
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFree-base amino acid-encoded microsystems for dual detection of narcotics and therapeutic antidote monitoring†
Sarvesh Kumar
Srivastava‡

Department of Health Technology, Technical University of Denmark (DTU), Denmark. E-mail: sksr@dtu.dk
First published on 9th May 2024
Abstract
In this proof-of-concept study, we demonstrate the simultaneous detection of narcotics and releasing of their respective antidote through real-time therapeutic drug monitoring (TDM). Our approach utilizes a free-base amino acid interface involving a two-step process. First, we create large surface area (4 × 4 mm) plasmonic gold nanostructures via a tryptophan (Trp) mediated supramolecular interface on pre-fabricated Si-nanopillars. Second, we protonate excess Trp (H+) to form a polymeric-Trp interface capable of entrapping drug particles like naloxone (OXN), an anti-opioid medication. This dual approach provides two distinct functionalities on a single Si-wafer substrate: (a) SERS-mediated detection of cocaine and heroin and (b) the evaluation of antidote drug (OXN) release kinetics using Raman spectroscopy. Combining supramolecular chemistry with microfabrication lays a strong foundation for developing highly ordered 3D nano/microstructures via a free-base amino acid chemistry towards next-generation theranostics.
Introduction
Therapeutic drug monitoring (TDM) plays a crucial role in ensuring the safe and effective use of medications by assessing pharmacokinetic parameters to prevent both under and overdosing.1 The misuse of opioids, often in high doses or in conjunction with other drugs, has led to a significant rise in addiction cases, resulting in severe drug-related organ failures, as well as combined liver and kidney disorders. Overdoses of opioids like cocaine and heroin can prove fatal unless a suitable antidote, such as naloxone (OXN), is administered promptly via injection.2 It's worth noting that approximately 45% of overdose patients experience adverse events after receiving naloxone.3 Therefore, the controlled release of rapidly dissolving therapeutics like naloxone, coupled with the simultaneous detection of narcotics, presents a fundamental challenge within the realm of microchip-based theranostics.We address this challenge by demonstrating the self-assembly of highly-branched plasmonically-active Au-nanocactus structures through a Trp-mediated supramolecular interface.4,5 The process involves forming Au-nanostructures, followed by a one-step protonation reaction that converts excess Trp into its polymeric form (poly-Trp). This results in the creation of two distinct surfaces on a Si-nanopillar substrate, facilitating (i) a highly-branched, plasmonic Au-interface for sensing, such as surface-enhanced Raman spectroscopy (SERS) and (ii) a poly-Trp matrix for real-time drug delivery kinetics monitoring. We explored the practical application of this switchable chemical interface for detecting narcotics (heroin and cocaine using SERS) and releasing an opioid antidote drug (naloxone using Raman spectroscopy). Overall, this study represents a novel “bottom-up” approach for the integrated fabrication of material interfaces with large surface areas, enabling both active sensing and drug delivery, all driven by a single free-base amino acid.6
In nature, complex structures are often formed through hierarchical assembly processes that impose specific boundary conditions at various length scales governed by kinetic controls.7,8 Amino acids and short peptides, typically composed of just two or three amino acids, contain sufficient chemical information to facilitate the assembly of organic nanomaterials with sequence-dependent properties.9,10,11 Jian et al. demonstrated the controlled formation of chiral, hierarchically organized architectures in calcium carbonate (vaterite) by introducing chiral acidic amino acids (Asp and Glu), where altering the amino-acid enantiomers induced a switch in chirality.12 In another study, Kumar et al. expanded on traditional supramolecular structure design by modifying or editing the supramolecular construct in situ, leading to the creation of conductive nanowires incorporating naphthalene di-amide in their core, with catalytic self-assembly encoded by amino acids (D- and L-Tyrosine).13 They demonstrated the formation of conductive nanowires, incorporating naphthalene di-amide in its core, with an amino-acid (D- and L-Tyrosine) encoded catalytic self-assembly. These studies, among others,14,15 show growing interest in function-driven molecular constructs. However, despite all the progress in the supramolecular assembly in water, the controlled covalent synthesis of large molecules and structures with distinct functions remains limited, particularly in fabricating functional theranostic microdevices and dual-action interfaces.
Results
In this study, we initiated a chemical reaction to create Au-nanocactus structures over a hydrophobic parafilm substrate (Scheme 1A). This assembly process created plasmonic Au nanoparticles, showcasing remarkable flexibility and precise control (Scheme 1B). Achieving such highly-ordered metallic nanostructures typically requires a complex fabrication approach, entailing sacrificial/supporting metal layers and cleanroom protocols, which demand significant resources and meticulous process control.16,17 Here, we demonstrated a facile fabrication scheme via a free-base amino acid mechanism, which is not limited to nanoparticle synthesis only but to an entire microdevice of several orders of magnitude (cm).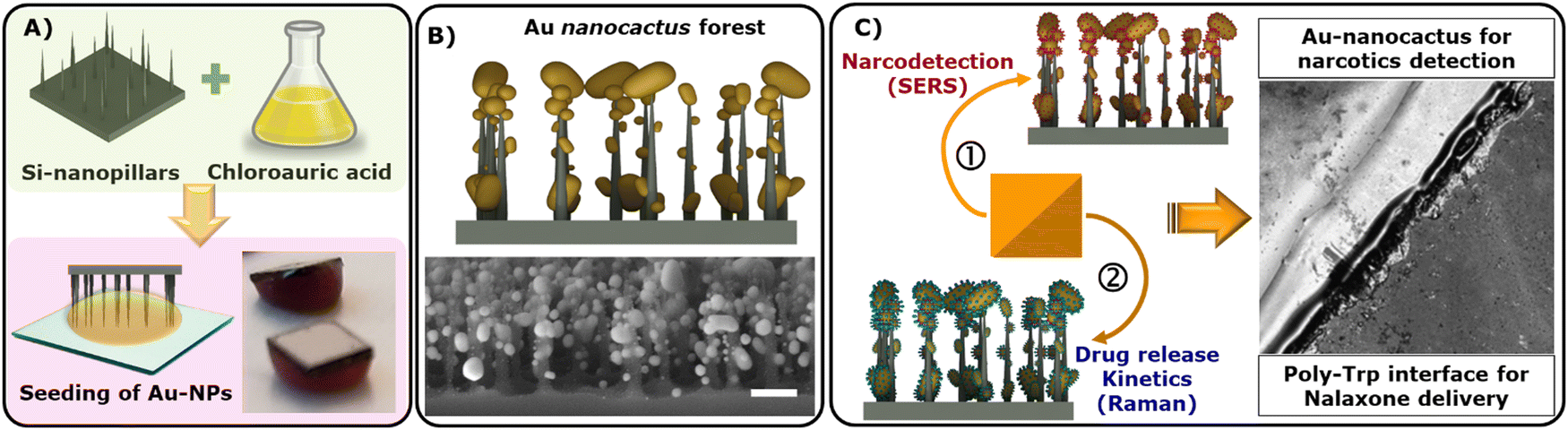 | ||
| Scheme 1 Designing of Au-theranostic platform: (A) formation of highly branched Au nanoparticles; (B) large surface area depicting Au particle decorated nanopillars (Au nanocactus) and; (C) a schematic representation highlighting dual-action Au nanocactus properties: SERS-based detection of a target drug and poly-Trp interface for drug release kinetics assessment (via Raman spectroscopy). Note that measurements were performed on two separate chips – one for detection (SERS) and the other for release kinetics. | ||
The resulting Au nanocactus (Au-NCs) were employed for SERS-based detection of opioids, including heroin and cocaine (Scheme 1C). Additionally, the integration of OXN with the poly-Trp matrix allowed us to monitor real-time drug release. To evaluate naloxone release, the OXN-poly-Trp complex was formed atop the Au nanocactus structures, which were then immersed in buffer solutions, with the release of naloxone (i.e., dissolution of the OXN crystal) being tracked by recording Raman scattering spectra from the NT complexes at the surface (Scheme 1C).
Fig. 1A illustrates the highly ordered growth of Au nanocrystals across a large region of the Si-substrate, spanning several millimeters. Numerous high-aspect ratio spikes formed within this closely packed structure, and these spikes could be employed as a SERS substrate.18 In Fig. 1B, we observe that the tips of the underlying Si nanopillars had the most significant Au-crystallization, measuring approximately 150 nm, followed by smaller Au-nanospikes ranging from 30 to 90 nm over the Si-substrate. This ‘star-like’ morphology is highly advantageous for plasmonic applications like SERS.19 Multicrystalline Au NPs produced a characteristic diffraction pattern, including Au(111), Au(200), Au(200), and Au(311), as observed through scattered area electron diffraction (SAED) microscopy in Fig. 1C.20 Importantly, Trp-mediated Au-nanocactus structures were found to be highly stable in both dry and wet states, as demonstrated in Fig. 1D.
 | ||
| Fig. 1 Self-templating of Au nanocrystals via Trp-encoded interface: (A) SEM image showing large surface area with uniform growth (top-down view); (B) highly branched Au-nanocactus formation; (C) SAED image of an Au nanocacti; and (D) SEM image of depicting the dry and wet state of the Au nanocactus in x–z plane. Scale bar: 1 μm. The wet state depicts wetting (5 μL H2O) of the substrate and drying off at room temperature before obtaining the SEM micrograph. | ||
The inability to achieve highly ordered metallic interfaces has been a significant limitation for wet-lab chemistry in MEMS-related microfabrication approaches. To address this limitation, Yan et al. proposed a peptide engineering-based method for branched synthesis of plasmonic gold nanoparticles, yielding coral-shaped nanoparticles with a plasmon resonance at 528 nm.21 Other synthesis and fabrication schemes have also been employed, including multi-cycled silver mirror reactions, enzyme-guided crystal growth, and liquid–liquid interfaces.22–25
Trp-mediated nanoparticle synthesis has been demonstrated by us and others before.4,5,26 Highly ordered structures were formed due to the stabilizing effect offered by indole (π electrons)27 and –NH3+ groups, creating a net positive charge around Au-core. The underlying nanoparticle growth mechanism can be explained as follows (Fig. 2A, step I): (i) Trp under physiological conditions (∼pH 7; aq. solution), attains a characteristic zwitter ion structure; (ii) pre-etched Si-nanopillar acts as a nucleation site where chloroauric acid gets reduced promoting larger crystal growth at the tips and; (iii) heterogeneous nucleation of Au atoms over the rest of Si-surface forms smaller Au-nanospikes. Highly ordered structures were formed as a result of the stabilizing effect offered by indole (π electrons)26,28 and –NH3+ groups, creating a net positive charge around Au-core. In our study, following the formation of Au nanoparticles, excess Trp was protonated, leading to poly-Trp formation (Fig. 2A, step II). This single-step modification allowed the trapping of naloxone within the poly-Trp interface, enabling controlled drug release assessment via Raman spectroscopy. The formation of poly-Trp was further confirmed through FT-IR analysis, as shown in Fig. 2B. Several characteristic peaks, including C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch (saturated) at 1672 cm−1, N–H bending vibration of a primary amine (–NH2) at 1580 cm−1, and others, supported the presence of poly-Trp. This was further supported by the presence of N–H wag (primary amine) and monosubstituted CH2 groups in the region of 750 cm−1 to 720 cm−1. Fig. 2C shows a TEM micrograph depicting an Au core with poly-Trp-OXN shell (inset showing the boundary layer).
O stretch (saturated) at 1672 cm−1, N–H bending vibration of a primary amine (–NH2) at 1580 cm−1, and others, supported the presence of poly-Trp. This was further supported by the presence of N–H wag (primary amine) and monosubstituted CH2 groups in the region of 750 cm−1 to 720 cm−1. Fig. 2C shows a TEM micrograph depicting an Au core with poly-Trp-OXN shell (inset showing the boundary layer).
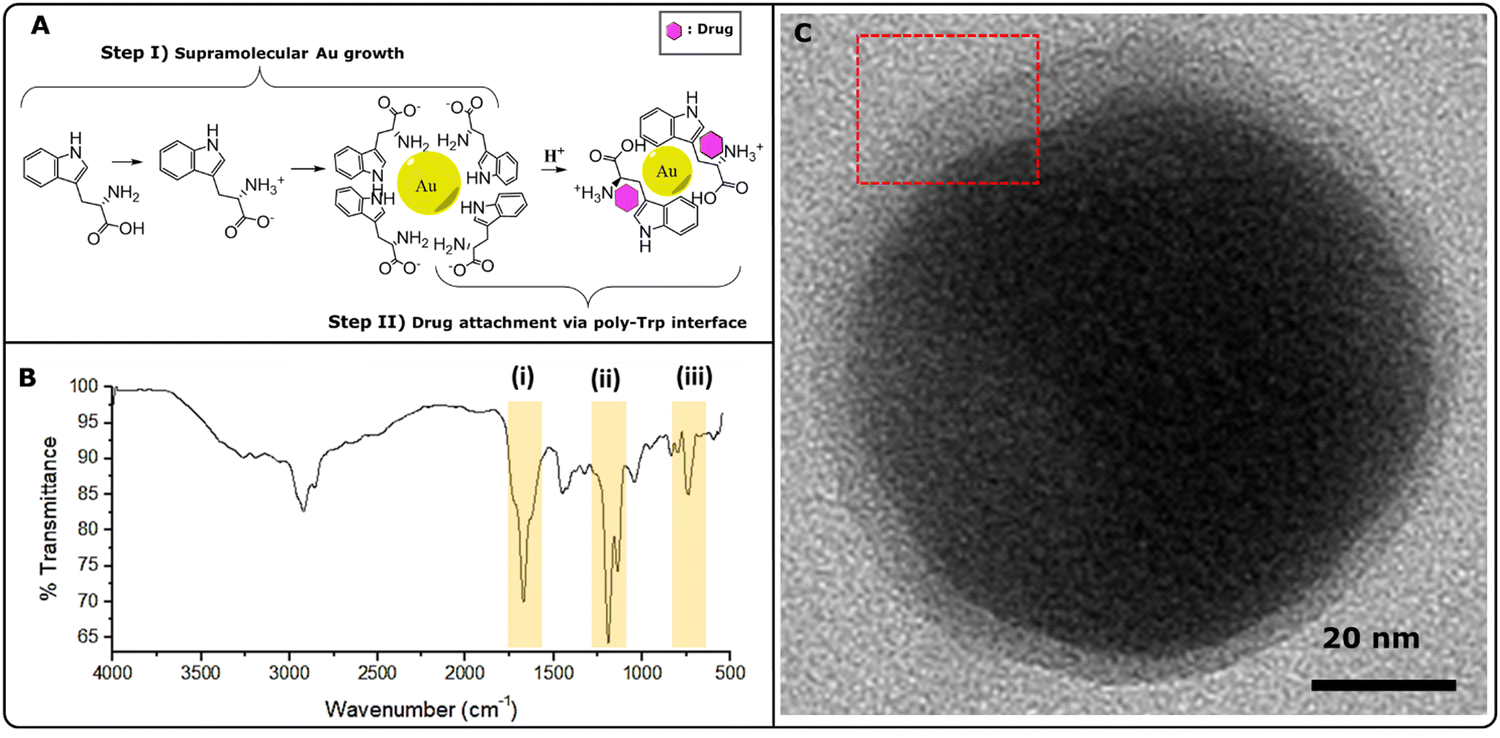 | ||
| Fig. 2 Designing of Au-theranostic assembly (A) molecular mechanism depicting Au-nanocacti formation (step I) and poly-Trp formation (step II) and (B) FT-IR spectra confirming the formation of poly-Tryptophan. (C) TEM micrograph depicting particle morphology of the poly-Trp interface with naloxone hydrochloride (inset showing the boundary layer). | ||
Fig. 3 illustrates the SERS-based detection of target drugs using Au nanocactus plasmonic structures synthesized via a Trp-mediated supramolecular interface. Fig. 3a shows vibrational peaks corresponding to cocaine, such as tropane ring stretch (840 cm−1 and 890 cm−1), phenyl ring breathing (1005 cm−1), stretching modes (1605 cm−1), and the ester carbonyl stretching mode (1722 cm−1).29 Similarly, heroin was detected with its characteristic vibrational peaks, including (630 cm−1) and (1200–1600 cm−1), as shown in Fig. 3b. Note that the characteristic peak of naloxone (633 cm−1 and 617 cm−1) was identified in its native state and confirmed when bound to the polymeric-Trp. The Trp-mediated Au NP interface exhibited minimal background interference, with a sharp peak at 523 cm−1 corresponding to the Si-substrate. Moving beyond the conventional notion of the limit of detection for a SERS substrate, we established a dual-action microdevice for real-time drug release assessment in vitro. However, it must be noted that given the heterogeneity of the chemical seeding process, together with the formation of the poly-Trp organic interface, dampening of the SERS signal is expected at the behest of imparting dual functionality for TDM monitoring via Raman. Furthermore, we observed that the SERS spectra of heroin appear to be of lower quality compared to those of cocaine. This difference could potentially be attributed to the aging or partially degraded condition of the heroin sample.
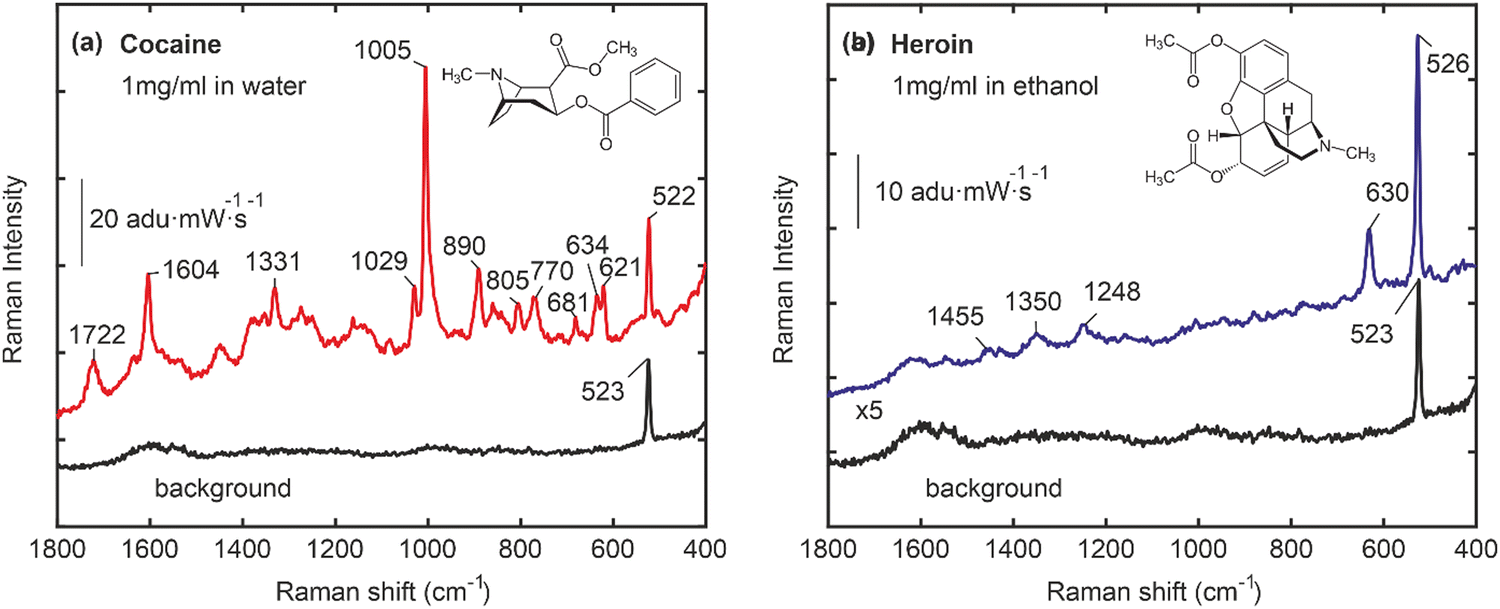 | ||
| Fig. 3 SERS spectra of bare Au nanocactus substrates, which can be utilized for the detection of (a) cocaine (1 mg mL−1 in ethanol) and (b) heroin (1 mg mL−1 in water). All spectra were collected using a 10× objective lens. The SERS signal collection time was 20 s, averaged 3 times. 1 μl droplets of heroin and cocaine solutions (1 mg mL−1) were deposited on Au nanocactus surfaces and let to dry prior to acquiring the spectra. | ||
The release of naloxone from the naloxone-poly-Trp complex (NT) complex on the Au nanocactus surface was verified using Raman spectroscopy (see Fig. 4). The results in Fig. 4(a) indicate that the Raman spectral fingerprint of naloxone powder closely resembles that of the produced NT complex, suggesting that the conjugation process preserves the original structure of naloxone. Upon exposure to 30 μl of PBS, the NT complex gradually dissolves, releasing naloxone into the PBS, as depicted in Fig. 4(b)–(g). At t ≈ 20 min, naloxone is fully released into the PBS, and the final Raman spectra of naloxone in the PBS solution can be used to estimate its concentration.
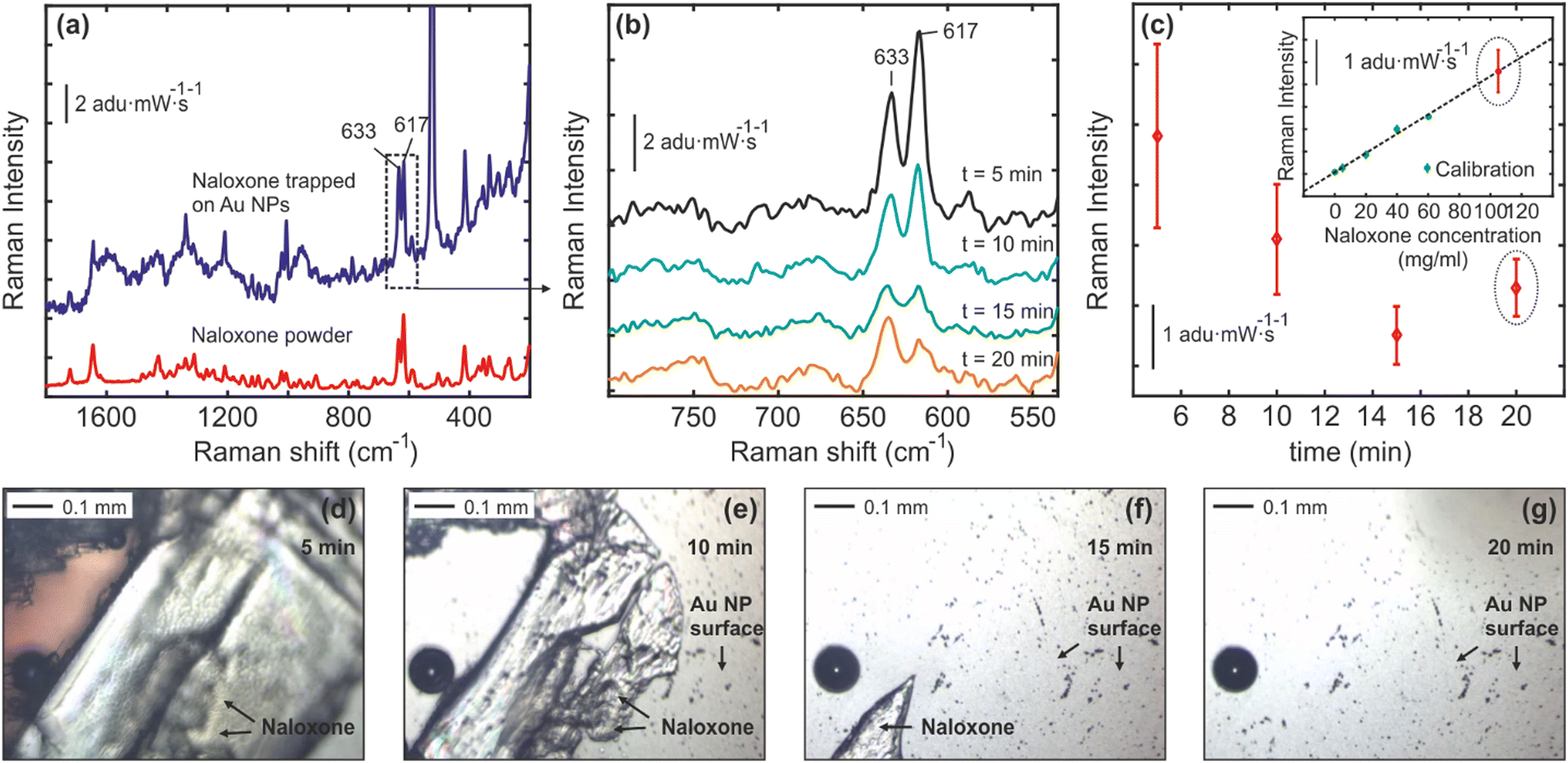 | ||
| Fig. 4 (a) Raman scattering spectra measured from naloxone powder and naloxone-tryptophane complexes (crystals) immobilized on the Au nanocactus surfaces. (b)–(c) The Raman scattering spectra recorded at different time intervals (t = 0–20 min) after 30 μl of PBS were deposited on the NT-Au nanocactus surface. In (b) averaged Raman spectra of three different measurements measured from three different Au nanocactus substrates (3 chips) are shown. In (c), the intensity change of the characteristic naloxone Raman vibrational mode at 637 cm−1vs. time indicates that naloxone crystals are gradually dissolved in PBS. The inset in (c) illustrates the intensity behavior of the same Raman mode (637 cm−1) for different naloxone concentrations in PBS (calibration curve). The naloxone Raman spectra at t = 20 min were recorded from a PBS solution after naloxone crystals were fully dissolved, and plotted together with the calibration curve in order to estimate the final naloxone concentration in PBS. (d)–(g) Optical images of naloxone crystals on the Au nanocactus surface exposed to the 30 μl PBS medium at t ≈ 0, 10, 15 and 20 min. | ||
The OXN-bound portion of the chip was investigated from real-time OXN release in a biological buffer medium. The characteristic peak of naloxone (633 cm−1 and 617 cm−1) was identified in its native state and was also confirmed when bound to the polymeric-Trp (ESI† Fig. S1). Raman measurements were made as a function of concentration, corresponding to OXN peak, demonstrating sustained release kinetics as shown in Fig. 4. Measurements (triplicate) confirmed that ∼110 mg mL−1 OXN (275 mM) was successfully released over a period of 20 minutes. This is similar to our starting concentration (250 mM; 1 mg/10 μl), highlighting the complete release of OXN with the poly-Trp matrix. It should be noted that naloxone hydrochloride dissolves rapidly in aqueous buffers (like PBS). However, upon being incorporated with the poly-Trp matrix, its releases stabilize, and associated release kinetics can be accessed over time, as demonstrated in Fig. 4b. The OXN concentration (∼110 mg mL−1) in our study is significant, as it aligns with reports of controlled-release oxycodone overdose needing large naloxone doses (over 100 mg in under 12 hours) for reversal.30 The three common routes of OXN administration are oral, IV, and intranasal. While oral dosing is uncommon due to significant first-pass metabolism, a dosage of 1000–3000 mg has shown efficacy over 6–24 hours. Remarkably, despite the poor bioavailability of intranasal naloxone dosage (0.4–1 mg mL−1), numerous reports of its clinical efficacy equal to or surpassing that of IV administration.31,32 This underlines why administering naloxone poses a challenge, as it involves balancing conflicting outcomes, which include reversing opioid toxicity while also preventing opioid withdrawal syndrome.
Fig. 4(c) shows the averaged Raman spectra of three different measurements measured from three different Au nanocactus substrates (from 3 chips). In Fig. 4(c), the intensity change of the characteristic naloxone Raman vibrational mode at 637 cm−1vs. time indicates that naloxone crystals are gradually dissolved in PBS. The inset in Fig. 4(c) illustrates the intensity behavior of the same Raman mode (637 cm−1) for different naloxone concentrations in PBS (calibration curve). The naloxone Raman spectra at t = 20 min were recorded from a PBS solution after naloxone crystals were fully dissolved and plotted with the calibration curve to estimate the final naloxone concentration in PBS. Fig. 4(d)–(g) depicts optical images of naloxone crystals on the Au nanocactus surface exposed to the 30 μl PBS medium at t ≈ 0, 10, 15 and 20 min. This serves as a visual confirmation for decreasing OXN intensity due to dissolution in the PBS solution over a period of time (20 minutes, as depicted in Fig. 4b).
Numerous studies have explored amino-acid mediated synthesis of nanoparticles in solution, including plasmonically active Au NPs.33 Additionally, a wide range of SERS substrates have been developed, even for ultra-low analyte detection.23,34–36 Likewise, a plethora of SERS substrates have been reported, even for ultra-low analyte detection.37,38 However, to the best of our knowledge, there exists no study demonstrating the designing of dual-activity: (1) capability to detect drugs (cocaine and heroin) via SERS, and (2) release kinetics of an antidote drug (naloxone) via Raman spectroscopy. The ability to design a dual-action fabrication scheme facilitating the physical ordering of nanostructures and a switchable chemical interface for drug delivery is vital. Likewise, “while supramolecular assembly is clearly an area of much interest, most work is still focussed on structure, rather than function”.39 Here, we have amalgamated the fundamentals of supramolecular chemistry and bio-conjugation chemistry, together with Raman-based modality for theranostics, for a clinically translatable proof-of-concept application. Unfortunately, there's not enough research on antidote kinetics, like OXN, with high fentanyl doses, and there are few controlled trials guiding the correct initial naloxone dose based on the specific opioid and dose exposure, making it hard for non-medical individuals to use naloxone correctly.40 Also, real-world data from take-home naloxone programs is limited because it doesn’t track what happens with every dispensed naloxone dose.41,42 This highlights the importance of developing dual-action TDM microsystems, as discussed in this study.
In conclusion, the ability to design a dual-action fabrication scheme, enabling both the physical ordering of nanostructures and the creation of a switchable chemical interface, holds significant promise. Combining supramolecular self-assembly with bio-conjugated interfaces in a single MEMS assembly creates new opportunities, particularly in theranostics and TDM for low therapeutic window drugs like chemotherapeutics and antibiotics, as demonstrated in our study. The next generation of this technology shall focus on designing closed-loop systems that can identify specific bio/chemical markers and trigger the release of suitable therapeutic/remedial agents with real-time kinetics assessment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SKS would like to thank H.C. Ørsted COFUND for funding. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement no. 713683. Also, Tomas Rindzevicius (IDUN, DTU Health Technology, DTU Denmark) procured the heroin solution for performing SERS measurements and related data analyses. Cocaine sample (cocaine solution) was obtained internally for technological exploration. Marlitt Viehrig (IDUN, DTU Health Technology, DTU Denmark) for manuscript feedback.References
- C. Liu, S. Weber, R. Peng, L. Wu, W. Zhang, P. B. Luppa, J. Popp and D. Cialla-May, TrAC, Trends Anal. Chem., 2023, 164, 117094 CrossRef CAS.
- E. La Van Dorp, A. Yassen and A. Dahan, Expert Opin. Drug Saf., 2007, 6, 125–132 CrossRef PubMed.
- I. Buajordet, A. C. Næss, D. Jacobsen and O. Brørs, Eur. J. Emerg. Med., 2004, 11, 19–23 CrossRef PubMed.
- S. K. Srivastava, T. Hasegawa, R. Yamada, C. Ogino, M. Mizuhata and A. Kondo, RSC Adv., 2013, 3, 18367 RSC.
- S. K. Srivastava, J. S. del Rio, C. K. O’Sullivan, C. Ogino and A. Kondo, RSC Adv., 2014, 4, 48458–48464 RSC.
- M. T. Klug, N. M. Dorval Courchesne, Y. E. Lee, D. S. Yun, J. Qi, N. C. Heldman, P. T. Hammond, N. X. Fang and A. M. Belcher, Chem. Mater., 2017, 29, 6993–7001 CrossRef CAS.
- C. Dahmann and K. Basler, Trends Genet., 1999, 15, 320–326 CrossRef CAS PubMed.
- A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12096–12101 CrossRef CAS PubMed.
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel and E. Gazit, Nat. Chem. Biol., 2012, 8, 701–706 CrossRef CAS PubMed.
- H. S. Jang, J. H. Lee, Y. S. Park, Y. O. Kim, J. Park, T. Y. Yang, K. Jin, J. Lee, S. Park, J. M. You, K. W. Jeong, A. Shin, I. S. Oh, M. K. Kwon, Y. Il Kim, H. H. Cho, H. N. Han, Y. Kim, Y. H. Chang, S. R. Paik, K. T. Nam and Y. S. Lee, Nat. Commun., 2014, 5, 3665 CrossRef PubMed.
- Z. F. Wu, Z. Wang, Y. Zhang, Y. L. Ma, C. Y. He, H. Li, L. Chen, Q. S. Huo, L. Wang and Z. Q. Li, Sci. Rep., 2016, 6, 22412 CrossRef CAS PubMed.
- W. Jiang, M. S. Pacella, D. Athanasiadou, V. Nelea, H. Vali, R. M. Hazen, J. J. Gray and M. D. McKee, Nat. Commun., 2017, 8, 15066 CrossRef CAS PubMed.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 CrossRef CAS PubMed.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817–823 CrossRef CAS PubMed.
- J. Boekhoven, J. M. Poolman, C. Maity, F. Li, L. Van Der Mee, C. B. Minkenberg, E. Mendes, J. H. Van Esch and R. Eelkema, Nat. Chem., 2013, 5, 433–437 CrossRef CAS PubMed.
- M. S. Schmidt, J. Hübner and A. Boisen, Adv. Mater., 2012, 24, OP11–OP18 CAS.
- H. Le-The, E. Berenschot, R. M. Tiggelaar, N. R. Tas, A. van den Berg and J. C. T. Eijkel, Microsyst. Nanoeng., 2018, 4, 4 CrossRef PubMed.
- J. Yang, M. Palla, F. G. Bosco, T. Rindzevicius, T. S. Alstrøm, M. S. Schmidt, A. Boisen, J. Ju and Q. Lin, ACS Nano, 2013, 7, 5350–5359 CrossRef CAS PubMed.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CrossRef CAS PubMed.
- S. Liang, C. Li, C. Zhang, Y. Chen, L. Xu, C. Bao, X. Wang, G. Liu, F. Zhang and D. Cui, Theranostics, 2015, 5, 970–984 CrossRef CAS PubMed.
- F. Yan, L. Liu, T. R. Walsh, Y. Gong, P. Z. El-Khoury, Y. Zhang, Z. Zhu, J. J. De Yoreo, M. H. Engelhard, X. Zhang and C. L. Chen, Nat. Commun., 2018, 9, 2327 CrossRef PubMed.
- L. Huang, J. Wan, X. Wei, Y. Liu, J. Huang, X. Sun, R. Zhang, D. D. Gurav, V. Vedarethinam, Y. Li, R. Chen and K. Qian, Nat. Commun., 2017, 8, 220 CrossRef PubMed.
- L. Rodríguez-Lorenzo, R. De La Rica, R. A. Álvarez-Puebla, L. M. Liz-Marzán and M. M. Stevens, Nat. Mater., 2012, 11, 604–607 CrossRef PubMed.
- M. P. Cecchini, V. A. Turek, J. Paget, A. A. Kornyshev and J. B. Edel, Nat. Mater., 2013, 12, 165–171 CrossRef CAS PubMed.
- Y. Wang, X. Yang, T. Liu, Z. Li, D. Leskauskas, G. Liu and J. B. Matson, J. Am. Chem. Soc., 2020, 142, 9158–9162 CrossRef CAS PubMed.
- Q. Zou and X. Yan, Chem. – Eur. J., 2018, 24, 755–761 CrossRef CAS PubMed.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS PubMed.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS PubMed.
- C. A. F. De Oliveira Penido, M. T. T. Pacheco, I. K. Lednev and L. Silveira, J. Raman Spectrosc., 2016, 47, 28–38 CrossRef.
- A. B. Schneir, T. F. Vadeboncoeur, S. R. Offerman, J. D. Barry, B. T. Ly, S. R. Williams and R. F. Clark, Ann. Emerg. Med., 2002, 40, 425–428 CrossRef PubMed.
- A. M. Sabzghabaee, N. Eizadi-Mood, A. Yaraghi and S. Zandifar, Arch. Med. Sci., 2014, 2, 309–314 CrossRef PubMed.
- M. A. Merlin, M. Saybolt, R. Kapitanyan, S. M. Alter, J. Jeges, J. Liu, S. Calabrese, K. O. Rynn, R. Perritt and P. W. Pryor, Am. J. Emerg. Med., 2010, 28, 296–303 CrossRef PubMed.
- H. E. Lee, H. Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–364 CrossRef CAS PubMed.
- M. P. Cecchini, V. A. Turek, J. Paget, A. A. Kornyshev and J. B. Edel, Nat. Mater., 2013, 12, 165–171 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- M. Li, X. Wu, J. Zhou, Q. Kong and C. Li, J. Colloid Interface Sci., 2016, 467, 115–120 CrossRef CAS PubMed.
- C. A. F. De Oliveira Penido, M. T. T. Pacheco, I. K. Lednev and L. Silveira, J. Raman Spectrosc., 2016, 47, 28–38 CrossRef.
- P. Chakraborty and E. Gazit, ChemNanoMat, 2018, 4, 730–740 CrossRef CAS PubMed.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 CrossRef CAS PubMed.
- R. Rzasa Lynn and J. Galinkin, Ther. Adv. Drug Saf., 2018, 9, 63–88 CrossRef CAS PubMed.
- A. Dahan, T. S. Franko, J. W. Carroll, D. S. Craig, C. Crow, J. L. Galinkin, J. C. Garrity, J. Peterson and D. B. Rausch, Front. Public Health, 2024, 12 DOI:10.3389/fpubh.2024.1346109.
- T. I. Saari, J. Strang and O. Dale, Clin. Pharmacokinet., 2024, 63, 397–422 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00824j |
| ‡ Present address: Centre for Biomedical Engineering, Indian Institute of Technology Delhi, Delhi, New Delhi, India. |
| This journal is © The Royal Society of Chemistry 2024 |