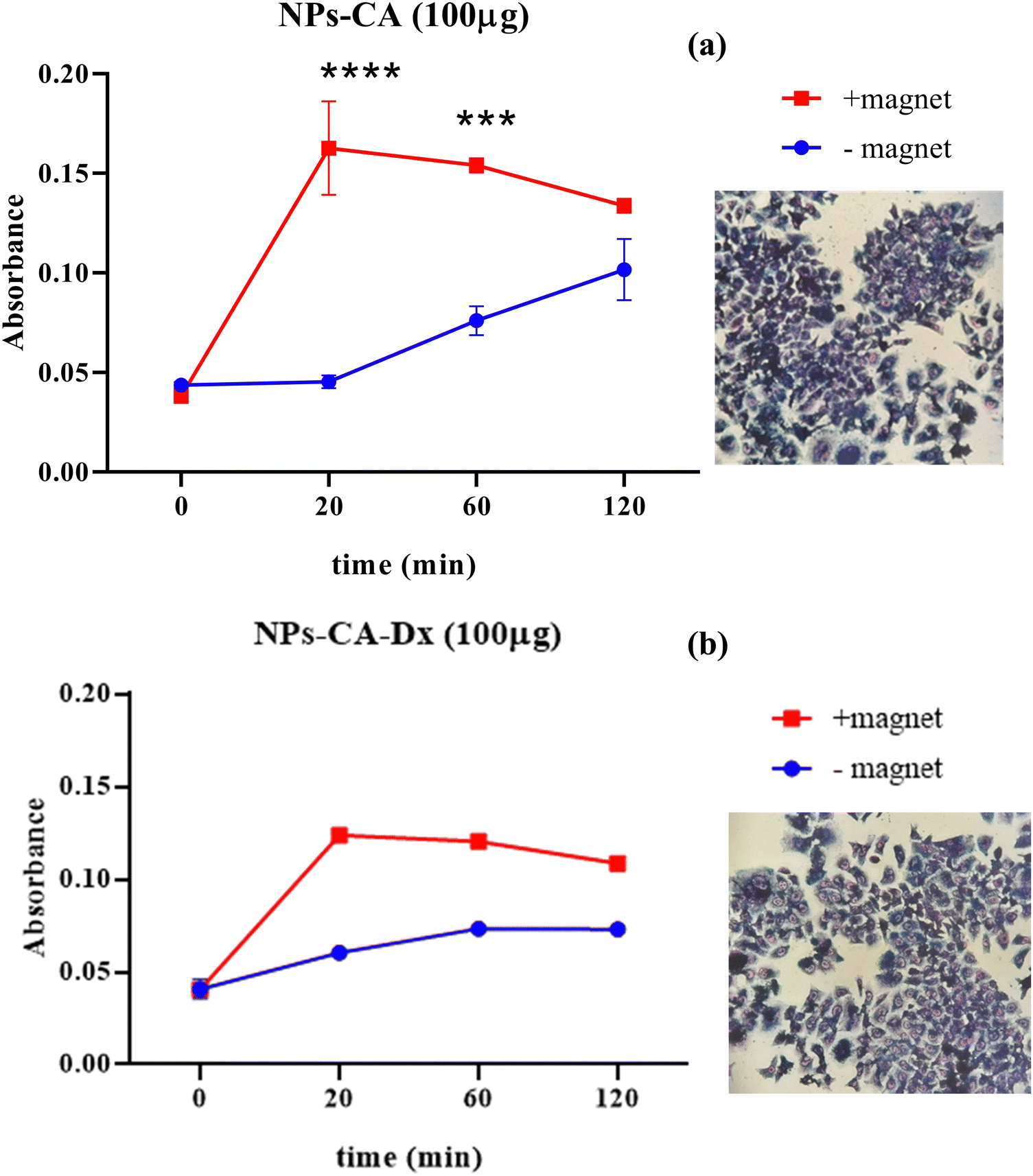 | ||
| Fig. 12 Kinetic response of cellular uptake in the presence and absence of a magnetic field, after treatment with 100 μg mL−1 of (a) NPs-CA and (b) NPs-CA-Dx for t = 0, 20, 60 and 120 min. | ||
3.2.6.1 Assessment of GFP–mRNA binding on the magnetic nanoparticles. The binding of GFP–mRNA onto the magnetic nanoparticles was assessed using native gel electrophoresis. The GFP–mRNA bearing SPIONs were diluted with loading buffer before undergoing electrophoresis in a 1% w/v agarose gel (Fig. S13, ESI†). Our intention was to assess the binding efficiency of GFP–mRNA onto SPIONs and not the estimation of the molecular weight of GFP–mRNA. A denaturing agarose gel would have changed the binding efficiency. The results indicated the successful GFP–mRNA binding on all the NPs tested. It is however noted, that lanes 9–12 appeared more intense indicating that the NPs-CA-Dx sample possesses a greater binding capacity for the nucleic acid. Worth mentioning is also the fact that besides the presence of dextran, and in contrast to chitosan (lane 1 vs. 2), the presence of citric acid also promoted the binding of the nucleic acid (lane 3 vs. lane 11) which was attributed to the effective dispersion of the NPs in the aqueous medium. In conclusion from the above results, on the cell toxicity, solution stability, and mRNA binding, the NPs-Ch, NPs-CA-Ch, NPs-Dx and NPs-CA-Dx SPIONs were selected to further study the mRNA expression in transfected T47D cells.
3.2.6.2 Assessment of the GFP–mRNA expression. The translational efficiency of the NPs-ChGFPmRNA, NPs-DxGFPmRNA, NPs-CA-ChGFPmRNA and NPs-CA-DxGFPmRNA SPIONs was assessed on T47D cells. The commercially available magnetic nanoparticles, polyMag (Magnetofection™, OZ Biosciences), conjugated with GFP–mRNA were used as a positive control for this experiment. The percentage of cells expressing the fluorescent protein and the fluorescence intensity of the cells were measured to quantify both the endocytosis of the nanocarriers and the translational efficiency of the gene. As depicted in Fig. 13, the expression of GFP was observed for all the SPIONs after 72 h of incubation, with the higher expression being observed for the NPs-CA-Dx nanocarriers (a 2.5 fold increase compared to the control case). Furthermore, the latter nanocarrier exhibited superior GFP–mRNA expression compared to PolyMag. Notably, no expression was observed for any of the other three magnetic carriers at 24 and 48 h of incubation. This observation might be related to slower release of the mRNA from the carriers along with increased mRNA stability.
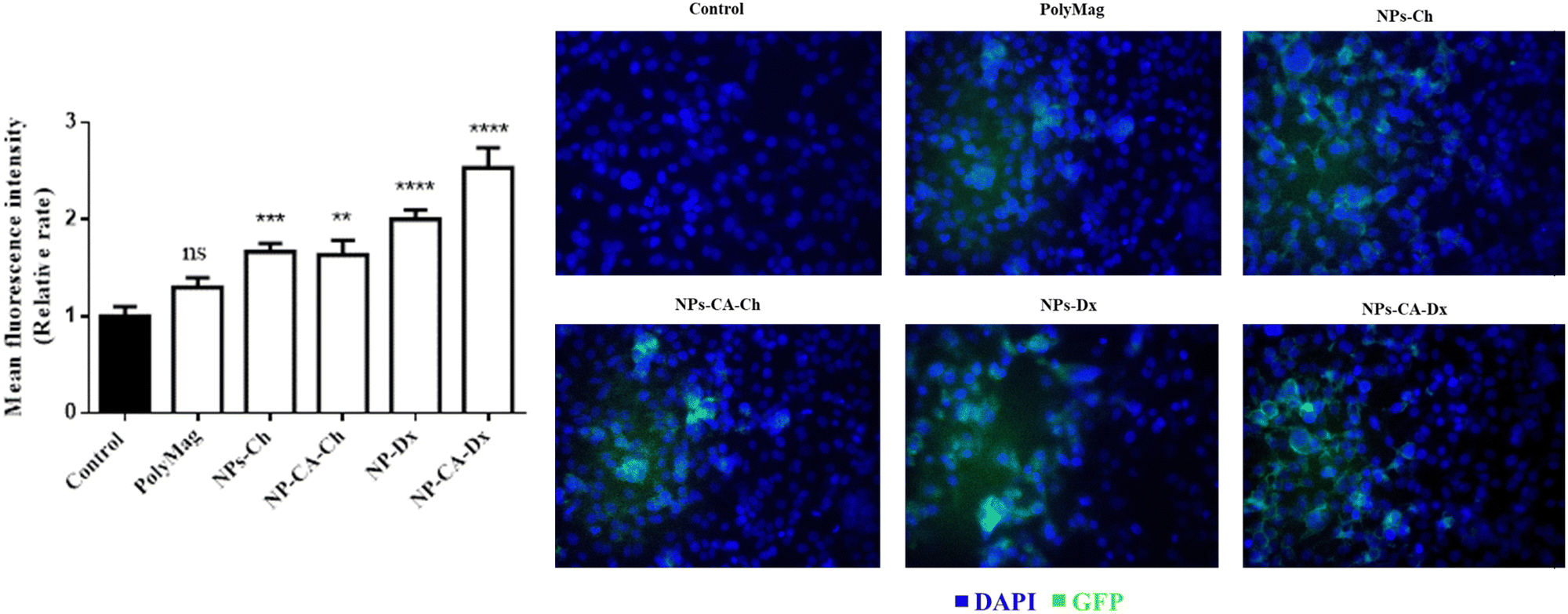 | ||
| Fig. 13 Quantification of mean fluorescence intensity by ImageJ software for the expression of GFP after 72 h of transfection of the T47D cells. | ||
The superior efficiency of the NPs-CA-DxGFPmRNA nanocarrier in the expression of GFP, was attributed to a combination of factors, including the amount of the polysaccharide coating on the NPs, the size and the dispersion stability of the NPs in the aqueous medium, the surface charge of the carriers as well as the covalent type of GFP–mRNA binding. The improved dispersion stability of the NPs-CA-Dx SPIONs in the aqueous media, which resulted in the formation of smaller sized agglomerates, evidenced by DLS, provided a higher surface area for the polysaccharide coating, and therefore, a higher polymer content and enhanced availability of the aldehyde groups of the Ox-dextran to react with the amino groups of GFP–mRNA via covalent imine bond formation. On the other hand, the type of gene binding (ionic vs. covalent) can also significantly affect the gene release mechanism and GFP expression. As shown in Fig. 13, in contrast to the NPs-Ch and NPs-CA-Ch samples, the NPs-Dx and NPs-CA-Dx nanocarriers exhibited an enhanced fluorescent signal, indicating higher protein expression. Cationic nanocarriers is a well-known and extensively studied family of gene transfer agents, however, there are still important open questions concerning the binding stability of the gene complex and the release mechanism of mRNA from the vectors after endosomal uptake. Recent research has shown that the acidic conditions prevalent in the endosome and lysosomes (pH 4.2–6.2) rather favor the complexation of the negatively charged mRNA with the cationic carrier prohibiting the release of gene.34 On the other hand, acid-sensitive bonds, such as the imine bonds employed in this study to bind the gene onto the dextran coated SPIONs, can be easily cleaved under the specific acidic conditions, effectively releasing the bound mRNA, which subsequently escapes into the nucleus. Therefore, the proposed gene delivery nanocarriers present superior gene binding and release properties and could function as more effective gene vectors compared to existing technologies (i.e., cationic Polymag). Moreover, this is a proof of principle study, while current research in our groups involves the functionalization of the optimum nanocarrier with mRNA that translates the production of proteins/antibodies with therapeutic properties such as trastuzumab in Her2+ breast cancer. Finally, the gene vector developed herein, could serve as a promising chemical platform for the enlightening of various cellular functions/mechanism related to the gene release from the endo-lysosomal compartments that remain unclear today.
4. Conclusions
In the current investigation, two distinct types of superparamagnetic nanocarriers coated with natural polysaccharide derivatives, namely Ox-dextran and Q-chitosan, bearing aldehyde and cationic quaternary ammonium salt groups, respectively, were successfully developed. The two types of vectors enabled the distinct nucleic acid binding onto the hybrid SPIONs, namely covalent and ionic, which were evaluated for efficient mRNA-delivery. The nanocarriers were subjected to a selection/rejection criterion for further translational studies based on their safety and effectiveness for use in vitro and their potential in future in vivo pre-clinical applications. These criteria included the: (a) dispersibility in solution/biological fluids, (b) minimal or no effect on cell survival, (c) significant intracellular uptake, (d) ability to bind mRNA and transport it into the cells, and (e) transport and expression of mRNA. The results showed that the biological behavior of the carriers was closely related to their structural and physicochemical profile, with the carriers bearing citric acid exhibiting increased stability and good dispersion in solution, while chitosan-coated particles showed inferior dispersibility in solution. Dextran-coated particles bearing citric acid exhibited the most favorable properties in terms of dispersion stability and polymer coating.Cell survival results on 2D cultures indicated that concentrations of 1 and 10 μg mL−1 of the SPIONs were not toxic to the cells for most carriers used, while higher concentrations of 100 and 1000 μg mL−1 decreased the cell viability in a time- and concentration-dependent manner. In contrast, 3D culture studies showed that the cytotoxicity did not depend on the concentration of the NPs in the first 24 h, but became both time- and concentration-dependent thereafter, contributing to cell cytotoxicity at 48 h. Comparison of the results suggested that the cytotoxicity of the SPIONs in 2D cultures was lower compared to 3D cultures, inferring the possible sensitivity of the beads to monolayer cultures. Morphology and adhesion studies confirmed that all the carriers can be used at concentrations up to 10 μg mL−1.
Furthermore, the effect of a magnetic field on the uptake of the nanoparticles by the cells was shown to accelerate endocytosis of the vectors. The optimal exposure time for the highest endocytosis was found between 20 and 30 min, which aligns with the recommended exposure times of commercially available magnetic carriers (e.g., Magnetofection™, OZ Biosciences). Faster endocytosis of the vectors has several advantages, such as to avoid degradation of the mRNA in the extracellular space before its uptake by the cells. Future work in our groups involve a more detailed experimental assessment of the carriers’ biological behavior in the presence of the magnetic field, which is of great importance for their performance in translational experiments. The impact of the NPs on cell migration was also evaluated using an in vitro scratch migration assay, which suggested that the employed SPIONs may affect the cell motility.
Finally, the mRNA binding capacity of the hybrid SPIONs showed that all four carriers (NPs-Ch, NPs-CA-Ch, NPs-Dx and NPs-CA-Dx) exhibited effective binding of the GFP–mRNA, with NPs-CA-Dx presenting the higher binding capacity for the nucleic acid. The Ox-dextran-based nanocarriers were also shown to promote the nucleic acid expression of GFP protein in translational studies. The primary factors that augmented the effectiveness of the NPs-CA-Dx nanocarrier were (1) the smaller size of the agglomerates and the higher polymer content and (2) the type of GFP–mRNA binding via the formation of acid-labile Schiff base linkages between the gene and Ox-dextran. The carrier developed in the current study can be further functionalized with mRNA that translates to proteins/antibodies with therapeutic properties as in the case of trastuzumab antibody in Her2+ breast cancer.
In conclusion, the proposed delivery system shows great promise as a chemical platform for gene delivery and expression in future in vivo preclinical studies and sets the basis for the tissue-specific delivery of mRNA(s) with therapeutic properties, such as trastuzumab mRNA in breast cancer cells, which is ongoing work in our labs.
Author contributions
Conceptualization (OT, MP, LN, MV, CC); methodology (OT, MP, GK, MV, CC); validation (OT, MP, GK, EP, RP, VZ, TCP, MV, CC); formal analysis (OT, MP, GK, EP, RP, MV, CC); investigation (OT, MP, GK, VZ, TCP, MV, CC); rresources (CS); data curation (OT, MP, GK, EP, RP); writing – original draft (OT, MP, GK, MV, CC); writing – review & editing (OT, MP, GK, MV, CC); visualization (OT, MP, GK, EP, RP); supervision (MV, CC); project administration (CC); funding acquisition (AO).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research has been co-financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code: T1EDK-02775). “The publication of the article in OA mode was financially supported by HEAL-Link”.References
- G. Cannon, P. Gupta, F. Gomes, J. Kerner, W. Parra, E. Weiderpass, J. Kim, M. Moor, C. Sutcliffe and S. Sutcliffe, ICCC-4 Working Group. Prevention of cancer and non-communicable diseases, Asian Pac. J. Cancer Prev., 2012, 13(4 Suppl.), 3–11 Search PubMed.
- P. Kanavos, The rising burden of cancer in the developing world, Ann. Oncol., 2006, 17(Suppl. 8), viii15–viii23, DOI:10.1093/annonc/mdl983.
- Z. Tao, A. Shi, C. Lu, T. Song, Z. Zhang and J. Zhao, Breast Cancer: Epidemiology and Etiology, Cell Biochem. Biophys., 2015, 72(2), 333–338, DOI:10.1007/s12013-014-0459-6.
- T. A. Moo, R. Sanford, C. Dang and M. Morrow, Overview of Breast Cancer Therapy, PET Clin., 2018, 13(3), 339–354, DOI:10.1016/j.cpet.2018.02.006.
- F. A. Fisusi and E. O. Akala, Drug Combinations in Breast Cancer Therapy, Pharm. Nanotechnol., 2019, 7(1), 3–23, DOI:10.2174/2211738507666190122111224.
- N. Harbeck, F. Penault-Llorca, J. Cortes, M. Gnant and N. Houssami, Breast cancer, Nat. Rev. Dis. Primers, 2019, 5, 66, DOI:10.1038/s41572-019-0111-2.
- J. J. Tao, K. Visvanathan and A. C. Wolff, Long term side effects of adjuvant chemotherapy in patients with early breast cancer, Breast, 2015, 24(Suppl. 2(2)), 149–153, DOI:10.1016/j.breast.2015.07.035.
- J. C. Burnett and J. J. Rossi, RNA-based therapeutics: current progress and future prospects, Chem. Biol., 2012, 19(1), 60–71, DOI:10.1016/j.chembiol.2011.12.008.
- A. R. MacLeod and S. T. Crooke, RNA Therapeutics in Oncology: Advances, Challenges, and Future Directions, J. Clin. Pharmacol., 2017, 57(Suppl. 10), S43–S59, DOI:10.1002/jcph.957.
- U. Sahin, K. Karikó and Ö. Türeci, mRNA-based therapeutics-developing a new class of drugs, Nat. Rev. Drug Discovery, 2014, 13, 759–780, DOI:10.1038/nrd4278.
- R. K. Oldham, Monoclonal antibodies in cancer therapy, J. Clin. Oncol., 1983, 1(9), 582–590, DOI:10.1200/JCO.1983.1.9.582.
- J. Capdevila, E. Elez, T. Macarulla, F. J. Ramos, M. Ruiz-Echarri and J. Tabernero, Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment, Cancer Treat. Rev., 2009, 35(4), 354–363, DOI:10.1016/j.ctrv.2009.02.001.
- C. L. Vogel, M. A. Cobleigh, D. Tripathy, J. C. Gutheil, L. N. Harris, L. Fehrenbacher, D. J. Slamon, M. Murphy, W. F. R. Novotny, M. Burchmore, S. Shak and S. J. Stewart, First-Line Herceptin® Monotherapy in Metastatic Breast Cancer, Oncology, 2001, 61(2), 37–42, DOI:10.1159/000055400.
- J. Klastersky, Adverse effects of the humanized antibodies used as cancer therapeutics, Curr. Opin. Oncol., 2006, 18(4), 316–320, DOI:10.1097/01.cco.0000228734.32261.62.
- M. Ramamoorth and A. Narvekar, Non-viral vectors in gene therapy- an overview, J. Clin. Diagn. Res., 2015, 9(1), GE01–GE06, DOI:10.7860/JCDR/2015/10443.5394.
- A. Zakeri, M. Amin, J. Kouhbanani, N. Beheshtkhoo, V. Beigi, S. M. Mousavi, S. Ali, R. Hashemi, K. Zade, A. M. Amani, A. Savardashtaki, E. Mirzaei, S. Jahandideh, A. Movahedpour, V. Beigi, S. M. Mousavi, S. Ali, R. Hashemi, A. K. Zade, M. Amani, A. Savardashtaki, E. Mirzaei, S. Jahandideh and A. Movahedpour, Polyethylenimine-based nanocarriers in co-delivery of drug and gene: a developing horizon, Nano Rev. Exp., 2018, 9, 1488497, DOI:10.1080/20022727.2018.1488497.
- A. P. Pandey and K. K. Sawant, Polyethylenimine: A versatile, multifunctional non-viral vector for nucleic acid delivery, Mater. Sci. Eng., C, 2016, 68, 904–918, DOI:10.1016/j.msec.2016.07.066.
- J. Karlsson, K. R. Rhodes, J. J. Green and S. Y. Tzeng, Poly(beta-amino ester)s as gene delivery vehicles: challenges and opportunities, Expert Opin. Drug Delivery, 2021, 17, 1395–1410, DOI:10.1080/17425247.2020.1796628.
- M. Borhaninia, M. Zahiri, K. Abnous, S. M. Taghdisi, M. Ramezani and M. Alibolandi, Self-targeted hyaluronic acid-b-poly (β-amino ester) pH-switchable polymersome for guided doxorubicin delivery to metastatic breast cancer, Int. J. Biol. Macromol., 2023, 248, 125882, DOI:10.1016/j.ijbiomac.2023.125882.
- F. Abedi-Gaballu, G. Dehghan, M. Ghaffari, R. Yekta, S. Abbaspour-Ravasjani, B. Baradaran, J. Ezzati Nazhad Dolatabadi and M. R. Hamblin, PAMAM dendrimers as efficient drug and gene delivery nanosystems for cancer therapy, Appl. Mater. Today, 2018, 12, 177–190, DOI:10.1016/j.apmt.2018.05.002.
- N. Bono, C. Pennetta, M. C. Bellucci, A. Sganappa, C. Malloggi, G. Tedeschi, G. Candiani and A. Volonterio, Role of Generation on Successful DNA Delivery of PAMAM − (Guanidino) Neomycin Conjugates, Appl. Mater. Today, 2019, 12, 177–190, DOI:10.1021/acsomega.8b02757.
- A. M. Beagan, A. A. Alghamdi, S. S. Lahmadi, M. A. Halwani, M. S. Almeataq, A. N. Alhazaa, K. M. Alotaibi and A. M. Alswieleh, Folic Acid-Terminated Poly(2-Diethyl Amino Ethyl Methacrylate) Brush-Gated Magnetic Mesoporous Nanoparticles as a Smart Drug Delivery System, Polymers, 2020, 13(1), 59, DOI:10.3390/polym13010059.
- T. K. Georgiou, M. Vamvakaki, L. A. Phylactou and C. S. Patrickios, Synthesis, Characterization, and Evaluation as Transfection Reagents of Double-Hydrophilic Star Copolymers: Effect of Star Architecture, Biomacromolecules, 2005, 6(6), 2990–2997, DOI:10.1021/bm050307w.
- J. M. Dang and K. W. Leong, Natural polymers for gene delivery and tissue engineering, Adv. Drug Delivery Rev., 2006, 58, 487–499, DOI:10.1016/j.addr.2006.03.001.
- V. Sagar, S. Tavakol, S. Moghassemi, A. Dadashzadeh, J. D. Schneible, I. Fatemi, A. Shirvani, A. Zarrabi and F. Azedi, Chitosan: A versatile bio-platform for breast cancer theranostics, J. Controlled Release, 2022, 341, 733–752, DOI:10.1016/j.jconrel.2021.12.012.
- H. Ishikawa, Y. Nakamura, J. Jo and Y. Tabata, Biomaterials Gelatin nanospheres incorporating siRNA for controlled intracellular release, Biomaterials, 2012, 33, 9097–9104, DOI:10.1016/j.biomaterials.2012.08.032.
- S. Guan and J. Rosenecker, Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems, Gene Ther., 2017, 24, 133–143, DOI:10.1038/gt.2017.5.
- A. P. Singh, A. Biswas and A. Shukla, et al., Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles, Signal Transduction Targeted Ther., 2019, 4, 33, DOI:10.1038/s41392-019-0068-3.
- M. J. Mitchell, M. M. Billingsley and R. M. Haley, et al., Engineering precision nanoparticles for drug delivery, Nat. Rev. Drug Discovery, 2021, 20, 101–124, DOI:10.1038/s41573-020-0090-8.
- M. Vallet-Regí, M. Colilla, I. Izquierdo-Barba and M. Manzano, Mesoporous silica nanoparticles for drug delivery: Current insights, Molecules, 2018, 23, 1–19, DOI:10.3390/molecules23010047.
- F. Tang, L. Li and D. Chen, Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery, Adv. Mater., 2012, 24, 1504–1534, DOI:10.1002/adma.201104763.
- M. Fan, Y. Han, S. Gao, H. Yan, L. Cao, Z. Li, X. J. Liang and J. Zhang, Ultrasmall gold nanoparticles in cancer diagnosis and therapy, Theranostics, 2020, 10, 494–4957, DOI:10.7150/thno.42471.
- S. H. Tseng, M. Y. Chou and I. M. Chu, Cetuximab-conjugated iron oxide nanoparticles for cancer imaging and therapy, Int. J. Nanomed., 2015, 10, 3663–3685, DOI:10.2147/IJN.S80134.
- Z. Wu and T. Li, Nanoparticle-Mediated Cytoplasmic Delivery of Messenger RNA Vaccines: Challenges and Future Perspectives, Pharm. Res., 2021, 38, 473–478, DOI:10.1007/s11095-021-03015-x.
- J. Dulińska-Litewka, A. Łazarczyk, P. Hałubiec, O. Szafrański, K. Karnas and A. Karewicz, Superparamagnetic Iron Oxide Nanoparticles-Current and Prospective Medical Applications, Materials, 2019, 12(4), 617, DOI:10.3390/ma12040617.
- C. Janko, T. Ratschker and K. Nguyen, et al., Functionalized Superparamagnetic Iron Oxide Nanoparticles (SPIONs) as Platform for the Targeted Multimodal Tumor Therapy, Front. Oncol., 2019, 9, 59, DOI:10.3389/fonc.2019.00059.
- L. Zhu, Z. Zhou, H. Mao and L. Yang, Magnetic nanoparticles for precision oncology: theranostic magnetic iron oxide nanoparticles for image-guided and targeted cancer therapy, Nanomedicine, 2017, 12(1), 73–87, DOI:10.2217/nnm-2016-0316.
- S. K. Murthy, Nanoparticles in modern medicine: state of the art and future challenges, Int. J. Nanomed., 2007, 2(2), 129–141 CAS.
- S. D. Anderson, V. V. Gwenin and C. D. Gwenin, Magnetic Functionalized Nanoparticles for Biomedical, Drug Delivery and Imaging Applications, Nanoscale Res. Lett., 2019, 14, 188, DOI:10.1186/s11671-019-3019-6.
- I. Khan, K. Saeed and I. Khan, Nanoparticles: Properties, applications and toxicities, Arabian J. Chem., 2019, 12(7), 908–931, DOI:10.1016/j.arabjc.2017.05.011.
- A. C. Anselmo and S. Mitragotri, Nanoparticles in the clinic, Bioeng. Transl. Med., 2016, 1(1), 10–29, DOI:10.1002/btm2.10003.
- A. C. Anselmo and S. Mitragotri, Nanoparticles in the clinic: An update, Bioeng. Transl. Med., 2019, 4(3), e10143, DOI:10.1002/btm2.10143.
- H. Huang, W. Feng, Y. Chen and J. Shi, Inorganic nanoparticles in clinical trials and translations, Nano Today, 2020, 35, 100972, DOI:10.1016/j.nantod.2020.100972.
- S. Hua, M. B. C. de Matos, J. M. Metselaar and G. Storm, Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization, Front. Pharmacol., 2018, 9, 790, DOI:10.3389/fphar.2018.00790.
- L. Nalbandian, E. Patrikiadou, V. Zaspalis, A. Patrikidou, E. Hatzidaki and C. N. Papandreou, Magnetic Nanoparticles in Medical Diagnostic Applications: Synthesis, Characterization and Proteins Conjugation, Curr. Nanosci., 2015, 12, 455–468, DOI:10.2174/1573413712666151210230002.
- S. Palanisamy and Y. M. Wang, Superparamagnetic iron oxide nanoparticulate system: Synthesis, targeting, drug delivery and therapy in cancer, Dalton Trans., 2019, 48, 9490–9515, 10.1039/c9dt00459a.
- S. Uthaman, S. J. Lee, K. Cherukula, C. S. Cho and I. K. Park, Polysaccharide-coated magnetic nanoparticles for imaging and gene therapy, Biomed. Res. Int., 2015, 959175, DOI:10.1155/2015/959175.
- M. P. A. Ferreira, V. Talman, G. Torrieri, D. Liu, G. Marques, K. Moslova, Z. Liu, J. F. Pinto, J. Hirvonen, H. Ruskoaho and H. A. Santos, Dual-Drug Delivery Using Dextran-Functionalized Nanoparticles Targeting Cardiac Fibroblasts for Cellular Reprogramming, Adv. Funct. Mater., 2018, 28, 1705134, DOI:10.1002/adfm.201705134.
- C. Hu, L. Long, J. Cao, S. Zhang and Y. Wang, Dual-crosslinked mussel-inspired smart hydrogels with enhanced antibacterial and angiogenic properties for chronic infected diabetic wound treatment via pH-responsive quick cargo release, Chem. Eng. J., 2021, 411, 128564, DOI:10.1016/j.cej.2021.128564.
- F. Shamsipour, A. H. Zarnani, R. Ghods, M. Chamankhah, F. Forouzesh, S. Vafaei, A. A. Bayat, M. M. Akhondi, M. Ali Oghabian and M. Jeddi-Tehrani, Conjugation of Monoclonal Antibodies to Super Paramagnetic Iron Oxide Nanoparticles for Detection of her2/neu Antigen on Breast Cancer Cell Lines, Avicenna J. Med. Biotechnol., 2009, 1, 27–31 CAS.
- M. Psarrou, A. Mitraki and M. Vamvakaki, Stimuli-Responsive Polysaccharide Hydrogels and Their Composites for Wound Healing Applications, Polymers, 2023, 15(4), 986, DOI:10.3390/polym15040986.
- X. N. Pham, T. P. Nguyen, T. N. Pham, T. T. N. Tran and T. V. T. Tran, Synthesis and characterization of chitosan-coated magnetite nanoparticles and their application in curcumin drug delivery, Adv. Nat. Sci. Nanosci. Nanotechnol., 2016, 7(4), 045010, DOI:10.1088/2043-6262/7/4/045010.
- I. Khmara, O. Strbak, V. Zavisova, M. Koneracka, M. Kubovcikova, I. Antal, V. Kavecansky, D. Lucanska, D. Dobrota and P. Kopcansky, Chitosan-stabilized iron oxide nanoparticles for magnetic resonance imaging, J. Magn. Magn. Mater., 2019, 474, 319–325, DOI:10.1016/j.jmmm.2018.11.026.
- B. F. M. Kievit, O. Veiseh, N. Bhattarai, C. Fang, W. Gunn, D. Lee, R. G. Ellenbogen and J. M. Olson, PEI-PEG-Chitosan Copolymer Coated Iron Oxide Nanoparticles for Safe Gene Delivery: synthesis, complexation, and transfection, Adv. Funct. Mater., 2009, 19(14), 2244–2251, DOI:10.1002/adfm.200801844.
- L. A. Frank, G. R. Onzi, A. S. Morawski, A. R. Pohlmann, S. S. Guterres and R. V. Contri, Chitosan as a coating material for nanoparticles intended for biomedical applications, React. Funct. Polym., 2020, 147, 104459, DOI:10.1016/j.reactfunctpolym.2019.104459.
- R. P. Dhavale, R. P. Dhavale, S. C. Sahoo, P. Kollu, S. U. Jadhav, P. S. Patil, T. D. Dongale and A. D. Chougale, Patil, Chitosan coated magnetic nanoparticles as carriers of anticancer drug Telmisartan: pH-responsive controlled drug release and cytotoxicity studies, J. Phys. Chem. Solids, 2021, 148, 109749, DOI:10.1016/j.jpcs.2020.109749.
- Y. Cao, Y. F. Tan, Y. S. Wong, M. W. J. Liew and S. Venkatraman, Recent advances in chitosan-based carriers for gene delivery, Mar. Drugs, 2019, 17(6), 381, DOI:10.3390/md17060381.
- V. Bansal, P. K. Sharma, N. Sharma, O. P. Pal and R. Malviya, Applications of Chitosan and Chitosan Derivatives in Drug Delivery, Biol. Res., 2011, 5, 28–37, DOI:10.1007/12.
- Y. Wen, Z. Tan, F. Sun, L. Sheng, X. Zhang and F. Yao, Synthesis and characterization of quaternized carboxymethyl chitosan/poly(amidoamine) dendrimer core-shell nanoparticles, Mater. Sci. Eng., C, 2012, 32, 2026–2036, DOI:10.1016/j.msec.2012.05.019.
- Y. Pakzad, M. Fathi, Y. Omidi, M. Mozafari and A. Zamanian, Synthesis and characterization of timolol maleate-loaded quaternized chitosan-based thermosensitive hydrogel: A transparent topical ocular delivery system for the treatment of glaucoma, Int. J. Biol. Macromol., 2020, 159, 117–128, DOI:10.1016/j.ijbiomac.2020.04.274.
- K. Y. Chen and S. Y. Zeng, Fabrication of Quaternized Chitosan Nanoparticles Using Tripolyphosphate/Genipin Dual Cross-Linkers as a Protein Delivery System, Polymers, 2018, 10(11), 1226, DOI:10.3390/polym10111226.
- L. Nalbandian, E. Patrikiadou, V. Zaspalis, A. Patrikidou, E. Hatzidaki and C. N. Papandreou, Magnetic Nanoparticles in Medical Diagnostic Applications: Synthesis, Characterization and Proteins Conjugation, Curr. Nanosci., 2016, 12(4), 455–468, DOI:10.2174/1573413712666151210230002.
- N. Artzi, T. Shazly, C. Crespo, A. B. Ramos, H. K. Chenault and E. R. Edelman, Characterization of star adhesive sealants based on PEG/Dextran hydrogels, Macromol. Biosci., 2009, 9, 754–765, DOI:10.1002/mabi.200800355.
- V. Vasilopoulos, M. Pitou, I. Fekas, R. Papi, A. Ouranidis, E. Pavlidou, P. Patsalas and T. Choli-Papadopoulou, Graphene-Wrapped Copper Nanoparticles: An Antimicrobial and Biocompatible Nanomaterial with Valuable Properties for Medical Uses, ACS Omega, 2020, 5(41), 26329–26334, DOI:10.1021/acsomega.0c00834.
- O. Tsave and A. Salifoglou, Biomimetic activity of soluble, well-defined, aqueous Ti(IV)-citrate species toward adipogenesis. An in vitro study, J. Inorg. Biochem., 2021, 214, 111290, DOI:10.1016/j.jinorgbio.
- C. T. Rueden, J. Schindelin and M. C. Hiner, et al., ImageJ2: ImageJ for the next generation of scientific image data, BMC Bioinf., 2017, 18, 529, DOI:10.1186/s12859-017-1934-z.
- W. Zhang, S. Cao, S. Liang, C. H. Tan, B. Luo, X. Xu and P. E. Saw, Differently Charged Super-Paramagnetic Iron Oxide Nanoparticles Preferentially Induced M1-Like Phenotype of Macrophages, Front. Bioeng. Biotechnol., 2020, 8, 537, DOI:10.3389/fbioe.2020.00537.
- A. Makridis, M. Tziomaki, K. Topouridou, M. P. Yavropoulou, J. G. Yovos, O. Kalogirou, T. Samaras and M. Angelakeris, A novel strategy combining magnetic particle hyperthermia pulses with enhanced performance binary ferrite carriers for effective in vitro manipulation of primary human osteogenic sarcoma cells, Int. J. Hyperthermia, 2016, 32(7), 778–785, DOI:10.1080/02656736.2016.1216183.
- O. N. Shebanova and P. Lazor, Raman study of magnetite (Fe3O4):laser-induced thermal effects and oxidation, J. Raman Spectrosc., 2003, 34, 845–852, DOI:10.1002/jrs.1056.
- A. M. Jubb and H. C. Allen, Supporting Information “Vibrational Spectroscopic Characterization of Hematite, Maghemite, and Magnetite Thin Films Produced by Vapor Deposition”, ACS Appl. Mater. Interfaces, 2010, 2(10), 2804–2812, DOI:10.1021/am1004943.
- Y. Wan, B. Peppley, K. A. M. Creber and V. T. Bui, Anion-exchange membranes composed of quaternized-chitosan derivatives for alkaline fuel cells, J. Power Sources, 2010, 195, 3785–3793, DOI:10.1016/j.jpowsour.2009.11.123.
- W. Tan, Q. Li, F. Dong, Q. Chen and Z. Guo, Preparation and Characterization of Novel Cationic Chitosan Derivatives Bearing Quaternary Ammonium and Phosphonium Salts and Assessment of Their Antifungal Properties, Molecules, 2017, 22(9), 1438, DOI:10.3390/molecules22091438.
- T. Chivangkul, S. Pengprecha, P. Padungros, K. Siraleartmukul, S. Prasongsuk and N. Muangsin, Enhanced water-solubility and mucoadhesion of N,N,N-trimethyl-N-gluconate-N-homocysteine thiolactone chitosan, Carbohydr. Polym., 2014, 108, 224–231, DOI:10.1016/j.carbpol.2014.02.078.
- J. Maia, R. A. Carvalho, J. F. J. Coelho, P. N. Simões and M. H. Gil, Insight on the periodate oxidation of dextran and its structural vicissitudes, Polymer, 2011, 52, 258–265, DOI:10.1016/j.polymer.2010.11.058.
- J. Maia, L. Ferreira, R. Carvalho, M. A. Ramos and M. H. Gil, Synthesis and characterization of new injectable and degradable dextran-based hydrogels, Polymer, 2005, 46, 9604–9614, DOI:10.1016/j.polymer.2005.07.089.
- S. Zhang, T. Lü, D. Qi, Z. Cao, D. Zhang and H. Zhao, Synthesis of quaternized chitosan-coated magnetic nanoparticles for oil-water separation, Mater. Lett., 2017, 191, 128–131, DOI:10.1016/j.matlet.2016.12.092.
- J. L. Arias, L. H. Reddy and P. Couvreur, Fe3O4/chitosan nanocomposite for magnetic drug targeting to cancer, J. Mater. Chem., 2012, 22, 7622–7632, 10.1039/c2jm15339d.
- Y. Xie, D. Liu, C. Cai, X. Chen, Y. Zhou, L. Wu, Y. Sun, H. Dai, X. Kong and P. Liu, Size-dependent cytotoxicity of Fe3O4 nanoparticles induced by biphasic regulation of oxidative stress in different human hepatoma cells, Int. J. Nanomed., 2016, 29(11), 3557–3570, DOI:10.2147/IJN.S105575.
- R. Samavini, C. Sandaruwan, M. De Silva, G. Priyadarshana, N. Kottegoda and V. Karunaratne, Effect of Citric Acid Surface Modification on Solubility of Hydroxyapatite Nanoparticles, J. Agric. Food Chem., 2018, 66(13), 3330–3337, DOI:10.1021/acs.jafc.7b05544.
- K. Nguyen, B. Nuß, M. Mühlberger, H. Unterweger, R. P. Friedrich, C. Alexiou and C. Janko, Superparamagnetic Iron Oxide Nanoparticles Carrying Chemotherapeutics Improve Drug Efficacy in Monolayer and Spheroid Cell Culture by Enabling Active Accumulation, Nanomaterials, 2020, 10(8), 1577, DOI:10.3390/nano10081577.
- U. De Simone, M. Roccio, L. Gribaldo, A. Spinillo, F. Caloni and T. Coccini, Human 3D Cultures as Models for Evaluating Magnetic Nanoparticle CNS Cytotoxicity after Short- and Repeated Long-Term Exposure, Int. J. Mol. Sci., 2018, 19(7), 1993, DOI:10.3390/ijms19071993.
- A. A. Khalili and M. R. Ahmad, A Review of Cell Adhesion Studies for Biomedical and Biological Applications, Int. J. Mol. Sci., 2015, 16(8), 18149–18184, DOI:10.3390/ijms160818149.
- M. M. Montt-Guevara, J. E. Shortrede, M. S. Giretti, A. Giannini, P. Mannella, E. Russo, A. D. Genazzani and T. Simoncini, Androgens Regulate T47D Cells Motility and Invasion through Actin Cytoskeleton Remodeling, Front. Endocrinol., 2016, 7, 136, DOI:10.3389/fendo.2016.00136.
- X. Li, Z. Wei, H. Lv, L. Wu, Y. Cui, H. Yao, J. Li, H. Zhang, B. Yang and J. Jiang, Iron oxide nanoparticles promote the migration of mesenchymal stem cells to injury sites, Int. J. Nanomed., 2019, 14, 573–589, DOI:10.2147/IJN.S184920.
- A. R. Tsiapla, V. Uzunova, T. Oreshkova, M. Angelakeris, T. Samaras, O. Kalogirou and R. Tzoneva, Cell Behavioral Changes after the Application of Magneto-Mechanical Activation to Normal and Cancer Cells, Magnetochemistry, 2022, 8(21), 21, DOI:10.3390/magnetochemistry8020021.
- M. Balk, T. Haus, J. Band, H. Unterweger, E. Schreiber, R. P. Friedrich, C. Alexiou and A. O. Gostian, Cellular SPION Uptake and Toxicity in Various Head and Neck Cancer Cell Lines, Nanomaterials, 2021, 11(3), 726, DOI:10.3390/nano11030726.
- V. Prigiobbe, S. Ko, C. Huh and S. L. Bryant, Measuring and modeling the magnetic settling of superparamagnetic nanoparticle dispersions, J. Colloid Interface Sci., 2015, 447, 58–67, DOI:10.1016/j.jcis.2015.01.056.
Footnotes |
| † Electronic supplementary information (ESI) available: Raman, 1H NMR, and FTIR spectra, TEM images, XRD diagram, 3D culture images, cell adhesion and morphology images and EC50 measurements. See DOI: https://doi.org/10.1039/d4ma00010b |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2024 |