
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the cycling performance of MgH2–LiBH4 based solid-state batteries via stacking pressure tailoring†
Xueye
Zhuang
,
Haoliang
Chen
,
Shiman
He
*,
Long
Hu
,
Hui
Wang
* and
Renzong
Hu
 *
*
School of Materials Science and Engineering, Guangdong Provincial Key Laboratory of Advanced Energy Storage Materials, South China University of Technology, Guangzhou, 510641, China. E-mail: heshm@scut.edu.cn; mehwang@scut.edu.cn; msrenzonghu@scut.edu.cn
First published on 19th April 2024
Abstract
MgH2 has been considered a promising anode material for all-solid-state batteries (ASSBs) due to its high theoretical capacity, low voltage plateau (0.5 V vs. Li+/Li), and minimal voltage hysteresis. Herein, by applying 25 MPa stacking pressure, the interfacial issues caused by the volume expansion of MgH2 electrodes can be well suppressed, which ultimately improves the electrochemical performance of the MgH2 anode material. The MgH2 half-cell achieves a high capacity of 1212 mA h g−1 after 200 cycles at 0.5C. When assembled with a S cathode, the Mg–LiH|LiBH4|S full cell delivers a high specific capacity of 1300 mA h g−1 at 0.1C and stably operates over 100 cycles. This work provides a facile strategy to improve the performance of the MgH2 anode in ASSB devices via stacking pressure tailoring.
All-solid-state batteries (ASSBs), without the flammability and leakage risks of organic electrolytes, as well as with high energy density and thermal stability, are considered to be the next generation of high-performance electrochemical energy storage systems.1–3 Currently, research on ASSBs is mainly focused on designing electrolytes with high ionic conductivity and wide electrochemical windows.4–6 However, the maintenance of good interfacial contact during battery operation is also vital for the practical application of ASSBs.
MgH2, as a highly viable anode material for high-energy-density ASSBs,7,8 possesses a high theoretical capacity (2038 mA h g−1), low voltage plateau (0.5 V vs. Li+/Li), and minimal voltage hysteresis (<0.1 V).9–11 Recent advancements have shown that MgH2 is well-suited with LiBH4 solid-state electrolytes, which can effectively avoid side reactions occurring in organic electrolyte systems and also enhance H− diffusion during lithiation. However, MgH2 still faces the issues of rapid capacity degradation and poor rate performance in the LiBH4 solid-state battery systems due to the large volume expansion.12,13 Several strategies have been adopted to respond to the above challenges. Compositing MgH2 with oxides, such as Nb2O514 or CoO,15 can reduce the diffusion activation energy of Li+ and H−, thus decreasing the charge/discharge hysteresis and improving the cycling stability. Additionally, innovative structural designs, like graphene-loaded MgH216 or composition with vapor-grown carbon nanofiber (VCGF),11 have been shown to enhance the material conductivity and mitigate volumetric strain.
Although a reasonable material structure design can significantly improve the battery performance, the complicated preparation processes could increase the cost of battery fabrication, which is not conducive to large-scale preparation and promotion. Besides material design strategies, the stacking pressure of ASSBs is also critical for the battery performance.17–19 The strategy of adjusting the stacking pressure for ASSBs possesses the advantages of lower cost and good performance consistency. A suitable stacking pressure can homogenize the deposition and stripping of alkali metal and alleviate the interfacial failure caused by the volume expansion effect.20 The effect of stacking pressure on the performance of ASSBs with chloride or sulfide solid-state electrolytes has been reported.21–23 However, it has not been systematically investigated in LiBH4 electrolyte-based ASSBs.
In this study, the impact of applied stacking pressure on the performance of the MgH2 electrode in LiBH4-based ASSBs is explored. Symmetric cells with a stacking pressure of 25 MPa are confirmed as the best condition for Li plating/stripping reaction over an extended duration. The battery with a MgH2 electrode at 25 MPa exhibits a highly reversible capacity of 1884 mA h g−1, and stable cycling for over 200 cycles. Additionally, an intact electrolyte–electrode interface without cracks is observed for the MgH2-based half-cell with 25 MPa after cycling. Finally, when combined with a sulfur–carbon nanotube composite (S@CNTs) cathode material, the Mg–LiH|LiBH4|S full cell achieves a high discharge capacity of 1300 mA h g−1 and an average discharge voltage of 1.5 V. We offer a facile strategy to improve the cycling stability and rate performance of MgH2 electrodes in LiBH4-based ASSBs by applying suitable stacking pressure.
Before adopting MgH2 anode materials in ASSBs, the Li|LiBH4|Li symmetric cells were conducted to investigate the effect of stacking pressure on Li deposition/stripping reactions. The stacking pressures of the batteries were controlled by a pressure pump with a digital pressure sensor, and the schematic diagram of the instrument is shown in Fig. S1 (ESI†). The symmetric cell with a stacking pressure of 25 MPa (Fig. 1a) delivered a Li plating/stripping polarization of ∼18 mV over 2000 hours at a current density of 0.2 mA cm−2. However, the symmetric cell with 5 MPa exhibited a much higher polarization of 30 mV, and the polarization gradually increased and the cell failed after 660 h. Moreover, the symmetric cell with 50 MPa also failed after cycling for 800 hours.
 | ||
| Fig. 1 (a) Time-voltage profiles of Li symmetric cells with different stacking pressures at 0.2 mA cm−2, where the insets are the enlarged profiles. SEM images of electrolyte surface morphology: (b) pristine state; (c)–(e) the ex situ electrolyte after cycling with the stacking pressure of (c) 5 MPa, (d) 25 MPa and (e) 50 MPa. | ||
The scanning electron microscope (SEM) images in Fig. 1b–e showed the surface morphologies of the LiBH4 electrolyte sheets before and after cycling. The surface of the LiBH4 electrolyte was roughened and part of the electrolyte particle was crushed after cycling with 5 MPa and 50 MPa. On the contrary, a smooth surface without obvious particle fragmentation was observed in the electrolyte with 25 MPa after cycling for 2000 h. The cross-sectional SEM images and the corresponding energy dispersive spectroscopy (EDS) mappings were adopted to further evaluate the lithium plating/stripping behaviours in the Li|LiBH4|Li symmetric cells at different pressures. The interface morphologies were clearly displayed in Fig. S2–S4 (ESI†) with yellow line markings. Noticeable gaps were found in the cell with 5 MPa (yellow circle in Fig. S2d, ESI†), while electrolyte cracking and fragments also appeared in the cell with 50 MPa. Both of the above cases can cause excessive current per unit area and uneven deposition of lithium, resulting in lithium dendrite growth.24–26 The continuous interface morphologies of the cells with 25 MPa revealed the uniform Li plating/stripping processes. The electrochemical impedance spectra (EIS) of the batteries at different pressures before and after cycling are shown in Fig. S5 (ESI†). The dramatically decreased resistance of the battery with 25 MPa revealed its improved interface contact during electrochemical reaction. However, the cell with 5 MPa still maintained a large interfacial resistance after cycling, further illustrating the poor contact of the electrolyte and lithium metal. Moreover, the disordered EIS data of the cycled battery with 50 MPa indicated that the batteries have failed with such a high pressure. These results reveal that the cell with 25 MPa is more favourable for long-term Li plating/stripping reactions, providing a basis for the following measurements of ASSBs for MgH2 anode materials.
The commercially purchased MgH2 was milled to refine the particle size by planetary ball milling. The XRD pattern showed the typical MgH2 diffraction peaks in Fig. S6 (ESI†). There is no significant change of the diffraction peaks before and after ball milling, indicating the stable tetragonal crystal structure of MgH2 during ball milling treatment. According to the SEM images (Fig. S7, ESI†), the particle size of MgH2 decreased from tens of micrometers to hundreds of nanometers or even tens of nanometers after ball milling. Smaller particle size can narrow the lithium diffusion paths, and also alleviate the volume expansion effect. Subsequently, the refined MgH2 materials were mixed with lithium borohydride and conductive carbon to prepare the electrode tablets. The XRD pattern in Fig. S8 (ESI†) showed an orthorhombic LiBH4 phase and MgH2 phase in the electrode tablet. And the corresponding EDS mappings in Fig. S9 (ESI†) exhibited that the MgH2 was dispersed in the LiBH4 and carbon materials, revealing the homogeneous mixing of the electrode materials.
The charge/discharge curves of MgH2-based electrodes at different stacking pressures under 0.1C (1C = 2038 mA g−1) are shown in Fig. 2a and Fig. S10 (ESI†). The half-cell exhibited a reversible specific capacity of 1818.9 mA h g−1 and a coulombic efficiency (CE) of 95.3% at the first cycle with 25 MPa stacking pressure. However, the same battery configurations with both 5 and 50 MPa showed comparatively lower CEs (91.2% and 94.7%, respectively) at the first cycle. Furthermore, the voltage gap between the lithiation and delithiation reactions at 25 MPa was calculated as ∼90 mV by the dQ/dE curve (Fig. S11, ESI†), which is much smaller than other conversion-type anodes in LiBH4-based ASSBs.27 The battery with 25 MPa (Fig. 2b) still maintained a high capacity of 1443.2 mA h g−1, with a capacity retained of 68.5% after 200 cycles. On the contrary, the capacity retention of the battery with 5 MPa was only 28.6% after 100 cycles, which could be attributed to the poor surface contact caused by volume expansion and contraction of the MgH2 electrodes during the charge and discharge processes. Moreover, an excessively high stacking pressure may accelerate the growth of Li dendrites and increase the risk of short circuits, leading to short cycling life. The batteries were easy to fail with a high stacking pressure of 50 MPa. Thus, the following measurements and analysis mainly focused on the batteries with 5 and 25 MPa. Fig. S12 (ESI†) shows the cycling performance under high current density of 0.5C. The battery with 5 MPa failed in less than 30 cycles, while it still maintained a specific capacity of 1212.0 mA h g−1 after 200 cycles, with a high capacity retention of 68.9% at 25 MPa. Fig. S13 (ESI†) shows repeatability testing of the cycling performances for Li|LiBH4|MgH2 cells under different pressures at 0.1C and 0.5C. The close cycling performances and capacity retentions indicate the reliability of our experiments.
 | ||
| Fig. 2 (a) Galvanostatic charge–discharge profiles of the MgH2 electrode with 25 MPa at 0.1C. (b) Cycling stability with different stacking pressures at 0.1C. (c) Rate performance of the MgH2 electrodes at various current densities from 0.1C to 1C. (d) Cycling performance comparison of the MgH2 electrode and previously reported metal hydride-based anodes in ASSBs. | ||
Fig. 2c displays the rate performances of MgH2 electrodes with different stacking pressures. The capacity of the battery with 5 MPa dramatically decreased with the increase of the current density from 0.1C to 1C, and it only showed a capacity of 1101.5 mA h g−1 when the current density returned to 0.1C. On the contrary, the battery with 25 MPa still possessed a high specific capacity of 758.7 mA h g−1 at 1C, and a specific capacity of 1857.5 mA h g−1 when the current density went back to 0.1C (Fig. S14, ESI†). The excellent rate performance of the battery demonstrates the strong tolerance of MgH2 toward fast charge and discharge processes with 25 MPa stacking pressure. Fig. 2d exhibits a graph that compares the cycling life and retention capacity of our MgH2 electrode and the reported hydride electrodes. By applying the appropriate stacking pressure, we have successfully realized a competitively high-capacity and long-cycle MgH2-based ASSBs. It is attractive that our MgH2 based ASSBs can achieve such high performance with a facile strategy, providing a prospect for further improvement by combining with other suitable material modification.
The phase changing of the MgH2 electrode at different charged and discharged states was examined by the ex situ XRD in Fig. 3a and b. The disappearance of MgH2 and the generation of the Mg and LiH diffraction peaks after being discharged to 0.3 V reveal the conversion-type reaction mechanism of the MgH2 anode materials. The regeneration of MgH2 after being charged to 1 V suggests the revertability of the reaction. Based on the electrochemical reaction mechanism of the MgH2 materials, the volume expansion and contraction ratio can be calculated as ∼85%.9,28 Therefore, the volume change of the MgH2 electrodes can be intuitively observed by the cross-section SEM images. As shown in Fig. 3c–e and Fig. S15, S16 (ESI†), obvious cracks appear at the electrode/electrolyte interface in the batteries with 5 MPa, which lead to the reduction of the effective area between the electrode and electrolyte, consequently impeding the lithium diffusion. On the contrary, there is no noticeable crack at the interface of the MgH2 electrode under 25 MPa. The intact interface makes a continuous lithium diffusion pathway, thus prolonging the stability of the battery. Moreover, the electrochemical impedance spectrum (EIS) exhibited that the impedance decreased from 180.9 Ω to 93.6 Ω after applying the stack pressure from 5 MPa to 25 MPa (Fig. S17, ESI†). The influence of the stacking pressure on the electrode–electrolyte interface was illustrated by the schematic diagrams in Fig. 3f. Abundant interspace was formed between the MgH2 electrode and LiBH4 electrolyte due to the large volume expansion of MgH2. The battery with low stacking pressure of 5 MPa failed to fill the interspace, leading to poor interface contact and inhomogeneous ion diffusion channels. On the contrary, with an appropriate stacking pressure of 25 MPa, the interspace can be filled immediately by releasing the inner pressure of the batteries, which can form an intact interface and homogeneous ion diffusion channels, thus resulting in more stable cycling performance.
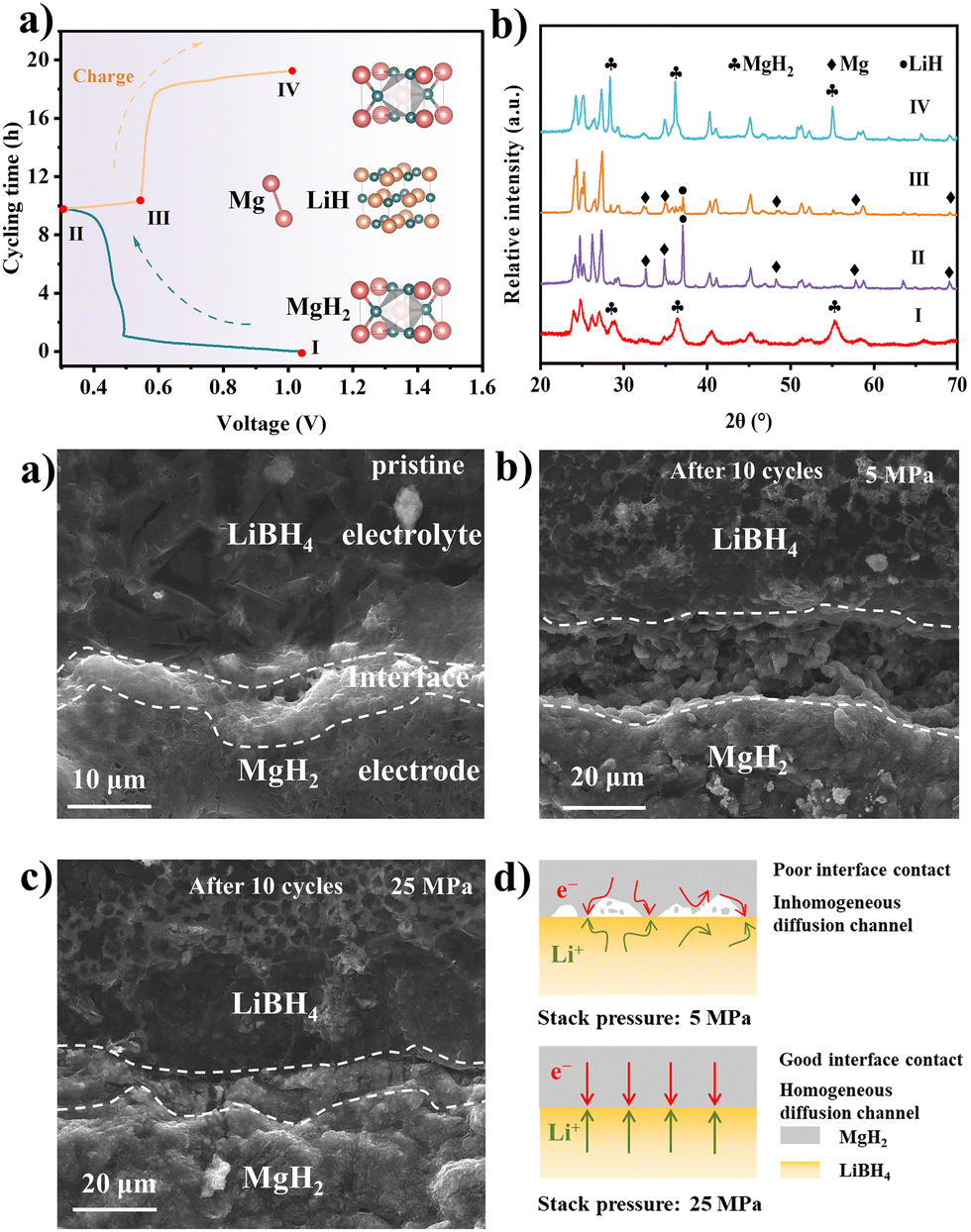 | ||
| Fig. 3 (a) Evolution of the voltage along with cycling time for the MgH2 electrode at 0.1C at the first cycle. (b) XRD patterns of the MgH2 electrode at different charged and discharged states. Cross-sectional SEM images of the interface between MgH2 and the LiBH4 solid electrolyte: (c) initial state, (d) after 10 cycles with 5 MPa and (e) 25 MPa at 0.1C. (f) Schematic diagram of a MgH2 electrode operated with different stacking pressures. | ||
To verify the application of the MgH2 anode in full batteries, the Mg–LiH|LiBH4|S batteries were investigated by introducing a sulfur@carbon nanotube (CNT) composite as the cathode material. The XRD pattern (Fig. 4a) of the S@CNT cathode exhibited the typical diffraction peaks of S8 structure and a distinct (002) peak of CNTs. The schematic diagram in Fig. 4b showed the configuration of Mg–LiH|LiBH4|S batteries, in which the LiBH4 electrolyte was uniformly mixed with MgH2 anode and S cathode to improve the Li ion diffusion within the solid-state electrodes. In order to assemble the Mg–LiH|LiBH4|S full cell, the MgH2‖Li half cell was firstly pre-lithiated by discharging to 0.3 V; then, the lithium electrodes were uncovered and replaced with S cathodes to assemble the full cells. The Mg–LiH|LiBH4|S battery achieved an initial capacity of 1300 mA h g−1 and discharged voltage plateaus at ∼1.5 V. The energy density can be calculated up to 381 W h kg−1 based on the mass of S and MgH2 (Fig. 4c and d), revealing the bright application prospects of the Mg–LiH|LiBH4|S battery system. Furthermore, the Mg–LiH|LiBH4|S full cell with 25 MPa still maintained a capacity of 404 mA h g−1 after 100 cycles, which provided a facile strategy for enabling long cycling life of ASSBs via stacking pressure tailoring. Fig. S18 (ESI†) presented again the cycling performance of the of Mg–LiH|LiBH4|S cells under 25 MPa pressure at 0.1C. The close capacity retention validates the reliability of our experiments.
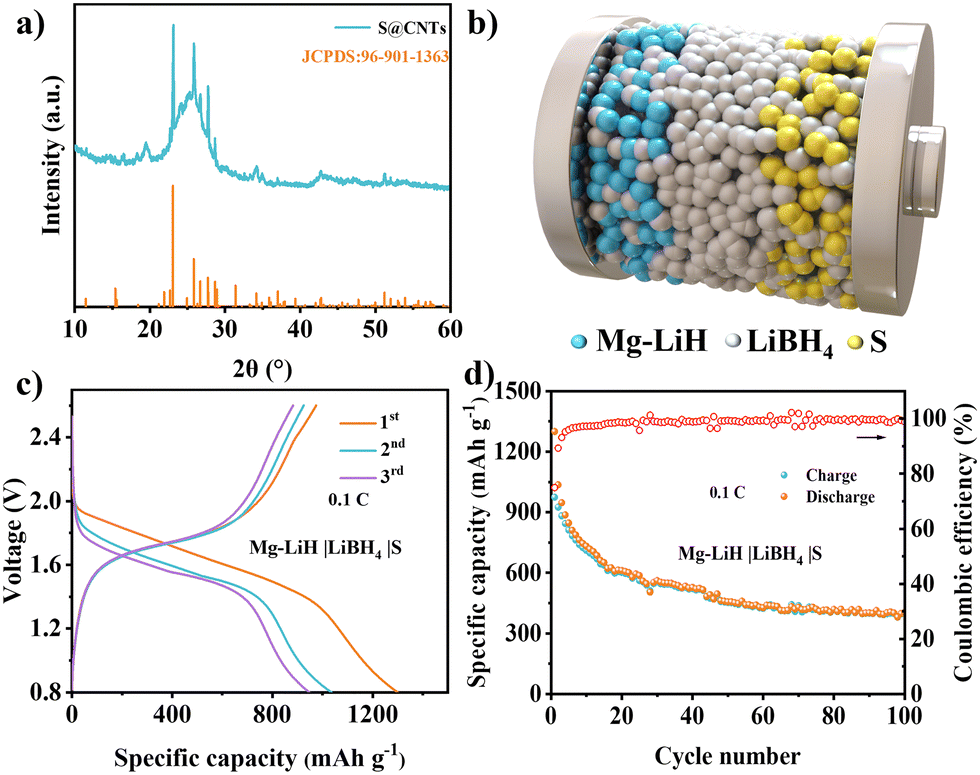 | ||
| Fig. 4 (a) XRD pattern of S@CNTs. (b) Schematic diagram of the Mg–LiH|LiBH4|S full battery. (c) Discharge–charge curve at the 1st, 2nd and 3rd cycle. (d) Cycling stability of the Mg–LiH|LiBH4|S battery at 0.1C. | ||
In conclusion, the influence of stacking pressure on the electrochemical performance of LiBH4-based ASSBs is initially confirmed by the symmetric cell. The MgH2 half cell with 25 MPa achieves a high capacity of 1212 mA h g−1 after 200 cycles at 0.5C, which is much better than those of other stacking pressures. The cracks at the electrode–electrolyte interface caused by the volume expansion of the MgH2 electrode in the charged and discharged processes can be well suppressed by impressing the batteries with 25 MPa, achieving an intact interface between the electrolyte and electrode. The intact interface makes a continuous lithium diffusion pathway, thus prolonging the stability of the battery. Finally, by combining with the sulfur cathode, the Mg–LiH|LiBH4|S full cell shows a capacity of 1300 mA h g−1 at 0.1C. This work provides a facile strategy to improve the performance of the MgH2 anode and other ASSB devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51831009, 52071144, and 52231009), Guangdong Basic and Applied Basic Research Foundation (2023B1515040011, 2023G008), Guangzhou Applied Basic Research Foundation (2023A04J1566), and Guangzhou Key Research and Development Program (no. 202103040001).References
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2(4), 16103 CrossRef CAS.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121(3), 1623–1669 CrossRef CAS PubMed.
- A. M. Bates, Y. Preger, L. Torres-Castro, K. L. Harrison, S. J. Harris and J. Hewson, Joule, 2022, 6(4), 742–755 CrossRef CAS.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5(3), 229–252 CrossRef CAS.
- S. Xia, X. Wu, Z. Zhang, Y. Cui and W. Liu, Chemistry, 2019, 5(4), 753–785 CrossRef CAS.
- Z.-Y. Wang, C.-Z. Zhao, S. Sun, Y.-K. Liu, Z.-X. Wang, S. Li, R. Zhang, H. Yuan and J.-Q. Huang, Matter, 2023, 6(4), 1096–1124 CrossRef CAS.
- R. Mohtadi and S.-I. Orimo, Nat. Rev. Mater., 2016, 2(3), 16091 CrossRef.
- Y. Huang, P. Gao, T. Zhang, X. Zhang, G. Xia, F. Fang, D. Sun, Z. Guo and X. Yu, Small, 2023, 19(26), e2207210 CrossRef PubMed.
- Y. Oumellal, A. Rougier, G. A. Nazri, J. M. Tarascon and L. Aymard, Nat. Mater., 2008, 7(11), 916–921 CrossRef CAS PubMed.
- P. Gao, S. Ju, Z. Liu, G. Xia, D. Sun and X. Yu, ACS Nano, 2022, 16, 8040 CrossRef CAS PubMed.
- L. Zeng, T. Ichikawa, K. Kawahito, H. Miyaoka and Y. Kojima, ACS Appl. Mater. Interfaces, 2017, 9(3), 2261–2266 CrossRef CAS PubMed.
- L. Aymard, Y. Oumellal and J. P. Bonnet, Beilstein J. Nanotechnol., 2015, 6, 1821–1839 CrossRef CAS PubMed.
- S. Brutti, S. Panero, A. Paolone, S. Gatto, D. Meggiolaro, F. Vitucci, J. Manzi, D. Munaò, L. Silvestri, L. Farina and P. Reale, Challenges, 2017, 8(1), 8 CrossRef.
- L. Zeng, K. Kawahito, S. Ikeda, T. Ichikawa, H. Miyaoka and Y. Kojima, Chem. Commun., 2015, 51(48), 9773–9776 RSC.
- A. El Kharbachi, H. Uesato, H. Kawai, S. Wenner, H. Miyaoka, M. H. Sørby, H. Fjellvåg, T. Ichikawa and B. C. Hauback, RSC Adv., 2018, 8(41), 23468–23474 RSC.
- Y. Lv, X. Zhang, W. Chen, S. Ju, Z. Liu, G. Xia, T. Ichikawa, T. Zhang and X. Yu, J. Mater. Sci. Technol., 2023, 155, 47–53 CrossRef.
- J. Sang, B. Tang, Y. Qiu, Y. Fang, K. Pan and Z. Zhou, Energy Environ. Mater., 2023, e12670 CrossRef.
- F. Zhang, Y. Guo, L. Zhang, P. Jia, X. Liu, P. Qiu, H. Zhang and J. Huang, eTransportation, 2023, 100220 CrossRef.
- C. Fang, B. Lu, G. Pawar, M. Zhang, D. Cheng, S. Chen, M. Ceja, J.-M. Doux, H. Musrock, M. Cai, B. Liaw and Y. S. Meng, Nat. Energy, 2021, 6(10), 987–994 CrossRef CAS.
- J. M. Doux, H. Nguyen, D. H. S. Tan, A. Banerjee, X. Wang, E. A. Wu, C. Jo, H. Yang and Y. S. Meng, Adv. Energy Mater., 2020, 10, 1903253 CrossRef CAS.
- C. Lee, S. Y. Han, J. A. Lewis, P. P. Shetty, D. Yeh, Y. Liu, E. Klein, H.-W. Lee and M. T. Mcdowell, ACS Energy Lett., 2021, 6(9), 3261–3269 CrossRef CAS.
- J.-M. Doux, Y. Yang, D. H. S. Tan, H. Nguyen, E. A. Wu, X. Wang, A. Banerjee and Y. S. Meng, J. Mater. Chem. A, 2020, 8(10), 5049–5055 RSC.
- B. Liu, S. D. Pu, C. Doerrer, D. Spencer Jolly, R. A. House, D. L. R. Melvin, P. Adamson, P. S. Grant, X. Gao and P. G. Bruce, SusMat., 2023, 3(5), 721–728 CrossRef CAS.
- D. Cao, X. Sun, Q. Li, A. Natan, P. Xiang and H. Zhu, Matter, 2020, 3(1), 57–94 CrossRef.
- S. Yu and D. J. Siegel, ACS Appl. Mater. Interfaces, 2018, 10(44), 38151–38158 CrossRef CAS PubMed.
- S. Yu and D. J. Siegel, Chem. Mater., 2017, 29(22), 9639–9647 CrossRef CAS.
- Y. Pang, Y. Liu, J. Yang, S. Zheng and C. Wang, Mater. Today Nano, 2022, 100194 CrossRef CAS.
- S. Brutti, G. Mulas, E. Piciollo, S. Panero and P. Reale, J. Mater. Chem., 2012, 22(29), 14531 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00177j |
| This journal is © The Royal Society of Chemistry 2024 |