
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iodine solid sorbent design: a literature review of the critical criteria for consideration†‡
Brian J.
Riley
 *a,
Joshua R.
Turner
*b,
Joanna
McFarlane
c,
Saehwa
Chong
a,
Krista
Carlson
d and
Josef
Matyáš
a
*a,
Joshua R.
Turner
*b,
Joanna
McFarlane
c,
Saehwa
Chong
a,
Krista
Carlson
d and
Josef
Matyáš
a
aPacific Northwest National Laboratory, Richland, WA, USA. E-mail: brian.riley@pnnl.gov; Tel: +1 509 372-4651
bNational Nuclear Laboratory, Sellafield, Cumbria, UK. E-mail: joshua.r.turner@uknnl.com
cOak Ridge national Laboratory, Oak Ridge, TN, USA
dUniversity of Nevada Reno, Reno, NV, USA
First published on 23rd October 2024
Abstract
Designing sorbents for iodine capture in different conditions requires selection and optimization of a large and diverse range of variables. These variables fall into general categories (or features) of sorbent activity, sorbent stability, and the fate of the loaded material in terms of the disposal (waste form) options available. To illustrate, silver-loaded, high-porosity sorbents make for maximized iodine capture and less pressure drop in a column-based sorption system approach, however, this high porosity can lead to less mechanically stable sorbents. Additionally, waste forms containing silver must also be compliant with additional criteria for hazardous waste disposal. Thus, all these aspects must be considered simultaneously when selecting a sorbent for utilization under specific conditions. Information is given for different types of sorbent design considerations for different operating conditions and some emphasis is also given on promising alternatives for silver as the active (chemisorption-based) getter metal. Discussion is given around demonstrated options for waste forms for different metal-iodide compounds.
1. Introduction
The effective capture and immobilization of radioiodine from nuclear processes is a very important topic from an environmental preservation perspective and in regard to the protection of human health from deleterious effects of radioiodine uptake by the thyroid.1,2 Over the past several decades, a wealth of literature has been produced from theoretical and experimental studies to identify and evaluate capture and treatment processes for managing radioiodine, understand the optimal parameters for capture systems, and find long-term storage solutions for these radioisotopes. Of most interest are the radioisotopes of short-lived 131I with a half-life (t1/2) of 8.02 days and long-lived 129I with a t1/2 of 1.57 × 107 y, which both decay through β− emission to Xe isotopes (i.e., 129I → 129Xe + β− and 131I → 131Xe + β−, respectively). It is likely that different iodine management solutions would be needed to capture 131I vs. 129I due to radiation tolerance and decay heat issues associated with 131I. Due to the short t1/2 of 131I, it is most relevant in nuclear accident scenarios or in applications where it is directly released from nuclear fission and requires a short-term solution, e.g., within the headspace of a molten salt reactor (MSR).Among the options for radioiodine capture, the majority can be classified as either liquid scrubbing approaches or solid sorbent approaches. The most commonly discussed liquid scrubbing options are aqueous caustic scrubber solutions3–8 whereby iodine entering the solution is converted into an ionic form. Other liquid scrubbing options include Iodox,9,10 Mercurex,9,11,12 and electrolytic scrubbing,13,14 which are discussed in previous reviews.8–10,15 For solid sorbents, the primary materials studied include metal-functionalized or metal-containing porous substrates (e.g., zeolites, silicic acid, alumina, aerogels, xerogels, metal organic frameworks or MOFs),10,16–40 but materials such as activated carbon have also been examined.41–43 The primary difference between the metal-functionalization and metal-containing (or metal-loaded) descriptors is that metal functionalization is where the metals are loaded onto the surfaces of a scaffold or into pores/channels of a porous material (e.g., Ag+ added to thiol tethers on the surfaces and then reduced to Ag0 as is done with Ag-functionalized silica aerogel),25 whereas an example of metal containing would be the metal node of a MOF. The functionalization process can include tethers between the scaffold and the getter, which is described below in more detail (see Section 7, Sorbent functionalization). It should also be noted that “metal” does not necessarily mean the element is at zero-valent charge and could also refer to ionic forms. Covalent organic frameworks (COFs),44–46 MOFs,38–40,47 and porous organic polymers (POPs)48–50 will not be discussed here in detail as they have been discussed elsewhere, but are mentioned to provide a broader list of options for readers.
The capture processes by which solid sorbents can immobilize a species or series of species can be thought of in terms of size exclusion mechanisms (molecular sieving), weak physical interaction mechanisms (physisorption), stronger chemical bonding capture mechanisms (chemisorption), or mixtures of these. The size exclusion mechanism is often referred to as a molecular sieving approach whereby species with sizes too large to fit through pores in a sorbent are not allowed through due to size restrictions; species that are small enough can enter into the sorbent where they can remain stationary. The weak physical mechanism is referred to as physisorption whereby interactions like van der Waals forces can hold an adsorbate to the surfaces of a sorbent. Species that are physisorbed can often desorb with small changes to the loaded sorbent like minimal heating.51 The chemical bonding capture mechanism is referred to as chemisorption, which is notably stronger than physisorption, whereby chemical bonds are made between the adsorbate and the sorbent.
For metal-containing and metal-functionalized sorbents, iodine is often captured by a chemisorption process by which the metal reacts with the iodine species to form a stable metal-iodide compound (MIx). The most commonly studied metal for the chemisorption-type solid sorbent for iodine capture is silver (e.g., Ag+, Ag0), but other metals have been demonstrated and show promise, such as Cu52–56 and Bi.56–61 Silver-based materials have also been the most widely implemented chemisorption-type solid sorbent options in various nuclear facilities across the world including in the U.S., France, Russia, United Kingdom, Italy, and Germany.62
For activated carbon, iodine capture often occurs primarily through physisorption51,63 with chemisorption achieved through impregnation or functionalization with organic species that enable binding with iodine within a specific form.64 As discussed later in the article, the different physical and chemical properties of the various forms of iodine present within nuclear-related processes dictate the sorption mechanism. Since the sorbent could be operated under a variety of conditions (including temperature, incoming gas flowrates, sorbate speciation and concentrations) or stored for an extended time period before processing, some of these conditions can result in release of physisorbed iodine before or during transformation into a waste form suitable for disposal. This is why chemisorption-type sorbents are generally preferred over physisorption-type sorbents.
Literature reviews of radioiodine capture technologies have been given in recent works.10,59,62,65–69 Materials capturing iodine in different forms through physisorption or a mixture of chemisorption and physisorption have also been studied and reviewed elsewhere.50,70 Instead of these approaches, while related, this review intends to fill the need to discuss the various categories that must be considered when designing a solid sorbent for radioiodine capture in realistic conditions and is a more detailed extension to other recent reviews.71,72 Additional unique aspects of this review are the discussion regarding realistic processing environments and updated waste form options based on more recent published works.
At a minimum, iodine solid sorbents should be designed considering the following categories (or features), which are summarized in Fig. 1 and below in the following sections as follows: (a) target analyte in the off-gas environment (see Section 2; Fig. 1a), (b) sorbent requirements (see Section 3; Fig. 1b), (c) getter selection (see Section 4; Fig. 1c), (d) scaffold selection (see Section 5; Fig. 1d), (e) getter addition (see Section 6; Fig. 1e), (f) sorbent functionalization (see Section 7; Fig. 1f), (g) sorbent loading (see Section 8; Fig. 1g), and (h) waste form options for loaded materials (see Section 9; Fig. 1h). Within these general categories, several subcategories exist for preferred criteria/features that should also be considered. While the following sections each focus on one of the general categories listed above, due to the interrelationships of these categories, several sections overlap with other sections and the reader will be referred to other sections at times for additional information.
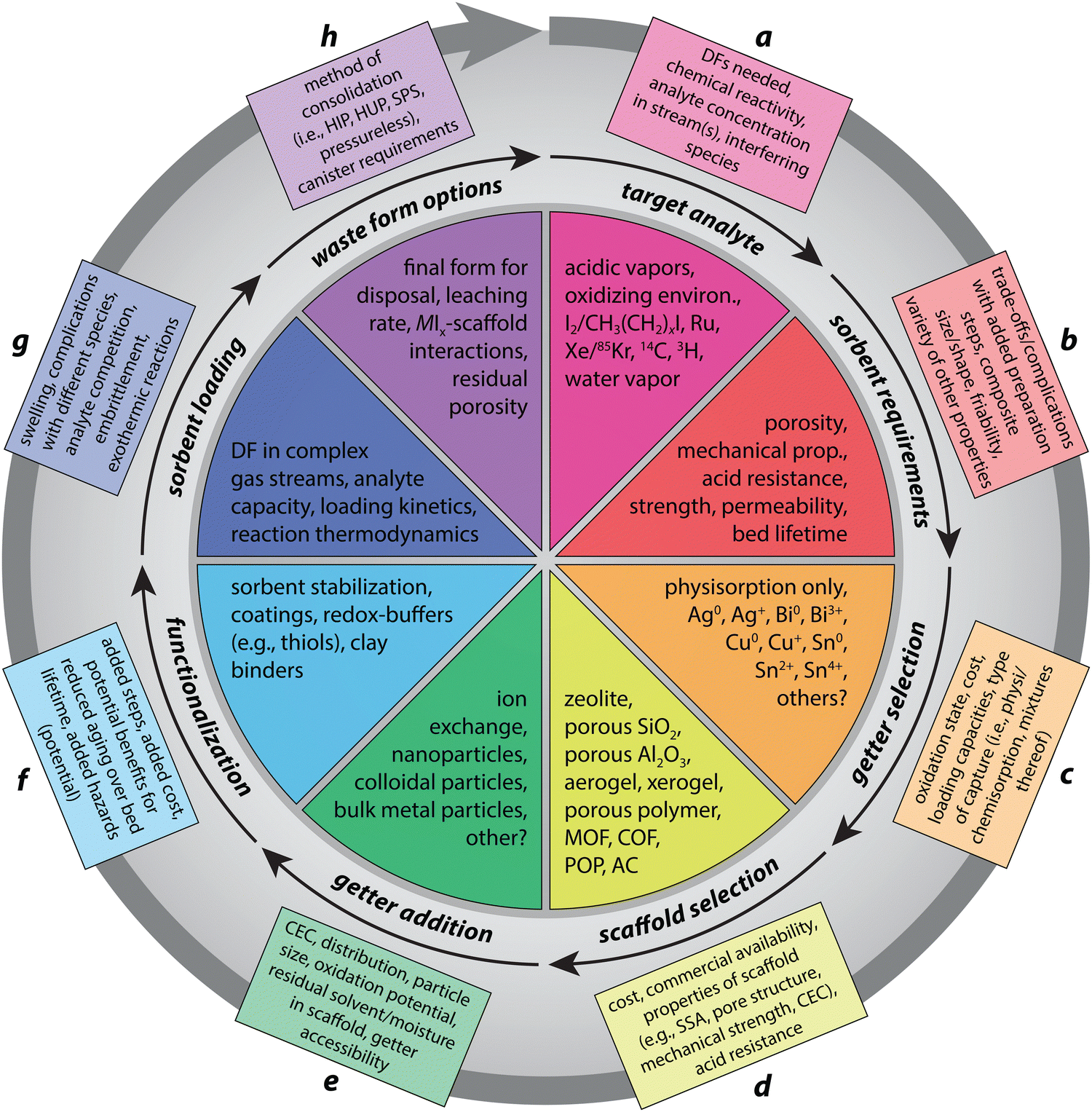 | ||
| Fig. 1 Description of solid sorbent design categories to consider including (a) target analyte in the off-gas environment (Section 2), (b) sorbent requirements (Section 3), (c) getter selection (Section 4), (d) scaffold selection (Section 5), (e) getter addition (Section 6), (f) sorbent functionalization (Section 7), (g) sorbent loading (Section 8), and (h) waste form options for loaded materials (Section 9). More details are provided below for these different categories. | ||
2. Target analyte in the off-gas environment
As mentioned in Fig. 1a, several different aspects need to be considered when attempting to capture a target analyte from a stream of interest. Considering iodine capture specifically, aspects to evaluate include the form of iodine (e.g., inorganic, organic), whether oxidizing conditions are present, the temperature of the stream, and the acidic vapor content of the stream.2.1. Forms of iodine and selectivity
Considering the target analyte, changes to the sorbent could be required if the iodine form is inorganic (e.g., I2, ICl) versus organic [e.g., CH3I, CH3(CH2)xI where x = 1–11],73 as capture performances might not necessarily be consistent between these different scenarios. The getter metal selection (discussed in Section 4, Getter selection) is a critical step in sorbent design because different metals will behave differently in changing conditions. The kinetics of reactions between different metals and various forms of iodine are dissimilar, as well as the iodine loading capacities, which are closely related to the metal oxidation state in the metal-iodide compound (MIx). Also, loading capacities for chemisorbing sorbents are not directly tied to the original speciation because that speciation is changed by chemisorption processes.To assess competitive gas adsorption on surfaces, calculations with the ideal adsorbed solution theory (IAST) can be used with single-component isotherms as well as Henry's Law where the concentration of the dissolved gas (C) is equal to the product of the Henry's Law constant (kH) and the partial pressure (P) of the gas. The selectivity of a sorbent towards the target analyte (a) over a competing species (b) is defined in eqn (2) as Sa,b, where xa and ya denote the mole fractions of species a and b, respectively, in the adsorbed phase, and xb and yb are the mole fractions of species a and b in the bulk phase, respectively. This property and associated requirements will influence the preprocessing steps required to purify the stream, separate chemical species that might interfere, and determine the target element concentration compared to undesired elements.
| C = kH·P | (1) |
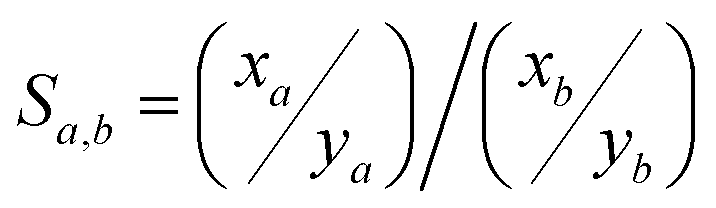 | (2) |
2.2. Highly oxidizing conditions
Some off-gas systems will create highly oxidizing environments and these conditions can accelerate aging of a sorbent (e.g., convert Ag0 to AgNO3). Sorbent performance (i.e., “shelf life”) under oxidizing conditions is often assessed in aging studies.20,74 Examples of oxidizing species include O2(g), NO(g), and NO2(g). Another example is advanced voloxidation used in nuclear fuel recycling, where upwards of 40% NO2 (in air) could be present along with the volatile and semi-volatile fission products.75 These fission products will include iodine isotopes in the form of I2(g), 3H2(g) (tritium) associated with NOx, 14C in the form of oxides, and various isotopes of Xe and Kr (noble gases).76The main goals of voloxidation are two-fold.75,77 First, it can be used to volatilize tritium and 129I from the used fuel with high enough efficiency so that these can be captured in the voloxidation gas stream to meet regulatory requirements and eliminate the need to manage or capture the remaining tritium and iodine that contaminate downstream dissolution and separation processes and waste streams. While this is likely possible with tritium, it remains unproven for iodine. Second, it can make the used fuel easier to dissolve by chemically separating it from cladding, oxidizing or nitrating the fuel, and chemically pulverizing the fuel. In particular, the capture of iodine and 3H2(g) released during voloxidation and subsequent capture on functionalized zeolites has been studied for mitigation of gas release during fuel recycling.78 The capture of iodine on Ag-based sorbents is complicated by the reaction of Ag0/Ag+ with the NO2 recirculating through the voloxidation reactor.79
These discussion points and experiment results emphasize that the stability of the sorbents will need to be tested under conditions of each specific process to which they are being applied. Alternatively, preprocessing strategies could allow the sorbents to maintain their performance. Examples of this include capturing NO2 from the off-gas upstream of the Ag-functionalized sorbent bed or destroying it using selective catalytic reduction with NH3 and a catalyst.80 Two additional examples that can degrade sorbent performance due to how they oxidize metal getters in sorbents include H2O and HNO3 (described in Section 2.4 below).
2.3. High-temperature conditions
Some off-gas applications will require that sorbents be functional at high to very high temperatures such as in the case of voloxidation (T ≈ 450–600 °C)75 and MSRs off-gas streams (T > 400 °C).81 Although MSRs operate at T > 600 °C, the off-gas will be cooler, especially if decay heat is removed by a 2-day hold up at the front end of the off-gas system.81,82 The off-gas byproducts of MSRs constitute the pressure boundary of the reactor and thus will be exposed to volatile fission products as well as aerosols of salt materials; additional information on MSR-specific off-gas capture requirements and concepts are provided elsewhere.81–83 This complex mixture will require several stages of filtration to confine the fission products until they can be safely contained for disposal or reuse; an example of the latter is 85Kr.84 The primary Xe radioisotopes (i.e., 133Xe, 133mXe, 135Xe, 135mXe, 137Xe, 139Xe) are sufficiently short lived (days to seconds),82 so that after ≈ 3 months, they could be released into the environment.Due to the 129I present, within the U.S., iodine must be trapped in the off-gas system prior to release to the environment.85 Standard filtration can be used, such as trapping on NaF sorbents. Novel approaches, such as a molten hydroxide scrubber86 (see example in Fig. S1, ESI‡)82,87 have been proposed that may alleviate the potential for clogging of a solid sorbent bed by removing particulates, mists, and aerosols upstream of the solid sorbents; this requirement poses a drawback for solid sorbents. A device such as a molten hydroxide scrubber would allow for the capture of iodine, along with other acidic gases that will evolve from the chloride-based or fluoride-based MSR. Entrained aerosols and the gases would flow through the scrubber within the cover case in the opposite direction to the liquid hydroxide eutectic. The wetted interior surface area is increased by a highly porous packed bed, facilitating uptake of gases and trapping of particles. A prototype of the molten hydroxide scrubber has been tested and an additional test of a small-scale device with recirculating NaOH-KOH is planned with a photograph of the prototype scrubber shown in Fig. 2. The chemistry of the iodine in the scrubber has been studied by Raman and Fourier transform infrared spectroscopies and shows that the iodine would be captured as an IO3− species.88 The development of this device is in an early stage and fundamental questions remain. For instance, the potential effect of radiolysis on iodine speciation has not been tested, and thus, this device would need to be coupled downstream with a trap that could remove residual halides (i.e., using a solid sorbent bed) along with any H2O or products of radiolysis evolved from the scrubber. Other considerations include the residual carbonate and residual H2O in the molten salt eutectic and their interaction with I2 and other acidic gases.88
 | ||
| Fig. 2 Molten hydroxide (NaOH-KOH eutectic) showing interior before insulation. The external salt storage tank is shown on the left. This figure was modified from the original by McFarlane et al.89 and reprinted with permission. | ||
2.4. Acidic conditions
Many nuclear process off-gas streams can contain acids of different types. Highly acidic vapors in off-gas environments can degrade (chemically attack) a sorbent as well, which can result in scaffold degradation and/or loss of sorbent performance.10,90 Acids include HNO3 that evolves from fuel dissolution during reprocessing as well as HF, HCl, and HI. Although a lower concentration of HNO3 is expected to have a lesser impact on Ag-functionalized materials than in NO2 voloxidation, it still has been highlighted as a potential concern.72,79 Small amounts of 3HCl and 3HF can be liberated from MSRs into the off-gas stream during operation. In the case of MSRs, iodine will most likely react with the most abundant halide in the salt mixture, producing ICl91 or IFx (x = 5, 7)92 in the off-gas rather than I2(g). Cations, such as Cs, will remain in the salt.93Note that acidic gases can also be generated during salt purification for molten salt reactors, which, in the case of chloride-based salts, often uses carbochlorination to remove water.94 The hydrogen from the stripped H2O molecules reacts with Cl− to generate volatile HCl(g). For fluoride salt purification, HF in excess is passed through the system, again reacting with residual OH− and H2O.95 In the laboratory, these gases are removed by bubbling them through NaOH(aq) traps; however, the processing of salt for the reactor prior to irradiation will not generate iodine.
Many of the passive substrates utilized in the current generations of advanced off-gas sorbents for halide capture are silica based, which would be readily attacked by HF vapors, specifically. However, CaCO3 or Na2CO3 are possible halide traps, as is soda lime. The I2(g), ICl(g), and IFx(g) (x = 5 or 7) species should also be sorbed along with the residual acidic gases.96 Innovative ways of removing halides include counterflowing liquid systems or a metal filter/trap such as Cu for scrubbing Cl2(g) (as CuCl2) and I2(g) (as CuI).97 Sorbents, such as a formaldehyde polymerizer, have been used to absorb HF residue in shielded facilities.98 However, these sorbents are unlikely to be robust towards radiolysis and the solid cake that forms may plug an off-gas filter; thus, these may be inappropriate for such an off-gas system.
In early work looking at silver zeolites, researchers noted that many were not acid resistant. One material in particular that worked well for high-acid streams was Norton Zeolon,10 which was a large-port mordenite (LPM), defined by the effective aperture (0.8 nm),99 loaded with Ag (AgZ). When Norton Company went out of business, IONEX Corporation purchased the LPM inventory, but now that this stock has been depleted and other natural feedstocks for these materials have been difficult to locate (as of 2024).100 Thus, it is likely that current acid-resistant mordenites will require replacement with a new material (e.g., a new zeolite), unless additional natural stores are located.100 This shortage of the commercial AgZ supply was one of the drivers for this paper: to help identify the necessary boxes to check when designing a new iodine sorbent from scratch.
3. General sorbent requirements
As mentioned in Fig. 1b, several requirements must be met for implementing these different capture technologies and Table 1 provides a summary of these criteria. The criteria fall into wide ranges of subcategories, including stability (i.e., radiation, mechanical/robustness, thermal, chemical), performance (e.g., capacity, reactivity, regeneration ability, pressure drop), physical properties (e.g., specific surface area or SSA, specific heat capacity, thermal conductivity, bulk density), and ease of implementation (e.g., process complexity, energy consumption, environmental safety and health concerns associated with the sorbent, and commercial availability). Additional criteria not included in Fig. 1 but discussed in previous reviews72,101 are listed Table 1 and described briefly within this section.| Category | Propertya | Abbrev. | Property unit | Desired trend |
|---|---|---|---|---|
| a TRL denotes technology readiness level; DF denotes decontamination factor; reactivity denotes the sorbent reacting with the analyte but also with other contaminants in the stream. b This pertains to regulations for storage, handling, disposal, and implementation of material based on Environmental protection Agency (EPA) [e.g., Resource Conservation and Recovery Act or RCRA102 elements including Ag, As, Ba, Cd, Cr, Hg, Pb, and Se] and National Fire Protection Association (NFPA) ratings. c In most conditions, low specific heat capacity would be preferred as materials with high specific heat capacity could result in overheating of (or prevent heat removal from) the sorbent bed. | ||||
| Stability | Radiation stability | S rad | %Drad (in mass%) | High (low %Drad) |
| Mechanical stability/robustness | S mech | %Dmech < 50 μg m−3 | High (low %Dmech) | |
| Thermal stability | S therm | %Dtherm | High (low %Dtherm) | |
| Chemical stability | S Chem | %Dchem | High (low %Dchem) | |
| Performance | Saturation capacity | SC | mol m−3, g g−1, or mg g−1 | High |
| Selectivity for target analyte(s) | S a,b | mole frac., mass frac. | High | |
| DF for sorbent-analyte system | DF | [Ia]/[Ib] (unitless) | High | |
| Reactivity (compatibility) | R | — | Depends | |
| Regeneration ability | — | N reg | High | |
| Co-adsorbed species (potential) | — | mol kg−1 | Low | |
| Flexibility and pretreatment | — | – | High | |
| Pressure drop across bed | P d | Pa m−1 | Low | |
| Bed volume | V bed | m3 | Low | |
| Physical properties | Specific surface area | SSA | m2 g−1 | Medium–High |
| Specific heat capacity | c | J kg−1 K−1 | Dependsc | |
| Thermal conductivity | k | W m−1 K−1 | High | |
| Bulk density | ρ b | kg m−3 | High | |
| Ease of implementation | Process complexity | — | – | Low |
| Energy consumption | — | kW mol−1 | Low | |
| Environmental safety and health | — | Low | ||
| TRL of sorbent system | TRL | — | Medium to High | |
| Commercial availability | — | — | High | |
| Time to commercialization | t c | years | Low | |
| Cost of sorbent material | — | $ kg−1, $ m−3 | Low | |
| Operating cost | — | $ Ci−1 | Low | |
3.1. Sorbent stability
The stability of the sorbent system is essential to consider from several vantage points including radiation stability (Srad), mechanical stability (Smech; robustness and resistance to particle attrition), thermal stability (Stherm), and chemical stability (Schem), all of which can be quantified as the percentage of degradation (%Di) of the “i”-th property over increased sorbent exposure time, i.e., %Drad, %Dmech, %Dtherm, and %Dchem, respectively. Increased radiation exposure is expected as a sorbent bed is implemented for longer and longer times in a facility, and degradation could introduce unwanted changes into the sorbent compromising performance.The mechanical stability can be quantified as the amounts of fines generated in the sorbent bed system, i.e., <420 μm with losses to the off-gas stream of <50 μg m−3.72 Mechanical attrition of the sorbent bed can be caused by several things including high volumes of gas entering the sorbent bed (causing high or variable Pd), vibrations within the sorbent bed, volumetric (density) changes between the unloaded and loaded sorbent, and unexpected fines entering into the sorbents. One of the biggest trade-offs regarding sorbent mechanical integrity is increased particle attrition with increased SSA where more porous sorbents can be less mechanically robust. The chemical stability can vary significantly for a given sorbent in different conditions (e.g., oxidizing streams, acidic streams) where performance drops can be observed after sorbent aging.20,25,74,102–104 Several of these concepts were discussed previously in Section 2 (Target analyte in the off-gas environment).
Thermal stability of the sorbent bed system can be an issue if the MIx compound formed after iodine loading has a low boiling temperature (Tb) near to that of the off-gas capture process (i.e., SnI4) or if the MIx compound is reactive with other materials present in the system. Also, the thermal stability of the base sorbent material, disregarding the active gettering species (e.g., Ag+, Ag0), has to be considered within this category as some scaffolds (e.g., aerogels, xerogels) might collapse at T > 200 °C, where MSR off gas scrubbing systems could be in operation.
One of the most important aspects of chemical stability has to do with the performance of the sorbent in streams contaminated with species that are well known to degrade sorbent performance such as highly oxidizing gases (e.g., NOx) and acidic species (e.g., HNO3, HCl); these concepts were discussed in previous sections (see Section 2, Target analyte in the off-gas environment). Another term to consider within the context of the Schem parameter is the reactivity of the sorbent. However, the difference with this parameter has to do with the measure of the reactivity between the sorbent and the materials of construction as well as other off-gas stream components.
3.2. Sorbent performance
Sorbent performance covers a wide range of different metrics including capacity, selectivity, decontamination factor (DF), reactivity, flexibility, pressure drop, and bed volume. The selectivity concept was described in Section 2.1 (Forms of iodine and selectivity). The concept of sorbent loading is often discussed in terms of the mass of loaded iodine (mI) per mass of initial sorbent (ms) (i.e., mIms−1), usually reported as mg g−1 or g g−1; this is described in more detail in Section 5.2 (Active scaffolds). The DF concept is described in more detail below in Section 8 (Sorbent loading).Another important aspect to consider is the regeneration ability of the sorbents, especially in cases where significant cost savings could be realized. Moving iodine from an Ag-based sorbent over to something less expensive and with higher loading would also likely save on storage costs in a repository. If the sorbent system could be regenerated in place without being removed from the off-gas capture facility, that would also be of interest. In a study by Thomas et al.,19 I2(g) saturation tests were conducted with NaX, AgX, PbX, and CdX to evaluate the maximum iodine concentrations under static loading conditions at 150 °C in sorbent beds with a face velocity of 1 cm s−1. After the loading tests, the sorbent beds were weighed and then purged with air (in the absence of iodine) for up to 120 h at 150 °C until a constant mass was obtained. These results show large variations in the chemisorbed fraction between the different materials with the AgX being the best performer (Table 2). It also shows that the total iodine mass loadings were very similar for AgX, CdX, and NaX. The PbX showed a much lower total loading with nearly all mass gains being attributed to physisorption-type capture processes, whereby iodine was loosely bonded and readily released.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C)19
°C)19
| Sorbent | Saturated (mg g−1) | Physisorbed (mg g−1) | Chemisorbed (mg g−1) | ρ d (g cm−3) | AE100°C (m%) |
|---|---|---|---|---|---|
| AgX | 349 | 135 | 214 | 0.85 | — |
| CdX | 374 | 329 | 45 | 0.71 | 99.992% |
| CuX | — | — | — | — | 4.96% |
| NaX | 364 | 334 | 30 | 0.85 | 80.69% |
| PbX | 179 | 153 | 26 | 0.61 | 56.88% |
| ZnX | — | — | — | — | 4.17% |
In the study by Thomas et al.,19 the PbX was demonstrated as an ineffective sorbent for I2(g), but it was evaluated as a sorbent for HI(g) that was removed from AgZ through a 100% H2(g) purge at 500 °C and captured on PbX at an optimal temperature of 100–150 °C.10 The reactions are shown below in eqn (3) and (4) and provide several advantages to using AgZ as the sole sorbent: (1) it allows for utilization of Ag-based sorbents for the primary capture process, which perform well in acidic environments, (2) it allows for final storage on a less expensive (i.e., Pb vs. Ag) and higher loaded (i.e., 317–408 mg g−1 on PbX vs. ∼113–137 mg g−1 on AgZ) sorbent with good chemical durability (see Section 9, Waste form options), and (3) it provides an opportunity for AgZ recycle.10,19,105 These steps could minimize the overall waste requiring disposal. The utilization of an Ag-based primary sorbent is ideal because Ag is well known to tightly chemisorb iodine and the AgZ zeolite sorbent does well in acidic gas conditions.10 The preliminary design of this bed is to have parallel sets of AgZ + PbX beds where one bed set could be regenerated while the other one was in use. In this process, which is shown in Fig. 3, chemisorbed I (i.e., as AgI) would come off as HI and react with the PbX to form PbI2. Also, the kinetics of iodine removal from AgZ was noted as being 10× faster than the iodine capture kinetics from typical dissolver off-gas conditions. The authors noted the improved effect of I2(g) capture when the Ag was reduced from the as-made material to Ag0 from 69 mg g−1 to 124 ± 17 mg g−1.10 The regeneration process for Ag0Z showed good I2(g) capture afterwards for the first eight cycles (117–140 mg g−1) with performance drops after the eighth cycle (90–100 mg g−1).
| 2 AgI + H2(g) → 2 HI(g) + 2 Ag0 | (3) |
| PbO + 2 HI(g) → PbI2 + H2O | (4) |
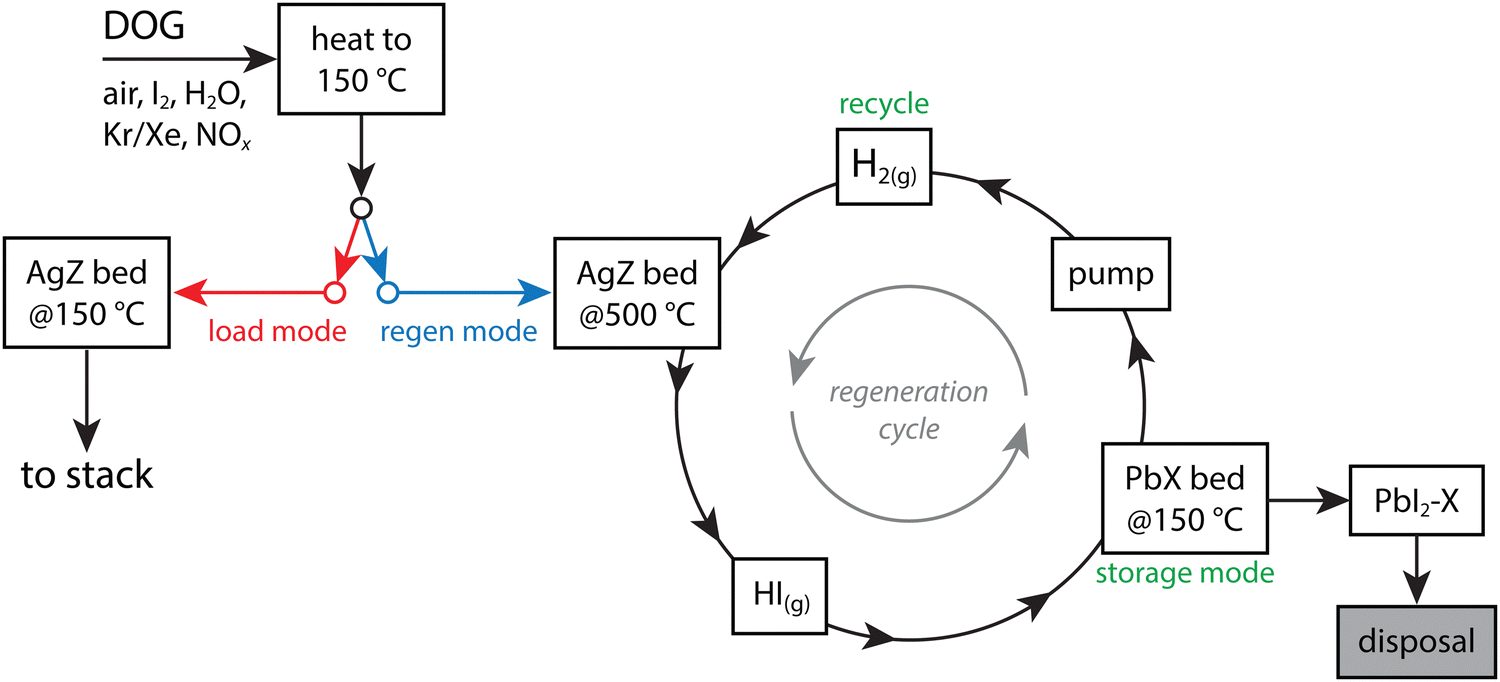 | ||
| Fig. 3 Process flow diagram showing an AgZ + PbX from Thomas et al.19 showing how the process has an I2(g) load mode and a regeneration mode where the iodine-loaded AgZ bed is heated under an H2(g) purge, HI(g) comes off and reacts with a PbX bed to form PbI2 [see eqn (4)] for disposal, and recycling the regenerated AgZ bed. This drawing was modified from the original by Thomas et al.19 | ||
In a study by Maeck et al.,16 a comparison between AgX and PbX showed significant changes in the adsorption efficiencies of elemental iodine at a function of bed temperature. While PbX was not effective at capturing CH3I, experimental results showed that 97% of the I2(g) was captured with PbX at relative humidities (RH) up to 90% when beds were at elevated temperatures tested up to 60 °C (see Fig. 4). The higher the bed temperatures, the better the PbX performed, and this was attributed to the increase in the reaction rate rather than the reduction in the RH within the packed bed. The proposition of the authors was to apply their gained knowledge to implement a multi-sorbent bed where Ag-zeolites could be used to capture organic iodide (e.g., CH3I) and the less expensive option of Pb-zeolite could be used for capture of the elemental iodide, as described above.
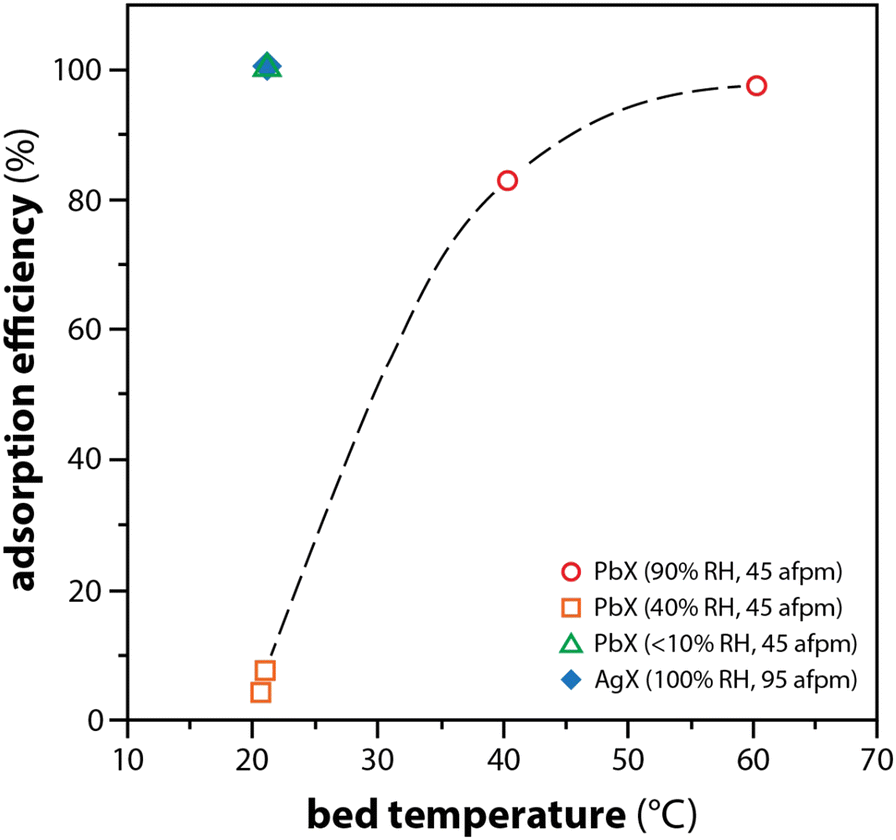 | ||
| Fig. 4 Comparison of I2(g) adsorption efficiency as a function of temperature for PbX and Ag. AFPM refers to actual feet per minute (face velocity). This graphic was modified from the original by Maeck and Pence16 and Staples et al.106 | ||
3.3. Physical properties
The SSA of the sorbent is typically reported in m2 g−1 using nitrogen adsorption/desorption isotherms using the Brunauer–Emmett–Teller method.107 Determining the true SSA value of a sorbent requires that the sorbent be degassed before the measurement, and the optimal time and temperature for the degassing process varies widely from sorbent to sorbent.The specific heat capacity (c, in J kg−1 K−1) is important to consider based on the heat of reaction during the sorbent-loading process. The heat of reaction for iodine capture should be negligible regardless of parameters like c and k (see Table 1) unless sorption/desorption of some other species (e.g., H2O) are present at much higher concentrations, e.g., in the dissolver off gas (DOG; discussed in Section 8 on Sorbent loading). Materials with high c values do not dissipate heat quickly whereas those with low c values can. Thus, if the heat of reaction generated during sorbent loading generates a lot of heat due to exothermic reactions, it is possible that sorbent systems that have high c values could further increase the sorbent bed temperature, thus causing unwanted changes to the sorbent system. While these effects might be miniscule at small sorbent bed volumes, in an actual facility, bed volumes could be very large compounding the issue further. The k (in W m−1 K−1) is also an important consideration that goes along with c. Higher k values in the loaded material will allow for the reaction heat to be dissipated faster.
The bulk density (ρb) is of interest because higher-ρb sorbents of equivalent size and shape to lower-ρb sorbents occupy less volume in an off-gas sorbent column (with equivalent SSA and SC and other parameters being equal). Thus, ρb needs to be considered both for sorbent bed implementation and the storage volume of the loaded sorbent material.
3.4. Ease of implementation
Some solid sorbent materials are already available commercially in large volumes (e.g., silver zeolites, activated carbons) or engineered forms of the precursor materials can be purchased in large volumes for subsequent functionalization (e.g., molecular sieves). On the other end of the spectrum, most sorbent options require a custom synthesis process where base materials are not available commercially [e.g., metal-exchanged aluminosilicate xerogels,21 polyacrylonitrile (PAN) composites108,109]. However, it is possible that some of the more complicated and/or expensive sorbents to produce will show advantages that justify the added requirement of longer lead times and higher fabrication costs. In this regard, additional functionalizations can be added to the sorbent to further improve sorption performance such as acid-resistant or reduction–oxidation (redox)-buffers such as thiol (–SH) groups (see Section 7, Sorbent functionalization).24,25 It should also be noted here that some applications will require that certain elements not be included in the sorbent to prevent complications during disposal in the U.S. as controlled by the Environmental Protection Agency through the Resource Conservation and Recovery Act (RCRA),110 which includes Ag, As, Ba, Cd, Cr, Hg, Pb, and Se.The commercial availability, time to commercialization (tc), cost of sorbent material, and the operating cost of the technology all need to be considered as well. Commercial iodine sorbents are available for various applications and range in cost, particle (or granule) size, volume availability, kinetics of adsorption, environmental stability, temperatures stability, and mechanical stability. In contrast with this, technology readiness levels (TRL) remain low to very low for most sorbent options. In cases where sorbents are commercially available, sorbent cost (i.e., $ kg−1 of sorbent, $ m−3 of sorbent) and the volume availabilities can vary by several orders of magnitude. Materials that include precious metals (e.g., Ag-based sorbents) tend to cost more than those that do not, and the getter loading can also influence the cost. In cases where sorbent technologies appear very promising, it is possible to commercialize the materials, but tc can vary significantly depending upon several factors. The operating cost of the sorbent technology could include a range of things, including the operating lifetime of the bed (how often it needs to be replaced) and how tight the operating parameter tolerances are for the material (e.g., temperature, competing species).
The TRL level of the sorbent technology is of utmost importance when implementing within a nuclear facility.111 The TRL chart is shown in Fig. 5 and ranges from TRL-1 (low) to TRL-9 (maximum). Prior to implementation in a nuclear facility, certain metrics are required to be met that include demonstration at different scales (i.e., bench-scale → pilot-scale → full-scale). The cost for each subsequently larger-scale demonstration can increase exponentially and is likely the factor limiting TRL jumps for emerging and promising new technologies.
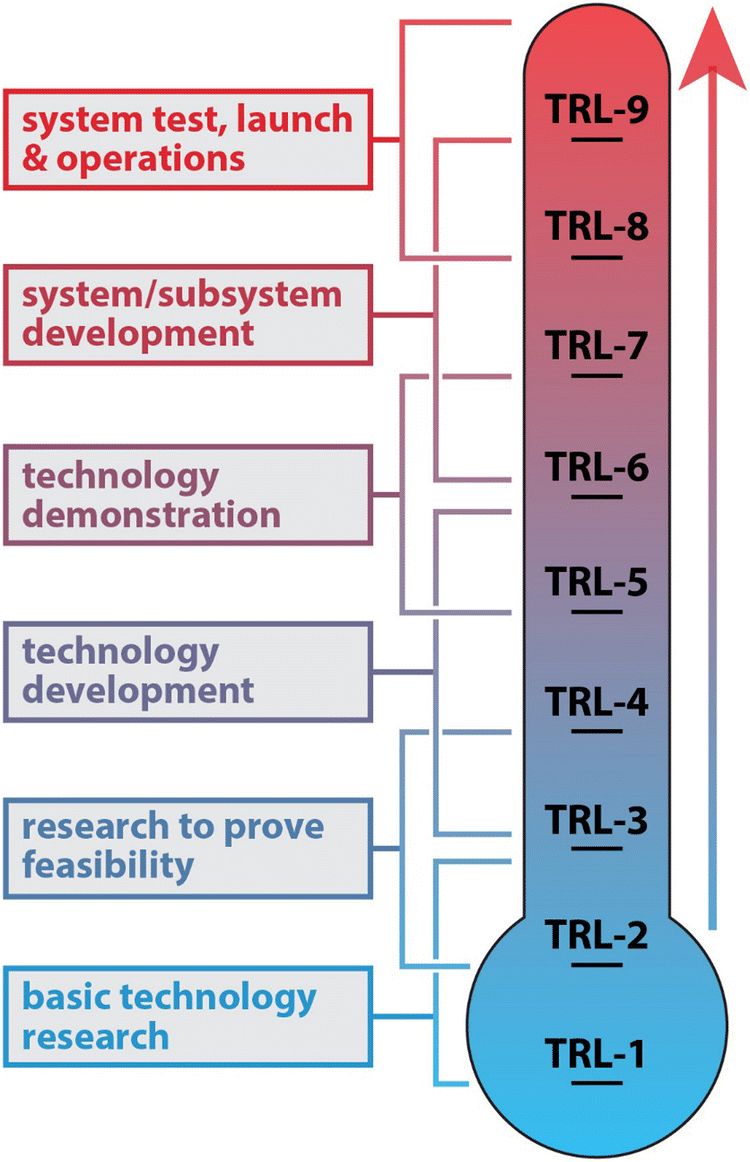 | ||
| Fig. 5 Technology readiness level (TRL) diagram.112,113 | ||
Additional considerations include sorbent robustness, flexibility and pretreatment, process complexity, energy consumption, and environmental safety and health. The robustness pertains to the variation in operating parameters tolerated without deleterious effects or might be better defined as the tolerance of the sorbent technology to upset process conditions. The flexibility and pretreatment aspects pertain to the range of processing conditions where the technology can be implemented when a wider operating envelope is ideal and less stringent product requirements are required (e.g., pre-purification, narrow temperature operation ranges, changes to the sorbent under varying process conditions). The process complexity pertains to the number and type of control systems and unit operations required to implement the technology, as well as the effects of cost, volume, and footprint of the treatment system on the overall cost of the technology. The energy consumption of system implementation should ideally be as low as reasonably possible. The environmental safety and health impacts of a sorption technology can include many different factors such as the toxicity of sorbent system or production of hazardous chemicals from implementation of the sorbent. Several of the more promising iodine gettering technologies proposed include the use of elements (e.g., Ag, Pb) or compounds (e.g., AgI, PbI2)19 that have disposal restriction requirements in the U.S. EPA under RCRA (40 CFR 261),110 which was discussed earlier within this section.
4. Getter selection
As mentioned in Fig. 1c, several aspects to getter selection must be considered when designing a sorbent for iodine that include the need for chemisorption, physisorption (for easier desorption), or a combination thereof as well as which metals are desired (for various reasons). After the getter and the support are selected, the method of adding the getter to the support is not a trivial process and requires some engineering. While some commercially available Ag-containing sorbents require ion exchange processes (e.g., IONEX Ag-400 AgX),114 others are likely produced using easier methods, including soaking the porous substrate in Ag-containing solutions (such as aqueous AgNO3 solution) and then drying, e.g., Ag-loaded silica (e.g., Ag-KTB, Ag-KTC)10,15,19,115,116 and Ag-loaded alumina (i.e., Clariant AC-6120).15,37,117,118 It is plausible to utilize similar processes to load these same base supports with different metals using different metal salts. However, for sorbents where the getter metal is incorporated into the sorbent structure through a process of ion exchange and/or surface functionalization, the overall process can quickly become more time consuming, more costly, and generate more waste. Examples of this are preparing metal-exchanged zeolites or loading metal nanoparticles onto porous and passive substrates,21,22,55,119,120 which can require several steps.4.1. Thermodynamics considerations and general properties of MIx compounds
For an MIx compound to form under the conditions of the desired application(s), the thermodynamic requirements must be met including a favorable Gibbs free energy of formation (ΔGf) in comparison to the formation of alternative compounds [e.g., metal oxides (MOy), other M-halides]. Considering MIx compounds that show spontaneous formation probabilities (i.e., ΔGf < 0), the formations of the equivalent MOy complexes are often even more spontaneous (i.e., ΔGf,MOy < ΔGf,MIx) under the relevant conditions and, actually, very few elements show the opposite (i.e., ΔGf,MOy > ΔGf,MIx) as summarized in Fig. 6. Several that do show the relationship of ΔGf,MOy > ΔGf,MIx, even if only at some temperatures in the range of 25–500 °C, include many that would not be plausible to implement in an actual capture process: lanthanides (i.e., Eu2+) due to costs, alkalis (i.e., Na+, K+, Rb+, Cs+) and alkaline earths (i.e., Ba2+, Sr2+) due to reactivity and water solubility of MIx compound, hazardous metals (e.g., Ag+, Ba2+, Hg+, Hg2+, Pb2+, Tl+) due to disposal restrictions, and precious metals (e.g., Pd2+, Pt2+, Pt4+) due to costs.121,122 Even though the formation of a given MIx compound might be spontaneous, it is unclear whether or not this would form in the presence of oxygen in the stream. It is possible that the MIx compound could form through modification of the capture environment. Also, just because an element is hazardous, does not preclude it from being considered if the MIx compound (e.g., PbI2)10 passes the toxicity characteristic leaching procedure.123 Finally, the formation of an MOxIy compound (e.g., AgIO3, BiOI) might also be favored after initial capture as MIx due to long-term stability, which is discussed more in Section 4.2 (Physical and chemical properties).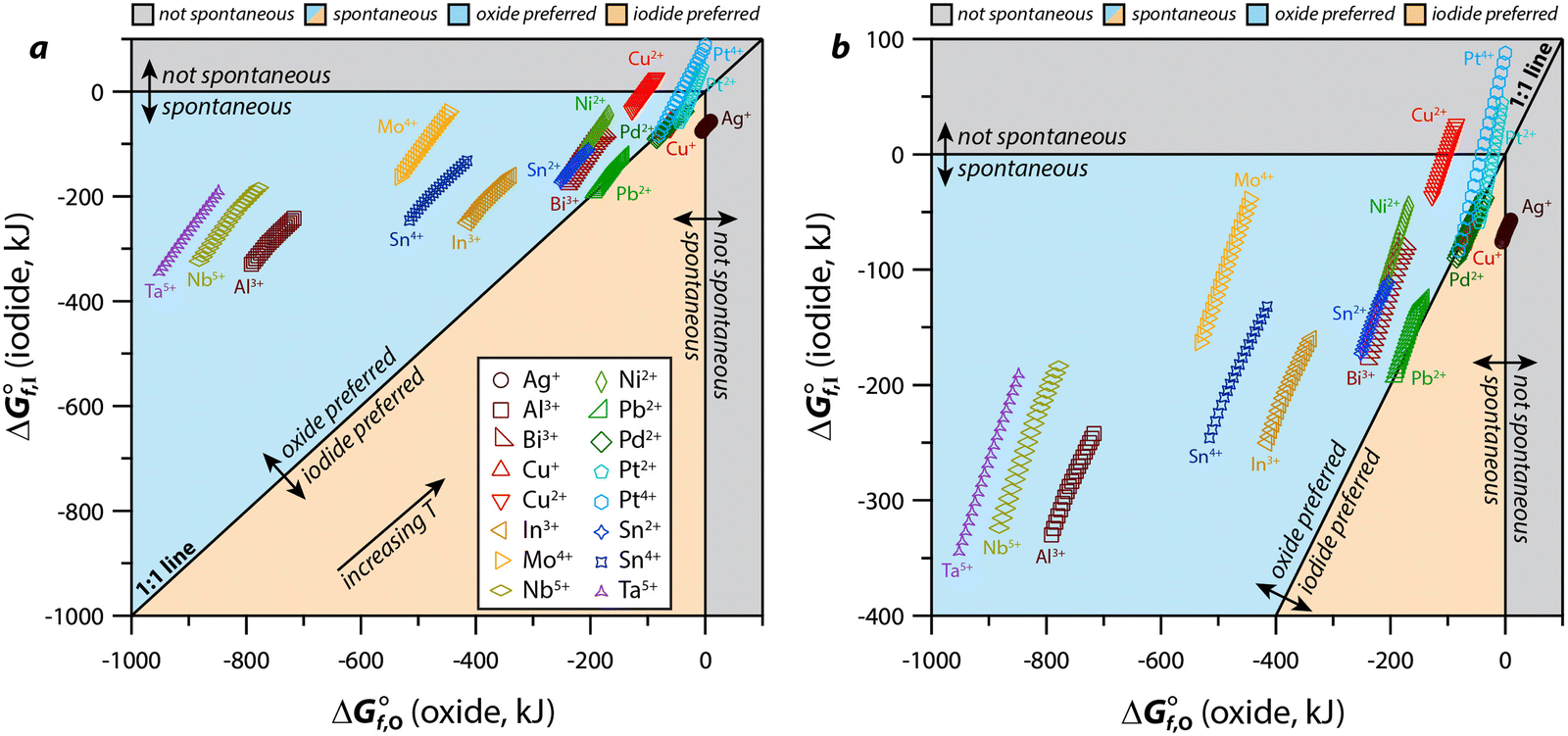 | ||
| Fig. 6 Comparison of Gibbs free energy of formation for metal oxides (ΔG°f,o) and metal iodides (ΔG°f,I) based on thermodynamic data from HSC Chemistry including the (a) the full data range and (b) the magnified data range based on where the data fall. The oxidation states chosen for this graphical representation are based off those available in the software for the base metal of interest. The 45° line represents the boundary between the preference of formation of the oxide over the iodide (above the line) or formation of the iodide over the oxide (below the line); these preferences are based on a lower Gibbs value for one species or the other. For all species, data is presented for multiple temperatures (T = 25–500 °C) where the higher temperatures are upwards and towards the right. These figures were modified from the originals and reprinted with permission from Riley et al.121 and Riley et al.122 Copyright 2020 and 2021 American Chemical Society. | ||
4.2. Physical and chemical properties (unloaded and loaded)
Another aspect to consider for implementing a specific getter metal onto (or into) a support for demonstrating iodine capture includes evaluating the properties of the metal and the intended MIx (or MIxOy) compound that will form under the expected conditions. The main properties of interest in this category include some physical properties of both the metal (M) and the MIx compound, such as the melting temperatures [i.e., Tm(M), Tm(MIx)], boiling temperatures [i.e., Tb(M), Tb(MIx)] (volatility), and hygroscopicity. The list also includes the molar ratio of I:M in the MIx compound (i.e., MIx = xI/M, e.g., TaI5 is 5), which drastically affects the overall waste loading in the final material so it can have a large impact on other properties down the line.
Examples of several MIx compounds (including BiOI) are summarized in Fig. 7 based on the I/Mn+ molar ratio and the iodine concentration in the MIx compound. This graphical representation shows which getters have been evaluated extensively, moderately, and those that are still in need of additional exploration, but they are ordered in terms of the total mass capture capacity based solely on the MIx compound where the addition of a support or scaffold only dilutes the iodine loading potential. The use of metals that have a higher I:M molar ratio means that one could capture more moles of iodine with the same number of moles of metal in comparison to a getter system with a lower I:M value. Those that have been deemed not viable in the past might be viable if sorbent recycling can be implemented (see Section 3.2, Sorbent performance, where dual-bed systems are discussed).19
 | ||
| Fig. 7 Summary of the iodine concentration (mass%) for different MIxOy compounds evaluated in the literature. This figure was modified from the original by Riley and Carlson71 and reprinted with permission. Copyright 2022 Frontiers. | ||
The Tm and Tb values are important properties as they can vary widely across MIx compounds. In some cases, thermal stability of the MIx compound is lower than that of the M alone so formation of the MIx can destabilize the sorbent (e.g., InI3, SnI2, SnI4).122 While the formation of InI3 showed very favorable in recent experiments,122 this compound readily deliquesces in room humidity air. Additionally, the decomposition temperature and Tb of the MIx compound are important because the common approach to iodine waste form production is using some type of hot-pressing process whereby MIx compounds could be broken down resulting in evolution of the iodine present and thus failing at immobilizing the iodine initially captured.
4.3. Other considerations
The many reviews have included discussions on metal-based getters used for gaseous iodine capture.10,50,59,62,65–70,124–126 As discussed above, some getters might show promise based on idealized thermodynamic calculations, however, practical implementation of these materials in an actual sorbent system might be limited to due to commercial feasibility (e.g., availability, cost, hazardousness) and/or performance feasibility (e.g., sustained capture in realistic off-gas environments).65,127 As thermodynamic and kinetics considerations of specific getters have been extensively covered in the other reviews cited above, this section will dive into performance feasibility discussing the use of promising getters in real-world systems.After down-selecting metal getter candidates based on commercial feasibility, the main performance challenge to overcome for many nuclear applications is sustained iodine capture in highly oxidizing conditions (e.g., in NOx) with competing species present [e.g., Cl2(g)]. Sorbents containing silver have been the most widely studied because the formation of a MIx compound is more thermodynamically stable than the corresponding oxide. Historially, AgZ (or Ag0Z) is considered the baseline sorbent for the capture of radioiodine by the U.S. Department of Energy because of its moderately high TRL, good capture efficiency in a wide range of realistic off-gas environments, moderate SSA, commercial availability, and low solubility of AgI in water (after iodine loading). In Europe and Japan, silver nitrate (AgNO3)-impregnated silica and alumina have been successfully used to remove elemental iodide and alkyl iodides from fuel reprocessing streams.10,15,19,37,115–118 These sorbents have been reported to be 3 to 10 times less expensive than zeolite.15 In either case, silver can be easily oxidized by the presence of O2, NO, NO2, and H2O in the environment. If oxygen is present, a silver iodate can form through the following reaction shown in eqn (5).126 For additional discussion on this topic, see Section 2.2 (Highly oxidizing conditions) above.
| 2 Ag0(s) + I2(g) + 1.5 O2(g) → AgI(s) + AgIO3(s) | (5) |
Lu et al.128 reported an interaction between silver oxide nanoparticles with iodine to form β-AgI [eqn (6)]. Wiechert et al.20 conducted aging studies on reduced silver mordenite (Ag0Z) exposed to NO and NO2 environments for up to 6 months. For this material and gas stream, any Ag2O that formed was just an intermediate product on the path to AgNO3 formation. Here, AgNO3 was attributed as a significant contributor to the adsorption of iodine; however, aged samples had <4% I2(g) capture capacity after 6 mo. This was attributed to a series of processes that led to the oxidation of Ag0 and the migration of the resulting ionic Ag into the mordenite channels and exchanging at protonated binding sites (i.e., return to its original state prior to reduction), in combination with the aggregation of residual nitrates within these channels, inhibiting diffusion of I2(g) into the crystal. Similar studies were conducted at Oak Ridge National Laboratory (ORNL).74
| 2 Ag2O(s) + 2 I2(g) → 4 AgI(s) + O2(g) | (6) |
As discussed previously, all reactions are highly dependent upon other species in the gas stream, which can include acidic gases, organic iodides [e.g., CH3(CH2)xI], water, and oxidizing gases, all of which will interfere with the gettering performance of a solid sorbent bed. While isolated studies are critical to understanding the underlying physical and chemical processes, dynamic studies that combine all the potential species in gas streams are still needed to fully understand the behavior of the sorbent in a realistic gas stream. Alternative metals to Ag are actively being explored due to low sustained iodine capture performance of Ag0Z/AgZ in extreme environments (e.g., NOx)129 and because Ag is an expensive metal whose disposition is controlled in the U.S. by the EPA under RCRA (see Table 1).110
Bismuth-based (Bi0/Bi3+) materials are one of the leading candidates to replace silver due to environmental and economic advantages with the additional benefit of having many stable oxyiodide (i.e., BiIxOy) compounds, e.g., BiOI, Bi5O7I, Bi4O5I2. While the formation of BiI3 is desirable due to the high I/M ratio, its high sensitivity to the environment and low thermal stability will likely require an additional step after iodine capture to prevent the release of I2(g) through a conversion to BiOI shown in eqn (7).130 Wagner et al.131 observed the decomposition of BiI3 to BiI or BiOI at room temperature depending upon the environment. No change was observed in the surface of BiI3 stored in an inert gas or in O2. When stored for a few months in a dynamic vacuum, BiI appeared on the surface, while BiOI was found to form after only a few hours in humidity. Rapid decomposition of BiI3 to BiIxOy compounds also occurs between 200–500 °C in eqn (7) and (8).132–134
| 2 BiI3(s) + O2(g) → 2 BiOI + 2 I2(g) | (7) |
| 5 BiIO(s) + O2(g) → Bi5O7I + 2 I2(g) | (8) |
To address the stability issue, Yang et al.57 reacted BiI3 captured by a bismuth-embedded SBA-15 mesoporous silica with Bi2O3 to convert to the chemically durable oxyiodide compound of Bi5O7I. While a reasonable solution at the bench-scale, this process is less feasible when it comes to deployment at an industrial scale due to the cost and complexity of the additional infrastructure required. To eliminate the need for a second step, the reaction to directly produce an oxynitride during capture has been explored. Also, the iodine loading in Bi5O7I is rather low (9.9 mass%).
Baskaran et al.104 used NO2 to examine the effect of the oxide layer on iodine capture capacity of bismuth nanoparticles (Bi-NPs) and bismuth nanoparticle-loaded silica xerogels (TEO-5). Exposure to 1 v/v% NO2 at 150 °C before iodine loading led to the formation of α-Bi2O3 phase or amorphous Bi2O3, which is thought to form through eqn (9) and (10). The bismuth in both sorbents was completely oxidized to Bi2O3 after 6 h; however, the rate of aging in the TEO-5 was slower than the Bi-NPs due to the protective silica matrix (Fig. 8). Shielding of the getter with the matrix using sacrificial coatings or redox-buffers (e.g., functionalization layers) are possibilities to prolong the life of the sorbent, which is discussed in greater detail in Section 7 (Sorbent functionalization). After exposure to iodine, the phases that were present depended on the original concentration of the Bi2O3. If the Bi:Bi2O3 molar ratio was >1, both BiI3 and BiOI were present with BiOI formed through reactions shown in eqn (11) and (12).
| 8 Bi(s) + 6 NO2(g) → 4 Bi2O3 + 3 N2(g) | (9) |
| 2 Bi(s) + 3 NO2(g) → Bi2O3 + 3 NO(g) | (10) |
| Bi2O3(s) + Bi(s) + 1.5 I2(g) → 3 BiOI | (11) |
| BiI3 + Bi2O3 → 3 BiOI | (12) |
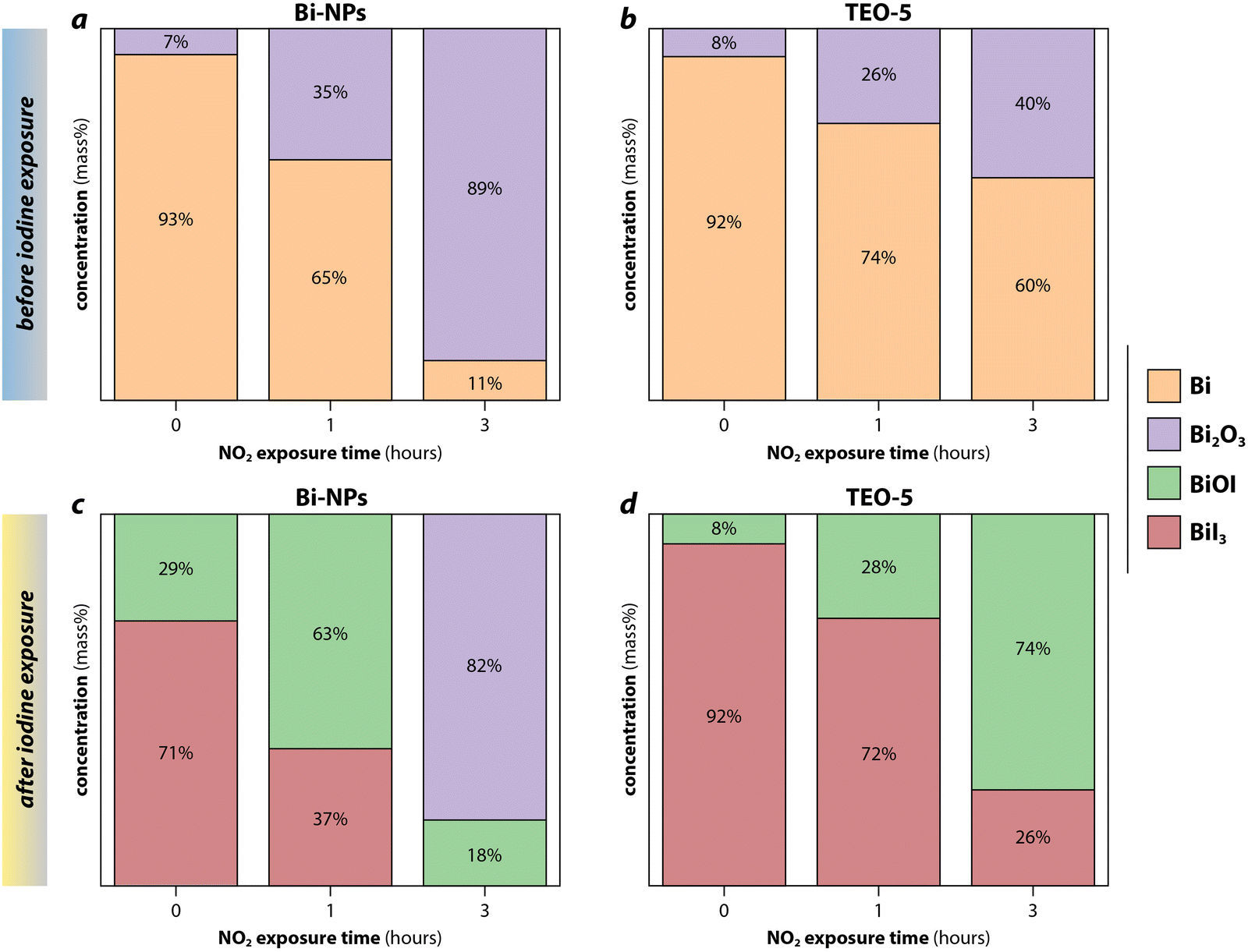 | ||
| Fig. 8 Results from Rietveld refinement applied on X-ray diffraction data of (a) bismuth nanoparticles (Bi-NPs) and (b) silica xerogels (TEO-5) loaded with Bi-NPs after 0 h, 1 h, and 3 h of 1 v/v% NO2 aging, and the same samples after subsequent iodine exposure (c) Bi-NPs and (d) TEO-5. The bismuth-compounds formed after aged samples were exposed to iodine is dependent upon the Bi:Bi2O3 ratio. When Bi2O3 was greater than 50% after aging, as for 3 h shown in (a), only BiOI and Bi2O3 were present after iodine exposure, as shown in (c). For aged samples with less than 50% Bi2O3, BiI3, and BiOI were present as shown in (c) 0-h and 1-h aged Bi-NPs and (d) 0–3 h aged TEO-5. This figure was recreated from Baskaran et al.105 and reprinted with permission. Copyright 2022 Elsevier. | ||
At Bi:Bi2O3 ≪ 1, the remaining Bi was not sufficient to form a bismuth-iodine compound, leaving a high concentration of unreacted Bi2O3.104 Understanding that oxidizing conditions are common in capture environments and when a plant is idling, a secondary aging was performed where iodine loaded samples were exposed to NO2. All BiI3 converted to BiOI and no Bi2O3 was observed; however, for TEO-5, the large volume increase as Bi changed to Bi2O3 and the iodide led to mechanical instability of the sorbent. Therefore, for this process to be feasible, a sorbent with high mechanical resilience must be formed.
Reda et al.135 recently reported on a material that solves both these issues through the formation of a composite of Bi2O3 doped graphitic carbon nitride nanosheets and bismuth-pillared interlayered clay (Bi2O3@g-CNN-PILC). Chemisorption of iodine was suggested to occur through two routes: (1) a direct reaction of I2 with Bi2O3/Bi to produce BiI3 and BiOI, and (2) a Lewis acid–base reaction between the I2 and a lone electron pair associated with the nitrogen species, leading to the formation of polyiodide anions. High sorption values were reported (830–1537 mg g−1); however, these values appear to be from both chemisorption and physisorption (attributed to pore filling). Another highly promising aspect of this material is the potential for regeneration. An ethanol soak was used to release iodine from the sorbent between iodine reloading. While the iodine capture capacity reduced from 830 mg g−1 to 725 mg g−1 after six cycles, it is still much higher than the capture performance of an unaged Bi-impregnated Al/Cu oxide-pillared montmorillonite clay (485 mg g−1),136 Ag-mordenite (170 mg g−1),15 and Bi5@mordenite (538 mg g−1).120 Additional studies on Bi2O3@g-CNN-PILC are warranted to determine the loading as a result of chemisorption, if the material can be tailored for greater chemisorption, and if different regeneration processes can be developed that limit aging and could be easily implemented in a facility.
Copper has received attention as another alternative to Ag for iodine capture and immobilization.52,54–56,108,122 The kinetics of copper-iodine reactions appear to be slower than that of silver-iodine,122 but the formed CuI compound has promising properties (see Fig. 7).53,108,130 Copper has been utilized in iodine capture studies in the form of Cu-exchanged zeolites,55 copper-exchanged aluminosilicate aerogels and xerogels,121 Bi-impregnated Al/Cu oxide-pillared montmorillonite clays,136 Cu2S in PAN beads,109 as well Cu0 in different forms.108,122 Many of these options show high loading potential and the cost of copper implementation is notably lower than that of Ag with the added benefit of the lower environmental toxicity. The main limitation with copper is the likelihood of oxidation or unwanted side reactions in oxidizing environments due to the lower nobility of Cu compared to some other candidate metals like Ag.
Developing sorbents that will overcome these performance and aging challenges is an exciting and worthwhile quest. However, the baseline (or most proven) sorbent technologies are attractive as they tend to be readily available (or easily scaled if not currently mass produced), cost effective, and, in the case of 129I, have the potential to provide long-term immobilization in a chemically durable waste form (e.g., as AgI).130,137 An alternative approach to solving these challenges, is to examine the regeneration and recycling of sorbents using a primary capture bed and a secondary bed for off-loading that iodine onto a different sorbent bed with higher loading capacity (discussed in Section 3.2, Sorbent performance).
5. Scaffold selection
Once the getter is selected, the scaffold (which can also be referred to as the support or substrate) for that getter is of utmost importance. As is mentioned in Fig. 1d, a variety of different options are available for implementation, including zeolites, porous ceramics, aerogels, xerogels, porous polymers, and organic substrates (e.g., MOFs). In general, the key categories to consider, which are summarized below, include the form and general properties of the scaffold, the chemical compatibility, and the ability for the scaffold to be active (actively be used in iodine capture) or passive (it does not capture). Some of the topics covered in this section were also covered briefly in Section 3 (General sorbent requirements).The support has to meet several process requirements, including a form that provides a minimal pressure drop across a sorbent bed (e.g., a packed bed of porous beads), high chemical compatibility with the stream (e.g., it cannot degrade), high mechanical integrity (e.g., it should not undergo particle attrition when in operation), and the ability for getter loading (e.g., passive support with active getter or active support consisting of the getter). The iodine loading in a chemisorption-based sorbent is directly related to the getter loading in the support. While the getter loading should be maximized, there is a threshold maximum before the getter loading is too high to where iodine loading processes will negatively affect the loaded sorbent (e.g., due to sorbent swelling and particle attrition) or where the getter utilization is incomplete138 resulting in higher operating costs. The SSA is also of utmost importance when trying to maintain porosity to allow accessibility of the active sites. The pore structure plays a crucial role as well because pores of different sizes and structures affect the sorption kinetics and capacity.
5.1. Form and general properties of the scaffold
When considering a packed bed assembly for off-gas capture, the pressure drop (Pd) across the bed volume is an important aspect of the system design. The Pd value is based off the restrictive flow influenced on the bed by the sorbent packing density, open pore volume, pore volume accessibility (path tortuosity), and the open volume between the sorbent particles in the bed. Different particle geometries will result in different packing densities, e.g., cylinders/extrudates, beads, berl saddles, irregular particles. The leading forms under consideration in the U.S. for nuclear applications are engineered forms of zeolite-based sorbents that are cylindrical or spherical in shape (e.g., AgZ/Ag0Z, AC-6120). If a granular sorbent is implemented, the open volume between the granules can be calculated based on packing of uniform shapes. Higher Pd values require higher fan energy and higher vacuum downstream, and larger sorbent bed cross-sectional areas (or multiple beds in parallel) if process pressures upstream of the sorbent bed need to be negative.Large variations in reported SSA values for sorbents result from moisture trapped in the material during actual operation, and any physical property changes that occur during the measurements, e.g., pore structure collapse due to thermal stresses. While higher SSA typically means higher sorption, it also tends to reduce mechanical integrity, so this property can have trade-offs.
Combining ρb, k, and the specific heat (Cp), the thermal diffusivity (α) of the material can be calculated using eqn (13) below. This α value combines these different material properties to determine how fast the temperature will change in a material as a function of temperature changes within its surrounding environment. In some instances, similar chemical species might be present in an off-gas stream where the target is iodine, and these unwanted species could be co-adsorbed by the sorbent along with the target analyte. Examples of these species include other halide gases like Cl2(g). Selecting a sorbent that can perform efficiently in the presences of these materials is essential based on the Sa,b values between the target analyte and these competing species (see eqn (2) and Section 2.1, Forms of iodine and selectivity).
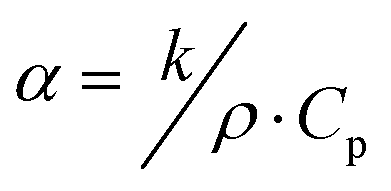 | (13) |
The bed volume metric has to do with the total amount of sorbent material required to effectively capture and immobilize the iodine phase. This metric ties back to other metrics including the saturation capacity (SC) of the sorbent, the sorbent packing density, and the sorbent ρb.
5.2. Active scaffolds
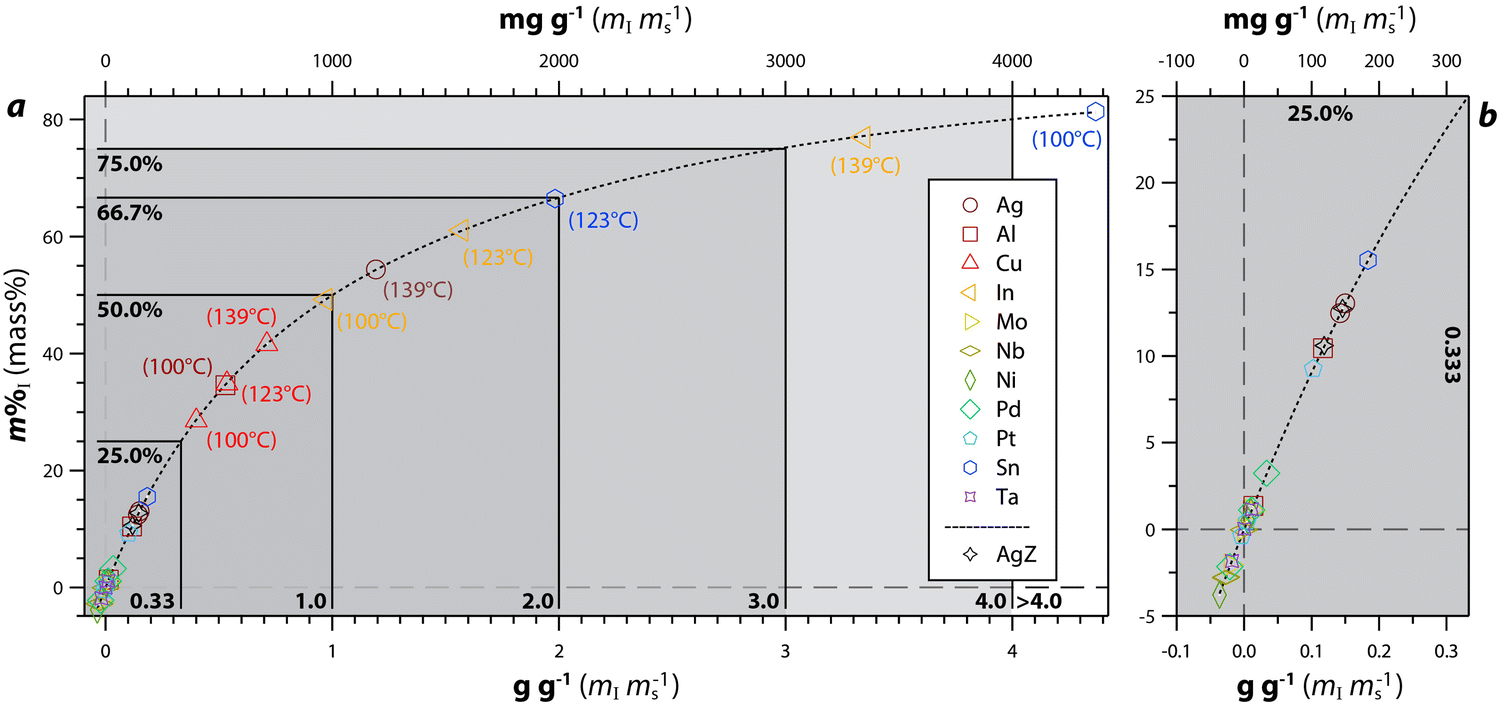 | ||
| Fig. 9 Data for 100 °C, 123 °C, and 139 °C experiments showing measured iodine (i.e., m%I or mass% via EDS) vs. g g−1 and mg g−1 along with a double exponential fit including (a) the full plot and (b) the magnified region of ≤25 m%I and ≤0.33 g g−1 (≤ 333 mg g−1). The highest performing samples are labeled, and the legend is the same for both plots. Reprinted with permission from Riley et al.122 Copyright 2021 American Chemical Society. | ||
5.3. Passive scaffolds
Passive scaffolds are the most heavily studied types of iodine sorbents as they provide many of the desirable characteristics described above. Several different types of passive scaffolds can be utilized to support active getters. The most studied type of passive substrates for radioiodine capture are different types of zeolites but metal-loaded ceramics, aerogels, xerogels, carbons, and metal–organic frameworks are also being developed.:Al molar ratio), unit cell sizes, cation exchange capacities (CEC), acid resistances, and porosities. The aluminosilicate zeolites contain negatively charged frameworks built from AlO45− and SiO44− tetrahedra, which are charge balanced by cations (e.g., H+, Na+) that reside within the cages and channels within these frameworks.10,16,18,19,143,144,148–150
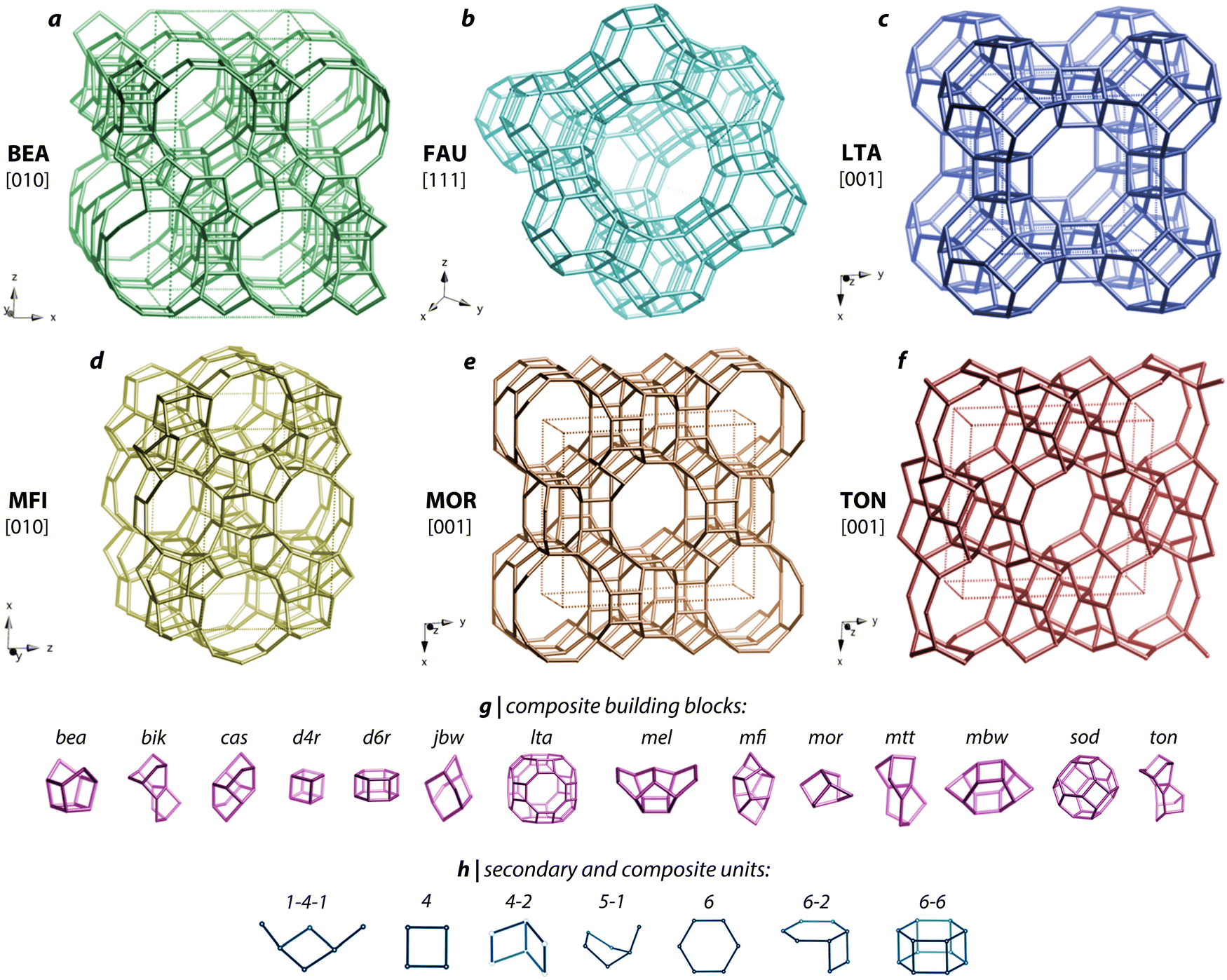 | ||
| Fig. 10 Crystal structure schematics for (a) BEA, (b) FAU, (c) LFA, (d) MFI, (e) MOR, and (f) TON zeolites. The main unit cell structures are shown at the top and (g) composite building blocks as well as (h) secondary and composite units present within these zeolites are shown below the main structures. Reproduced in part with permission from Riley et al.119 Copyright 2022 American Chemical Society. Reproduced in part with permission from Baerlocher et al.151,152 | ||
While several zeolites are available commercially, many are not and require laboratory syntheses to produce. Some of the different chemistries are shown in Fig. 11. The Si:Al molar ratio of MOR is typically 5:1, while the different FAU-type zeolites are classified by lower ratios of Si/Al < 2 for FAU-X and Si/Al >2 for FAU-Y.
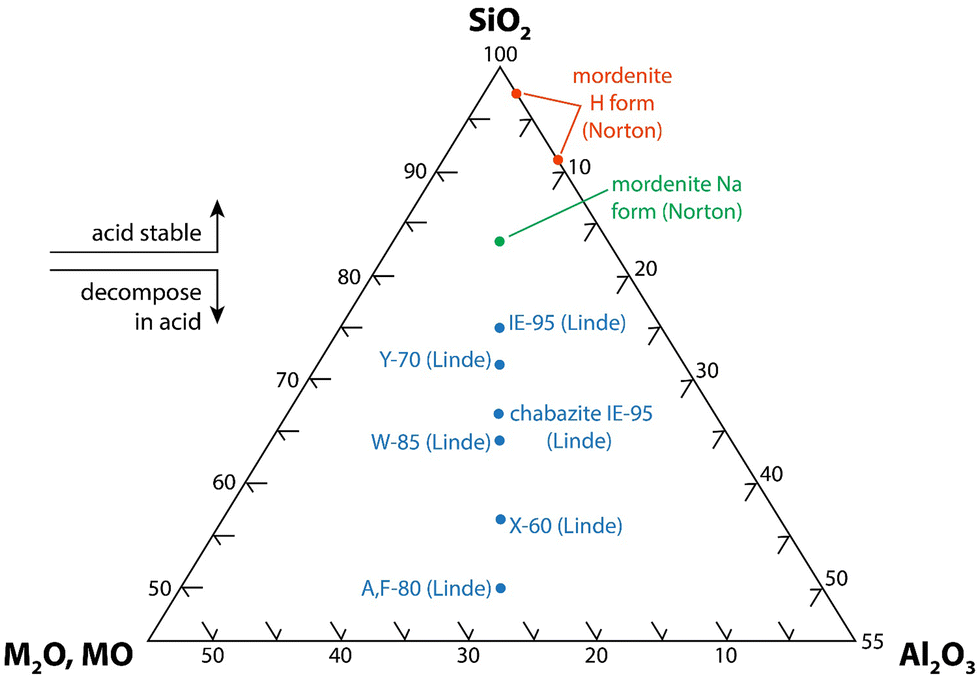 | ||
| Fig. 11 SiO2-Al2O3-MxO (M = alkali or alkaline earth, x = 1 or 2) ternary diagram for zeolite minerals that shows the acid stability8 (this drawing was modified from ORNL drawing 78-15097152). | ||
In a study conducted by Pence et al.,18 several different types of metal-exchanged zeolites were evaluated under similar conditions. Several different materials were considered including Cd, Cu, Hg, Pb, Tl, as well as mixed rare-earth cations through ion exchanging base zeolite materials. Most of these do not perform well under increasing relative humidities, even though many do show high I2(g) capture efficiencies. The results from CuX and PbX experiments are summarized in Table 3. A commercial Y-zeolite loaded with Hg2+ ions (from Union Carbide Corporation and referred to as Hg2+Y) was shown to be the best of the metal-exchanged zeolites tested with >99.9% efficiency under 23 cm s−1 face velocity in a water-saturated atmosphere at 22 °C. They reported that the only metal-exchanged zeolites that showed appreciable capture in experiments with CH3I instead of I2(g) were the Ag-zeolites. The highest performing non-Ag zeolite was Hg2+Y, where an efficiency of 33.8% was documented at 125 °C under 90% RH at 23 cm s−1 face velocity. In a separate study reported by Staples et al.,106 5-cm deep beds were packed with metal-exchanged faujasite zeolites including CdX, CuX, NaX, PbX, and ZnX, and I2(g) capture efficiencies were evaluated at different bed temperatures (i.e., 100 °C or 200 °C) and different RH values (i.e., 1.5% or 8.5%); these data are also summarized in Table 3. For these tests, experimental details included a 1.4 mg I2 g−1 substrate loading at 99% efficiency, 90 mg I2 m−3 airborne concentration, a 60-min pretest purge, a 60-min test period, and a 15-min post-test purge. From this study, the effects of humidity and temperature on the I2(g) adsorption efficiencies are very apparent.
| Material | c I (mg m−3) | T (°C) | V f (cm s−1) | RH (%) | AET (%) | Ref. |
|---|---|---|---|---|---|---|
| CuX | 5 | 22 | 23 | 98+ | 47.2 | 18 |
| CuX | 33 | 90 | 46 | 98+ | 54.0 | 18 |
| CuX | 75 | 90 | 23 | 98+ | 59.1 | 18 |
| CuX | 70 | 90 | 23 | <5 | 99.1 | 18 |
| PbX | 5 | 22 | 23 | 98+ | 35.7 | 18 |
| PbX | 45 | 22 | 23 | <5 | 99.9+ | 18 |
| PbX | 56 | 22 | 48 | 98+ | 5.2 | 18 |
| PbX | 25 | 90 | 46 | 98+ | 51.2 | 18 |
| PbX | 58 | 90 | 23 | 98+ | 67.2 | 18 |
| PbX | 69 | 90 | 23 | <5 | 99.9+ | 18 |
| CdX | 90 | 100 | 25 | 1.5 | 99.986 | 106 |
| CdX | 90 | 100 | 25 | 8.5 | 99.992 | 106 |
| CdX | 90 | 200 | 25 | 1.5 | 99.836 | 106 |
| CdX | 90 | 200 | 25 | 8.5 | 99.985 | 106 |
| CuX | 90 | 100 | 25 | 1.5 | 31.67 | 106 |
| CuX | 90 | 100 | 25 | 8.5 | 4.96 | 106 |
| CuX | 90 | 200 | 25 | 1.5 | 0.45 | 106 |
| CuX | 90 | 200 | 25 | 8.5 | 0.50 | 106 |
| NaX | 90 | 100 | 25 | 1.5 | 99.86 | 106 |
| NaX | 90 | 100 | 25 | 8.5 | 80.69 | 106 |
| NaX | 90 | 200 | 25 | 1.5 | 99.93 | 106 |
| NaX | 90 | 200 | 25 | 8.5 | 64.39 | 106 |
| PbX | 90 | 100 | 25 | 1.5 | 93.45 | 106 |
| PbX | 90 | 100 | 25 | 8.5 | 56.88 | 106 |
| PbX | 90 | 200 | 25 | 1.5 | 88.15 | 106 |
| PbX | 90 | 200 | 25 | 8.5 | 77.61 | 106 |
| ZnX | 90 | 100 | 25 | 1.5 | — | 106 |
| ZnX | 90 | 100 | 25 | 8.5 | 4.17 | 106 |
| ZnX | 90 | 200 | 25 | 1.5 | — | 106 |
| ZnX | 90 | 200 | 25 | 8.5 | 4.15 | 106 |
In a study by Yadav et al.,114 AgX (IONEX Ag-400) and AgZ (IONEX Ag-900) were evaluated for iodine capture and recyclability of Ag. Both were loaded with iodine in static conditions at 150 °C for 24 h. The iodine adsorption capacities of AgX and AgZ were 334 mg g−1 and ∼120 mg g−1, respectively. After iodine capture, the framework structures of AgX and AgZ collapse, and the SSA values were decreased by 91% for AgX and 58% for AgZ. Formation of AgI was observed for both AgX and AgZ, and the iodine adsorption capacities were directly related to the initial Ag amounts in the sorbents.
Aside from carbon-based COFs, MOFs, and POPs, the activated carbon class of scaffolds is one of the most studied sorbent materials after zeolites due to ease of preparation in many forms (e.g., pellets, fibers, foam), good chemical stability, decent thermal stability, high SSA values, and (oftentimes) very low cost. Additionally, the pore structure of these materials is easily tunable through variations in the carbonization methods. The supports from activated carbon are easily made from naturally occurring carbonaceous materials and can trap iodine via physisorption due to weak van der Waals forces.15,50,67,153,154,156 Oxygen-rich micropores were reported to greatly enhance the adsorption capacity of iodine on carbon derived from cellulose diacetate using hydrothermal carbonization followed by KOH activation.157 While physisorption performance typically decreased at high temperatures and is negatively affected by the presence of water and NOx,15 the presence of oxygen-rich structures (i.e., C24O12H6, C24O15) mitigated these effects.157 As the carbon itself only interacts with the iodine through physisorption, surface functionalization (see Section 7, Sorbent functionalization) must be performed to enable chemisorption. While only physisorption is undesirable, it has the potential to enhance chemisorption for getters with lower iodine capture kinetics if the carbon matrix acts as a concentrator.
Yang et al.63 explored how the density, adsorption coefficient, pore volume, and structure impacted the iodine sorption of activated carbon fibers (ACFs). Commercially available ACFs underwent treatments with KOH (0.1% KOH solution at 40 °C for 24 h) and/or were microwaved (385 W for 1 min) to alter the pore structure of the parent material. After modification with KOH (ACF-K), the SSA values increased from 1253 m2 g−1 to 1363 m2 g−1 and the maximum saturated adsorption of iodine increased from 1675 m2 g−1 to 1803 m2 g−1. Interestingly, the microwave treatment (ACF-W) had a surface area of only 1286 m2 g−1 but the highest iodine adsorption of 1958 m2 g−1. While the overall pore volume and volume of micropores of the ACF-W was lower than the ACF-K, the rate of adsorption and initial desorption was faster, indicating that the pore structure played a significant role in the iodine adsorption kinetics.
A challenge with many of these microporous carbon sorbents is their low mechanical stability, which can lead to the release of particulate matter containing iodine during operation. One solution to this challenge is to carbonize a mechanically stable monolith. The use of a polycondensate containing melamine (C3H6N6), formaldehyde (CH2O), and polyoxymethylene ([CH2O]n) with an antioxidation agent foam has been reported to produce a nonfriable, carbon monolith.158,159 Although it shows a lower SSA than other carbon-based sorbents, it is minimally friable and can be easily functionalized using electrodeposition or hydrothermal methods.
6. Getter addition
As mentioned in Fig. 1e, getters can be added to scaffolds using a variety of approaches, including as colloidal (or nano) particles, as bulk metal particles, through ion exchange, added as aqueous salts that are dried within the scaffold, as well as other methods. Several of these are described below in different capacities.6.1. Metal-loaded silica and alumina
Ding et al.161 studied Bi0 loaded silica with different morphologies, including plates, rods, and spheres that were investigated for iodine capture at 75 °C, 130 °C, and 200 °C. The molar ratio of Bi0 to silica was ∼0.3. Exposing Bi0-loaded silica to gaseous iodine resulted in the formation of BiI3 by chemisorption, and physisorption was more dependent upon the morphologies. The plate form of Bi0-loaded silica showed the highest adsorption capacity of 960 mg g−1. Adsorption at higher temperatures lowered the total iodine loading but increased the ratio of chemisorption to physisorption.Reda et al.136 studied Bi-impregnated AlCu oxide-pillared montmorillonite that was synthesized and evaluated for iodine capture. The Bi loadings of 5 mass%, 10 mass%, or 20 mass% were produced using Bi(NO3)3·5H2O solution, and the resulting AlCu oxide pillars showed mostly Bi with a small amount of Bi2O3. The iodine adsorption was conducted at 75 °C for 48 h. The highest adsorption capacity of 485 mg g−1 was observed for the 20% Bi-impregnated AlCu oxide pillars. Both BiI3 and BiOI phases were present in the iodine captured AlCu oxide pillars.
Wilhelm and Schuttelkopf162,163 investigated the iodine removal efficiencies of AC-6120, Ag-KTB, Ag-KTC, JFM1 using commercial products by Süd-Chemie in Germany; as mentioned above, some of these appear to be AgNO3-impregnated. The main differences in these materials were the Ag contents, which ranged from 6–12 mass% (g Ag per g of sorbent). Depending upon the sorbent bed depth and residence time, >99.99% of iodine removal efficiency for Ag-KTC under standard conditions was observed.162 Iodine loading of 40 mg g−1 I2 or CH3I for Ag-KTB (Ag-impregnated silica gel) was obtained.163 In a study by Kikuchi et al.,164 the removal efficiencies of methyl iodide (CH3I) using Ag-impregnated alumina and zeolite were evaluated as a functions of the Ag-loading, and RH. At 90% RH, alumina with 10 mass% Ag showed a removal efficiency of >95%. The results demonstrated that Ag-impregnated alumina was more efficient for iodine removal even with a smaller amount of Ag loading compared to the Ag-impregnated zeolite.
6.2. Metal-loaded zeolites
Getters are loaded into zeolites through ion exchange from parent materials (e.g., molecular sieves). Additionally, other processes could be used such as functionalization of the zeolite to add additional getters to the exterior surfaces of the sorbent. Zeolites have been discussed throughout various sections within the paper and will not be elaborated upon in this subsection.6.3. Metal-loaded aerogel or xerogel substrates
Different scaffold systems in the form of aerogels and xerogels (broadly referred to as “gels”) have been evaluated in this area with different metals loaded. The scaffolds evaluated include silica aerogels and aluminosilicate aerogels and xerogels. Matyas et al.25 investigated the aging effect on iodine capture for the silver-functionalized silica aerogel (Ag0-aerogel) using H2O and NOx at 150 °C. To load the Ag+ onto the silica aerogel, the ions were tethered to thiol groups (see Section 7, Sorbent functionalization) and then reduced to Ag0 under a hydrogen atmosphere to create Ag0-aerogel. Exposure of Ag0-aerogel to water and NOx gas resulted in oxidation of thiol to sulfate and growth of silver particles, and these results reduced the iodine sorption capacity of Ag0-aerogel. Compared to AgZ, Ag0-aerogel was more resistant to H2O and NOx gas. One hypothesized solution to minimize the aging issue was exposing the Ag0-aerogel to a reducing gas stream for a short period of time to convert sulfate to sulfide.In studies by Riley et al.21,23,121 and Chong et al.,22,165 the authors demonstrated ion-exchangeable sodium aluminosilicate (i.e., nominally NaAlSiO4) aerogels and xerogels for hosting different getter metal ions including Ag+, Cs+, Cu+, Cu2+, Fe3+, K+, Li+, Rb+, Sb3+, Sn2+, and Sn4+ through ion exchanges with Na+ in the as-made gels.121 For ion exchange processes, most of the ions were dissolved in water starting from nitrates or using colloidal SnO2; in addition, three other materials were dissolved in formamide as the solvent, including Sn(II) acetate (for Sn2+), Sn(IV) acetate (for Sn4+), and potassium antimony tartrate (for Sb3+). Following cation exchange, many of the samples showed high exchange rates with reduction in Na+, but the most effective ions were Ag+, Cu2+ from Cu(NO3)2 (not from CuSO4), K+, and Rb+. Iodine uptake experiments performed at 150 °C showed very high loadings for the Sn4+ sample (12bA-Sn4+ + I; 61 mass%), Ag+ (12bA-Ag + I; 42 mass%), Sn2+ (12bA-Sn2+ + I; 41 mass%), and Sb3+ (11bX-Sb + I; 38 mass%) – see Fig. 12.122 In addition to just metal ions, the ions can be reduced through heating in gases such as H2/Ar to generate metal nanoparticles, which was demonstrated for reducing Ag+ to Ag0 in both aluminosilicate aerogels and xerogels generating Ag0 crystals and agglomerates on the order of <10 nm and >20 nm.21,23 A similar approach could be used to generate nanoparticles of other metal ions as well.
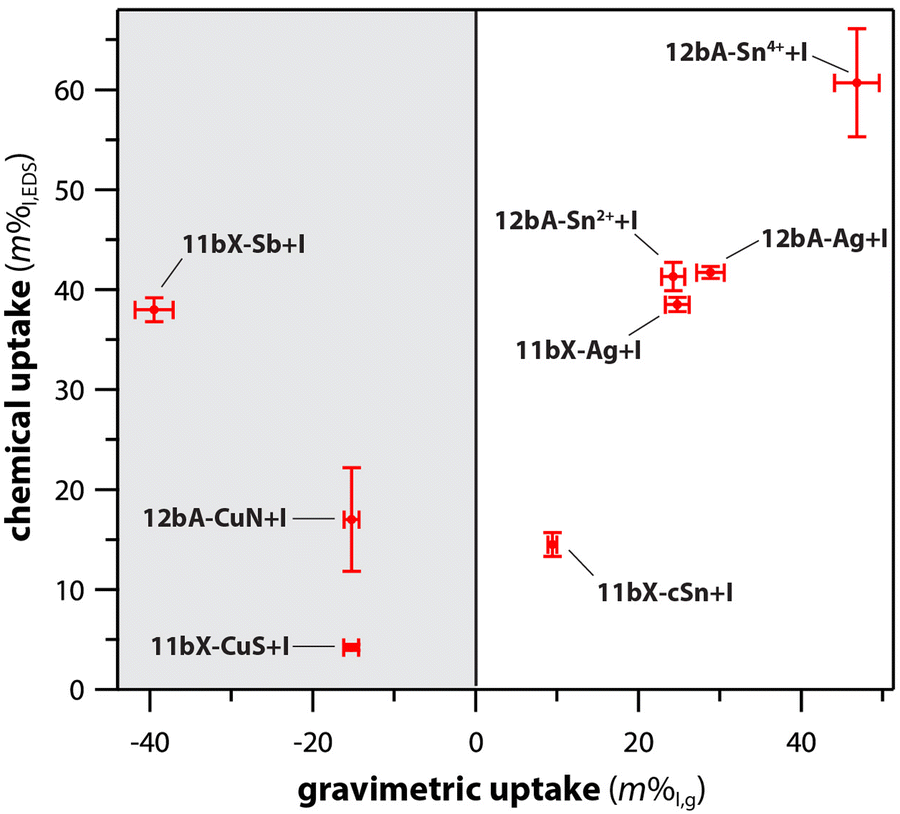 | ||
| Fig. 12 Comparison of chemical uptake (composition measured with EDS) and gravimetric (mass change) for different ion-exchanged aluminosilicate aerogels (#bA-) and xerogels (#bX-) containing Ag, Cu, Sb, and Sn. Reproduced with permission from Riley et al.122 Copyright 2020 American Chemical Society. | ||
6.4. Metal sulfides
A range of metal sulfides (MSx) have been investigated for iodine capture, which was briefly discussed in both Section 5.2.2 (Chalcogel sorbents) and Section 5.3.3 (Polymer-based sorbents). These materials have been evaluated in the form of aerogels, xerogels, and MSx compounds embedded in PAN matrices where iodine reactions can result in the formation of MIx compounds such as BiI3, KI, SbI3, SnI4, and SnI4(S8)2 (see Table 4).140,160,166,167 These materials have an inherent affinity for iodine without the addition of metal getters where all sulfides tested to date show high iodine loadings under a variety of conditions (e.g., temperature, iodine concentration, atmosphere). This inherent iodine affinity is attributed to the chemical interactions between the chalcogen (e.g., S) and the I2(g) molecules, and this is explained by Pearson's Hard Soft Acid Base (HSAB) principle. Based on the HSAB principle, sulfur is characterized as a soft Lewis base with a chemical hardness (ηs) value of 4.12 and I2(g) is classified as a soft Lewis acid with a ηs value of 3.4.168–170 It was postulated168 that Se-based and Te-based chalcogels might show an even greater affinity for I2(g) based on ηs being even closer to that of I2(g), but these would be more expensive, more toxic, and more difficult to produce for a variety of reasons. While chalcogenide aerogels and xerogels (called chalcogels)171 show promise, producing these materials is time consuming and costly, especially since many of the required precursors for synthesis are not commercially available. Thus, the concept of embedding commercially available MSx compounds in a porous matrix (e.g., PAN) is another approach that provides a variety of additional opportunities. This concept has been shown to provide high iodine loadings when evaluated with Ag2S,109 Bi2S3,109,160 Cu2S,109 and SnS2167 active getters in PAN beads (see Table 4).| Type | MSx compound | MIx compound | q e (mg g−1) | T (°C) | Ref. |
|---|---|---|---|---|---|
| Aerogel | Sn2S3 (100%) | SnI4, SnI4(S8)2 | 2155 | 25 | Riley et al.166 |
| Sb4Sn3S12 (100%) | SbI3 | 2000 | 60 | Subrahmanyam et al.140 | |
| Zn2Sn2S6 (100%) | SnI4 | 2250 | 60 | Subrahmanyam et al.140 | |
| KCoSx (100%) | KI | 1600 | 60 | Subrahmanyam et al.140 | |
| PAN composite | Ag2S (90%) | AgI | 826 | 130 | Riley et al.109 |
| Bi2S3 (70%) | BiI3 | 986 | 75 | Yu et al.160 | |
| Bi2S3 (80%) | BiI3 | 909 | 130 | Riley et al.109 | |
| Cu2S (80%) | CuI | 1095 | 130 | Riley et al.109 |
6.5. Organic sorbents
While many iodine capture studies have been done with organic sorbents like MOFs, COFs, and POPs,38–40,45,46,172–174 they are not the focus of the current paper, but this subsection was included for completion within this section. Their pore sizes and pore volumes can be tuned by different building blocks and linking topologies to accommodate iodine molecules at high concentrations utilizing molecular sieving approaches or intermolecular interactions, but chemisorption has also been demonstrated.1756.6. Carbon-based sorbents
This section covers activated carbons, charcoal, carbon nanofibers, and carbon foams. Carbon-based sorbents can be functionalized with organic and inorganic species to enable chemisorption. The choice between the two depends upon the capture environment, iodine species, and disposal pathway after capture.Organic compounds can be used to chemically bind organic iodide species typically through an SN2 reaction.64,176 In this reaction, the nucleophile (species rich in electron density) attacked a tetrahedral (sp3) carbon bearing group (leaving group) to remove an electron pair. The SN2 reaction occurs as the nitrogen atom in the nucleophilic amine 1,4-diazabicyclo[2.2.2]octane (DABCO) has a lone pair of electrons, which can cause heterolytic cleavage of the carbon–iodine bond. Charcoal functionalized with DABCO was used to capture volatile organic iodine compounds relevant for radiological release scenarios (e.g., CH3I, C2H5I, C3H7I, and chloromethyl iodide). DABCO was selected as it could easily convert the organic iodide to a stable compound that does not easily revert and/or leave the pores of the impregnated charcoal. The organic iodine compounds reacted with DABCO as shown in Fig. 13. The sorption mechanism of compounds with greater steric hinderance were slower to react in the SN2 reaction. In addition to the rate at which the organic iodide species is converted to the chemically bound compound, the number of carbon groups attached to the carbon on the leaving group also impacts the retention in the charcoal. Organic iodide species must initially physically adsorb to the charcoal before they are able to chemically react with the DABCO. Compounds C2H5I and C3H7I have higher boiling points than CH3I and are therefore more likely to be more successfully physisorbed than CH3I. These more stable physisorbed precursor states lead to less mobility and longer retention in the charcoal.
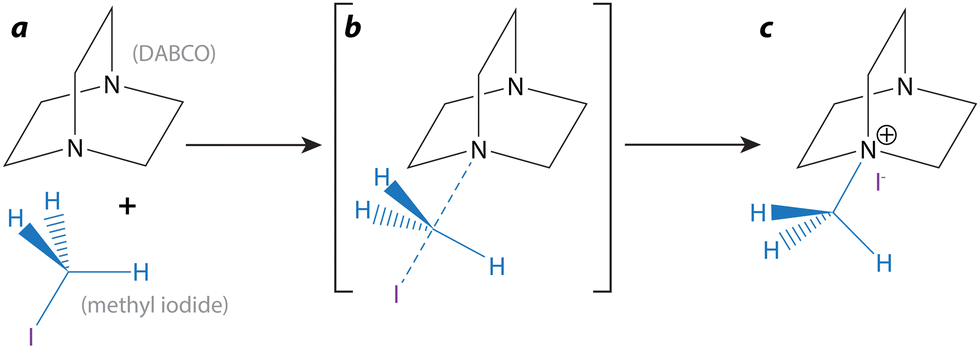 | ||
| Fig. 13 The reaction of CH3I with DABCO showing (a) the initial forms of each, (b) the intermediate bonding environment, and (c) the final form.176 This figure was reprinted with permission from Aneheim et al.176 Copyright 2018 Elsevier. | ||
Tian et al.177 synthesized Bi0-coated, Bi2O3-coated, and Bi0 + Bi2O3-coated carbon nanofibers for iodine capture. Iodine adsorption tests were performed at 200 °C for 4 h, and Bi0-coated carbon nanofiber showed the highest loading of 732 mg g−1, whereas the Bi2O3-coated sample showed the lowest loading of 364 mg g−1. Formation of BiI3 and BiOI were observed on the carbon nanofibers after iodine adsorption. However, a small amount of polyiodide anions including I5− and I3− formed due to charge transfer between carbon fibers and iodine molecules.
Baskaran et al.158 reported on the formation of bismuth-functionalized carbon foams (CF). The CF was produced by carbonizing commercially available (and inexpensive) melamine foam. The highly porous CF can be compressed but was not friable and does not decompose until 550 °C (no combustion). Bismuth was deposited onto the CF using electrodeposition. Physisorption of iodine on the CF was found to decrease with increased metal loading. As mentioned above, intermediate physisorption could act as an iodine concentrator and lead to enhanced chemisorption for metals, such as Bi, which have slower reaction kinetics. Under realistic conditions in which iodine concentrations are orders of magnitude lower than in the saturated conditions used for initial testing, it could be possible for chemisorption to dominate on the CFs, and to have similar levels of physisorption as the zeolites.
7. Sorbent functionalization
As mentioned in Fig. 1f, functionalization of the scaffold/support is an effective concept in the development of high-performance sorbents. Functionalization, in general, is the process of modifying pore surfaces of a scaffold/substrate using functional groups with the goal of enhancing sorption performance. Tailoring surface chemistry increases the loading capacity and selectivity of the sorbent toward specific elements of interest and improves uptake kinetics. A common method to incorporate metal ions on different supports are through organic tethers between the passive scaffold and the active getter. For example, Matyas et al.24,25 synthesized Ag-functionalized silica aerogels in three steps: (1) the silica aerogel was functionalized with propylthiol monolayer using (3-mercaptopropyl)trimethoxysilane (3-MPTMS) under supercritical CO2 conditions; (2) the thiolated silica aerogel was then treated at room temperature with a 3.94% AgNO3 solution (5:1 deionized water to methanol, by volume); and (3) silver nanoparticles were produced on porous aerogel surfaces by reduction of silver thiolate adduct ions using 2.7%H2 in Ar at 165 °C for 2 h. This method allowed control of the size, distribution, and concentration of silver nanoparticles within the final sorbent. In this case, the functional propylthiol monolayers acted as tethers to hold the Ag0 nanoparticles in place.
Similarly, Riley et al.21 synthesized Ag-functionalized Na–Al–Si–O and Al–Si–O aerogels by soaking the base aerogels in aqueous AgNO3 solutions as mentioned above in the Matyas et al.24,25 studies, followed by drying and Ag+ reduction under H2/Ar to form silver nanoparticles within the aerogel matrix. Subsequent studies were performed using the same type of material but produced in xerogel form instead of aerogel form,23 where the xerogels had more structural rigidity and maintained higher SSA values during subsequent processing steps after the base Na–Al–Si–O gel was produced. In both of these studies by Riley et al.,21,23 thiolation was performed to assist in silver loading but also to add these thiol groups to the surfaces. A description of the full process including thiolation, silver loading, silver reduction, and iodine loading using substrates thiolated with 3-MPTMS is depicted in Fig. 14.
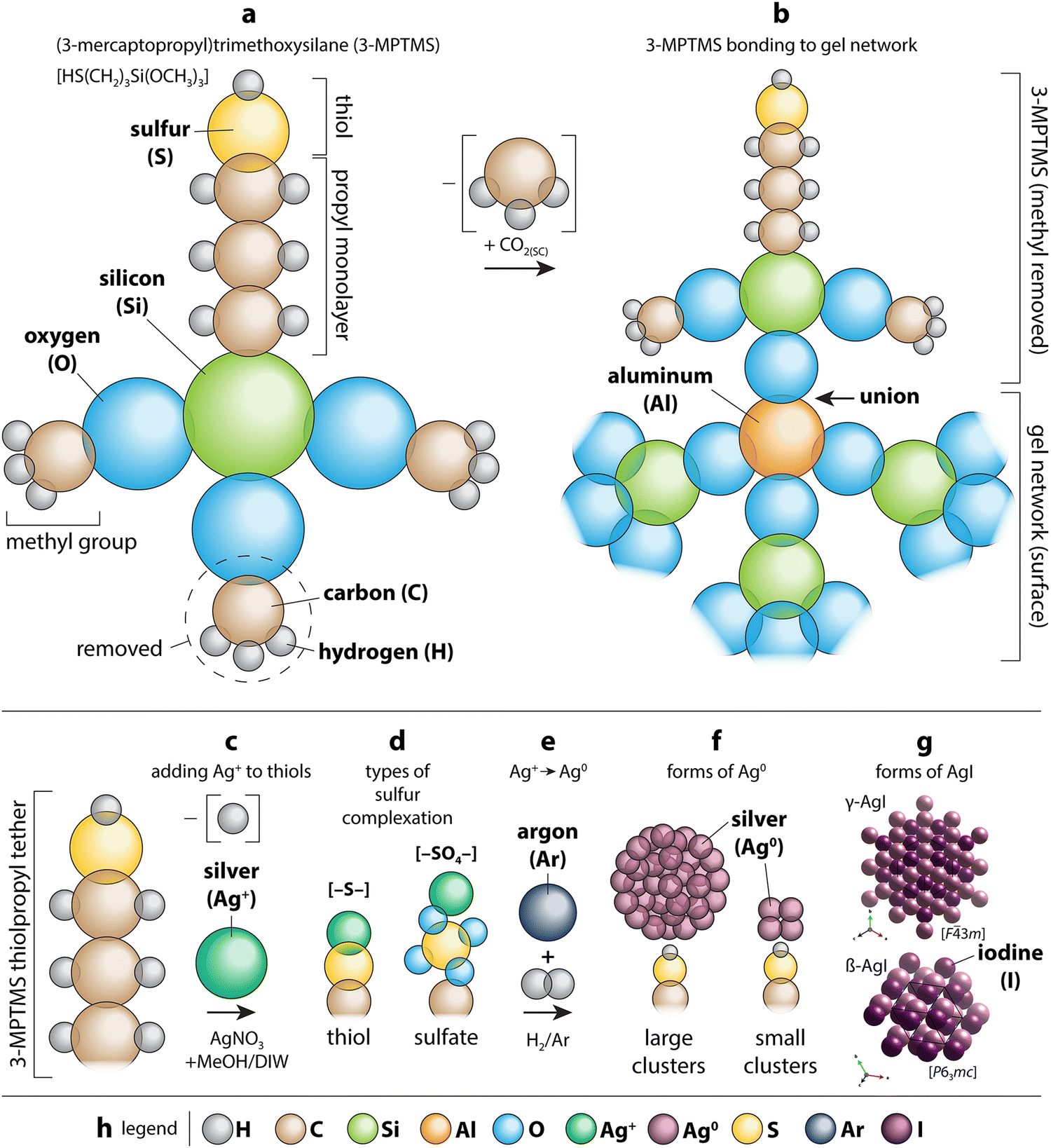 | ||
| Fig. 14 Summary of how (3-mercaptopropyl)trimethoxysilane [i.e., 3-MPTMS shown in (a)] interacts with the aluminosilicate xerogel network [shown in (b)] showing the union between the 3-MPTMS and the xerogel. (c) Summary of how Ag+ interacts with the thiol group, (d) sulfur complexation, (e) how Ag+ is reduced to Ag0, (f) how the Ag0 crystals form around the thiol group and on the surface of the gel as confirmed by various characterizations described in the text, (g) the forms of AgI that are created upon iodine chemisorption, and (h) the legend showing which colors represent which elements. Atoms are not drawn to scale. Reproduced from Riley et al.23 and reprinted with permission. Copyright 2022 American Chemical Society. | ||
Based on the work by Matyas et al.25 discussed above, X-ray photoelectron spectroscopy analysis of thiolated Ag-functionalized silica aerogels before and after exposure in iodine atmosphere at 150 °C showed that the oxidation of these materials tended to form more oxidized sulfur species [i.e., SO42− or S6+; see eqn (14)] over the oxidation of Ag0 to Ag+ [see eqn (15)]. This showed that adding this type of thiol functionalization layer not only aided in the tethering of silver clusters to the silica aerogel surfaces, but it also provided a redox buffer for the silver by preventing the silver oxidation reaction and thereby increasing the longevity of these sorbents at high temperatures considering that reduced forms of silver tend to react with iodine more effectively than oxidized forms.21,23
| S[2−] + 2 O2(g) → S[6+]O42− | (14) |
| 4 Ag0 + O2(g) → 2 Ag2O | (15) |
Lee et al.178 synthesized Ag-containing hydrophobic aluminosilicate aerogels by ion exchange of Na+ for Ag+. The prepared Na+AlSi-OH gels were exposed to 0.5 M AgNO3 at a fixed Ag:Na molar ratio of 6.5 to match the Na content in Na1.05AlSiO4.025 and Na1.3Al1.1SiO4.3. Produced Ag+AlSi-OH hydrogels were washed with purified water and filtered to remove NaNO3, their surface modified with silylating agents, and silver ions reduced to silver nanoparticles during ambient pressure drying.
8. Sorbent loading
As mentioned in Fig. 1g, sorbent loading includes discussion of the reaction thermodynamics and loading kinetics of iodide formation within the sorbent as well as other key factors. The saturation capacity (SC; see Table 1) for a sorbent to capture a target analyte is defined as the total amount of that analyte that can be loaded onto the sorbent. The units for this property can vary depending on several factors but can include moles of analyte per volume of total starting sorbent (mol m−3), grams of analyte loaded per gram of starting sorbent (g g−1 or Qe), or milligrams of analyte per gram of starting sorbent (mg g−1 or qe) at equilibrium. The terms P, E, and DF for a sorbent technology are defined in eqn (16) and (17) below where P and E denote bed penetration (material getting through the bed) and bed efficiency (what is captured by the bed), respectively, and DF was previously defined.| P = 1 − E | (16) |
| DF = 1/(1 − E) = 1/P | (17) |
In the U.S., regulations179–183 require an overall plant DF of ∼2000 for 129I, i.e., >3000 for the DOG, >1000 for the vessel ventilation off-gas (VOG), >1000 for the cell off-gas (COG), and >1000 for the melter off-gas (MOG).184–186 As an example, a DF of 200 for a sorbent technology means that 99.5% of 129I was captured in the process where P and E denote the amount of a species getting through the sorbent (remaining uncaptured) and the amount captured on a mass fraction basis, respectively. The relationships between P, E, and DF are also shown graphically in Fig. 15.
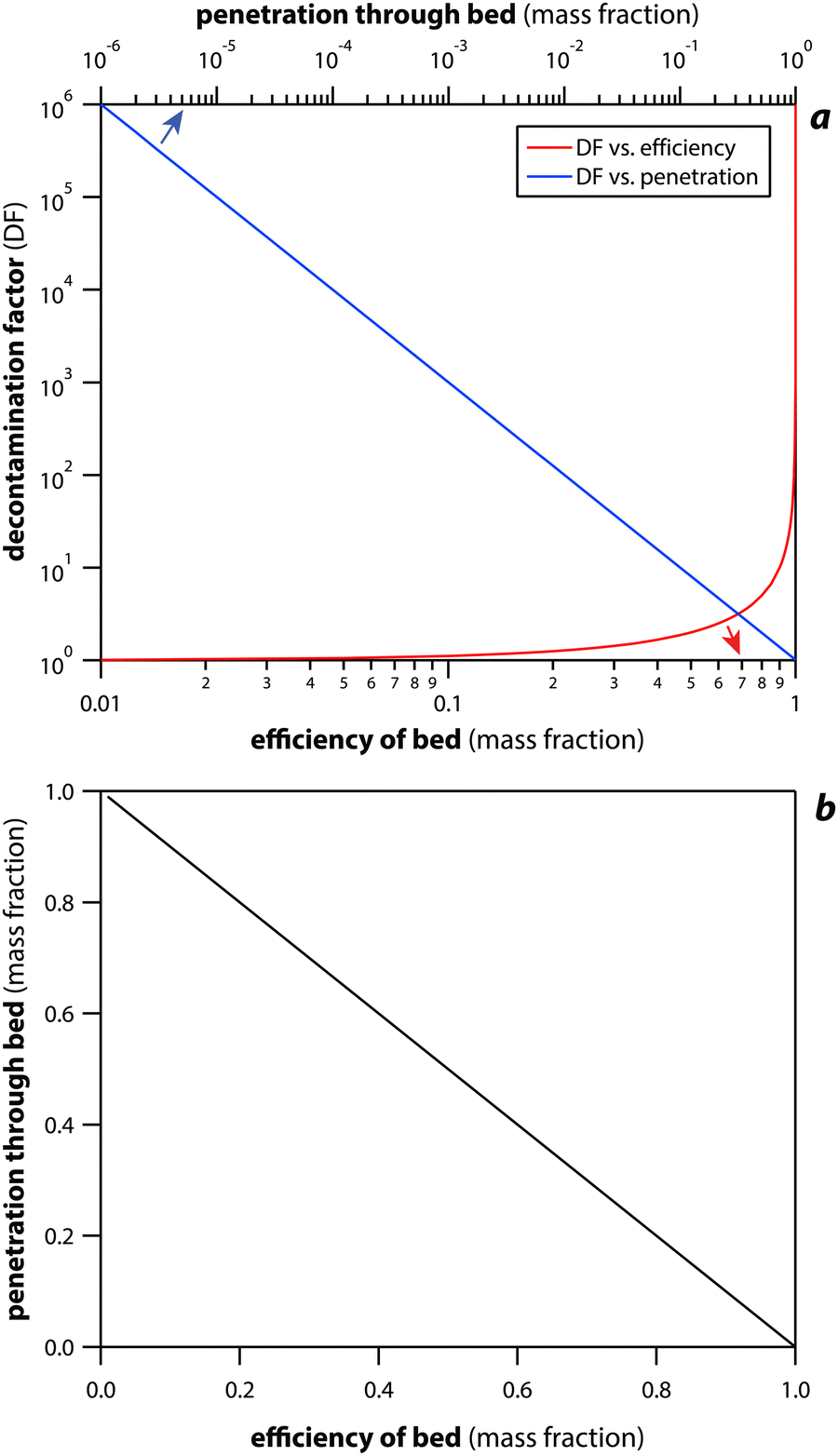 | ||
| Fig. 15 Summary showing the relationships between DF, bed efficiency (to 0.999999), and bed penetration (to 0.000001), including (a) DF vs. both bed efficiency and bed penetration (all axes are log10) as well as (b) bed penetration vs. bed efficiency. | ||
While these relationships between P, E, and DF are valid, theoretically, a bed that is at least as deep as the mass transfer zone would have (essentially) infinite DF and 100% capture of the target analyte. Experimentally, this has been impossible to demonstrate because of limited detection limits of the measurement techniques for iodine species.187 So, it seems impossible to use P, E, and/or DF as metrics for a sorbent, because in a properly designed system, every sorbent should perform the same achieving infinite DF even though that cannot be measured. What would differentiate sorbents are other parameters also addressed in this paper. While the capacity described above applies for determining loading for chemisorption-based sorbents, it does not apply as well to physisorption-based sorbents. For physisorption-based sorbents, the capacity depends on the concentration of the sorbate in the gas stream, according to its isotherm under the operating conditions.
9. Waste form options
As mentioned in Fig. 1h, the entire sorbent process is concluded by the final fate of the loaded sorbent (or final form of the MIxOy compound), i.e., the waste form for long-term disposal. This section discusses general considerations (Section 9.1) that have to be made when preparing a loaded sorbent for disposal as well as direct consolidation of loaded materials (Section 9.2) and pretreatment followed by consolidation into waste forms (Section 9.3).9.1. General discussion
Two of the properties that make sorbents effective to maximize gas-solid reactions, i.e., high porosity and SSA, become liabilities when disposing the material where minimizing porosity reduces the rate of dissolution of equivalent sample mass. This means that some type of process is recommended between the loading step and the disposal step to reduce overall sorbent volume but also reduce the SSA available for leaching. In addition, these sorbents typically do not have suitable mechanical properties for long-term surface storage and transport. Therefore, a conversion process is required to produce a durable waste form that can satisfy both mechanical and the long-term durability requirements. Conceptually, this can be broken down into two approaches. The first approach is about minimizing any post-capture treatment prior to waste form conversion. In the second approach, the iodine can be separated from the capture material and processed into a waste form that better incorporates the iodine into the structure either increasing overall chemical durability or enabling a greater amount of iodine to be immobilized. Results thus far show that the latter approach enables disposal options that are more chemically durable and mechanically stable,137 but this is at the expense of increased process complexity. Relative to the conversion of AgI-based sorbents into waste forms, which have been substantially reviewed elsewhere,62,68,188 there are fewer examples of alternative metal (non-Ag) sorbents being converted. Thus, an emphasis on alternative waste forms is presented within this section.The simplest waste form approach is to directly dispose the MIx compound.189 Conceptually, this is the simplest iodine waste form, but the majority of iodine capture concepts would require the MIx compound to be recovered from the sorbent after loading. Additional processes can be used to increase the density of the material and reduce porosity, such as hot isostatic pressing (HIP) or spark plasma sintering (SPS).54,138,190,191 Metal iodides (CuI, PbI2, HgI2) and iodates have been considered for direct disposal, taking into account solubility, hydrolysis, redox stability, radiation stability, and anion exchange reactions.189,192 Similar to AgI, these data strongly suggested that the direct disposal of these materials will result in an unstable material within typical reducing deep geological disposal scenarios. Of the materials compared, AgI remains one of the more stable iodides alongside CuI and a mixture of bismuth oxyiodide (BiIxOy) and bismuth oxide (Bi5O7I + Bi2O3, see Section 4.3, Other considerations, for additional discussion on BiIxOy compounds). This combination is being used because the Bi2O3 suppresses the hydrolysis of the oxyiodide. The susceptibility of bismuth oxyiodide to attack from carbonate and chloride ions, both being potential groundwater components, is an additional challenge. The authors noted that, in general, the preferred disposal conditions for the metal iodides would be in cool, nonreducing, and low dissolved salt conditions. This is why many studies have focused on methods to either surround the metal iodide within a more durable matrix or to incorporate the iodine into a more stable structure (see Section 9.2 below). To aid the comparison, Table 5 shows selected physical properties of relevant metal iodides.
| Metal iodide (MIx) | Metal (M) | Ref(s). | |||||
|---|---|---|---|---|---|---|---|
| MIx | K sp (25 °C) | T m (°C) | ΔGf° (kJ) | M | E° (V) | Cost ($ kg−1) | |
| AgI | 8.3 × 10−17 | 559 | −66 | Ag | 0.800 | $772 | 192,195–197 |
| BiI3 | 8.1 × 10−19 | 409 | −149 | Bi | 0.308 | $9 | 192,195,198,199 |
| CuI | 1.1 × 10−12 | 588 | −70 | Cu | 0.518 | $8 | 192,195,200,201 |
| HgI2 | 2.0 × 10−24, 1.1 × 10−12 | 250 | −52 | Hg | 0.852 | $9 | 192,195,202,203 |
| PbI2 | 7.1 × 10−9 | 407 | −174 | Pb | −0.126 | $2 | 192,195,196,204 |
| PdI2 | 2.5 × 10−23 | 350 | −63 | Pd | 0.915 | $30700 |
205–207 |
| TlI | 6.5 × 10−8 | 440 | −125 | Tl | −0.336 | $7400 | 192,195,208,209 |
| NaI | 1.84 × 102 | 661 | −286 | Na | −2.714 | $9 | 196,210 |
In a rare example of MIx consolidation, HIP has been used to reduce the porosity in CuI by applying 100 MPa for 2 h at either 200 °C or 550 °C.54 At the 550 °C processing temperature, the resulting CuI achieved 83% of the theoretical density. During chemical durability studies, the resultant CuI is relatively stable in near neutral conditions but remains susceptible to reducing conditions, which releases iodine into solution more rapidly. An additional study by Oshiro et al.130 was done looking at consolidation of pure AgI, BiI3, BiOI, CuI, and SnI4 reagents with SPS followed by ASTM C1308211 leaching studies. The results showed that the AgI and CuI had superior chemical durability to the others, BiOI performed well, and BiI3 and SnI4 performed poorly. It is surprising that there have not been other reports on consolidating simple metal iodides to reach theoretical density. This would enable a fundamental study of the chemical durability of pure, dense MIx phases, which has yet to be carried out for most MIx compounds. Other reviews of iodine materials exist that provide greater details on material durability137 and provide examples of iodine waste forms,62,68,188 but the focus here is on highlighting the non-Ag based examples where materials have been consolidated into a waste form.
Whilst reducing conditions have been shown to accelerate dissolution of iodine, evidence exists that formation of the metallic Ag0 may occur as a result of Fe(II) reduction thereby passivating dissolution.211,212 The degree to which this mechanism slows dissolution has yet to be explored in detail and is often not observed in most reductive dissolution tests. This may be due to the nature of those tests, which are not carried out on long enough time scales.
9.2. Direct consolidation of capture materials into a waste form
The principle of direct consolidation is to convert the iodine-loaded sorbent into a waste form in as few steps as possible. Iodine is not separated from the sorbent during direct consolidation; instead, both the capture material and iodine are processed together in situ. Because of the prevalence of Ag-based capture materials, there has been comparatively few examples of alternative metals being directly consolidated into a waste form. Examples are provided below for different sample matrices.:Si ratio within the waste form.220 The result is that iodine is very mobile within the pore water of the waste form and is controlled by the solubility of the metal iodide/iodate. The vast majority of work relates to the incorporation of AgI, but there are two examples of alternatives being encapsulated in cement, i.e., zeolite containing Ba(IO3)2 and PbI2.9 The results showed a limited effect of the cement to retain the iodine, consistent with AgI work. It is unlikely that the use of different MIx compounds will result in improved waste performances relative to Ag-based systems.
:Al ratio in the starting zeolite was key to enabling maximum formation of sodalite whilst avoiding the formation of nepheline and amorphous phases. The closer it was to 1, the fewer unwanted phases were observed. A compromise between capture and waste form performance was established with Na-based capture materials leading to waste forms with fewer phases whilst sacrificing iodine capture performance (Na-X: 120 mg g−1) relative to Ag (Ag-X: 250 mg g−1). Additionally, in a study by Chong et al.,226 HIP was used to immobilize iodosodalite [Na8(AlSiO4)6I2] using different glass binders, which showed notably different chemical durabilities.
:1, the temperature was 150 °C, and the mixture was stirred in an autoclave. This yielded 91.3% sodalite, 1.3% cancrinite, 0.4% kaolinite, and 12.7% amorphous phases (by mass). In general, the lower the fraction of the non-sodalite phases, the more durable the sodalite waste form becomes.229
9.3. Pretreatment followed by consolidation into a waste form
Pretreatment involves the use of multiple chemical steps to convert the iodide into a suitable disposal form. It may include extraction of the iodine from the capture sorbent followed by further treatment to exchange cations or a next step. The main reasons for using a more involved processing step here would be to maximize waste incorporation and to create materials with higher durability, but an added opportunity is the recycle of the primary sorbent, which was discussed previous in Section 3.2 (Sorbent performance). The main drawback of a pretreatment step is the extra industrial processing required between capture and waste form conversion, which leads to additional costs. However, if this can have a benefit with respect to the disposal case, then the extra effort involved may be justified. A summary of reported waste forms that involve non-AgI-based iodine species is provided in Table 6.| Starting speciation | Waste form speciation | Ref(s) |
|---|---|---|
| NaI | Sodalite, cancrinite, glass | 114 |
| CsI + BiI3 | Cs3Bi2I9 (perovskite) | 233 |
| CuI | CuI | 54 |
| BiPbO2I | BiPbO2I + lead-boron-zinc glass matrix | 216 |
| KIO3/NH4IO3, CaI2 | Apatite: calcium phosphate | 234 |
| PbI2/NaI | Apatite: lead vanadate | 109,190,235–239 |
| IO3− | Apatite: calcium phosphate, A2NaIO6 Perovskite (A = Ba, Sr, Ca) | 240 |
| I2, MnI2 | Iodoboracite | 241,242 |
| CsI | Synroc-C | 243 |
| CaI2/NaI | Ceramic composite (zeolite, hydroxyapatite, and fluorohydroxyapatite) | 244 |
| 2 AgI(s) + Na2S(aq) → Ag2S(s) + 2 NaI(aq) | (18) |
| 3 Al2Si2O7(s) + 6 NaOH(aq) + 2 NaI(aq) (180 °C for 7 days) → Na8(AlSiO4)6I2(s) + 3 H2O | (19) |
A reaction of the resultant solutions, which contained both NaI and Ag2S with NaOH and Al2Si2O7, yielded sodalite, NaI, and some unknown phases. This example shows the practical challenges involved with recovering iodine from a capture material and converting it into a useful speciation that can be consolidated into a stable waste form. Due to impurities in the starting solution, the resulting waste form product was as pure as possible when starting from an idealized solution, as is often reported.
9.4. Wrap-up
The reason for investigating alternatives to disposal of AgI in waste forms is predicated on avoiding the disposal of an expensive and environmentally controlled metal. However, most alternatives suffer from a lack of thermodynamic stability in disposal environments and/or can also have similar toxicity drawbacks. Within the literature, two broad approaches to iodine waste forms research have been documented that include incorporation of the iodine into the waste form crystal structure (e.g., in sodalites, apatites, or perovskites) or where a MIx compound is encapsulated by a durable matrix (e.g., glass composite materials, cermet composites). Only a few reported examples exist of directly converting a non-AgI based capture material and producing a waste form including BiI3, BiOI, CuI, and SnI4 (see Section 9.1, General discussion).130 With increasing interest in Bi-based capture materials,58,59,61,120,257 it is important that these materials are developed with consideration of the final waste form. Also, it will be important to compare new waste forms to equivalent Ag-based waste forms when using the direct disposal methods. The study of iodine waste form durability has been inconsistent across the many options. This has made it challenging to compare the merits of different materials on a fair basis. Thus, one critical area where more work is needed is to find better methods for comparing different waste form technologies. It is still unclear which approach is optimal and thus research to look at both simplified waste form conversion and more involved post-capture pretreatment processes are important to provide a broader set of pathways for iodine disposal.A detailed review of durability is beyond the scope of this paper as it has been discussed in a recent review,137 but there are some important discussion points to consider when selecting a waste form. Standardized tests available to experimentalists often do not provide a good overview of material performance. The majority of work has been carried out at pH 7, but the stability of a particular waste form is often highly sensitive to pH and the redox chemistry of the system.137 Thus, to be confident in the long-term durability of a waste form, a broader understanding of the mechanism is required and is currently being assessed as part of a multi-laboratory effort in the U.S. (Pacific Northwest National Laboratory and Argonne National Laboratory) on a variety of different iodine waste forms.258–261 Examples of detailed studies include those on vanadate waste forms, where substantial work has been carried out to determine the different processes that affect leaching behaviors.232 Another example where detailed mechanistic studies have been carried out is in cementitious materials.195,262,263 The more future studies follow this approach, the easier it will be to evaluate the strengths and weaknesses of different options in head-to-head comparisons.231,264
10. Summary, conclusions, and recommendations
In summary, a variety of different selection criteria and features must be considered when designing new sorbent materials, which were highlighted in Fig. 1 with more detail provided in Table 1. These include the target analyte in the off-gas environment (Fig. 1a), general sorbent requirements (Fig. 1b), getter selection (Fig. 1c), scaffold selection (Fig. 1d), getter addition (the process by which the getter is added to the scaffold, Fig. 1e), sorbent functionalization (Fig. 1f), sorbent loading (Fig. 1g), and the final waste form (Fig. 1h). The overall list of sorbent requirements is extensive and includes a range of thermal, mechanical, structural, chemical, radiation stability, thermodynamic, and general (e.g., cost, availability) properties of the base material and the loaded material. In most cases, based on several inherent properties, many candidate materials could be removed from the pool of “easy options” for immediate industrial-scale deployment due to high costs (e.g., precious metals), low commercial availabilities (e.g., aerogels, xerogels), and high environmental toxicities (e.g., those containing RCRA metals and/or low chemical durabilities).The most promising getter metal based on many decades of literature is Ag, where reduced silver (Ag0) appears to out-perform Ag+ when present in the same base sorbent. The main issues with using silver are that disposal of Ag-containing materials in the U.S. is controlled by the EPA under RCRA (40 CFR 261)110 and the cost is high compared to most other (non-precious) metals. Thus, alternative technologies are sought after to replace them. Promising chemisorption-based candidate sorbents include Bi and Cu.
When Bi0/Bi3+ reacts with iodine, it can form BiI3 or BiOxIy compounds; additionally, BiI3 can decompose to form BiOxIy compounds during loading experiments or after exposure at elevated temperatures in oxygen-containing atmospheres through I2(g) evolution. This complication might make implementation of a bismuth-containing sorbent difficult for capturing all available iodine without later desorbing some of it. Evidence also shows that additives (e.g., Bi2O3) and additional processing steps might be required to fully stabilize iodine-loaded Bi-based sorbents thereby lowering the overall iodine loading in the final form.
Copper shows promise as a replacement for Ag where CuI has similar thermal stability and chemical durability to AgI. Copper is much cheaper and more environmentally friendly than Ag, and it can be incorporated into a variety of porous scaffolds similarly to Ag. However, Cu tends to oxidize more readily than Ag being that Cu is less noble of a metal than Ag. Thus, while Ag-based materials show extreme promise, it is possible that utilizing these other metals (e.g., Bi, Cu), while having some shortfalls, might prove easier, cheaper, and more effective in the long run when considering the full life cycle of the sorbent from synthesis (preparation) to disposal.
Many iodine waste form options have been considered for long-term radioiodine disposal over the past several decades. The candidates can loosely be binned into either (1) direct consolidation of capture materials into a waste form or (2) pretreatment of the loaded material followed by consolidation into a waste form. For the first category, the options documented here include the following: glass; low-temperature hydrating/polymerizing materials; conversion of alumina, silica, and/or zeolite materials; sodalite; and apatite. For the second category, the options discussed here included the following: ceramic composites; iodosodalite; apatite (i.e., lead vanadate and calcium phosphate); perovskites; glass, glass ceramics and glass composite materials; titanate ceramics; and iodoboracite. The ranges of iodine loadings, chemical durabilities, fabrication difficulties, mechanical properties, and thermal properties of these different options vary extensively.
When designing a sorbent, the cradle-to-grave processes need to be considered holistically. The purpose of this paper is to guide researchers towards a more comprehensive checklist of applicable and critical criteria to consider when developing new iodine sorbent materials and iodine waste forms as well as advancing the TRL of existing materials.
Author contributions
BJR – conceptualization, funding acquisition, data curation, visualization, methodology, writing – original draft, writing – review and editing. JRT – conceptualization, methodology, writing – original draft, writing – review and editing. JM – conceptualization, funding acquisition, writing – original draft, writing – review and editing. SC – writing – original draft, writing – review and editing. KC – writing – original draft, writing – review and editing. JM – writing – original draft, writing – review and editing.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Pacific Northwest National Laboratory (PNNL) is operated by Battelle Memorial Institute for the DOE under contract DE-AC05-76RL01830. PNNL and ORNL contributions were funded through the United States Department of Energy, Office of Nuclear Energy's (DOE-NE) Material Recovery and Waste Form Development Campaign under the Nuclear Fuel Cycle and Supply Chain (NFCSC) Program. PNNL and ORNL authors thank Kimberly Gray (DOE-NE) and Ken Marsden (Idaho National Laboratory) for programmatic support. KC's contributions to this effort were supported by the United States Department of Energy (DOE) Nuclear Energy University Program (NEUP) under contract DE-NE0009317, and the US Nuclear Regulatory Commission (USNRC) under contract 31310022M015. BR thanks Sheila Riley for her help in editing the document.References
- L. B. Zablotska, E. Ron, A. V. Rozhko, M. Hatch, O. N. Polyanskaya, A. V. Brenner, J. Lubin, G. N. Romanov, R. J. McConnell, P. O'Kane, V. V. Evseenko, V. V. Drozdovitch, N. Luckyanov, V. F. Minenko, A. Bouville and V. B. Masyakin, Br. J. Cancer, 2011, 104, 181–187 CrossRef CAS PubMed.
- S. Tokonami, M. Hosoda, S. Akiba, A. Sorimachi, I. Kashiwakura and M. Balonov, Sci. Rep., 2012, 2, 507 CrossRef PubMed.
- R. A. Brown, J. D. Christian and T. R. Thomas, Airborne radionuclide waste-management reference document, Report ENICO-1133, Exxon Nuclear Idaho Co., Inc., Idaho Falls, ID, 1983.
- L. L. Burger and R. D. Scheele, HWVP Iodine Trap Evaluation, Report PNNL-14860, Pacific Northwest National Laboratory, Richland, WA, 2004.
- H. A. C. McKay, P. Miquel and I. F. White, Radioact. Waste Manage., 1982, 5–172 CAS.
- R. A. Brown, J. D. Christian and T. R. Thomas, Airborne Radionuclide Waste Management, Report ENICO-1132, Exxon Nuclear Idaho Company, Idaho Falls, ID, 1983.
- R. McNabney and A. M. Lyon, The removal of iodine from gas streams by reaction with silver in packed towers, Report 0099-8524, U.S. Atomic Energy Commission, Richland, WA, 1949.
- R. T. Jubin, Airborne waste management technology applicable for use in reprocessing plants for control of iodine and other off-gas constituents, Report ORNL/TM-10477, Oak Ridge National Laboratory, Oak Ridge, TN, 1988.
- L. E. Trevorrow, G. F. Vandegrift, V. M. Kolba and M. J. Steindler, Compatibility of Technologies with regulations in the waste management of H-3, I-129, C-14, and Kr-85. Part I. Initial information base, Report ANL-83-57, Argonne National Laboratory Argonne, IL, 1983.
- D. W. Holladay, A literature survey: Methods for the removal of iodine species from off-gases and liquid waste streams of nuclear power and nuclear fuel reprocessing plants, with emphasis on solid sorbents, Report ORNL/TM-6350, Oak Ridge National Laboratory, Oak Ridge, TN, 1979.
- G. E. R. Collard, D. Hennart, J. Van Dooren and W. R. A. Goosens, Proceedings of the 16th DOE Nuclear Air Cleaning Conference, 552–564, 1980.
- R. Stromatt, Removal of Radioiodine from PUREX Off-Gases with Nitric Acid and Nitric Acid-Mercuric Nitrate Solutions, Report USAEC HW-55735, Hanford Atomic Products Operation, Richland, WA, 1958.
- J. C. Mailen and D. E. Horner, Nucl. Technol., 1975, 30, 317–324 CrossRef.
- D. E. Horner, J. C. Mailen and F. A. Posey, Electrolytic trapping of iodine from process gas streams, US. Pat., US4004993A, 1977 Search PubMed.
- D. R. Haefner and T. J. Tranter, Methods of Gas Phase Capture of Iodine from Fuel Reprocessing Off-Gas: A Literature Survey, Report INL/EXT-07-12299, Idaho National Laboratory, Idaho Falls, ID, 2007.
- W. J. Maeck and D. T. Pence, 11th AEC Air Cleaning Conference, CONF-700816, 1970.
- D. T. Pence, F. A. Duce and W. J. Maeck, 11th AEC Air Cleaning Conference, CONF 700816, 1970.
- D. T. Pence, F. A. Duce and W. J. Maeck, 12th AEC Air Cleaning Conference, CONF 720823, 1972.
- T. R. Thomas, L. P. Murphy, B. A. Staples and J. T. Nichols, Airborne elemental iodine loading capacities of metal zeolites and a method for recycling silver zeolite, Report ICP-1119, Idaho National Laboratory, Idaho Falls, ID, 1977.
- A. I. Wiechert, A. P. Ladshaw, J. Moon, C. W. Abney, Y. Nan, S. Choi, J. Liu, L. L. Tavlarides, C. Tsouris and S. Yiacoumi, ACS Appl. Mater. Interface, 2020, 12, 49680–49693 CrossRef CAS.
- B. J. Riley, J. O. Kroll, J. A. Peterson, J. Matyáš, M. J. Olszta, X. Li and J. D. Vienna, ACS Appl. Mater. Interfaces, 2017, 9, 32907–32919 CrossRef CAS.
- S. Chong, B. J. Riley, W. Kuang and M. J. Olszta, ACS Omega, 2021, 6, 11628–11638 CrossRef CAS PubMed.
- B. J. Riley, S. Chong, J. Marcial, N. Lahiri, M. K. Bera, S. Lee, T. Wu, K. Kruska and J. Matyáš, ACS Appl. Nano Mater., 2022, 5, 9478–9494 CrossRef CAS.
- J. Matyáš, G. E. Fryxell, B. J. Busche, K. Wallace and L. S. Fifield, Functionalized silica aerogels: Advanced materials to capture and immobilize radioactive iodine, in Ceramic Materials for Energy Applications, ed. H. Lin, Y. Katoh, K. M. Fox, I. Belharouak, S. Widjaja and D. Singh, John Wiley & Sons, Inc., Hoboken, NJ, US, 2011, pp. 23–33 Search PubMed.
- J. Matyáš, E. S. Ilton and L. Kovařík, RSC Adv., 2018, 8, 31843–31852 RSC.
- H. Mineo, M. Gotoh, M. Iizuka, S. Fujisaki and G. Uchiyama, J. Nucl. Sci. Technol., 2002, 39, 241–247 CrossRef CAS.
- F. J. Herrmann, B. Herrmann, V. Hoeflich, C. Beyer and J. Furrer, 24th DOE/NRC Nucl. Air Cleaning Conf., NUREG-CP-0153, 609–617, 1997.
- F. J. Herrmann, B. Herrmann, K. D. Kuhn, A. Van Schoor, M. Weishaupt, J. Furrer and W. Knoch, 24th DOE/NRC Nucl. Air Cleaning Conf., NUREG-CP-0153, 618–627, 1997.
- F. J. Herrmann, V. Motoi, B. Herrmann, D. Fang, L. Finsterwalder, K. D. Kuhn, A. van Schoor, C. Beyer, J. Furrer and W. Knoch, 22nd DOE/NRC Nucl. Air Cleaning Conf., NUREG/CP-0130, 75–90, 1993.
- F. J. Herrmann, V. Motoi, B. Herrmann, A. Van Schoor, D. Fang and H. Fies, 21st DOE/NRC Nucl. Air Cleaning Conf., CONF-900813, 222–233, 1991.
- O. Kindel, V. Hoeflich, F. J. Herrmann and P. Patzelt, J. Radioanal. Nucl. Chem., 1993, 176, 251–259 CrossRef CAS.
- Y. Kondo, Y. Sugimoto, Y. Hirose, T. Fukasawa, J. Furrer, F. J. Herrmann and W. Knoch, 22nd DOE/NRC Nucl. Air Cleaning Conf., NUREG/CP-0130, 118–127, 1993.
- E. Henrich, F. J. Herrmann, W. Weinlaender, P. Dedaldechamp, J. P. Goumandy and A. Leseur, Reaktortag., [Fachvortr.], 1976, 351–354 CAS.
- E. Henrich and R. Hüfner, 16th DOE Nucl. Air Cleaning Conf., CONF-801038, 597–611, 1981.
- E. Henrich, H. Schmieder, W. Roesch and F. Weirich, 16th DOE Nucl. Air Cleaning Conf., CONF-801038, 612–627, 1981.
- J. Furrer, R. Kaempffer, F. J. Herrmann, B. Nemes and V. Motoi, KFK-Nachr., 1988, 20, 102–103 CAS.
- J. A. Jordan, Nuclear Engineering, MS thesis, University of Tennessee, Knoxville, 2018.
- M.-H. Zeng, Q.-X. Wang, Y.-X. Tan, S. Hu, H.-X. Zhao, L.-S. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561–2563 CrossRef CAS PubMed.
- J. T. Hughes, D. F. Sava, T. M. Nenoff and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 16256–16259 CrossRef CAS PubMed.
- D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Zhao, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2013, 25, 2591–2596 CAS.
- C. M. González-García, J. F. González and S. Román, Fuel Proc. Technol., 2011, 92, 247–252 CrossRef.
- A. J. Juhola, Carbon, 1975, 13, 437–442 CrossRef CAS.
- J. Zhou, S. Hao, L. Gao and Y. Zhang, Ann. Nucl. Energy, 2014, 72, 237–241 CrossRef CAS.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- Y. Xie, T. Pan, Q. Lei, C. Chen, X. Dong, Y. Yuan, W. A. Maksoud, L. Zhao, L. Cavallo, I. Pinnau and Y. Han, Nat. Commun., 2022, 13, 2878 CrossRef CAS PubMed.
- L. Zhang, J. Li, H. Zhang, Y. Liu, Y. Cui, F. Jin, K. Wang, G. Liu, Y. Zhao and Y. Zeng, Chem. Commum., 2021, 57, 5558–5561 RSC.
- B. Valizadeh, T. N. Nguyen, B. Smit and K. C. Stylianou, Adv. Funct. Mater., 2018, 28, 1801596 CrossRef.
- X. Qian, B. Wang, Z.-Q. Zhu, H.-X. Sun, F. Ren, P. Mu, C. Ma, W.-D. Liang and A. Li, J. Hazard. Mater., 2017, 338, 224–232 CrossRef CAS.
- P. Wang, Q. Xu, Z. Li, W. Jiang, Q. Jiang and D. Jiang, Adv. Mater., 2018, 30, 1801991 CrossRef.
- W. Xie, D. Cui, S.-R. Zhang, Y.-H. Xu and D.-L. Jiang, Mater. Horiz., 2019, 6, 1571–1595 RSC.
- S. Chong, B. J. Riley, K. Baskaran, S. Sullivan, L. G. El Khoury, K. Carlson, R. M. Asmussen and M. S. Fountain, New J. Chem., 2024, 48, 9880–9884 RSC.
- M. M. Metalidi, V. I. Beznosyuk, N. N. Kalinin, A. B. Kolyadin and Y. S. Fedorov, Radiochemistry, 2009, 51, 409–411 CrossRef CAS.
- H. Kitagawa, H. Ohtsu and M. Kawano, Angew. Chem., Int. Ed., 2013, 52, 12395–12399 CrossRef CAS PubMed.
- E. R. Vance, C. Grant, I. Karatchevtseva, Z. Aly, A. Stopic, J. Harrison, G. Thorogood, H. Wong and D. J. Gregg, J. Nucl. Mater., 2018, 505, 143–148 CrossRef CAS.
- J. Zhou, Q. Chen, T. Li, T. Lan, P. Bai, F. Liu, Z. Yuan, W. Zheng, W. Yan and T. Yan, Inorg. Chem., 2022, 61, 7746–7753 CrossRef CAS PubMed.
- B. J. Riley, N. C. Canfield, S. Chong and J. V. Crum, ACS Omega, 2024, 9, 34661–34674 CrossRef CAS PubMed.
- J. H. Yang, Y.-J. Cho, J. M. Shin and M.-S. Yim, J. Nucl. Mater., 2015, 465, 556–564 CrossRef CAS.
- J. H. Yang, J. M. Shin, J. J. Park, G. I. Park and M. S. Yim, J. Nucl. Mater., 2015, 457, 1–8 CrossRef CAS.
- A. Tesfay Reda, M. Pan, D. Zhang and X. Xu, J. Environ. Chem. Eng., 2021, 9, 105279 CrossRef CAS.
- Z. Tian, T.-S. Chee, L. Zhu, T. Duan, X. Zhang, L. Lei and C. Xiao, J. Hazard. Mater., 2021, 417, 125978 CrossRef CAS.
- Y. Hao, Z. Tian, C. Liu and C. Xiao, Front. Chem., 2023, 11, 1122484 CrossRef CAS.
- B. J. Riley, J. D. Vienna, D. M. Strachan, J. S. McCloy and J. L. Jerden Jr, J. Nucl. Mater., 2016, 470, 307–326 CrossRef CAS.
- X. Yang, H. Wu and D. Xie, J. Radioanal. Nucl. Chem., 2023, 332, 4427–4439 CrossRef CAS.
- M. Chebbi, C. Monsanglant-Louvet, P. Parent, C. Gerente, L. L. Coq and B. M. Mokili, Carbon Trends, 2022, 7, 100164 CrossRef CAS.
- T. J. Robshaw, J. Turner, S. Kearney, B. Walkley, C. A. Sharrad and M. D. Ogden, SN Appl. Sci., 2021, 3, 843 CrossRef CAS.
- S. U. Nandanwar, K. Coldsnow, V. Utgikar, P. Sabharwall and D. Eric Aston, Chem. Eng. J., 2016, 306, 369–381 CrossRef CAS.
- J. Huve, A. Ryzhikov, H. Nouali, V. Lalia, G. Augé and T. J. Daou, RSC Adv., 2018, 8, 29248–29273 RSC.
- R. Pénélope, L. Campayo, M. Fournier, A. Gossard and A. Grandjean, J. Nucl. Mater., 2022, 563, 153635 CrossRef.
- T. Pan, K. Yang, X. Dong and Y. Han, J. Mater. Chem. A, 2023, 11, 5460–5475 RSC.
- S.-T. Wang, Y.-J. Liu, C.-Y. Zhang, F. Yang, W.-H. Fang and J. Zhang, Chem Eur. J., 2023, 29, e202202638 CrossRef CAS PubMed.
- B. J. Riley and K. Carlson, Front. Chem., 2022, 10, 969303 CrossRef CAS PubMed.
- S. H. Bruffey, R. T. Jubin, D. Strachan, N. Soelberg, B. B. Spencer and B. J. Riley, Performance Criteria for Capture and/or Immobilization Technologies (Revision 1), Report ORNL/SPR-2020/1583, Oak Ridge National Laboratory, Oak Ridge, TN, 2020.
- S. H. Bruffey, B. B. Spencer, D. M. Strachan, R. T. Jubin, N. Soelberg and B. J. Riley, A Literature Survey to Identify Potentially Problematic Volatile Iodine-Bearing Species Present in Off-Gas Streams, Report FCR&D-MRWFD-2015-000421, ORNL-SPR-2015/290, Oak Ridge National Laboratory, Oak Ridge, TN, 2015.
- S. H. Bruffey, K. K. Patton, J. F. Walker Jr. and R. T. Jubin, Complete NO and NO2 Aging Study for AgZ, Report FCRD-MRWFD-2015-000631, ORNL/SPR-2015/128, Oak Ridge National Laboratory, Oak Ridge, TN, 2015.
- J. A. Johnson, C. J. Rawn, B. B. Spencer, R. A. Meisner and G. D. Del Cul, J. Nucl. Mater., 2017, 490, 211–215 CrossRef CAS.
- R. T. Jubin and B. B. Spencer, Evaluation of Tritium Management Approaches on Tritium Waste Volumes in Reprocessing Plants, Report ORNL/TM-2017/308, Oak Ridge National Laboratory, Oak Ridge, TN, 2017.
- J. H. Goode, Voloxidation: removal of volatile fission products from spent LMFBR fuels, Report ORNL-TM-3723, Oak Ridge National Laboratory, Oak Ridge, TN, 1970.
- B. B. Spencer, S. H. Bruffey and R. T. Jubin, Test Plan to Demonstrate Removal of Iodine and Tritium from Simulated Nuclear Fuel Recycle Plant Off-Gas Streams Using Adsorption Processes, Report ORNL/LTR-2013/542, Oak Ridge National Laboratory, Oak Ridge, TN, 2013.
- A. T. Greaney, S. H. Bruffey and R. T. Jubin, Effect of NOx and water variations on iodine loading of AgZ, Report ORNL/SPR-2020/1581, Oak Ridge National Laboratory, Oak Ridge, TN, 2020.
- L. Han, S. Cai, M. Gao, J.-Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- H. B. Andrews, J. McFarlane, A. S. Chapel, N. D. B. Ezell, D. E. Holcomb, D. de Wet, M. S. Greenwood, K. G. Myhre, S. A. Bryan, A. Lines, B. J. Riley, H. M. Felmy and P. W. Humrickhouse, Nucl. Eng. Des., 2021, 385, 111529 CrossRef CAS.
- B. J. Riley, J. McFarlane, G. D. DelCul, J. D. Vienna, C. I. Contescu and C. W. Forsberg, Nucl. Eng. Des., 2019, 345, 94–109 CrossRef CAS.
- J. McFarlane, B. J. Riley, D. E. Holcomb, A. Lines, H. B. Andrews, S. A. Bryan, A. S. Chapel, N. D. B. Ezell, H. M. Felmy, M. S. Greenwood, P. W. Humrickhouse and K. G. Myhre, Molten Salt Reactor Engineering Study for Off-Gas Management, Report ORNL/TM-2020/1602, PNNL-30159, Oak Ridge National Laboratory, Oak Ridge, TN, 2020.
- J. A. Peterson, M. MacDonell, L. Haroun, F. Monette, R. D. Hildebrand and A. Taboas, “Krypton” in Radiological and Chemical Fact Sheets to Support Health Risk Analyses for Contaminated Areas Reporthttps://remm.hhs.gov/ANL_ContaminantFactSheets_All_070418.pdf, Argonne National Laboratory, Environmental Science Division, Lemont, IL, 2007.
- 40 CFR 190, Environmental radiation protection requirements for normal operations of activities in the uranium fuel cycle, Environmental Protection Agency, Washington, D.C.
- L. D. Trowbridge, L. M. Toth and E. D. Collins, Molten Hydroxide Trapping Process for Radioiodine, Report ORNL/TM-2002/247, Oak Ridge National Laboratory, Oak Ridge, TN, 2003.
- B. J. Riley, J. Mcfarlane, G. DelCul, J. D. Vienna, C. I. Contescu, L. M. Hay, A. V. Savino and H. E. Adkins, Identification of Potential Waste Processing and Waste Form Options for Molten Salt Reactors, Pacific Northwest National Laboratory, Richland, WA, 2018.
- H. M. Felmy, A. J. Clifford, A. S. Medina, R. M. Cox, J. M. Wilson, A. M. Lines and S. A. Bryan, Environ. Sci. Technol., 2021, 55, 3898–3908 CrossRef CAS PubMed.
- J. McFarlane, D. Orea, K. Robb, B. Hunter, K. R. Johnson, H. Andrews, K. Lee and R. Salko, Bubble Mass Transport Measurement in the Large Scale Test Loops at Oak Ridge National Laboratory, Report ORNL/TM-2024/3579, Oak Ridge National Laboratory, Oak Ridge, TN, 2024.
- L. C. Lewis, Evaluation of Adsorbents for Purification of Noble Gases in Dissolver Off-Gas, Report IN-1402, Idaho Nuclear Corporation, National Reactor Testing Station, Idaho Falls, ID, 1970.
- K. D. Hughey, A. M. Bradley, R. G. Tonkyn, H. M. Felmy, T. A. Blake, S. A. Bryan, T. J. Johnson and A. M. Lines, J. Phys. Chem. A, 2020, 124, 9578–9588 CrossRef CAS PubMed.
- C. J. Adams, Inorg. Nucl. Chem. Lett., 1974, 10, 831–835 CrossRef CAS.
- O. Beneš, E. Capelli, N. Morelová, J. Y. Colle, A. Tosolin, T. Wiss, B. Cremer and R. J. M. Konings, Phys. Chem. Chem. Phys., 2021, 23, 9512–9523 RSC.
- J. M. Kurley, P. W. Halstenberg, A. McAlister, S. Raiman, S. Dai and R. T. Mayes, RSC Adv., 2019, 9, 25602–25608 RSC.
- G. Yoder Jr., K. R. Robb, E. Dominguez-Ontiveros, D. K. Felde, D. L. Fugate and D. E. Holcomb, Start-up Operation Experience with a Liquid Fluoride Salt Forced Convection Loop, Report ORNL/TM-2023/2978, Oak Ridge National Laboratory, 2023.
- R. V. Ostvald, V. V. Shagalov, I. I. Zherin, G. N. Amelina, V. F. Usov, A. I. Rudnikov and O. B. Gromov, J. Fluor. Chem., 2009, 130, 108–116 CrossRef CAS.
- M.-Y. Wey, C.-Y. Lu, H.-H. Tseng and C.-H. Fu, J. Air Waste Manage. Assoc., 2002, 52, 449–458 CrossRef CAS PubMed.
- S. F. Marsh and K. K. S. Pillay, Effects of Ionizing Radiation on Modern Ion Exchange Materials, Report LA-12655-MS, Los Alamos National Laboratory, Los Alamos, NM, 1993.
- F. Raatz, C. Marcilly and E. Freund, Zeolites, 1985, 5, 329–333 CrossRef CAS.
- D. Porrey, personal communication, December 2023.
- R. T. Jubin, S. H. Bruffey, D. Strachan, N. Soelberg, B. B. Spencer and B. J. Riley, Performance Criteria for Capture and/or Immobilization Technologies, Report FCRD-MRWFD-2016-000533, ORNL/TM-2016-73, INL/EXT-16-38035, Oak Ridge National Laboratory, Oak Ridge, TN, 2016.
- Y. Nan, S. Choi, A. P. Ladshaw, S. Yiacoumi, C. Tsouris, D. W. DePaoli and L. L. Tavlarides, Trans. Am. Nucl. Soc., 2017, 116, 130–132 Search PubMed.
- Z. Shen, A. I. Wiechert, S. Choi, L. L. Tavlarides, C. Tsouris and S. Yiacoumi, Ind. Eng. Chem. Res., 2022, 61, 14393–14401 CrossRef CAS.
- K. Baskaran, C. Elliott, M. Ali, J. Moon, J. Beland, D. Cohrs, S. Chong, B. J. Riley, D. Chidambaram and K. Carlson, J. Hazard. Mater., 2023, 446, 130644 CrossRef CAS.
- L. P. Murphy, B. A. Staples and T. R. Thomas, Development of Ag0Z for bulk129I removal from nuclear fuel reprocessing plants and PbX for129I storage, Report ICP-1135, Allied Chemical Corporation, Idaho Falls, ID, 1977.
- B. A. Staples, L. P. Murphy and T. R. Thomas, 14th ERDA Air Cleaning Conference (CONF-760822), 363–380, 1976.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- S. Chong, B. J. Riley, R. M. Asmussen, A. Fujii Yamagata, J. Marcial, S. Lee and C. A. Burns, ACS Appl. Polym. Mater., 2022, 4, 9040–9051 CrossRef CAS.
- B. J. Riley, S. Chong and N. L. Canfield, New J. Chem., 2024, 48, 3352–3356 RSC.
- 40 CFR 261, Identification and listing of hazardous waste, Environmental Protection Agency, Washington, D.C.
- Technology Readiness Assessment (TRA) Guidance, US Department of Defense, US Department of Defense, Washington, DC.
- P. Johansson, C. Johansson and O. Isaksson, CIRP IPS2 Conference, 379–385, 2010.
- J. C. Mankins, Technology Readiness Levels, NASA Advanced Concepts Office, 1995.
- A. Yadav, S. Chong, B. J. Riley, J. S. McCloy and A. Goel, Ind. Eng. Chem. Res., 2023, 62, 3635–3646 CrossRef CAS.
- J. Wang, M. Li, Y. Feng, Y. Liu and J. Liu, Nucl. Mater. Energy, 2022, 33, 101270 CrossRef CAS.
- J. Tang, M. Ye, Y. Rao, X. Ding and Z. Tang, Chinese J. Nucl. Sci. Technol., 1991, 11, 67–72 Search PubMed.
- J. Wilhelm and H. Mahnau, 13th AEC Air Cleaning Conference 863–875, 1975.
- G. O. Modolo, R, Global 1995 - ANS International Conference on Evaluation of Emerging Nuclear Fuel Cycle Systems, CONF-950919, 1995.
- B. J. Riley, S. Chong, J. Schmid, J. Marcial, E. T. Nienhuis, M. K. Bera, S. Lee, N. L. Canfield, S. Kim, M. A. Derewinski and R. K. Motkuri, ACS Appl. Mater. Interfaces, 2022, 14, 18439–18452 CrossRef CAS PubMed.
- A. Al-Mamoori, M. Alsalbokh, S. Lawson, A. A. Rownaghi and F. Rezaei, Chem. Eng. J., 2020, 391, 123583 CrossRef CAS.
- B. J. Riley, S. Chong, M. J. Olszta and J. A. Peterson, ACS Appl. Mater. Interfaces, 2020, 12, 19682–19692 CrossRef CAS PubMed.
- B. J. Riley, S. Chong and C. L. Beck, Ind. Eng. Chem. Res., 2021, 60, 17162–17173 CrossRef CAS.
- EPA, SW-846 Test Method 1311: Toxicity Characteristic Leaching Procedure (TCLP), Report SW-846 Test Method 1311, Environmental Protection Agency, Washington, D.C., 1992.
- R. C. Moore, C. I. Pearce, J. W. Morad, S. Chatterjee, T. G. Levitskaia, R. M. Asmussen, A. R. Lawter, J. J. Neeway, N. P. Qafoku, M. J. Rigali, S. A. Saslow, J. E. Szecsody, P. K. Thallapally, G. Wang and V. L. Freedman, Sci. Total Environ., 2020, 716, 132820 CrossRef CAS PubMed.
- J. F. Kurisingal, H. Yun and C. S. Hong, J. Hazard. Mater., 2023, 458, 131835 CrossRef CAS PubMed.
- J. Wang, D. Fan, C. Jiang and L. Lu, Nano Today, 2021, 36, 101034 CrossRef CAS.
- C. Muhire, A. Tesfay Reda, D. Zhang, X. Xu and C. Cui, Chem. Eng. J., 2022, 431, 133816 CrossRef CAS.
- Y. Lu, H. Liu, R. Gao, S. Xiao, M. Zhang, Y. Yin, S. Wang, J. Li and D. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 29179–29185 CrossRef CAS.
- S. H. Bruffey and R. T. Jubin, Initial Evaluation of Effects of NOxon Iodine and Methyl Iodide Loading of AgZ and Aerogels, Report FCRD-MRWFD-2015-000426, ORNL/SPR-2015/125, Oak Ridge National Laboratory, Oak Ridge, TN, 2015.
- J. M. Oshiro, A. Fujii Yamagata, S. Sharma, R. M. Asmussen, S. Chong, B. J. Riley, J. V. Crum, J. A. Silverstein and J. Lian, Ind. Eng. Chem. Res., 2024, under review Search PubMed.
- B. Wagner, A. Huttner, D. Bischof, A. Engel, G. Witte and J. Heine, Langmuir, 2020, 36, 6458–6464 CrossRef CAS PubMed.
- J. H. Yang, H. S. Park, D.-H. Ahn and M.-S. Yim, J. Nucl. Mater., 2016, 480, 150–158 CrossRef CAS.
- H. Zou, F. Yi, M. Song, X. Wang, L. Bian, W. Li, N. Pan and X. Jiang, J. Hazard. Mater., 2019, 365, 81–87 CrossRef CAS PubMed.
- H. Zou, J. Guo, M. Song, F. Yi, X. Wang, N. Pan, H. Li, W. Li and L. Bian, Prog. Nucl. Energy, 2021, 135, 103705 CrossRef CAS.
- A. Tesfay Reda, D. Zhang, X. Xu and S. Xu, Sep. Purif. Technol., 2022, 292, 120994 CrossRef CAS.
- A. Tesfay Reda, D. Zhang, X. Xu, M. Pan, C. Chang, C. Muhire, X. Liu and S. Jiayi, Sep. Purif. Technol., 2021, 270, 118848 CrossRef CAS.
- J. T. Reiser, A. R. Lawter, N. A. Avalos, J. Bonnett, B. J. Riley, S. Chong, N. Canfield, S. A. Saslow, A. Bourchy and R. M. Asmussen, Chem. Eng. J. Adv., 2022, 11, 100300 CrossRef CAS.
- B. J. Riley, S. Chong, M. Zhao and J. Lian, Ind. Eng. Chem. Res., 2023, 62, 8779–8792 CrossRef CAS.
- B. J. Riley, J. Chun, W. Um, W. C. Lepry, J. Matyas, M. J. Olszta, X. Li, K. Polychronopoulou and M. G. Kanatzidis, Environ. Sci. Technol., 2013, 47, 7540–7547 CrossRef CAS PubMed.
- K. S. Subrahmanyam, D. Sarma, C. D. Malliakas, K. Polychronopoulou, B. J. Riley, D. A. Pierce, J. Chun and M. G. Kanatzidis, Chem. Mater., 2015, 27, 2619–2626 CrossRef CAS.
- B. J. Riley and S. Chong, in An Introduction to Aluminosilicates, ed. N. R. Blevins, Nova Science Publishers, 2020, ch. 7 Search PubMed.
- W. J. Maeck, D. T. Pence and J. H. Keller, A Highly Efficient Inorganic Adsorber for Airborne Iodine Species (Silver Zeolite Development Studies), Report IN-1224, Idaho Nuclear Corporation - National Reactor Testing Station, Idaho Falls, ID, 1968.
- S. H. Bruffey, R. T. Jubin and J. A. Jordan, Fundamental Aspects of Zeolite Waste Form Production by Hot Isostatic Pressing, Report FCRD-MRWFD-2016-000267, ORNL/SR-2016/759, Oak Ridge National Laboratory, Oak Ridge, TN, 2016.
- T. Ayadi, M. Badawi, L. Cantrel and S. Lebègue, Mol. Syst. Des. Eng., 2022, 7, 422–433 RSC.
- S. S. Metwally and M. F. Attallah, J. Mol. Liq., 2019, 290, 111237 CrossRef CAS.
- T. C. T. Pham, S. Docao, I. C. Hwang, M. K. Song, D. Y. Choi, D. Moon, P. Oleynikov and K. B. Yoon, Energy Environ. Sci., 2016, 9, 1050–1062 RSC.
- Z. Tian, T.-S. Chee, R. Meng, Y. Hao, X. Zhou, B. Ma, L. Zhu, T. Duan and C. Xiao, Environ. Funct. Mater., 2022, 1, 92–104 Search PubMed.
- S. H. Bruffey and R. T. Jubin, Recommend HIP Conditions for AgZ, Report FCRD-MRWFD-2015-000423, ORNL/SPR-2015/503, Oak Ridge National Laboratory, Oak Ridge, TN, 2015.
- S. Chibani, M. Chebbi, S. Lebègue, T. Bučko and M. Badawi, J. Chem. Phys., 2016, 144 Search PubMed.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier, Oxford, 6th revised edn, 2007 Search PubMed.
- C. Baerlocher, Database of Zeolite Structures, https://www.iza-structure.org/databases/, (accessed 12/20/2023).
- Norton Company, Chemical Process Products Division, Akron, OH.
- D. Banerjee, X. Chen, S. S. Lobanov, A. M. Plonka, X. Chan, J. A. Daly, T. Kim, P. K. Thallapally and J. B. Parise, ACS Appl. Mater. Interfaces, 2018, 10, 10622–10626 CrossRef CAS PubMed.
- P. Chen, X. He, M. Pang, X. Dong, S. Zhao and W. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 20429–20439 CrossRef CAS PubMed.
- Q.-M. Zhang, T.-L. Zhai, Z. Wang, G. Cheng, H. Ma, Q.-P. Zhang, Y.-H. Zhao, B. Tan and C. Zhang, Adv. Mater. Interface, 2019, 6, 1900249 CrossRef.
- X. Yang, D. Xie, W. Wang, S. Li, Z. Tang and S. Dai, Chem. Eng. J., 2023, 454, 140365 CrossRef CAS.
- Z. Xu, Q. Zhang, P. Lin, Y. Gao, Y. Wen, K. Li and L. Li, Mater. Chem. Phys., 2022, 285, 126193 CrossRef CAS.
- K. Baskaran, M. Ali, B. J. Riley, I. Zharov and K. Carlson, ACS Mater. Lett., 2022, 4, 1780–1786 CrossRef CAS.
- S. Chen, G. He, H. Hu, S. Jin, Y. Zhou, Y. He, S. He, F. Zhao and H. Hou, Energy Environ. Sci., 2013, 6, 2435–2439 RSC.
- Q. Yu, X. Jiang, Z. Cheng, Y. Liao, Q. Pu and M. Duan, New J. Chem., 2020, 44, 16759–16768 RSC.
- Y. Ding, W. Fan, Q. Xian, H. Dan, L. Zhu and T. Duan, Chem. Eng. J., 2023, 451, 138887 CrossRef CAS.
- J. G. Wilhelm and H. Schuettelkopf, Eleventh AEC Air Cleaning Conference, 568 (CONF-700816), 1970.
- J. G. Wilhelm and H. Schuettelkopf, 12th AEC Air Cleaning Conference, 540 (CONF-720823), 1972.
- M. Kikuchi, M. Kitamura, H. Yusa and S. Horiuchi, Nucl. Eng. Des., 1978, 47, 283–287 CrossRef CAS.
- S. Chong, B. J. Riley, J. A. Peterson, M. J. Olszta and Z. J. Nelson, ACS Appl. Mater. Interfaces, 2020, 12, 26127–26136 CrossRef CAS PubMed.
- B. J. Riley, D. Pierce, A. J. Chun, J. Matyáš, W. C. Lepry, T. Garn, J. Law and M. G. Kanatzidis, Environ. Sci. Technol., 2014, 48, 5832–5839 CrossRef CAS PubMed.
- Q. Yu, X. Jiang and M. Duan, Soc. Sci. Res. Net., 2021 DOI:10.2139/ssrn.3978461.
- B. J. Riley, J. Chun, J. V. Ryan, J. Matyáš, X. S. Li, D. W. Matson, S. K. Sundaram, D. M. Strachan and J. D. Vienna, RSC Adv, 2011, 1, 1704–1715 RSC.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- S. Bag, I. U. Arachchige and M. G. Kanatzidis, J. Mater. Chem., 2008, 18, 3628–3632 RSC.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- J. Wang, C. Wang, H. Wang, B. Jin, P. Zhang, L. Li and S. Miao, Microporous Mesoporous Mater., 2021, 310, 110596 CrossRef CAS.
- Z. Yan, Y. Yuan, Y. Tian, D. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12733–12737 CrossRef CAS PubMed.
- T. C. Schäfer, J. Becker, M. T. Seuffert, D. Heuler, A. E. Sedykh and K. Müller-Buschbaum, Chem. – Eur. J., 2022, 28, e202104171 CrossRef PubMed.
- E. Aneheim, D. Bernin and M. R. S. Foreman, Nucl. Eng. Des., 2018, 328, 228–240 CrossRef CAS.
- Z. Tian, T.-S. Chee, X. Zhang, L. Lei and C. Xiao, Chem. Eng. J., 2021, 412, 128687 CrossRef CAS.
- J. W. Lee, S. M. Hong, J. H. Lee and Y. Z. Cho, J. Nucl. Mater., 2021, 557, 153309 CrossRef CAS.
- 40 CFR 190.10, Environmental Protection Agency, Washington, DC.
- 40 CFR 61.92, Environmental Protection Agency, Washington, DC.
- 40 CFR 191.13, Environmental Protection Agency, Washington, DC.
- 10 CFR 61.41, Nuclear Regulatory Commission, Washington, DC.
- 10 CFR 20, Nuclear Regulatory Commission, Washington, DC.
- R. Jubin, N. Soelberg, D. Strachan and G. Ilas, Fuel Age Impacts on Gaseous Fission Product Capture during Separations, Report FCRD-SWF-2012-000089, PNNL-22550, Oak Ridge National Laboratory, Oak Ridge, TN, 2012.
- N. R. Soelberg, T. G. Garn, M. R. Greenhalgh, J. D. Law, R. Jubin, D. M. Strachan and P. K. Thallapally, Sci. Technol. Nucl. Install., 2013, 1–12 Search PubMed.
- J. D. Vienna, E. D. Collins, J. V. Crum, W. L. Ebert, S. M. Frank, T. G. Garn, D. Gombert, R. Jones, R. T. Jubin, V. Maio, J. C. Marra, J. Matyáš, T. M. Nenoff, B. J. Riley, G. J. Sevigny, N. Soelberg, D. Strachan, P. K. Thallapally and J. H. Westsik Jr., Closed Fuel Cycle Waste Treatment Strategy, Report FCRD-MRWFD-2015-000674, Rev. 0, PNNL-24114, Pacific Northwest National Laboratory, Richland, WA, 2015.
- B. J. Riley, C. L. Beck, J. S. Evarts, S. Chong, A. M. Lines, H. M. Felmy, J. McFarlane, H. B. Andrews, S. A. Bryan, K. C. McHugh, H. S. Cunningham, R. M. Asmussen, J. A. Dhas, Z. Zhu, J. V. Crum, S. D. Shen, J. S. McCloy and Z. M. Heiden, AIP Adv., 2024, 14, 080701 CrossRef CAS.
- R. M. Asmussen, J. Turner, S. Chong and B. J. Riley, Front. Chem., 2022, 10 Search PubMed.
- P. Taylor, A review of methods for immobilizing iodine-129 arising from a nuclear fuel recycle plant, with emphasis on waste-form chemistry, Report AECL-10163, Atomic Energy of Canada Ltd. Whiteshell Nuclear Research Establishment, Pinawa, MB (Canada), 1990.
- L. Campayo, S. Le Gallet, D. Perret, E. Courtois, C. Cau Dit Coumes, Y. Grin and F. Bernard, J. Nucl. Mater., 2015, 457, 63–71 CrossRef CAS.
- S. Le Gallet, L. Campayo, E. Courtois, S. Hoffmann, Y. Grin, F. Bernard and F. Bart, J. Nucl. Mater., 2010, 400, 251–256 CrossRef CAS.
- L. L. Burger, R. D. Scheele and K. D. Wiemers, Selection of a form for fixation of iodine-129, Report PNL-4045, Pacific Northwest National Laboratory, Richland, WA, 1981.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, CRC Press, New York, 1985 Search PubMed.
- W. H. Freeman, Appendix H - Standard Reduction Potentials, 2010, AP20–AP27, https://www.csun.edu/~hcchm003/321/Ered.pdf.
- R. D. Scheele, L. L. Burger and K. D. Wiemers, Geochemical Behavior of Disposed Radioactive Waste, American Chemical Society, 1984, vol. 246, ch. 24, pp. 373–387 Search PubMed.
- A. Seidell, Solubilities of Inorganic and Organic Compounds: A Compilation of Quantitative Solubility Data from the Periodical Literature, D. Van Nostrand Company, Inc, Second edn, 1919 Search PubMed.
- Silver price, https://ycharts.com/, (accessed 2/14/2024).
- M. V. Moroz and M. V. Prokhorenko, Inorg. Mater., 2016, 52, 765–769 CrossRef CAS.
- Argus, Bismuth prices, https://www.argusmedia.com/metals-platform/metal/minor-and-specialty-metals-bismuth, (accessed 2/14/2024).
- Chemister, Copper(I) iodide, https://chemister.ru/Database/properties-en.php?dbid=1&id=701, (accessed 2/14/2024).
- Copper price, https://markets.businessinsider.com/commodities/copper-price, (accessed 2/14/2024).
- S. Baily, What to Know About Mercury (II) Iodide, https://www.azom.com/article.aspx?ArticleID=8447, (accessed 2/14/2024).
- Mercury Price, https://www.indexbox.io/search/mercury-price-the-uk/).
- Lead price, https://ycharts.com/, (accessed 2/14/2024).
- P. Patnaik, Handbook of Inorganic Chemicals, McGraw-Hill, New York, 2003 Search PubMed.
- B. Baeyens and I. G. McKinley, A PHREEQE Database for Pd, Ni and Se, Report 88-28, Nationale Genossenschaft für die Lagerung radioaktiver Abfalie, Baden, Switzerland, 1989.
- Palladium prices, https://www.metalsdaily.com/live-prices/palladium/, (accessed 2/14/2024).
- D. M. Babanly, L. F. Mashadieva and M. B. Babanly, Inorg. Mater., 2017, 53, 519–524 CrossRef CAS.
- Chemical elements by market price, http://www.leonland.de/elements_by_price/en/list, (accessed 2/14/2024).
- Alkali Metals; Sodium Price, https://www.indexbox.io/search/alkali-metals-sodium-price-the-uk/, (accessed 2/14/2024).
- ASTM, ASTM C1308-21, Standard Test Method for Accelerated Leach Test for Measuring Contaminant Releases From Solidified Waste, Report ASTM C1308-21, American Society for Testing and Materials International, ASTM International Book of Standards Volume 12.01, West Conshohocken, PA, 2021.
- Y. Inagaki, T. Imamura, K. Idemitsu, T. Arima, O. Kato, T. Nishimura and H. Asano, J. Nucl. Sci. Technol., 2008, 45, 859–866 CrossRef CAS.
- J. Marcial, B. J. Riley, A. A. Kruger, C. E. Lonergan and J. D. Vienna, J. Hazard. Mater., 2024, 461, 132437 CrossRef CAS PubMed.
- T. J. Garino, T. M. Nenoff, J. L. Krumhansl and D. X. Rademacher, J. Am. Ceram. Soc., 2011, 94, 2412–2419 CrossRef CAS.
- A. Mukunoki, T. Chiba, Y. Suzuki, S. Uehara, H. Asano and T. Nishimura, 11th Int. Conf. Rad. Waste Manag. Environ. Remed., 459–464, 2007.
- A. Mukunoki, T. Chiba, Y. Suzuki, K. Yamaguchi, T. Sakuragi and T. Nanba, 12th International Conference on Environmental Remediation and Radioactive Waste Management, ICEM2009, 329–334, 2009.
- D. I. Kaplan, K. A. Price, C. Xu, D. Li, P. Lin, W. Xing, R. Nichols, K. Schwehr, J. C. Seaman, T. Ohnuki, N. Chen and P. H. Santschi, Environ. Int., 2019, 126, 576–584 CrossRef CAS PubMed.
- B. Guo, Y. Xiong, W. Chen, S. A. Saslow, N. Kozai, T. Ohnuki, I. Dabo and K. Sasaki, J. Hazard. Mater., 2020, 389, 121880 CrossRef CAS PubMed.
- N. M. Avalos, T. Varga, S. T. Mergelsberg, J. A. Silverstein and S. A. Saslow, Appl. Geochem., 2021, 125, 104863 CrossRef CAS.
- N. D. M. Evans, Cem. Concr. Res., 2008, 38, 543–553 CrossRef CAS.
- N. C. Hyatt, J. A. Hriljac, A. Choudhry, L. Malpass, G. P. Sheppard and E. R. Maddrell, MRS Online Proc. Lib., 2003, 807, 212–217 Search PubMed.
- S. H. Bruffey, J. A. Jordan, R. T. Jubin, M. L. Parks and T. R. Watkins, Hot Isostatic Pressing of Engineered Forms of I-AgZ, Report NTRD-MRWFD-2017-000412, ORNL/SR-2017/707, Oak Ridge National Laboratory, Oak Ridge, TN, 2017.
- S. H. Bruffey and R. T. Jubin, Preparation of Four Large Format Hot Isostatically Pressed I-AgZ Waste Form Samples for Performance Testing, Report NTRD-MRWFD-2018-000198; ORNL/SPR-2018/1026, Oak Ridge National Laboratory, Oak Ridge, TN, 2018.
- R. T. Jubin, S. H. Bruffey and K. K. Patton, Expanded Analysis of Hot Isostatic Pressed Iodine-Loaded Silver-Exchanged Mordenite, Report FCRD-SWF-2014-000278; ORNL/LTR-2014/476, Oak Ridge National Laboratory Oak Ridge, TN, 2014.
- H. An, S. Kweon, S. Park, J. Lee, H.-K. Min and M. B. Park, Nanomaterials, 2020, 10, 2157 CrossRef CAS PubMed.
- S. Chong, B. J. Riley, R. M. Asmussen, A. R. Lawter, S. H. Bruffey, J. Nam, J. S. McCloy and J. V. Crum, J. Nucl. Mater., 2020, 538, 152222 CrossRef CAS.
- D. L. Bollinger, J. Erickson, J. M. Bussey and J. S. McCloy, MRS Adv., 2022, 7, 110–116 CrossRef CAS.
- A. J. Lere-Adams, M. C. Dixon Wilkins, D. Bollinger, S. Stariha, R. Farzana, P. Dayal, D. J. Gregg, S. Chong, B. J. Riley, Z. M. Heiden and J. S. McCloy, J. Nucl. Mater., 2024, 591, 154938 CrossRef CAS.
- J. S. McCloy, N. Stone-Weiss and D. L. Bollinger, MRS Adv., 2023, 8, 261–266 CrossRef CAS.
- T. Yao, F. Lu, H. Sun, J. Wang, R. C. Ewing and J. Lian, J. Am. Ceram. Soc., 2014, 97, 2409–2412 CrossRef CAS.
- Z. Zhang, A. Heath, K. T. Valsaraj, W. L. Ebert, T. Yao, J. Lian and J. Wang, RSC Adv., 2018, 8, 3951–3957 RSC.
- Z. Zhang, W. L. Ebert, T. Yao, J. Lian, K. T. Valsaraj and J. Wang, ACS Earth Space Chem., 2019, 3, 452–462 CrossRef CAS.
- H. Zhang, Y. Xu, Q. Sun, J. Dong, Y. Lu, B. Zhang and W. Jie, Cryst. Eng. Commun., 2018, 20, 4935–4941 RSC.
- M. Hassan and H. J. Ryu, J. Nucl. Mater., 2019, 514, 84–89 CrossRef CAS.
- E. V. Johnstone, D. J. Bailey, S. Lawson, M. C. Stennett, C. L. Corkhill, M. Kim, J. Heo, D. Matsumura and N. C. Hyatt, RSC Adv., 2020, 10, 25116–25124 RSC.
- E. V. Johnstone, D. J. Bailey, M. C. Stennett, J. Heo and N. C. Hyatt, RSC Adv., 2017, 7, 49004–49009 RSC.
- C. Guy, F. Audubert, J.-E. Lartigue, C. Latrille, T. Advocat and C. Fillet, C. R. Phys., 2002, 3, 827–837 CrossRef CAS.
- E. R. Maddrell and P. K. Abraitis, MRS Proc., 2003, 807, 565–570 CrossRef.
- F. Audubert and J. Lartigue, Atalante, P4.13-11-14, 2000.
- S. E. O’Sullivan, E. Montoya, S.-K. Sun, J. George, C. Kirk, M. C. Dixon Wilkins, P. F. Weck, E. Kim, K. S. Knight and N. C. Hyatt, Inorg. Chem., 2020, 59, 18407–18419 CrossRef PubMed.
- X.-L. Wang, Y.-Z. Huang and L.-M. Wu, Inorg. Chem., 2013, 52, 9–11 CrossRef CAS PubMed.
- F. Kubel and O. Crottaz, Z. Kristallogr., 1996, 211, 926 CAS.
- E. R. Vance and J. S. Hartman, Mater. Res. Soc., 1999, 556, 41 CrossRef CAS.
- Y. Watanabe, T. Ikoma, H. Yamada, Y. Suetsugu, Y. Komatsu, G. W. Stevens, Y. Moriyoshi and J. Tanaka, ACS Appl. Mater. Interfaces, 2009, 1, 1579–1584 CrossRef CAS PubMed.
- J. S. Evarts, S. Chong, J. M. Oshiro, B. J. Riley, R. M. Asmussen and J. S. McCloy, Ind. Eng. Chem. Res., 2024, 63, 6003–6023 CrossRef CAS.
- M. C. Stennett, I. J. Pinnock and N. C. Hyatt, J. Nucl. Mater., 2011, 414, 352–359 CrossRef CAS.
- Y. Suetsugu, J. Nucl. Mater., 2014, 454, 223–229 CrossRef CAS.
- F. Lu, T. Yao, J. Xu, J. Wang, S. Scott, Z. Dong, R. C. Ewing and J. Lian, RSC Adv., 2014, 4, 38718–38725 RSC.
- S. M. Scott, W. Zhu and J. Lian, Trans. Am. Nucl. Soc., 2017, 292–293 Search PubMed.
- B. J. Riley, M. J. Schweiger, D.-S. Kim, W. W. Lukens, B. D. Williams, C. Iovin, C. P. Rodriguez, N. R. Overman, M. E. Bowden, D. R. Dixon, J. V. Crum, J. S. McCloy and A. A. Kruger, J. Nucl. Mater., 2014, 452, 178–188 CrossRef CAS.
- P. R. Hrma, Retention of Halogens in Waste Glass, Report PNNL-19361, Pacific Northwest National Laboratory, Richland, WA, 2010.
- H. Fujihara, T. Murase, T. Nisli, K. Noshita, T. Yoshida and M. Matsuda, Mater. Res. Soc., 1999, 556, 375 CrossRef CAS.
- A. Mukunoki, T. Chiba, Y. Benino and T. Sakuragi, Prog. Nucl. Energy, 2016, 91, 339–344 CrossRef CAS.
- C. W. Lee, J.-Y. Pyo, H.-S. Park, J. H. Yang and J. Heo, J. Nucl. Mater., 2017, 492, 239–243 CrossRef CAS.
- C. W. Lee, J. Kang, Y. K. Kwon, W. Um and J. Heo, J. Nucl. Fuel Cycle Waste Technol., 2021, 19, 323–330 CrossRef.
- S.-S. Shin, G.-Y. Kim, B. Lee, J.-H. Yang, Y.-S. Son, J.-H. Choi, J.-Y. Pyo, K. R. Lee, H.-S. Park and H. W. Kang, J. Non-Cryst. Solids, 2024, 624, 122728 CrossRef CAS.
- E. A. Cordova, V. Garayburu-Caruso, C. I. Pearce, K. J. Cantrell, J. W. Morad, E. C. Gillispie, B. J. Riley, F. C. Colon, T. G. Levitskaia, S. A. Saslow, O. Qafoku, C. T. Resch, M. J. Rigali, J. E. Szecsody, S. M. Heald, M. Balasubramanian, P. Meyers and V. L. Freedman, ACS Appl. Mater. Interfaces, 2020, 12, 26113–26126 CrossRef CAS PubMed.
- V. K. Gattu, S. Stariha, E. Lee, J. A. Fortner and W. L. Ebert, Degradation Tests with Iodide Waste Forms: FY20 Status Report, Report ANL/CFCT-20/22, Argonne National Laboratory, Lemont, IL, 2020.
- S. A. Stariha, V. K. Gattu and W. L. Ebert, Durability Tests with Iodide Waste Form Materials, Report ANL/CFCT-22/50, Argonne National Laboratory, Lemont, IL, 2022.
- A. Lawter, J. Bonnett, N. Avalos, N. Canfield and R. M. Asmussen, Method Sensitivity Analysis for Iodine Waste Form Durability, Report PNNL-29098, Pacific Northwest National Laboratory, Richland, WA, 2019.
- A. R. Lawter, N. M. Avalos, J. F. Bonnett, N. Canfield, M. E. Bowden, L. M. Seymour, S. A. Saslow, A. L. Bourchy and R. M. Asmussen, Iodine Waste Form Corrosion: PNNL Summary Testing Report, Report PNNL-30127, Pacific Northwest National Laboratory, Richland, WA, 2020.
- M. Barnes, D. Roy and L. Wakeley, in Advances in Ceramics, ed. G. Wicks and W. Ross, American Ceramic Society, Columbus, OH, 1983, vol. 8, pp. 406–412 Search PubMed.
- A. Fujii Yamagata, S. A. Saslow, J. J. Neeway, T. Varga, L. R. Reno, Z. Zhu, K. A. Rod, B. R. Johnson, J. A. Silverstein, J. H. Westsik, G. L. Smith and R. M. Asmussen, J. Environ. Radioact., 2022, 244–245, 106824 CrossRef CAS PubMed.
- Z. Zhang, L. Gustin, W. Xie, J. Lian, K. T. Valsaraj and J. Wang, J. Nucl. Mater., 2019, 525, 161–170 CrossRef CAS.
Footnotes |
| † This manuscript has been authored in part by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d4ma00266k |
| This journal is © The Royal Society of Chemistry 2024 |