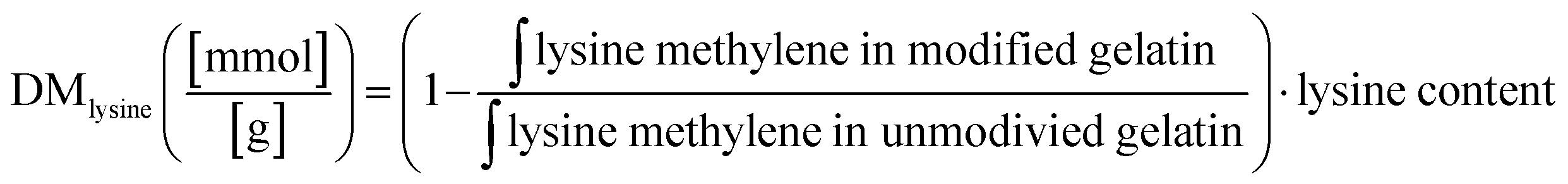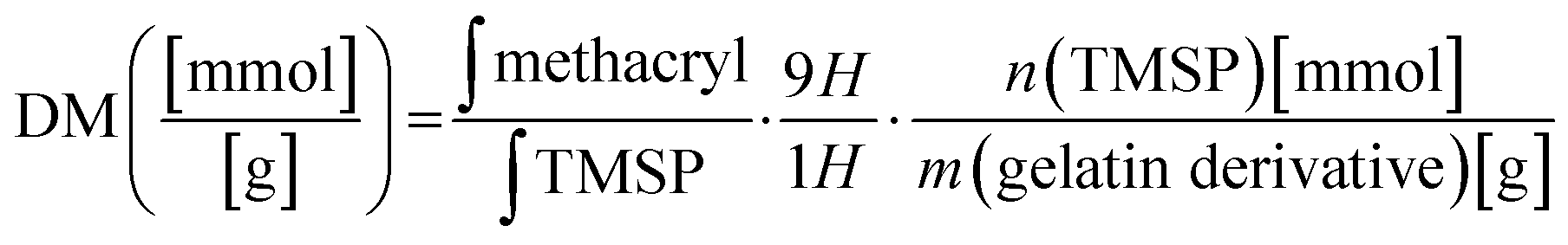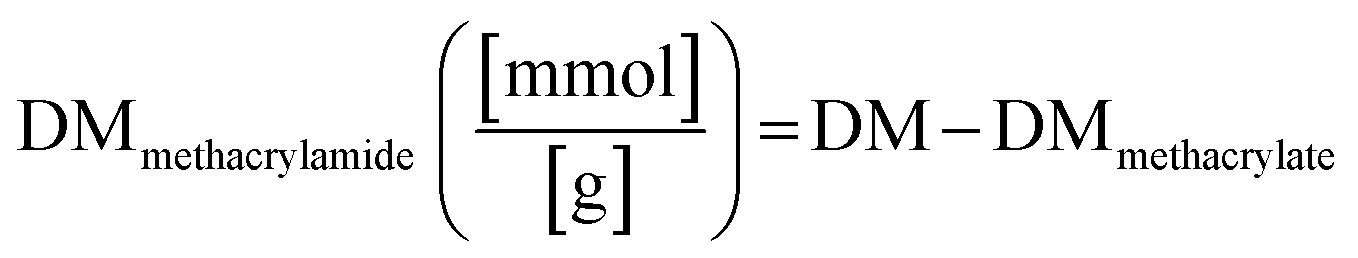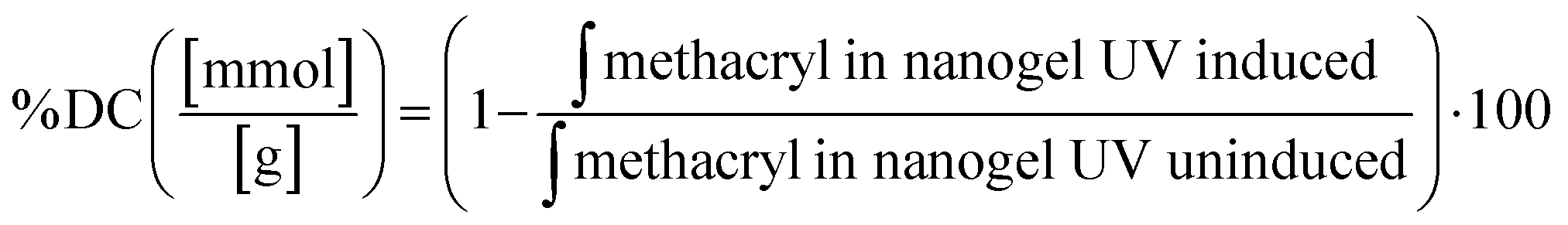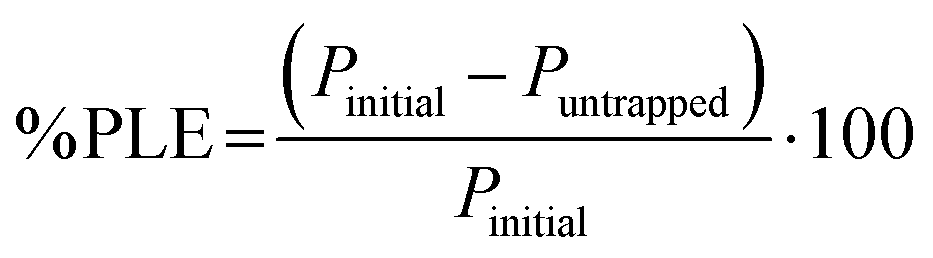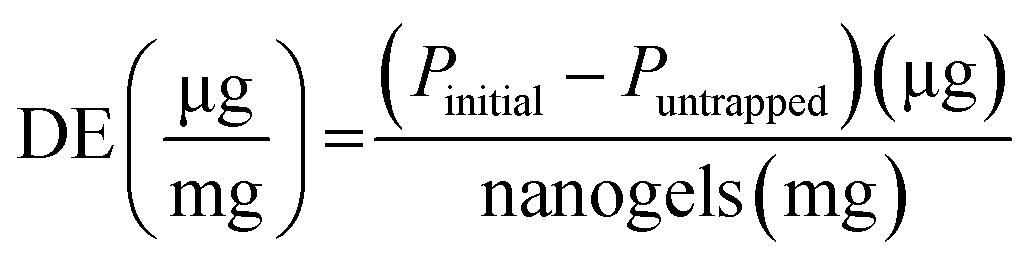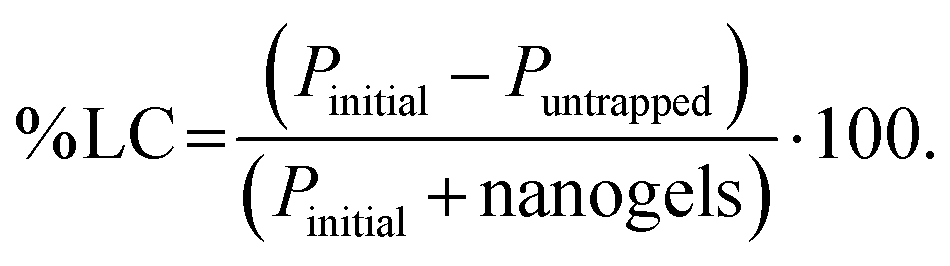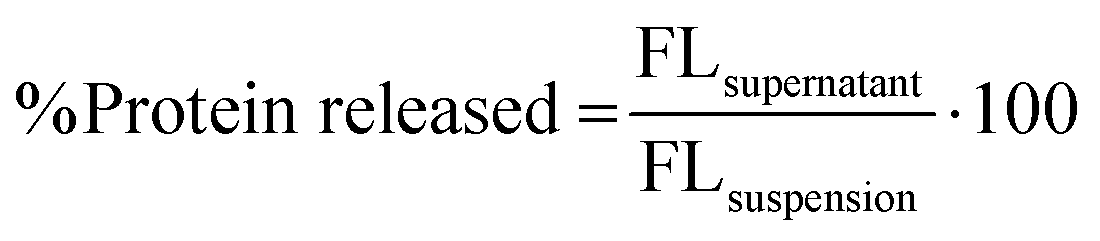DOI:
10.1039/D4MA00183D
(Paper)
Mater. Adv., 2024,
5, 5527-5542
Harnessing nanoreactors: gelatin nanogels for human therapeutic protein delivery†
Received
22nd February 2024
, Accepted 20th May 2024
First published on 21st May 2024
Abstract
Nanogels, polymeric nano-hydrogels suspended in an aqueous solution, have emerged as potential vehicles for transporting therapeutic proteins. These systems offer high protein loading capacity and a tunable gel matrix for controlled protein encapsulation and release. In this study, we designed and fabricated nanogels via a nanoreactor method (i.e., water droplets in organic solvent), followed by radical photopolymerization, and investigated the tunable swelling properties of the nanogels. Our results demonstrated that nanogels with less modified gelatin had a higher degree of swelling capacity and larger mesh size. Interestingly, we found that the initial size of the nanogels was solely dependent on the nanoreactor condition rather than the modified gelatin, the gelling biomaterial, signifying the importance of nanoreactor control in particle size determination. Nanogels showcased high protein loading capacity and rapid response to changes in salt condition, pH, and temperature, thereby accelerating the rates of protein release. This study demonstrated the tunable swelling properties, high protein loading and rapid release ability of nanogels triggered by internal/external modulators. Therefore, the nanogels developed in this study present a versatile platform for protein delivery, offering enhanced protein absorption and release capabilities.
1. Introduction
Proteins represent a major class of pharmaceuticals on the market due to their diverse physiological roles, such as biocatalysts, receptors, membrane channels, macromolecule carriers, and immune response agents, and are widely applied as therapeutics, diagnostics, and vaccines.1,2 Numerous food and drug administration (FDA)-approved protein-based products have been introduced to the current market,3–5 and protein therapeutics are used for target-specific medical treatments of a broad range of diseases caused by protein deficiency or insufficiency.2 Despite the recent advances in protein therapeutics development, however, proteins have inherent limitations as pharmaceuticals because proteins often undergo degradation via proteolysis, and some proteins, such as antibodies show low penetration ability through the membranes, such as blood–brain barrier.6 Moreover, protein localization to the target lesions can be insufficient due to the molecular size and binding affinity of the proteins.7 Consequently, there is an unequivocal demand for the development of protein carriers that can protect and deliver the protein therapeutic payloads to the target lesions while ensuring drug efficacy and clinical safety.
Various nanotechnology-based therapeutic approaches, including nanogels (hydrogel particles in nanoscale), have been widely investigated to enhance drug delivery efficacy.8,9 Nanogels provide two main advantages in protein delivery over non-hydrogel-based nanoparticles. First, its highly water-absorbing mesh structure and nanoscale size confer excellent colloidal stability and biocompatibility. Nanogel-based delivery carriers improve drug stability by preventing them from chemical destruction and enzymatic degradation.10 In addition, the porosity of nanogels enhances drug-loading capacity and enables controlled drug release, which is a challenge to accomplish in conventional large porosity hydrogels.11 Second and most importantly, the gelling biomaterials that nanogels comprise possess stimuli-responsive properties (i.e., swelling and shrink states) as delivery carriers. The tunable swelling capacity is instrumental in controlling the amount of therapeutics that can be entrapped in a gel matrix and subsequently released through the gel network.12
The balance between the inner and outer osmotic pressures in the gel phase regulates their swelling ability. Any alterations in osmotic pressures induce the transition between gel swelling and shrinking states. Structural modulation of gels can be achieved by modifying external parameters such as pH and salt concentrations, as well as internal parameters derived from gelling biomaterials, including porosity and surface charge. These are of particular significance in drug delivery applications as ‘controllable’ parameters, as they allow for the entrapment and subsequent release of payloads at a rate governed by such external and internal modulations. Hence, the swelling capacity of nanogels was of interest in our study for the entrapment and release of recombinant proteins. The protein diffusion rate through the matrix is dependent on the mesh size of the nanogels matrix rather than the particle size of nanogels.13 However, the substantial surface-to-volume mass ratio of nanogels increases the protein loading capacity. By modulating the degree of photopolymerization of gelling biomaterial, we offer a practical method to control the swelling capacity and, in turn, control protein binding and release. Furthermore, the optimized nanoreactor system plays a pivotal role in controlling the size of the nanogels and increasing protein loading capacity.
In this study, we designed a protein delivery carrier for rapid and complete protein release (Scheme 1). We utilized this new system to deliver Filaggrin (FLG), a human skin protein known for its essential role in maintaining human skin barrier integrity. Recent studies on the FLG treatment for skin barrier reconstruction revealed that the recombinant FLG, once internalized in the cytoplasm of keratinocytes, was likely processed for skin restoration.14 However, FLGs were denatured and proteolyzed during the application and had a poor ability to penetrate biological barriers, which reduced the bioavailability.6 To overcome the challenge, we entrapped the protein in a nanogel delivery system, allowing its polyionic complex formation, which provides protein stabilization and sustainable release from the nanogels.
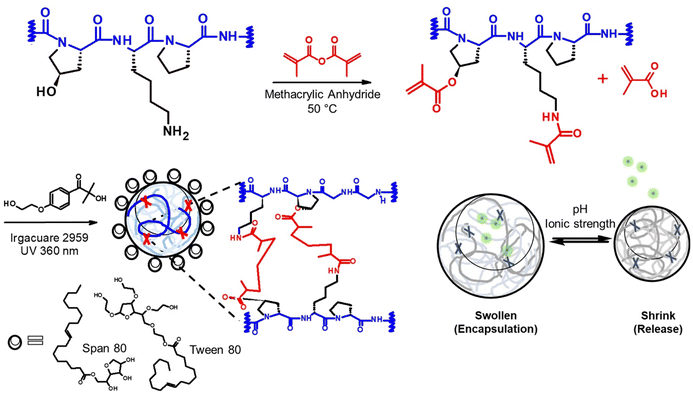 |
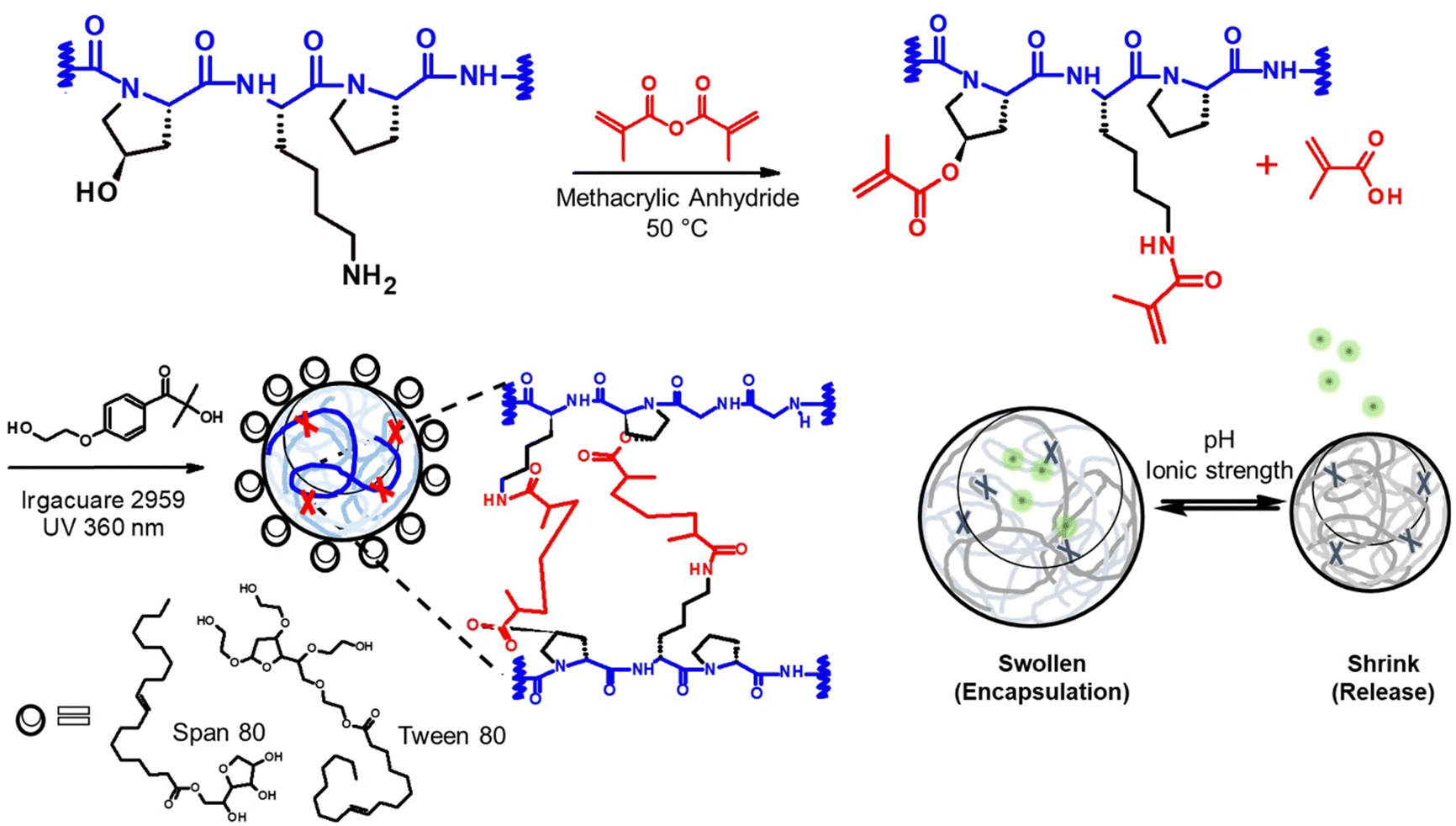
| | Scheme 1 The schematic representation of nanogels structure formation and the stimuli-responsive property. (upper) The synthesis of gelatin derivative, the modified gelatin reacted with methacrylic anhydride. (lower) Nanogel structure created by methacryloylation reaction and the swelling property of nanogels. | |
2. Results and discussion
2.1. Gelatin derivatives
2.1.1. Gelatin methacryloyl (GelMA), the modified gelling biomaterial.
Gelatin is one of the well-characterized biomaterials inspired by its unique properties, such as thermo-reversibility, simple fabrication methods, and forming of polyion complexes with charged bioactive compounds. We utilized gelatin to fabricate nanogels to entrap human skin protein as a model therapeutic agent. Gelatin can be chemically modified using crosslinkable functional groups such as glutaraldehyde and methacrylic functions. In this study, chemically modified gelatin, gelatin methacryloyl (GelMA) was synthesized and analyzed as a precursor polymer. GelMA forms the three-dimensional gel network when exposed to UV light via crosslinking the methacryloyl substituents in gelatin side chains. This reaction can be conducted under mild conditions (i.e., temperature between 40 to 50 °C, aqueous condition, neutral pH). The degree of methacryloylation (DM) was controlled by the molar ratio of methacryloyl substituents to reactive side groups in the reaction mixture, as shown in Table S1 and Fig. S1 (ESI†). The pH of the reaction solution was set to pH 7.4 to increase the reactivity of amino and hydroxyl groups and thus allow a higher degree of substitution.15
2.1.2. Determining the degree of methacryloylation.
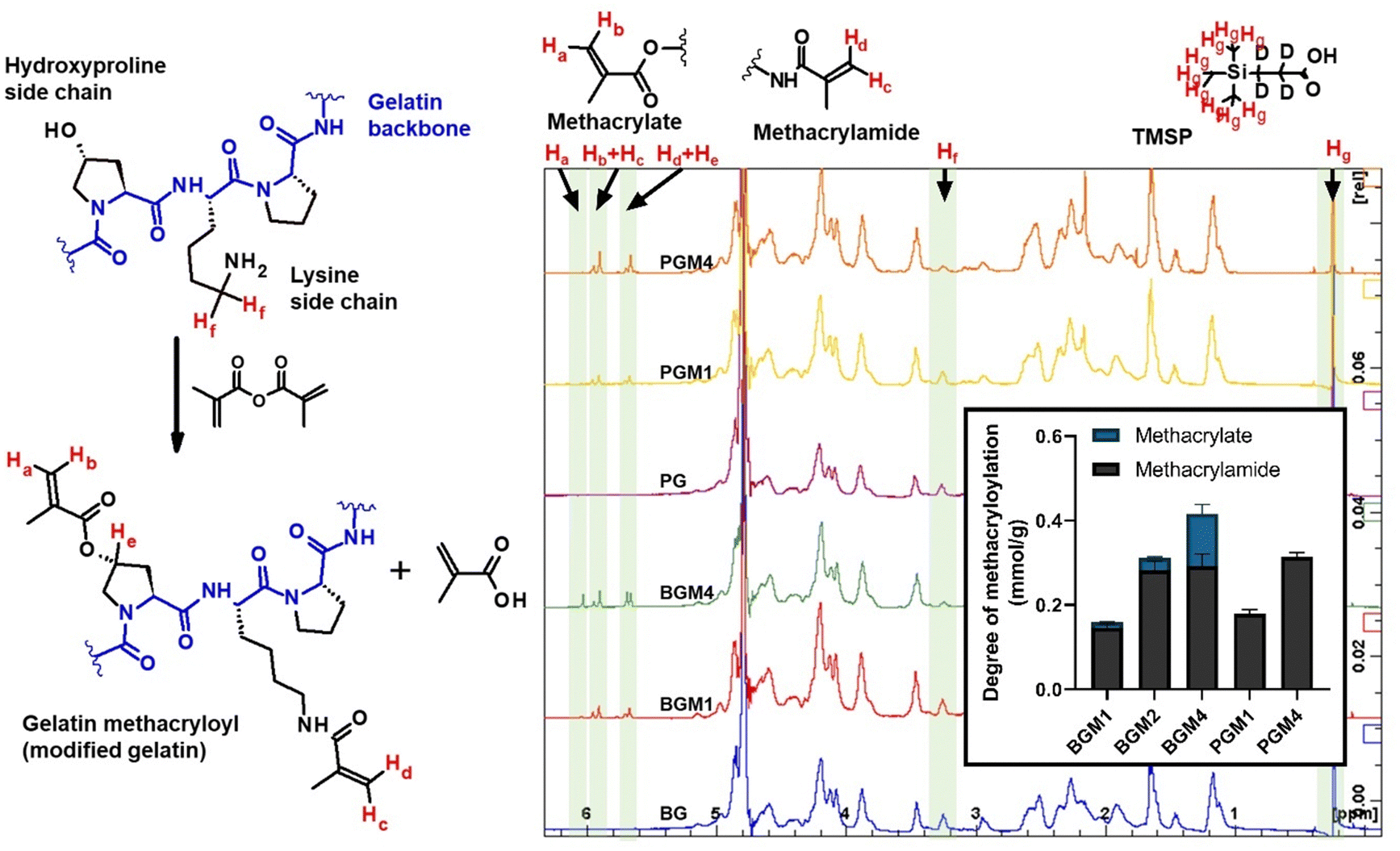
Although the quantitative methods to determine the DM in gelatin have been reported,16 these methods were only able to measure the methacrylamide group bound to the lysine and hydroxylysine side chain in gelatin without considering the amount of the methacryl group bound to hydroxyl groups in the overall DM assessment. Because the reacted amount of methacrylate group was relatively low compared to the methacrylamide substitution, no accurate method was available to determine each methacrylate and methacrylamide bound to the gelatin side chain. Claaßen et al. identified the methacryl groups bound to hydroxyl groups and amino groups in gelatin using 2D NMR, introducing an accurate method to determine each methacryl group bounded to different functional groups in the gelatin side chain.17 The reaction between methacryl and amino groups was more abundant, and the methacrylate formed almost after the completion of methacrylate substitution in the lysine and hydroxyl side chain.17,18 However, these study results were limited to the methacryloylation of porcine gelatin. The relative reactivity of methacryl groups on functional groups in the gelatin side chain may vary based on the molar ratio of hydroxyl/amino functional groups. Moreover, the DM quantitative method has largely depended on the lysine content in reference. In order to acquire accurate DM quantification results, we examined two types of gelatin, each containing different amino acid compositions. The different molar ratios in total hydroxyl groups and amino groups in type A (porcine, PGM) and B (bovine BGM) gelatin showed the different reactivity of methacrylate over methacrylamide in gelatin. The DM of modified gelatin was calculated using eqn (1) based on a decreased amount of unmodified lysine after gelatin methacyloylation (Fig. S1A, ESI†). DM increased proportionally by increasing the excessive amount of methacrylic anhydride (MAA) added to gelatin. GelMA from type A gelatin (PGM) shows relatively higher DM than the GelMA from type B gelatin (BGM). This can be explained for two reasons: first, this quantification method is based on the methacryl groups bound to amino groups, so the portion of methacryl groups bound to hydroxyl groups (i.e., methacrylate groups) was not included in DM. BG has a higher amount of free hydroxyl group in the side chain, where the methacryl groups are more likely to react. Thus, the portion of methacryl groups bound to the hydroxyl group has to be included in the total DM calculation. Second, the signal of methylene protons neighboring to unmodified amino groups (δ = 2.7–2.9) used in our calculation was superimposed, making it difficult to obtain an accurate value of DM (Fig. 1). The difference of DM in PG and BG resulted from its amino acid composition in gelatin, especially the total functional group contents (Fig. S1B, ESI†). BG has a notably higher amount of hydroxy groups per gram of gelatin, while the measured amount of amino groups per gram of gelatin was similar in both PG (0.34 mmol g−1) and BG (0.31 mmol g−1), which is in agreement with the 0.35 mmol g−1 of amino groups in gelatin previously reported.19
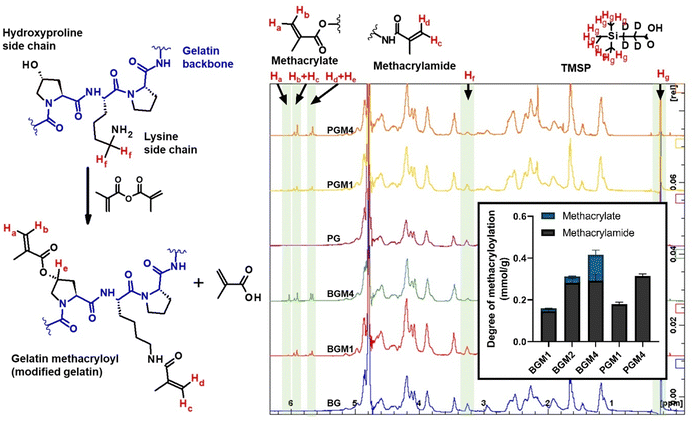 |
| | Fig. 1 Proton NMR spectra of gelatin and gelatin derivatives with different DM. The signal of protons was highlighted as follows: Ha–Hd: acrylic protons of methacryl groups; Hf: two methylene protons in unmodified lysine residues, Hg: nine protons in TMSP, He: a proton in methacryl-modified hydroxyproline residues. DM of gelatin derivates: BGM1: type B gelatin (BG) treated with the equal molar MAA to amino groups; BGM2: BG treated with 2× excessive molar MAA to amino groups; BGM4: BG treated with 4× excessive molar MAA to amino groups; PGM1: type A gelatin (PG) treated with the equal molar MAA to amino groups; PGM4: PG treated with 4× excessive molar MAA to amino groups. | |
The proton NMR spectra and the structure of the reactive functional groups in the gelatin side chain and the substituted methacryl moiety to amino acid residues are depicted in Fig. 1. MAA reacts not only with free amino groups in lysine and hydroxylysine but also with free hydroxyl groups in hydroxyproline, tyrosine, threonine, and serine, although MAA is more reactive to amino groups.18 Integrating the signal of methacrylate rather than that of methylene protons neighboring unmodified lysine gives a more accurate DM substitution, which includes both methacrylate and methacrylamide functions in gelatin derivatives. The proton NMR spectra of gelatin derivatives in deuterium oxide (including trimethylsilylpropanoic acid, TMSP) were obtained, and the DM of gelatin derivatives was calculated using eqn (2)–(4). When the methacrylic function of MAA reacts to the ε-amino group in the lysine side chain, the integral of the methylene protons signal decreases (represented “Hf” in Fig. 1). This signal was used to calculate DM using eqn (1). When the methacryl group of MAA reacts to the hydroxyl group in the gelatin side chain, the integral of the proton signal increases (represented “Ha” in Fig. 1). If the reaction is specific to the hydroxyl groups in hydroxyproline, then the superimposed signal will appear (represented “Hd + He” where “Hd” is the signal of acrylic protons in methacrylamide). These superimposed signals, Hb + Hc, and Hd + He were identified using 2D NMR.17 The integral of signal “Hb + Hc” and “Ha” was used to calculate the degree of methacryloylation (DM; DMmethacrylate + DMmethacrylamide) and the degree of methacrylate (DMmethacrylate), respectively, using eqn (2) and (3). TMSP was used as an internal standard to obtain the accurate number of methacryl functions that reacted per gelatin mass during the gelatin derivatization reaction.
2.1.3. Gelatin type and methacryloylation reaction.
The excessive amount of MAA added in the gelatin solution, and the degree of substitution in the gelatin side chain were highly correlated (Fig. 1, inset). For instance, the BGM4 and PGM4, which were treated with a high dose of MAA, showed the highest degree of substitution. The slightly higher amount of methacrylamide in PGM1 and PGM4 than in BGM1 and BGM4 is because PG has a higher amount of free amino groups than that of BG.20 However, the methacrylate groups were not detected in PGM, which aligns with the previous study reporting the dominance of methacrylamide formation over methacrylate during the gelatin methcryloylation process.17 In our study, methacryl groups favorably reacted to amino groups when amino groups were available, showing that the degree of methacrylamide (i.e., substituted amino groups) in PGM1 (0.179 mmol g−1) and PGM4 (0.314 mmol g−1) was below the total free amino groups in PG (0.341 mmol g−1), and the methacrylate groups (i.e., substituted hydroxyl groups) were not detected. In contrast, methacrylate was detected in BGM, indicating MAA reacted with free hydroxyl groups even in the presence of free amino groups. The degree of methacrylamide in BGM1 (0.146 mmol g−1), BGM2 (0.283 mmol g−1), and BGM4 (0.291 mmol g−1) was below the total free amino groups in BG (0.315 mmol g−1). This infers that the higher amount of free hydroxyl groups presented in BG (1.075 mmol g−1) made it a more accessible substrate, and the free MAA reacted to hydroxyl groups in BG.
2.2. Nanoreactor
Our study leveraged a water-in-oil nanoemulsion system, wherein we introduced water droplets that serve as nanoreactors to contain photocrosslinkable polymer chains, specifically PGM and BGM. The nanoreactor plays a significant role as confined “aqueous nanocontainers” during the UV irradiation process. In order to control the size of resulting photopolymerized nanogels precisely, it is critical to ensure the stability and size distribution of water droplets in the emulsion both before and during the crosslinking reaction (Fig. 2). The photopolymerization method allowed the creation of a controllable and well-structured gelatin polymer network by crosslinking a gelatin derivative chain with methacrylic moiety. However, to create a “nano-sized” gelatin polymer network (scale down hydrogels to nanogels), the crosslinkable gelatin derivative chain had to be confined in aqueous droplets before photopolymerization. The nanoparticle status in each process is depicted schematically in Fig. 2(A). Initially, GelMA chains are confined in water droplets called “nanoreactors,” and at this entrapped polymer stage, the GelMA polymers are yet to crosslink and reversibly form a physical gel network. In the next stage, UV irradiation induces photocrosslinking of GelMA polymers within the nanoreactors, resulting in the formation of an irreversible chemical gel network. This ensures that the nano-size is retained in an aqueous solution even after surfactant and oil removal. Upon resuspending GelMA in an aqueous phase, the gel absorbs water and swells in the solution. At this stage, nanogels can be lyophilized and stored in a −20 °C freezer. The lyophilized nanogels can fully recover the size in swollen status when resuspended in an aqueous solution. Fig. 2(B) and (C) show the change in the size distribution of water droplets in each process. The hydrodynamic diameter (dH) of photocrosslinked nanogel was identical to that of entrapped polymers (precursor polymer nanodroplets before photopolymerization), indicating the nanoreactors in the water-in-oil nanoemulsion system remained at a controlled size (mid-range polydispersed) after UV irradiation. The discrepancy of dH between photocrosslinked gel and hydrate gel infers that nanogels confined in nanoreactors did not reach the swelling equilibrium, but when in the aqueous phase, they swelled freely and showed larger hydrodynamic diameters.
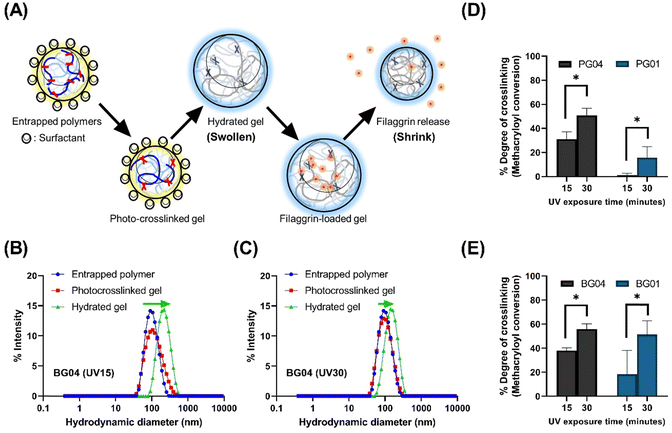 |
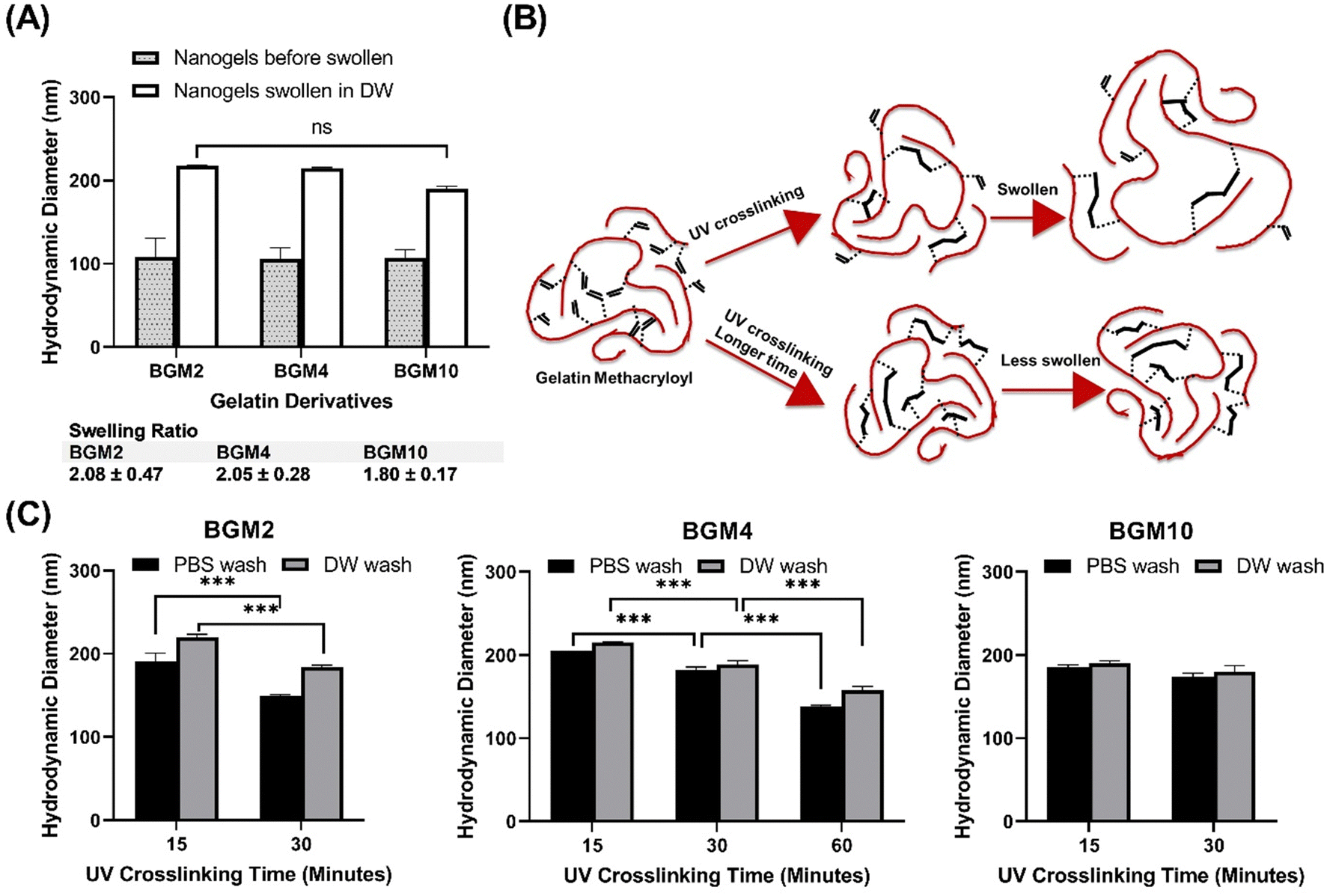
| | Fig. 2 (A) The schematic representation of the nanogels stages. Entrapped polymers: crosslinkable polymer confined in nanoreactor; photo-crosslinked gel: crosslinked gel upon UV irradiation; hydrated gel: nanogels swollen in aqueous solution. (B) Changes in dH of BG04 nanogel with fewer number of incident photons (total energy) by shorter UV exposure time of 15 min: more swollen nanogels in PBS solution. (C) Changes in dH of BG04 nanogel that was subjected to longer UV exposure time of 30 min: less swollen nanogels in PBS solution. (D) The crosslinking degree of nanogels induced by the conversion of methacryloyl groups on type A gelatin side chains. PG04: nanogels made of PGM4; PG01: nanogels made of PGM1. (E) The crosslinking degree of nanogels induced by the conversion of methacryloyl groups on type B gelatin side chains. BG04: nanogels made of BGM4; BG01: nanogels made of BGM1. | |
Alterations in dH of hydrated gel formed from photocrosslinked nanogels swollen in the aqueous solution are regulated by irradiation conditions. Photocrosslinked BG04 nanogels, subject to high photon incidence, showed identical dH values to those of the gels undergoing lesser photon exposure, indicating the photopolymerization process in the nanoreactor did not affect dH. However, the different levels of size increase in hydrated gels demonstrated that the swelling ability of nanogels can be tuned by the modulation of the number of incident photons resulting from the different UV irradiation times. Gels subjected to prolonged UV exposure exhibited a higher degree of crosslinking, which might limit the subsequent swelling of hydrated gels (Fig. 2(D) and (E) and Fig. S2, ESI†).
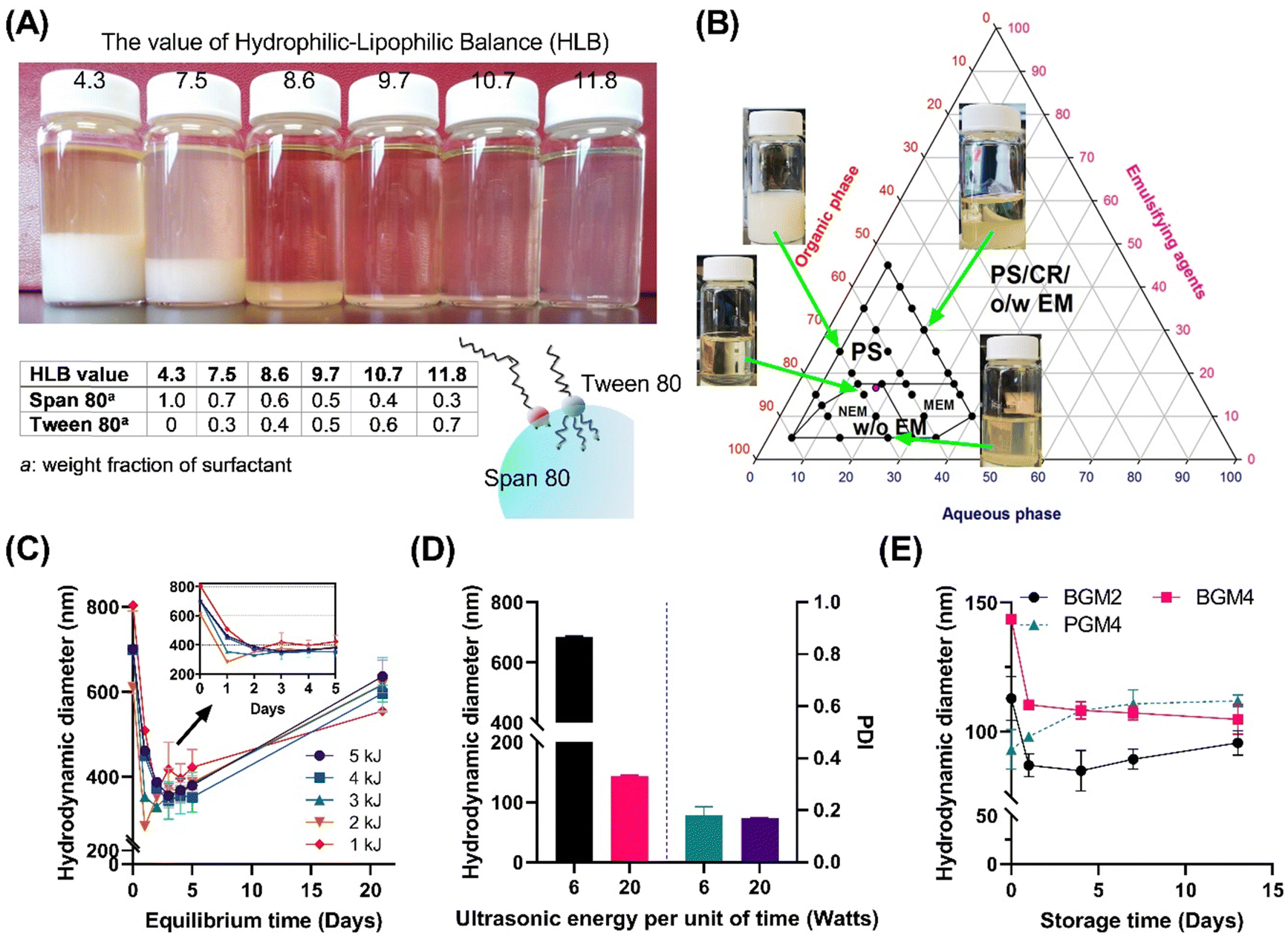
The inversed emulsion (water-in-oil emulsion) was chosen to create a nanoreactor system as it is a broadly applicable method to confine a variety of hydrophilic polymers. However, it is relatively less stable compared to thermodynamically stable emulsions (oil-in-water emulsion), and potential phase separation can occur due to the increased particle attraction driven by the polarity of the organic phase dispersion medium. This necessitates a meticulous optimization of emulsion composition and processing conditions. Furthermore, the emulsion had to be visually transparent to minimize incident beam loss through reflection or refraction by droplets during UV irradiation. Since surfactants significantly contribute to stabilizing two immiscible phases on the water–oil interface, screening for the optimal surfactant mixing ratio is critical for enhancing emulsion stability.21 We first stabilized the nanoemulsion by determining the optimal hydrophilic–lipophilic balance (HLB) of the surfactant mixture (Fig. 3). Tween 80 (hydrophilic surfactant, HLB = 15) and Span 80 (hydrophobic surfactant, HLB = 4.3) were mixed in various ratios (HLBmix) (Fig. 3(A)). The HLBmix value of surfactant mixture with various weight ratios was calculated using eqn (5). The optimal HLB was determined when the resulting nanoemulsion showed narrow size distribution by dynamic light scattering (DLS) and formed a visually transparent emulsion without phase separation after 12 h of equilibrium. The optimal value (HLBmix = 10.7) for this particular emulsion system was selected for the rest of the study.
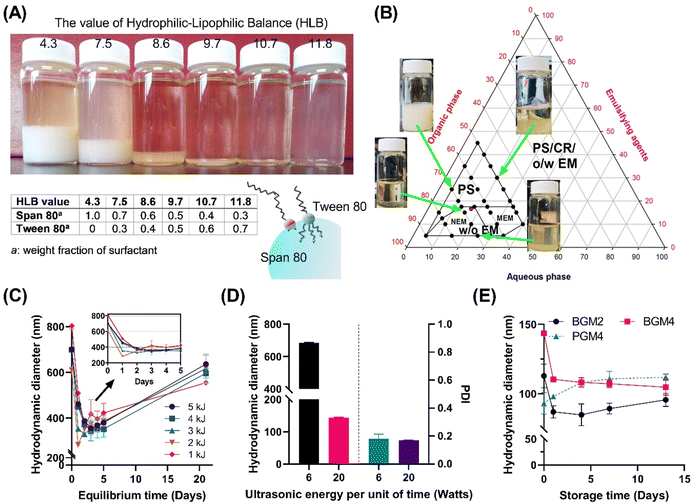 |
| | Fig. 3 (A) The effect of the hydrophilic–lipophilic balance of surfactant mixture on reversed nanoemulsion stability (phase separation and turbidity). (B) optimization of stable water-in-oil nanoemulsion formation region. PS: phase separation (liquid–liquid phase separation, sedimentation); CR: crystallization; O/W EM: oil-in-water emulsion; W/O EM: water-in-oil emulsion; NEM: nanoemulsion (dH < 250 nm); MEM: microemulsion (dH > 250 nm). (C) changes in dH of nanoemulsion during the equilibrium time. (D) The effect of ultrasonic energy input on dH and PDI. (E) Nanoemulsion stability: change in hydrodynamic diameter by time after nanoemulsion formation processing. | |
A ternary phase diagram study was conducted to determine the optimal water, oil, and surfactant volume ratio to form stable nanodroplets (Fig. 3(B)). The marked area indicates the formation region of homogeneous nanoemulsions without sedimentation. A surfactant mixture was dissolved in n-octane and mixed with an aqueous solution containing crosslinkable polymer, followed by high-speed homogenization and ultrasonication to form stable water-in-oil nanoemulsions. More than 15% mass ratio of surfactants in the system made surfactant sedimentation. Stable and homogeneous nanoemulsions only formed under specific water, oil, and surfactant weight ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1. The stability of the nanoemulsion was determined by the change in hydrodynamic diameters of nanoparticles in n-octane (Fig. 3(C)). Over the initial 24 h, nanoparticles displayed a decrease in size (equilibrium stage) while demonstrating stable size distribution (polydispersity index < 0.3) for up to 5 days, indicating the formation of stable nanoemulsions. This finding suggests that the UV crosslinking procedure can be conducted within a 2–5 days period following 24 h equilibrium stage to create photocrosslinked nanogels exhibiting a small hydrodynamic diameter with narrow size distribution. Next, we examined the effect of high ultrasonication energy on nanoemulsion stability (Fig. 3(D)). A higher energy input per unit of time (20 Watts) appeared to reducing the droplet size to under 200 nm. In contrast, the low energy input per unit of time (6 Watts) facilitated nanoemulsion forming with a narrow polydispersity index (PDI) but rather larger particle size. We obtained nanoemulsions with well-controlled distribution showing excellent emulsion stability over 2 weeks (Fig. 3(E)). We found that the initial diameter of the emulsion nanodrops, ranging from 80 to 120 nm, critically influenced the photopolymerization process as nanoreactors. This initial diameter was a major parameter dictating the final size of the non-swollen nanogels in an aqueous solution. For instance, gel sizes derived from different GelMA were nearly identical before swelling because the initial dH of the nanoreactor determined the gel size.
:4:1. The stability of the nanoemulsion was determined by the change in hydrodynamic diameters of nanoparticles in n-octane (Fig. 3(C)). Over the initial 24 h, nanoparticles displayed a decrease in size (equilibrium stage) while demonstrating stable size distribution (polydispersity index < 0.3) for up to 5 days, indicating the formation of stable nanoemulsions. This finding suggests that the UV crosslinking procedure can be conducted within a 2–5 days period following 24 h equilibrium stage to create photocrosslinked nanogels exhibiting a small hydrodynamic diameter with narrow size distribution. Next, we examined the effect of high ultrasonication energy on nanoemulsion stability (Fig. 3(D)). A higher energy input per unit of time (20 Watts) appeared to reducing the droplet size to under 200 nm. In contrast, the low energy input per unit of time (6 Watts) facilitated nanoemulsion forming with a narrow polydispersity index (PDI) but rather larger particle size. We obtained nanoemulsions with well-controlled distribution showing excellent emulsion stability over 2 weeks (Fig. 3(E)). We found that the initial diameter of the emulsion nanodrops, ranging from 80 to 120 nm, critically influenced the photopolymerization process as nanoreactors. This initial diameter was a major parameter dictating the final size of the non-swollen nanogels in an aqueous solution. For instance, gel sizes derived from different GelMA were nearly identical before swelling because the initial dH of the nanoreactor determined the gel size.
2.3. Nanogels swelling capacity control
Controlling the swelling capacity of nanogels is essential for protein binding and release control in drug delivery applications. Previous research by Messager et al. identified the degree of polymer substitution and the conversion degree as crucial parameters to control the swelling ability of nanogels.13
This study aimed to identify the factors determining the degree of swelling. We selected the DM in the gelatin derivatives side chain and the degree of conversion (DC) as modulators of the swelling ability. DC reflects an increased level of crosslinking density via photopolymerization, subject to the number of incident photons exposed to the photocrosslinkable moieties in GelMA, and can be tuned by UV irradiation power and time. We prepared nanogels, demonstrating various swelling degrees through controlled structure photocrosslinking. We assessed two internal parameters of nanogels that can be adjusted during the fabrication process to control swelling capacity: (1) DM in gelatin derivatives and (2) DC to gauge the effectiveness in the modulation of nanogels for protein delivery carriers. Fig. 4(A) depicts the size difference between nanogels confined in a nanoreactor after UV irradiation and nanogels fully swollen in an aqueous phase. We observed that the photocrosslinked GelMA with various DM consistently showed increasing dH when nanogels were dispersed in an aqueous phase. The swelling ratio varied in nanogels with different DM levels. As anticipated, nanogels with low DM (i.e., BGM2) had a high swelling ability with a swelling ratio of 2.08 ± 0.47, whereas BGM10 showed a relatively low swelling ratio (1.80 ± 0.17). This result suggests that DM is a modulation factor controlling the swelling properties of nanogels. However, while we observed a decreasing trend in dH with higher DM nanogels, the effect of DM on swelling capacity was not significant in our study. This can be attributed to the fact that BGM2 and BGM10 only have a slight difference in concentration of methacryl groups, 0.3123 mmol g−1 and 0.3522 mmol g−1, respectively, available for crosslinking via photopolymerization. This insignificant difference in DM may not be sufficient to induce a noticeable macroscopic scale change in nanogels’ properties, such as the swelling capacity. Fig. 4(B) and (C) represent the swollen nanogels with various degrees of conversion and the impact of DC on swelling capacity, respectively. Nanogels exposed to UV for longer durations showed lower swelling ability. The more nanogels were crosslinked, the less it was swollen in an aqueous solution, which aligns with the results reported by Messager et al. The swelling capacity was also influenced by the external factors, such as the presence of salt in the solution. For instance, the dH of nanogels under various DM and irradiation conditions was consistently reduced in PBS ([NaCl] = 0.2 M) compared to in deionized water. Gelatin derivatives possess a high number of electrolyte groups in the side chain that allows them to be highly charged at pH levels below and above its isoelectric point (pI, PG: 7.0–9.5 and BG: 4.7–5.3). Nanogels made of these polyelectrolytes are charged in solution when electrolyte groups dissociate in an aqueous environment. They swell less in PBS with electrolytes because the swelling status is determined by the osmotic pressure exerted by free counterions within the nanogels matrix. In the presence of an electrolyte, the osmotic pressure difference is reduced, and the swelling degree decreases. These changes in the swollen state can be analyzed by measuring the dH of nanogels by DLS. The external parameters that modify the swelling and shrinking state of nanogels, which affect protein binding and release profiles, will be discussed later. The modulation of the photopolymerization condition altered the crosslinking density of nanogels, which offered a significant change in the degree of swelling.
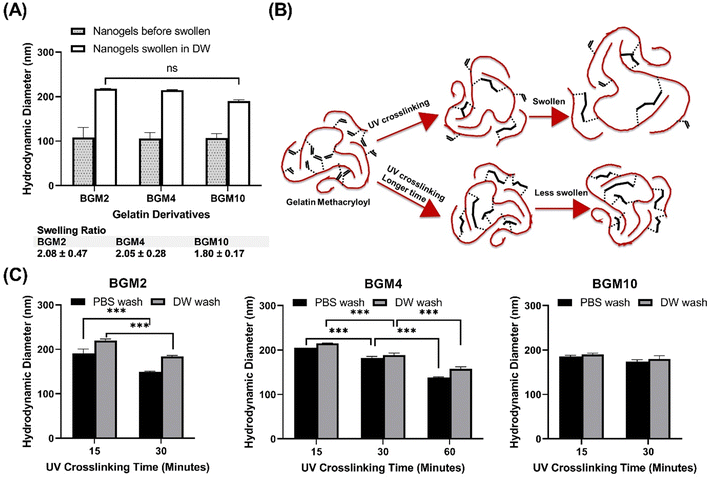 |
| | Fig. 4 (A) Swelling ability modulation by DM. (B) Schematic representation of gelatin derivatives swelling property. The degree of conversion can be tuned by the number of incident photons exposed to crosslinkable GelMA using different UV irradiation exposure time. (C) Swelling ability governed by the number of incident photons (total energy) during the photopolymerization process. | |
The morphology of nanogels was analyzed using transmission electron microscopy (TEM) on a copper grid in the dry state (5 mg mL−1). Given the low electron density of these soft material particles, the dried nanogels were stained with a negative contrast agent before TEM analysis. The nanogels exhibited mid-range polydispersed, spherical morphology with narrow size distribution (Fig. S3, ESI†). The size of nanogels (<200 nm) observed in the TEM image analysis corresponded well with the result obtained by DLS. The black arrows in the TEM images indicate the encapsulated recombinant protein, FLG in the nanogel matrix. A slight aggregation of nanogels was observed in the TEM image. To further understand the microstructure of the nanogels matrix, we obtained scanning electron microscopy (SEM) images (Fig. S4, ESI†). Both BGM4 and PGM4 demonstrated a highly porous structure, implying that recombinant proteins can be entrapped in the porous gel matrix and merely bound to the surface of nanogels.
The swelling degree of nanogels can be influenced by both internal and external parameters. Internally, the degree of swelling can be controlled by manipulating the photopolymerization conditions, including irradiation power and time, and the DM of gelatin derivatives. These aspects can be precisely tuned during the nanogel synthesis process, allowing us to regulate the swelling ability of nanogels finely. This is essential for achieving controlled protein release. External factors, such as the presence of salt and pH changes, also play crucial roles in the swelling and shrinking behavior of nanogels, consequently altering the protein release rate from the crosslinked nanogels matrix. Importantly, gelatin derives its composition and properties from the collagen source and extraction method used.22 Since the nanogels matrix consists of modified gelatin chains, studying the surface charge of gelatin in solution is important to understand the behavior of nanogels in that solution. The ζ-potential serves as an effective measurable parameter for determining the surface charge of polymers in solution. This charge can be significantly influenced by the presence of salt and pH changes, as demonstrated in Fig. S5 (ESI†). In deionized water (DW, pH adjusted to 7.4), PG and BG are oppositely charged. However, in the presence of salt in sodium phosphate buffer (PB, pH adjusted to 7.4), both PG and BG exhibited weak charges, indicating an increased interaction between the mobile electrolyte in solution and the polyelectrolytes in gelatin. It is important to note that PG has the weakest charge (close to 0) at pH 7.4 or above, whereas BG has the weakest charge at pH 5.8 or below because of the different pI's (PG: 7.0–9.0 and BG: 4.7–5.2). This difference could result in gelatin precipitation due to decreased suspension stability from the reduced repulsion forces between polymers. To illustrate this, the PGM nanogels in a pH 7.4 solution demonstrated a higher evolution rate in dH and PDI than those of BGM nanogels over 5 days of storage (Fig. S6, ESI†). This result correlated with the result in Fig. S5 (ESI†), showing that the ζ-potential of PG at pH 7.4 is almost neutral, implying an unstable state of PGM suspension at this pH. For the remainder of this study, we used the deionized water (DW, pH 7.4), PB at pH 5.8 (an acidic physiological pH similar to that of the healthy stratum corneum of the skin), and PB at pH 7.4 (a slightly alkaline physiological pH similar to blood) was used to mimic the environmental condition when protein-loaded nanogels are administered either topically or intravenously.
2.4. Stimuli-responsive nanogels matrix
The colloidal stability of hydrated nanogels was evaluated prior to lyophilization. Over 5 days of storage at 25 °C, nanogels’ dH and PDI increased over time in an aqueous condition. To prevent nanogels size changes and GelMA polymer degradation, the fully hydrated nanogels in solution (10 mg mL−1) were filtered (0.45 μm syringe filter), lyophilized, and stored at −80 °C until use. The resuspended nanogels recovered their original dH in a swollen state (nearly 100%), which corresponds to the dH of the hydrated nanogels prior to lyophilization (Fig. 5). We evaluated the degree of shrinking of the nanogels exposed to different solutions by comparing their dH and fully swollen size in deionized water (labeled as ‘DW7.4’ in Fig. 5). As previously discussed, the mobile counterions, when introduced to the nanogels matrix, reduced the osmotic pressure of the nanogels, consequently decreasing the swollen degree. We observed that the degree of swelling of both PGM4 and BGM4 nanogels decreased (or nanogels shrank) in response to the presence of salt (Fig. 5(A)). The BGM4 was particularly sensitive to the PB 5.8 solution, and its dH was reduced to about 80% of its maximum dH in a swollen state, as the pH was close to the pI of BG. PGM4 was less sensitive at pH 5.8. PGM was negatively charged in both DW and physiological fluid, as opposed to BG, which was positively charged in PB with pH ranging from 5.8–7.4. This change in ζ-potential derived from the gelatin derivatization process: methacryl groups, which substituted the majority of free amino groups in PGM, remained un-crosslinked during the photopolymerization process, causing PGM to be negatively charged (Fig. 5(A)). The presence of salt greatly altered ζ-potential in both PGM and BGM. The effect of salt on the nanogels was observed (Fig. 5(B)). BGM2, which possesses more free hydroxyl groups than BGM10, can be affected by the counterions presence in the PB buffer. BG becomes neutrally charged and unstable at pH 5.8, which is close to the pI of PB. The nanogels with various DMs exhibited a high degree of shrinking. Less crosslinked nanogels (i.e., those with more of free hydroxyl groups) were more responsive to external conditions such as salt concentration and changes in pH (Fig. 5(C)).
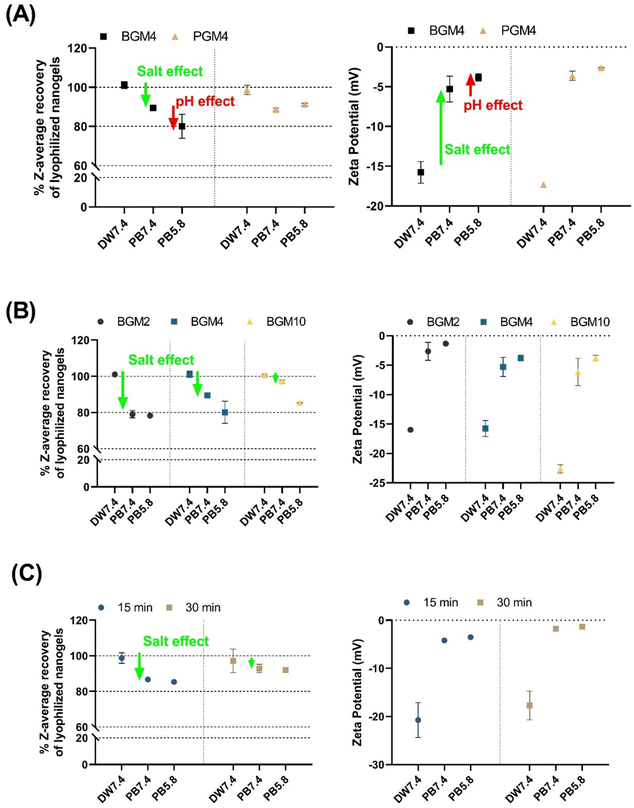 |
| | Fig. 5 The effect of salt and pH on recovery% of lyophilized nanogels. (A) Z-average recovery% and zeta potential of nanogels made of BGM4 and PGM4. (B) Z-average recovery% and zeta potential in nanogels made of BGM with different degree of methacryloylation (DM). (C) Z-average recovery% and zeta potential in nanogels made of BGM4 with different UV exposure time (i.e., degree of crosslinking). DW7.4: deionized water pH adjusted to 7.4, PB7.4: sodium phosphate buffer pH adjusted to 7.4, PB5.8: sodium phosphate buffer pH adjusted to 5.8. | |
2.5. Recombinant protein synthesis and loading
In order to explore the protein loading efficiency and nanogels–protein binding, we applied a fluorescent protein (mNeonGreen)-fused recombinant FLG (mNG-FLG) (Fig. 6). Lyophilized nanogels were rehydrated in DW containing mNG-FLG (0.5 mg mL−1) for 24 h, allowing protein absorption onto the nanogels. Changes in dH and ζ-potential of the nanogels before and after protein loading were then studied. The concentration of the nanogels in the solution was 5 mg mL−1, and the resulting mixture was diluted (5 –10×) in DW to determine the size distribution of the protein-loaded nanogels by DLS. Nanogels showed a significantly increased swelling degree of PGM4/BGM4 nanogels after protein loading (Fig. S7A, ESI†). Since we used DW to exclude other environmental effects (salt, pH) during protein absorption onto the nanogels, the increased dH could be attributed to protein–nanogels interaction. However, interestingly, these protein–nanogels interactions did not alter the ζ-potential of PGM4/BGM4 nanogels (Fig. S7B, ESI†). This was an unexpected result as we had hypothesized that the main attractive force between the protein and nanogels would be due to electrostatic interaction. The result indicated that the porous hydrogel matrix could provide a favorable environment for steric interaction, serving as another main attractive force between proteins. Considering that the dH of nanogels was about 200 nm and the mNG-FLG protein diameter is roughly 2–3 nm based on its molecular weight (62 kDa), we could conclude that the highly structured yet porous matrix of nanogels, enhanced by photopolymerization, provided a favorable environment to capture and stabilize proteins. This result corresponds well with the observation in the protein release study discussed in the next section. The released amount of mNG-FLG protein from the nanogels did not increase during the initial 24 h of incubation when the protein-loaded nanogels were rehydrated in DW. Another explanation for the unchanged ζ-potential of the nanogels before and after protein loading is the loading capacity of mNG-FLG to nanogels shown in Fig. S8C (ESI†). The total mass percentage of protein in protein-loaded nanogels was about 2–4%, which varied depending on the loading condition (Fig. S8, ESI†), indicating that the effect of protein on the overall ζ-potential of the nanogels would be minor in the protein–nanogels complex. FLG (pI ≈ 7.4) is weakly charged in DW. Based on the results shown in Fig. S8A–C (ESI†), we decided on the loading concentration of mNG-FLG (500 μg mL−1) and nanogels concentration (5 and 10 mg mL−1). The concentration of 2 mg mL11 for nanogels was excluded even though it showed comparable values in protein absorption and loading capacity (Fig. S8B and C, ESI†) because of the low protein loading efficiency and low protein mass per volume of the protein–nanogels complex, which was difficult to detect.
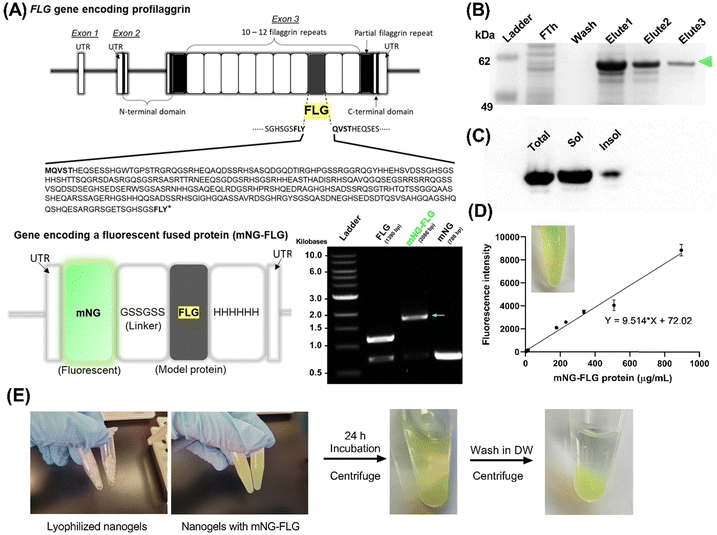 |
| | Fig. 6 Preparation of therapeutic recombinant protein for nanogel–protein binding study: (A) design of a fluorescent fused recombinant protein. (B) Gel electrophoresis of IMAC-purified mNG-FLG protein. FTh: flow-through. (C) Western blot of mNG-FLG in total cell protein, soluble protein, and insoluble protein. (D) Determination of mNG fluorescence intensity which is proportional to protein concentration of purified mNG-FLG (detectable range using the linear regression model: 15–900 μg mL−1). (E) mNG-FLG protein encapsulation to prepare mNG-FLG-loaded nanogels. | |
2.6. Nanogels release study
Next, we aimed to identify the parameters controlling mNG-FLG release kinetics from the nanogels matrix. The modulation of environmental conditions, such as pH, salt, and temperature, led to changes in the protein release rate from the nanogels’ matrix, thereby resulting in stimuli-responsive nanogels. In addition, we investigated protein release profile differences between the nanogels made of different sources (PGM4 and BGM4). In our prior nanogel characterization studies, PGM4 and BGM4 exhibited different macroscopic behaviors in solution (e.g., degree of swelling and shrinking under various pH and salt conditions) and reactivity to methacryl groups, which are derived from their different amino acid composition. The degree of protein absorption of the nanogels and whether this could influence the protein release rate from the nanogels matrix was also investigated, in particular considering high and low amounts of absorbed protein per gram nanogels. The results allowed us to understand the effect of the amount of loaded protein in the nanogels on the release rate of protein from the matrix as a function of time.
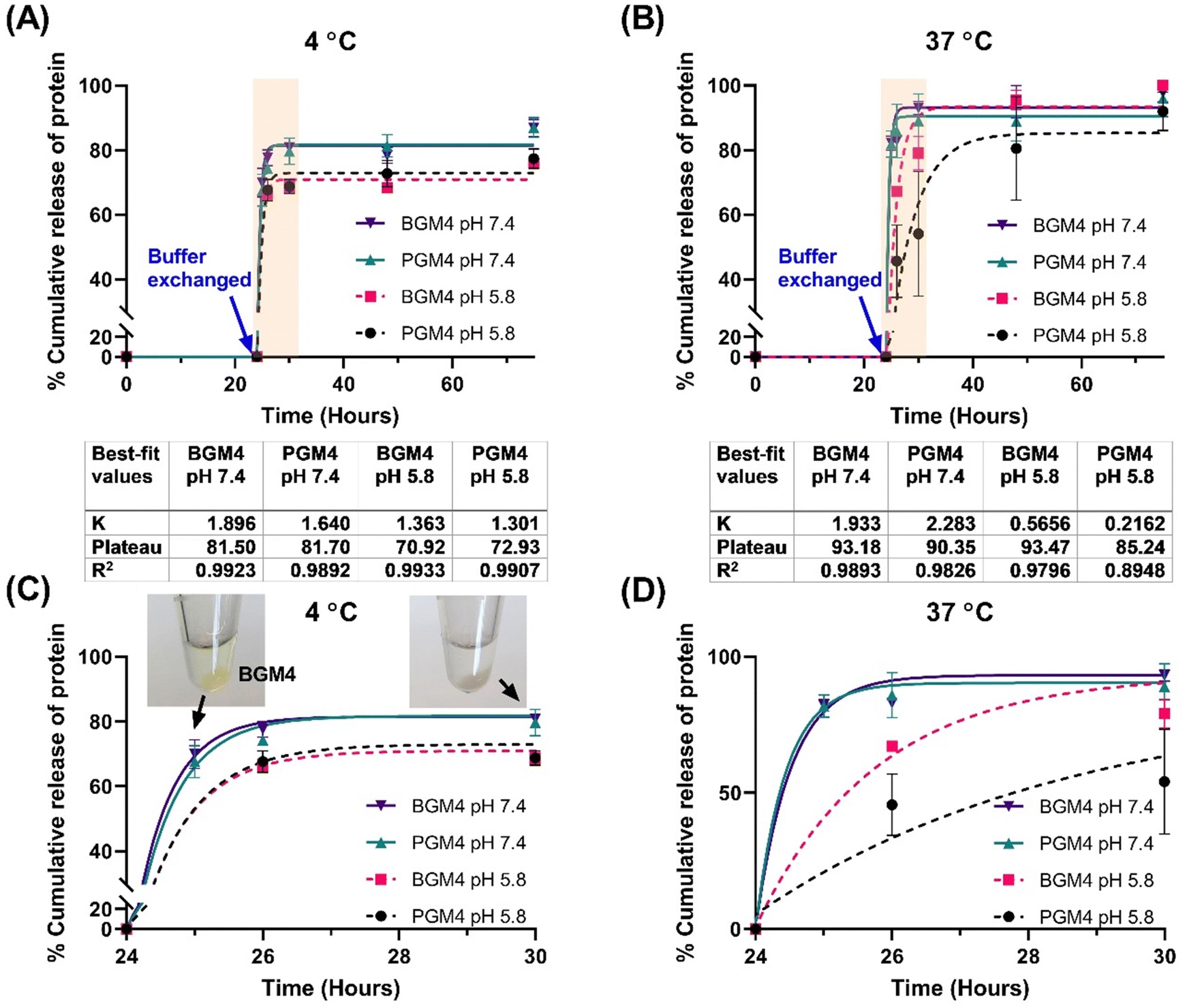
Four parameters were selected that could potentially govern protein release from the crosslinked matrix of nanogels: (1) increasing salt concentration by changing the buffer from DW to PB (both solution pH is 7.4), (2) modulation pH by comparing PB adjusted to pH 7.4 and pH 5.8, (3) Temperature effect (4 °C representing storage temperature and 37 °C representing physiological condition), and (4) different degree of protein absorption (absorbed protein per gram nanogels).
As a drug release model, we utilized a single-phase exponential decay model to analyze mNG-FLG protein release from the nanogels, as the protein release rate was rapid in the initial 6 h, followed by a significant decrease and reached a plateau. A second release phase, referred to as slow release, was not observed following the first release phase (burst release). Since it was a short period (3–4 days) protein release observation, it is conceivable that a longer period observation (over two weeks) may present a release profile that fits into other nonlinear kinetics models. Mumcuoglu et al. reported a similar observation in their growth factor release study from the microspheres, where the initial 48 h release data fitted a single-phase exponential decay model, whereas release curves over two weeks fitted better into a two-phase decay model. Our results were aligned well with Mumcuoglu et al., with the protein release over 75 h fitting well into the single-phase exponential decay model (R2 > 0.979 except for PGM4 incubated at 37 °C in PB solution (pH 5.8)) (Fig. 7). The presence of salt in the nanogels caused a decrease in the degree of swelling (nanogels matrix shrinking), which initiated protein release when the buffer was changed. This was evident from the stable interaction between nanogels and the protein, as indicated by the lack of protein release from the nanogels matrix during the initial 24 h of incubation in DW. Changes in pH were another determinant of protein release. It affected the surface charge on gelatin which regulated protein–matrix interaction. PGM4/BGM4 nanogels at pH 5.8 showed a relatively lower protein release rate constant (K) than the nanogels at pH 7.4, indicating slower protein dissociation from the nanogel matrix. Furthermore, more proteins remained in the nanogels matrix at pH 5.8 after the protein release reached an equilibrium state (protein release rate became close to 0). Temperature also played a significant role in protein release. At 4 °C, proteins in the nanogel matrix were released quickly and reached equilibrium status fast (2–4 h), but about 18% of protein remained in the nanogels at pH 7.4 and 30% at pH 5.8, respectively. At 37 °C, over 90% of the proteins were rapidly released at pH 7.4, while more than 85% of proteins were released, albeit slowly, at pH 5.8. The nanogels with a low degree of protein absorption on their matrix showed the same trend in protein release (Fig. S9, ESI†). These findings underscore the important influence of environmental conditions and the nanogel composition on protein release kinetics, thus providing insights for the design of controlled drug delivery systems.
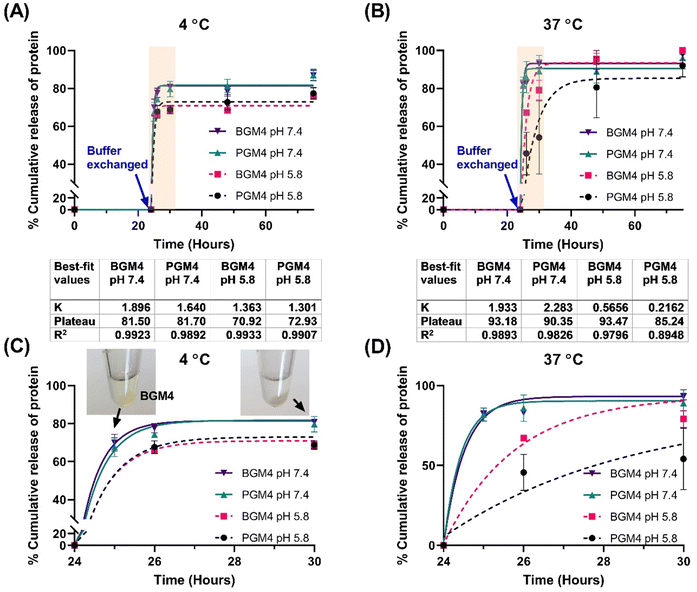 |
| | Fig. 7 Nanogels with high degree of protein absorption resulted from the high protein loading condition (deionized water with 0.5 mg mL−1 of protein and 5 mg mL−1 of lyophilized nanogels). (A) The (mNG-FLG)high-PGM4 nanogels and (mNG-FLG)high-BGM4 nanogels incubated either in PB (pH 7.4) or in PB (pH 5.8) at 4 °C. In the first 24 h, the nanogels were incubated in deionized water and then changed to PB. (B) The (mNG-FLG)high-PGM4 nanogels and (mNG-FLG)high-BGM4 nanogels incubated either in PB (pH 7.4) or in PB (pH 5.8) at 37 °C. In the first 24 h, the nanogels were incubated in deionized water and then changed to PB. (C) The protein release profile from PGM4/BGM4 nanogels in the first 6 h after buffer change at 4 °C. (D) The protein release profile from PGM4/BGM4 nanogels in the first 6 h after buffer change at 37 °C. | |
3. Conclusion
Nanogels are versatile biomaterials that can be transformed into effective vehicles for protein transport. In this study, we investigated the utilization of nanogels as tunable biomaterials, focusing on the construction and optimization of the nanogel matrix to facilitate controlled protein transport and release. Our analysis considered three critical parameters to shape the three-dimensional gel network of nanogels – the degree of methacryloylation, the type of gelatin polymer, and the degree of crosslinking density within the nanogels. Our results unambiguously showed that both the diameter of nanogels and the crosslinking density play a decisive role in dictating the interaction dynamics between proteins and nanogels. In our pursuit of creating nanosized gel particles, we successfully implemented a hybrid approach combining photopolymerization and reversed nanoemulsion methods. We optimized the photopolymerization process to control nanogel fabrication by modulating UV exposure time, forming smaller sizes of nanogels with higher crosslinking density at prolonged time of UV exposure. Furthermore, through careful titration of methacrylic anhydrate during gelatin solution preparation, we achieved varying degrees of methacryloylation of the gelatin, resulting in a diverse set of nanogels. We employed various characterization techniques, including, DLS, NMR, TEM, and UV/Vis spectroscopy to investigate the nanogels’ size distribution, ζ-potential, morphology, and protein load/release. Our model protein was a fluorescent protein-fused therapeutic filaggrin, which was synthesized and tested within our nanogels system. Our study delivered strong evidence that we could leverage nanogel matrix crosslinking density to modulate the release kinetics of therapeutic proteins. This approach provides a robust platform for controlled release of protein therapeutics, paving the way for targeted drug delivery and tissue engineering applications. Also, the results highlight the promise of nanogels as transformative tools in the field of engineered biomaterials with broader implications in the field of controlled delivery of therapeutics.
4. Experimental section/methods
4.1. Materials
Porcine gelatin (type A, 300 bloom) and bovine gelatin (type B, 225 bloom) were purchased from Sigma-Aldrich (St. Louis, MO). Gelatin contains 0.78–0.8 mmol (type A), 1–1.15 mmol (type B) of free hydroxyl groups per gram gelatin and 0.35 mmol of amino groups in side-chain per gram gelatin.19 Type A and type B gelatin have similar content of amino groups in the lysine and hydroxylysine side chains. The isoelectric point of gelatin is 7.0–9.0 (type A) and 4.7–5.2 (type B), respectively.22 Polyoxyethylene sorbitan monooleate (Tween 80), sorbitan monooleate (Span 80), n-octane, acetone, tetrahydrofuran (THF), methacrylic anhydride (MAA), the radical initiator 1-[4-(2-hydroxyethoxy)phenyl]-2-hydroxy-2-methyl-1-propan-1-one (Irgacure 2959), and 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid sodium salt (TMSP) were purchased Sigma-Aldrich (St. Louis, MO). Dialysis membrane (MWCO 6–8 kDa) was purchased from Spectrum Laboratories (Rancho Dominguez, CA).
4.2. Synthesis of gelatin derivatives
Methacryloylation of gelatin was carried out using the previously published protocols.16,19 Briefly, gelatin (12.5 g) was dissolved in 100 mL phosphate-buffered saline (PBS, pH 7.4) at 40–42 °C. The pH of gelatin solution was adjusted to pH 7.4 using 3 M NaOH after the gelatin was completely dissolved. MAA was added dropwise into the solution while vigorously stirring at 40 °C. A different amount of MAA was added to give a 1, 2, 4, or 10 molar excess of MAA to free amino groups of gelatin based on the content of 0.35 mmol amino groups per gram of gelatin (Table S1, ESI†).19 The degree of methacryloylation (DM) was known to be affected by reaction time, pH, and concentration of MAA added.15 After a 2-hour reaction, the solution was transferred to the dialysis tubing (MWCO 6–8 kDa) and dialyzed for 2–4 days against deionized water at 42 °C. After dialysis, GelMA was lyophilized and stored at −80 °C until further use. We calculated DM using 1H NMR spectra obtained from 400 MHz NMR spectrometer at 323.15 K (AVIII 400, Bruker, Germany).
4.3. Determination of methacryloylation degree
4.3.1. Degree of methacryloylation by unmodified lysine content.
The degree of methacryloylation (DM) is the extent of substitution of free amine groups and hydroxyl groups in gelatin as the methacryloyl moieties reacted to free amine groups (majority) and hydroxyl groups (minority).17,18 The gelatin and lyophilized gelatin derivatives of 15 mg were dissolved in 1 mL of deuterium oxide. Before the interpretation, the chemical shift scale was adjusted to the residual solvent signal (4.79 ppm). The phase correction was applied to spectra and baselines were corrected before integrating the signals of interest. The chemical shift of methylene protons of neighboring the lysine amino acid (δ = 3.2–2.9) was used to determine the degree of substitution. The degree of methacryloylation was calculated by eqn (1):| |  | (1) |
4.3.2. Degree of methacryloylation by direct methacryl substitution.
The method of quantification of substitution of GelMA was previously described.17 The gelatin and lyophilized gelatin derivatives of 15 mg were dissolved in 1 mL deuterium oxide with trimethylsilyl propanoic acid (TMSP) as the internal reference in the NMR spectrum (1 mg mL−1). 1H NMR spectra were recorded at 323.15 K with 32 scans. The chemical shift scale was adjusted and the phase and baselines were corrected before integrating the signals of interest. The signal of protons resulting from TMSP was used as a reference in each spectrum. The degree of methacryloylation (DM) is defined as the molar amount of substituted methacryl groups per gram of gelatin derivatives. The area under curve of methacryl bound to hydroxyl group and amino group (δ = 5.9–5.5) and the integral of the TMSP signal area are inserted in eqn (2):| | 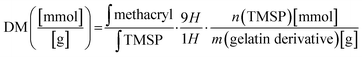 | (2) |
The degree of methacrylate (DMmethacrylate) is defined as the molar amount of substituted methacrylate groups per gram of gelatin derivatives. To obtain DMmethacrylate, the integral of the signal area of methacryl bound to hydroxyl group (δ = 6.3–5.9) and the integral of the TMSP signal area are inserted in eqn (3):
| | 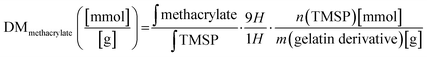 | (3) |
The degree of methacrylate (DMmethacrylamide) is defined as the molar amount of substituted methacrylamide groups per gram of gelatin derivatives. DMmethacrylamide is calculated by eqn (4):
| | 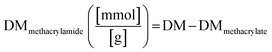 | (4) |
4.4. Fabrication of the nanoreactor
To transform the gelatin derivatives-based hydrogel into gel nanoparticles, the nanoreactor system was applied for nanogels synthesis. The method of gel nanoparticle synthesis was described previously.21 The precursor polymer GelMA is confined within water droplets in the water-in-oil (inversed emulsion) system, which functions as a nanoreactor during photo-polymerization. Each volume composition of nanoemulsion was experimentally determined to create homogenous and stable nanodroplets in the system. The HLB value of the surfactant mixture was calculated by eqn (5):23| | | HLBmix = fA·HLBA + fb·HLBB | (5) |
where HLBA and HLBB are the HLB values of surfactant A and B, respectively, and fA and fB are the weight fraction of surfactant A and B, respectively.
Briefly, 5 g mixture of surfactants is dissolved in 20 mL of n-octane for the organic phase, which functions as a continuous phase in the inversed emulsion system. GelMA (50 g mL−1) and photoinitiator Irgacure 2959 (10 g mL−1) was dissolved in PBS solution at 60 °C. The aqueous solution is added to the organic solution. The volume percentage of Irgacure 2959 in the nanoreactor system was 0.167% (w/v). The two immiscible phases were mixed and homogenized for one minute using a high-speed homogenizer (T-18 Ultra-Turrax, IKA, Wilmington, NC) at 20000 rpm to make a crude emulsion. The mixture was then further homogenized using an ultrasonicator for 10 min on ice with different ultrasonic energy per unit time (6–20 W) to create nano-sized polymeric gel particles in the solution.
4.5. Synthesis of nanogels
The mixing ratio of water/oil/surfactant (1:4:1) was experimentally determined to create fine and homogenous nanodroplets in the inversed emulsion system. The mixture of Tween 80 and Span 80 (weight ratio of 3:2) was used as a surfactant to achieve hydrophilic–hydrophobic balance for fine-size nanoparticles.23,24 The resulting inversed emulsion was placed at RT overnight to reach equilibrium. After equilibrium, 7 mL of the suspension was UVA (280–480 nm) exposed using the Greenspot curing system (American Ultraviolet) at 12–15 W cm−2 for 15–60 min at 25 °C (RT) to enable the photocrosslinking of nanogels. The photopolymerized gel nanoparticles were collected in the form of a precipitate by the addition of 15 mL of tetrahydrofuran (THF) and centrifuge at 12000g for 15 min. The supernatant solvent THF containing n-octane, photoinitiator, and surfactant was decanted. Nanogels in the precipitate were dried in a fume hood overnight with continuing ventilation to remove a trace of THF. Dried nanogels were resuspended in 10 mL of deionized water. The gel suspension was homogenized using an ultrasonicator at 4 °C until the mixture became visually transparent without precipitates. Any possible dust obtained during the process and nanogels aggregate were cleared from the nanogels suspension by filtering with a 0.45 μm filter. The resulting nanogels were lyophilized and stored at −20 °C until further use.
4.6. Characterization of nanogels
4.6.1. Dynamic light scattering.
Hydrodynamic diameter (DH), polydispersity index (PDI), and ζ-potential of nanogels were measured using the dynamic light scattering (DLS) at the different processing points during the nanogel preparation to understand the degree of swelling ability and the electrostatic interaction between nanogel matrix and protein. All measurements were performed using a nano-size analyzer (Malvern Zetasizer Nano ZS, Malvern Instruments, UK). All electrophoretic light scattering (ELS) measurements were obtained three times at 25 °C after the thermal equilibration time for 2 min. The changes in ζ-potential of each sample dilute (1 mg ml−1) in different pH and salt condition was studied by DLS to understand the physicochemical characteristics of nanogels in an aqueous solution.
4.6.2. Determining degree of conversion (degree of crosslinking) of nanogel matrix.
The degree of crosslinking of methacryl moiety in the nanogel matrix after UV irradiation was determined by 1H NMR. The lyophilized nanogel of 15 mg was dissolved in 1 mL of deuterium oxide. 1H NMR spectra were recorded at 323.15 K with 16 scans. The chemical shift scale was adjusted and the phase and baselines were corrected before integrating the signals of interest. The integral signal of phenylalanine was used as a reference in each spectrum. The integral of the signal area of methacryl bound to hydroxyl group and amino group (δ = 5.9–5.5) is inserted in eqn (6):| | 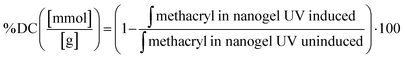 | (6) |
4.6.3. Transmission electron microscopy.
The morphology of spherical gel nanoparticles was characterized using the transmission electron microscope (JEM-1400 TEM, JEOL USA, Peabody, MA). The diluted nanogels sample (2 mg mL−1) was used for the TEM imaging analysis as described: the nanogels were dropped onto a copper carbon-coated grid and dried. The excess is carefully wicked off with membrane filter paper. The grid was treated with a negative staining solution uranyl acetate to enhance the contrast. The images were acquired with 120 kV of the accelerating voltage and 25000× – 80000× magnification.
4.7. Filaggrin recombinant protein absorbing nanogels
4.7.1. Preparation of recombinant plasmid and protein expression.
The recombinant green fluorescent-fused filaggrin was synthesized for nanogels–protein binding study. The mNeonGreen (mNG) was used as a fluorescent reporter protein.25 The preparation method of the recombinant plasmid for a monomeric filaggrin protein expression was previously described.26 The recombinant plasmid pETBlue1-mNG-FLG was assembled to quantify the amount of filaggrin protein encapsulated in nanogels. Briefly, the sequence of nucleotide encoding mNG was inserted into the N-terminal of a monomeric filaggrin translational region in the pETBlue1-FLG plasmid following the standard Gibson assembly protocol used previously.26 The E. Coli Rosetta-gami B (DE3) pLacI cell was transformed with the new expression vector pETBlue1-mNG-FLG. The transformed cells were grown in LB media, induced with IPTG (at OD600 0.5), harvested at the exponential growth phase, and lysed using a sonicator. The mNG-FLG protein was separated from cell lysate by centrifugation and purified using the Ni-NTA IMAC chromatography.
4.7.2. Western blot.
Before the lysate centrifugation, 5 μL of the sample was taken for total cell protein. After the lysate centrifugation, the protein in the supernatant was taken as a soluble fraction. The remaining pellet was resuspended with an equal volume of water as the supernatant volume was removed, and 5 μL was taken as an insoluble fraction for analysis. After completion of electrophoresis, the unstained gel underwent western blot analysis using the Power Blotter system following the manufacturer's protocol (Thermo, Waltham, MA). For antibody treatment, 2 μL of 1 mg mL−1 HRP HisTag antibody was added in 20 mL TBS buffer (dilution in 1:10000) and treated blotted membrane for 1 hour at RT on a rocking shaker. The western blotting signal was obtained and visualized in the iBright Imaging system (Thermo, Waltham, MA).
4.7.3. Fluorescence intensity quantification.
The protein content and the relative fluorescence intensity on a series of mNG-FLG dilutions were measured. Briefly, the purified green fluorescent-tagged filaggrin in solution was diluted to make 8 different variations in concentration (dilution factor 1 to 200) to ensure the values were in the range of the linear regression model. A standard calibration curve with bovine serum albumin dilutions was made using the Bradford assay. The protein content on a series of mNG-FLG protein dilutions was calculated by the standard curve. The fluorescence intensity from the same protein dilution was quantified using the UV/Vis spectrophotometer with excitation at 485 nm and emission at 528 nm with 20 nm bandwidth. These measured values were plotted in a linear regression model to estimate the mNG-FLG protein amount by its fluorescence intensity.
4.8. Filaggrin encapsulated nanogels
4.8.1. Binding ability quantification.
The protein binding capacity of nanogels was studied using different nanogels concentrations (2, 5, 10 mg mL−1). For the optimal protein loading dose, two different protein concentration in deionized water (0.5, 1 mg mL−1) was incubated at 4 °C for 24 h with nanogels concentration 5 mg mL−1. After the completion of 24 h incubation, the mixture was centrifuged at 12000g for 25 min to separate the free protein from the protein–nanogels complex. The protein in the supernatant was collected and analyzed for free protein content using a UV/Vis microplate. The protein binding was determined by subtracting the amount of protein collected from the supernatant that was not entrapped in nanogels. % Protein loading efficiency (% PLE), degree of entrapment (DE, defined as absorbed protein per g nanogels), and % loading capacity (% LC) was obtained using eqn (7)–(9):| |  | (7) |
| | 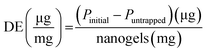 | (8) |
| | 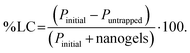 | (9) |
where Pinitial is the initial amount of protein applied to nanogels and Puntrapped is the amount of free protein unbound to nanogels.
4.9. Protein release study
The experimental method of protein release was adapted from.27 The purified mNG-FLG protein in deionized water with a concentration of 0.5 mg mL−1 was added on top of lyophilized nanogels in a 1.5 mL centrifugal tube and incubated for 24 h at 4 °C to allow protein absorption during the complete nanogels swelling. The resulting solution was centrifuged at 12000g for 25 min. The free protein in the supernatant was decanted and the nanogel–protein complex in the pellet was resuspended in fresh deionized water. After second centrifugation at 12000g for 25 min to remove a trace of unbound proteins, the nanogel–protein complex in the pellet was incubated in 0.2 M sodium phosphate buffer (pH 7.4/pH 5.8) at 37 °C/4 °C. The amount of protein released from the nanogels matrix was measured at different time intervals up to 52 h. The cumulative amount of released protein was calculated using eqn (10):| | 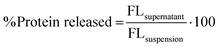 | (10) |
where FLsupernatant is the fluorescence intensity from the fluorescent protein in the supernatant after centrifugation and FLsuspension is the fluorescence intensity from the fluorescent protein in nanogel suspension (i.e., total protein) before centrifugation.
The data of the cumulative amount of released protein was plotted in a nonlinear regression (curve fit) model. As a drug release model, one phase exponential decay model was applied to experimental data. The protein dissociation rate (K) and the protein concentration in the plateau were obtained using eqn (11):
| | | (Y = Span·exp−K·X + Plateau) | (11) |
where
K is the dissociation rate constant,
X is time (h) and
Y is concentration (μg mL
−1).
Y starts equal to span + plateau and decreases to plateau with a rate constant
K.
Author contributions
J. K. and Y.-C. K. conceived the project. J. K. and Y.-C. K. designed and conceptualized experiments. J. K. prepared and performed experiments and acquired data. J. K. analyzed and interpreted data. J. K. wrote the original manuscript. J. K., C. E. C., and Y.-C. K. revised and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the Louisiana Board of Regent (RCS, grant no. LEQSF(2020-23)RD-A-01) and the USDA National Institute of Food and Agriculture (HATCH, accession no. 1021535, project no. LAB94414).
References
- P. J. Carter, Introduction to current and future protein therapeutics: a protein engineering perspective, Exp. Cell Res., 2011, 317(9), 1261–1269 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Protein therapeutics: a summary and pharmacological classification, Nat. Rev. Drug Discovery, 2008, 7(1), 21–39 CrossRef CAS PubMed.
- R. M. Lieser, D. Yur, M. O. Sullivan and W. Chen, Site-specific bioconjugation approaches for enhanced delivery of protein therapeutics and protein drug carriers, Bioconjugate Chem., 2020, 31(10), 2272–2282 CrossRef CAS PubMed.
- R. M. Lu, Y. C. Hwang, I. J. Liu, C. C. Lee, H. Z. Tsai, H. J. Li and H. C. Wu, Development of therapeutic antibodies for the treatment of diseases, J. Biomed. Sci., 2020, 27(1), 1 CrossRef CAS PubMed.
- A. Szlachcic, M. Zakrzewska and J. Otlewski, Longer action means better drug: tuning up protein therapeutics, Biotechnol. Adv., 2011, 29(4), 436–441 CrossRef CAS PubMed.
- L. A. Lampson, Monoclonal antibodies in neuro-oncology: getting past the blood-brain barrier, MAbs, 2011, 3(2), 153–160 CrossRef PubMed.
- M. M. Schmidt and K. D. Wittrup, A modeling analysis of the effects of molecular size and binding affinity on tumor targeting, Mol. Cancer Ther., 2009, 8(10), 2861–2871 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, Impact of nanotechnology on drug delivery, ACS Nano, 2009, 3(1), 16–20 CrossRef CAS PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nanotechnology in drug delivery and tissue engineering: from discovery to applications, Nano Lett., 2010, 10(9), 3223–3230 CrossRef CAS PubMed.
- F. U. Din, W. Aman, I. Ullah, O. S. Qureshi, O. Mustapha, S. Shafique and A. Zeb, Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors, Int. J. Nanomed., 2017, 12, 7291–7309 CrossRef PubMed.
- T. R. Hoare and D. S. Kohane, Hydrogels in drug delivery: progress and challenges, Polymer, 2008, 49(8), 1993–2007 CrossRef CAS.
- J. Li and D. J. Mooney, Designing hydrogels for controlled drug delivery, Nat. Rev. Mater., 2016, 1(12), 16071 CrossRef CAS PubMed.
- L. Messager, N. Portecop, E. Hachet, V. Lapeyre, I. Pignot-Paintrand, B. Catargi, R. Auzély-Velty and V. Ravaine, Photochemical crosslinking of hyaluronic acid confined in nanoemulsions: towards nanogels with a controlled structure, J. Mater. Chem. B, 2013, 1(27), 3369–3379 RSC.
- T. E. Stout, T. McFarland, J. C. Mitchell, B. Appukuttan and J. Timothy Stout, Recombinant filaggrin is internalized and processed to correct filaggrin deficiency, J. Invest. Dermatol., 2014, 134(2), 423–429 CrossRef CAS PubMed.
- K. Yue, G. Trujillo-de Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Synthesis, properties, and biomedical applications of gelatin methacryloyl (GelMA) hydrogels, Biomaterials, 2015, 73, 254–271 CrossRef CAS PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Cell-laden microengineered gelatin methacrylate hydrogels, Biomaterials, 2010, 31(21), 5536–5544 CrossRef CAS PubMed.
- C. Claaßen, M. H. Claaßen, V. Truffault, L. Sewald, G. E. M. Tovar, K. Borchers and A. Southan, Quantification of substitution of gelatin methacryloyl: best practice and current pitfalls, Biomacromolecules, 2018, 19(1), 42–52 CrossRef PubMed.
- K. Yue, X. Li, K. Schrobback, A. Sheikhi, N. Annabi, J. Leijten, W. Zhang, Y. S. Zhang, D. W. Hutmacher, T. J. Klein and A. Khademhosseini, Structural analysis of photocrosslinkable methacryloyl-modified protein derivatives, Biomaterials, 2017, 139, 163–171 CrossRef CAS PubMed.
- A. I. Van Den Bulcke, B. Bogdanov, N. De Rooze, E. H. Schacht, M. Cornelissen and H. Berghmans, Structural and rheological properties of methacrylamide modified gelatin hydrogels, Biomacromolecules, 2000, 1(1), 31–38 CrossRef CAS PubMed.
- R. M. H. R. Nhari, Y. b C. Man, A. Ismail and N. I. S. Anuar, In Chemical and functional properties of bovine and porcine skin gelatin, Int. Food Res. J., 2011, 18(2), 787–791 Search PubMed.
- J. Kim, R. Gauvin, H. J. Yoon, J.-H. Kim, S.-M. Kwon, H. J. Park, S. H. Baek, J. M. Cha and H. Bae, Skin penetration-inducing gelatin methacryloyl nanogels for transdermal macromolecule delivery, Macromol. Res., 2016, 24(12), 1115–1125 CrossRef CAS.
- S. M. Ahsan and C. M. Rao, The role of surface charge in the desolvation process of gelatin: implications in nanoparticle synthesis and modulation of drug release, Int. J. Nanomed., 2017, 12, 795–808 CrossRef CAS PubMed.
- J. H. Kim, J. A. Ko, J. T. Kim, D. S. Cha, J. H. Cho, H. J. Park and G. H. Shin, Preparation of a capsaicin-loaded nanoemulsion for improving skin penetration, J. Agric. Food Chem., 2014, 62(3), 725–732 CrossRef CAS PubMed.
- G. Lv, F. Wang, W. Cai and X. Zhang, Characterization of the addition of lipophilic Span 80 to the hydrophilic Tween 80-stabilized emulsions, Colloids Surf., A, 2014, 447, 8–13 CrossRef CAS.
- C. E. Copeland, J. Kim, P. L. Copeland, C. J. Heitmeier and Y. C. Kwon, Characterizing a new fluorescent protein for a low limit of detection sensing in the cell-free system, ACS Synth. Biol., 2022, 11(8), 2800–2810 CrossRef CAS PubMed.
- J. Kim, C. E. Copeland, K. Seki, B. Vögeli and Y. C. Kwon, Tuning the cell-free protein synthesis system for biomanufacturing of monomeric human filaggrin, Front. Bioeng. Biotechnol., 2020, 8, 590341 CrossRef PubMed.
- D. Mumcuoglu, L. de Miguel, S. Jekhmane, C. Siverino, J. Nickel, T. D. Mueller, J. P. van Leeuwen, G. J. van Osch and S. G. Kluijtmans, Collagen I derived recombinant protein microspheres as novel delivery vehicles for bone morphogenetic protein-2, Mater. Sci. Eng., C, 2018, 84, 271–280 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00183d |
| ‡ Current address: Alnylam Pharmaceuticals, Inc., Cambridge, MA 02142, USA; Department of Chemical Engineering, Stanford University, Stanford, CA 94306, USA. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Caroline E.
Copeland‡
a and
Yong-Chan
Kwon
a,
Caroline E.
Copeland‡
a and
Yong-Chan
Kwon