
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel approach to enhancing the anticancer efficacy of methyl jasmonate with PEG-incorporated cationic polymers†
Nishant
Kumar
 ab,
Pratibha
Singh
ac,
Dandan
Zhao
a,
Robin
Rajan
*a and
Kazuaki
Matsumura
*a
ab,
Pratibha
Singh
ac,
Dandan
Zhao
a,
Robin
Rajan
*a and
Kazuaki
Matsumura
*a
aSchool of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: robin@jaist.ac.jp; mkazuaki@jaist.ac.jp
bResearch Center for Macomolcules and Biomaterials, Data-driven Polymer Design Group, National Institute for Materials Science, Tsukuba, Japan
cDepartment of Chemistry, The University of Manchester, Manchester, UK
First published on 24th May 2024
Abstract
Methyl jasmonate (MJ) is a known hexokinase-II inhibitor with notable anticancer efficacy; however, its polymerisation for use as an anticancer agent has not been reported. Using an innovative synthetic methodology, we explored the conjugation of MJ to a cationic polymer recognised for its potent anticancer properties. This conjugation markedly enhanced both the solubility and cytotoxicity of MJ against cancer cells, exhibiting an efficacy approximately 10-fold higher than that of MJ alone. Furthermore, we introduced PEG-based copolymers, incorporating the jasmonate-based monomer and cationic co-monomer, which demonstrated substantial anticancer activity (IC50 = 9 μg mL−1 for B16F10). This was evidenced by low IC50 values while maintaining increased selectivity (IC50 = 1000 μg mL−1) towards normal cell lines. The newly synthesised polymers represent a substantial advancement in targeted cancer therapy by leveraging the synergistic potential of combining anticancer agents.
Introduction
Cancer poses a severe threat to humankind and is one of the major causes of mortality worldwide, according to the World Health Organization.1 The design and discovery of effective molecules for cancer treatment remain persistent and ongoing challenges. Numerous treatments, such as radiation therapy, chemotherapy, and various drugs, are currently employed; however, their harmful side effects, ability to induce aggressive drug resistance, low aqueous solubility, rapid clearance from the body, and off-target toxicity present major roadblocks as cancer treatment regimens.2–6 Hence, there is an urgent need to develop effective cancer treatments and drugs with few side effects at a low cost. For improved selectivity, drug design must satisfy the condition that the molecular target must be distinct from normal cells.Warburg et al. demonstrated that cancer cells differ from normal cells in their energy consumption.7 Specifically, cancer cells predominantly utilise the glycolytic pathway for energy production at a high rate, rather than the tricarboxylic acid cycle, even in the presence of abundant oxygen, resulting in increased lactate production and local acidification within cancerous cells.8 The phosphorylation of glucose to glucose-6-phosphate, the first step in glycolysis, is catalysed by the enzyme hexokinase-II (HK-II), which is overexpressed in cancer cells when compared with that in normal cells.9,10 Therefore, the energy consumption of cancer cells can be prevented by inhibiting the HK-II enzyme during glycolysis and could be an effective and attractive target for anticancer drug development.11 Methyl jasmonate (MJ) is a well-known inhibitor of HK-II; the therapeutic potential of MJ and its derivatives against cancer have been investigated both in vivo and in vitro.12,13 In normal eukaryotic cells, phosphatidylethanolamine and phosphatidylserine are predominantly located in the inner leaflet of the cell membrane, whereas choline-containing phospholipids, sphingomyelin, and phosphatidylcholine are mainly present in the outer leaflet. Furthermore, phosphatidylserine, O-glycosylated mucins,3,4 sialylated gangliosides,5 and heparan sulfates6 are overexpressed in cancer cell membranes. Hence, understanding the anionic nature of cancer cells may contribute to the high selectivity and potency of chemotherapeutic agents.
Based on the aforementioned accumulated evidence, our proposed strategy involves combining the pharmacophoric moieties of bioactive substances such as MJ with cationic macromolecules or polymerising them to generate a new hybrid compound with improved affinity and efficacy when compared with that of the parent drug, representing a novel approach in drug design. Research on anticancer polymers is comparatively new and has shown promising results in cancer treatment, with only a few studies reporting the use of polymers as an anticancer agent.14–17 In the current study, we utilised the polymer itself as the drug, not merely as a drug carrier. Additionally, the polymerisation of MJ, a bioactive compound, has not been reported elsewhere and is regarded as an innovation in drug design.
Herein, we designed and synthesised novel modified monomers based on MJ. These synthesised monomers were co-polymerised with varying amounts of the cationic monomer 3-(acrylamidopropyl)trimethylammonium chloride (AMPTMA). Modifications of the MJ-based monomers and their copolymers were confirmed using proton nuclear magnetic resonance (NMR) spectroscopy. Subsequently, their anticancer activity was evaluated against several cancer cell lines (HepG2, Colon 26, and B16F10). Additionally, to assess polymer selectivity, the polymers were tested using a normal cell line, i.e., primary human dermal fibroblasts (HDFs).
Experimental
Materials
MJ, lithium hydroxide monohydrate (LiOH·H2O), 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (RAFT agent), and azobisisobutyronitrile (AIBN; initiator) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received. A 75% aqueous solution of the corresponding monomers, AMPTMA, 2-hydroxethyl methacrylate (HEMA), and 4-dimethylaminopyridine (DMAP), was purchased from TCI Japan. The coupling agent 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) was purchased from Wako Chemicals, Japan. All solvents were of reagent grade and were used without further purification.Monomer synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Subsequently, LiOH·H2O (2 mmol) was added, and the reaction was stirred for 1 h at 25 °C. After 1 h, the reaction mixture was diluted with 30 mL of water. Then, 30 mL of hexane was added, followed by acidification with 1 N HCl. Finally, the product was extracted with ethyl acetate, dried with Na2SO4, and the solvent was evaporated to yield a light yellow oil as the final product.
:3 mixture of ethyl acetate and hexane.
:1). Subsequently, LiOH·H2O (2 mmol) was added, and the reaction was stirred for 1 h at 25 °C. After 1 h, the reaction mixture was diluted with 30 mL of water. Then, 30 mL of hexane was added, followed by acidification with 1 N HCl. Finally, the product was extracted with ethyl acetate, dried with Na2SO4, and the solvent was evaporated to yield a light yellow oil as the final product.
:3 mixture of ethyl acetate and hexane.
Copolymer synthesis of JA-HEMA and AMPTMA
We synthesised a series of copolymers (Scheme 1) by varying the proportion of AMPTMA used. For example, a JA-HEMA : AMPTMA (1:2) copolymer was synthesised by adding JA-HEMA (1 g, 3.1 mmol), AMPTMA (1.28 g, 6.202 mmol), RAFT agent (22.62 mg, 0.062 mmol), and AIBN (2.71 mg, 0.012 mmol) to a round-bottomed flask, followed by the addition of 25 mL of DMF. The resulting solution was then purged with nitrogen gas for 30 min and stirred at 70 °C for 24 h. The obtained random copolymer was purified via dialysis using distilled water (4 days) and a 3500 MWCO dialysis membrane (Spectra/Por Dialysis Membrane), followed by lyophilisation.
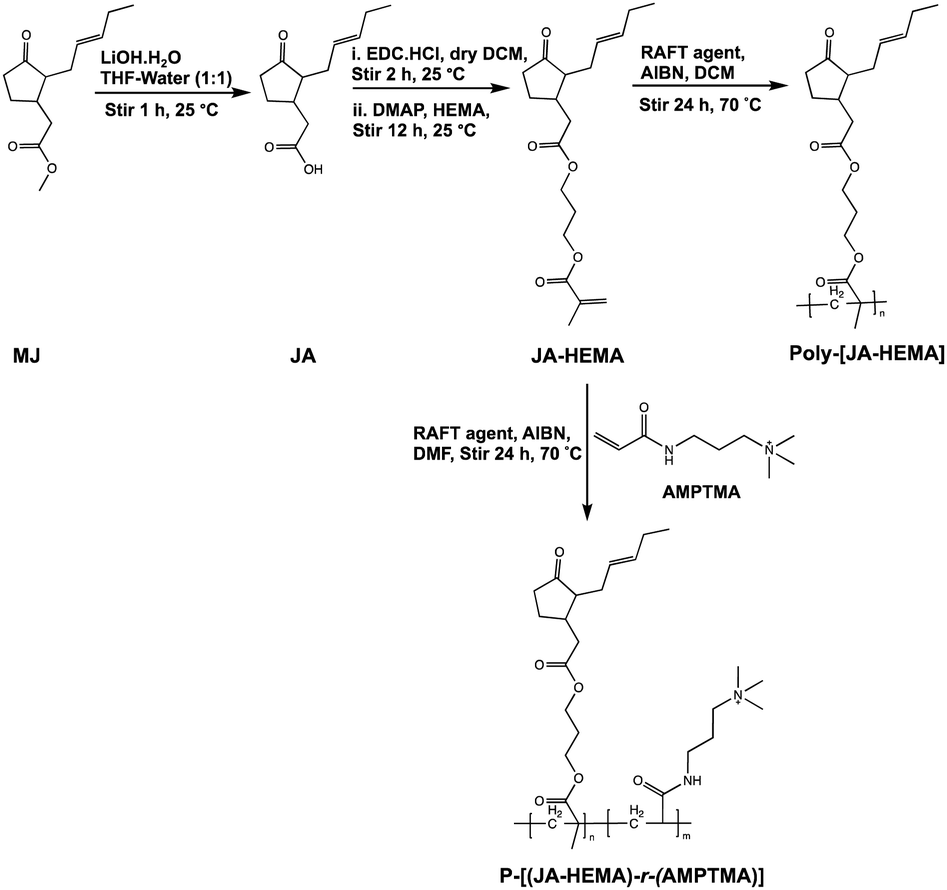 | ||
| Scheme 1 Synthesis of methyl jasmonate-modified JA-HEMA monomer and its subsequent homo and copolymer with AMPTMA. | ||
PEG-based copolymers of JA-HEMA and AMPTMA
:1) copolymer, JA-HEMA (0.931 mmol), PEG-CTA (RAFT agent, 0.0931 mmol), AMPTMA (0.931 mmol), and AIBN (0.0186 mmol) were dissolved in DMF (25 mL) in a round-bottom flask. The resulting solution was then purged with nitrogen gas for 30 min and stirred at 70 °C for 24 h under an inert gas atmosphere. The obtained random copolymer was purified via dialysis using distilled water (4 days) and a 3500 MWCO dialysis membrane (Spectra/Por Dialysis Membrane), followed by lyophilisation.
Polymer characterisation
1H-NMR spectra of the obtained polymers were recorded using a Bruker Avance NEO 400 spectrometer (400 MHz). The chemical shifts were referenced based on the solvent peak (δ = 4.79 ppm for D2O). The molecular weight and distribution (polydispersity index, PDI) of the polymers were determined via gel-permeation chromatography (GPC; waters e2695) using an Ultrahydrogel 250 column and 2414 refractive index detector at 50 °C. A 10% methanol-PBS solution (pH 7.4) was used as the mobile phase (flow rate: 1 mL min−1), and a PEG standard was used as the standard. Zeta potential measurements were performed using a Zetasizer Nano-ZS instrument (Malvern, UK) using disposable folded capillary cells (DTS1070) with a scattering angle of 173°.Cell culture
Cancer cells (HepG2, Colon 26, and B16F10) were acquired from the American Type Culture Collection. Primary HDFs were purchased from Cell Applications (San Diego, CA, USA). All cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) supplemented with 10% foetal bovine serum (Invitrogen, Singapore) at 37 °C in a CO2 incubator in a humidified atmosphere. Cells were subcultured after reaching appropriate confluency using a 0.25% [w/v] trypsin solution containing 0.02% [w/v] ethylenediaminetetraacetic acid in PBS to detach the cells.In vitro cytotoxicity assay
The cytotoxicity of synthesised polymers against cancer and normal cell lines was examined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, 3 × 103 cells suspended in 100 μL of the respective culture media were seeded per well of a 96-well plate and incubated for 24 h at 37 °C in a humidified atmosphere with 5% CO2. After 24 h, the spent culture medium was replaced with fresh medium containing various concentrations of polymers (2–1000 μg mL−1) and incubated for 48 h at 37 °C. Subsequently, 100 μL of the MTT working solution (300 μg mL−1 in DMEM) was added to the cells. The cells were then incubated for 3 h, following which the MTT-containing medium was replaced with 100 μL of DMSO. Finally, the absorbance of each well was measured at 540 nm using a microplate reader (Infinite 200 Pro, Infinite M Nano, Tecan). The cell experiments were independently repeated three times.Lactate dehydrogenase (LDH) leakage assay
In brief, HepG2 cells were initially seeded onto 96-well flat-bottomed polystyrene cell culture plates at a density of 3 × 103 cells per well and allowed to adhere for 24 h. Subsequently, the culture medium was replaced with various concentrations of the polymer P8, followed by a 48-h incubation period. After completion of the incubation period, 10 μL of the Lysis Buffer from the Cytotoxicity LDH assay Kit-WST (Dojindo) was added to four wells, which served as positive controls, indicating 100% lysis, and incubated for 30 min at 37 °C, in accordance with the manufacturer's protocol. Following incubation, 100 μL of the working solution was added to each well and further incubated for 30 min at room temperature (25 °C). The reaction was then terminated by adding 50 μL of the stop solution, and the absorbance was measured at 490 nm using the Infinite 200Pro or Infinite M Nano Tecan microplate reader. The percentage of LDH leakage was determined relative to cells undergoing 100% lysis. Each assay was conducted independently and performed in triplicate.Statistical analysis
All data values are expressed as the mean ± standard deviation (SD). All experiments were performed in quadruplicate. For data comparisons, an ordinary two-way analysis of variance was performed, followed by Tukey's and Šídák's multiple comparison tests. Differences were considered statistically significant at P < 0.05.Results and discussion
Synthesis of JA-HEMA monomer and its homopolymer
The monomers were synthesised according to Scheme 1. Initially, the ester groups of MJ were modified to form JA. The formation of JA was confirmed via1H-NMR, as evidenced by the disappearance of the ester methyl group (δ = 3.5 ppm) (Fig. S1 and S2, ESI†). The synthesised JA was esterified with the hydroxyl group of HEMA and purified using column chromatography. The formation of JA-HEMA was confirmed via1H-NMR (Fig. S3, ESI†). The appearance of the olefinic double bond peaks (δ = 5.8–6.1 ppm) indicated the successful modification of JA with the HEMA group. Subsequently, the obtained monomer, JA-HEMA, was subjected to RAFT polymerisation, and the resulting polymer structure was confirmed using proton NMR. However, the obtained homopolymer was insoluble in water, possibly due to the presence of large hydrophobic moieties. To enhance its aqueous solubility, the JA-HEMA monomer was copolymerised with the cationic monomer AMPTMA.Copolymerisation with cationic monomer AMPTMA
A cationic monomer, AMPTMA, was introduced to balance the aqueous solubility of the homopolymer JA-HEMA. Notably, introducing a cationic moiety could lead to a macromolecular therapeutic effect and increased selectivity towards cancer cells.14 To this end, a series of random copolymers P-[(JA-HEMA)-r-(AMPTMA)] were synthesised by varying the amount of AMPTMA from 1:1 mol% to 1:3 mol% (Scheme 1). The successful formation of these copolymers was confirmed by the disappearance of double bond peaks (δ = 5.8–6.1 ppm), corresponding to the acrylate protons in both JA-HEMA and the AMPTMA (Fig. S4, ESI†). The respective amounts of JA-HEMA and AMPTMA incorporated into each copolymer were calculated by comparing the integral value of the methyl proton peak of JA-HEMA (δ = 0.8 ppm) with that of the methylene peak of AMPTMA (δ = 3.0–3.5 ppm) in the 1H-NMR spectra (Fig. S5, ESI† and Table 1). Fig. S6 (ESI†) presents a comparison of the copolymers containing different amounts of AMPTMA. The incorporation of AMPTMA was controlled by altering the initial feed amount. The number-average molecular weight (Mn), weight-average molecular weight (Mw), and PDI values of these copolymers were determined using GPC. The PDI values were within an acceptable range (1.1–1.6; Table 1).
| S. no. | Polymer | Composition | Zeta potentiala (mV) | Added (JA-HEMA:AMPTMA) |
Obtained (JA-HEMA:AMPTMA)b |
M n × 103,c | M w/Mn | IC50 for HepG2d (μg mL−1) | IC50 for HDFd (μg mL−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined in PBS. b By NMR. c By GPC. d By MTT assay; N.D.: not determined. | |||||||||
| 1. | MJ | Methyl jasmonate | — | — | — | — | — | 313 | 498 |
| 2. | JA | Jasmonic acid | — | — | — | — | — | 300 | 267 |
| 3. | P0 | PAMPTMA100 | 20.2 ± 0.47 | 100 | 100 | 9.5 | 1.15 | N.D. | 187 |
| 4. | P1 | P-[(JA-HEMA)50-r-(AMPTMA)100] | 21.6 ± 1.07 | 1:2 |
1:5 |
14.1 | 1.19 | 65 | 14 |
| 5. | P2 | P-[(JA-HEMA)50-r-(AMPTMA)150] | 20.4 ± 0.17 | 1:3 |
1:8 |
23.0 | 1.56 | 84 | 11 |
| 6. | P3 | P-[(JA-HEMA)50-r-(AMPTMA)200] | 9.54 ± 0.86 | 1:4 |
1:25 |
21.5 | 1.59 | 219 | 20 |
| 7. | P4 | P-[(JA-HEMA)25-r-(AMPTMA)50] | 18.6 ± 1.96 | 1:2 |
1:22 |
21.3 | 2.03 | 68 | 13 |
| 8. | P5 | P-[(JA-HEMA)15-r-(AMPTMA)50] | 12.8 ± 0.85 | 1:3.3 |
1:6 |
13.7 | 1.45 | N.D. | 37 |
In vitro cytotoxicity determination
The anticancer activity of the synthesised polymers was assessed using the MTT assay. Cancer cells exhibit more negative charges than normal cells owing to the overexpression of phospholipids on their surface.18 Initially, the cytotoxic effects of MA and JA were evaluated against the HepG2 cancer cell line. Both MJ and JA exhibited modest anticancer activities, with IC50 values of 313 and 300 μg mL−1, respectively (Fig. 1 and Table 1). The cytotoxicity of the cationic homopolymer AMPTMA (P0) was determined; however, it lacked any significant anticancer activity against HepG2 cells (IC50 = N.D.) (Fig. 1 and Table 1). This suggests that the cationic polymer alone exerts insufficient anticancer activity.14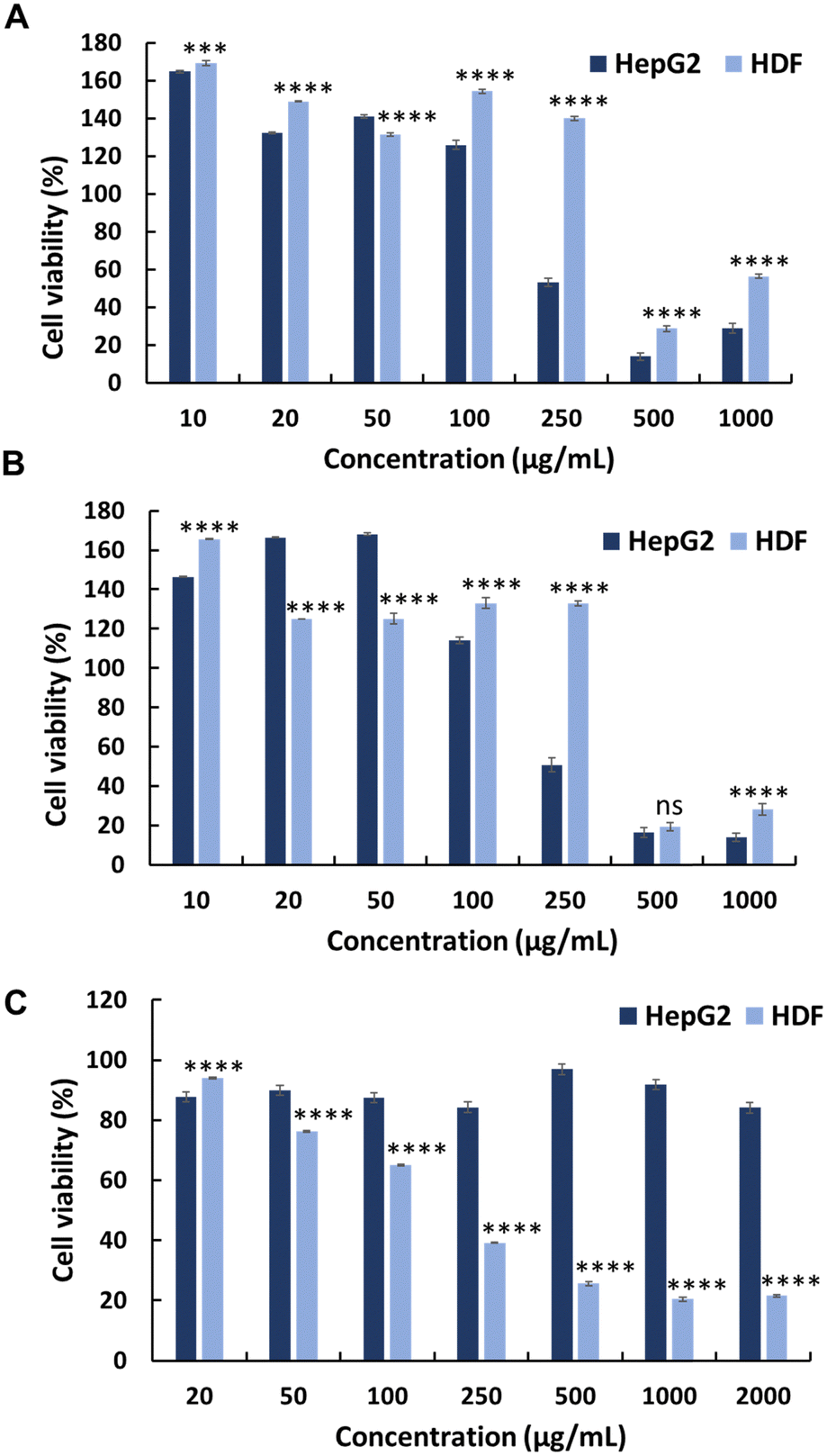 | ||
| Fig. 1 Anticancer activity of the precursors towards the HepG2 and normal HDF cell lines: (A) methyl jasmonate, (B) jasmonic acid, (C) and the AMPTMA homopolymer (P0). Statistical analysis was performed using 2-way ANOVA with Šídák's multiple comparison test (n = 4), P < 0.05. | ||
Conversely, modifying MJ to the JA-HEMA monomer and subsequent copolymerisation with the cationic monomer AMPTMA enhanced anticancer activity. Copolymers with varying amounts of AMPTMA monomers were synthesised, and their formation was confirmed via1H-NMR (Fig. S5 and S6, ESI† and Table 1). The P1 copolymer, containing two equivalents of AMPTMA to JA-HEMA, demonstrated promising anticancer activity with a low IC50 (65 μg mL−1) against the HepG2 cancer cell line (Fig. 2). This synergistic effect had better efficacy than the previously reported MJ derivatives, with IC50 values ranging from 6–9 mM (1.43 mg mL−1 for MJ) against the A549 lung cancer cell line.19 We hypothesised that the anticancer activity was enhanced owing to the inhibitory effect of MJ on cellular HK-II, considering the high expression of HK-II in cancer cells. HK-II reportedly binds to the voltage-dependent anion channel, an integral membrane protein of the mitochondrial outer membrane, to prevent mitochondria-mediated apoptotic cell death.11,19,20 However, as the amount of AMPTMA in the copolymers increased from P1 to P3 (Fig. 2 and Table 1), the anticancer activity of the resulting compound decreased. We hypothesised that this reduced cytotoxicity could be attributed to the decreased proportion of JA-HEMA in the copolymer, leading to the reduced inhibition of the HK-II enzyme and, consequently, anticancer activity.19
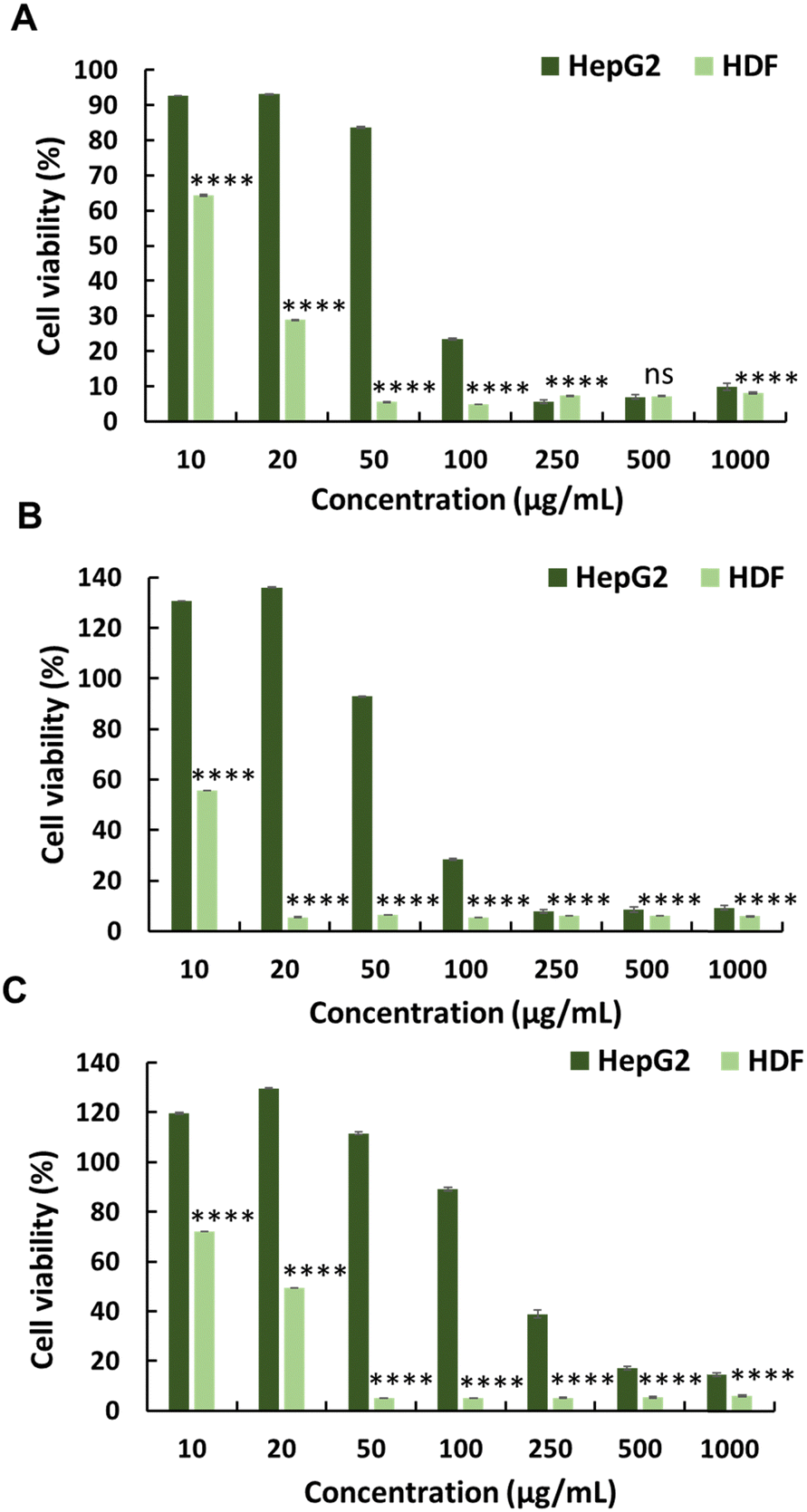 | ||
| Fig. 2 Anticancer activity of the copolymers towards the cancer cell line HepG2 and normal HDF cell lines: (A) copolymer P1, (B) P2, and (C) P3. Statistical analysis was performed using 2-way ANOVA with Šídák's multiple comparison test (n = 4), P < 0.05. | ||
To evaluate the selectivity of the synthesised polymers towards normal cell lines, the cytotoxicity of polymers (P0–P3) was evaluated against HDFs (Fig. 1, 2 and Table 1). All synthesised polymers exhibited high cytotoxicity towards the HDF cell line, as indicated by their markedly low IC50 values (IC50 = 14–20 μg mL−1). We believe that this undesired cytotoxicity could be attributed to the high cationic charge resulting from the long chain lengths of AMPTMA and JA-HEMA. Consequently, new polymers with reduced chain lengths were synthesised for both JA-HEMA and AMPTMA (P4–P5). Polymer P4 exhibited increased cytotoxicity against the HepG2 cancer cell line (IC50 = 68 μg mL−1); however, it still lacked selectivity against normal cells (Fig. S7, ESI† and Table 1). Similarly, polymer P5, comprising even shorter chains, demonstrated less cytotoxicity towards the cancer cell line; however, suitable selectivity towards HDFs was not achieved (Fig. S7, ESI† and Table 1).
Synthesis and characterisation of PEG-based copolymers
Given that the AMPTMA-based random copolymers failed to exhibit sufficient selectivity, we synthesised PEG-based polymers because the biocompatibility of PEG is well established.21 PEG reportedly enhances hydrophilicity and prolongs blood circulation time.22–24 Accordingly, we synthesised PEG-based copolymers of JA-HEMA and AMPTMA with varying amounts of JA-HEMA and AMPTMA (Scheme 2). Copolymers P6–P8 were synthesised by maintaining a constant amount of AMPTMA monomer (DP-10) and progressively increasing the chain length of the JA-HEMA monomer from 10 to 30 units (theoretical DP). Additionally, other copolymers were synthesised by maintaining a constant amount of JA-HEMA (DP-10) and gradually increasing the amount of the AMPTMA monomer (Table 2). Initially, PEG-CTA, which served as the macro-RAFT agent, was synthesised (Step a, Scheme 2), and its formation was confirmed via1H-NMR (Fig. S8, ESI†). Following its successful synthesis, other copolymers were prepared by varying the amounts of both AMPTMA and JA-HEMA (Step b, Scheme 2) (Table 2), and their formation was confirmed via1H-NMR (Fig. S9, ESI†). The presence of JA-HEMA in the synthesised polymer was confirmed by the methyl proton of JA-HEMA (δ = 0.8 ppm), the olefinic double bond (δ = 5.2–5.4 ppm); the characteristic peak of the quaternary methyl protons of AMPTMA (δ = 3.2–3.4 ppm) indicated the presence of AMPTMA (Fig. S9, ESI†). Thus, copolymers containing all these components (PEG, JA-HEMA, and AMPTMA) were successfully synthesised. The Mn and Mw values of these compounds were determined via GPC. The PDI values were detected within the desirable range (1.0–1.1; Table 2). Following the successful synthesis of these copolymers, in vitro cytotoxicity experiments were performed.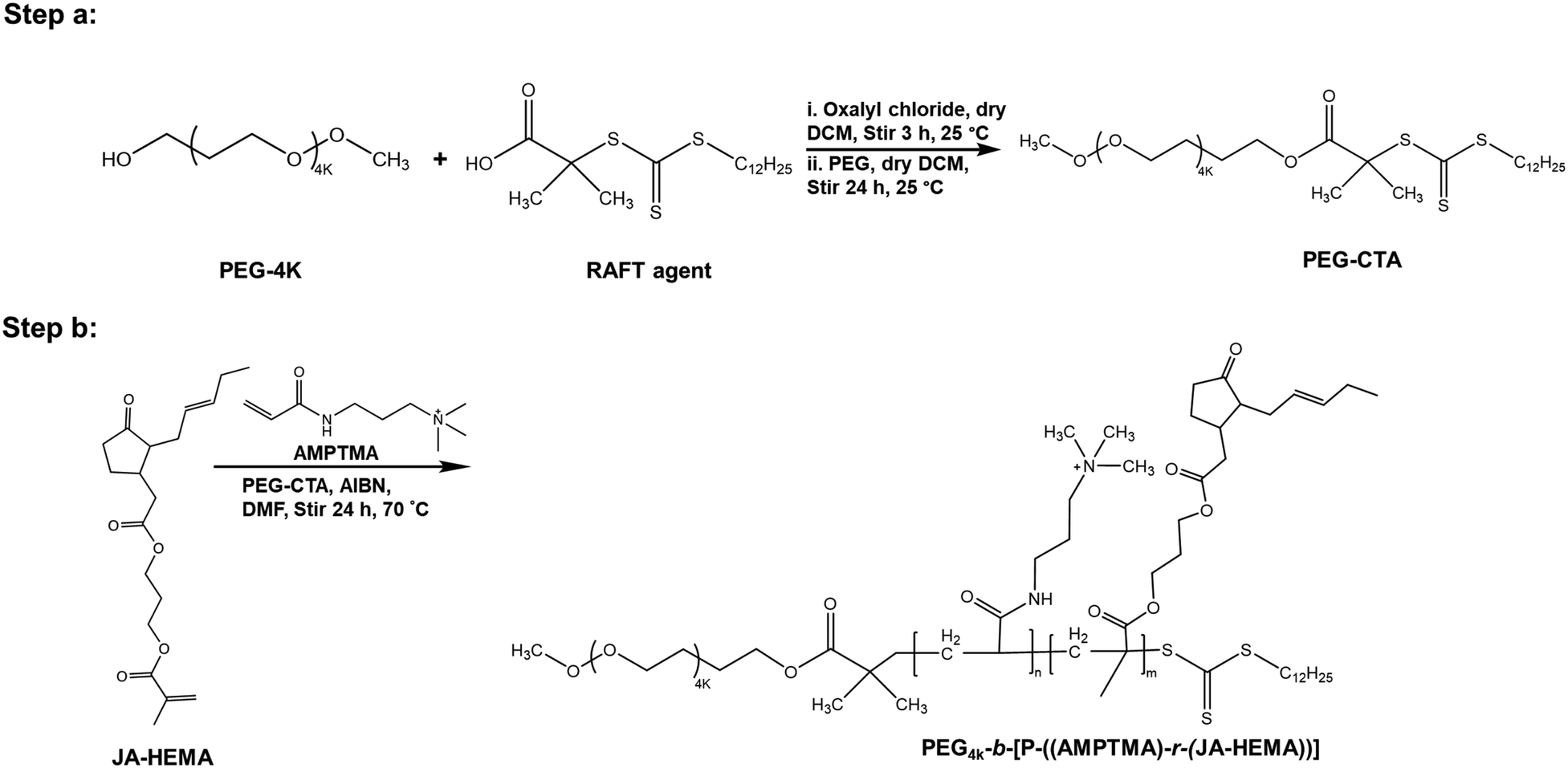 | ||
| Scheme 2 Synthesis of PEG-CTA-based copolymers of JA-HEMA and AMPTMA. | ||
| S. no. | Polymer | Zeta potentiala (mV) | M n × 103,b | M w/Mn | IC50 for HepG2c (μg mL−1) | IC50 for colon 26 (μg mL−1) | IC50 B16F10c (μg mL−1) | IC50 for HDFc (μg mL−1) | |
|---|---|---|---|---|---|---|---|---|---|
| a Determined in PBS. b By GPC. c By MTT assay, and N.D.: not determined. | |||||||||
| 1. | MJ | Methyl jasmonate | — | — | — | 313 | 390 | 373 | 498 |
| 2. | JA | Jasmonic acid | — | — | — | 300 | 639 | 663 | 267 |
| 3. | P6 | PEG4k-b-[P-((JA-HEMA)10-r-(AMPTMA)10)] | −2.69 ± 0.22 | 7.10 | 1.01 | 641 | 201 | 62 | 859 |
| 4. | P7 | PEG4k-b-[P-((JA-HEMA)20-r-(AMPTMA)10)] | −1.32 ± 0.45 | 7.34 | 1.02 | N.D. | N.D. | 28 | N.D. |
| 5. | P8 | PEG4k-b-[P-((JA-HEMA)30-r-(AMPTMA)10)] | −1.86 ± 0.61 | 7.18 | 1.01 | 503 | N.D. | 9 | N.D. |
| 6. | P9 | PEG4k-b-[P-((JA-HEMA)10-r-(AMPTMA)20)] | 0.91 ± 0.26 | 8.31 | 1.06 | N.D. | 191 | 35 | 1000 |
| 7. | P10 | PEG4k-b-[P-((JA-HEMA)10-r-(AMPTMA)30)] | 2.35 ± 0.42 | 9.33 | 1.11 | N.D. | 425 | 32 | 894 |
In vitro cytotoxicity of PEG-based copolymers
The in vitro anticancer activity of the synthesised PEG-based copolymers was evaluated against the HepG2, Colon 26, and B16F10 cancer cell lines. The P6 copolymer exhibited enhanced anticancer activity against HepG2 (IC50 = 641 μg mL−1) (Fig. 3A) and superior anticancer activity against Colon 26 (Fig. 3B) and B16F10 (Fig. 3C) cells, with IC50 values of 201 and 62 μg mL−1, respectively. This increase in cytotoxicity suggests a synergistic effect between MJ and the cationic polymer. Enhancing the JA-HEMA monomer content in P7 and P8 further improved the anticancer activity of the copolymer against B16F10 cells, a more malignant cancer cell line (IC50 = 28 μg mL−1 and 9 μg mL−1, respectively) (Fig. 3C). However, contrasting results were obtained in HepG2 and Colon 26 cancer cell lines, where the anticancer activity decreased with increasing JA-HEMA content (Table 2).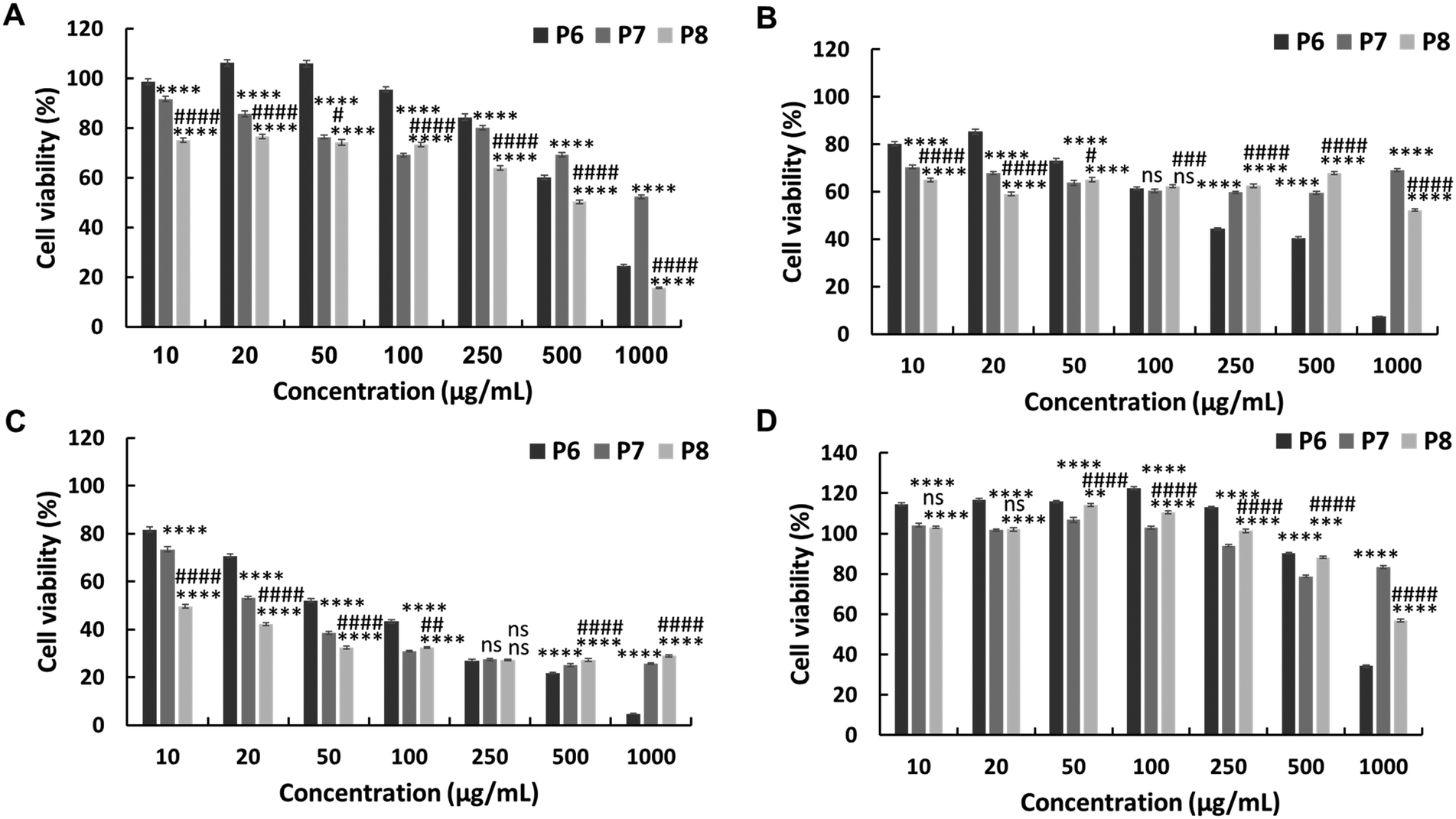 | ||
| Fig. 3 In vitro cytotoxicity of the PEG-based copolymers containing various amounts of JA-HEMA against (A) HepG2 cells, (B) Colon 26, (C) B16F10 cells, and (D) normal HDF cells. Statistical analysis was performed using two-way ANOVA using Tukey's multiple comparison tests (n = 4), P < 0.05. P6 vs. P7 or P8 is represented by *, P7 vs. P8 is represented by #. | ||
However, upon testing these copolymers on a normal cell line (HDF), all polymers exhibited significantly higher selectivities than the non-PEG-based copolymers. The P6 polymer showed greater selectivity, with a higher IC50 (859 μg mL−1), than MJ and JA alone (Fig. 3D, Table 2). Similarly, P7 and P8 exhibited increased selectivity, with no IC50 up to 1000 μg mL−1 of the polymer concentration. This enhanced selectivity could be attributed to the overexpression of the HK-II enzyme in cancer cells when compared with that in normal cells, which results in the greater selectivity of MJ-containing polymers towards cancer cells.19 Additionally, the PEG block serves to shield the cationic core, enhancing blood circulation times and extending passive tumour accumulation, while the cationic block binds and disrupts the cellular membrane, leading to enhanced selectivity.15,25
Additionally, we synthesised PEG-based copolymers by maintaining a constant amount of JA-HEMA and progressively increasing the AMPTMA concentration (P9 and P10). These copolymers were found to be effective against cancer cells (Table 2). Both P9 and P10 did not show significant anticancer activity against HepG2 cells at polymer concentrations up to 1000 μg mL−1 (Fig. 4a). However, when tested against Colon 26 (Fig. 4b) and B16F10 cells (Fig. 4c), these polymers exerted notable anticancer activity with lower IC50 values (Table 2). Polymer P9 demonstrated good selectivity against the HDF cell line with an IC50 of almost 1000 μg mL−1. Conversely, selectivity towards HDF cells was slightly reduced from P9 to P10 (IC50 of P10 = 894 μg mL−1), which may be attributed to the increased cationic chain length (Fig. 4d).
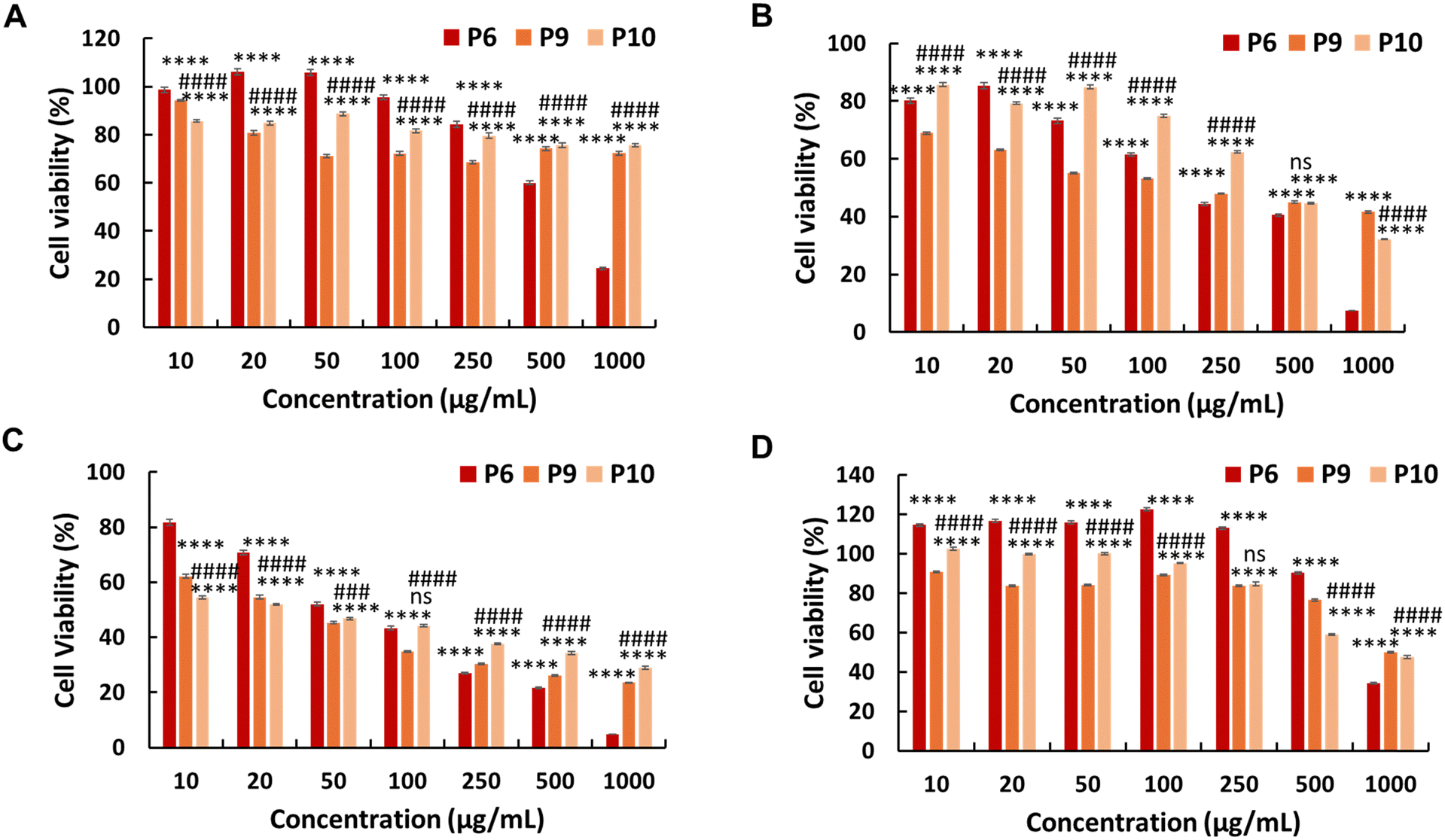 | ||
| Fig. 4 In vitro cytotoxicity of the PEG-based copolymers containing various amounts of AMPTMA against (A) HepG2 cells, (B) Colon 26 cells, (C) B16F10 cells, and (D) normal HDF cells. Statistical analysis was performed using two-way ANOVA using Tukey's multiple comparison tests (n = 4), P < 0.05. P6 vs. P9 or P10 is represented by *, P9 vs. P10 is represented by #. | ||
To elucidate the mechanism of action of the polymers, we investigated their impact on cancer cell membranes. Initially, we conducted an LDH leakage assay utilising the selected polymer, P8, to gain insight into its membrane-disruptive properties. The assessment involved quantifying the extracellular release of the cytoplasmic enzyme LDH from HepG2 cells treated with the polymers for 48 h. Surprisingly, even at elevated polymer concentrations, minimal LDH leakage was observed with the selected polymer (Fig. S10, ESI†), suggesting that cell membrane permeability may not be the primary mechanism of action. However, to validate these findings and gain a deeper understanding of the underlying mechanisms, further comprehensive studies are warranted, which we plan to undertake in future investigations.
Overall, while the PEG-based copolymers exhibited reduced cytotoxicity towards cancer cells when compared with the P-[(JA-HEMA)-r-(AMPTMA)] copolymers, an enhancement in selectivity towards normal cells was observed. This increased selectivity was likely due to enhanced biocompatibility resulting from the incorporation of PEG. Although these polymers were not as effective against HepG2 cancer cells, they demonstrated significant efficacy against the more malignant Colon 26 and B16F10 cancer cells. Therefore, these polymers were confirmed to be both effective and selective against malignant cancer cells. This promising efficacy against highly malignant cell lines underscores the potential of these polymers as cornerstones for the development of targeted and less toxic anticancer therapies, paving the way for more refined and effective cancer treatment strategies in the future.
Conclusions
In this study, we successfully synthesised an MJ monomer and subsequently copolymerised it with a cationic co-monomer. The resultant copolymers demonstrated enhanced solubility and cytotoxicity, with an almost 10-fold increase in effectiveness against cancer cells (IC50 = 65 μg mL−1). Consequently, we synthesised PEG-based copolymers of JA-HEMA (an MJ analogue) and AMPTMA. These synthesised polymers were highly effective against cancer cells, exhibiting very low IC50 values, reaching 9 μg mL−1. Importantly, all PEG-based copolymers exhibited increased selectivity towards normal cell lines, indicating their selective action against cancer cells. Moreover, the homopolymer PAMPTMA displayed no cytotoxicity towards cancer cell lines, underscoring the cytotoxic effect of MJ. To the best of our knowledge, this is the first study to report the polymerisation of the bioactive compound MJ, which represents a novel approach to anticancer drug design. The cytotoxicity of MJ analogues synthesised in this study was significantly amplified by approximately 10–12-fold and showed enhanced selectivity, marking a major advancement in the field of anticancer drug development. Further investigations are currently underway to elucidate the underlying mechanisms and evaluate the effectiveness of these compounds in vivo.Further research endeavours are imperative for a thorough evaluation of the efficacy, safety, and therapeutic potential of anticancer polymers. Moreover, the effectiveness and safety of anticancer polymers need to be validated through rigorous clinical trials. These investigations are crucial steps towards advancing our understanding of the therapeutic utility of these polymers and their translational potential in clinical settings.
Author contributions
Nishant Kumar: data curation, methodology, investigation, visualisation, writing-original draft, writing-review, and editing. Pratibha: investigation. Dandan Zhao: investigation, Robin Rajan: writing-review and editing, supervision, project administration, and conceptualisation. Kazuaki Matsumura: writing-review and editing, supervision, project administration, conceptualisation, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NK would like to thank Dr Naito Masanobu, Deputy Director, Research Centre for Macromolecules and Biomaterials Field Data-Driven Polymer Design Group, Sengen Site, National Institute for Materials Science, Tsukuba, Japan.References
- N. Kumar, S. Fazal, E. Miyako, K. Matsumura and R. Rajan, Mater. Today, 2021, 51, 317–349 CrossRef CAS.
- A. Ganoth, K. C. Merimi and D. Peer, Expert Opin. Drug Delivery, 2015, 12, 223–238 CrossRef CAS PubMed.
- S. Mignani, M. Bryszewska, B. Klajnert-Maculewicz, M. Zablocka and J.-P. Majoral, Biomacromolecules, 2015, 16, 1–27 CrossRef CAS PubMed.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- Y. D. Livney and Y. G. Assaraf, Adv. Drug Delivery Rev., 2013, 65, 1716–1730 CrossRef CAS PubMed.
- E. K. Rowinsky, E. A. Eisenhauer, V. Chaudhry, S. G. Arbuck and R. C. Donehower, Semin. Oncol., 1993, 20, 1–15 CAS.
- V. Bhardwaj, N. Rizvi, M. B. Lai, J. C. Lai and A. Bhushan, Anticancer Res., 2010, 30, 743 CAS.
- A. M. Otto, Cancer metab., 2016, 4, 5 CrossRef PubMed.
- F. Bao, K. Yang, C. Wu, S. Gao, P. Wang, L. Chen and H. Li, Fitoterapia, 2018, 125, 123–129 CrossRef CAS PubMed.
- Z. Chen, H. Zhang, W. Lu and P. Huang, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 553–560 CrossRef CAS PubMed.
- H. Lin, J. Zeng, R. Xie, M. J. Schulz, R. Tedesco, J. Qu, K. F. Erhard, J. F. Mack, K. Raha, A. R. Rendina, L. M. Szewczuk, P. M. Kratz, A. J. Jurewicz, T. Cecconie, S. Martens, P. J. McDevitt, J. D. Martin, S. B. Chen, Y. Jiang, L. Nickels, B. J. Schwartz, A. Smallwood, B. Zhao, N. Campobasso, Y. Qian, J. Briand, C. M. Rominger, C. Oleykowski, M. A. Hardwicke and J. I. Luengo, ACS Med. Chem. Lett., 2016, 7, 217–222 CrossRef CAS PubMed.
- H. T. Dang, H. J. Lee, E. S. Yoo, J. Hong, B. Bao, J. S. Choi and J. H. Jung, Bioorg. Med. Chem., 2008, 16, 10228–10235 CrossRef CAS PubMed.
- S. Liu, W. H. Wang, Y. L. Dang, Y. Fu and R. Sang, Tetrahedron Lett., 2012, 53, 4235–4239 CrossRef CAS.
- N. Kumar, K. Oqmhula, K. Hongo, K. Takagi, S.-I. Yusa, R. Rajan and K. Matsumura, J. Mater. Chem. B, 2023, 11, 1456–1468 RSC.
- N. H. Park, W. Cheng, F. Lai, C. Yang, P. Florez de Sessions, B. Periaswamy, C. Wenhan Chu, S. Bianco, S. Liu, S. Venkataraman, Q. Chen, Y. Y. Yang and J. L. Hedrick, J. Am. Chem. Soc., 2018, 140, 4244–4252 CrossRef CAS PubMed.
- H. Takahashi, K. Yumoto, K. Yasuhara, E. T. Nadres, Y. Kikuchi, L. Buttitta, R. S. Taichman and K. Kuroda, Journal, 2019, 9, 1096 Search PubMed.
- N. Kumar, T. Nakaji-Hirabayashi, M. Kato, K. Matsumura and R. Rajan, Biomacromolecules, 2024, 25, 1481–1490 CrossRef CAS PubMed.
- I. Dobrzyńska, B. Szachowicz-Petelska, S. Sulkowski and Z. Figaszewski, Mol. Cell. Biochem., 2005, 276, 113–119 CrossRef PubMed.
- B. O. Sucu, O. S. Ipek, S. O. Kurtulus, B. E. Yazici, N. Karakas and M. Guzel, Bioorg. Chem., 2019, 91, 103146 CrossRef CAS PubMed.
- D. Xu, J. Jin, H. Yu, Z. Zhao, D. Ma, C. Zhang and H. Jiang, J. Exp. Clin. Cancer Res., 2017, 36, 44 CrossRef PubMed.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61, 1177–1188 CrossRef CAS PubMed.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- S. Chen, K. Yang, R. G. Tuguntaev, A. Mozhi, J. Zhang, P. C. Wang and X.-J. Liang, Nanomedicine, 2016, 12, 269–286 CrossRef CAS PubMed.
- G. Zhong, C. Yang, S. Liu, Y. Zheng, W. Lou, J. Y. Teo, C. Bao, W. Cheng, J. P. K. Tan, S. Gao, N. Park, S. Venkataraman, Y. Huang, M. H. Tan, X. Wang, J. L. Hedrick, W. Fan and Y. Y. Yang, Biomaterials, 2019, 199, 76–87 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00146j |
| This journal is © The Royal Society of Chemistry 2024 |