
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A water-stable copper-based nanosized metal–organic framework for discriminative sensing of amines†
Pablo
Álvarez García
 a,
Enrique
Álvarez Rubiera
a,
David
Gutiérrez Armayor
a,
Candela
Melendreras
b,
Ana Belen
Soldado
b,
José Manuel
Costa
b,
Elena
Lastra
*a and
Fco. J.
García Alonso
*ac
a,
Enrique
Álvarez Rubiera
a,
David
Gutiérrez Armayor
a,
Candela
Melendreras
b,
Ana Belen
Soldado
b,
José Manuel
Costa
b,
Elena
Lastra
*a and
Fco. J.
García Alonso
*ac
aDepartamento de Química Orgánica e Inorgánica, Facultad de Química, Avda. Julian Claveria 8, Universidad de Oviedo, 33006 Oviedo, Principado de Asturias, Spain. E-mail: elb@uniovi.es; fjga@uniovi.es
bDepartamento de Química Física y Analítica, Facultad de Química, Avda. Julian Claveria 8, Universidad de Oviedo, 33006 Oviedo, Principado de Asturias, Spain
cCentro de Investigación en Nanomateriales y Nanotecnologia, Universidad de Oviedo-CSIC, 33940 El Entrego, Spain
First published on 25th June 2024
Abstract
The ability to design new metal–organic frameworks (MOFs) from different functional groups and different metals can lead to multifunctional materials with a discrimination capacity between chemically similar structures. Furthermore, MOFs produced at the nanoscale (nano-MOFs) appear to be innovative and highly promising materials for the development of improved bioanalytical applications (e.g. chemical sensing). Here, a water-stable green copper-based nano-MOF, [Cu(DHBDC)(H2O)2−x(DMF)x] (1), was prepared at the nanoscale size using polyacrylic acid at 5 °C in order to obtain nanocrystals. The resulting nano-MOF displays an intense fluorescence behaviour, where the presence of strong basic aliphatic amines with a low cone angle leads to “turn-off” of the emission at a rate depending on their nature. This new nanomaterial has proven to be useful as a discriminative sensor of amines either in a vapor phase via colorimetric detection or in water solution via luminescence analysis.
Introduction
Metal–organic frameworks (MOFs), also known as coordination polymers (CPs), are a kind of crystalline porous materials possessing high porosity (up to 90% free volume) and a Langmuir surface greater than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 g−1.1 Their unique chemical (sorptive, chiral, and catalytic) and physical (optical, electrical, electronic, and magnetic) properties2 make them valuable for a broad range of applications such as removal of toxics and pollutants, gas adsorption, sensors, catalysis, drug delivery, bioimaging, and biomedical and antimicrobial activities. Also, they are components in the fabrication of elements for batteries, supercapacitors and fuel cells.3
000 m2 g−1.1 Their unique chemical (sorptive, chiral, and catalytic) and physical (optical, electrical, electronic, and magnetic) properties2 make them valuable for a broad range of applications such as removal of toxics and pollutants, gas adsorption, sensors, catalysis, drug delivery, bioimaging, and biomedical and antimicrobial activities. Also, they are components in the fabrication of elements for batteries, supercapacitors and fuel cells.3
In addition, the customizable pore sizes/nature and/or the installation of specific recognition groups in the MOF structure allow for modulation of their affinity toward distinct classes of chemicals, which is of high value in the development of improved analytical and bioanalytical applications.
MOFs are constructed from metal ions or clusters bridged by polydentate organic groups to form one-, two-, or three-dimensional infinite networks.4 The used metals are broadly distributed across the periodic table5 and most of the linking ligands are neutral N-donor heterocycles or anionic polytopic carboxylates,6 although more exotic species like phosphonate7 or even “edible”8 ligands have also been satisfactorily utilized. In the past decades, the number of papers in the field has been growing incessantly9 and, accordingly, the design of MOFs has improved significantly.10
Thus, apart from the different methods employed in their preparation,11 some other factors have been studied,10 namely, the nature of solvents, the concentration and ratio of reactants, the stirring conditions, and the presence of additives.
Despite the efforts being made to prepare novel MOFs with new compositions and structures, only a reduced number of MOFs have been commercialized. This is because the directly synthesized MOFs are usually hindered by physical and chemical limitations, leading to poor performance in practical uses.1 Fortunately, it has been found that certain MOFs can be modified post-synthetically by altering the properties of the pristine material and, consequently, obtaining new MOFs better adapted to realistic applications.12
The preparation of nanosized metal–organic frameworks, n-MOFs, has attracted permanent interest as they seem to offer new applications or better performance than their macroscopic counterparts in various research areas. For example, nanosized MOFs have higher surface-to-volume ratios in comparison with their larger counterparts, showing faster diffusion rates and therefore faster responses to interactions with other species. Although many laboratories have developed particular recipes to synthetize them, only recently,13 a general hypothesis has been offered regarding the conditions that facilitate their formation.
Among MOF materials, the carboxylate linker, 2,5-dihydroxy-1,4-benzenedicarboxylic acid, is one of the most utilized.14 This ligand can act as a fully deprotonated linker (2,5-dioxy-1,4-benzenedicarboxylate), DOBDC−4, giving rise to a whole family of MOFs, named MOF-74 (or CPO-27), [M2 (DOBDC)(solvent)x]·y(solvent), where M are divalent metal cations.15 The same ligand can also act as a dihydroxy-terephthalate diphenol linker, 2,5-dihydroxy-1,4-bencenodicarboxilate, DHBDC−2, originating [M(DHBDC)] or [M2(DHBDC))3] MOFs for di- or trivalent metal cations.16 Surprisingly, although Cu-MOF-74 was synthesized a long time ago,17 only recently have [Cu(DHBDC))(DMF)2]·DMF, [Cu(DHBDC))(DMF)(H2O)2]·DMF and Cu(DHBDC))(H2O) been prepared.18 In any case, it should be emphasized that the resulting materials were synthesized at the micro-size scale and it would be desirable, for sensing purposes, to reduce the particle size to the nanoscale. In fact, moving to the nanoscopic size implies increasing significantly the surface-to-volume ratio of the MOF, increasing the exposed active sites and improving the diffusion of the analytes towards these active sites.13,19
Herein, we present the synthesis of a light green new nano-MOF, namely [Cu(DHBDC)(H2O)2−x(DMF)x] (1). The procedure reported here led to the formation of nearly spherical particles with dimensions of only a few nanometers in diameter. The resulting nano-MOFs can be used as an efficient dual fluorescent-colorimetric discriminative sensor of amines.
Optical sensing of amines is a matter of permanent interest,20 and several MOF materials have been demonstrated to be appropriate for this purpose.21 However, the number of MOFs able to use simultaneously luminescence quenching in solution and vapochromism to detect amines is scarce. In this context, a Mg-MOF bearing naphthalenediimide ligands was demonstrated to be able to detect organic alkyl amines by photoluminescence quenching and visual color changes.22 Furthermore, the quenching luminescence efficiency that undergoes an Eu-MOF containing 1,1′-bis(4-carboxyphenyl)-(4,4′-bipyridinium) ligands in the presence of organic amines in MeOH follow the order of amine sizes. It also changes its color in the presence of amine vapors.23 A Zn-MOF that contains encapsulated naphthalene can serve as a luminescent (in EtOH) and colorimetric probe for the detection of strongly basic organic amines, the quenching by trimethylamine is slower due to its bulky size24 and a Zr MOF UiO-67 containing electron-deficient bipyridinium sites shows fast and reversible vapochromism and luminescence quenching in response to alkylamines.25 Moreover, solid samples of the Zr MOF (UiO-67-DQ–PsO), where DQ is the electron-deficient diquat unit and PsO is the electron-rich 1-pyrenesulfonate anion, exhibit vapochromism and luminescence quenching in the presence of alkylamines.26 Ammonia, aniline and small primary aliphatic amines induce vapochromism in an Eu-MOF with a tripyridinium–tricarboxylate linker, while t-butylamine and aniline produce its luminescence quenching.27 Various post-synthetic derivatives of a Zr-bipy MOF exhibit vapochromism and fluorescence quenching in the presence of ammonia and different amines.28 However, it must be highlighted that none of these previously reported examples use nano-MOFs for amine sensing.
Intriguingly, only a few members of the large family of MOFs containing DOBDC have been used as ammonia16,29 or amine sensors.30 The new n-MOF [Cu(DHBDC)(H2O)2−x(DMF)x] (1), here synthesized and characterized, is found to be very sensitive to the presence of strong basic amines with a low cone angle both in the vapor phase and in solution.
Results and discussion
Synthesis and characterization of [Cu(DHBDC)(H2O)2−x(DMF)x]
In the first steps of the synthesis of the bright red Cu-MOF-74, we detected the presence of a greenish material. This prompted us to change the reaction conditions in order to isolate it. After several attempts, we found out that by lowering the temperature to 5 °C, using a ligand to copper ratio of 1.5/1 and N,N-dimethylformamide as the solvent, we were able to prepare a new nano-MOF 1.Thus, by stirring 2,5-dihydroxyterephthalic acid with Cu(CH3COO)2·H2O using DMF as the solvent, at 5 °C for 72 h, in the presence of poly(acrylic acid) (PAA),13 we obtained a green solid. This crude was washed successively with water and ethanol to obtain light green nanoparticles of [Cu(DHBDC)(H2O)2−x(DMF)x] (1). The average size of the n-MOF 1 particles was found to be 35 nm in diameter, as shown in the TEM micrographs (see Fig. 1).
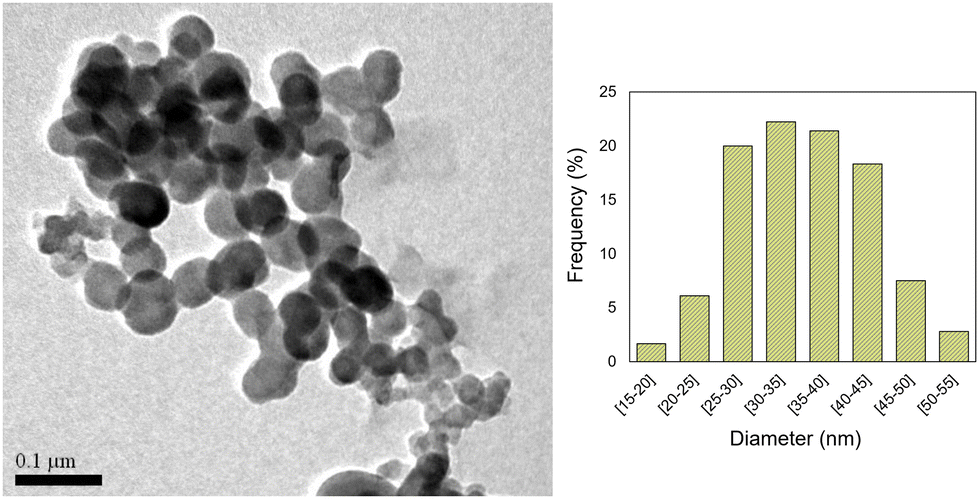 | ||
| Fig. 1 Transmission electron microscopy (TEM) image and histogram size distribution plot of 1. | ||
Nanoparticles of 1 are air stable in water solutions as evidenced by IR DRX and TEM (see Fig. S1–S5, ESI†). This property is remarkable since only a small portion of MOFs is known to be free from problems with water stability.31 In particular, several copper carboxylate MOFs are stable under humidity only for short periods of time.32 Additional studies on the stability of Cu-MOFs support the need for enhanced stability in these complexes.33–35
When the reaction was carried out without PAA, the size of the particles increased (see Fig. S6, ESI†) but the obtained IR and DRX were identical. When intermediate amounts of PAA were used, particles of different sizes were obtained mixed together. The ζ potential of 1 was determined to be −11 mV (see Fig. S7, ESI†), which is in accordance with the presence of carboxylic acid groups of the linker on its external surface. It seems to be big enough to ensure the dispersion of the small NPs of the nano-MOF 1 in water for several hours without any observable aggregation.
The infrared spectrum of 1 (see Fig. S1, ESI†) shows bands in the 3500–3000 cm−1 region attributable to the νO–H, and νC–H stretching vibrations. The signals between 1631 and 1300 cm−1 correspond to the symmetric and asymmetric stretching νCOO vibrations of the carboxylate groups and the stretching νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrations of the benzene ring of the linkers and their wavenumbers and relative intensities constitute a characteristic pattern of the infrared spectrum of a MOF with a determined set of benzenedicarboxylate ligands. The peak at 1241 cm−1 can be attributed to the stretching νC–O vibration of the carboxylate groups.
C vibrations of the benzene ring of the linkers and their wavenumbers and relative intensities constitute a characteristic pattern of the infrared spectrum of a MOF with a determined set of benzenedicarboxylate ligands. The peak at 1241 cm−1 can be attributed to the stretching νC–O vibration of the carboxylate groups.
The band at 1631 cm−1 suggests the ligand is coordinated only through the carboxylate groups, (DHBDC)36 which is also in accordance with a Cu/DHBDC ratio of 1/1 as indicated in the elemental analysis.
The elemental analysis of 1 (see the experimental section) and its TGA (see the ESI,† Fig. S8) strongly support the proposed molecular stoichiometry.
The powder X-ray diffractogram of [Cu(DHBDC)(H2O)2−x(DMF)x] (1) (Fig. 4) evidences its low crystallinity probably due to the small size of the obtained nanoparticles. We were able to obtain crystals by slowly adding a solution of 2,5-dihydroxy-1,4-bencenedicarboxylic acid onto a DMF solution of Cu(CH3COO)2·H2O, contained in a jacketed Schlenk tube at 5 °C, but the DRX diffractogram is quite different to that obtained for nano-MOF 1 and therefore, its structure is not that of the MOF we are using in our experiments. This is not surprising since ref. 18 shows that for MOFs with the [Cu(DHBDC)(DMF)x(H2O)y] stoichiometry, the structures are very dependent on x and y values and the Cu coordination sphere varies drastically from one MOF to another. Furthermore, our diffractogram is completely different from those of the related MOFs of [Cu(DHBDC)(DMF)2]·DMF, [Cu(DHBDC)(DMF)(H2O)2]·DMF and Cu(DHBDC)(H2O) and it does not contain any mixture of them (see Fig. S4 of ref. 18).
On the other hand, n-MOF 1 was found to exhibit an intense fluorescence emission when dispersed in water. In fact, it shows a strong UV absorption band at 350 nm with a luminescence emission centred at 530 nm (see Fig. S9, ESI†). Interestingly, the resulting n-MOF exhibited a luminescence behaviour showing a high Stokes shift allowing easy separation between excitation and emission photons, which proves highly valuable in the case of designing simple optoelectronic instrumentation.
When trying to functionalize n-MOF 1, we noticed that the product was very reactive, and in particular the light green colour of its dispersion in water vanishes in the presence of amines. This observation prompted us to study the phenomenon more carefully. Thus, we observed that the fluorescence of the aqueous solutions of n-MOF 1 fade out in the presence of amines and the colour of the solid samples of 1 changes when exposed to amine vapours. In photoluminescence sensors, the MOF serves as a transducer and a recognition material. The photoluminescence of MOFs can be attributed to various sources,37 being the most significant: (i) the luminescence characteristics of organic linkers (typically extended π-conjugation systems with rigid structures), (ii) metal-based emissions, like those found in lanthanide-based MOFs, (iii) metal-to-ligand charge transfer (e.g. that found in Cu(I) or Ag(I) based MOFs), (iv) ligand-to-metal charge transfer, like that occurring in Zn(II)/Cd(II) and carboxylate ligand-based MOFs, (v) ligand-to-ligand charge transfer, also known as the antennae effect, which involves the indirect excitation of the metal (e.g., in the case of MOFs with absorbing ligands such as π- and σ-bonded antenna ligands) and emitting lanthanide ions and (vi) fluorophores loaded as guest molecules in the MOF pores, like guest-centered emission and guest-sensitization.
The vast majority of luminescent metal-type MOFs can be classified into two groups: lanthanide-based MOFs (Ln-MOFs) and transition metal-based MOFs.38 Zn- and Cd-MOFs are the most frequently reported luminescent transition metal-based MOFs because d10 metal ions have multiple coordination numbers and geometries and glow when attached to functional ligands. Transition metals typically do not possess intrinsic luminescence; instead, they participate in ligand-to-metal charge transfer (commonly reported in Zn- and Cd-MOFs) or metal-to-ligand charge transfer processes (commonly detected in Cu- and Ag-MOFs) to modulate the MOF emission.39
Fluorescence sensing experiments
Amines are key intermediates in industrial organic synthesis, but they are also environmental pollutants and harmful to human health. They are present in household cleaning products, refrigerant gas, pesticides, plastics, dyes, and other chemicals. In addition, amines are markers for food freshness and medical diagnosis. A major and ongoing hazard to the environment, amines ‘corrosive nature’, can also cause lung damage, respiratory issues, and skin irritation, among other health issues.40 Consequently, it is necessary to detect amines at low concentration levels. The presence of amines is commonly determined using complicated, time consuming and expensive centralized analytical techniques. In contrast, simpler and low-cost sensing methods like those based on luminescence measurements are becoming popular due to their simplicity, high sensitivity and capabilities for developing portable devices for in situ measurements.In this context, MOFs are being used as amine sensors in the solid, solution, or vapor phase by the change of color, luminescence quenching or enhancement or even by fluorescence shifts.30
Thus, fluorescence sensing experiments were conducted in a systematic way where the emission spectra of the n-MOF were recorded in an aqueous medium, followed by the measurement every two minutes (until ½ h) of the fluorescence intensity of a water dispersion of 0.005 mg mL−1 in the presence of the amine under scrutiny at a 1 mM concentration. The sensing studies were carried out using a selection of different primary, secondary and tertiary amines, namely, ethylamine (EA), propylamine (PA), aniline (Ani), diethylamine (DEA), piperidine (Pip), triethylamine (TEA), and pyridine (Py) (see Fig. 2).
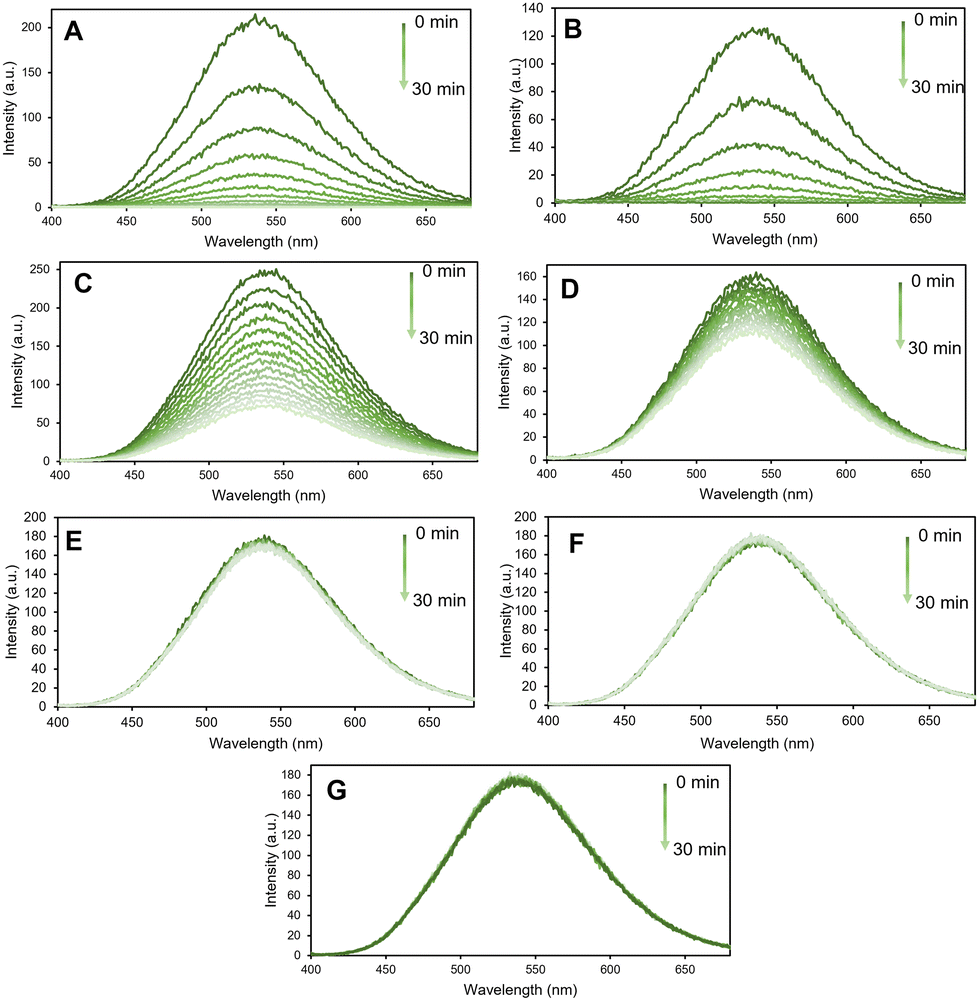 | ||
| Fig. 2 Evolution with the time of the fluorescence spectra of a water dispersion of 0.005 mg mL−1 after the addition of 0.001 M of: (A) propylamine (PA), (B) ethylamine (EA), (C) piperidine (Pip), (D) diethylamine (DEA), (E) trimethylamine (TEA), (F) aniline (Ani), and (G) pyridine (Py). | ||
Interestingly, the n-MOF materials’ fluorescence intensity was quenched in less than 30 minutes by more than 90% when 1 mM of PA (primary amine) was present in the media. Conversely, the fluorescence quenching degree produced by Pip and DEA is lower compared with that by PA. The effect of the presence of other amines, such as pyridine (Py), on the fluorescence of n-MOF 1 was found to be negligible (see Fig. 2).
The results obtained allowed the different amines to be ordered according to the rate of nano-MOF fluorescence quenching as follows: EA > PA > Pip > DEA > TEA ≈ Py ≈ Ani. This sequence suggests that the basicity of the amines plays an important role in the observed quenching rate (see the pKa of their conjugated acids in Table 1) although this relationship is modulated by their steric hindrance (see their cone angles in Table 1).
| Amine | EA | PA | Pip | DEA | TEA | Py | Ani |
|---|---|---|---|---|---|---|---|
| pKa | 10.65 | 10.54 | 11.12 | 10.84 | 10.75 | 5.23 | 4.87 |
| C.A. | 106 | 106 | 121 | 125 | 150 | — | 111 |
Thus, amines with a stronger electron-donating ability can quench the fluorescence intensity of the MOFs quickly, while the less basic pyridine and aniline practically do not alter the fluorescence of 1 after 30 minutes (under the chosen experimental conditions). Furthermore, PA and EA have exactly the same cone angle (106), but EA is a little bit more basic (pKa = 10.65) than PA (pKa = 10.54) and EA is, also, a little bit faster than PA in quenching the fluorescence of 1. Similarly, Pip and DEA have cone angles very close (121 and 125, respectively) but the pKa value of Pip (11.12) is higher than that of DEA (10.84) and the quenching of Pip is faster than the quenching of DEA. On the other hand, in the series EA, DEA and TEA (of roughly the same basicity), TEA, with a cone angle of 150°, is as inactive as Py in fluorescence quenching; EA, with a cone angle of 106°, is the most active; and DEA, with a cone angle of 125°, lies in between.
In general, the sensing of amines by MOFs in solution can be carried out either by enhancement or by quenching the fluorescence of the MOFs or, occasionally, by enhancement by one type of amine and quenching by a different group of amines.42 MOF sensors that experience fluorescence quenching in the presence of amines in water solutions are scarce. One of them43 is only sensitive to aromatic amines. Another one44 detects amines with a very different electron donor ability, the less basic amines are those with lower luminescence quenching ability, while another two26,45 are very sensitive to alkyl amines, but there is no detectable order between them. In any case, none of them detect the presence of amines by quenching speed, as we do.
Vapochromic behaviour of 1 towards amines
The effect on the luminescence of solid samples of 1, when treated with vapors of all the selected amines previously mentioned, was also investigated. Amines are known to be highly volatile and mostly released into the environment as gas or vapor media. Thus, it becomes essential to design a sensing material that can rapidly detect volatile organic amines.Taking into account the discoloration of the water dispersion of 1 in the presence of primary amines we wondered if the presence of amine vapors would also modify the solid samples of 1. This would be of great value in the eventual design of vapochromic probes able to detect amines in their vapor phase with a clear color change of the sensor material. Keeping this in mind, the as-synthesized n-MOF 1 crystals were deposited into a small petri dish placed inside a closed container and exposed to different amine vapors for 30 minutes.
In Fig. 3, pictures of a solid sample of the starting material 1 and those of the samples of 1 after 30 min in contact with the vapors of aniline (Ani), pyridine (Py), triethylamine (TEA), piperidine (Pip), propylamine (PA), ethylamine (EA) and diethylamine (DEA) are shown. Although aniline and pyridine do not appear to affect very much the n-MOF colour, interestingly the other tested amines produced a clear color change in the deposited n-MOF 1, from green to light (TEA and DEA) or dark brown (Pip, PA and EA). This shows that n-MOF 1 has vapochromic properties.
 | ||
| Fig. 3 Pictures of a sample solid of 1 before (MOF picture) and after being exposed for 30 min to vapors of aniline (Ani), pyridine (Py), triethylamine (TEA), piperidine (Pip), propylamine (PA), ethylamine (EA) and diethylamine (DEA). | ||
Monitorization of the process
The effect of the vapor of these amines on a sample of n-MOF 1 can be studied by comparing the powder X-ray diffractograms of the resulting samples with that of the starting material, and all of them are included in Fig. 4. They exhibit an increasing loss of crystallinity from the one of the aniline derivative to the diffractogram of the piperidine derivative; the peaks in the diffractograms are always the same (see Fig. 4), indicating that a crystalline core remains along the decomposition processes.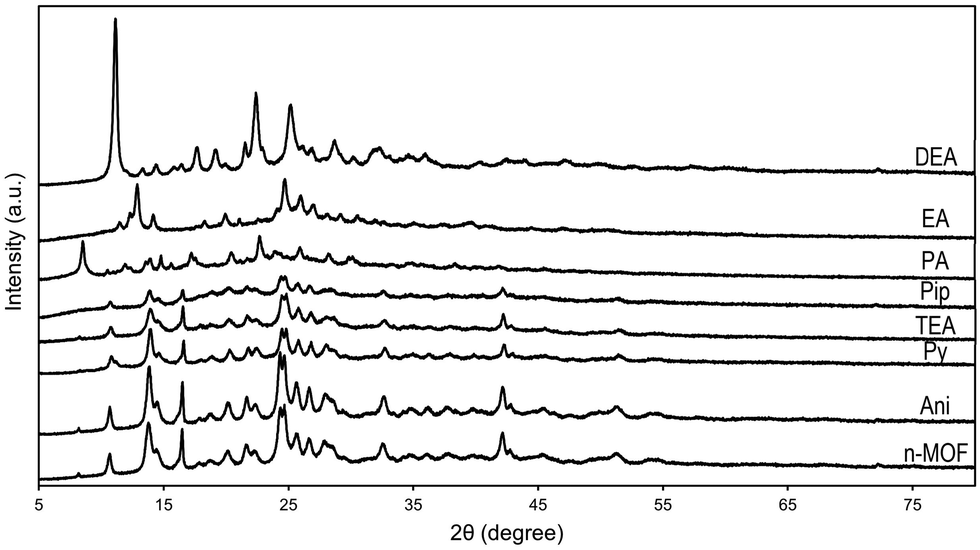 | ||
| Fig. 4 Powder X-ray diffractograms of 1 before (MOF) and after being exposed for 30 min to vapors of aniline (Ani), pyridine (Py), triethylamine (TEA), piperidine (Pip), propylamine (PA), ethylamine (EA) and diethylamine (DEA). | ||
In contrast, the diffractograms corresponding to the final products of the reactions between n-MOF 1 and PA, EA and DEA indicate that new crystalline (unknown) materials have been formed.
In order to get deep inside these processes, IR studies on the resulting products were carried out. The infrared spectra of the samples of [Cu(DHBDC)(H2O)2−x(DMF)x] 1 before and after the solid was exposed to the amine vapors were recorded and are shown in Fig. 5. The IR spectra of the samples treated with aniline and triethylamine vapors are coincident with that of the starting material (see Fig. 5), confirming that there is no reaction of n-MOF 1 with aniline. In the case of TEA, the change of the color and the loss of crystallinity (see Fig. 3 and 4) clearly indicate that the amine reacts with 1, but the infrared spectrum does not provide any evidence about the nature of these reaction products.
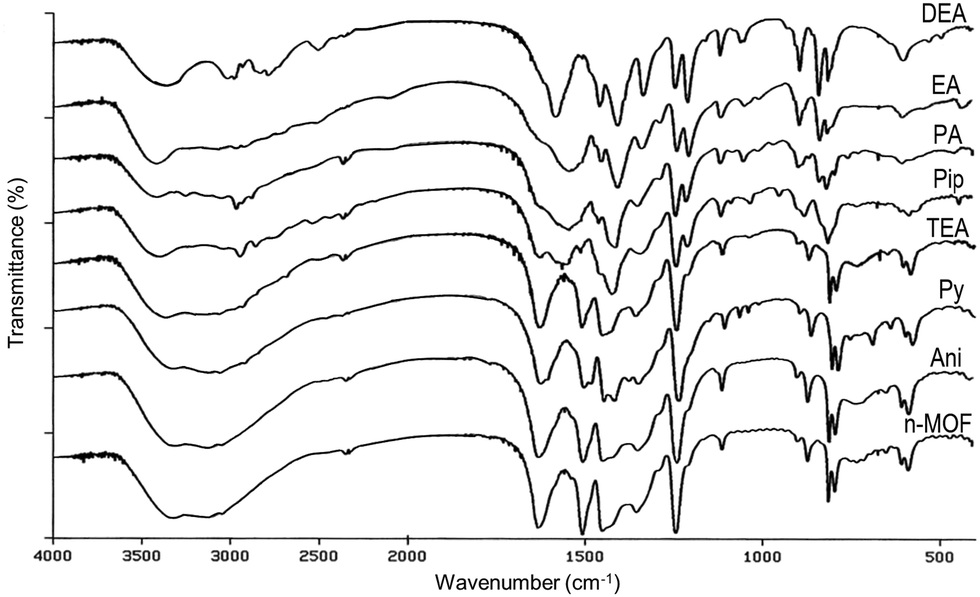 | ||
| Fig. 5 FTIR spectra of 1 before (MOF) and after being exposed for 30 min to vapors of aniline (Ani), pyridine (Py), triethylamine (TEA), piperidine (Pip), propylamine (PA), ethylamine (EA) and diethylamine (DEA). | ||
The IR spectrum of the pyridine derivative is almost coincident with that of 1, but it has some extra weak peaks. The new absorptions could be assigned to a coordinated pyridine Cu-py (1486, 1070, 1046, and 693 cm−1),46 pyH+ (1607, 1379 and 757 cm−1)47 and, tentatively, to DHBDC− (C8H5O6)− (1419 cm−1), which could be the anionic counterion of the pyridinium cation. In fact, a band at 1419 cm−1 is present in the IR spectrum of 2,5-dihydroxy-1,4-benzenedicarboxylic acid (solid) after being exposed to propylamine vapor due to the formation of (C3H7NH3)+ (C8H5O6)−. On the other hand, this is the expected product since the reaction of 2,5-dihydroxy-1,4-bencenedicarboxylic acid with ammonia gives (NH4)+(C8H5O6)−.
The IR spectra of the other amine derivatives (Pip, PA, EA and DEA) increasingly differ from that of n-MOF 1, suggesting that the reaction of the amines with 1 destroys more and more the starting material, a process also detected in the corresponding PXRD diffractograms.
On the other hand, the IR spectra of PA, EA and DEA derivatives show some similarities. Thus, they contain bands characteristic of the alkylammonium salt cations,47,48 including a broad band at about 3400 cm−1 due to N–H stretching (see Fig. 5 and the ESI†). Their counterion should be the anion (C8H5O6)− as indicated by the presence of a peak at about 1414 cm−1. Interestingly, their spectra exhibit bands close to the characteristic peaks of Cu-MOF-74,49 namely, 1552 (s), 1419 (s), 1249 (w), 1187 and 828 (m) cm−1, but their PXRD diffractogram differs from that of Cu-MOF-7450 These coincidences suggest the formation of new products containing the moiety DOBDC, (C8H2O6)−4, as the ligand, also detected by the lower intensity of the band at 1631 cm−1, due to the (C8H4O6)−2 ligand.
Experimental section
Materials and methods
Polyacrylic acid, copper(II) acetate monohydrate (≥99.0%), 2,5-dihydroxyterephthalic acid (98%), and N,N-dimethylformamide (≥99.8%) were obtained from Sigma-Aldrich (Spain) and ethanol (96% vol) was purchased from VWR chemicals.FT-IR measurements were conducted utilizing a Spectrum RXI FT-IR spectrometer by PerkinElmer (USA), employing KBr pressed disks. An MET JEOL 1011 apparatus, operating at 100 kV, was used to capture transmission electron microscopy (TEM) images on a copper grid. ImageJ software was utilized for size estimation by measuring the largest dimension of individual particles. X-ray powder diffraction (XRPD) data for the particles were collected at room temperature (RT) using CuKα 1,2 radiation (λ = 1:54056 Å and 1.54439 Å) with a Bragg–Brentano reflection setup. This was conducted on a PHILIPS X’PERT PRO Panalytical diffractometer within a 2θ range spanning 5–80°, employing a step size of 0.0167. Thermogravimetric analysis (TGA) and elemental analysis were performed using a TG-DSC analyzer, SETSYS Evolution 1750 (Setaram), and a Carlo Erba 1108 elemental analyzer, respectively. These analyses were conducted in collaboration with the C.A.C.T.I. at the University of Vigo. Copper analysis was performed through elemental mass spectrometry (i.e. ICP-MS analysis). For this purpose, nano-MOF samples were dried and digested using aqua regia (30 min in an ultrasonic bath). Copper (Cu) quantification was performed in the resulting solution using an ICP-MS/MS equipped with a triple quadrupole (Agilent 8900, Agilent Technologies, Japan). The operation conditions were optimized daily using a tuning solution. Cu isotopes were detected using an “on-mass mode” (63Cu+, 65Cu+). Helium was introduced into the collision/reaction cell at a flow rate of 5 mL min−1. The dwell time for each of the targeted isotopes was 1 s. Cu was quantified using an external calibration prepared using certified 1000 mg L−1 Cu standard solution (Merck, Germany). All fluorescence measurements were carried out using a Varian Cary Eclipse Fluorescence Spectrometer (Agilent, Germany), equipped with a xenon discharge lamp and a photomultiplier tube detector (Model R-298). Fluorescence spectra were recorded with a fixed excitation wavelength of 350 nm and with excitation and emission slit widths of 5 nm. The measurements were performed at room temperature and atmospheric pressure, employing quartz fluorescence cells.
Synthesis of [Cu(DHBDC)(H2O)2−xDMFx] (1)
For MOF synthesis, dissolve 0.5300 g of polyacrylic acid (PAA) (7.35 mmol) in 10 mL of DMF. After dissolving the PAA, 0.3000 g of 2,5-dihydroxyterephthalic acid (DHBDC) (1.47 mmol) is added to the vial containing the PAA and shaken until dissolved. At the same time, 0.2000 g of copper(II) acetate monohydrate (CuAc2·H2O) (0.99 mmol) is dissolved in 10 mL of DMF in a Jacketed glass Schlenk flask. The DHBDC + PAA mixture is slowly added to the Jacketed glass Schlenk and stirred vigorously for about 5 minutes. During the addition, a chocolate brown color is observed in the area where the drop falls, which disperses upon stirring. When all the ligands have been added, the entire content of the Jacketed Schlenk is observed to become brown, but after a few minutes, it turns a bright light green color in a perfectly transparent solution. The mixture is stirred for 72 h at 5 °C. The resulting product is a very intense light pastel green color, and is centrifuged in two 10 mL portions at 9158 g for 10 min. Each portion is washed with 5 mL of DMF and re-centrifuged twice. The centrifugate is washed with 10 mL of milli-Q water and re-centrifuged, repeating this process one more time. Upon washing with water, the pastel colour of the compound is slightly lost in favor of a slightly darker color. Finally, the contents of both tubes are suspended in one in 10 mL of ethanol, centrifuged and dried under vacuum. Anal. calcd for Cu(DHBDC)(H2O)1.87(DMF)0.13: C, 33.27; H, 2.88; N, 0.61; Cu, 20,9. Found: C, 32.21; H, 2.95; N, 0.53; Cu, 17,20.Conclusions
In summary, a novel copper-based nano-MOF was synthesized and investigated in detail for discriminative sensing of amines. The synthetic method proposed here produces nano-MOFs with a fairly homogeneous size and morphology. Furthermore, the produced nano-MOF maintains its chemical stability in aqueous media, which is of utmost importance if it is intended to be used for bio-applications. The resulting frameworks are efficient luminescent probes for discriminative detection of different amines via “turn-off” fluorescence emission. The solid n-MOF 1 does not react with aniline, the less basic of the used amines, and reacts moderately with py, also a very weak base. The attack of the other amines destroys the starting material, although in the case of PA, EA and DEA, new crystalline unknown products are formed. The mechanism of these reactions could have some similarities. Initially, the amines would replace the coordinated water entering the copper coordination sphere, then the amines extract the carboxylic ligand giving the salts (ammonium)+ (C8H5O6)−, releasing OH− anions which could eventually favor the generation of the (2,5-dioxy-1,4-benzenedicarboxylate) tetra-anion ligand. This new nanomaterial could contribute to the development of basic detection systems for harmful species, which could be used for environmental control and food quality regulation.Author contributions
Pablo Álvarez García: investigation, methodology, data curation, and formal analysis. Enrique Álvarez Rubiera: investigation and methodology. David Gutiérrez Armayor: investigation and methodology. Candela Melendreras: investigation, methodology, data curation, and formal analysis. Ana Belen Soldado: investigation, supervision, and validation. José Manuel Costa: investigation, supervision, validation, and writing – review and editing. Elena Lastra: investigation, supervision, and writing – review and editing. Fco. J. García Alonso: funding acquisition, supervision, writing – original draft, and writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Principado de Asturias (Spain) through the project AYUD/2021/51323 and from the Ministry of Science and Innovation (Spain) through the projects Ref. PID2022-142323NB-I00, PID2020-115204RB-I00 and PID2020-117282RB-I00 is gratefully acknowledged. We acknowledge Larry R. Falvello from the Instituto de Nanociencia y Materiales de Aragón for their helpful discussions and for crystal determination of related compounds, Laura Roces for PXRD data acquisition, Borja Moreira for the ICP-MS analysis and Beatriz Ramajo for BET experiments and fruitful discussions. Candela Melendreras acknowledges her PhD grant (PAPI-22-TESIS-2) from the Plan Propio de la Universidad de Oviedo.References
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC; I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- M. Safaei, M. M. Foroughi, N. Ebrahimpoor, S. Jahani, A. Omidi and M. Khatami, Trac-Trends Anal. Chem., 2019, 118, 401–425 CrossRef CAS; J. Yang and Y.-W. Yang, Small, 2020, 16, 1906846 CrossRef PubMed.
- Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC; X. Shi, Y. Shan, M. Du and H. Pang, Coord. Chem. Rev., 2021, 444, 214060 CrossRef CAS; D. Bradshaw, S. El-Hankari and L. Lupica-Spagnoloa, Chem. Soc. Rev., 2014, 43, 5431–5443 RSC.
- S. L. Griffin and N. R. Champness, Coord. Chem. Rev., 2020, 414, 213295 CrossRef CAS.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. GentleIII, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- K. J. Gagnon, P. P. Houston and A. Clearfield, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS PubMed.
- D. Lv, W. Nong and Y. Guan, Coord. Chem. Rev., 2022, 450, 214234 CrossRef CAS.
- W. Fan, X. Zhang, Z. Kang, X. Liu and D. Sun, Coord. Chem. Rev., 2021, 440, 213968 CrossRef.
- B. Seoane, S. Castellanos, A. Dikhtiarenko, F. Kapteijn and J. Gascon, Coord. Chem. Rev., 2016, 307, 147–187 CrossRef CAS.
- S. Mubarak, D. Dhamodharan, P. N. P. Ghoderao and H.-S. Byun, Coord. Chem. Rev., 2022, 471, 214741 CrossRef CAS.
- J. Lei, R. Qian, P. Ling, L. Cui and H. Ju, Trac-Trends Anal. Chem., 2014, 58, 71–78 CrossRef CAS.
- C. R. Marshall, S. A. Staudhammer and C. K. Brozek, Chem. Sci., 2019, 10, 9396–9408 RSC.
- S. Abednatanzi, M. Najafi, P. G. Derakhshandeh and P. Van Der Voort, Coord. Chem. Rev., 2022, 451, 214259 CrossRef CAS.
- T. Xiao and D. Liu, Micropor. Mesopor. Mat., 2019, 283, 88–103 CrossRef CAS; H. Kim and C. S. Hong, CrystEngComm., 2021, 23, 1377–1387 RSC.
- N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dinca, J. Am. Chem. Soc., 2013, 135, 13326–13329 CrossRef CAS PubMed; S. Hussain, X. Chen, W. T. A. Harrison, S. Ahmad Sharif, S. J. Su, S. Muhammad and S. Li, RSC Adv., 2020, 10, 12841–12850 RSC.
- R. Sanz, F. Martinez, G. Orcajo, L. Wojtas and D. Briones, Dalton Trans., 2013, 42, 2392–2398 RSC.
- L. Shao, F. Fan, X. Dai, H. Fu, W. Li, W. Qi, F.-B. Meng and Y. Fu, Chem. Mater., 2022, 34, 5356–5365 CrossRef CAS; J. Han, L. Shao, H. Chen, H. Zhou, B. Zhang, Y. Zhang, H. Yuan, Y. Chen, J. Zhou and Y. Fu, Inorg. Chem., 2022, 61, 7173–7179 CrossRef PubMed.
- C.-S. Liu, J. Li and H. Pang, Coord. Chem. Rev., 2020, 410, 213222 CrossRef CAS.
- Z. Li, S. Hou, H. Zhang, Q. Song, S. Wang and H. Guo, Adv. Agrochem., 2023, 2, 79–87 CrossRef CAS.
- T. Du, L. Huang, J. Wang, J. Sun, W. Zhang and J. Wang, Trends Food Sci. Tech., 2021, 111, 716–730 CrossRef CAS.
- A. Mallick, B. Garai, M. A. Addicoat, P. S. Petkov, T. Heine and R. Banerjee, Chem. Sci., 2015, 6, 1420 RSC.
- J.-J. Liu, Y.-F. Guan, M.-J. Lin, C.-C. Huang and W.-X. Dai, Cryst. Growth Des., 2015, 15, 5040–5046 CrossRef CAS.
- J.-J. Liu, Y.-B. Shan, C.-R. Fan, M.-J. Lin, C.-C. Huang and W.-X. Dai, Inorg. Chem., 2016, 55, 3680–3684 CrossRef CAS PubMed.
- N.-N. Yang, W. Sun, F.-G. Xi, Q. Sui, L.-J. Chen and E.-Q. Gao, Chem. Commun., 2017, 53, 1747–1750 RSC.
- N.-N. Yang, L.-J. Zhou, P. Li, Q. Sui and E.-Q. Gao, Chem. Sci., 2019, 10, 3307–3314 RSC.
- M.-Y. Guo, P. Li, S.-L. Yang, R. Bu, X.-Q. Piao and E.-Q. Gao, ACS Appl. Mater. Interfaces, 2020, 12, 43958–43966 CrossRef CAS PubMed.
- X.-Y. Liu, X.-M. Yin, S.-L. Yang, L. Zhang, R. Bu and E.-Q. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 20380–20387 CrossRef CAS PubMed.
- Y.-P. Li, S.-N. Li, Y.-C. Jiang, M.-C. Hu and Q.-G. Zhai, Chem. Commun., 2018, 54, 9789–9792 RSC.
- J. Othong, J. Boonmak, N. Wannarit, F. Kielar, T. Puangmali, W. Phanchai and S. Youngme, Sensor Actuat. B-Chem., 2021, 343, 130066 CrossRef CAS; S.-L. Yao, H. Xu, T.-F. Zheng, S.-J. Liu, J.-L. Chen and H.-R. Wen, Cryst. Growth Des., 2021, 21, 5765–5772 CrossRef.
- T. Rasheed, Chemosphere, 2023, 313, 137607 CrossRef CAS PubMed.
- N. R. Dhumal, M. P. Singh, J. A. Anderson, J. Kiefer and H. J. Kim, J. Phys. Chem. C, 2016, 120, 17323–17333 CrossRef.
- J. B. DeCoste, G. W. Peterson, B. J. Schindler, K. L. Killops, M. A. Browe and J. J. Mahle, J. Mater. Chem. A, 2013, 1, 11922–11932 RSC.
- L. A. Brandner, M. Linares-Moreau, G. Zhou, H. Amenitsch, S. Dal Zilio, Z. Huang, C. Doonan and P. Falcaro, Chem. Sci., 2023, 14, 12056–12067 RSC.
- B. D. Dhanapala, D. L. Maglich and M. E. Anderson, Langmuir, 2023, 39(34), 12196–12205 CrossRef CAS PubMed.
- A. D. Pournara, A. Douvali, S. Diamantis, G. S. Papaefstathiou, A. G. Hatzidimitriou, S. Kaziannis, C. Kosmidis, T. Lazarides and M. J. Manos, Polyhedron, 2018, 151, 401–406 CrossRef CAS.
- A. Zuliani, N. Khiar and C. Carrillo-Carrión, Anal. Bioanal. Chem., 2023, 415, 2005–2023 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- L. Shi, N. Li, D. Wang, M. Fan, S. Zhang and Z. Gong, Trac-Trends Anal. Chem., 2021, 134, 116131 CrossRef CAS.
- Section 8: Analytical Chemistry. CRC Handbook of Chemistry and Physics 84th Edition. CRC Press 2003–2004.
- A. L. Seligson and W. C. Trogler, J. Am. Chem. Soc., 1991, 113, 2520–2527 CrossRef CAS.
- P.-M. Chuang, Y.-J. Tu and J.-Y. Wu, Sensor Actuat. B Chem., 2022, 336, 131967 CrossRef.
- Y. Tang, H. Huang, B. Peng, Y. Chang, Y. Li and C. Zhong, J. Mater. Chem. A, 2020, 8, 16542–16550 RSC.
- I. Ahmed, H. J. Lee and S. H. Jhung, J. Mol. Liq., 2021, 344, 117765 CrossRef CAS.
- H. Huang, W. Gao, X.-M. Zhang, A.-M. Zhoua and J.-P. Liu, CrystEngComm, 2019, 21, 694–702 RSC.
- M. Goldstein, E. F. Mooney, A. Anderson and H. A. Gebbie, Spectrochim. Acta, 1965, 21, 105–117 CrossRef CAS.
- https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi (see on november 9, 2023).
- F. Lambarki, A. Ouasri, H. Zouihri and A. Rhandour, J. Mol. Struct., 2017, 1142, 275–284 CrossRef CAS; T. Dhanabal, G. Amirthaganesan and M. Dhandapani, Optik, 2013, 124, 6877–6881 CrossRef.
- H. Zheng, D. Wang, X. Sun, S. Jiang, Y. Liu, D. Zhang and L. Zhang, Chem. Eng. J., 2021, 411, 128524 CrossRef CAS; J. G. Flores, E. Sánchez-González, A. Gutiérrez-Alejandre, J. Aguilar-Pliego, A. Martínez, T. Jurado-Vázquez, E. Lima, E. González-Zamora, M. Díaz-García, M. Sánchez-Sánchez and I. A. Ibarra, Dalton Trans., 2018, 47, 4639–4645 RSC.
- J. Cai, Y. Li, M. Zhang and Z. Li, Inorg. Chem., 2019, 58, 7997–8002 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectrum and TGA thermogram of 1 and FTIR spectra data (KBr pellets, cm−1) of 1 before and after its reaction with different amine vapors (PDF file). See DOI: https://doi.org/10.1039/d4ma00204k |
| This journal is © The Royal Society of Chemistry 2024 |